

Abstract
Introduction
Recent advances in genetic analysis have led to the discovery of novel genetic subtypes of precursor B‐cell acute lymphoblastic leukemia (B‐ALL) with prognostic relevance. In this study, we studied a cohort of pediatric B‐ALL patients to retrospectively determine the incidence of patients harboring novel genetic subtypes, as well as their outcome.
Methods
B‐ALL patients (N = 190) diagnosed in a single Korean hospital were included in the study. Patients' medical records were reviewed for data on established genetic abnormalities and outcome. CRLF2 expression was analyzed by quantitative RT‐PCR. Anchored multiplex PCR‐based enrichment was used to detect fusions and point mutations in 81 ALL‐related genes.
Results
Incidence of established recurrent genetic subtypes was as follows: high hyperdiploidy (21.6%), ETV6‐RUNX1 (21.6%), BCR‐ABL1 (7.9%), KMT2A rearrangement (7.4%) TCF3‐PBX1/TCF3‐HLF (7.4%), and hypodiploidy (1.1%). Incidence of new genetic subtypes was as follows: BCR‐ABL1‐like (13.2%), ETV6‐RUNX1‐like (2.1%), EWSR1‐ZNF384 (1.1%), and iAMP21 (1.1%). Median age at diagnosis of BCR‐ABL1‐like ALL was 6.8 years. According to type of genetic abnormality, BCR‐ABL1‐like ALL was divided into ABL class (12%), CRLF2 class (8%), JAK‐STAT class (12%), and RAS class (68%). The 5‐year event‐free survival (EFS) of BCR‐ABL1‐like patients was significantly inferior to non‐BCR‐ABL1‐like low‐ and standard‐risk patients (71.5 ± 9.1% vs 92.5 ± 3.2%, P = .001) and comparable to non‐BCR‐ABL1‐like high (75.2 ± 6.2%) and very high‐risk patients (56.8 ± 7.4%). All four ETV6‐RUNX1‐like patients survived event‐free.
Conclusion
Analogous to previous studies, incidence of BCR‐ABL1‐like ALL in our cohort was 13.2% with outcome comparable to high and very high‐risk patients. A significantly high number of RAS class mutations was a distinct feature of our BCR‐ABL1‐like ALL group.
Keywords: acute lymphoblastic leukemia, BCR-ABL1-like, RAS mutation
Incidence of BCR‐ABL1‐like ALL in a Korean pediatric cohort was 13.2% with outcome comparable to high and very high‐risk patients. A significantly high number of RAS class mutations was a feature that uniquely identified our BCR‐ABL1‐like ALL group.

1. INTRODUCTION
Recurrent genetic abnormalities in precursor B‐cell acute lymphoblastic leukemia (B‐ALL) are used to define disease subtype and predict patient prognosis. Studies aiming to identify genetic lesions in B‐ALL patients without established genetic abnormalities led to the discovery of BCR‐ABL1‐like ALL. Initial studies defined this subtype as having a gene expression profile similar to BCR‐ABL1 (+) ALL, a high incidence of IKZF1 deletions, and overall poor prognosis. 1 , 2 Subsequent studies showed that the majority of BCR‐ABL1‐like ALL patients harbor kinase‐activating abnormalities, including ABL‐class fusions, CRLF2 rearrangements, JAK2 fusions, other mutations activating the JAK‐STAT pathway, and mutations of the RAS pathway. 3 , 4 , 5 , 6 In addition, recent studies identified new genetic subtypes, including those characterized by rearrangements of DUX4, MEF2D, and ZNF384. 7 , 8 , 9
Identification of these newly proposed subtypes is important not only because the subtypes correlate with clinical parameters including outcomes, but also because they reveal therapeutic targets that may allow a reduction in morbidity and mortality related to conventional chemotherapy. However, optimum diagnostic strategy of these new genetic subtypes in the clinic lacks consensus, and several practical considerations remain.
In this study, we determined the genetic characteristics, including the incidence of patients harboring new genetic subtypes, as well as outcome in a cohort of pediatric B‐ALL treated at a single Korean hospital. In addition, we tried to outline the optimum laboratory methodology required for accurate identification of new genetic subtypes.
