

Abstract
Epigenetic regulators, when genomically altered, may become driver oncogenes that mediate otherwise unexplained pro-oncogenic changes lacking a clear genetic stimulus, such as activation of the WNT/β-catenin pathway in melanoma. This study identifies previously unrecognized recurrent activating mutations in the G9a histone methyltransferase gene, as well as G9a genomic copy gains in ~26% of human melanomas, which collectively drive tumor growth and an immunologically sterile microenvironment beyond melanoma. Furthermore, the WNT pathway is identified as a key tumorigenic target of G9a gain-of-function, via suppression of the WNT antagonist DKK1. Importantly, genetic or pharmacologic suppression of mutated or amplified G9a using multiple in vitro and in vivo models demonstrate that G9a is a druggable target for therapeutic intervention in melanoma and other cancers harboring G9a genomic aberrations.
Introduction
The identification and targeting of genomically altered oncogenic drivers remains a compelling therapeutic strategy for otherwise incurable cancers. Disruption of the epigenetic landscape is a relatively common event in cancer, often due to genetic alterations of epigenetic regulatory genes (1). One epigenetic modifier that undergoes somatic recurrent activating oncogenic mutations is enhancer of zeste homolog 2 (EZH2), which can silence expression of target genes (including tumor suppressors) through H3K27 tri-methylation (2). Recurrent mutations of EZH2 have been observed within its SET domain, which is well conserved across SET domain-containing histone methyltransferases (HMTs) and is essential for their enzymatic activity (3–5). The SET domain-containing HMTs Mixed Lineage Leukemia 1 (MLL1) (6), MLL3 (7), and NSD2 (8) are also targeted by gain-of-function genetic alterations that engender oncogenic properties.
Another histone methyl transferase, G9a (gene name Euchromatic Histone lysine MethylTransferase 2, EHMT2), encodes a primary SET domain-containing enzyme that can catalyze mono- and di-methylation of histone H3K9 in a heterodimeric complex with G9a-like protein (GLP) (9). G9a plays critical roles in multiple developmental processes and cell fate decisions through modulation of H3K9me2 levels (10). Genome-wide analysis suggests that H3K9me2 is functionally linked with transcriptional repression (11). Multiple studies have reported elevated G9a expression in various cancers and suggested functional linkages with malignant behaviors of cancer cells (e.g., aberrant proliferation, chemoresistance, and metastasis) by silencing tumor suppressors (12) and/or activating survival genes (13) or epithelial-to-mesenchymal transition (EMT) programs (14). In addition, recent functional studies have implicated G9a’s oncogenic role in MYC-driven tumorigenesis (15). However, genomic alterations of G9a that could directly trigger oncogenesis have not been previously identified. Here we report the occurrence of recurrent activating mutations within the SET domain of G9a and demonstrate their oncogenic function. We further find that genomic copy gains of G9a are relatively common in melanomas and other malignancies, and they display very similar oncogenic activity in vitro and in vivo. G9a is found to function through repression of DKK1, a negative regulator of the WNT pathway, an important developmental pathway heavily implicated in numerous malignancies including some in which its overactivity has lacked prior mechanistic explanation.
Results
G9a is recurrently mutated and amplified in melanoma patients
We interrogated publicly available whole-exome sequencing data for human melanomas and identified 6 cases harboring recurrent G9a point mutations at glycine 1069 (Fig.1A): four cases with G1069L and two cases with G1069W (p=8.45e-13). The recurrently mutated site, glycine 1069, resides within the highly conserved SET methyltransferase domain (Fig. 1A and B; Supplementary Fig. S1A and S1B; Supplementary Table S1) and aligns two residues from the corresponding location of activating point mutations in the SET domain of EZH2 (catalytic site Y641, Fig. 1B) (4,5). Furthermore, analysis of all downloadable copy number datasets from TCGA melanomas using GISTIC revealed a significant copy number gain (q-value=7.65e-17) at the 6p21 locus (chr6: 30,950,307–33,085,850), which encompasses the G9a gene (Fig. 1C). Comparable statistically significant amplifications of validated oncogenes known to be recurrently mutated or focally amplified in melanoma, such as MITF (3p13) (16), SETDB1 (1q21) (17), and NEDD9 (6p24) (18), were also observed in the same datasets (Fig. 1C). These observations are consistent with the possibility of a gain of function role for G9a in melanoma.
Figure 1. G9a recurrent mutations G1069L/W enhance catalytic activity and melanomagenesis.
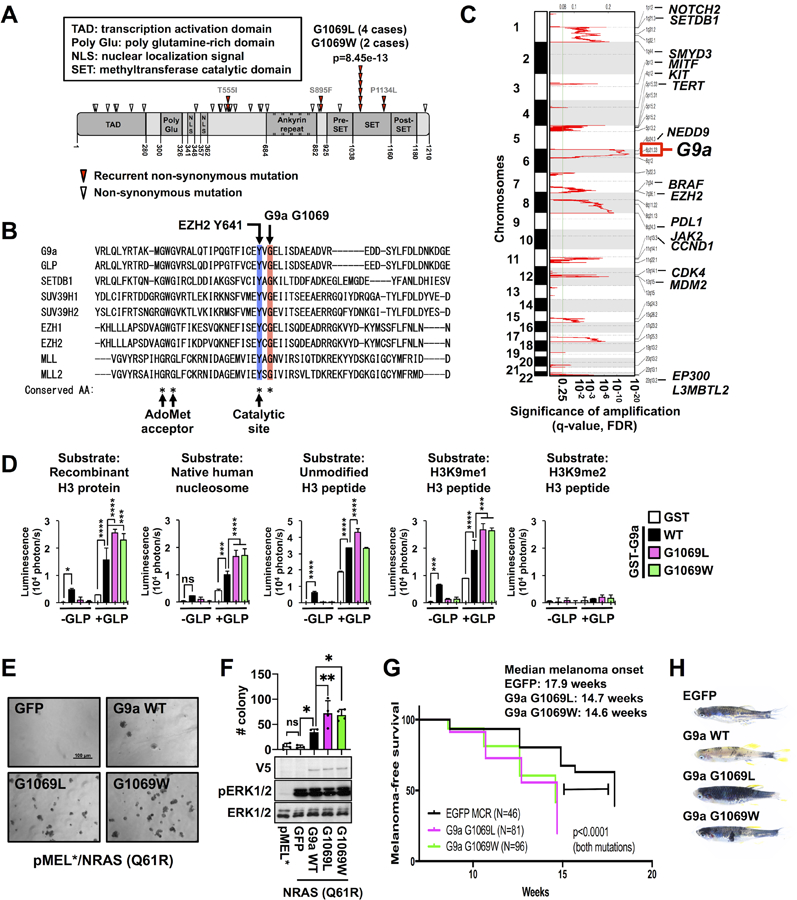
(A) Domain architecture of human G9a and mutations reported in 16 publicly available whole exome sequence datasets of patient-derived melanomas (2034 cases). Red arrowheads indicate recurrent nonsynonymous mutations. (B) Alignment of a portion of the human G9a SET domain with 8 different SET domain-containing histone methyltransferases. The blue and red columns indicate the highly conserved catalytic site tyrosine (e.g., EZH2 Y641 or G9a Y1067) and glycine (e.g., G9a G1069 or EZH2 G643), respectively. (C) GISTIC analysis (see Methods) revealed significant regions of recurrent focal chromosomal copy number gain/amplification among TCGA human melanomas. (D) In vitro methyltransferase assay using recombinant human G9a wild type (WT) and mutants in the presence or absence of recombinant GLP protein with different substrates: recombinant H3 protein, native human nucleosome, unmodified H3 tail peptide (1–16), monomethylated H3K9 (H3K9me1) peptide, and dimethylated H3K9 (H3K9me2) peptide. Data represent mean ± SD (n=4, representative of two independent experiments). (E and F) Representative images of soft agar culture (E) and colony numbers (F, top) and western blots (F, bottom) of pMEL* (left lane) and pMEL* transduced with NRASQ61R and either GFP, G9a WT, G9a G1069L, or G9a G1069W. Data with error bars represent mean ± SD of 3–4 replicates from a representative of 3 independent experiments. Western blots show expression of V5-tagged G9a WT and mutants, as well as total- and phospho-ERK1/2, a downstream target of NRAS. (G) Kaplan-Meier plot showing melanoma-free survival of BRAFV600E;tp53−/− zebrafish injected with G9a G1069 mutant (pink and green) or EGFP (control, black) miniCoopR constructs. P-values were calculated by the log-rank (Mantel-Cox) test. The experiments were repeated twice independently by two different operator, and a representative cohort is shown. (H) Representative images of the zebrafish injected with EGFP, G9a WT, G9a G1069L, or G1069W miniCoopR. P-values were calculated by one-way ANOVA with the Holm-Šidák correction for multiple pairwise comparisons (D and F). *p<0.05, ***p<0.001, ****p<0.0001.
G9a G1069 mutants complexed with GLP enhance H3K9 methylation levels and promote melanoma development
In order to directly determine the functional effect of the G1069L/W point mutations, we tested the in vitro catalytic activity of wild-type G9a and the G1069L and G1069W mutants. In the absence of its binding partner GLP, G9a showed significant catalytic activity on several substrates, but neither G9a G1069L nor G9a G1069W displayed significant activity in the absence of GLP (Fig. 1D). We next co-incubated G9a with GLP, which is reported to synergistically increase catalytic activity (19). The G1069L and G1069W mutations do not affect binding potential of G9a with GLP (Supplementary Fig. S1C). However, in the presence of the GLP binding partner, we found that the G9a G1069L and G1069W mutants enhanced H3K9 methylation to a significantly greater degree than wild type G9a (Fig. 1D; Supplementary Fig. S1D). Along with this functional difference, we found that, in the absence of GLP, the G9a G1069L/W mutants bound to H3K9-monomethylated H3 tail peptides with increased efficiency compared to wild type G9a (Supplementary Fig. S1E). A possible explanation for the functional difference warranting further investigation is that tighter binding of the mutants to H3 peptides impairs binding or proper positioning of methyl donor S-adenosylmethionine in the G9a active site, which can be rectified and enhanced by interaction with GLP.
H3K27 methylation was not increased by the G9a G1069L/W mutants compared to wild type G9a (Supplementary Fig. S1D), suggesting that the mutations specifically enhance dimethylation of H3K9 without extending substrate specificity to the target of the related EZH2 enzyme. Consistent with these findings, overexpression of the G9a G1069L/W mutants in the human melanoma cell line UACC62 increased H3K9me2 levels more than overexpression of wild type G9a (Supplementary Fig. S1F and S1G).
Next, we sought to investigate a functional relationship between the G9a G1069L/W mutants and melanoma development in established in vitro and in vivo assays. First, we tested the impact of these mutations on immortalized human melanocytes (16) (hereafter termed pMEL*) expressing NRASQ61R. These cells exhibited significantly more anchorage-independent growth after addition of G9a WT or the G1069L or G1069W mutant; however, the effects of the mutants were significantly greater than that of wild type G9a (Fig. 1E and F). Similarly, proliferation of UACC62 melanoma cells was significantly increased by overexpression of G9a and further enhanced by the G1069L and G1069W mutants (Supplementary Fig. S1H). These growth advantages were fully reversed by the G9a/GLP inhibitor UNC0638 (Supplementary Fig. S1I), providing initial evidence that G9a/GLP inhibitors might be effective in targeting G9a mutated melanomas.
