

Abstract
Peyronie’s disease (PD) is a superficial fibrosing disorder that causes penile deformity and can interfere with sexual intercourse and reproduction, as well as diminish quality of life. While the exact mechanism of PD is still being investigated, there is likely a genetic component to the predisposition to penile plaque formation. Ultimately, however, perturbations in normal wound healing and aberrant deposition of extracellular matrix components lead to fibrotic tissue deposition. Fibrosis in PD is regulated by a complex pathway of inflammatory and fibrotic mediators. Current clinical care standards for PD address symptoms, as there are currently no treatments for PD that address its cause or progression of the disorder. In this review, we provide an overview of the known inflammatory and fibrotic mediators of PD and how these are related to the pathophysiology of other human superficial fibrosing disorders, including Dupuytren’s disease and Ledderhose disease.
Keywords: Peyronie’s Disease, Penile induration, Dupuytren’s Contracture, Ledderhose disease, fibrosis, inflammation, Transforming Growth Factor beta, cytokines
Introduction
Peyronie’s disease (PD) is a superficial fibrosing disease of the penis that can result in penile deformity. This condition may limit the ability to have sexual intercourse and reproduce, ultimately impacting a man’s quality of life. PD was initially thought to be rare, but given the sensitive nature of the condition, there is a potential for underreporting. More recent reports estimate a prevalence of 1–13%[1,2]. PD most commonly affects men in their fifth decade of life and beyond, but has been reported in younger men as well [1,3,4].
Perturbations in normal wound healing lead to fibrosis associated with PD. While an inflammatory or traumatic trigger is often thought to initiate PD, patients infrequently remember such insults. While normal wound healing after an inciting event leads to reorganization of the extracellular matrix (ECM) in tissue repair, an imbalance in fibrosis and fibrinolysis can result in fibrotic plaque formation [5]. The mediators that regulate collagen deposition and degradation are key players in the development of penile plaques. While the pathogenesis of PD to date is thought to be mediated via signaling primarily through the Transforming Growth Factor β (TGF-β) pathway, the true etiology of PD may be multifactorial, involving a number of other pathways. A genetic component of PD likely also exists, with a variant of PD showing autosomal dominant inheritance in pedigree studies [6]. This may further impact disease onset and progression in a subset of affected patients, resulting in variable risk of disease across the population. Additionally, familial aggregation of PD among specific histocompatibility antigens (HLA) has been suggested, including HLA-B7 [7].
Current PD therapies alleviate symptoms of PD, such as penile curvature, but do not address the underlying causes of onset and progression of the disease. As such, having a more complete understanding of the molecular mediators and pathways that result in PD and other superficial fibrosing disorders is highly desirable. There is significant pathophysiologic and genetic overlap of PD with other superficial fibrosing disorders including Dupuytren’s Disease (DD), a palmar fibromatosis, and Ledderhose disease (LD), which is a plantar fibromatosis. Much of what is known about the pathophysiology of PD has been correlated with DD and these diseases are often studied together. However, corellations with other human fibrotic conditions such as systemic sclerosis have been limited. In this review, we provide an overview of the known inflammatory and fibrotic mediators of PD pathogenesis and how these relate to other fibrosing disorders.
Association of Peyronie’s Disease with Human Fibrosing Disorders
Fibrosis can affect any organ system in the body. These diseases can be systemic (systemic sclerosis or nephrogenic systemic fibrosis) or organ-specific (cardiac fibrosis, diabetic or hypertensive nephropathy, pulmonary fibrosis, liver cirrhosis) [8,9]. The inciting mechanisms for these conditions vary, but common inflammatory pathways that contribute to fibrosis have been implicated in the pathogenesis of a number of these conditions as well as in the development of PD.
There is significant mechanistic and molecular overlap of PD with superficial fibrosing conditions including DD and LD. DD is reported in approximately 1% of adults in the United States; however, the prevalence is estimated to be 7% when including self-reported symptoms [10]. DD and PD are often seen concurrently at higher rates than either disease is individually observed in the general population. In a cohort of 415 men with PD, 22% had DD [11]. Similarly, in a cohort of 85 men with DD, 26% had PD-like symptoms [12]. PD and DD also share a similar molecular etiology. A number of studies have implicated the TGF-β signaling pathway, fibroblast growth factor (FGF), platelet derived growth factor (PDGF), and a number of matrix metalloproteinases (MMPs), a family of endopeptidases that degrade collagen and are involved in ECM turnover, in the development of fibrosis in DD [13–18]. A prior study explored common mRNA expression profiles using DNA microarrays in tissue samples from patients with PD, DD, and control tissue from affected sites [19]. Qian et al. observed upregulation of MMP-2 and MMP-9, as well as thymosins (TM), which are MMP activators, specifically TMβ10 and TMβ4, in both PD and DD tissues[19]. There is also a suggested association between PD and LD, which is a rare entity (fewer than 200,000 cases described in the U.S.) that can occur concurrently in patients with polyfibromatosis [20]. There are no studies reporting rates of concurrent PD and LD. Genome studies have identified risk-conferring polymorphisms for DD and LD, similar to PD, although none of these have been causally linked to these conditions [21,22].
There have also been reports linking PD and organ confined and systemic fibrotic diseases. PD has been reported concurrently with idiopathic pulmonary fibrosis [23], Paget’s disease of the bone [24], and retroperitoneal fibrosis [25]. PD has also been associated with systemic fibrotic conditions such as scleroderma [26], polyfibromatosis [27], and systemic sclerosis [28,29]. However, in a larger study of 148 men with PD, Ventimiglia et al. did not find an association between PD with systemic sclerosis [30]. More recently, Pastuszak et al. reported the association of fibrotic conditions in nearly 9,000 men with PD, 200,000 men with ED, and 90,000 healthy controls within a large claims database cohort study. The authors identified an increased risk of keloids in patients with PD compared to controls [31]. In the same study, there was a reduced risk of liver fibrosis among PD patients compared to healthy controls, and the study did not find significant associations between PD and any of the conditions examined in prior studies.
Mediators of the Inflammatory and Fibrotic Response in Peyronie’s Disease:
Microtrauma to the penile tunica albuginea has traditionally been thought to be the most common trigger for plaque development in PD. Repetitive stress and destabilization of the tunica albuginea may lead to delamination of the fascial layer and cell damage. The resulting release of inflammatory mediators into the extracellular space in turn triggers a complex cascade leading to wound healing [32]. TGF-β is one of the key profibrotic mediators that leads to deposition of early ECM. Early deposition of ECM components causes platelet aggregation and clot formation. Concomitantly, platelet degranulation results in vasodilation and vascular permeability. Resident myofibroblasts and epithelial cells secrete metalloproteinases that increase basement cell membrane permeability, and the local production of inflammatory mediators leads to recruitment of additional inflammatory cells, including neutrophils, macrophages, and leukocytes, to the site of injury. Together, these inflammatory cells facilitate tissue repair and wound healing.