2. PATIENTS AND METHODS
2.1. Study group
Patients diagnosed with B‐ALL and treated at the Department of Pediatrics in Seoul St. Mary's Hospital from March 2009 to September 2015 were included in the study (n = 190, male 116 (61.1%)). The study received institutional review board approval from Seoul St. Mary's Hospital, The Catholic University of Korea (IRB No. KC17SESI0717). Diagnosis of ALL was done according to the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues based on bone marrow (BM) pathology, immunophenotyping, cytogenetics, and molecular genetics. 10 Presence of established, recurrent genetic abnormalities of pediatric B‐ALL (ETV6‐RUNX1, BCR‐ABL1, E2A‐PBX1, KMT2A rearrangements, high hyperdiploidy, hypodiploidy) in the patients was diagnosed using reverse transcription polymerase chain reaction (RT‐PCR), fluorescence in situ hybridization (FISH) and Giemsa band karyotyping, using methods reported previously. 11 High hyperdiploidy was defined as a karyotype with 51‐65 chromosomes, while hypodiploidy was diagnosed with <45 chromosomes. All patients were risk classified and treated according to an institutional protocol, the details of which have been reported previously. 12 Clinical data, including age and initial white blood cell (WBC) count at diagnosis, National Cancer Institute/Rome (NCI) risk group, 13 and institutional risk group 12 were recorded.
2.2. Targeted next‐generation sequencing based on anchored multiplex PCR
From the overall study group, 127 patients were classified as B‐ALL with the six recurrent genetic abnormalities (Table 1). High hyperdiploidy and ETV6‐RUNX1 were most common, with both detected in 41 patients (21.6%). BCR‐ABL1 was detected in 15 patients (7.9%), followed by KMT2A rearrangement (n = 14, 7.4%), TCF3‐PBX1/TCF3‐HLF (n = 14, 7.4%), and hypodiploidy (n = 2, 1.1%). Among 63 patients without recurrent genetic abnormalities, 49 patients with remaining samples underwent further genetic analysis including anchored multiplex PCR (AMP) (Figure S1). First, we measured CRLF2 gene expression by quantitative RT‐PCR (RT‐qPCR) as described in a previous report. 14 The relative expression levels were estimated using the 2−ΔΔCt method. Then, AMP was performed to determine gene expression, fusions, exon‐skipping and point mutations associated with the BCR‐ABL1‐like subtype using the Archer FusionPlex ALL kit (ArcherDX). Briefly, reverse transcription using random primers was performed for synthesis of cDNA, followed by end repair and adenylation steps. The cleanup of cDNA using Agencourt® AMPure® XP beads and ligation of molecular barcode (MBC) adapters and universal primer sites were performed. The MBC adapter‐attached cDNA was amplified by the GSP1 primer pool and primer complementary to universal primer site, and the second PCR using GSP2 primer pool was performed. The libraries were quantitated using a KAPA Universal Library Quantification Kit (Kapa Biosystems), then normalized and loaded to NextSeq (Illumina). Data were analyzed by Archer® Analysis version 5.1.7 (ArcherDX).
TABLE 1.
Genetic classification of the study group
| 190 (%) | |
|---|---|
| Recurrent genetic abnormalities | 127 (66.8) |
| High hyperdiploidy | 41 (21.6) |
| ETV6‐RUNX1 | 41 (21.6) |
| BCR‐ABL1 | 15 (7.9) |
| KMT2A rearrangement | 14 (7.4) |
| TCF3‐PBX1 | 14 (7.4) a |
| Hypodiploidy | 2 (1.1) |
| Other | 63 (33.2) b |
| iAMP21 | 2 (1.1) |
| BCR‐ABL1‐like ALL | 25 (13.2) |
| ETV6‐RUNX1‐like ALL | 4 (2.1) |
| EWSR1‐ZNF384 | 2 (1.1) |
| B‐other | 16 (8.4) |
Abbreviation; iAMP21, intrachromosomal amplification of chromosome 21.
