

Abstract
Purpose:
Application of allogeneic hematopoietic cell transplantation (allo-HCT) for patients with hematological disorders is limited by the development of graft-versus-host-disease (GVHD). Separation of GVHD and graft-versus-leukemia (GVL) remains a great challenge in the field. We investigated the contribution of individual pathways involved in the complement cascade in GVH and GVL responses in order to identify specific targets by which to separate these two processes.
Experimental Design:
We used multiple preclinical murine and human-to-mouse xenograft models involving allo-HCT recipients lacking components of the AP or CP/LP to dissect the role of each individual pathway in GVHD pathogenesis and the GVL effect. For translational purposes, we used the AP-specific complement inhibitor, CR2-fH, which localizes in injured target organs to allow specific blockade of complement activation at sites of inflammation.
Results:
Complement deposition was evident in intestines of mice and patients with GVHD. In a preclinical setting, ablation of the AP, but not the CL/LP, significantly improved GVHD outcomes. Complement activation through the AP in host hematopoietic cells, and specifically dendritic cells (DCs), was required for GVHD progression. AP-deficiency in recipients decreased donor T-cell migration and Th1/Th2 differentiation, while increasing the generation of regulatory T-cells (Tregs). This was due to decreased activation and stimulatory activity of recipient DCs in GVHD target organs. Treatment with CR2-fH effectively prevented GVHD while preserving GVL activity.
Conclusion:
This study highlights the AP as a new therapeutic target to prevent GVHD and tumor relapse after allo-HCT. Targeting the AP by CR2-fH represents a promising therapeutic approach for GVHD treatment.
Keywords: Complement, Alternative Complement Pathway, GVHD, GVL, Hematopoietic Stem Cell Transplantation
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is a potentially curative option for a variety of benign and malignant hematopoietic disorders (1,2). The therapeutic benefit of allo-HCT is attributable to the graft-versus leukemia effect (GVL). However, graft-versus-host disease (GVHD) development remains a significant cause of transplant-related mortality and morbidity (3). As T-cells in the donor graft mediate the GVL effect and GVHD concurrently, treatment of GVHD without compromising GVL activity is a major challenge in the field. Current immunosuppressive medications used to prevent or treat GVHD often impair the T cell- mediated GVL effect and increase the chance of relapse from primary malignancy.
Complement activation is involved in the pathogenesis of GVHD (4–7). Complement proteins secreted from antigen presenting cells (APCs), such as dendritic cells (DCs), regulate the expansion of naïve and primed effector T-cells after allo-HCT (6,8,9). These findings suggest that local complement activation may be important for GVHD pathogenicity, as circulating complement in the serum does not correlate with GVHD. With regard to potential complement effector mechanisms, our previous data demonstrated that systemic blockade of complement anaphylatoxin molecules (C3a and C5a) severely impaired GVL activity (5). However, no attempt has been made to target complement activation in target organs to control GVHD and tumor relapse after allo-HCT.
Complement activation is essential for priming naïve T-cells and their subsequent differentiation into Th1/Th17 in both the peripheral blood and lymphoid organs (10). Absence of intracellular C3 or C3aR/C5aR signaling on T-cells or APCs induces the generation of Tr-1(11,12) and Tregs (13–15). Blockade of CD46, C3aR or C5aR ameliorates T-cell alloreactivity in xenograft GVHD models (11,14,15). However, C3−/− T-cells have a comparable capacity to induce GVHD as compared to WT in murine GVHD models (16). Targeting C3 in transplant recipients showed a modest effect on GVHD severity (17). The aforementioned evidence indicates that specific inhibiton of local complement signaling is required to control GVHD.
The complement system can be activated via three pathways: the classical (CP), lectin (LP), or alternative (AP) (13,18). The AP can both spontaneously activate as well as amplify the CP and LP, and can potentially contribute up to 80% of total complement activation (19). Thus, the AP is important for inflicting maximum injury during the pathogenesis of autoimmune diseases (19–22). Among them, inflammatory bowel disease shares certain pathological similarities with intestinal GVHD (23); a potent catalyst for systemic GVHD development. This evidence provides rationale for blocking the AP in GVHD target organs in order to effectively control GVHD.
In the current study, using patient samples and murine models of allo-HCT, we demonstrate that complement activation specifically through the AP in injured target organs plays an essential role in GVHD after allo-HCT. To inhibit complement activation in GVHD target organs, we used a complement inhibitor comprised of an AP-specific inhibitory domain (fH) linked to complement receptor 2 (CR2) that selectively binds the membrane bound complement activation product C3d. CR2-fH effectively reduced GVHD while preserving the GVL effect after allo-HCT in multiple murine and human-to-mouse models of GVHD/GVL. Importantly, a humanized version of CR2-fH is currently available and was evaluated in a clinical trial (NCT01335165). Hence, the current study has substantial translational potential for patients undergoing allo-HCT.
Materials and Methods
Human subjects
Tissue samples were collected from patients undergoing allo-HCT. Colon samples were collected from patients for diagnostic purposes, and the tissue blocks used in this study were selected from a sample repository. All the patient’s specimens used in this study are de-identified. This study was approved by the Medical University of South Carolina Institutional Review Board (IRB) and was conducted in accordance with International Ethical Guidelines for Biomedical Research Involving Human Subjects, Good Clinical Practice guidelines, the Declaration of Helsinki, and local laws.
Animals and Reagents
fB−/− and C1q/MBL−/− mice on C57BL/6 background were bred and housed at the Medical University of South Carolina (MUSC). FVB (H-2q, CD45.1), C57BL/6 (H-2b, CD45.2), B6 Ly5.1 (H-2b, CD45.1), B6D2F1 (H-2b/d, CD45.2), C3H-SW (H-2b, CD45.1), and BALB/c (H-2d) mice were purchased from the National Cancer Institute or Charles River lab. Animals were maintained in pathogen-free facilities in the American Association for Laboratory Animal Care–accredited Animal Resource Center at MUSC. All animal procedures were approved by the Institutional Animal Care and Use Committee of MUSC. CR2-fH was generated in Dr. Tomlinson’s lab at MUSC.