We also tested the impact of these mutants in a BRAFV600E;p53−/− zebrafish melanoma model. We used the miniCoopR transgenic system (17) to express the G9a G1069L/W mutants and wild type G9a and found that both mutants significantly accelerated melanoma onset compared with EGFP control (Fig. 1G). Unexpectedly, the role of wild type G9a could not be evaluated in zebrafish melanomas because its overexpression resulted in a developmental deficiency of melanocytes compared with control and G9a mutant-expressing zebrafish (Fig. 1H), a phenotype that might be related to the difference in enzymatic function of wild type G9a vs. the point mutants (see Fig. 1D). Since the G9a plasmids were injected into single cell embryos, one possibility is that developing melanocytes with low levels of endogenous zebrafish G9a and GLP homologs will express an excess of human G9a monomer. In the case of wild type G9a, which has activity as a monomer in vitro, the excess monomer may methylate H3 at inappropriate sites and cause aberrant gene repression or induction that impairs development of melanocyte progenitor cells. On the other hand, excess mutant G9a monomer will be inactive in melanocyte progenitor cells in zebrafish embryos if it behaves as it does in vitro, allowing melanocyte development to proceed until mutant human G9a/zebrafish GLP dimers exert their tumorigenic effects.
Further confirmation of the oncogenic function of G9a was provided by a conventional transformation assay in NIH3T3 cells showing copious focus formation by G9a wild type- and mutant-transduced cells (Supplementary Fig. S1J). Together, the in vitro and in vivo results suggest that G9a is a novel melanoma oncogene and the G9a recurrent mutations at G1069 could be driver mutations for development of melanoma.
G9a is required for melanomagenesis and growth in G9a-gained melanomas
Along with recurrent mutations, copy number gains/amplifications may be drivers of tumor development in different malignancies with potential for therapeutic targeting. To interrogate candidate genes within the smallest recurrently amplified amplicon in the 6p21 locus, 287 TCGA melanomas with both RNA-seq and SNP array data were analyzed. Amplification of the 6p21 locus is associated with >1.75-fold increased expression of only 4 out of 119 RefSeq genes within the 6p21 amplicon relative to unamplified melanomas; one of these 4 overexpressed genes is G9a (Supplementary Fig. S2A). Functional validation using multiple shRNA hairpins for each candidate gene (CCHCR1, G9a, ZBTB12, or RNF5) revealed that only knockdown of G9a consistently suppressed the growth of 6p21-amplified melanoma cell line Hs944T (Supplementary Fig. S2B–S2D). Moreover, of the 4 candidate genes, only G9a expression correlates significantly with poorer prognosis among TCGA melanoma patients (Supplementary Fig. S2E).
Analyses of TCGA SNP array datasets indicate that 25.8% of melanomas harbor 3 or more G9a copies (Fig. 2A). Because SNP arrays are only semiquantitative with respect to copy number, we measured G9a copy numbers using genomic quantitative PCR in 19 melanoma cell lines, including Hs944T cells, which are reported in the Cancer Cell Line Encyclopedia database to carry G9a amplification (Supplementary Fig. S3A). All of the G9a alleles in the 19 melanoma cell lines we utilized are wild type. G9a protein levels that we determined by Western blotting correlated significantly with copy number (Supplementary Fig. S3B–S3D). Consistently, G9a mRNA expression levels in G9a copy number-gained and -amplified melanomas are significantly higher than that in G9a diploid melanomas in the TCGA melanoma dataset (Supplementary Fig. S3E). Important for the functional assessment of G9a activity, western blot analyses indicated that 4 melanoma cell lines with 3 or more copies of G9a contained significantly higher overall H3K9me2 levels compared with G9a-unamplified melanoma cell lines or primary human melanocytes (Fig. 2B; Supplementary Fig. S3F). Importantly, G9a knockdown selectively suppressed proliferation (Fig. 2C) and anchorage-independent growth (Supplementary Fig. S3, G and H) of the G9a-gained or -amplified melanoma lines. In LOX-IMVI, a G9a WT/diploid melanoma cell line expressing a high level of G9a (Supplementary Fig. S3C), the growth rate was strongly suppressed by longer treatment (7 days) with G9a knockdown, an effect that was not seen in other G9a WT/diploid melanoma cell lines (Supplementary Fig. S3I), suggesting that G9a-high melanoma cells are consistently sensitive to G9a inhibition, but the molecular kinetics may vary somewhat between G9a-diploid melanoma cells with relatively high G9a expression and G9a-gained/amplified cells. Conversely, G9a overexpression significantly enhanced anchorage-independent growth of M14, a G9a-heterogygous loss melanoma cell line (Supplementary Fig. S3J). Consistent with the genetic findings, G9a-gained/H3K9me2-high melanoma cells are highly sensitive to the G9a/GLP inhibitors UNC0638 and BIX01294 compared with G9a-unamplified/H3K9me2-low melanoma cells and primary human melanocytes (Fig. 2D; Supplementary Table S2). The antiproliferative effect of UNC0638 was strongly associated with accumulation of LC3B-II, an autophagy marker (Fig. 2E), as reported previously (13). Moreover, following treatment with an autophagy inhibitor, bafilomycin A1, accumulation of LC3B-I and -II was strongly promoted in M14 cells overexpressing wild type G9a, and the elevation was further enhanced by expression of the oncogenic G1069L/W mutants (Supplementary Fig. S3K), suggesting that genetic G9a dysregulation confers sensitivity to autophagy inhibitors. Taken together, these data suggest that G9a is required for growth and represents a potential therapeutic target in not only melanomas with G9a point mutations, but also in a larger subset of G9a copy number-gained melanomas (about 26% of TCGA melanomas, Fig. 2A).
Figure 2. G9a is required for growth in G9a copy number-gained melanoma cells.
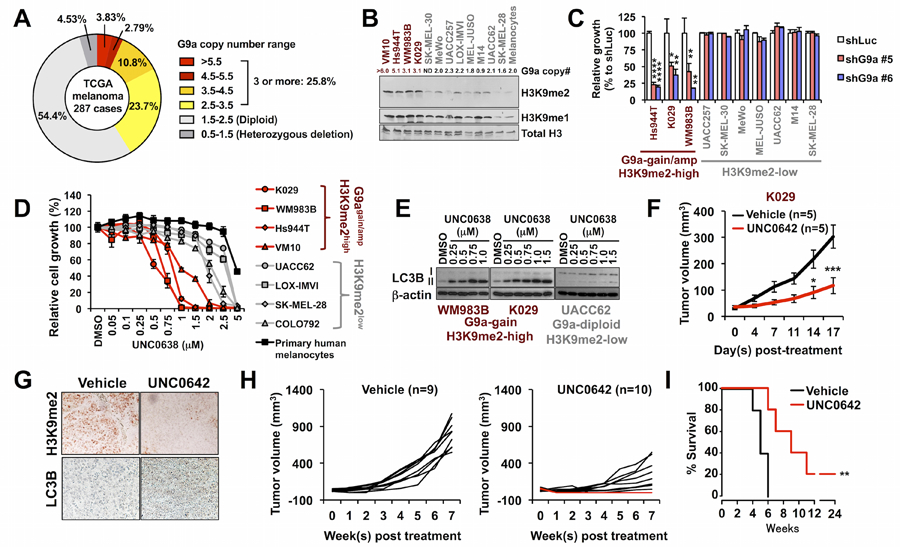
(A) Proportions of TCGA melanomas with different G9a copy numbers. (B) Representative western blot of H3K9me2 in melanoma cell lines and primary human melanocytes. The numbers indicate qPCR-determined G9a copy numbers in G9a-gained/amplified (red) and -unamplified (black) melanoma lines and melanocytes; ND, not determined). The experiment was repeated four times independently (refer to Supplementary Fig. S3F). (C) Colony formation of G9a-gained or -amplified/H3K9me2-high and H3K9me2-low melanoma cell lines with G9a knockdown or control shRNA (shLuc). Data represent mean +/− SD of triplicates. P-values were calculated by one-way ANOVA with the Holm-Šidák correction for multiple pairwise comparisons. *p<0.05, **p<0.01, ****p<0.0001 vs. shLuc. (D) Dose-dependent growth inhibition by UNC0638 in melanoma cell lines and primary human melanocytes. Data represent mean +/− SD of triplicates from at least two independent experiments. (E) Western blotting for autophagy marker LC3B in G9a-gained melanoma cell lines WM983B and K029, and G9a-unamplified melanoma cell line UACC62. Cells were treated with UNC0638 at the indicated concentrations for 72h. Representative images from two-independent experiments were shown. (F) In vivo effect of potent G9a/GLP inhibitor UNC0642 (2.5 mg/kg) on growth of xenografts from G9a-gained/H3K9me2-high melanoma cell line K029. *p<0.05, ***p<0.001 by repeated measures two-way ANOVA with the Holm-Šidák correction for multiple pairwise comparisons of the two groups at each time point. N=5/group. (G) Representative immunohistochemistry images (40X) of H3K9me2 (top) and LC3B (bottom) in K029 melanoma xenograft tissue samples from mice treated with vehicle or UNC0642. (H) Growth of individual WM983B xenograft tumors in mice treated daily with vehicle (10% DMSO/PBS) (left, n=9) or UNC0642 (2.5 mg/kg) (right, n=10). Red lines (2/10) indicate complete tumor regressions. (I) Kaplan-Meier survival curves of WM983B-xenografted mice treated with vehicle or UNC0642 (2.5 mg/kg). **p<0.01 by the log-rank (Mantel-Cox) test.
We also tested an additional G9a/GLP inhibitor, UNC0642, with improved potency and pharmacokinetics over UNC0638 in vivo (20). This inhibitor strongly suppressed the growth of xenografted tumors of the melanoma cell line K029, which contain 3 copies of the G9a gene (Fig. 2F; Supplementary Fig. S4A). UNC0642 treatment was associated with decreased H3K9me2 and increased LC3B levels, at well tolerated drug dosing (Fig. 2G; Supplementary Fig. S4B). UNC0642 induced complete regression of 20–25% of xenografts from G9a-gained WM983B and G9a-amplified Hs944T melanoma cells (Fig. 2H; Supplementary Fig. S4C and S4D) and significantly extended survival (Fig. 2I; Supplementary Fig. S4E). Similar results were observed in pMEL* cells expressing BRAFV600E and G9a (pMEL*/BRAF/G9a) (16), which exhibit anchorage independent growth (Supplementary Fig. S4F and S4G) similar to pMEL*/NRAS/G9a (refer to Fig. 1D–F), as well as G9a-dependent tumor growth in vivo (Supplementary Fig. S4H–S4K). In contrast, the antiproliferative effect of UNC0642 was not seen in G9a diploid/wild type melanoma cell line UACC62 in vivo (Supplementary Fig. S4L).
G9a stimulates MITF expression in melanoma through canonical WNT/β-catenin signaling
To elucidate potential mechanisms by which G9a regulates proliferation and melanomagenesis, genome-wide RNA sequencing was performed in G9a-amplified Hs944T melanoma cells with and without G9a knockdown. Unexpectedly, microphthalmia-associated transcription factor (MITF), a master regulator of melanocyte development and survival that is also an amplified or mutated melanoma oncogene (16,21) was downregulated by G9a knockdown in the Hs944T cells (Fig. 3A). Consistent with this, several MITF target genes were significantly downregulated in Hs944T cells upon G9a knockdown (Fig. 3A; Supplementary Fig. S5A). MITF and its target gene MLANA were also downregulated consistently by G9a knockdown in G9a-gained WM983B and K029 melanoma cell lines (Fig. 3B; Supplementary Fig. S5B), but not in multiple G9a diploid melanoma lines (Fig. 3B; Supplementary Fig. S5C). Note that we observed unanticipated upregulation of MITF and MLANA upon G9a knockdown in some G9a diploid or heterozygous loss melanoma cell lines, Mel-juso and M14 (Supplementary Fig. S5C), suggesting differential functions of G9a or feedback regulation of G9a by MITF in these melanoma cells. Furthermore, reductions of MITF protein and H3K9me2 levels upon G9a knockdown or G9a/GLP inhibition were observed in G9a-amplified/gained melanoma cell lines, but not in G9a diploid UACC62 cells (Fig. 3C and D; Supplementary Fig. S5D). G9a copy number is positively correlated with MITF expression in the TCGA melanoma dataset (Supplementary Fig. S5E). Ectopic MITF was able to partially rescue the growth defect induced by G9a knockdown or inhibition in G9a-gained and -amplified melanoma cells, but did not affect the growth of G9a diploid/wild type melanoma cells with or without G9a knockdown (Fig. 3E; Supplementary Fig. S5F–S5H), suggesting that G9a controls survival and growth of G9a-gained/amplified melanomas through the stimulation of MITF expression.