Including one TCF3‐HLF.
Including 14 cases with suboptimal RNA.
2.3. Confirmatory tests for gene fusion by RT‐PCR and FISH
RT‐PCR and FISH were performed to confirm the fusion genes detected by AMP NGS. RT‐PCR was carried out using primers that were previously described in other studies and designed by Primer3 (http://bioinfo.ut.ee/primer3‐0.4.0/). 4 Amplification conditions were as follows; initial denaturation at 95°C for 5 minutes, 25 PCR cycles, denaturation at 95°C for 30 seconds, annealing at 58°C for 30 seconds, extension at 72°C for 90 seconds and hold at 4°C.
In addition, we underwent FISH as an alternative method to identify gene fusions, using ABL1 break‐apart probe (Cytocell Ltd), JAK2 break‐apart probe (Cytocell Ltd), IGH break‐apart probe (Cytocell Ltd) and P2RY8 deletion probe (Cytocell Ltd).
2.4. Confirmatory tests for gene mutation by massive parallel sequencing
We validated genetic mutations identified from AMP NGS with those from massive parallel sequencing. DNA was extracted from patients’ BM aspirates using Wizard® Genomic DNA Purification kit (Promega). We used a clinically targeted panel (Customized St. Mary's hematology NGS panel; ThermoFisher Scientific) and the Ion Torrent Sequencer, S5XL. Base calling and alignment of the sequences to reference genome hg19 were performed on the Ion Torrent Suite Software (Version 5.8.0). Variant calling was done using Ion Reporter software (Version 5.6). Elaborated sequence data in FASTQ format were adjusted and annotated according to the hg19 human reference genome.
2.5. Study objectives
The initial objective of the study was to retrospectively determine the incidence of established genetic abnormalities, as well as novel genetic subtypes such as BCR‐ABL1‐like ALL in our study group. The subsequent objective was to analyze clinical parameters at diagnosis and EFS of patients with novel genetic subtypes.
2.6. Statistical analysis
Comparisons of median age and WBC count at diagnosis, and ALL risk group classification between genetic subgroups was done with the Mann‐Whitney test and Pearson's chi‐square test, respectively. Patients with RAS class mutations comprised a significant subgroup of BCR‐ABL1‐like ALL patients. Clinical and treatment response‐based parameters predicting the presence of RAS class mutation were analyzed by logistic regression. EFS and OS were determined by Kaplan‐Meier method, with comparisons of EFS done with log‐rank test. EFS was defined as the time from diagnosis to last follow‐up in CR, or first event (relapse, primary refractory disease, death, or secondary malignancy). OS was defined as the time from diagnosis to last follow‐up or death from any cause. The date of last follow‐up was 31 September 2019. P value < .05 was considered significant.
3. RESULTS
3.1. Genetic classification of the study group
Among 49 patients who lacked established recurrent genetic abnormalities, we identified two patients with intrachromosomal amplification of chromosome 21 (iAMP 21) by FISH. Gene fusion was detected in 18 patients; RANBP2‐ABL1 (n = 1), EBF1‐PDGFRB (n = 2), IGH‐CRLF2 (n = 2), other IGH rearrangement (n = 2), EWSR1‐ZNF384 (n = 2), PAX5 rearrangement (n = 6), TCF3 rearrangement (n = 1), PICALM‐ME3 (n = 1), and ETV6‐EP400 (n = 1) (Figure 1).
FIGURE 1.