Myofibroblasts are central in the inflammatory response and subsequent development of fibrosis in PD. These mesenchymal cells produce and respond to a number of factors that mediate wound healing and fibrosis, and an imbalance can lead to aberrant collagen deposition. At the cellular level, aberrations in the levels of the growth factor and cytokines in the inflammatory response are associated with overstimulation of myofibroblast proliferation and myofibroblast latency, as well as inhibition of myofibroblast apoptosis [8,33]. Additionally, it has been suggested that appropriate levels of these mediators in response to cell injury may also contribute to fibrosis through an intrinsic hypersensitivity by myofibroblasts to these stimuli [9]. There are a number of key inflammatory mediators, cytokines, and growth factors in this process that are reviewed below (Figure 1).
Figure 1: Inflammatory and Fibrotic Response after Cell Damage in Peyronie’s Disease.
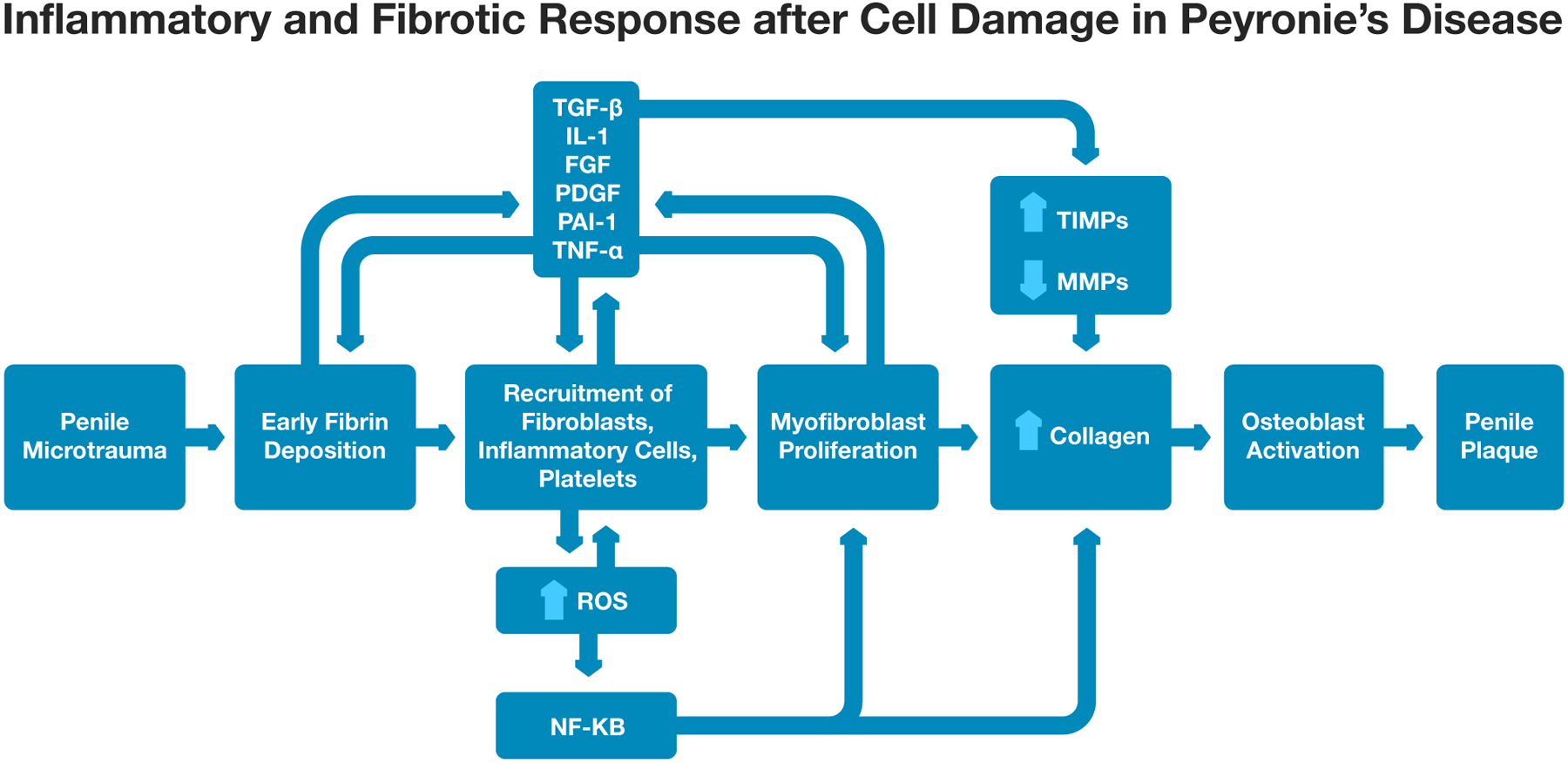
“Abbreviations: TGF-β - Transforming Growth Factor-β; IL-1 – Interleukin-1; FGF – Fibroblast Growth Factor; PDGF – Platelet Derived Growth Factor; PAI-1 - Plasminogen Activator Inhibitor 1; TNF- Tumor Necrosis Factor-α; ROS – Reactive Oxygen Species; NF-ΚB - nuclear factor kappa-light-chain-enhancer of activated B cells; MMP - matrix metalloproteinase; TIMP - Tissue Inhibitor of MMP.”
Transforming Growth Factor-β (TGF-β)
Transforming Growth Factor-β (TGF-β) is an early mediator produced in response to cell injury and stimulates myofibroblast proliferation and collagen production for early ECM deposition [34–37,5,38,39]. TGF-β also leads to chemotaxis of cells involved in inflammation and fibrosis including neutrophils, macrophages, monocytes, and lymphocytes [34,35,5]. Importantly, a rat model of PD developed via injection of cytomodulin into the penile tunica albuginea induced TGF-β1 expression [36]. Different concentrations of cytomodulin were injected into the tunicae albugineae of 18 rats, with 6 rats treated with saline as controls. Penile tissue was then obtained 3 days, 2 weeks and 6 weeks after treatment and histochemical analyses were performed. Histologic changes included increased elastin deposition, presence of a chronic inflammatory infiltrate composed of small lymphocytes and histocytes, and deposition of disorganized collagen bundles. Histologic evaluation was not performed on control rats. Immunoblot analyses demonstrated significantly higher levels of TGF-β1 mRNA levels compared to controls after 3 days and 2 weeks. This small animal study suggested a role for TGF-β in the pathogenesis of PD.