Figure 3. G9a stimulates MITF expression in melanoma through canonical WNT/β-catenin signaling.
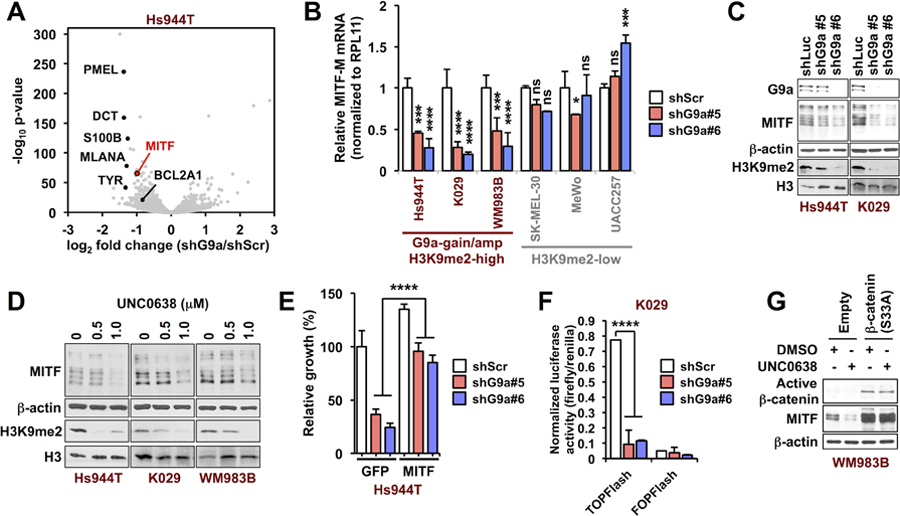
(A) Volcano plot showing genes that are significantly altered by G9a knockdown in G9a-amplified Hs944T cells. The one red and six black dots indicate MITF and several of its target genes. The whole transcriptome RNA-seq was performed in duplicate. (B) qRT-PCR for MITF-M upon G9a knockdown in G9a-amplified, -gained, and G9a diploid melanoma cells. Data represent mean +/− SD of triplicates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 vs. shScr in the same cell line by two-way ANOVA with the Holm-Šidák correction for pairwise comparisons. (See also Supplementary Fig. S4, A–C) (C and D) Western blots of MITF and H3K9me2 72h after (C) G9a knockdown and (D) pharmacological inhibition in G9a-amplified (Hs944T) or -gained (WM983B and K029) melanoma cells. Representative images from at least two independent experiments are shown. (E) Rescue of G9a-amplified or gained melanoma cells from G9a knockdown by ectopic MITF overexpression. ****p<0.0001 by two-way ANOVA after normalizing to eliminate the difference between the shScr groups, with the Holm-Šidák correction for multiple pairwise comparisons. The data represent mean +/− SD from triplicates. (F) TOP/FOPFlash transcriptional activity 72 h after G9a knockdown in G9a-gained K029 melanoma cells. FOPFlash is a control luciferase reporter with mutant TCF/LEF-binding sites. Data represent mean +/− SD of 3–4 replicates from three independent experiments. (G) Western blots of MITF and non-phosphorylated (active) β-catenin in G9a-gained WM983B melanoma cells expressing constitutively active β-catenin (S33A) or empty vector, following incubation with UNC0638 (750 nM) for 72h. P-values were calculated by one-way ANOVA with the Holm-Šidák correction for multiple pairwise comparisons.
Accumulating evidence has shown that G9a represses transcription through H3K9 dimethylation (9,11), implying that G9a is likely to elevate MITF levels indirectly in melanomas containing extra G9a copies. Several signaling pathways are known to regulate expression of MITF, and genomic dysregulation of these pathways might contribute to development of melanoma (22). To identify G9a-regulated pathways that could contribute to MITF downregulation by G9a knockdown, our RNA-seq data were analyzed by gene set enrichment analysis (GSEA). GSEA analysis with the KEGG pathway gene sets revealed enrichment of MITF-related genes in Hs944T-shScr (control) cells, including KEGG_TYROSINE_METABOLISM and KEGG_MELANOGENESIS (Supplementary Table S3). GSEA further revealed that target genes of p300 (complexes with CBP) and TCF4 (complexes with LEF1/β-catenin), both of which are key transcriptional co-factors for MITF expression (23,24), are significantly enriched among the genes downregulated by G9a knockdown (Supplementary Fig. S6A). This suggests that suppression of TCF/LEF/β-catenin or p300/CBP may be involved in MITF downregulation. Furthermore, WNT/β-catenin-upregulated genes are significantly enriched in Hs944T-shScr cells (Supplementary Fig. S6B). Activation of canonical WNT signaling has been shown to play a vital role in melanocytic development through targeting MITF (24,25). These observations suggest that G9a may activate the canonical WNT pathway by repressing known WNT antagonists. Consistent with this possibility, β-catenin target gene expression, TOPFlash luciferase activity (β-catenin-activated, TCF/LEF-dependent transcription), and nuclear β-catenin expression were all significantly decreased by G9a knockdown or G9a/GLP inhibitor UNC0638 in G9a-gained and -amplified melanoma cells (Fig. 3F and Supplementary Fig. S6C, and S6D). TOPFlash luciferase activity was not affected by G9a knockdown in G9a diploid UACC62 cells (Supplementary Fig. S6E). Importantly, MITF downregulation by UNC0638 was fully reversed by ectopic expression of constitutively active β-catenin [β-catenin (S33A)] in G9a-gained WM938B cells (Fig. 3G). Basal MITF expression was also upregulated by β-catenin (S33A) in G9a-diploid UACC62 cells, but was not affected by G9a/GLP inhibitor UNC0638 (Supplementary Fig. S6F). Therefore, G9a stimulates MITF expression through canonical WNT/β-catenin signaling in G9a copy-gained melanoma cells.
G9a activates the WNT-MITF axis by repressing WNT antagonist DKK1
To identify G9a’s target gene that can potentially repress the WNT signaling and MITF expression, we comprehensively analyzed our RNA-seq data, including G9a overexpression in pMEL*/BRAF cells and G9a knockdown in Hs944T cells. DKK1 is consistently down- and up-regulated by G9a overexpression in pMEL*/BRAF cells and G9a knockdown in Hs944T cells, respectively (Fig. 4A). We then examined a publicly available G9a ChIP-seq data set from colon cancer initiating cells (GSE82131) and found that the putative DKK1 promoter region is occupied by G9a (Fig. 4B), which we also observed by G9a ChIP-qPCR in Hs944T, but not in UACC62, melanoma cells (Fig. 4C). Furthermore, ChIP-qPCR revealed that G9a inhibition with UNC0638 reduced H3K9 dimethylation at the DKK1 promoter in Hs944T cells and increased occupancy by phosphorylated-RNApol II (pSer5, marker of active transcription) (Fig. 4D), suggesting that G9a directly represses DKK1 transcription through H3K9me2 histone modification. Consistent with the finding from the genome-wide analysis, expression of DKK1 mRNA and protein was induced by either G9a knockdown or UNC0638 drug treatment in G9a-amplified/gained melanoma cells (Fig. 4E; Supplementary Fig. S6G–S6I). Conversely, DKK1 mRNA and protein expression was repressed, while MITF, its target TRPM1, WNT target CCND1, and pigmentation increased upon G9a overexpression in G9a-unamplified melanoma cells (Supplementary Fig. S7A–S7D). G9a overexpression also suppressed DKK1 protein levels in G9a-heterogygous loss M14 melanoma cells, along with upregulation of MITF and increased β-catenin activity and H3K9 dimethylation (Supplementary Fig. S7E).
Figure 4. G9a stimulates WNT/β-catenin and subsequent MITF expression by repressing WNT antagonist DKK1 in melanoma.
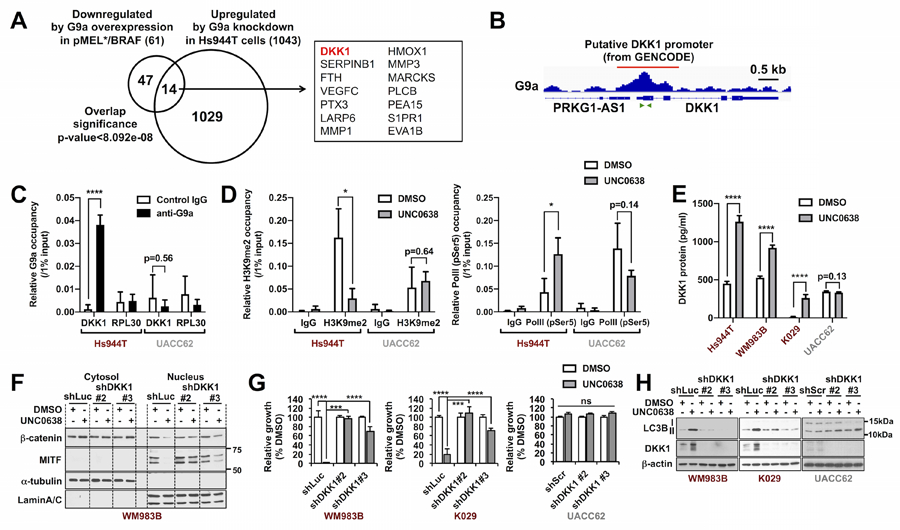
(A) Venn diagram shows genes that are downregulated by G9a overexpression in pMEL*/BRAF and upregulated by G9a knockdown in Hs944T, respectively (adjusted p-value<0.05). The 14 candidate target genes that overlap in the two datasets are shown in the box. (B) Snapshot image of G9a ChIP-seq peak in colon cancer initiating cells (GSE82131) at the putative DKK1 promoter region (from GENCODE). The publicly available dataset was visualized by IGV (ver_2.3.55). Green arrows indicate the primer set used for ChIP-qPCR in subsequent Figures 4C and 4D. (C) G9a ChIP-qPCR for DKK1 promoter in Hs944T and UACC62 cells. RPL30 (human RPL30 gene body (exon 3)) serves as a negative control. (n=3 from two independent experiments). (D) ChIP-qPCR of (left) H3K9me2 and (right) phosphorylated-RNA-polymerase II (pSer5) at the DKK1 promoter region in Hs944T cells. Cells were treated with DMSO or UNC0638 (500 nM) for 72 h and subjected to H3K9me2-ChIP or Pol II (pSer5)-ChIP. (n=3–4 from two independent experiments). (E) ELISA for secreted DKK1 levels after UNC0638 (500 nM) for 72 h in G9a-amplified (Hs944T), -gained (WM983B and K029), and G9a diploid (UACC62) melanoma cells. Data represent mean +/− SD of 3–4 replicates. (F) Western blots of cytosolic and nuclear β-catenin and MITF expression in UNC0638-treated WM983B-shLuc and -shDKK1 cells. α-tubulin and LaminA/C served as internal controls for the cytosolic and nuclear fractions, respectively. Representative images from one of two independent experiments are shown. (G and H) Growth measured by CellTiter-Glo assay (n=4) (G) and western blot of autophagy marker LC3B (representative images from one of two independent experiments are shown) (H) in WM983B, K029 and UAC62 cells stably expressing shLuc or shDKK1 hairpins, after 750 nM UNC0638 treatment for 72 h. P-values were calculated by unpaired, two-tailed T tests with the Holm-Šidák correction for multiple comparisons (C, D and E) or by two-way ANOVA with the Holm-Šidák correction for multiple pairwise comparisons (G). *p<0.05, ***p<0.001, ****p<0.0001.
