

Abstract
Aim:
Computer-aided drug design approaches were applied to identify chalcones with antiplasmodial activity.
Methodology:
The virtual screening was performed as follows: structural standardization of in-house database of chalcones; identification of potential Plasmodium falciparum protein targets for the chalcones; homology modeling of the predicted P. falciparum targets; molecular docking studies; and in vitro experimental validation.
Results:
Using these models, we prioritized 16 chalcones with potential antiplasmodial activity, for further experimental evaluation. Among them, LabMol-86 and LabMol-87 showed potent in vitro antiplasmodial activity against P. falciparum, while LabMol-63 and LabMol-73 were potent inhibitors of Plasmodium berghei progression into mosquito stages.
Conclusion:
Our results encourage the exploration of chalcones in hit-to-lead optimization studies for tackling malaria.
Keywords: : drug design, experimental validation, molecular docking, pharmacophore, Plasmodium falciparum, virtual screening
Graphical abstract

Malaria is a serious worldwide health problem caused by parasites of the genus Plasmodium and transmitted to humans through the bites of infected female Anopheles mosquitoes. Despite extensive public health interventions, malaria continues to be a major global health challenge, causing expressive morbidity and mortality, as well as significant socio-economic losses to the affected countries. In 2016, over 216 million malaria cases were reported in 91 countries, causing approximately 445,000 deaths [1].
Current control efforts rely on the elimination of malaria parasites using artemisinin-based combination therapies. However, the efficacy of front-line artemisinin-based combination therapies is threatened by the emergence and spread of drug resistant Plasmodium strains [2–4]. All these highlight the compelling need for discovery of new drugs that act on novel targets for tackling Malaria.
Future efforts to the design and discovery of new antimalarial drugs can be inspired on exploration of the natural-product-derived scaffolds, such as chalcones (1,3-diaryl-2-propen-1-ones). Recently, we have demonstrated that a series of 5-heteroaryl chalcone compounds presented in vitro antituberculosis [5] and antileishmanial [6] activity. Previous studies also found that diverse chalcone-like compounds show potent antiplasmodial activity. For instance, Singh and co-workers synthesized a series of piperazine‐linked 4‐aminoquinoline‐chalcone/ferrocenyl‐chalcone conjugates with in vitro EC50 values from 0.41 to 2.38 μM against asexual blood stages of Plasmodium falciparum [7]. Smit and N’Da developed a series of 4-aminoquinolinyl-chalcone amides with EC50 values ranging between 0.04–0.5 μM and 0.07–1.8 μM against sensitive and resistant strains, respectively. They demonstrated moderate to high selective activity toward the parasitic cells in the presence of mammalian cells [8]. Sharma and co-workers developed a series of stilbene-chalcone hybrids that block the progression of the parasite life cycle at the ring or the trophozoite stages at submicromolar concentrations. Further, authors showed that stilbene-chalcone hybrids cause chromatin condensation, DNA fragmentation and loss of mitochondrial membrane potential in P. falciparum, thereby suggesting their ability to cause apoptosis [9]. A review of the chalcone activities is available in reference [10].
The broad biological profile of chalcones led us to apply computer-aided drug design approaches to identify chalcones with promising antimalarial activity. To achieve this goal, we pursued the following specific aims: to perform structural standardization of an in-house database of chalcone and chalcone-like compounds synthetized by Gomes and coworkers [5]; to build a pool of potential P. falciparum protein targets for the investigated chalcones based on substructure search analysis and structure-based pharmacophores; to build homology models and perform structural refinement of predicted P. falciparum targets; to perform molecular docking studies with the chalcones and the predicted targets; and to perform experimental validation of the best scored chalcones against asexual blood stages of P. falciparum and Plasmodium berghei sexual stages. The overall study design is shown in Figure 1.
Figure 1. . General workflow with the main steps of this study.

Materials & methods
Computational
Structural standardization
An in-house collection of 28 chalcones and chalcone-like compounds previously synthetized by Gomes and coworkers [5] was carefully standardized using the software Standardizer v.16.9.5.0 (ChemAxon, Budapest, Hungary; www.chemaxon.com) according to the protocols proposed by Fourches and colleagues [11–13]. Briefly, explicit hydrogens were added, whereas polymers, salts, metals, organometallic compounds and mixtures were removed. In addition, specific chemotypes such as aromatic rings and nitro groups were normalized. Subsequently, compounds were imported to Maestro workspace v.9.3 and their 3D structures and tautomeric and protonation states were predicted using LigPrep 2.5 (Schrödinger, LCC, NY, USA).