GVHD/GVL models
As previously describled (5), recipient mice were lethally irradiated at 700 cGy for BALB/c and 1000-1200 cGy (2 split doses, 3-hour interval) for B6 recipient mice using an X-RAD 320 irradiator (Precision X-Ray). Within 24 hours of irradiation, Balb/c or B6 recipient mice were transplanted with 5.0×106 T-cell depleted bone marrow cells (TCD-BM) from B6 or FVB donors with or without T cells (0.5-1×106/mouse). Recipient survival was monitored throughout the experiment. Body weight loss was monitored twice per week and clinical signs of GVHD were monitored once per week and include posture, skin damage, hair loss, ruffled fur, diarrhea, and decreased activity(5) (24). For GVL experiments, tumor cells were injected intravenously (i.v.) on the same day of transplantation. In cases of luciferase transduced acute myeloid leukemia C1498 (2000 cells/mouse), tumor burden was estimated with bioluminescent imaging (BLI) using Xenogen-IVIS 200 in vivo Imaging System (Perkin-Elmer, Waltham, MA). MLL-AF9-GFP+ tumors were identified via the percentages of GFP+ cells in peripheral blood using flow cytometry. For human-to-mouse xenograft models, sublethally irradiated NSG-A2(+) mice were transplanted with HLA-A2(−) PBMCs (15×106). Recipient survival and GVHD severity demonstrated by recipient weight loss were monitored throughout experiment.
Flow Cytometry
As described previously (5,25), the following antibodies were used for cell-surface staining: anti–CD4 (clone RM4-5, BD Biosciences), anti–CD8 (clone 53-6.7, BD Biosciences), anti–H-2q (KH114, Biolegend), anti–CXCR3-biotin (CXCR3-173, eBioscience); and anti–CCR6-AF647 (BioLegend, clone 29-2L17), anti-FasL (MFL3, BD Biosciences), anti-PD-1 (MFL3, eBioscience), anti-NK1.1 (PK136, eBioscience), anti-CD44 (IM7, Biolegend), anti-CD62L (MEL-14, eBioscience), anti-TCRβ (H57-597 BD Biosciences), anti-CD11b (M1/70, eBioscience). Biotinylated Abs were detected using APCcy7 (BD Biosciences, catalog 554063) or PEcy7 (BD Biosciences, catalog 557598) conjugated to streptavidin. To measure intracellular cytokines, cells were stimulated for 4-5 h at 37°C with PMA (100 ng/ml, Sigma-Aldrich) and ionomycin (100 ng/mL; Calbiochem, EMD) in the presence of GolgiStop (BD Biosciences). Fix and permeabilization were performed using Cytofix/Cytoperm Plus (BD Biosciences), followed by staining with the appropriate antibodies including anti–IFNγ (clone XMG1.2, eBioscience), anti–IL-17 (clone TC11-18H10.1, BioLegend), anti–IL-4 (clone 11B11, BD Biosciences), anti–IL-5 (clone TRFK5, eBioscience), anti–FOXP3, (clone FJK-16s, eBioscience), anti-Ki67 (16A8, Biolegend), and anti–pS6-AF467 (Cell Signaling Technology, clone D57.2.2E). Live/dead yellow cell staining kit (catalog L-34968) and CFSE (catalog C1157) were purchased from Invitrogen. Apoptosis was measured by Annexin V kit (BD Biosciences, San Jose, CA). XenoLight CF750 labeling kit was procured from Caliper. Data were analyzed with FlowJo software (Tree Star, OR). Blood was collected from recipients 14 days after BMT, and serum cytokines quantified using a cytometric bead assay kit (BD Biosciences, catalog 560485) (25).
Bone-Marrow-Derived DCs
DCs were generated from the BM of 8- to 12-week old mice as described previously (5). BM cells were flushed from the femurs and tibias with RPMI containing 1% FCS, 100 U/mL of penicillin/streptomycin, and 2mM L-glutamine. The single cell suspension was then filtered through a nylon mesh strainer (70 mm; BD Biosciences), washing twice with the same medium. BM cells (10x106/petri dish) were differentiated in the presence of GM-CSF (20 ng/mL, PeproTech, Rocky Hill, NJ) in complete culture medium (RPMI containing 10% FBS, 100 U/mL of penicillin/streptomycin, 2mM l-glutamine, and 50 mM b-mercaptoethanol) for 6 days. Half of the medium was replaced with an equal volume of GM-CSF, containing culture medium on the day 3. Immature DCs were stimulated with lipopolysaccharide (LPS) (25 ng/ml, Sigma Aldrich) for 12 h.
Immunofluorescence (IF)
A similar approach was used to stain 10 μm sections as outlined previously (26). Primary antibodies used for these experiments were against murine antigens; anti-C3d (Goat Ab, R&D), anti-CD3e, anti-fB (abcam). Directly conjugated antibodies used were used: anti-C3d-FITC (MP Biomedicals). Secondary antibodies were: Donkey anti-Goat-Alexa fluor 488nm and 555nm, Donkey anti-rabbit-Alexa fluor 488nm, 555nm and 647nm, Donkey anti-rat Alexa fluor 488nm and 555nm, DAPI (Vector Labs) were used as nuclear stains. Super-resolution imaging was performed using a Zeiss LSM 880 confocal microscope (Zeiss. Uniform field sizes of 240x240x40 μm dimensions were imaged with a 40x water objective. High-resolution microglial fields were imaged with a 63X oil objective. Individual tracks were used for each channel with sequential imaging to avoid channel bleeding. 3D-rendering was performed using ZEN-black software (Zeiss).