To determine whether DKK1 is required for the observed WNT signaling inactivation, MITF downregulation, and growth inhibition in UNC0638-treated G9a-gained/amplified melanoma cells, two individual shRNAs targeting DKK1 were tested. In WM983B and Hs944T cells, UNC0638-induced reductions of nuclear β-catenin and MITF, as well as upregulation of DKK1 mRNA and downregulation of MITF RNA, were fully and largely reversed by shDKK1#2 and shDKK1#3, respectively (Fig. 4F and Supplementary Fig. S7, F and G). In G9a diploid/wild type UACC62 cells, on the other hand, both cytosolic and nuclear β-catenin and MITF protein levels were not affected by UNC0638 with or without DKK1 knockdown (Supplementary Fig. S7G). Moreover, the growth inhibitory effects of UNC0638 on G9a-gained WM983B and K029 melanoma cells were correspondingly completely or largely reversed by shDKK1#2 and shDKK1#3, respectively (Fig. 4G), as was upregulation of LC3B-II by UNC0638 (Fig. 4H), however neither of those effects of UNC0638 were observed in UACC62 melanoma cells with or without DKK1 knockdown (Fig. 4G and H). Conversely, overexpression of DKK1 in G9a-gained and -amplified melanoma cells was sufficient to decrease active β-catenin and MITF levels as well as growth (Supplementary Fig. S7, H and I). Of note, the G9a G1069L and G1069W mutants have stronger impacts on the repression of DKK1 and the induction of MITF than wild-type G9a (Supplementary Fig. S7, J and K). Taken together, these results suggest that G9a-mediated repression of DKK1 induces activation of the WNT/β-catenin-MITF axis and thereby enhances the growth potential of G9a copy number-gained or mutated melanomas.
G9a-DKK1-WNT axis is conserved across multiple cancer types beyond melanoma
Almost one third of primary human melanoma specimens have been reported to display nuclear β-catenin accumulation without evidence of somatic mutations within the β-catenin gene (26) or other WNT pathway-related genes (27). G9a copy number gains correlate significantly with higher WNT signature scores (p=0.0060, Supplementary Fig. S8A) and occur mutually exclusively with other known genetic alterations within the β-catenin destruction complex, such as loss-of-function mutations or deletions in negative regulators of the WNT pathway (APC, AXIN1, and FAT1 (28)) and gain-of-function mutations in β-catenin (Supplementary Fig. S8B). Our study links G9a-mediated epigenetic silencing of DKK1 with aberrant WNT/β-catenin activation in melanoma cells and implies that G9a genetic alterations may account for such activation in some melanomas that do not harbor intrinsic WNT pathway somatic mutations that lead more directly to β-catenin accumulation.
The WNT signaling pathway has been strongly implicated in tumorigenesis of a wide variety of malignancies beyond melanoma (27), prompting us to examine potential relationships to G9a. We observed the same G9a-DKK1 inverse correlation in CCLE melanoma and multiple non-melanoma cancer cell panels, including lung, colon, pancreatic, glioma, and sarcoma (Fig. 5A). Furthermore, GSEA revealed significant positive correlations between G9a and multiple WNT target gene signatures in melanoma and multiple non-melanoma cancer cell lines (Supplementary Fig. S8C). In particular, all of the CCLE cell line panels that showed inverse G9a-DKK1 correlations displayed positive correlations with the SANSOM_APC_TARGETS_REQUIRE_MYC gene set (Fig. 5B). Intriguingly, consistent with the strong susceptibility of G9a-amplified melanoma to G9a inhibition (Fig. 2), sensitivity to G9a inhibitor BIX-01294 among 325 cancer cell lines in the Cancer Therapeutics Response Portal (http://portals.broadinstitute.org/ctrp.v2.2/), including melanoma, lung cancer, colon cancer, glioma, pancreatic cancer, and sarcoma, correlates significantly with G9a mRNA level and copy number (Fig. 5C). Several CCLE non-melanoma cell line panels that did not show a significant correlation between G9a and DKK1 expression still show strong correlations between G9a expression and multiple WNT target signatures (Supplementary Fig. S8D), suggesting G9a might activate the WNT signaling pathway through other mechanisms in these cancer types, such as suppression of other WNT antagonists. These bioinformatic analyses suggest that the G9a-WNT signaling axis is highly conserved and G9a potentially contributes to tumorigenesis by activating WNT signaling in a variety of cancers, not limited to G9a-amplified or -mutated melanomas.
Figure 5. G9a-DKK1-WNT pathway is highly conserved across cancers and associated with a ‘cold’ tumor immune microenvironment.
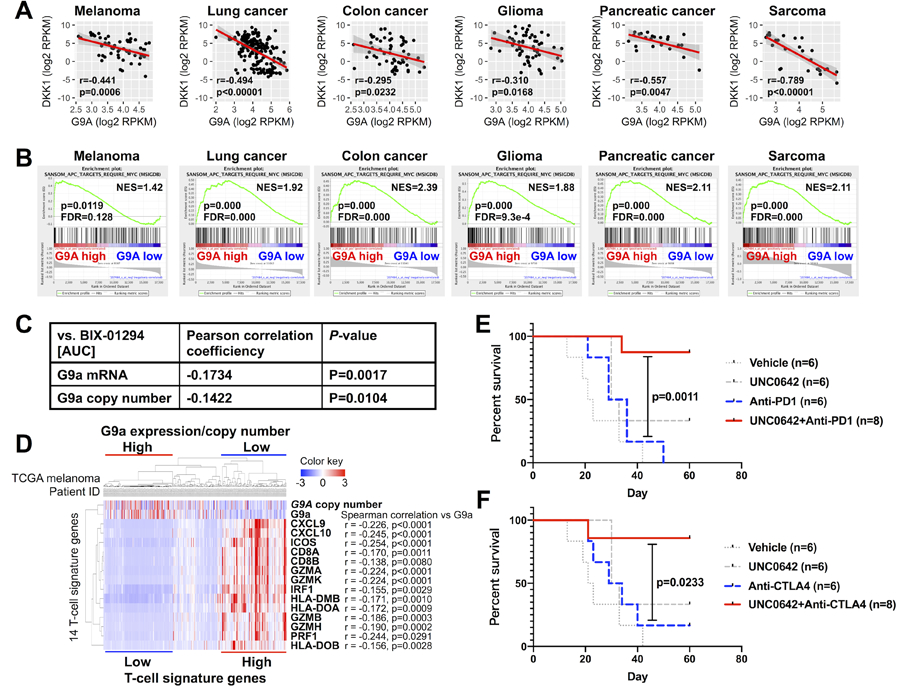
(A) Correlations between mRNA expression levels of G9a and DKK1 in the indicated CCLE cancer cell line datasets. Each dot indicates one cell line in the dataset. (B) GSEA showing correlations between G9a expression and the WNT target gene set SANSOM_APC_TARGETS_REQUIRE_MYC (from MSigDB), in the same CCLE datasets as in (A). (C) Correlation between sensitivity to G9a inhibitor BIX-01294 (area-under-the-curve metric) and G9a mRNA level or copy number across cancers available in the CTRPv2 dataset. (D) Hierarchical clustering of 367 TCGA melanoma patients with average linkage by G9a copy number/expression and Spranger T-cell signature genes. Correlations between G9a expression and each T-cell signature gene were analyzed by Spearman’s rank correlation. (E and F) Kaplan-Meier plots showing overall survival of mice harboring D4M.3A.3-UV3 tumors and treated with vehicle (n=6; gray dotted line), UNC0642 (5 mg/kg) (n=6; gray dashed line), anti-PD-1 (F) or anti-CTLA-4 (G) (n=6; gray dashed line), or combination therapy (UNC0642 + either anti-PD-1 or anti-CTLA-4) (n=8; black solid line). P-values were calculated by the log-rank (Mantel-Cox) test.
Recently, various molecular and/or genetic alterations in specific cancer cell-intrinsic oncogenic pathways have been reported to affect the degree of T cell infiltration into a given tumor, which correlates with response rate to immune-based therapeutics (29). In melanoma, active β-catenin was implicated in a poorly immunogenic or ‘cold’ tumor immune microenvironment (e.g., poor recruitment of CD8+ T-cells) and resistance to immune checkpoint (30). On the other hand, another study utilizing a murine engineered melanoma model did not observe the same correlation (31). We therefore examined this question for G9a/WNT activated tumors. We found that G9a expression and copy number gain correlate inversely with T-cell signatures [both Spranger T-cell signature (30) and Ayers expanded immune signature (32)] in the TCGA melanoma dataset (Fig. 5D; Supplementary Fig. S8E, S9A and S9B). The correlations of MITF with some of the immune signature genes, in particular Th1 cytokines/chemokines (e.g., CXCL9, CXCL10, CXCL11, IFNG and STAT1), are weaker than those of G9a with these genes (Fig. 5D and Supplementary Fig. S9A). We observed some melanoma cases with G9a amplification that express low MITF along with low T-cell signatures, suggesting that G9a-induced immune suppression may be mediated by WNT/β-catenin (upstream of MITF), but not by MITF. Also, in non-melanoma cancers, inverse correlations of G9a and CD8+ T-cell infiltration are observed (Supplementary Fig. S9B), and are consistent with the functional immune suppressive role of G9a reported in bladder cancer (33).
To further interrogate this question with an independent melanoma dataset, 276 primary melanoma specimens obtained from Northern England (the Leeds Melanoma Cohort- LMC) were molecularly annotated (see Methods) and analyzed for G9a genomic copy number, G9a expression, immune inflammatory signature, and patient outcomes. G9a copy number correlated positively with G9a gene expression, R=0.4, P=4.4×10−13 (Supplementary Fig. S10A). This observation reassured us that further analyses focusing only on copy number alterations were justifiable. Participants whose tumors had high G9a copy numbers (highest quartile, N=70) had significantly worse prognoses compared to those with low G9a copy number tumors (lowest quartile, N=69): HR=2.5, P=0.001, 95% CI 1.4–3.9 (Supplementary Fig. S10B). Six immunologically different clusters (Consensus Immune Clusters - CICs) were previously reported among the LMC tumors (34), using the immune gene list adapted from Bindea et al. (35). One of these clusters (CIC 4) was a subset of tumors characterized as “cold”. CIC4 was depleted of immune signals (imputed T cell, dendritic cell, and cytotoxic cell scores), had significantly increased WNT/β-catenin pathway signaling and the worst survival. On the contrary, CIC2 was identified as immune rich, with reduced WNT/β-catenin signaling and the best prognosis (CIC 2). We therefore tested if tumors with a high G9a copy number were associated with these clusters. Indeed, we found that 69% of CIC4 tumors (“cold”/high β-catenin subgroup) had a high G9a copy number, a higher percentage than in all other subgroups, Chi2 p=0.017 (Supplementary Fig. S10C). We also performed a whole transcriptome comparison between tumors with high and low G9a copy number, to identify genes/pathways that are differentially expressed between these two tumor groups. Among the pathways that were significantly more highly expressed in G9a high tumors, Wnt signaling was agnostically identified as a top correlate (FDR=0.001) (Supplementary Fig. S10D). In a separate whole transcriptome comparison between tumors with high G9a copy numbers (highest quartile, N=70) and all of the other tumors in the Leeds cohort (2nd, 3rd, and 4th quartiles, N=206), Wnt signaling was again identified as a top correlate (FDR = 2.19 × 10−6) (Supplementary Table S4).