Substructure search analysis
Aiming to build a pool of potential P. falciparum protein targets for the investigated compounds, a substructure search was carried in ChEMBL database [14] using 1,3-diayl-2-propen-1-one as query. During the substructure search, only compounds with experimental IC50, Ki or Kd data ≤10 μM obtained from biochemical assays were considered for further analyses.
Structure-based pharmacophore screening
In parallel, the 3D structures of 28 studied chalcones were submitted to PharmMapper (lilab.ecust.edu.cn/pharmmapper/) [15,16], a web server that predicts potential biological activities using structure-based pharmacophores. This server automatically ranks the best-fitted biological targets for a given small molecule against all the experimentally determined 3D structures of proteins available on PharmTargetDB. The screening was carried out using 300 conformations for each chalcone, and the number of predicted targets was set to 300. Other parameters were kept as default.
Protein alignment
Considering the limited number of parasite protein targets in PharmMapper, protein alignment tools were applied to screening using the underlying assumption that targets sharing structure similarity (orthology) have enhanced the probability of sharing the same ligands [17–19]. Here, the top scored PharmMapper targets were aligned against P. falciparum proteome using Basic Local Alignment Search Tool (BLAST) implemented in PlasmoDB (http://plasmodb.org/plasmo/) [20]. BLAST finds regions of similarity between biological sequences. The program compares protein sequences to sequence databases and calculates the sequential identity. We considered the P. falciparum targets for further evaluation only in cases where there was sequential identity ≥55% with the predicted targets.
Homology modeling
The amino acid sequence of P. falciparum prioritized proteins were retrieved from the UniProt database [21] and used as target for homology modeling in the SWISS-MODEL server (https://swissmodel.expasy.org/) [22,23]. Then, the built 3D protein models were exported to the GalaxyWEB server (http://galaxy.seoklab.org/) [24], which refines loop and terminus regions by ab initio modeling. Further, the refined models were exported to SAVES server (http://services.mbi.ucla.edu/SAVES/) and their overall stereochemical and structural quality were checked according to PROCHECK [25,26], ERRAT [27] and VERIFY-3D [28,29] scores. The PROCHECK checks stereochemical quality of a protein structure by analyzing residue by residue geometry and overall structure geometry [25,26]. ERRAT analyzes the statistics of nonbonded interactions between different atom types and plots the value of the error function versus position of a 9-residue sliding window, calculated by a comparison with statistics from highly refined structures [27]. Finally, the VERIFY-3D determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) by assigning a structural class based on its location and environment (α, β, loop, polar, nonpolar, etc.) and comparing the results with good structures [28,29].
Molecular docking
The molecular docking and preparation of molecular structures were carried out on the Maestro workspace (Schrödinger LLC), with many tools, software and modules. The 3D structures of prioritized P. falciparum targets were processed using the Protein Preparation Wizard. For protein preparation, hydrogen atoms were added to the proteins, and bond orders and formal charges were adjusted. The protonation state of polar amino acids were predicted by PROPKA v.3.1 (Schrödinger LLC) [30] at neutral pHs. Proteases were prepared at acid pH conditions, meaning that the protonation states of amino acid residues were predicted at the same pH conditions of the standard enzyme assays carried out for proteases. Subsequently, full-atom protein structure minimization using the OPLS-2005 force field was carried out [31]. Before docking studies, grids were established to each protein ruled by a box space of 10 × 10 × 10 Å3, and fixing the box on the geometrical center of active sites using the receptor grid generation panel of the Glide v.5.8 [32]. More grid details are available in Supplementary Table 1. Then, molecular docking calculations were carried out using Glide XP (extra precision) mode.
Experimental evaluation
Materials
Investigated chalcones were previously synthesized by Gomes and coworkers [5]. It is important to mention that all chemical structures were experimentally confirmed using NMR spectroscopy (1H) at 400 MHz, (13C) at 100 MHz, infrared spectroscopy and mass spectrometry. High-performance liquid chromatography analysis confirmed a minimum purity of >96% for all studied chalcones (see details in [5]). The culture medium and chloroquine were purchased from Sigma Aldrich®.