Statistical analysis
Data were analyzed using Prism GraphPad (version 7). As described previously (5,25), comparisons between two groups were calculated using a two-tailed Student t test. Clinical scores and body weight loss were compared using a nonparametric Mann-Whitney U test. The log-rank (Mantel-Cox) test was utilized to analyze survival data. A p value less than 0.05 was considered significant.
Results
Complement activation via the alternative pathway (AP) plays a central role in GVHD pathobiology after allo-HCT
To examine the importance of complement activation in GVHD target organs post allo-HCT, we initially measured complement C3d deposition in the skin, lung, liver, small intestine (SI) and large intestine (LI). We found that C3d deposition was evident in injured target organs of allo-HCT recipients 7d post-BMT compared to recipients that did not receive T cells (Fig. 1A). We next examined complement gene expression in the intestines of transplanted recipients using Nanostring technology. Expression of genes encoding the central complement protein (C3) and AP proteins (fD, fB, fP), were significantly upregulated in GVHD recipients compared to those without GVHD (Fig. 1B). In contrast, there was no difference in the expression of genes encoding CP or LP proteins (Fig. 1B). We also observed increased expression of genes encoding inflammatory proteins (IFNγ, IL-6, TNFα), cytolytic activity (GrzmB), and cell death (fas) in the intestines of mice with GVHD (Fig. 1C). These results implicate the AP as the central complement regulator in GVHD target organs. In order to determine if the AP specifically played an augmented role in GVHD, we next performed allo-HCT using recipient mice deficient for AP (fB−/−) or CP/LP (C1q/MBL−/− ). Immunological phenotypes were similar across different mouse strains (Supplementary Fig. S1). We found that ablating the AP caused reductions in gene expression of molecules associated with inflammation, cytolysis, and cell death to levels comparable to recipients without GVHD (Fig. 1D). We observed that GVHD severity was drastically reduced in fb−/− recipients, as indicated by improved survival and reduced GVHD clinical signs (Fig. 1 E–F). Interestingly, the absence of CP/LP had no significant effect on GVHD severity in MHC-mismatched (FVB→B6) models of allo-HCT, supporting the notion that the AP played a more important role than the CP/LP in GVHD (Fig. 1E). To confirm this, we tested an MHC-matched (C3H.SW→B6) model, in which we observed that ablation of CP/LP resulted in accelerated GVHD compared to WT recipients (Fig. 1F).
Figure 1. Complement activation via the AP has a central role in GVHD pathobiology.

(A) Lethally irradiated Balb/c mice were transplanted with TCD-BM (5x106/mouse) and with or without T-cells (0.75x106/mouse) from B6 mice. GVHD target organs, including skin, lung, liver, small intestine and colon were collected and stained for anti-C3d and DAPI; (B-D) Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with BM (5x106/mouse) and with or without T-cells (0.6x106/mouse) from FVB donors. RNA was isolated from large intestine 7 days after HCT and analyzed with Nanostring nCounter System using the Mouse Immunology Codeset. (B, C) Heat map indicating the relative expression of genes coding for components of central and/or individual complement pathways in the colons of WT (GVHD) and BM alone (Non-GVHD) recipients. (D) Heat map indicating relative expression of genes coding for pro-inflammatory cytokine or cytotoxicity markers in the colons of WT, fB−/−, or C1q/MBL−/− recipients. (E) Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (0.6x106/mouse) from FVB donors. Survival and clinical score were monitored throughout the experiment. (F) Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (2.0 x106/mouse) from C3H-SW donors. Survival and clinical score were monitored throughout the experiment. (G) Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (2.0x106/mouse) from FVB donor mice. IHC staining for C3d was performed on GVHD target organ sections including the skin, lung, liver, small and large intestine on day 7 after HCT. A graphical summary of C3d deposition in target organs is shown. (H) CBA analysis of serum cytokine levels in recipients 7 days after HCT. The log-rank (Mantel-Cox) test (left panels E,F), nonparametric Mann-Whitney U test (right panels E,F) and unpaired t test (H) were used to compare between groups. Data are mean ± S.E.M. and *p < 0.05, **p < 0.01, ***p < 0.001.
In the absence of AP in recipients (FVB-B6 model), complement deposition was markedly decreased in GVHD target organs including liver, lung, and large intestine (Fig. 1G). We also observed that serum pro-inflammatory cytokines, including TNFα and IFNγ were diminished in fB−/− recipients after allo-HCT (Fig. 1H). These results indicate a crucial role for the AP, but not the CP/LP, in GVHD development after allo-HCT. As intracellular complement activation can affect T-cell alloreactivity (27), we investigated whether complement generated from donor T cells contributes to GVHD pathogenicity after allo-HCT. We therefore transplanted T cells from WT, fB−/− or C1q/MBL−/− donors into allogeneic recipients. No difference was observed in GVHD severity (Supplementary Fig. S2), suggesting a negligible role for intracellular complement activation in T cell pathogenicity in the induction of GVHD.
Complement activation in recipient hematopoietic cells is crucial for the initiation of GVHD after allo-HCT
Complement activation via the AP can occur in both hematopoietic and non-hematopoietic recipient cells. Hence, we investigated whether the source of complement generation is important for GVHD development after allo-HCT. To this end, we generated three types of bone marrow chimeric mice (BM chimeras), where the AP was absent in hematopoietic, non-hematopoietic, or present in both compartments (Supplementary Fig. S3A–B). Using BM chimeras as recipients, we observed that severe GVHD was evident in chimeras where fB was present in the recipient hematopoietic system [FVB (WT B6 fb−/−)], while significantly reduced GVHD was observed in the chimeras where fB was absent in the recipient hematopoietic system [FVB (fb−/− WT B6)] (Supplementary Fig. S3A–B). Thus, complement activation through the AP in the recipient hematopoietic, but not nonhematopoietic, compartment plays an important role in the induction of GVHD.
Complement activation through the AP in recipient dendritic cells (DCs) is critical for GVHD development post-HCT.