Genetic and clinical characteristics of patients. Data are shown for 25 patients with BCR‐ABL1‐like ALL, 4 with ETV6‐RUNX1‐like ALL and 13 other B‐ALL. ABL1r, ABL1 rearrangement; FLT3r, FLT3 rearrangement; NCI, National Cancer Institute/Rome
We also detected single nucleotide variations and small insertion/deletions by FusionPlex® ALL. Mutations activating JAK‐STAT pathways were identified in three patients including IL7R (n = 1) and FLT3 (n = 2). Interestingly, RAS pathway mutations were most commonly identified (n = 17); NRAS (n = 8), KRAS (n = 3), KRAS and NRAS (n = 1), NRAS and NF1 (n = 1), PTPN11 (n = 3), and NF1 (n = 1) (Table S1). For the three patients with PTPN11 mutations, genetic study of complete remission BM samples was negative, indicating that the mutations were somatic status. Clinical and treatment response‐based factors (patient gender, age at diagnosis, initial WBC count, NCI risk group, prephase steroid response of peripheral blasts) did not significantly predict the presence of RAS mutations (data not shown). Exon skipping was also detected in ETV6 (n = 6), RUNX1 (n = 7), EBF1 (n = 3), and IKZF1 (n = 7).
We grouped patients carrying genetic abnormalities associated with BCR‐ABL1‐like ALL including ABL‐class fusions, CRLF2 rearrangements, JAK‐STAT pathway mutations and RAS pathway mutations, and classified them as BCR‐ABL1‐like ALL (n = 25, 13.2% of B‐ALL) (Table 2). We classified patients as ETV6‐RUNX1‐like ALL when both ETV6 and IKZF1 rearrangements were identified (n = 4, 2.1% of B‐ALL). The remaining 16 patients (8.4%) were grouped as B‐other ALL (Table 1).
TABLE 2.
Sentinel genetic lesions of 25 BCR‐ABL1‐like ALL
| Genetic lesions | No (%) |
|---|---|
| ABL‐class fusions | 3 (12) |
| EBF1‐PDGFRB | 2 (8) |
| RANBP2‐ABL1 | 1 (4) |
| CRLF2 rearrangements | 2 (8) |
| IGH‐CRLF2 | 2 (8) |
| JAK‐STAT pathway mutations | 3 (12) |
| IL7R mutation | 1 (4) |
| FLT3 mutation | 2 (8) |
| RAS pathway mutations | 17 (68) |
| NRAS | 8 (32) |
| KRAS | 3 (12) |
| NRAS and KRAS | 1 (4) |
| NRAS and NF1 | 1 (4) |
| PTPN11 | 3 (12) |
| NF1 | 1 (4) |
3.2. Gene expression of the study group
We compared gene expression between BCR‐ABL1‐like ALL, ETV6‐RUNX1‐like ALL and other B‐ALL. The expression of the CRLF2, PDGFRB and IRF8 genes was higher in BCR‐ABL1‐like ALL, while expression of the SH2B3, NTRK3, SOX11 genes was lower in BCR‐ABL1‐like ALL (Figure S2). Then, we analyzed and compared gene expression within the BCR‐ABL1‐like ALL group of patients, according to type of genetic abnormality. Because each group consisted of a limited number of patients, there was no significant difference in gene expression among the patients. CRLF2 expression was highest in patients with CRLF2 rearrangement. In patients with ABL‐class rearrangement, the SEMA6A and EBF1 genes showed high expression (Figure S3).
3.3. Clinical characteristics and outcome of new subgroups
The median age at diagnosis of the BCR‐ABL1‐like ALL subgroup was 6.1 years (range: 0.6‐18), and did not differ significantly from that of the non‐BCR‐ABL1‐like ALL subgroup. The median WBC count of the BCR‐ABL1‐like ALL subgroup was 18.98 × 109/L (range: 0.89‐161.61 × 109/L), and again did not show any clear difference from that of the non‐BCR‐ABL1‐like ALL subgroup. A significantly higher proportion of BCR‐ABL1‐like ALL patients were classified as NCI high risk than non‐BCR‐ABL1‐like ALL patients. Also, a greater proportion of patients was classified as either high or very high‐risk according to institutional classification criteria among BCR‐ABL1‐like ALL patients than among non‐BCR‐ABL1‐like ALL patients (Table 3).
TABLE 3.