P. falciparum culture
P. falciparum 3D7 strain was continuously cultured using the method described by Trager and Jensen [33]. Briefly, the parasites were maintained at 37°C under an atmosphere of reduced oxygen (5% CO2, 5% O2 and 90% N2) in RPMI-1640 culture medium supplemented with 0.05 mg/ml gentamycin and 10% A+ human serum. Washed erythrocytes (blood group O+) were added to obtain a 5% of hematocrit and parasitemia was maintained between 2 and 5%. Synchronic cultures at ring stage were obtained by two consecutive treatments, at 48-h interval with a 5% solution of D-sorbitol [34].
Determination of P. falciparum growth inhibition
The in vitro antiplasmodial activity was measured using the fluorescent SYBR Green I®-based 96 microplate assay [35]. Briefly, synchronized 3D7 parasites (>90% ring forms) were diluted to 0.5% parasitemia with 2% hematocrit and incubated with prioritized chalcones in 96-well culture plates. After 72 h of growth period, the plates were frozen at -20°C for 24 h. For measurement of parasite growth, the plates were thawed and 100 μl of homogenized parasite culture was transferred into 96-well black plates with 100 μl of lysis buffer (20 mM Tris, 5 mM EDTA, 0.008% wt/vol saponin, 0.08% vol/vol Triton X-100 and 0.4 μl/ml of SYBR Green). After 1 h in darkness, the fluorescence was read with a plate reader at 490 nm excitation and 540 nm emission (CLARIOstar, Labtech BMG, Ortenberg, Germany). Parasite growth inhibition was calculated relative to the control (infected erythrocytes without compounds). Chloroquine was used as positive control.
Inhibition of P. berghei sexual stage progression into mosquito stages
This research protocol was approved by the Ethics Committee of the Institute of Biomedical Sciences – University of Sao Paulo, protocol number 132/2014-CEUA. C57BL/6 mice were infected with a P. berghei line expressing nano luciferase (nLuc) specifically under the control of an ookinete promoter (Ookluc line) [36]. Five days after infection, mice with gametocytemia above 0.4% were used as blood donors for experiments of in vitro conversion of gametocytes to ookinetes. Conversion assays consisted of 4 μl of infected blood dispensed into 80 μl of ookinete medium [37] at 21°C previously mixed with DMSO control or 10 μM of the prioritized chalcones in a 96-well plate. The assays were incubated at 21°C for 24 h and one volume of nLuc substrate (Nano-Glo, Promega, Madison, WI, USA) was added to each well, incubated for 5 min at 37°C, and the nLuc signal in Relative Light Units was detected using a plate luminometer. The nLuc detection using this model corresponds directly to the number of ookinetes formed in each well. The percent of conversion inhibition was calculated considering the detected signal in DMSO control wells being 100%.
Statistical analysis
Dose response and EC50 determination were calculated only for P. falciparum inhibition, by plotting the percentage parasite growth against log-transformed drug concentration and using a nonlinear regression in GraphPad Prism v. 5.01. The results were expressed as mean ± SD of three independent assays.
Results & discussion
Substructure search analysis
We performed a substructure searching in ChEMBL database [14] aiming to identify potential P. falciparum protein targets for chalcone and chalcone-like compounds. Consequently, only compounds with IC50, Ki or Kd data ≤10 μM obtained from biochemical/enzymatic assays were considered for further analyses. This chemical substructure search yielded chalcones or chalcone-like compounds reported on the literature are active against six extensively studied P. falciparum proteins: FP2, TCP, ENR, Plm2, protein kinase PfMRK and NMT, indicating the potential of chalcone-based compounds act as antimalarial agents. The biological profile of known chalcones in P. falciparum is presented in Supplementary Table 2.
Structure-based pharmacophore screening
In parallel, a set of 28 previously synthesized chalcones and chalcones-like compounds was fitted against thousands of structure-based pharmacophores using PharmMapper server. The list of top ranked targets for two representative compounds (LabMol-86 and LabMol-87) is presented in Supplementary Table 3. Then, the predicted targets with Z’-score higher than 0 were selected as potential chalcone targets. Z’-score is a better descriptor for ranked targets generated from the molecule’s fit score and a library score matrix calculated beforehand. This score does not only apply to the pharmacophore matching but also consider normal distribution that a randomly given molecule’s fit score may follow.
Considering the limited number of parasite targets in PharmMapper database, protein alignment tools were also applied to screening using the underlying assumption that targets sharing structure similarity (orthology) have enhanced the probability of sharing the same ligands [17–19]. Therefore, top Z’-scored targets were aligned against P. falciparum proteome using BLAST implemented in PlasmoDB [20]. We considered the P. falciparum targets for further evaluation only in cases where there was sequential identity ≥55% with the predicted targets. As a result, two potential P. falciparum targets were prioritized, the proteins OAT and PK5.