In another publicly available clinical melanoma dataset, higher G9a mRNA expression is significantly associated with worse response to anti-CTLA4 therapy (36) (Figure S10E). Also, G9a expression shows a tendency to be inversely associated with median survival rate in response to anti-PD1 in melanoma patients in two studies (37,38) (Fig. S10F, G9a high vs. low: 542 days vs 718 days). Thus, G9a expression can be a predictable biomarker for the response to immune checkpoint blockade. Finally, to examine the functional impact of a G9a/GLP inhibitor on the response to immune checkpoint blockade, we tested combinatorial therapies of UNC0642 with either anti-PD1 or anti-CTLA4 in a syngeneic mouse melanoma model using the G9a wild type D4M.3A.3-UV3 cell line (see Methods). The G9a inhibitor significantly increased complete regression rates to either anti-PD1 or anti-CTLA4 and extended survival in the mouse model (Fig. 5E and F; Supplementary Figure S10G and S10H). This result raises the potential that G9a inhibition could improve clinical responses to those immune checkpoint inhibitors in patients with melanoma.
Discussion
The relevance of genetic alterations in epigenetic modulators in cancer has been emphasized by discoveries of high-frequency mutations and copy number changes (39), suggesting the involvement of epigenetic dysregualtion in cancer development. In melanoma, H3K9 methylation/demethylation is likely a key epigenetic modifier of transformation from melanocytes to malignant melanoma (17,40). G9a is a major H3K9me1/2 histone methyltransferase of euchromatin and is often upregulated in different types of cancers. It has also been suggested to mediate aberrant proliferation and metastasis in multiple cancers, however, genomic abnormalities that could activate G9a’s oncogenic activity have not previously been identified. Our present study provides evidence that genetic modifications of G9a, including mutations within the SET domain and copy number gain/amplification, cause elevated global H3K9me2 levels and accelerate melanomagenesis in conjunction with BRAF(V600E) and NRAS(Q61R) both in vitro and vivo. These data strongly support the model that G9a-G1069 mutations and G9a copy number gain are drivers of melanomagenesis.
Recurrent gain-of-function mutations within the SET domain have been reported to constitutively activate enzymatic activity alone and/or in epigenetic regulatory complexes. For instance, EZH2 Y641, a key component of the catalytic center for the methyltransferase reaction, has been found to be mutated in diffuse large B cell lymphoma, follicular lymphoma, and melanoma, and promotes oncogenic events in association with high H3K27 trimethylation levels at target genes of polycomb repressive complex 2 (PRC2) (5). In human G9a, the corresponding catalytic tyrosine site, Y1067, is located within the same active site domain as the G1069 residue. G1069 is located adjacent to the histone binding pocket and probably does not physically interact with the histone tail, as shown in a study using structural model analysis of H3K9 HMTs (41). However, replacement of this glycine by a larger hydrophobic non-polar residue (Leu or Trp) that faces the histone-binding pocket is likely to enhance the hydrophobicity of this active site pocket, thereby potentially affecting activity and/or histone binding potential.
Due to the change in the histone binding pocket of G9a caused by the G1069 recurrent mutations, the mutant proteins have lost basal catalytic activity, but can induce higher H3K9 methylation than wild type G9a when complexed with binding partner GLP in biochemical assays and in melanoma cells. Similar observations have been made with another SET domain-containing methyltransferase mutant, MLL1 S3865F, the activity of which is also stimulated by its binding partners WDR5/RBBP5/ASH2L (WRA) via an allosteric mechanism (6). In addition, MLL3 Y4884C exhibits higher catalytic activity in the WRA complex than wild type MLL3 complexed with WRA (7). Besides the SET domain-containing methyltransferases, the catalytically inactive DNA methyltransferase-like protein DNMT3L interacts with the catalytic domain of DNMT3A and specifically recruits the DNMT3A-DHMT3L heteromeric complex to unmethylated H3K4, demonstrating that DNMT3L has dual functions of binding the unmethylated histone tail and activating DNA methyltransferases (42). The SET domains of G9a and GLP are required for heterodimer formation (19), and the G9a G1069L/W mutations do not disturb the interaction with GLP (Supplementary Fig. S1C). Therefore, the G9a G1069L/W mutations may induce higher levels of H3K9 methylation due to altered binding potential to histone tails (H3K9me0/1/2 modified histone tails) (Supplementary Fig. S1E) and/or allosteric mechanisms within the GLP-containing complex.
In addition to the somatic recurrent mutations of G9a, we identified G9a copy gains in a significant proportion of TCGA melanomas (3 or more copies of G9a in 25.8% of TCGA melanoma patients). We recapitulated this in primary melanoma tumors from the Leeds Melanoma Cohort and showed that copy number is associated with gene expression in that cohort. Furthermore, in an agnostic interrogation of genes differentially expressed in the G9a high vs. G9a low primary tumors, WNT signaling is one of the most strongly upregulated pathways in the G9a high tumors. Our analysis revealed that one or more extra copies of G9a are strongly associated with higher global H3K9me2 levels and dependence on G9a for survival in melanoma cells. Future studies should examine whether elevated H3K9me2 levels predict sensitivity to agents targeting G9a. More recently, frequent G9a copy number gains have also been in hepatocellular carcinoma (50% with 3 copies and 10% with 4 or more copies), and HCCs that express high levels of G9a are dependent on its activity (12), suggesting that G9a-targeted therapy could be applicable for patients with non-melanoma cancers.
There has been a long-standing question of how the WNT/β-catenin pathway is activated in the many melanomas that lack intrinsic pathway mutations (26). Here we find that G9a-mediated DKK1 silencing activates the WNT/β-catenin-MITF axis to promote melanomagenesis. On the other hand, genetic dysregulation of EZH2 has recently been reported to activate WNT/β-catenin signaling and metastasis by promoting cilium disassembly and subsequent nuclear translocation of β-catenin (43). GISTIC analysis revealed that copy number gain/amplification of G9a (chromosome 6p21) and EZH2 (chromosome 7q34) genomic loci preferentially occur in NRAS- and BRAF-mutated melanoma subsets, respectively (44), suggesting mutual exclusivity of G9a and EZH2 gain/amplification in melanoma patients. Another epigenetic modifier, BRCA1-associated protein-1 (BAP1), is frequently somatically inactivated in cutaneous melanoma, uveal melanoma, renal cell carcinoma and malignant mesothelioma, and highly-penetrant germline BAP1 mutations predispose to those malignancies (45). As for the reported tumor suppressive mechanism, BAP1 can antagonize EZH2/PRC2 or RING1B (RNF2)/PRC1 in a tissue specific manner (46,47). For instance, while loss of Bap1 activates intrinsic apoptosis in several mouse cell types (hepatocytes, keratinocytes, fibroblasts, and embryonic stem cells) in an RNF2-dependent fashion, the Bap1 loss enhances proliferation of melanocytes in association with upregulation of lineage-specific oncogenes MITF and BCL2, independently of RNF2 (48). Therefore, these epigenetic modifiers may share a common endpoint of stimulating WNT signaling and MITF in melanoma.
While recent advances in immunotherapy have dramatically improved clinical prognosis of melanoma, substantial proportions of patients exhibit treatment resistance (49). Beyond its oncogenic potential in melanoma, WNT/β-catenin signaling confers multiple aspects of malignant phenotypes, including metastasis (50), acquired resistance to BRAF inhibitor (51), and immune evasion (30). Intriguingly, G9a inhibitor was synergistic with immune checkpoint blockades in a murine melanoma model (Fig. 5E and 5F). Taken together, these studies identify G9a as a recurrently mutated and gain-of-function oncogene in melanoma, and also demonstrate its functional role in stimulating WNT-mediated oncogenicity, a behavior that appears to be shared among melanoma and multiple non-melanoma malignancies. Attempts at targeting the WNT pathway pharmacologically have been underway and will be important to develop further. In addition, given the druggability of G9a, this pathway could represent a new therapeutic opportunity both for direct targeting and potentially to enhance immunotherapy efficacy for certain cancers.
Methods
Whole exome sequencing datasets
The mutation annotation files of The Cancer Genome Atlas (TCGA) and 15 publicly available whole exome sequencing datasets were downloaded from (https://gdac.broadinstitute.org) (See Supplementary Table S1). Non-synonymous G9a mutations were counted in each dataset and the total cases found in the 16 datasets are summarized in Figure 1A. To evaluate whether the frequency of non-synonymous mutations at G9a G1069 is significantly higher than would be expected if the mutation were neutral (median mutation rate of melanoma:14.4 coding mutations per megabase (39), we computed a one-sided p-value using the dbinom function (Poisson distribution model) in the R statistical software as described previously (52).
GISTIC and G9a copy number analysis
All downloadable batches (180, 198, 206, 240, 262, 277, 291, 316, 332, 358, 388, 393, 408 and 416) of level 3 processed SNP 6.0 array datasets of Skin Cutaneous Melanoma (SKCM) were obtained from the legacy database of The Cancer Genome Atlas (https://tcga-data.nci.nih.gov/docs/publications/tcga/). All of the SNP array data were compiled in one segmentation file and used for further Genomic Identification of Significant Targets in Cancer (GISTIC) analysis. GISTIC analysis was carried out by the GISTIC 2.0 pipeline (GenePattern, https://genepattern.broadinstitute.org/).
The putative G9a copy number data of 287 TCGA human melanomas were obtained from cBioportal (http://www.cbioportal.org). Based on their analysis, the melanomas were ordered according to the G9a copy number (regardless of focality of the G9a gain or amplification) and the proportion of melanomas harboring G9a copy number gain (3 or more G9a copies) and amplification (4 or more copies) were tallied.
The G9a copy number of melanoma cell lines was determined by genomic DNA quantitative PCR (qPCR). Genomic DNAs of melanoma cells and primary human melanocytes were isolated using DNeasy Blood & Tissue Kit (Qiagen). Primers used for copy number analysis are shown in Supplementary Table S5. The comparative cycle threshold method was used to quantify copy numbers in the samples. Results were normalized to the repetitive transposable element LINE-1 as described previously (16). The relative copy number level was normalized to normal genomic DNA from primary human melanocytes as calibrator.
Protein alignment and visualization
Amino acid sequences of SET domains of histone methyltransferases were obtained from NCBI (https://www.ncbi.nlm.nih.gov) and aligned using the ClustalX algorithm. The co-crystal structure of G9a and H3 peptide was obtained from the RCSB Protein Data Bank (PDB: 5jin) and visualized using JSmol (https://www.rcsb.org).
Plasmid and mutagenesis
pLenti CMV GFP Blast (659–1) was a gift from Eric Campeau & Paul Kaufman (Addgene plasmid # 17445) and pLenti6-MK1-EHMT2-V5 was a gift from Bernard Futscher (Addgene plasmid # 31113). Mutagenesis was performed using a QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA) with specific primer pairs (Supplementary Table S5), resulting in mutation of G1069 to L or W in G9a. The resulting mutant sequences were confirmed by conventional Sanger sequencing at the MGH DNA core. The full length human G9a wild-type and mutant cDNAs were amplified from the pLenti6-MK1-EHMT2-V5 vector and cloned into pGEX6p1 (GE Healthcare) using the primers indicated in Supplementary Table S5. G9a WT and G1069 mutant cDNAs were also cloned into the pENTR-D/TOPO cloning vector (Thermo Fisher) and subsequently used to establish MiniCoopR vectors for the zebrafish melanoma model as described below (see Zebrafish melanoma model and MiniCoopR system). GFP and MITF were respectively cloned into the pCW45 lentiviral expression vector (Dana-Farber/Harvard Cancer Center DNA Resource Core) as described previously (53). Human DKK1 cDNA was amplified from discarded human foreskin and cloned into the pCW45 vector. pLenti-hygro-hTERT, pLenti-hygro-CDK4(R24C), and pLenti-hygro-NRASQ61R were gifts from Ryo Murakami (Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School). All pLKO.1-shRNA constructs were obtained from the Molecular Profiling Laboratory (Massachusetts General Hospital Center for Cancer Research). pMD2.G and psPAX2 were gifts from Didier Trono (Addgene plasmid # 12259 and 12260).