Comparison of clinical characteristics of the BCR‐ABL1‐like ALL and non‐BCR‐ABL1‐like ALL subgroups
|
BCR‐ABL1‐like ALL (N = 25) |
Non‐BCR‐ABL1‐like ALL (N = 165) |
P value | |
|---|---|---|---|
| Median age at diagnosis (range) | 6.1 y (0.6‐18.0) | 5.4 y (0.2‐17.1) | 0.311 |
| Median WBC count at diagnosis (range) | 18.98 × 109/L (0.89‐161.61) | 12.70 × 109/L (1.21‐726.93) | 0.332 |
| NCI risk group (%) | 0.011 | ||
| Standard | 7 (28) | 92 (56) | |
| High | 18 (72) | 73 (44) | |
| Overall risk group (%) b , a | 0.033 | ||
| Low | 0 (0) | 44 (27) | |
| Standard | 5 (20) | 24 (15) | |
| High | 10 (40) | 49 (30) | |
| Very high | 10 (40) | 48 (29) |
Abbreviations: NCI, National Cancer Institute; WBC, white blood cell.
Based on institutional risk group criteria.
The 5‐year EFS of the BCR‐ABL1‐like ALL subgroup was 71.5 ± 9.1% (18/25). There was no significant difference in EFS and OS for BCR‐ABL1‐like ALL patients with or without RAS pathway mutations (Figure S4). Events included six patients with relapsed disease and one patient who died of acute respiratory distress syndrome during remission induction chemotherapy. Median time to event was 21.5 months (range: 0.9‐46 months). Three of the six relapsed patients currently survive disease‐free, two patients after receiving chemotherapy and allogeneic hematopoietic cell transplantation (HCT) and one patient after receiving chemotherapy and scheduled for allogeneic HCT, resulting in a 5‐year OS for the BCR‐ABL1‐like ALL subgroup of 83.6 ± 7.5% (21/25).
Two of the six BCR‐ABL1‐like ALL patients who relapsed were standard risk according to institutional criteria, with the remainder being high or very high‐risk. Hence, for the standard‐risk group as a whole (N = 29), all of the 24 non‐BCR‐ABL1‐like ALL standard‐risk patients survive event‐free, while two of the five BCR‐ABL1‐like ALL patients experienced an event (5‐year EFS of 100% vs 53.3%, P < .001).
The 5‐year EFS of the BCR‐ABL1‐like ALL subgroup was significantly lower than that of the non‐BCR‐ABL1‐like ALL low‐ and standard‐risk patients (92.5 ± 3.2%, P = .001), and was similar to the 5‐year EFS of 75.2 ± 6.2% found for non‐BCR‐ABL1‐like ALL high‐risk patients (Figure 2). Also, no significant difference was found in the 5‐year EFS between BCR‐ABL1‐like ALL patients and non‐BCR‐ABL1‐like ALL very high‐risk patients (56.8 ± 7.4%, P = .367).
FIGURE 2.
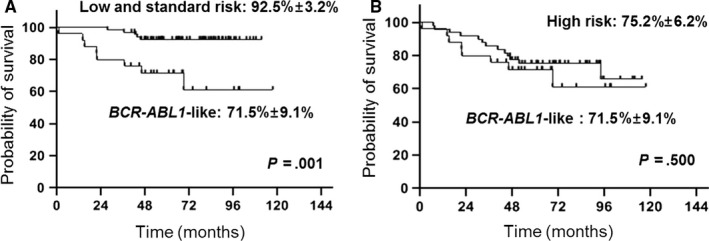
Comparison of 5‐y EFS of BCR‐ABL1‐like ALL subgroup and non‐BCR‐ABL1‐like low and standard risk subgroup (A), and non‐BCR‐ABL1‐like high risk subgroup (B)
ETV6‐RUNX1‐like patients did not show significant difference in clinical characteristics including age and WBC count compared with other B‐ALL patients (data not shown). All 6 ETV6‐RUNX1‐like patients survived event‐free.