Homology modeling
The 3D structures of protein kinase PfMRK, NMT and TCP were not available on the Protein Data Bank at the time this work was conducted. Consequently, homology models were built by comparing the P. falciparum primary sequences with similar sequences of other proteins (templates) for which 3D structures were available. After the homology modeling, the loops and terminus regions were structurally refined. The details of homology modeling and protein refinement are presented in Supplementary Table 4 & Supplementary Figure 1.
Validation of the refined models was done for various levels of structural quality. Analysis of dihedral angles phi against psi of amino acid residues (stereochemical quality obtained from PROCHECK analysis) revealed that 86.1−95.0% of residues of the modeled proteins are within the most favored regions, 4.7−12.9% of residues are within the additional allowed regions, only 0.0−0.3% of residues are within the generously allowed regions, and only 0.0−0.6% of residues are within the disallowed regions. The residues in disallowed regions are out of binding site not disturbing the active site. The analysis of models also indicated good protein environments for the nonbonded interactions between different atom types (ERRAT scores ranging between 81.6 and 93.6) and acceptable atomic coordinates for the structures (VERIFY-3D scores ranging between 67.3 and 91.2). So, the overall stereochemistry and conformation characteristics of atoms, and the compatible chemical interaction environment indicate that generated models could be useful to prospective molecular docking studies.
Molecular docking
After target pool prioritization, a virtual screening was carried out using molecular docking aiming to investigate which protein of P. falciparum could interact with the in-house collection of chalcones. As we can see in Supplementary Table 5, docking scores demonstrate that 16 studied compounds (LabMol-63, -64, -69, -70, -72, -73, -74, -77, -80, -82, -86, -88, -93, -95) have considerable affinity to PK5, Plm2 and/or NMT. This allowed us to assume that the investigated compounds may have antiplasmodial activity.
Experimental validation
Considering the docking results, the top 16 scored chalcones (in-house database) and chloroquine (positive control) were tested against asexual blood stages of P. falciparum 3D7 strain (Supplementary Table 6). Table 1 shows the docking scores and experimental results for the best six experimental hits, including the EC50 for P. falciparum 3D7 strain, selectivity indices (SI) calculated using the cytotoxicity in mammalian cells (Vero cells and macrophages), as well as P. berghei % conversion inhibition. Four out of selected compounds (LabMol-77, -86, -87, and -93) showed good antiplasmodial activity, with EC50 <5 μM (Table 1). Among the active hits, LabMol-86 and LabMol-87 were the most promising ones, with EC50 values of 2.2 and 2.7 μM, respectively. Dose-response curves for the active hits are presented in Supplementary Figure 2. Remaining tested compounds were considered inactive, with EC50 values >5 μM. In addition to P. falciparum growth inhibition assay, active hits demonstrated low cytotoxicity in mammalian cells (Table 1), with SI ranging between 17 and 45.4 for Vero cells and modest SI for macrophages. Further, the top 16 scored chalcones were also tested at 10 μM against P. berghei conversion into ookinetes, which are formed in vitro through gametocyte activation, gamete formation and fertilization (Supplementary Table 6). Two chalcones, LabMol-63 and LabMol-73 inhibited conversion by 60.8 and 96.0%, respectively (Table 1). However, the fact that the tested chalcones cannot fully clear all asexual and sexual stage parasitemia do not make them unfeasible hits, since drug combinations in clinical trials may require multiple exposures [38].
Table 1. . Docking scores and experimental results for the best antiplasmodial hits.
| Compound | Docking scores (Kcal/mol) | In vitro assays (μM) | |||||
|---|---|---|---|---|---|---|---|
| PK5 | Plm2 | NMT | Plasmodium falciparum 3D7 EC50 | SI† | SI‡ | Plasmodium berghei % conversion inhibition (10 μM) | |
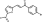 LabMol-63 |
-7.6 | -7.0 | -6.8 | >5 | N.D. | N.D. | 60.8 |
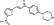 LabMol-73 |
-7.4 | -6.1 | -7.8 | >5 | N.D. | N.D. | 96.0 |
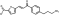 LabMol-77 |
-7.6 | -8.8 | -7.2 | 4.4 ± 0.59 | 17.2 | 11.1 | 4.2 |
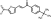 LabMol-86 |
-7.6 | -7.6 | -7.8 | 2.2 ± 0.58 | >45.4 | 3.9 | 0.0 |
 LabMol-87 |
-8.7 | -8.8 | -6.2 | 2.7 ± 0.41 | 30.1 | 6.3 | 7.5 |
 LabMol-93 |
-7.7 | -7.3 | -5.1 | 4.0 ± 0.92 | >25.0 | 2.1 | 0.1 |
| Chloroquine | – | – | – | 0.011 ± 0.0006 | N.D. | N.D. | – |
Selectivity index obtained for Vero cells [5].