TGF-β also alters production of other key mediators in the inflammatory and wound healing response. It inhibits production of mediators involved in collagenolysis (MMP-1, MMP-8, and MMP-13) and fibrinolysis (plasminogen activator inhibitor-1, PAI-1) [40–42]. Collagen turnover and normal wound healing are regulated by various MMPs that affect cell survival, gene expression, and continued myofibroblast activation [40,41]. In PD, MMPs are over-inhibited, leading to excess collagen deposition within the ECM, which then leads to plaque formation [43,44]. TGF-β can also increase production of reactive oxygen species (ROS), and has been implicated in plaque calcification as well through osteoblast differentiation [45,46].
Interleukin-1 (IL-1)
Interleukin-1 (IL-1) is a pro-inflammatory cytokine produced by macrophages. Early proliferation of resident myofibroblasts and macrophages that are recruited to the site of injury release IL-1, which functions as a potent chemotactic mediator that further recruits additional fibroblasts and stimulates collagen production [47,44]. Similar to TGF-β, IL-1 also decreases production of MMPs 1, 2, 8, 9, 10, and 13 by fibroblasts that lead to increased collagen deposition [48,49]. IL-1 promotes inducible nitric oxide synthase (iNOS) production, which is involved in management of free radicals. Upregulation of iNOS prolongs wound healing and predisposes to fibrosis [50].
Fibroblast Growth Factor (FGF)
Fibroblast growth factor (FGF) is produced by fibroblasts and attracts other fibroblasts to the injury site, inducing their proliferation [51,49]. FGF also increases production of certain MMPs, including MMP-1 and MMP-9, and Tissue Inhibitor of MMP-1 (TIMP-1), ultimately leading to over-deposition of fibrin [51,49]. TIMP-1 balances the activity of MMPs and is involved in normal wound healing, but also plays a role in promotion of fibrosis and plaque formation [52]. The increased fibrin deposition can then induce further TGF-β expression.
Platelet-Derived Growth Factor (PDGF)
Platelet-Derived Growth Factor (PDGF) is a profibrotic cytokine produced by platelets and macrophages in response to exposure to ECM components. PDGF is a chemotactic agent that recruits fibroblasts to the site of injury and leads to fibroblast proliferation [39,44]. It can also stimulate TIMP synthesis. Additionally, PDGF has been linked to plaque calcification via its ability to recruit osteoblasts and induce osteoblast differentiation [46].
Plasminogen Activator Inhibitor 1 (PAI-1)
Plasminogen Activator Inhibitor 1 (PAI-1), like PDGF, is also a platelet product that is released in response to fibrin deposition. In patients with PD, tunica albuginea plaque samples demonstrated increased PAI-1 expression compared to controls [53]. PAI-1 inhibits fibrinolysis and is also a potent chemotactic agent, recruiting fibroblasts and inflammatory cells to the site of expression. Additionally, PAI-1 inhibits MMP-3 and MMP-9 and alters collagenolysis, promoting excess collagen deposition [44,42].
Reactive Oxygen Species (ROS)
Although mechanical trauma is thought to be the most common inciting trigger for fibrosis in PD, hypoxia and ROS are associated triggers that have been identified in other fibrotic disease pathways. Hypoxia inducible factor-1α (HIF-1α) is a key mediator following hypoxia-induced injury and has been implicated in the activation of profibrotic gene expression and secretion of growth factors such as TGF-β [54,55]. Excessive production of ROS has been implicated in the pathogenesis of idiopathic pulmonary fibrosis and liver fibrosis [56,57,45].
There is growing evidence to suggest that oxidative stress also has an important role in the development of erectile dysfunction and subsequently PD [5]. Overproduction of ROS reduces the concentration of nitric oxide that is available for cavernosal smooth muscle relaxation and can lead to long-term endothelial dysfunction. Repetitive stress and penile destabilization, which is more likely in men with ED, can lead to the development of PD.
In addition to the growth factor and cytokine production that occurs after an initial cellular insult, an “oxidative burst” also occurs concurrently in the early stages of tissue remodeling [58]. This “oxidative burst” is a rapid release of ROS, including superoxide radicals, by macrophages and neutrophils. Several nicotinamide adenine dinucleotide phosphate (NADPH) oxidase isoforms are upregulated in neutrophils and macrophages within 24–48 hours after the initial cellular insult and are largely responsible for initiation of the oxidative burst [59,58]. The excess production of ROS activates Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB) which then regulates production of TGF-β, fibrin, collagen, and other profibrotic mediators [5].
Tumor Necrosis Factor-α (TNF-α)
Tumor Necrosis Factor-α (TNF-α) is produced by macrophages in response to tissue injury, and directly stimulates fibroblast proliferation and iNOS production, leading to prolonged wound healing and fibrosis [5,44,50]. Excess ROS and TNF-α production leads to nitro-oxidative stress tissue damage and a cycle of abnormal collagen deposition.
Intracellular Pathways in Fibrosis
Activation of myofibroblasts in response to an inciting event is thought to be primarily induced by transforming growth factor-β (TGF-β) [34,60]. TGF-β is produced as an inactive peptide that is secreted into the extracellular space. TGF-β homodimers complex with arginine-glycine-aspartate-containing N-terminal binding peptide remain latent until several proteolytic activating events occur, activating TGF-β [60]. The active form of TGF-β binds the TGF-β receptor II, which is a serine/threonine transmembrane receptor kinase located on myofibroblasts, and initiates an intracellular signaling cascade which involves the Smad proteins (Smad2, Smad3, Smad4), and represents the canonical TGF-β/Smad fibrosis pathway [61]. Alternative TGF-β pathways mediated by non-Smad proteins including the PI3K/PAK-2/c-Abl pathway, the Rho/ROCK pathway, and the JNK/C-JUN pathway are alternate pathways resulting in fibrosis through TGF-β signaling (Figure 2).
Figure 2: Signaling Pathways in Fibrosis. TGF-β and BMP-mediated signaling pathways within fibroblasts.