4. DISCUSSION
Recent advances in genetic analysis have led to the identification of novel genetic subtypes of B‐ALL with prognostic relevance. In this study, we studied a cohort of pediatric B‐ALL patients to retrospectively determine the incidence of patients harboring new genetic subtypes, as well as their outcome. We successfully identified patients in the BCR‐ABL1‐like ALL category by detecting gene fusions and mutations. BCR‐ABL1‐like ALL comprised 13.2% of pediatric B‐ALL in our cohort, an incidence similar to that reported by previous studies. 2 , 15 A key distinguishing feature of BCR‐ABL1‐like ALL is aberrant signaling through cytokine and tyrosine kinase receptors. 4 Common genetic abnormalities include rearrangements of CRLF2, ABL‐class tyrosine kinase genes, and JAK2, as well as mutations activating JAK‐STAT and RAS signaling.
RAS pathway mutations had the highest frequency in our BCR‐ABL1‐like ALL patients (N = 17). Previous studies showed that patients with RAS mutations do not comprise a significant proportion of BCR‐ABL1‐like ALL patients, with incidence ranging from 3.6% to 6%. 4 , 5 , 16 , 17 In contrast, another study showed that BCR‐ABL1‐like ALL is one of the subtypes of ALL in which RAS mutations are more prevalent, with an incidence of clonal RAS mutations reported in approximately 30% of BCR‐ABL1‐like ALL patients. 18 Several studies of RAS mutation in Asian children with ALL showed a higher incidence than had been reported in patients of Western nations. 19 , 20 Similarly, patients with RAS mutations may form a significant subset of Asian patients with BCR‐ABL1‐like ALL. Recent studies based on Asian patients focused on the identification of kinase fusions, IKZF1 deletions, and JAK mutations associated with BCR‐ABL1‐like ALL. 21 , 22 Hence, further studies are necessary to ascertain potential ethnic differences in the role of RAS mutation in BCR‐ABL1‐like ALL.
In terms of clinical parameters, pediatric patients with BCR‐ABL1‐like ALL are likely to be older and have a high WBC count at diagnosis, resulting in a greater proportion of NCI high‐risk patients in the BCR‐ABL1‐like ALL category. 5 These clinical features, as well as the greater likelihood of having high levels of minimal residual disease after induction chemotherapy than other B‐ALL patients, 23 are key factors contributing to the overall poor outcome of BCR‐ABL1‐like ALL patients. In our study, a significantly higher proportion of BCR‐ABL1‐like ALL patients were classified as high risk according to both NCI and institutional classification criteria compared with non‐BCR‐ABL1‐like ALL patients.
The 5‐year EFS of the BCR‐ABL1‐like ALL subgroup was significantly lower than that of the non‐BCR‐ABL1‐like ALL low‐ and standard‐risk patients, and was similar to that of non‐BCR‐ABL1‐like ALL high‐risk patients. Inferior outcome of BCR‐ABL1‐like ALL patients compared with low‐ and standard‐risk patients is consistent with the majority of BCR‐ABL1‐like ALL patients being high risk according to both NCI and institutional criteria. However, two of the five standard‐risk BCR‐ABL1‐like ALL patients relapsed, while all of the 24 non‐BCR‐ABL1‐like ALL standard‐risk patients survive event‐free. One extensive study showed that standard‐risk BCR‐ABL1‐like ALL patients had significantly worse EFS than other standard‐risk patients, 17 indicating that the BCR‐ABL1‐like ALL genotype may influence the role of well‐established prognostic factors in pediatric ALL.
Gene expression profiling is the main method for diagnosis of BCR‐ABL1‐like ALL. However, it is not widely available in routine clinical practice. Several studies using an 8‐ or 15‐gene quantitative assay using TaqMan‐based low‐density array showed high correlation with gene expression profiling, 5 , 16 but the low‐density array is only available in a few clinical laboratories. 24 In this study, we screened BCR‐ABL1‐like ALL using AMP NGS. This method utilizes specific and universal primers, allowing for diagnosis of a translocation without the identification of both fusion partners. 25 When gene rearrangements and mutations were detected, the sentinel genetic abnormalities of BCR‐ABL1‐like ALL were confirmed by RT‐PCR, FISH or sequencing. Through this strategy, we identified sentinel gene fusions that may be used as therapeutic targets. A substantial number of clinical reports have shown that patients with kinase‐activating lesions associated with BCR‐ABL1‐like ALL are amenable to targeted therapy, with tyrosine kinase inhibitors for patients with ABL‐class fusions, and JAK inhibitors for those with JAK2 fusions or mutations. 26 , 27 , 28 , 29 , 30 Identification of sentinel genetic abnormalities associated with BCR‐ABL1‐like ALL may allow for novel and effective targeted therapy. Other recurrent genetic abnormalities including ETV6‐RUNX1‐like ALL and ZNF384 rearrangements were identified in this study. Although past studies have tried to characterize their incidence and clinical significance, 9 , 15 prognostic implications of these novel B‐ALL subtypes require further study.