Selectivity index obtained for macrophages [39]; EC50 and standard deviation were derived of three independent experiments; the data are expressed as mean ± SD of three independent assays.
Inhibition of ookinete stage of P. berghei.
N.D.: Not determined; SI: Selectivity index.
Structure–activity relationships
Based on the experimental results, we derived structure–activity relationships (SAR) rules to reveal the molecular substituents favorable and unfavorable for antiplasmodial activity (Figure 2). The information revealed by the SAR allowed us to derive the following rules: 5-nitrofuran or 5-nitrothiophene as ring B increases the activity; substitution of 5-nitroaryl ring B by benzene with dimethylamino, and methoxy in para-position decreases the activity; bulky groups and heterocyclic rings on para-position of ring A, for example, imidazole, cyclohexyl, phenyl or morpholine decrease the activity; halogen atoms (Br) in meta-position of ring A decrease the activity; and aliphatic groups on para-region of ring A, for example, methylthiole, tert-butyl or butyl increase the activity.
Figure 2. . Derived structure–activity relationship rules for chalcones and chalcone-like compounds with antiplasmodial activity.

Substituents inside green (dotted line in printed version) boxes are favorable to the antiplasmodial activity, whereas substituents in red (solid line in printed version) boxes decrease the activity.
Rationalizing antiplasmodial activity
Understanding the interaction pattern between the ligand and the protein target is essential for designing more potent and selective analogs. Here, molecular docking studies allowed us to rationalize the interaction in the best scored compound and best experimental hit (LabMol-87) with its predicted protein targets (Figure 3).
Figure 3. . Intermolecular interactions of LabMol-87 with PK5 (A & B), Plm-2 (C & D) and NMT (E & F).

In 3D representation plots (A, C & E), hydrogen bonds are presented as yellow-dashed lines, and the color code for hydrogen, oxygen, nitrogen and sulfur atoms are white, red, blue and yellow, respectively. The carbon atoms of the ligand are colored cyan. In 2D interaction diagrams (B, D & F), hydrogen bonds are presented as magenta arrows, ionic interaction as gradient lines (red to blue) and π–stacking interaction as green lines.
PK5 is a member of CDKs family [40]. The CDK activity of this kinase has been proved with an important role in parasite DNA replication based on the following considerations: it shows nuclear localization during DNA synthesis [41]; treatment with CDK inhibitors results in an impairment of DNA synthesis; and parasites treated with a DNA synthesis inhibitor display elevated PK5 kinase activity [42]. The GlideScore energy (-8.7 Kcal/mol) obtained from docking suggested that LabMol-87 could inhibit P. falciparum PK5 at low micromolar concentrations. The binding mode of LabMol-87 in ATP-binding site of PK5 can be generalized as follows (Figure 3A & B): butyl moiety and aromatic ring A interact with the hydrophobic pocket formed by Leu54, Phe79, Leu132 and Ala142, whereas the carbonyl and nitro groups form hydrogen bond interactions (represented as yellow dashed lines) with the amide backbone of Leu83 and side chain of Gln129, Lys87 and Lys88, respectively. Interestingly, Geyer and colleagues [43] showed that a series of chalcones selective inhibit a homologe CDK in P. falciparum known as PfMRK. According to these authors, the substitution of Phe143 within the binding site of PfMRK by a Leu132 may contribute to lack of affinity against PK5 [43]. However, we still consider PK5 an interesting target since the lowest volume of Leu132 could better accommodate our compounds in binding site, since they have more bulky substituents in para-position of ring A.