Given that recipient-derived DCs are the most important hematopoietic cells in terms of generating complement to activate donor T-cells in target-injured organs(9), we evaluated the contribution of AP-associated complement in recipient DCs to GVHD development post-HCT. BM-derived DCs (BMDCs) were differentiated from WT or fB−/− BM and matured via lipopolysaccharide (LPS) stimulation. Upon co-transfer of fB−/− DCs with donor grafts, GVHD mortality and morbidity was significantly reduced (Supplementary Fig. S3C–D). This data is indicatitive of a critical role for complement activation via AP in DCs during GVHD development.
Complement AP activation is essential for donor T cell migration, DC antigen presentation, and Th1/Tc1 differentiation in target organs during GVHD development
Donor T cell migration and activation in target organs is required for GVHD development(9). We observed a decreased number of donor lymphocytes in the intestine of fB−/− recipients (Fig. 2A). The expression levels of the intestinal-homing chemokine CCR9 was consistently lower in lymphocytes isolated from fB−/− recipients compared to WT controls (Fig. 2B). CD103+CD8+ T cells were previously reported to be pathogenic in the intestine (28). Hence, we examined this population. A lower percentage of donor-derived CD103+CD8+ cells were found in the intestines of fB−/− recipients (Fig. 2). Host AP deficiency reduced the frequency (Fig. 3D) and activation of pathogenic CD103+ DCs(29), as demonstrated by lower CD80 expression (Fig. 2E) and increased activation as well as frequency of the protective CD8+CD11c+ DC population (Fig. 2F). Local complement activation results in generation of effector molecules, C3a and C5a, which can differentiate naïve T-cells into Th1/Tc1 subsets and reduce Treg differentiation(27). Indeed, deficiency of recipient AP decreased Th1/Tc1 differentiation and increased Treg differentiation, which is demonstrated by reduced percentages of IFNƔ-secreting CD4+- and CD8+- (Fig. 3A–B) and an increased percentage of foxp3+ CD4+-H2q+ T-cells (Fig. 3C) in the liver of transplanted recipients. Further, Th2 differentiation was significantly reduced in the lung of fB−/− recipients indicated by a reduction in percentage of IL-4/5-secreting H2q+ T-cells (Fig. 3D). Notably, thymic function was preserved in fB−/− recipients, indicated by an increased number of CD4+CD8+ thymocytes in fB−/− recipients compared to WT recipients (Fig. 3E). On the other hand, we observed similar immunological characteristics between C1q/MBL and WT cohorts (Fig. 3A–B), indicating a vital role for AP activation in driving GVHD development after allo-HCT. Interestingly, in lymphoid organs of fB−/− recipients, we did not observe a significant difference in Th1/Tc1 differentiation (Supplementary Fig. S4A–B). While the percentage of splenic DC (Supplementary Fig. S4C) was significantly greater in fB−/− compared to WT recipient; the numbers of splenic DC were similar (Supplementary Fig. S4G). There was a smaller percentage of CD103+DCs in the spleen of fB−/− compared to WT recipients (Supplementary Fig. S4D). However, the number of these splenic DCs were comparable in different recipient types (Supplementary Fig. S4H). Both the number (Supplementary Fig. S4E) and percentage (Supplementary Fig. S4I) of splenic CD11b+DCs were remarkably increased in fB−/− compared to WT recipients. DCs also expressed a comparable level of co-stimulator receptor CD80 in both fB−/− and WT recipients (Supplementary Fig. S4F and J). Taken together, absence of the AP drives an anti-inflammatory phenotype of DCs and T-cells selectively in target organs, but not in lymphoid organs, after allo-HCT.
Figure 2. Host AP deficiency affects donor T-cell migration and recipient DC activation after HCT.
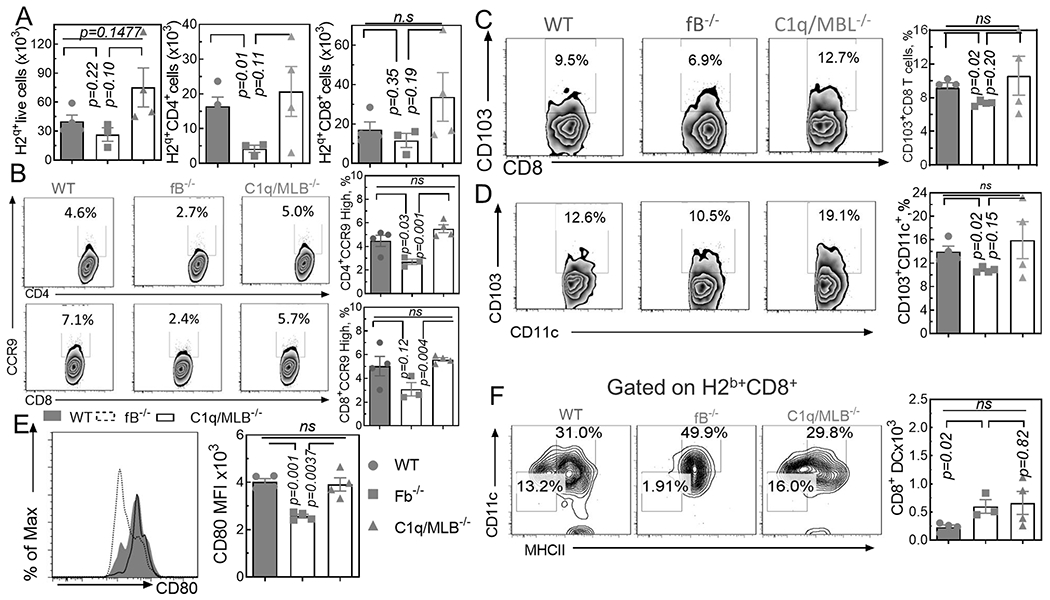
Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (2.0x106/mouse) from FVB donor mice. After 7 days, recipient intestinal lymphocytes were isolated and stained for analysis by flow cytometry. (A) Graphical summary of frequencies and absolute numbers of donor lymphocytes. H2q+(left), H2q+CD4+(center), H2q+CD8+(right) are shown; (B) Representative flow diagrams for frequencies of CCR9high cells among gated donor CD8+ cells. Graphical summary of donor CD4+ (top) and CD8+ (bottom) cells are shown; (C) Representative flow diagrams and graphical summary for frequencies of CD103+CD8+ among gated H2q+ cells; (D) Representative flow diagrams and graphical summary for frequencies of CD103+CD11c+ and H2b+CD11c+ among gated H2b+ cells; (E) Representative histogram and graphical summary of CD80 expression among H2b+CD11c+ cells (F) Representative flow diagrams of CD8+CD11c+MHCII+ among gated H2b+ cells and graphical summary of absolute numbers are shown. Unpaired two-tail t-test was used to compare between two individual groups. Data is presented as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 3. Host AP deficiency affects donor T-cell differentiation in recipient liver and lung as well as thymic function after HCT.