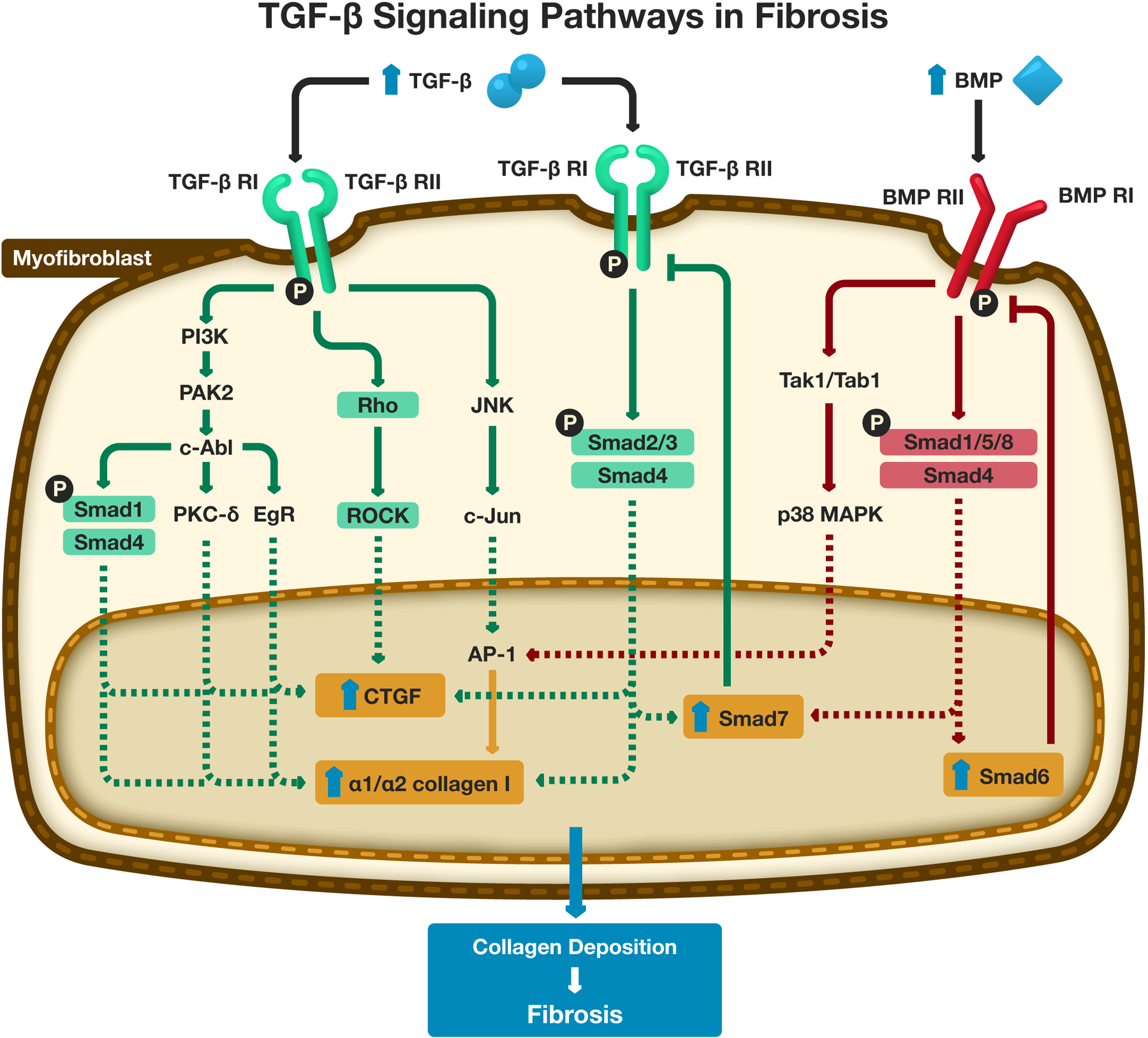
“Abbreviations: TGF-β - Transforming Growth Factor-β; TGF-β RI - Transforming Growth Factor-β Receptor I; TGF-β RII - Transforming Growth Factor-β Receptor II; BMP – Bone Morphogenetic Protein; BMP RI - Bone Morphogenetic Protein Receptor I; BMP RII - Bone Morphogenetic Protein Receptor II; PAK-2 - P21-activated kinase 2; c-Abl- c-Abelson kinase; PKC-δ - protein kinase C delta; EgR – Early growth response protein; ROCK – Rho associated protein kinase; CTGF – Connective Tissue Growth Factor; JNK - c-Jun N terminal kinase; AP-1 – Activator Protein −1; Tak1 - TGF-β-activated kinase 1; Tab1 - TGF-β activated kinase 1 binding protein 1; MAPK – mitogen activated protein kinase.
In the TGF-β/Smad-mediated pathways, the phosphorylated ligand-bound TGF-β receptor II phosphorylates TGF-β receptor I [60]. TGF-β receptor I consists of a family of activin-like kinases (Alk). The most important Alk associated with TGF-β receptor I is Alk-5. Activation of Alk-5 associated with TGF-β receptor I leads to phosphorylation of Smad2/Smad3 that complexes with Smad4. This complex moves across the nuclear membrane and acts as a transcription factor, activating the promotor for COL1A1 and COL1A2 which produces α1 and α2 chains of collagen I respectively. Additional targets include activation of the connective tissue growth factor (CTGF) promoter. Unfortunately, the complete set of targets is incompletely characterized.
The non-Smad signaling pathways are also important effectors of TGF-β, and are thought to be activated concurrently with the Smad signaling pathways [61]. However, it is uncertain how these pathways are selectively activated. The phosphorylated ligand-bound TGF-β receptor II complexes with Alk-1 and phosphorylates PI3K, which triggers activation of P21-activated kinase 2 (PAK2) followed by c-Abelson kinase (c-Abl). Activation of c-Abl is important given its activation of several downstream effectors including early growth response protein (EgR-1), protein kinase C delta (PKC-δ), and phosphorylation of Smad1. The phosphorylated Smad1 complexes with Smad4 and translocates to the nucleus. Together, downstream effects include increased transcription of CTGF, COL1A1 and COL1A2. Concurrently, ligand-bound TGF-β receptor II/Alk-5 complex activates c-Jun N terminal kinase (JNK), through another non-Smad mediated fibrotic pathway [61]. JNK phosphorylates c-Jun, which is an important transcription factor that is part of the Jun family of basic leucine zipper (b-ZIP) transcription factors. c-Jun activates other activator protein-1 (AP-1) transcription factors (including members of the Jun and Fos family) and promotes transcription of COL1A1 and COL1A2. Additional targets of AP-1 in the profibrotic response are incompletely characterized, although it is a known mediator for the recruitment of macrophage and regulatory T cells. Another concurrent non-Smad pathway is the Rho/ROCK pathway. The ligand-bound TGF-β receptor II/Alk-1 complex phosphorylates Rho protein. This in turn leads to activation of Rho associated protein kinase (ROCK). ROCK promotes transcription of CTGF which contributes to fibrosis [61].