The Plm2 is an important parasite aspartic protease involved in host hemoglobin hydrolysis. In addition to its hemoglobinase activity, Plm2 may directly digest the membrane and cytoskeletal proteins of the host presumably required for parasite egress [44,45]. In addition, a number of chalcone derivatives have been found to inhibit Plm2 at low micromolar concentrations [46,47], reinforcing our hypothesis that the active compounds could be inhibitors of this enzyme. The GlideScore energy (-8.8 Kcal/mol) obtained from the docking suggest that LabMol-87 also has considerable affinity with P. falciparum Plm2. The binding mode of LabMol-87 in the active site of Plm2 can be generalized as follows (Figure 3C & D): butyl group and aromatic ring A interact with the hydrophobic pocket formed by Tyr201, Phe235, Tyr239, Val206, Pro167 and Trp165, whereas the thiophen ring B interact with Ile156 and Thr238.
Finally, NMT is an attractive and druggable target against the blood stage of malaria infections. This enzyme catalyzes transfer of the lipid myristate from myristoyl coenzyme A to specific substrate proteins in parasite cell. Inhibition of this reaction may lead to catastrophic and irreversible failure to assemble the inner membrane complex, a critical subcellular organelle in the parasite life cycle [48]. The GlideScore energy (-6.2 Kcal/mol) suggest that LabMol-87 also have good affinity with P. falciparum NMT. As we can see in Figure 3E, the butyl group and the aromatic ring A of LabMol-87 interact with the hydrophobic pocket formed by Phe105, Tyr211, Phe226, Leu330 and Val363, whereas the carbonyl and nitro groups form hydrogen bond interactions (represented as yellow-dashed lines) with the amide backbone of Gly386 and the side chain of Tyr95, respectively.
The chalcones identified as inhibiting P. berghei conversion were different from those active against P. falciparum asexual blood stages. This may reflect species divergence of targets between P. falciparum and P. berghei. However, it may also reflect different susceptibilities between asexual and sexual stages. Previous studies found that >90% of antimalarial compounds active against P. falciparum asexual stages are inactive against sexual stages [49].
Conclusion
We have developed and applied a computer-aided drug design workflow to predict the antiplasmodial activity for a series of in-house chalcones and chalcone-like compounds. As a result, 16 chalcones were prioritized for biological evaluation. Two compounds, LabMol-86 and LabMol-87, showed potent activity against P. falciparum 3D7 asexual blood stages, with EC50 values around 2.5 μM. Both compounds also demonstrated low cytotoxicity in mammalian cells and thus high selectivity (SI >30). Moreover, we found that two different chalcones, LabMol-63 and LabMol-73, can inhibit P. berghei conversion into ookinetes, blocking the parasite progression into mosquito stages. Molecular docking studies provided insights into the binding modes of LabMol-87, the best experimental hit, into its predicted Plasmodium proteins, PK5, Plm2 and NMT. The analysis of interaction environment combined with generated SAR rules indicated that the presence of hydrophobic substituents such as butyl, tert-butyl and methylsulfanyl in the para-position of ring A, and 5-nitrofuran or 5-nitrothiophene as ring B are required for optimal interaction with the binding site and thus for the antiplasmodial activity.
Future perspective
Despite the urgent need in finding a new treatment for malaria and the relative success that has been recently achieved through approval of the tafenoquine for clinical use, defeating this disease may still be far reaching. The chalcones exemplify a promising scaffold for therapeutic intervention, especially in infectious diseases. However, the use of chalcones as starting points for drug design have been sparse and mainly from academic groups, thus their further development as marketed drugs has been stunted. Currently, no specific chalcone or chalcone-like compound is in clinical trials for malaria and to our knowledge are not being actively pursued by the pharma industry. However, based on the existing research, the limited chemical diversity of antimalarial drugs, and availability of lead compounds of other chemical classes, it is evident that there is need for new chemical scaffolds with novel mechanisms of action. So, the experimental validation of the mechanism of action and optimization of the biological profile may open future broad prospects of development of new drugs for malaria treatment and prophylaxis based on these compounds and their combinations.
Summary points.
Substructure search analysis and structure-based pharmacophore screening
Computer-aided drug design approaches were used to prioritize chalcones with potential antiplasmodial activity.
Experimental validation
Selected hits were tested in vitro against Plasmodium falciparum asexual blood stages and Plasmodium berghei sexual stages.
5-nitroheteroaryl chalcones with aliphatic groups on para-region of ring A were identified as promising antiplasmodial hits.
Two compounds, LabMol-86 and LabMol-87, showed potent antiplasmodial activity and low cytotoxicity.
Two compounds, LabMol-63 and LabMol-73, inhibited P. berghei progression into mosquito stages.
Molecular docking
Molecular docking provided insights into the binding modes of active compounds into its predicted Plasmodium targets: PK5, Plm2 and NMT.