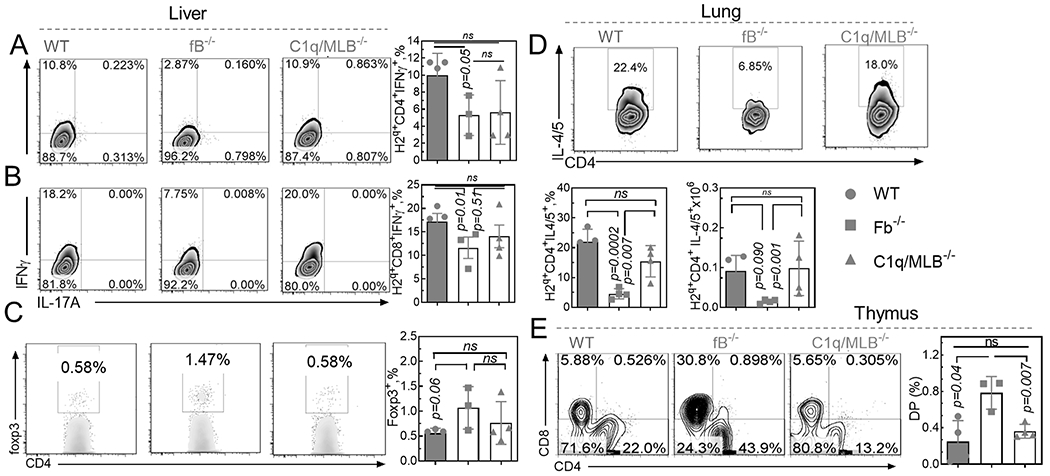
Lethally irradiated WT B6, fB−/−, or C1q/MBL−/− mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (2.0x106/mouse) from FVB donor mice. After 7 days, cells were isolated from recipient liver (A-C), lung (D) and thymus (E) and analyzed by flow cytometry. (A-C) Representative flow diagrams and graphical summary for frequencies of of H2q+CD4+IFN-γ+(A), H2q+CD8+IFN-γ+ (B), and H2q+CD4+foxp3+ (C)- in the liver are shown. (D) Representative flow diagrams and graphical summary for frequencies of H2q+CD4+IL-4/5+ cells in the lung are shown. (E) Representative flow diagrams and graphical summary for frequencies of CD4+CD8+ cells in the thymus of recipients are shown. Unpaired two-tail t-test was used to compare between groups. Data is presented as mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001.
Our results thus far indicate that activation of the AP is critical for GVHD pathogenicity. As such, we hypothesized that specific and site-targeted inhibition of the AP in GVHD target organs would effectively control GVHD. We utilized a fusion construct comprised of a complement-inhibitory domain (fH) attached to a complement receptor 2 (CR2) fragment in order to selectively bind C3d, a complement activation product that is deposited in GVHD target organs (Fig. 1A). In initial studies, we labeled the inhibitor with a fluorophore to perform a visual “mapping” of its distribution among target organs in recipient animals (Fig. 4A). Balb/c recipients were transplanted with T-cells from luciferase-transduced B6 donors. Migration of donor T-cells to the intestine was observed in allogeneic (GVHD), but not syngeneic (without GVHD) recipients 7d post allo-HCT (Fig. 4A). High fluorescence signal (CR2-fH) was detected in the colon (Fig. 4B), yet low signal was observed in lymphoid (spleen) and non-GVHD organs (heart) of GVHD recipients (Fig. 4B). The localization of CR2-fH in conjuction with donor lymphocytes to target organs suggests that CR2-fH preferentially affects complement generation from immune cells in GVHD target organs of transplanted recipients.
Figure 4. CR2-fH treatment effectively ameliorates GVHD and preserves the GVL effect.

Lethally irradiated B6 or BALB/c recipients were transplanted with WT B6 TCD-BM plus T-cells from luciferase-transduced B6 donors. On day 7 after HCT, labeled CR2-fH was injected via tail vein. (A) Experimental schematic (top) and representative BLI images (bottom) taken 9 days post-HCT are shown. (B) Representative images of labelled CR2-fH organ localization via Maestro system are shown. (C-E) Lethally irradiated B6 mice were transplanted with TCD-BM (5x106/mouse) with or without T-cells (0.6x106/mouse) from FVB donors and with or without luciferase-tranduced C1498 tumor cells. Recipient mice were treated with vehicle or CR2-fH every other day for 12 days. Survival (C) and clinical score (D) are shown. (E) Representative BLI images taken throughout the experiment are shown. The log-rank (Mantel-Cox) test (C), nonparametric Mann-Whitney U test (D) were used to compare between groups. Data is presented as mean ± S.E.M. and *p < 0.05, **p < 0.01, ***p < 0.001.