Bone Morphogenetic Proteins (BMPs) are members of the TGF-β family but act as a counterbalance to the profibrotic effects of most TGF-β family proteins (Figure 2). BMP-6, BMP-7, and BMP-9 are key antifibrotic effectors [60,62,63]. Similar to TGF-β, BMP signaling involves canonical Smad-dependent and non-canonical Smad-independent pathways (i.e. the p38 mitogen-activate protein kinase [MAPK] pathway) [64]. In the canonical Smad-dependent pathway, BMPs bind to BMP receptor II and phosphorylate BMP Receptor I. BMP Receptor I can include several activin-like kinases including Alk-1, 2, 3, and 6. This then leads to phosphorylation of Smad1/Smad5/Smad8 that then complexes with Smad4, with the resulting complex translocating to the nucleus and stimulating expression of Smad6 and Smad7. Smad6 provides negative feedback to BMP Receptor I to regulate phosphorylation of Smad1/Smad5/Smad8. Smad7 promotes degradation of the TGF-β Receptor I, which downregulates the TGF-β/Smad pathway and the JNK/c-Jun pathway. The non-canonical Smad-independent pathway is thought to promote collagen synthesis by myofibroblasts but has a role in stimulating osteoblast differentiation and calcification as well [65]. This pathway is mediated by activation of Tak1/Tab1 by a ligand bound BMP receptor II/BMP receptor I complex. Tak1/Tab1 activates the p38 MAPK pathway including transcriptional upregulation of certain genes including AP-1 in myofibroblasts and the Runt-related transcription factor 2 (Runx2) gene in osteoblasts, which controls osteoprogenitor cell differentiation. There is little known regarding the antifibrotic mechanisms of BMPs and their role in balancing the profibrotic mechanisms of TGF-β.
Other Molecular Pathways involved in Human Fibrotic Diseases
Although the TGF-β pathway has the most important role in fibrosis, there are other molecular pathways that have been identified in fibrosis for other organ systems [8,9,33]. The intracellular molecular pathways in PD have historically focused on the TGF-β pathway. However, a more complete understanding of the involvement of other pathways that can stimulate fibrosis may provide novel targets for therapeutics in the setting of PD. Systemic sclerosis is an acquired fibrotic disease characterized by uncontrolled deposition of ECM causing thickened dermis. The relative ease of obtaining human dermal tissue samples and human derived dermal fibroblast cell lines has made systemic sclerosis a model disease for the study of human fibrotic diseases. Several additional fibrotic signaling pathways contributing to fibrosis have been identified in systemic sclerosis, including the Notch, Sonic Hedgehog, and Wnt/β-catenin pathways. However, these pathways have not been extensively studied with regards to pathogenesis in PD. A genome wide association study identified an association between the single nucleotide polymorphism (SNP) rs4730775 in the Wnt2 gene and PD susceptibility, suggesting a role for the Wnt signaling pathway in PD [66]. Associations between the Notch and Sonic Hedgehog signaling pathways and PD have yet to be explored. Although there is some evidence suggesting roles for these three pathways in the pathogenesis of fibrosis for systemic sclerosis, our understanding of these signaling pathways and the crosstalk between pathways is limited.
The Notch signaling pathway was initially identified in Drosophila [67]. It involves binding of the Jagged-1 and Delta-like ligands to Notch membrane receptors that results in the release of the active Notch intracellular domain (NICD) [67]. This domain translocates to the cell nucleus where it enhances transcription of Hairy/Enhancer of Split (Hes-1). Activation of the Notch signaling pathway ultimately increases collagen production and promotes fibrosis although the nuclear targets of NICD have been incompletely identified. In an in vitro model of cultured dermal systemic sclerosis fibroblasts, stimulation with recombinant Jag-1 led to increased collagen synthesis and differentiation of resting fibroblasts to myofibroblasts [68].
The Sonic hedgehog (Shh) family of proteins has also been implicated in fibrosis. Shh binds Patched (Ptc), a transmembrane protein that stabilizes the Gli family of zinc finger transcription factors that enhance transcription of profibrotic genes [69]. Without the presence of Shh, Ptc is bound to Smoothened (Smo), a G protein-coupled receptor, in a latent state. In another systemic sclerosis model of cultured fibroblasts, overstimulation by Shh led to increased levels of Gli-2 and increased collagen synthesis [70]. Additionally, overexpression of both TGF-β and Shh in these models led to more pronounced myofibroblast differentiation and increased collagen production compared to TGF-β stimulation alone, suggesting a synergistic effect of both signaling pathways [70].
The Wnt/β-catenin pathways have also been implicated in fibrosis [71]. In the absence of Wnt, β-catenin forms an intricate complex which is tagged for ubiquitin-mediated degradation. In the presence of Wnt protein, the Frizzled (FZD) receptor and low density lipoprotein receptor related protein 1 (LRP1) co-receptor, bind Wnt and disrupt the degradation of β-catenin and promote β-catenin stabilization. β-catenin then moves to the nucleus where it binds T-cell factor/Lymphoid enhancer factor (TCF/LEF) to promote transcription of several genes including c-myc, c-jun, and MMP-7. Other downstream effectors regulate cell proliferation and cell death. In a mouse model of systemic sclerosis, Beyer et al. observed increased levels of β-catenin, Wnt-1 and Wnt-10b in dermal tissue samples obtained from patients with systemic sclerosis compared to healthy controls [72]. Mice fibroblast cultures with stabilization of β-catenin, developed dermal thickening and accumulation of collagen within 2 weeks.
Conclusions
Current knowledge of the pathogenesis of PD and related fibrosing disorders is limited. Although TGF-β is thought to be the primary mediator of fibrosis, there are multiple pathways, cytokines, and growth factors that are involved in fibrosis, and there are likely other pathways and mediators involved in human fibrotic conditions that remain to be identified. Identification of pathways predisposing to or leading to fibrosis in other organ-confined and systemic fibrotic conditions may lead to a better overall understanding of the common molecular alterations that lead to fibrosis in PD and additional insight into other benign urologic conditions such as erectile dysfunction. The development of novel preventative or disease modifying therapeutics will require appreciation of the breadth of signaling pathways leading to fibrosis in PD. Future treatments may be most successful by interfering at multiple points in the molecular mechanisms of fibrosis due to the redundancy within these pathways.
Acknowledgements:
A.W.P. is a National Institutes of Health K08 Scholar supported by a Mentored Career Development Award (K08DK115835-01) from the National Institute of Diabetes and Digestive and Kidney Diseases. This work is also supported in part through a Urology Care Foundation Rising Stars in Urology Award (to A.W.P.)
Footnotes
Conflicts of Interest:
DP Patel: None
MB Christensen: None
JM Hotaling: Dr. Hotaling receives research and fellowship support from Boston Scientific and Endo Pharmaceuticals. He holds leadership roles/equity interest in Nanonc, StreamDx, and Andro360 which are all early stage startup companies.