Lentivirus generation and infection
Lentivirus was generated in Lenti-X™ 293T cells (Clontech, 632180). The Lenti-X cells are transfected using 250 ng pMD2.G, 1250 ng psPAX2, and 1250 ng lentiviral expression vector in the presence with PEI (MW:25K). For infection with lentivirus, 0.1–1 ml of lentivirus-containing media was used in the presence of 8 μg/ml Polybrene (Sigma). Selection was performed the day after infection with puromycin (1 μg/ml) or blasticidin (5 μg/ml).
Preparation of GST-fused recombinant G9a
GST-tagged G9a (GST-G9a) wild-type and G1069 mutants were expressed in BL21 (DE3) competent cells (Clontech #C2527H) using pGEX6p-G9a constructs. Briefly, the day after transformation with pGEX6p-G9a, a single clone was expanded at 37°C until OD600 reached 0.4–0.6 and further cultured in the presence of 0.5 μM IPTG overnight at room temperature. The BL21 cells were then lysed by sonication in lysis buffer [100 mM NaH2PO4, 10 mM Tris-HCl (pH8.0)] supplemented with 1 mM lysozyme, 1 mM PMSF and protease inhibitors (Roche). Soluble proteins were collected by centrifugation (12,000 rpm, 10 min, 4°C) and applied to GST spin columns (GST Spin Purification Kit, Thermo Scientific Pierce) according to the manufacture’s instruction. The purified protein fractions were subsequently subjected to Amicon® Ultra 50K devices to concentrate GST-fused G9a proteins and replace the buffer with Mg2+- and Ca2+-free PBS. GST-G9a protein concentrations were determined by Bradford protein assay (Pierce) and Coomassie Brilliant Blue (CBB) staining. GST-G9a aliquots were stored at −80°C before use.
In vitro methyltransferase and pull-down assay
In vitro methyltransferase assays were performed using an MTase-Glo™ kit (Promega) according to the manufacturer’s instructions. 10 ng/well GST-G9a, 30 ng/well histone substrate [unmodified H3 peptide (Abcam, ab7228)], H3K9-modified peptides (Epigentek, R-1024, 1026, and 1028), recombinant human histone H3 (Abcam, ab198757), or human native nucleosome (Thermo Fisher, 141057)], and 2 μM S-adenosyl methionine (SAM) were incubated with or without recombinant human GLP (Sigma, SRP0383) in the reaction buffer (50 mM Tris-HCl, pH8.1, 5 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1 mM PMSF, and 1% DMSO) for 1 h at room temperature. After stopping the reaction, the luminescence readout was measured using an EnVision 2104 Multilabel Reader (PerkinElmer).
The pull-down assay for recombinant GST-G9a and histone H3 peptides was carried out using a Pull-Down Biotinylated Protein:Protein Interaction Kit (Thermo Fisher) with biotinylated histone H3 (1–21) or H3K9-methylated (me1 or me2) H3 tail peptides (Epigentek), according to the manufacturer’s protocol. After elution, histone H3-interacting GST-G9a WT and mutant proteins were visualized by immunoblot using anti-GST antibody (ab9085).
Protein sample preparation
After the in vitro methylation reaction of recombinant H3 protein as described above, an equal volume of 2x Laemmli sample buffer was added to the reaction mixture, which was subsequently used for western blotting and CBB staining.
Whole cell lysates were prepared using lysis buffer (25 mM HEPES pH7.7, 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100) supplemented with protease inhibitors. Nuclear and cytoplasmic proteins were fractionated using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, 78833) according to manufacturer’s protocols. Histone proteins were extracted by salt extraction buffer (50 mM Tris-HCl, pH7.6, 0.5M NaCl, 1 % deoxycholic acid, 1 % SDS and 2 mM EDTA) with protease inhibitors. Protein concentrations were quantified by the Bradford protein assay (Thermo Fisher, 23236).
Nuclear protein fractions were prepared using a Nuclear Complex Co-IP Kit (Activemotif). Briefly, after extraction of nuclear proteins, protein samples were pre-cleared with control IgG and Pierce Protein A/G UltraLink Resin (Life Technologies, 53133) with 0.25% BSA. Pre-cleared samples were incubated with 2 μg of anti-V5 antibody (Abcam, ab27671) or non-specific normal mouse IgG (Santa Cruz Biotechnology, sc-2025) at 4°C overnight and then rotated with Pierce Protein A/G UltraLink Resin at 4°C for 4h. The beads were washed three times and subsequently eluted according to the manufacturer’s protocol.
Western blotting
Protein samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked in 3% BSA buffer (10 mM Tris-HCl, pH7.4, 150 mM NaCl, 1 mM EDTA, 0.1% Tween-20 and 3% BSA). Primary antibodies used for western blotting were: anti-H3K9me1 (Cell Signaling, #14186), anti-H3K9me2 (Cell Signaling, #4658), anti-H3K9me3 (Wako Diagnostics/Chemicals, 309–34839), anti-H3K27me1 (Cell Signaling, #7693), anti-H3K27me2 (EMD Millipore, #04–821), anti-H3K27me3 (Abcam, ab6002), anti-total H3 (EMD Millipore, 06–755), anti-V5 (Abcam, ab27671), anti-phospho ERK1/2 (Cell Signaling, #4370), anti-ERK1/2 (Cell Signaling, #4695), anti-G9a (Cell Signaling, #3306), anti-GLP (Abcam, ab135487), anti-LC3B (Cell Signaling, #3868), anti-MITF (C5, in-house), anti-active β-catenin (Cell Signaling, #8814), β-catenin (Cell Signaling, #9587), anti-DKK1 (Santa Cruz, sc-374574), anti-β-actin (Santa Cruz, sc-47778), anti-α-tubilin (Sigma Aldrich, T9026), anti-Lamin A/C (Cell Signaling, #4777) and anti-Lamin B (Cell Signaling, #12586). Appropriate secondary antibodies were used in 5% skim milk/TBST buffer. Protein bands were visualized using Western lightning plus ECL (Perkin Elmer) and quantified using ImageJ software.
Zebrafish melanoma model and MiniCoopR system
Experiments were performed as published previously (17). In brief, p53/BRAF/Na one-cell embryos were injected with 20 ng/μl of control or experimental MiniCoopR (MCR) DNA along with tol2 RNA for integration. Control vectors expressed EGFP. Embryos were sorted for melanocyte rescue at 5 days post-fertilization to confirm vector integration. Equal numbers of melanocyte-rescued embryos were grown to adulthood. Twenty fish were raised per tank to control for density effects. Raised zebrafish were scored for the emergence of raised melanoma lesions as published (17).
Zebrafish were anesthetized in 0.16 g/L tricaine solution (MS-222) and oriented in an imaging mold (2% agarose in 1 × PBS). Zebrafish were photographed at 10 weeks post-fertilization via brightfield microscopy (Nikon DS-Ri2). Maximum backlight and LED illumination (NII-LED) settings were utilized to distinguish melanocytes from iridophores.
DKK1 ELISA
Concentrations of secreted DKK1 in culture supernatant were determined using a Human DKK1 Quantikine ELISA Kit (R&D systems, DKK100) according to the manufacturer’s protocols. Briefly, after lentivirus-mediated infections with shG9a- or G9a/V5-expressing vector and proper selections with antibiotics, equal numbers of infected cells were re-plated in 96-well plates. After 72 h of additional culture, the culture supernatants were harvested and subsequently subjected to ELISA. Also, culture supernatants were harvested from DMSO- and UNC0638-treated cells 72 h after treatment. All of the supernatant samples were stored at −80°C after removal of cell debris by centrifugation.
Melanoma cell lines and compounds
Hs944T, MeWo, SK-MEL-3 and SK-MEL-28 cells were obtained from ATCC. The WM983B cell line was kindly provided by Meenhard Herlyn (The WISTAR Institute). The K029 cell line was a gift from Dr. Stephen Hodi (DFCI). UACC257, UACC62, MALME3M, LOX-IMVI and M14 cells were obtained from the NCI, Frederick Cancer Division of Cancer Treatment and Diagnosis (DCTD) Tumor Cell Line Repository. SK-MEL-30 and SK-MEL-119 cells were from Memorial Sloan Kettering Cancer Center. MEL-JUSO and MEL-HO cells were from DSMZ. COLO792 cells were purchased from Sigma Aldrich. LB373-MEL cells were from Ludwig Institute of Cancer Research. The VM10 cell lines was established at the Institute of Cancer Research, Medical University of Vienna. UACC257, UACC62 and LOX-IMVI melanoma cell lines were cultured in RPMI 1640 supplemented with 1% penicillin/streptomycin/L-glutamine and 9% FBS in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The other melanoma cell lines were maintained in DMEM with 1% penicillin/streptomycin/L-glutamine and 9% FBS. Human primary neonatal melanocytes were prepared from discarded foreskins and maintained in TIVA medium (F12 medium with 1% penicillin/streptomycin/L-glutamine, 8 % FBS, 50 ng/ml TPA, 225 μM IBMX, 1 μM Na3VO4 and 1 μM dbcAMP). Most of the melanoma cell lines have been authenticated by our lab using ATCC’s STR profiling service. The following cell lines have not been authenticated because no STR profile information for them was found in any cancer cell line data bank: K029, SK-MEL-119 and VM10.
The C57BL/6 syngeneic mouse melanoma cell line D4M.3A was a gift from David Mullins (Dartmouth Geisel School of Medicine), and from it a single cell clone D4M.3A.3 was derived. D4M.3A.3-UV3 cells were generated by sequentially irradiating D4M.3A.3 cells in culture three times with 25 mJ/cm2 UVB followed by isolation and propagation of single cell clones from the surviving population (Lo et al, manuscript submitted). The UV3 clone was shown by whole exome sequencing to carry 87 mutations/Mb (including a synonymous G9a mutation: c.3322T>C), comparable to somatic mutation rates in human melanomas, and similar expression of G9a (120 transcripts/million), PD-L1, PD-1, and MHC class I and II relative to parental D4M.3A.3 cells (Lo et al, manuscript submitted). D4M.3A.3-UV3 cells were cultured in DMEM with 1% penicillin/streptomycin/L-glutamine and 10% FBS.
UNC0638 was purchased from Cayman Chemical (#10734) and reconstituted with DMSO. UNC0642 was provided from Dr. Jian Jin for in vivo experiments. Bafilomycin A1 was purchased from EMD Millipore.