Conclusion
Discovered hits are new scaffolds for further hit-to-lead optimization studies.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.future-science/doi/suppl/10.4155/fmc-2018-0255
Financial & competing interests disclosure
The authors thank Brazilian funding agencies, CNPq, CAPES, FAPESP and FAPEG for financial support and fellowships. CH Andrade and FTM Costa are productivity fellows of CNPq. EN Muratov gratefully thank the NIH (grants 1U01CA207160 and R01-GM114015). CH Andrade is supported by CNPq (grant 400760/2014-2) and FAPESP #2017/02353-9. FTM Costa is supported by FAPESP (grants #2012/16525-2, #2017/18611-7 and 2018/0700-4). DY Bargieri is supported by FAPESP (grant #2013/13119-6), Instituto Serrapilheira (grant #G-1709-16618) and CNPq (grant 405996/2016-0). J Calit, GG Cassiano and LT Ferreira were supported by FAPESP (Fellowships #2018/24878-9, #2017/20774-6, and #2017/02031-1, respectively). We are grateful to ChemAxon (https://chemaxon.com/) for providing academic license of their program. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
This research protocol was approved by the Ethics Committee of the Institute of Biomedical Sciences – University of Sao Paulo, protocol number 132/2014-CEUA.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.WHO. Malaria (2017). www.who.int/mediacentre/factsheets/fs094/en/ [Google Scholar]
- 2.Rogers WO, Sem R, Tero T. et al. Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar. J. 8(1), 10 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashley EA, Dhorda M, Fairhurst RM. et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371(5), 411–423 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witkowski B, Khim N, Chim P. et al. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in Western Cambodia. Antimicrob. Agents Chemother. 57(2), 914–923 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Structure–activity relationship rules and QSAR models were used to prioritize new heteroaryl chalcone compounds for synthesis and biological evaluation.
- 5.Gomes MNMN, Braga RCRC, Grzelak EMEM. et al. QSAR-driven design, synthesis and discovery of potent chalcone derivatives with antitubercular activity. Eur. J. Med. Chem. 137, 126–138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomes MNMN, Alcantara LM, Neves BJBJ. et al. Computer-aided discovery of two novel chalcone-like compounds active and selective against Leishmania infantum. Bioorganic Med. Chem. Lett. 27(11), 2459–2464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A, Rani A, Gut J, Rosenthal PJ, Kumar V. Piperazine-linked 4-aminoquinoline-chalcone/ferrocenyl-chalcone conjugates: synthesis and antiplasmodial evaluation. Chem. Biol. Drug Des. 90(4), 590–595 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Smit FJ. Synthesis, in vitro antimalarial activity and cytotoxicity of novel 4-aminoquinolinyl-chalcone amides. Bioorg. Med. Chem. 22(3), 1128–1138 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Sharma N, Mohanakrishnan D, Shard A. et al. Stilbene–chalcone hybrids: design, synthesis, and evaluation as a new class of antimalarial scaffolds that trigger cell death through stage specific apoptosis. J. Med. Chem. 55(1), 297–311 (2012). [DOI] [PubMed] [Google Scholar]; • Presents current methodological developments toward the design and synthesis of new chalcone derivatives and state-of-the-art medicinal chemistry strategies.