AW Pastuszak: Dr. Pastuszak is an advisor to Endo Pharmaceuticals, Boston Scientific, and Antares Pharmaceuticals, and a speaker for Endo Pharmaceuticals and Bayer AG. He is a consultant and receives research and fellowship support from Endo Pharmaceuticals. He holds a leadership position with Woven Health, of which he is also a founder.
As this is a review article, no research involving human participants and/or animals was carried out and no informed consent was needed.
References:
- 1.Dibenedetti DB, Nguyen D, Zografos L, Ziemiecki R, Zhou X (2011) A Population-Based Study of Peyronie’s Disease: Prevalence and Treatment Patterns in the United States. Advances in urology 2011:282503. doi: 10.1155/2011/282503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stuntz M, Perlaky A, des Vignes F, Kyriakides T, Glass D (2016) The Prevalence of Peyronie’s Disease in the United States: A Population-Based Study. PloS one 11 (2):e0150157. doi: 10.1371/journal.pone.0150157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarzer U, Sommer F, Klotz T, Braun M, Reifenrath B, Engelmann U (2001) The prevalence of Peyronie’s disease: results of a large survey. BJU international 88 (7):727–730 [DOI] [PubMed] [Google Scholar]
- 4.Lindsay MB, Schain DM, Grambsch P, Benson RC, Beard CM, Kurland LT (1991) The incidence of Peyronie’s disease in Rochester, Minnesota, 1950 through 1984. The Journal of urology 146 (4):1007–1009 [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez-Cadavid NF, Rajfer J (2005) Mechanisms of Disease: new insights into the cellular and molecular pathology of Peyronie’s disease. Nature clinical practice Urology 2 (6):291–297. doi: 10.1038/ncpuro0201 [DOI] [PubMed] [Google Scholar]
- 6.Nyberg LM Jr., Bias WB, Hochberg MC, Walsh PC (1982) Identification of an inherited form of Peyronie’s disease with autosomal dominant inheritance and association with Dupuytren’s contracture and histocompatibility B7 cross-reacting antigens. The Journal of urology 128 (1):48–51 [DOI] [PubMed] [Google Scholar]
- 7.Willscher MK, Cwazka WF, Novicki DE (1979) The association of histocompatibility antigens of the B7 cross-reacting group with Peyronie’s disease. The Journal of urology 122 (1):34–35 [DOI] [PubMed] [Google Scholar]
- 8.Rockey DC, Bell PD, Hill JA (2015) Fibrosis--A Common Pathway to Organ Injury and Failure. The New England journal of medicine 373 (1):96. doi: 10.1056/NEJMc1504848 [DOI] [PubMed] [Google Scholar]
- 9.Rosenbloom J, Castro SV, Jimenez SA (2010) Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Annals of internal medicine 152 (3):159–166. doi: 10.7326/0003-4819-152-3-201002020-00007 [DOI] [PubMed] [Google Scholar]
- 10.Dibenedetti DB, Nguyen D, Zografos L, Ziemiecki R, Zhou X (2011) Prevalence, incidence, and treatments of Dupuytren’s disease in the United States: results from a population-based study. Hand (New York, NY) 6 (2):149–158. doi: 10.1007/s11552-010-9306-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nugteren HM, Nijman JM, de Jong IJ, van Driel MF (2011) The association between Peyronie’s and Dupuytren’s disease. International journal of impotence research 23 (4):142–145. doi: 10.1038/ijir.2011.18 [DOI] [PubMed] [Google Scholar]
- 12.Shindel AW, Sweet G, Thieu W, Durbin-Johnson B, Rothschild J, Szabo R (2017) Prevalence of Peyronie’s Disease-Like Symptoms in Men Presenting With Dupuytren Contractures. Sexual medicine 5 (3):e135–e141. doi: 10.1016/j.esxm.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alioto RJ, Rosier RN, Burton RI, Puzas JE (1994) Comparative effects of growth factors on fibroblasts of Dupuytren’s tissue and normal palmar fascia. The Journal of hand surgery 19 (3):442–452 [DOI] [PubMed] [Google Scholar]
- 14.Badalamente MA, Hurst LC, Grandia SK, Sampson SP (1992) Platelet-derived growth factor in Dupuytren’s disease. The Journal of hand surgery 17 (2):317–323 [DOI] [PubMed] [Google Scholar]
- 15.Badalamente MA, Sampson SP, Hurst LC, Dowd A, Miyasaka K (1996) The role of transforming growth factor beta in Dupuytren’s disease. The Journal of hand surgery 21 (2):210–215. doi: 10.1016/s0363-5023(96)80102-x [DOI] [PubMed] [Google Scholar]
- 16.Bayat A, Alansar A, Hajeer AH, Shah M, Watson JS, Stanley JK, Ferguson MW, Ollier WE (2002) Genetic susceptibility in Dupuytren’s disease: lack of association of a novel transforming growth factor beta(2) polymorphism in Dupuytren’s disease. Journal of hand surgery (Edinburgh, Scotland) 27 (1):47–49. doi: 10.1054/jhsb.2001.0689 [DOI] [PubMed] [Google Scholar]
- 17.Berndt A, Kosmehl H, Mandel U, Gabler U, Luo X, Celeda D, Zardi L, Katenkamp D (1995) TGF beta and bFGF synthesis and localization in Dupuytren’s disease (nodular palmar fibromatosis) relative to cellular activity, myofibroblast phenotype and oncofetal variants of fibronectin. The Histochemical journal 27 (12):1014–1020 [PubMed] [Google Scholar]
- 18.Gonzalez AM, Buscaglia M, Fox R, Isacchi A, Sarmientos P, Farris J, Ong M, Martineau D, Lappi DA, Baird A (1992) Basic fibroblast growth factor in Dupuytren’s contracture. The American journal of pathology 141 (3):661–671 [PMC free article] [PubMed] [Google Scholar]
- 19.Qian A, Meals RA, Rajfer J, Gonzalez-Cadavid NF (2004) Comparison of gene expression profiles between Peyronie’s disease and Dupuytren’s contracture. Urology 64 (2):399–404. doi: 10.1016/j.urology.2004.04.006 [DOI] [PubMed] [Google Scholar]
- 20.Bilgutay AN, Pastuszak AW (2015) PEYRONIE’S DISEASE: A REVIEW OF ETIOLOGY, DIAGNOSIS, AND MANAGEMENT. Current sexual health reports 7 (2):117–131. doi: 10.1007/s11930-015-0045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SK, Ioannidis JPA, Ahmed MA, Avins AL, Kleimeyer JP, Fredericson M, Dragoo JL (2018) Two Genetic Variants Associated with Plantar Fascial Disorders. International journal of sports medicine 39 (4):314–321. doi: 10.1055/s-0044-100280 [DOI] [PubMed] [Google Scholar]