Soft agar assay using primary human melanocytes and melanoma cells
Primary human melanocytes were immortalized by simultaneous lentivirus-mediated infections with pLenti-hTERT, pLenti-CDK4(R24C) and pLenti-p53DD (gifts from Ryo Murakami), followed by hygromycin selection for 3 days and culture for an extended period of time (>30 days) in TIVA media with hygromycin. The resulting polyclonal populations of pMEL/hTERT/CDK4(R24C) cells were termed pMEL* in this study. The pMEL* cells were infected with pLenti-GFP, -G9a WT, -G9a G1069L or -G9a G1069W. After selection with blasticidin for 1 week, these infected pMEL* cells were subsequently infected with pLenti-NRASQ61R and selected by growth-factor deprivation in F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin/L-glutamine. BRAFV600E-expressing pMEL* cells were established as described previously (16). Also, G9a-gained/amplified melanoma cell lines Hs944T and K029 were infected with shG9a hairpins, followed by puromycin selection for 5 days. Following these lentivirus infections, pMEL* and melanoma cells were subjected to a soft agar assay. Briefly, cells (5,000 cells/well in a 24-well plate) were resuspended in 0.1% agarose-containing DMEM with 10% FBS and 1% penicillin/streptomycin/L-glutamine and plated on bottom agar consisting of 0.75% agarose in DMEM. 21 days after culture in the soft agar, whole well images were obtained and analyzed for total colony numbers using CellProfiler software (Size: 5–1000, Circularity: 0.2–1).
Transformation assay
NIH3T3 cells were plated in 6-cm dishes (2 × 106 cells per well) and cultured until the confluency reached approximately 80–90%. The monolayer cells were then infected with control GFP, wild type G9a, or G1069L/W-mutated G9a lentiviral construct. A day after infection, the lentivirus medium was replaced with fresh regular culture medium and cultured for an additional 10 days. The medium was refreshed every other day. Finally, the cells were fixed with 4% PFA and colonies were visualized by staining with 0.05% crystal violet. Visible macroscopic colonies were counted manually.
Cell viability assay
The growth potential of melanoma cells was determined by colony formation assay. Briefly, 72 h after lentivirus infections with shRNAs, equal numbers (10,000 cells/well) of melanoma cells were re-plated in a 12-well plate and further cultured for 7 days. Cell number was estimated by crystal violet staining followed by extraction with 10% acetic acid and measurement at 595 nm using a spectrophotometer (FLUOstar, Omege, BMG LABTECH).
The effect of G9a inhibitor UNC0638 on cell viability was evaluated by CellTiter-Glo assay (Promega) and measurement of luminescence using an EnVision 2104 Multilabel Reader (PerkinElmer). Melanoma cells and primary human melanocytes were plated in 96-well black plates (2,000 cells/well) (Thermo Fisher, 07200565) and treated with titrated doses of UNC0638 (0 to 5 μM) for 72 h. IC50s of UNC0638 were calculated in GraphPad Prism.
In vivo xenograft and syngeneic tumor studies
Female hairless SCID mice (crl:SHO-Prkdcscid Hrhr) aged 5–8 weeks were purchased from Charles River Laboratories. Transformed pMEL* cells expressing BRAFV600E and either GFP or wild type G9a were inoculated subcutaneously at bilateral flank positions (1 × 106 cells in 100 μl PBS(–) per site). Palpable tumor establishment was monitored twice per week and terminated after 8 weeks. Mice harboring palpable pMEL*/BRAFV600E/G9a tumors were subsequently used to test the potency of UNC0642. For longitudinal tumor treatment studies, 5 × 106 K029, WM983B, Hs944T or UACC62 melanoma cells in 100 μl PBS(–) were injected subcutaneously into bilateral flanks. Once tumors reached 50 mm3, mice were randomly sorted into treatment and control groups ensuring similar initial tumor size. Mice were treated with 2.5 mg/kg UNC0642 or vehicle [10% DMSO/90% PBS(–)] 3 times per week. For syngeneic mouse models, eight-week-old female c57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). One million melanoma D4M.3A.3-UV3 cells in PBS were inoculated subcutaneously in the right flank. Vehicle or UNC0642 (5 mg/kg) was administrated intraperitoneally daily for the duration of the experiment, starting 6 days after tumor inoculation. Blocking antibodies, anti-PD-1 (a gift from Gordon Freeman, Dana-Farber Cancer Institute) and anti-CTLA-4 (BioXcell, BE0164, clone 9D9), were administrated intraperitoneally on days 7, 9, 11 at a dose of 200 μg per mouse. For survival studies, mice were sacrificed when tumors reached a maximum volume of 1000 mm3. All studies and procedures involving animal subjects were approved by the Institutional Animal Care and Use Committees (IACUC) of Massachusetts General Hospital and were conducted strictly in accordance with the approved animal handling protocols. Tumor volumes were measured using digital calipers and calculated by the following formula: volume (mm3) = (width2 x length)/2.
Immunohistochemistry
K029 tumors were harvested on Day 17 post-treatment with vehicle or UNC0642, and then were fixed and embedded with formalin and paraffin respectively. Tumor sections were cut at a depth of 5 microns by a microtome, then dried overnight in the oven. Tumor sections were deparaffinized and dehydrated following the standard procedure. Heat-induced antigen retrieval was performed. Immunohistochemical staining was performed by incubation of tumor sections with 1:200 diluted primary antibody for H3K9me2 (Abcam, ab1220) or LC-3B (Cell Signaling, #3868) at 4°C overnight, followed by incubation with 1:2000 HRP-linked secondary antibody for 30 minutes at room temperature. Staining results were revealed by applying AEC peroxidase substrate (Vector Laboratories, SK-4200). Hematoxylin-counterstained slides were mounted with coverslips, and staining results were analyzed using a Leica DMR microscope and Nikon NIS-Elements Imaging Software version 4.30.
RNA purification and quantitative RT-PCR (qRT-PCR)
RNA was isolated from melanoma cells at indicated time points using the RNeasy Plus Mini Kit (Qiagen). mRNA expression was determined using intron-spanning primers with SYBR FAST qPCR master mix (Kapa Biosystems). Expression was normalized to RPL11. The primers used for qRT-PCR are shown in Supplementary Table S5.
Whole transcriptome RNA sequencing (RNA-seq)
Total RNA was extracted from Hs944T melanoma cells 72 h after infection with pLKO.1-shScr or pLKO.1-shG9a#5, and from pMEL*/BRAFV600E cells 72h after infection with pLenti6-MK1-EHMT2-V5 or pLenti CMV GFP Blast (659–1). All RNA samples were submitted for Quality control (QC), cDNA synthesis, library construction, size selection and NGS sequencing at the Beijing Genomics Institute (BGI, Cambridge, USA). In brief, during the QC steps, an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System are used in quantification and qualification of the sample library. The multiplexed library was sequenced using an Illumina HiSeq 4000 system. Reads were aligned to the reference genome (hg19) by STAR 2.5.2. Reads were counted by HTSeq-0.6.1 using UCSC annotation, as downloaded from the Illumina iGenomes collection. Only reads with mapping score of 10 or more were counted. Differentially expressed genes were detected by DESeq2, using the Wald test.
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis was performed using the GSEA module of Genepattern (https://genepattern.broadinstitute.org/). For identifying pathways that are regulated by G9a, our RNA-seq data set was analyzed by GSEA with the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway gene sets.
For correlation between G9a expression and WNT signature gene sets, microarray data of CCLE cancer cell line panels (GSE36133) was analyzed using GSEA. The WNT signature gene set in melanomas was obtained from GSE32907. Briefly, 396 genes that are significantly upregulated by constitutively active β-catenin (β-cateninSTA) were identified using the Comparative Marker Selection module (Genepattern) and used as a WNT signature gene set, named WNT_BETA_CATENIN_MELANOMA, in this study. The WNT_BETA_CATENIN_MELANOMA signature gene set was validated in the GSE26656 dataset. Other curated WNT signature gene sets tested were obtained from MSigDB (http://software.broadinstitute.org/gsea/msigdb). G9a expression (probe ID: 207484_s_at) was used as a continuous label and applied to GSEA in accordance with gene set-based permutation and Pearson correlation analysis.
For correlation analysis of WNT signatures scores with genetic alterations within WNT pathways, the WNT_BETA_CATENIN_MELANOMA signature scores were computed by single-sample gene set enrichment analysis (ssGSEA) (54), which is able to estimate the degree of coordinated up- and down-regulation of a given gene set, in melanoma cell lines. Genetic profiles (somatic mutations and copy number variations) for all WNT pathway genes were obtained from the CCLE data repository (https://portals.broadinstitute.org/ccle/).
Enrichr (http://amp.pharm.mssm.edu/Enrichr/) was used to analyze the enrichment of downregulated genes by G9a knockdown (283 genes, log2 fold<−0.585, adjusted p-value<0.05) in annotated genesets (ChEA).
TOPFlash/FOPFlash luciferase assay
M50 Super 8x TOPFlash and M51 Super 8x FOPFlash (TOPFlash mutant) plasmids were gifts from Randall Moon (Addgene # 12456 and 12457, respectively). K029 melanoma cells were plated in 24-well plates (5 × 104 cells/well) the day before transfection. After shScr- or shG9a-mediated knockdown and subsequent selection with puromycin as described above, the K029 cells were transfected with TOPFlash or FOPFlash vector (0.8 μg/well) along with pRL-SV40 Renilla control (0.2 μg/well) using Lipofectamine 2000 transfection reagent (Life Technologies). At 72 h, luciferase readings were made using a Dual Luciferase Reporter Assay System (Promega). For testing UNC0638 in the TOPFlash/FOPFlash luciferase assay, 24 h after transfection with the TOPFlash or FOPFlash vector plus pRL-SV40, the transfection medium was replaced with fresh culture medium (10% FBS, 1% penicillin/streptomycin/L-glutamine) containing DMSO or 500 nM UNC0638. 48 h after additional culture in the presence of DMSO or UNC0638, the K029 cells were subjected to the Dual Luciferase Reporter Assay. Firefly luciferase values were normalized to Renilla luciferase values. Results reported are the average of three independent experiments done in duplicate.
TCGA survival and gene expression analysis
To test the clinical impact of G9a and other candidate genes within the 6p21 amplicon, TCGA melanoma patients were ordered according to each candidate gene and survival curves were drawn using OncoLnc (http://www.oncolnc.org).
The TCGA RNA-seq data was calculated by RSEM (obtained from cBioportal) and then used for the gene expression analysis in Figure 5D and Supplementary Fig. S9A and S9B.
Chromatin immunoprecipitation (ChIP)
ChIPed DNA samples were prepared from 50 million Hs944T cells treated with 500 nM UNC0638 or DMSO vehicle for 72 h as described previously (55). Immunoprecipitations were performed with anti-G9a rabbit antibody (Cell Signaling, #3306), anti-H3K9me2 mouse antibody (Abcam, ab1220), anti-phosphor-PolII (Ser5) antibody (Abcam, ab5131), and normal rabbit or mouse IgG (Santa Cruz, sc-2027 or sc-2025) as controls. qPCR assays were performed using primers specific for the human DKK1 putative promoter and the RPL30 gene body (see Supplementary Table S5). Ct values of ChIPed DNA samples were normalized to that of 1% Input. The data represent averages of at least three independent experiments.
Gene expression analysis in TCGA and Cancer Cell Line Encyclopedia (CCLE)
Log-transformed RPKM (Reads Per Kilobase of exon model per Million mapped reads) in melanoma cell lines and TCGA melanoma patients were obtained from CCLE and the Genome Data Analysis Center (GDAC). Gene expression data for 88 short-term-cultured melanoma samples were obtained from the Broad Melanoma Portal (http://www.broadinstitute.org/melanoma/branding/browseDataHome.jsf). Correlations between gene expression levels (e.g., G9a vs. DKK1) were calculated by Spearman’s rank correlation.
BIX-01294 sensitivity and G9a mRNA levels and copy number variations
CCLE gene expression data for G9a was obtained from GSE36133. Copy number data for G9a for all CCLE cell lines were obtained from the Broad Institute website (CCLE_copynumber_byGene_2013–12-03.txt.gz). G9a inhibitor sensitivity was inferred from the area under the curve (AUC) values obtained from the CTRP2.2 database, downloaded from the OCG data portal (https://ocg.cancer.gov/programs/ctd2/data-portal). For the cancer types in which correlations of G9a expression with DKK1 expression and WNT pathway signatures were found as described above, Pearson correlations of the AUC values for BIX-01294 with G9a expression and G9a copy number values were calculated using Morpheus software (https://software.broadinstitute.org/morpheus).