In summary, our cohort of BCR‐ABL1‐like ALL patients comprised 13% of B‐ALL, had inferior outcome compared with low‐ and standard‐risk patients, and had a high incidence of RAS class mutations. This is the first report to demonstrate the characteristics of BCR‐ABL1‐like ALL in Korean pediatric patients. Further analysis based on a greater number of BCR‐ABL1‐like ALL patients is necessary to confirm the key findings of our study.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization and design: YK, N‐GC, and MK. Patient data and samples: JWL, BC, SK, and P‐SJ. Experiments, collection and assembly of data: JL, HC, and GDL. Data analysis and interpretation: JWL, YK, HC, and MK. Manuscript writing and editing: JWL, N‐GC, YK, and MK.
Supporting information
Supinfo
ACKNOWLEDGMENTS
Financial support for this study was obtained from the Catholic Medical Center Research Foundation in the program year 2018. This study was also supported by a research fund from Seoul St. Mary’s Hospital, The Catholic University of Korea.
Lee JW, Kim Y, Cho B, et al. High incidence of RAS pathway mutations among sentinel genetic lesions of Korean pediatric BCR‐ABL1‐like acute lymphoblastic leukemia. Cancer Med. 2020;9:4632–4639. 10.1002/cam4.3099
Jae Wook Lee and Yonggoo Kim contributed equally.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome‐wide classification study. Lancet Oncol. 2009;10(2):125‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high‐risk B‐precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome‐wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116(23):4874‐4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts KG, Li Y, Payne‐Turner D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reshmi SC, Harvey RC, Roberts KG, et al. Targetable kinase gene fusions in high‐risk B‐ALL: a study from the Children's Oncology Group. Blood. 2017;129(25):3352‐3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tran TH, Harris MH, Nguyen JV, et al. Prognostic impact of kinase‐activating fusions and IKZF1 deletions in pediatric high‐risk B‐lineage acute lymphoblastic leukemia. Blood Adv. 2018;2(5):529‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lilljebjörn H, Henningsson R, Hyrenius‐Wittsten A, et al. Identification of ETV6‐RUNX1‐like and DUX4‐rearranged subtypes in paediatric B‐cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7(1):11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirabayashi S, Ohki K, Nakabayashi K, et al. ZNF384‐related fusion genes define a subgroup of childhood B‐cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102(1):118‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borowitz MJ, Chan JKC, Downing JR, Le Beau MM. B‐lymphoblastic leukaemia/lymphoma with recurrent genetic abnormalities In: Swerdlow SH, Camp E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, eds. WHO classification of tumours of haematopoietic and lymphoid tissues (4th revised edn). Lyon: IARC; 2017203–209. [Google Scholar]
- 11. Kim H‐J, Oh HJ, Lee JW, et al. Utility of a multiplex reverse transcriptase‐polymerase chain reaction assay (HemaVision) in the evaluation of genetic abnormalities in Korean children with acute leukemia: a single institution study. Korean J Pediatr. 2013;56(6):247‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee JW, Kim S‐K, Jang P‐S, et al. Treatment of children with acute lymphoblastic leukemia with risk group based intensification and omission of cranial irradiation: a Korean study of 295 patients. Pediatr Blood Cancer. 2016;63(11):1966‐1973. [DOI] [PubMed] [Google Scholar]
- 13. Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J Clin Oncol. 1996;14(1):18‐24. [DOI] [PubMed] [Google Scholar]
- 14. Palmi C, Vendramini E, Silvestri D, et al. Poor prognosis for P2RY8‐CRLF2 fusion but not for CRLF2 over‐expression in children with intermediate risk B‐cell precursor acute lymphoblastic leukemia. Leukemia. 2012;26(10):2245‐2253. [DOI] [PubMed] [Google Scholar]