- 22.Dolmans GH, de Bock GH, Werker PM (2012) Dupuytren diathesis and genetic risk. The Journal of hand surgery 37 (10):2106–2111. doi: 10.1016/j.jhsa.2012.07.017 [DOI] [PubMed] [Google Scholar]
- 23.Martinez MA, Ferrando D, Cordero PJ (1997) [Idiopathic pulmonary fibrosis and Peyronie’s disease]. Archivos de bronconeumologia 33 (10):549–550 [DOI] [PubMed] [Google Scholar]
- 24.Lyles KW, Gold DT, Newton RA, Parekh S, Shipp KM, Pieper CF, Krishan R, Carson CC 3rd (1997) Peyronie’s disease is associated with Paget’s disease of bone. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 12 (6):929–934. doi: 10.1359/jbmr.1997.12.6.929 [DOI] [PubMed] [Google Scholar]
- 25.Akbal C, Tanidir Y, Ozgen MB, Simsek F (2008) Erectile dysfunction and Peyronie’s disease in patient with retroperitoenal fibrosis. International urology and nephrology 40 (4):971–975. doi: 10.1007/s11255-008-9381-4 [DOI] [PubMed] [Google Scholar]
- 26.Chen TY, Zahran AR, Carrier S (2001) Penile curvature associated with scleroderma. Urology 58 (2):282. [DOI] [PubMed] [Google Scholar]
- 27.Chen DL, Chong AH, Green J, Orchard D, Williams R, Clemens L (2006) A novel case of polyfibromatosis and interstitial granulomatous dermatitis with arthritis. Journal of the American Academy of Dermatology 55 (2 Suppl):S32–37. doi: 10.1016/j.jaad.2006.02.038 [DOI] [PubMed] [Google Scholar]
- 28.Simeon CP, Fonollosa V, Vilardell M, Ordi J, Solans R, Lima J (1994) Impotence and Peyronie’s disease in systemic sclerosis. Clinical and experimental rheumatology 12 (4):464. [PubMed] [Google Scholar]
- 29.Ordi J, Selva A, Fonollosa V, Vilardell M, Jordana R, Tolosa C (1990) Peyronie’s disease in systemic sclerosis. Annals of the rheumatic diseases 49 (2):134–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ventimiglia E, Capogrosso P, Colicchia M, Boeri L, Serino A, La Croce G, Russo A, Capitanio U, Briganti A, Cantiello F, Mirone V, Damiano R, Montorsi F, Salonia A (2015) Peyronie’s disease and autoimmunity-a real-life clinical study and comprehensive review. The journal of sexual medicine 12 (4):1062–1069. doi: 10.1111/jsm.12825 [DOI] [PubMed] [Google Scholar]
- 31.Pastuszak AW, Rodriguez KM, Solomon ZJ, Kohn TP, Lipshultz LI, Eisenberg ML (2018) Increased Risk of Incident Disease in Men with Peyronie’s Disease: Analysis of U.S. Claims Data. The journal of sexual medicine 15 (6):894–901. doi: 10.1016/j.jsxm.2018.04.640 [DOI] [PubMed] [Google Scholar]
- 32.Lue TF (2002) Peyronie’s disease: an anatomically-based hypothesis and beyond. International journal of impotence research 14 (5):411–413. doi: 10.1038/sj.ijir.3900876 [DOI] [PubMed] [Google Scholar]
- 33.Wynn TA (2008) Cellular and molecular mechanisms of fibrosis. The Journal of pathology 214 (2):199–210. doi: 10.1002/path.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Border WA, Noble NA (1994) Transforming growth factor beta in tissue fibrosis. The New England journal of medicine 331 (19):1286–1292. doi: 10.1056/nejm199411103311907 [DOI] [PubMed] [Google Scholar]
- 35.Border WA, Ruoslahti E (1992) Transforming growth factor-beta in disease: the dark side of tissue repair. The Journal of clinical investigation 90 (1):1–7. doi: 10.1172/jci115821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El-Sakka AI, Hassoba HM, Chui RM, Bhatnagar RS, Dahiya R, Lue TF (1997) An animal model of Peyronie’s-like condition associated with an increase of transforming growth factor beta mRNA and protein expression. The Journal of urology 158 (6):2284–2290 [DOI] [PubMed] [Google Scholar]
- 37.El-Sakka AI, Hassoba HM, Pillarisetty RJ, Dahiya R, Lue TF (1997) Peyronie’s disease is associated with an increase in transforming growth factor-beta protein expression. The Journal of urology 158 (4):1391–1394 [PubMed] [Google Scholar]
- 38.Ryu JK, Kim WJ, Choi MJ, Park JM, Song KM, Kwon MH, Das ND, Kwon KD, Batbold D, Yin GN, Suh JK (2013) Inhibition of histone deacetylase 2 mitigates profibrotic TGF-beta1 responses in fibroblasts derived from Peyronie’s plaque. Asian journal of andrology 15 (5):640–645. doi: 10.1038/aja.2013.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez-Cadavid NF, Magee TR, Ferrini M, Qian A, Vernet D, Rajfer J (2002) Gene expression in Peyronie’s disease. International journal of impotence research 14 (5):361–374. doi: 10.1038/sj.ijir.3900873 [DOI] [PubMed] [Google Scholar]
- 40.Giannandrea M, Parks WC (2014) Diverse functions of matrix metalloproteinases during fibrosis. Disease models & mechanisms 7 (2):193–203. doi: 10.1242/dmm.012062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pardo A, Selman M (2006) Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proceedings of the American Thoracic Society 3 (4):383–388. doi: 10.1513/pats.200601-012TK [DOI] [PubMed] [Google Scholar]
- 42.Van de Water L (1997) Mechanisms by which fibrin and fibronectin appear in healing wounds: implications for Peyronie’s disease. The Journal of urology 157 (1):306–310 [DOI] [PubMed] [Google Scholar]
- 43.El-Sakka AI, Salabas E, Dincer M, Kadioglu A (2013) The pathophysiology of Peyronie’s disease. Arab journal of urology 11 (3):272–277. doi: 10.1016/j.aju.2013.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreland RB, Nehra A (2002) Pathophysiology of Peyronie’s disease. International journal of impotence research 14 (5):406–410. doi: 10.1038/sj.ijir.3900875 [DOI] [PubMed] [Google Scholar]
- 45.Liu RM, Desai LP (2015) Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox biology 6:565–577. doi: 10.1016/j.redox.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vernet D, Nolazco G, Cantini L, Magee TR, Qian A, Rajfer J, Gonzalez-Cadavid NF (2005) Evidence that osteogenic progenitor cells in the human tunica albuginea may originate from stem cells: implications for peyronie disease. Biology of reproduction 73 (6):1199–1210. doi: 10.1095/biolreprod.105.041038 [DOI] [PubMed] [Google Scholar]