Local inhibition of AP with CR2-fH decreases GVHD damage in target organs yet spares the GVL effect
We next evaluated the effect of CR2-fH treatment on the GVL effect after allo-HCT. Given that CR2-fH has a tissue half-life of 2-3 d (30), we treated recipients (0.5 mg/mouse) with CR2-fH or vehicle (PBS) every other day for 14d post allo-HCT. We observed that CR2-fH treatment effectively reduced GVHD development (Fig. 4C–E). Furthermore, there was a significant reduction in number of recipients treated with CR2-fH that were observed with a leukemia relapse compared to PBS-treated controls (Fig. 4E). These results demonstrate that blocking the AP prevents GVHD while preserving GVL activity.
CR2-fH treatment maintains GVL activity via preserving the anti-tumor function of donor CD8+T-cells
To substantiate our observation, we examined the effect of CR2-fH on GVHD and GVL activity using a more aggressive and clinically relevant acute lymphoblastic leukemia, MLL-AF9. We observed that CR2-fH treatment dramatically improved recipient survival and decreased GVHD severity (Fig. 5A–B). Additionally, on contrary to tumor relapse observed in the recipients of BM and MLL-AF9, the percentages of MLL-AF9 was markedly reduced in the recipients of BM and MLL-AF9 plus T-cells regardless of CR2-fH treatment (Fig. 5C). Together, these evidiences indicate that CR2-fH treatment reduces GVHD severity while preserving GVL activity. Donor CD8 T cells play a dominant role in mediating the GVL effect via CTL activity(31). In this context, we explored the mechanisms by which CR2-fH treatment preserved GVL activity. At 30d post allo-HCT, the expression of key molecules driving CTL activity, granzyme B and perforin, were preserved with CR2-fH treatment (Fig. 5D), which correlates with preserved GVL activity upon CR2-fH treatment.
Figure 5. CR2-fH treatment maintains GVL activity against acute leukemia via preserving the donor CD8 CTL activity.

Lethally irradiated BALB/c recipients were transplanted with TCD-BM (5x106/mouse) with or without T-cells (0.75x106/mouse) from WT B6 donors and with or without MLL-AF9 tumor cells (10,000 MLL-AF9/mouse). CR2-fH and vehicle treatment were administered as described in Figure 4. (A,B) Recipient survival (A) and body weight loss (B) are shown. (C) Representative flow diagrams of CD11b+GFP+ (MLL-AF9) taken 30 days post-HCT from peripheral blood samples and stained for analysis by flow cytometry are shown. (D) Graphical summary for expression of cytolytic markers granzyme B (left) and perforin (right) on the surface of blood lymphocytes in vehicle and CR2-fH treated recipients. Log-rank (Mantel-Cox) (A) and unpaired two-tail t-test (D) was used to compare between groups. Data is presented as mean ± SEM.
Complement activation via AP manifests in the intestines of GVHD patients
To extend our findings towards clinical application, we analyzed complement deposition in the colon of patients either with GVHD or without GVHD based on pathologic diagnosis (Supplementary Table S1). We observed that the deposition of complement C3d was higher in biopsies of GVHD patients (Fig. 6A), and that increased C3d deposition was positively correlated with the number of CD3+ lymphocytes (Fig. 6A–B). We also found a trend indicating higher expression of fB in the intestines of GVHD patients (Fig. 6A–B). Together, these findings suggest that complement activation through the AP likely contributes to GVHD development in patients after HCT.
Figure 6. CR2-fH treatment reduces GVHD severity in human-to-mouse xenograft models.

(A-B) Paraffin-fixed patient colon samples were stained with anti-C3d, anti-fB, anti-CD3e and mounted with DAPI cover slips. Images were taken using Zeiss880 and analyzed by Zen 2.0. (A, B) Representative images of C3d (left; n=12) and fB (right; n=8) densities (A) and graphical summaries (B) are shown. (C-D) NSG-A2(+) mice were irradiated (280 cGy), treated with CR2-fH as described in Figure 5, and transfused with HLA-A2(−) PBMCs (15×106/mouse). (C) Recipient survival (top) and weight loss (bottom) are shown. (D) Representative flow diagrams of recipient blood samples collected on day 21 (top) and day 30 (bottom) post-transplant are shown. Unpaired two-tail t-test (B), log-rank (Mantel-Cox) test (C) were used to compare between groups. Data is presented as mean ± SEM; and *p < 0.05, **p < 0.01, ***p < 0.001.
CR2-fH treatment suppresses GVHD in xenograft models
To further enhance translational potential, we examined the effect of CR2-fH in a human-to-mouse xenograft model. Sublethally irradiated NSG-A2(+) mice were transplanted with HLA-A2(−) PBMCs and treated with vehicle or CR2-fH. We found that CR2-fH treatment sufficiently prevented GVHD progression in transplanted recipients. No recipient mortality was observed in transplanted recipients treated with CR2-fH (Fig. 6C). In addition, treated recipients had improved body weight maintenance and donor engraftment was not affected(Fig. 6D).
Discussion
Complement can be activated via three different pathways(19), but the role of each individual pathway in GVHD has not been extensively studied. Here, we show that specifically inhibiting the AP of complement activation reduces local inflammation within target organs. Further, these data represent a novel approach to treat GVHD while preserving the GVL effect. While a predominant role for the AP in the progression of various autoimmune diseases has been reported, we show for the first time that complement activation through the AP, specifically in target organs, also drives GVHD development after allo-HCT (Fig. 1E). It is not clear how the AP is activated, but intestinal damage caused by total body irradiation (TBI) conditioning induces translocation of LPS in the gut lumen and exacerbates GVHD severity (32). We found that the AP drives complement activation and GVHD via its activity in target organs (Fig. 1 D–G and Fig. 6A), but that effects on lymphoid organs were arbitrary. Indeed, AP deficiency decreased the capacity for alloantigen-presentation of DCs and reduced the generation of a pathogenic phenotype for DCs in target organs.