Leeds Melanoma Cohort analysis
Gene expression and copy number alteration data were collected from a cohort of 2184 primary melanoma patients (essentially treatment naive) recruited in the North of England (56,57). Transcriptomic data was generated for 703 tumors and pre-processed as previously described using the Illumina DASL whole genome array (34), accessible from the European Genome-Phenome Archive with accession number EGAS00001002922. The study participants provided written informed consent and the Leeds Melanoma Cohort Study received ethical approval from the Medical Research Ethics Committee (MREC 1/03/57) and the Patient Information Advisory Group (PIAG3–09(d)/2003).
Next-Generation Sequencing (NGS)-derived copy number alteration profiles were generated for 276 tumor samples among the 703 transcriptomic-profiled tumors as described by Filia et al. (58). Quality control of the data was amended afterwards. Briefly, the control germline DNA sequence data, which were obtained from the Phase 3 data of the 1000 Genomes Project (n=312) (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data/) that matched the experimental setup (Illumina platform, low coverage, paired end library layout) was used. In order to create bins or windows of size 10k across the genome, bamwindow (https://github.com/alastair-droop/bamwindow) was utilized. Blacklisted regions (those for which sequence data were unreliable) were identified and masked. Highly variable regions in the genome were identified using the QDNASeq package in R and were added to the blacklist. This pipeline empirically identified highly variable regions including common germline variations in the genome using the 312 germline controls (59). This step did not identify any large variation in the germline copy number in the G9a region. QDNASeq was also used to adjust the read counts from each valid window based on the interaction of GC content and mappability.
G9a copy number data was categorized to identify “High” and “Low” G9a tumors as first and fourth quartile, respectively. To test the correlations between G9a copy number and G9a expression, Spearman’s rank correlation was used. Survival analysis to assess the association of G9a copy number with melanoma-specific survival (MSS) was performed using a Cox proportional hazards model and the significance of this model was assessed by the likelihood ratio test. To test the differences in proportions of G9a low and high tumors, among the 6 Consensus Immune Clusters (CICs), chi-squared tests were used. Whole transcriptome differential gene expression levels between low and high G9a tumors were assessed using Mann-Whitney U tests with the Benjamini-Hochberg correction for multiple testing (FDR<0.05). Genes identified as significantly upregulated (z-score<0) or downregulated (z-score>0) were analyzed for pathway enrichment using Reactome FIViz software; significance of enriched pathways was denoted by FDR from hypergeometric tests. The volcano plot was produced using EnhancedVolcano package in R.
Statistical analysis
The statistical tests indicated in the figure legends were calculated using GraphPad Prism 7.0 and 8.0. P values < 0.05 were considered statistically significant.
Supplementary Material
Significance.
Oncogenic G9a abnormalities drive tumorigenesis and the ‘cold’ immune microenvironment by activating WNT signaling through DKK1 repression. These results reveal a key druggable mechanism for tumor development and identify strategies to restore ‘hot’ tumor immune microenvironments.
Acknowledgements
The authors thank Dr. Stephen Hodi (Dana-Farber Cancer Institute) for kindly providing the K029 melanoma cell line. We thank Ryo Murakami (Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School) for the pLenti-hygro-hTERT, hCDK4(R24C), p53DD, and NRASQ61R plasmids used for the immortalization of primary human melanocytes. We thank members of the Fisher laboratory for discussions and suggestions. The results presented here are fully or partially based upon data generated by the Cancer Target Discovery and Development (CTD2) Network (https://ocg.cancer.gov/programs/ctd2/data-portal) established by the National Cancer Institute’s Office of Cancer Genomics.
Funding: D.E.F. gratefully acknowledges grant support from the National Institutes of Health (5P01 CA163222, 1R01CA222871, 5R01AR072304 and 5R01 AR043369–22), the Melanoma Research Alliance, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. S.K. acknowledges support by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowships for Research Abroad. J.J. acknowledges support by grants R01HD088626, R01GM122749 and R01CA218600 from the National Institutes of Health. The Leeds research was funded by Cancer Research UK C588/A19167, C8216/A6129, and C588/A10721 and NIH CA83115. J.P, J.M.D and S.M. were funded by Horizon 2020 Research and Innovation Programme no. 641458 (MELGEN).
Footnotes
COI statement: S.K., Q.Y.W., and D.E.F. declare that parts of the work are the subject of a U.S. provisional patent application titled “Treatment of cancers having alterations within the SWI/SNF chromatin remodeling complex.” Dr. Fisher has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin darkening treatments that might be used for a broad set of human applications. Dr. Fisher’s interests were reviewed and are managed by Massachusetts General Hospital and Partners Healthcare in accordance with their conflict of interest policies.
Data and materials availability: RNA-seq data have been deposited in the NCBI GEO database with accession numbers GSE47427 and GSE147419 for G9a knockdown in Hs944T melanoma and G9a overexpression in pMEL*/BRAFV600E, respectively. Additional data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nature Medicine. 2016;22:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yap DB, Chu J, Berg T, Schapira M, Cheng S-WG, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu J-Y, Paulk J, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nature Medicine. 2016;22:632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weirich S, Kudithipudi S, Jeltsch A. Somatic cancer mutations in the MLL1 histone methyltransferase modulate its enzymatic activity and dependence on the WDR5/RBBP5/ASH2L complex. Molecular Oncology. 2017;11:373–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weirich S, Kudithipudi S, Kycia I, Jeltsch A. Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme. Clinical Epigenetics. 2015;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nature Genetics. 2013;45:1386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25:781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129:823–37. [DOI] [PubMed] [Google Scholar]
- 12.Wei L, Chiu DK-C, Tsang FH-C, Law C-T, Cheng CL-H, Au SL-K, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. Journal of Hepatology. 2017;67:758–69. [DOI] [PubMed] [Google Scholar]
- 13.Ding J, Li T, Wang X, Zhao E, Choi J-H, Yang L, et al. The Histone H3 Methyltransferase G9A Epigenetically Activates the Serine-Glycine Synthesis Pathway to Sustain Cancer Cell Survival and Proliferation. Cell Metabolism. 2013;18:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu WB, Shiah Y-J, Lourenco C, Mullen PJ, Dingar D, Redel C, et al. MYC Interacts with the G9a Histone Methyltransferase to Drive Transcriptional Repression and Tumorigenesis. Cancer Cell. 2018;34:579–595.e8. [DOI] [PubMed] [Google Scholar]
- 16.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–22. [DOI] [PubMed] [Google Scholar]
- 17.Ceol CJ, Houvras Y, Jane-Valbuena J, Bilodeau S, Orlando DA, Battisti V, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471:513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim M, Gans JD, Nogueira C, Wang A, Paik J-H, Feng B, et al. Comparative Oncogenomics Identifies NEDD9 as a Melanoma Metastasis Gene. Cell. 2006;125:1269–81. [DOI] [PubMed] [Google Scholar]
- 19.Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, et al. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005;19:815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang X-P, et al. Discovery of an in vivo Chemical Probe of the Lysine Methyltransferases G9a and GLP. Journal of Medicinal Chemistry. 2013;56:8931–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, Zismann V, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends in Molecular Medicine. 2006;12:406–14. [DOI] [PubMed] [Google Scholar]
- 23.Sato S, Roberts K, Gambino G, Cook A, Kouzarides T, Goding CR. CBP/p300 as a co-factor for the Microphthalmia transcription factor. Oncogene. 1997;14:3083–92. [DOI] [PubMed] [Google Scholar]
- 24.Takeda K, Yasumoto K, Takada R, Takada S, Watanabe K, Udono T, et al. Induction of Melanocyte-specific Microphthalmia-associated Transcription Factor by Wnt-3a. Journal of Biological Chemistry. 2000;275:14013–6. [DOI] [PubMed] [Google Scholar]
- 25.Dorsky RI, Raible DW, Moon RT. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000;14:158–62. [PMC free article] [PubMed] [Google Scholar]
- 26.Rimm DL, Caca K, Hu G, Harrison FB, Fearon ER. Frequent Nuclear/Cytoplasmic Localization of β-Catenin without Exon 3 Mutations in Malignant Melanoma. The American Journal of Pathology. 1999;154:325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris LGT, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan Ş, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nature Genetics. 2013;45:253–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nature Reviews Cancer. 2018;18:139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 31.Moreno BH, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, et al. Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res. 2016;4:845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segovia C, José-Enériz ES, Munera-Maravilla E, Martínez-Fernández M, Garate L, Miranda E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med. 2019;25:1073–81. [DOI] [PubMed] [Google Scholar]
- 34.Nsengimana J, Laye J, Filia A, O’Shea S, Muralidhar S, Poźniak J, et al. β-Catenin–mediated immune evasion pathway frequently operates in primary cutaneous melanomas. J Clin Invest. 2018;128:2048–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity. 2013;39:782–95. [DOI] [PubMed] [Google Scholar]
- 36.Allen EMV, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, et al. A Landscape of Driver Mutations in Melanoma. Cell. 2012;150:251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Y, Schleich K, Yue B, Ji S, Lohneis P, Kemper K, et al. Targeting the Senescence-Overriding Cooperative Activity of Structurally Unrelated H3K9 Demethylases in Melanoma. Cancer Cell. 2018;33:322–336.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, et al. Structural Biology of Human H3K9 Methyltransferases. PLoS One. 2010;5:e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zingg D, Debbache J, Peña-Hernández R, Antunes AT, Schaefer SM, Cheng PF, et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell. 2018;34:69–84.e14. [DOI] [PubMed] [Google Scholar]
- 44.Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campagne A, Lee M-K, Zielinski D, Michaud A, Le Corre S, Dingli F, et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat Commun. 2019;10:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He M, Chaurushiya MS, Webster JD, Kummerfeld S, Reja R, Chaudhuri S, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP. Science. 2019;364:283–285. [DOI] [PubMed] [Google Scholar]
- 49.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, et al. β-Catenin Signaling Controls Metastasis in Braf-Activated Pten-Deficient Melanomas. Cancer Cell. 2011;20:741–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinnberg T, Makino E, Krueger MA, Velic A, Macek B, Rothbauer U, et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine. 2016;8:132–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gartner JJ, Parker SCJ, Prickett TD, Dutton-Regester K, Stitzel ML, Lin JC, et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci U S A. 2013;110:13481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell. 2013;23:302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li J, Song JS, Bell RJA, Tran T-NT, Haq R, Liu H, et al. YY1 Regulates Melanocyte Development and Function by Cooperating with MITF. Bosenberg M, editor. PLoS Genetics. 2012;8:e1002688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newton-Bishop JA, Beswick S, Randerson-Moor J, Chang Y-M, Affleck P, Elliott F, et al. Serum 25-Hydroxyvitamin D3 Levels Are Associated With Breslow Thickness at Presentation and Survival From Melanoma. J Clin Oncol. 2009;27:5439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Newton-Bishop JA, Davies JR, Latheef F, Randerson-Moor J, Chan M, Gascoyne J, et al. 25-hydroxyvitamin D2/D3 levels and factors associated with systemic inflammation and melanoma survival in the Leeds Melanoma Cohort. Int J Cancer. 2015;136:2890–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Filia A, Droop A, Harland M, Thygesen H, Randerson-Moor J, Snowden H, et al. High-Resolution Copy Number Patterns From Clinically Relevant FFPE Material. Sci Rep. 2019;9:8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheinin I, Sie D, Bengtsson H, Wiel MA van de, Olshen AB, Thuijl HF van, et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014;24:2022–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.