- 47.Huleihel M, Douvdevani A, Segal S, Apte RN (1990) Regulation of interleukin 1 generation in immune-activated fibroblasts. European journal of immunology 20 (4):731–738. doi: 10.1002/eji.1830200404 [DOI] [PubMed] [Google Scholar]
- 48.Pryor JP, Ralph DJ (2002) Clinical presentations of Peyronie’s disease. International journal of impotence research 14 (5):414–417. doi: 10.1038/sj.ijir.3900877 [DOI] [PubMed] [Google Scholar]
- 49.Sasaki K, Hattori T, Fujisawa T, Takahashi K, Inoue H, Takigawa M (1998) Nitric oxide mediates interleukin-1-induced gene expression of matrix metalloproteinases and basic fibroblast growth factor in cultured rabbit articular chondrocytes. Journal of biochemistry 123 (3):431–439 [DOI] [PubMed] [Google Scholar]
- 50.Bivalacqua TJ, Champion HC, Hellstrom WJ (2002) Implications of nitric oxide synthase isoforms in the pathophysiology of Peyronie’s disease. International journal of impotence research 14 (5):345–352. doi: 10.1038/sj.ijir.3900872 [DOI] [PubMed] [Google Scholar]
- 51.Mulhall JP, Thom J, Lubrano T, Shankey TV (2001) Basic fibroblast growth factor expression in Peyronie’s disease. The Journal of urology 165 (2):419–423. doi: 10.1097/00005392-200102000-00016 [DOI] [PubMed] [Google Scholar]
- 52.Lambert E, Dasse E, Haye B, Petitfrere E (2004) TIMPs as multifacial proteins. Critical reviews in oncology/hematology 49 (3):187–198. doi: 10.1016/j.critrevonc.2003.09.008 [DOI] [PubMed] [Google Scholar]
- 53.Davila HH, Magee TR, Zuniga FI, Rajfer J, Gonzalez-Cadavid NF (2005) Peyronie’s disease associated with increase in plasminogen activator inhibitor in fibrotic plaque. Urology 65 (4):645–648. doi: 10.1016/j.urology.2005.01.010 [DOI] [PubMed] [Google Scholar]
- 54.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH (2007) Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. The Journal of clinical investigation 117 (12):3810–3820. doi: 10.1172/jci30487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C (2001) Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. The Journal of biological chemistry 276 (42):38527–38535. doi: 10.1074/jbc.M104536200 [DOI] [PubMed] [Google Scholar]
- 56.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J (2010) NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 65 (8):733–738. doi: 10.1136/thx.2009.113456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernandez-Rodriguez CM, Pinedo F, Huber H, Eferl R, Mikulits W, Fabregat I (2012) NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PloS one 7 (9):e45285. doi: 10.1371/journal.pone.0045285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poli G, Parola M (1997) Oxidative damage and fibrogenesis. Free radical biology & medicine 22 (1–2):287–305 [DOI] [PubMed] [Google Scholar]
- 59.Paulis G, Romano G, Paulis L, Barletta D (2017) Recent Pathophysiological Aspects of Peyronie’s Disease: Role of Free Radicals, Rationale, and Therapeutic Implications for Antioxidant Treatment-Literature Review. Advances in urology 2017:4653512. doi: 10.1155/2017/4653512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meng XM, Nikolic-Paterson DJ, Lan HY (2016) TGF-beta: the master regulator of fibrosis. Nature reviews Nephrology 12 (6):325–338. doi: 10.1038/nrneph.2016.48 [DOI] [PubMed] [Google Scholar]
- 61.Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425 (6958):577–584. doi: 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- 62.Miyazono K, Kusanagi K, Inoue H (2001) Divergence and convergence of TGF-beta/BMP signaling. Journal of cellular physiology 187 (3):265–276. doi: 10.1002/jcp.1080 [DOI] [PubMed] [Google Scholar]
- 63.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R (2003) BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature medicine 9 (7):964–968. doi: 10.1038/nm888 [DOI] [PubMed] [Google Scholar]
- 64.Chen G, Deng C, Li YP (2012) TGF-beta and BMP signaling in osteoblast differentiation and bone formation. International journal of biological sciences 8 (2):272–288. doi: 10.7150/ijbs.2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nohe A, Keating E, Knaus P, Petersen NO (2004) Signal transduction of bone morphogenetic protein receptors. Cellular signalling 16 (3):291–299 [DOI] [PubMed] [Google Scholar]
- 66.Dolmans GH, Werker PM, de Jong IJ, Nijman RJ, Wijmenga C, Ophoff RA (2012) WNT2 locus is involved in genetic susceptibility of Peyronie’s disease. The journal of sexual medicine 9 (5):1430–1434. doi: 10.1111/j.1743-6109.2012.02704.x [DOI] [PubMed] [Google Scholar]
- 67.Kavian N, Servettaz A, Weill B, Batteux F (2012) New insights into the mechanism of notch signalling in fibrosis. The open rheumatology journal 6:96–102. doi: 10.2174/1874312901206010096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, Beyer C, Zwerina J, Distler O, Schett G, Distler JH (2011) Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Annals of the rheumatic diseases 70 (7):1304–1310. doi: 10.1136/ard.2010.134742 [DOI] [PubMed] [Google Scholar]
- 69.Rohatgi R, Milenkovic L, Corcoran RB, Scott MP (2009) Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proceedings of the National Academy of Sciences of the United States of America 106 (9):3196–3201. doi: 10.1073/pnas.0813373106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horn A, Palumbo K, Cordazzo C, Dees C, Akhmetshina A, Tomcik M, Zerr P, Avouac J, Gusinde J, Zwerina J, Roudaut H, Traiffort E, Ruat M, Distler O, Schett G, Distler JH (2012) Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis and rheumatism 64 (8):2724–2733. doi: 10.1002/art.34444 [DOI] [PubMed] [Google Scholar]
- 71.Clevers H, Nusse R (2012) Wnt/beta-catenin signaling and disease. Cell 149 (6):1192–1205. doi: 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 72.Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, Sonnylal S, de Crombrugghe B, Taketo MM, Distler O, Schett G, Distler JH (2012) beta-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Annals of the rheumatic diseases 71 (5):761–767. doi: 10.1136/annrheumdis-2011-200568 [DOI] [PMC free article] [PubMed] [Google Scholar]