- 10.Gomes M, Muratov E, Pereira M. et al. Chalcone derivatives: promising starting points for drug design. Molecules 22(8), 1210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fourches D, Muratov E, Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 50(7), 1189–1204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fourches D, Muratov E, Tropsha A. Curation of chemogenomics data. Nat. Chem. Biol. 11(8), 535–535 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Fourches D, Muratov E, Tropsha A. Trust, but verify II: a practical guide to chemogenomics data curation. J. Chem. Inf. Model. 56(7), 1243–1252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaulton A, Bellis LJ, Bento AP. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40(D1), D1100–D1107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Ouyang S, Yu B. et al. PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 38(Suppl. 2), W609–W614 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Pan C, Gong J, Liu X, Li H. Enhancing the enrichment of pharmacophore-based target prediction for the polypharmacological profiles of drugs. J. Chem. Inf. Model. 56(6), 1175–1183 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Pollastri MPM, Campbell RRK. Target repurposing for neglected diseases. Future Med. Chem. 3(10), 1307–1315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rognan D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 152(1), 38–52 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klabunde T. Chemogenomic approaches to drug discovery: similar receptors bind similar ligands. Br. J. Pharmacol. 152(1), 5–7 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aurrecoechea C, Brestelli J, Brunk BP. et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37(Suppl. 1), D539–543 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apweiler R, Bairoch A, Wu CH. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32(Suppl. 1), D115–D119 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biasini M, Bienert S, Waterhouse A. et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42(W1), W252–W258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 4(1), 1–13 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 40(W1), W294–W297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26(2), 283–291 (1993). [Google Scholar]
- 26.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 8(4), 477–486 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 2(9), 1511–1519 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253(5016), 164–170 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature 356(6364), 83–85 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J. Chem. Theory Comput. 7(7), 2284–2295 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Banks JL, Beard HS, Cao Y. et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 26(16), 1752–1780 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friesner RA, Banks JL, Murphy RB. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47(7), 1739–1749 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 193(4254), 673–675 (1976). [DOI] [PubMed] [Google Scholar]
- 34.Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65(3), 418 (1979). [PubMed] [Google Scholar]
- 35.Hartwig CL, Ahmed AOA, Cooper RA, Stedman TT. SYBR Green I®-based parasite growth inhibition assay for measurement of antimalarial drug susceptibility in Plasmodium falciparum. : Methods in Malaria Research. Moll K, Kaneko A, Scherf A, Wahlgren M (Eds). EVIMalaR, Glasgow, Scotland, 122–129 (2013). [Google Scholar]
- 36.Calit J, Dobrescu I, Gaitán XA. et al. Screening the pathogen box for molecules active against Plasmodium sexual stages using a new nanoluciferase-based transgenic line of P. berghei identifies transmission-blocking compounds. Antimicrob. Agents Chemother. 62(11), e01053– 18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blagborough AM, Delves MJ, Ramakrishnan C, Lal K, Butcher G, Sinden RE. Assessing transmission blockade in Plasmodium spp. Methods Mol. Biol. 923, 577–600 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Burrows JN, Duparc S, Gutteridge WE. et al. New developments in anti-malarial target candidate and product profiles. Malar. J. 16(1), 26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomes MN, Alcântara LM, Neves BJ. et al. Computer-aided discovery of two novel chalcone-like compounds active and selective against Leishmania infantum. Bioorganic Med. Chem. Lett. 27(11), 2459–2464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deshmukh AS, Agarwal M, Mehra P. et al. Regulation of Plasmodium falciparum origin recognition complex subunit 1 (PfORC1) function through phosphorylation mediated by CDK-like kinase PK5. Mol. Microbiol. 98(1), 17–33 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Graeser R, Franklin RM, Kappes B. Mechanisms of activation of the cdc2-related kinase PfPK5 from Plasmodium falciparum. Mol. Biochem. Parasitol. 79(1), 125–127 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Graeser R, Wernli B, Franklin RM, Kappes B. Plasmodium falciparum protein kinase 5 and the malarial nuclear division cycles. Mol. Biochem. Parasitol. 82(1), 37–49 (1996). [DOI] [PubMed] [Google Scholar]; •• Reports inibition activity of chalcones against Plasmodium falciparum kinases.
- 43.Geyer JA, Keenan SM, Woodard CL. et al. Selective inhibition of Pfmrk, a Plasmodium falciparum CDK, by antimalarial 1,3-diaryl-2-propenones. Bioorg. Med. Chem. Lett. 19(7), 1982–1985 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Blackman MJ. Malarial proteases and host cell egress: an ‘emerging’ cascade. Cell. Microbiol. 10(10), 1925–1934 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moura PA, Dame JB, Fidock DA. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 53(12), 4968–4978 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tiwari HK, Upadhyay S, Upadhyay RK, Rawat M, Sharma A. Chalcones induced inhibition of plasmepsin II, a hemoglobin-degrading malarial aspartic protease from Plasmodium falciparum. J. Pharm. Res. 4(4), 1253–1258 (2011). [Google Scholar]; •• Reports inibition activity of alkoxylated chalcones against P. falciparum aspartic acid protease plasmepsin-2.
- 47.Sriwilaijaroen N, Liu M, Go ML, Wilairat P. Plasmepsin II inhibitory activity of alkoxylated and hydroxylated chalcones. Southeast Asian J. Trop. Med. Public Health 37(4), 607–612 (2006). [PubMed] [Google Scholar]
- 48.Wright MH, Clough B, Rackham MD. et al. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat. Chem. 6(2), 112–121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plouffe DM, Wree M, Du AY. et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe 19(1), 114–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.