- 15. Zaliova M, Stuchly J, Winkowska L, et al. Genomic landscape of pediatric B‐other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica. 2019;104(7):1396‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts KG, Gu Z, Payne‐Turner D, et al. High frequency and poor outcome of philadelphia chromosome‐like acute lymphoblastic leukemia in adults. J Clin Oncol. 2017;35(4):394‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts KG, Reshmi SC, Harvey RC, et al. Genomic and outcome analyses of Ph‐like ALL in NCI standard‐risk patients: a report from the Children's Oncology Group. Blood. 2018;132(8):815‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jerchel IS, Hoogkamer AQ, Ariës IM, et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B‐cell precursor acute lymphoblastic leukemia. Leukemia. 2018;32(4):931‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al‐Kzayer LFY, Sakashita K, Al‐Jadiry MF, et al. Analysis of KRAS and NRAS gene mutations in Arab Asian children with acute leukemia: high frequency of RAS mutations in acute lymphoblastic leukemia. Pediatr Blood Cancer. 2015;62(12):2157‐2161. [DOI] [PubMed] [Google Scholar]
- 20. Liang DC, Chen SH, Liu HC, et al. Mutational status of NRAS, KRAS, and PTPN11 genes is associated with genetic/cytogenetic features in children with B‐precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2018;65:e26786 10.1002/pbc.26786 [DOI] [PubMed] [Google Scholar]
- 21. Yano M, Imamura T, Asai D, et al. Identification of novel kinase fusion transcripts in paediatric B cell precursor acute lymphoblastic leukaemia with IKZF1 deletion. Br J Haematol. 2015;171(5):813‐817. [DOI] [PubMed] [Google Scholar]
- 22. Imamura T, Kiyokawa N, Kato M, et al. Characterization of pediatric Philadelphia‐negative B‐cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J. 2016;6:e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roberts KG, Pei D, Campana D, et al. Outcomes of children with BCR‐ABL1‐like acute lymphoblastic leukemia treated with risk‐directed therapy based on the levels of minimal residual disease. J. Clin Oncol. 2014;32(27):3012‐3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Siegele BJ, Nardi V. Laboratory testing in BCR‐ABL1‐like (Philadelphia‐like) B‐lymphoblastic leukemia/lymphoma. Am J Hematol. 2018;93(7):971‐977. [DOI] [PubMed] [Google Scholar]
- 25. Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next‐generation sequencing. Nat Med. 2014;20(12):1479‐1484. [DOI] [PubMed] [Google Scholar]
- 26. Weston BW, Hayden MA, Roberts KG, et al. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1‐PDGFRB‐positive acute lymphoblastic leukemia. J Clin Oncol. 2013;31(25):e413‐e416. [DOI] [PubMed] [Google Scholar]
- 27. Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E. Successful tyrosine kinase inhibitor therapy in a refractory B‐cell precursor acute lymphoblastic leukemia with EBF1‐PDGFRB fusion. Haematologica. 2013;98(11):e146‐e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobayashi K, Miyagawa N, Mitsui K, et al. TKI dasatinib monotherapy for a patient with Ph‐like ALL bearing ATF7IP/PDGFRB translocation. Pediatr Blood Cancer. 2015;62(6):1058‐1060. [DOI] [PubMed] [Google Scholar]
- 29. Mayfield JR, Czuchlewski DR, Gale JM, et al. Integration of ruxolitinib into dose‐intensified therapy targeted against a novel JAK2 F694L mutation in B‐precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2017;64(5):e26328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding YY, Stern JW, Jubelirer TF, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome‐like acute lymphoblastic leukemia with GOLGA5‐JAK2 fusion and induction failure. Haematologica. 2018;103(9):e427‐e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.