Although the role of DC-derived complement has been reported(6), it is not clear which pathway is the primary contributor to the complement deposition in GVHD target organs; in particular intestinal GVHD. In the current study, we demonstrated that complement activation through the AP is critical for the complement generated in injured target organs (Fig. 1G). Specifically, we have shown that complement effector proteins derived from host DCs play a dominate role in promoting donor T cell allogeneic responses and GVHD pathogenicity (Supplementary Fig S3 C–D). Our previous study indicates that complement regulates recipient DCs via modulating their survival during GVHD (5). The current study provides evidence that complement activation via the AP is important for DC’s ability to generate effector molecules. These deposited complement products are able to regulate the antigen presentation capacity of DCs, which is required for GVHD development.
Complement activation has been observed in non-hematopoietic cells, such as fibroblasts(33), endothelial(34), and epithelial cells (35). Despite their capacity to prime T cells, host hematopoietic APCs (DCs, B cells and macrophages) are required for maximum activation of donor T cells and subsequent GVHD induction after allo-HCT (9). Therefore, the current study suggests an important role for host DCs in amplifying complement activation and deposition in GVHD target/injured tissues.
Complement activation results in differentiation of naïve T-cells into Th1/Tc1 through the generation of the anaphylatoxins C3a and C5a (36), and the opsonin C3d (37). We found that, in the absence of the AP, donor T cells skew from Th1/Tc1-differentiation toward Treg generation in GVHD target organs (Fig. 3). Given Th1 cells are pathogenic and Tregs are suppressive in GVHD, T cell differentiation is an important factor that contributes to effect of the AP on GVHD development (38). Since stimulating DCs by complement-activating products (C3d, C3a, C5a) creates Th1/Th17 priming cytokines (39,40), the effect of AP generated complement products on DCs may precede the modulation of donor T cell immunity in GVHD recipients.
T cell migration to GVHD target organs is critical for GVHD development(41). As complement activation generates the chemotactic molecule C5a, which is important for lymphocyte migration to target organs(42), reduced levels of complement activation may lead to fewer infiltrating donor lymphocytes in target organs of AP-deficient recipients (Fig. 2A). In addition, the decreased expression of target organ-homing chemokines, such as CCR9 (43), on donor T-cells (Fig. 2B) could likely contribute to less T cell accumulation in the intestine of recipients with GVHD.
The intestine is a primary site of organ damage in GVHD, and intestinal injury also plays a role in amplifying systemic GVHD development(38). In the current study, this association is linked with GVHD and complement activation in the intestines in both mice and humans (Fig 6A, B). Our findings are consistent with a previous study, in which C3 deposition was seen in the skin of acute GVHD patients(37). Importantly, we provide compelling evidence that local complement activation, specifically in injured target organs, plays an important role in organ damage during GVHD development (Fig. 4A). Furthermore, our study shows that complement effector molecules involved in the pathogenesis of GVHD are generated mainly through the AP in recipients with intestinal GVHD (Fig. 4A). In contrast, we did not observe a significant role for the CP/LP in GVHD development. Consistently, immunological characteristics of GVHD-driven immune cells in target organs were similar between WT and C1q/MBL−/− recipient mice (Fig. 2, 3). Presumably this is due to the fact that complement activation through the CP/LP is dependent on the AP to propragate maximal damage in GVHD target organs (21).
Together, these data suggest that inhibition of the AP in GVHD target organs may provide a therapeutic option for controlling GVHD. Indeed, we found that pharmacologically targeting the AP during complement activation in GVHD target organs effectively protected against GVHD while preserving GVL activity after allo-HCT (Fig. 4C–E). We reasoned that complement inhibition did not affect the differentiation and expansion of donor T-cells in lymphoid organs, which may permit these T-cells to maintain their role in mediating the GVL effect after allo-HCT(44). In line with this notion, the immunological phenotype of DCs and T-cells were largely unaffected by AP deficiency in the lymphoid organs of transplanted recipients (Supplementary Fig. S4). Accordingly, the CTL activity of donor CD8+ T-cells(45), an important factor responsible for GVL activity after allo-HCT, is maintained during CR2-fH treatment.
In conclusion, we investigated the role of complement in GVHD/GVL responses after allo-HCT. Specifically, we demonstrate that complement activation, specifically through the host-derived AP activation, is critical for GVHD pathogenesis. Furthermore, specific and localized inhibition of the AP using a site-targeted inhibitor provided protection from GVHD while maintaining GVL activity after allo-HCT. Given that a humanized version of CR2-fH, termed TT30, has been in clinical trial for Paroxysmal nocturnal hemoglobinuria (PNH) patients (46), the current findings have high translational potential for patients undergoing allo-HCT.
Supplementary Material
Translational Relevance.
Allogeneic hematopoietic cell transplantation (allo-HCT) is a potentially curative immunotherapy for hematological malignancies derived from the T cell-mediated graft-versus-leukemia (GVL) effect; yet its therapeutic application is limited by the development of graft-versus-host-disease (GVHD). The complement pathway is part of the innate immune response and complement activation has been implicated in GVHD pathobiology. Complement can be activated via three different pathways: the classical (CP), lectin (LP) and alternative pathway (AP). Here, we found that pharmacological blockade of complement activation, specifically the alternative pathway (AP), effectively ameliorates GVHD severity. Importantly, GVL activity post allo-HCT was largely preserved against multiple lines of leukemia and lymphoma. A humanized version of the complement AP inhibitor, TT30, is currently available. Thus, our findings have great potential for clinical application in patients undergoing allo-HCT.
Acknowledgements.
Financial Support: This work is supported by NIH grants R01s HL140953 and R01HL137373 to X-Z. Yu. University of Central Florida (UCF) Start-up Grant # 2540-0715 to H. Nguyen. Institutional resources at the Medical University of South Carolina were supported by NIH support C06 RR15455 and P30 CA138313 grants (to Hollings Cancer Center).
We thank cell and molecular imaging, flow cytometry, and pathology cores at the Medical University of South Carolina (MUSC) for their valuable services. We also thank Dr. Angie Duong at MUSC for her coordination in obtaining patient biopsies.
Footnotes
Disclosure of Potential Conflicts of Interest. The authors have no conflicts of interest to declare.
REFERENCES
- 1.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol 2007;7(5):340–52 doi 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 2.Appelbaum FR. Haematopoietic cell transplantation as immunotherapy. Nature 2001;411(6835):385–9 doi 10.1038/35077251 35077251 [pii]. [DOI] [PubMed] [Google Scholar]
- 3.Pasquini MC, Wang Z, Horowitz MM, Gale RP. 2010. report from the Center for International Blood and Marrow Transplant Research (CIBMTR): current uses and outcomes of hematopoietic cell transplants for blood and bone marrow disorders. Clin Transpl 2010:87–105. [PubMed] [Google Scholar]
- 4.Tsoi MS, Storb R, Jones E, Weiden PL, Shulman H, Witherspoon R, et al. Deposition of IgM and complement at the dermoepidermal junction in acute and chronic cutaneous graft-vs-host disease in man. J Immunol 1978;120(5):1485–92. [PubMed] [Google Scholar]
- 5.Nguyen H, Kuril S, Bastian D, Kim J, Zhang M, Vaena SG, et al. Complement C3a and C5a receptors promote GVHD by suppressing mitophagy in recipient dendritic cells. JCI Insight 2018;3(24) doi 10.1172/jci.insight.121697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwan WH, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest 2012;122(6):2234–8 doi 10.1172/JCI61019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gloude NJ, Khandelwal P, Luebbering N, Lounder DT, Jodele S, Alder MN, et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood 2017;130(10):1259–66 doi 10.1182/blood-2017-05-782870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, Heeger PS. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J Immunol 2013;190(12):5921–5 doi 10.4049/jimmunol.1300847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraverty R, Sykes M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood 2007;110(1):9–17 doi 10.1182/blood-2006-12-022038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 2008;28(3):425–35 doi 10.1016/j.immuni.2008.02.001 S1074-7613(08)00071-X [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA, Laing A, et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol 2012;13(12):1213–21 doi 10.1038/ni.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 2016;352(6292):aad1210 doi 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Friec G, Kohl J, Kemper C. A complement a day keeps the Fox(p3) away. Nat Immunol 2013;14(2):110–2 doi 10.1038/ni.2515. [DOI] [PubMed] [Google Scholar]
- 14.Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 2015;42(6):1033–47 doi 10.1016/j.immuni.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant 2013;13(10):2530–9 doi 10.1111/ajt.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seignez A, Joly AL, Chaumonnot K, Hazoume A, Sanka M, Marcion G, et al. Serum Gp96 is a chaperone of complement-C3 during graft-versus-host disease. JCI Insight 2017;2(6):e90531 doi 10.1172/jci.insight.90531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Q, Li D, Carreno R, Patenia R, Tsai KY, Xydes-Smith M, et al. Complement component C3 mediates Th1/Th17 polarization in human T-cell activation and cutaneous GVHD. Bone Marrow Transplant 2014;49(7):972–6 doi 10.1038/bmt.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nat Rev Immunol 2012;12(6):431–42 doi 10.1038/nri3225 nri3225 [pii]. [DOI] [PubMed] [Google Scholar]
- 19.Chen M, Daha MR, Kallenberg CG. The complement system in systemic autoimmune disease. J Autoimmun 2010;34(3):J276–86 doi 10.1016/j.jaut.2009.11.014. [DOI] [PubMed] [Google Scholar]
- 20.Arroyave CM, Wilson MR, Tan EM. Serum factors activating the alternative complement pathway in autoimmune disease: description of two different factors from patients with systemic lupus erythematosus. J Immunol 1976;116(3):821–6. [PubMed] [Google Scholar]
- 21.Manickam B, Jha P, Matta B, Liu J, Bora PS, Bora NS. Inhibition of complement alternative pathway suppresses experimental autoimmune anterior uveitis by modulating T cell responses. J Biol Chem 2011;286(10):8472–80 doi 10.1074/jbc.M110.197616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noris M, Donadelli R, Remuzzi G. Autoimmune abnormalities of the alternative complement pathway in membranoproliferative glomerulonephritis and C3 glomerulopathy. Pediatr Nephrol 2018. doi 10.1007/s00467-018-3989-0. [DOI] [PubMed] [Google Scholar]
- 23.Naymagon S, Naymagon L, Wong SY, Ko HM, Renteria A, Levine J, et al. Acute graft-versus-host disease of the gut: considerations for the gastroenterologist. Nat Rev Gastroenterol Hepatol 2017;14(12):711–26 doi 10.1038/nrgastro.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 1996;88(8):3230–9. [PubMed] [Google Scholar]
- 25.Nguyen HD, Chatterjee S, Haarberg KM, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest 2016;126(4):1337–52 doi 10.1172/JCI82587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alawieh A, Langley EF, Weber S, Adkins D, Tomlinson S. Identifying the role of complement in triggering neuroinflammation after traumatic brain injury. J Neurosci 2018. doi 10.1523/JNEUROSCI.2197-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West EE, Kolev M, Kemper C. Complement and the Regulation of T Cell Responses. Annu Rev Immunol 2018;36:309–38 doi 10.1146/annurev-immunol-042617-053245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou S, Ueta H, Xu XD, Shi C, Matsuno K. Predominant donor CD103+CD8+ T cell infiltration into the gut epithelium during acute GvHD: a role of gut lymph nodes. Int Immunol 2008;20(3):385–94 doi 10.1093/intimm/dxm153. [DOI] [PubMed] [Google Scholar]
- 29.Koyama M, Cheong M, Markey KA, Gartlan KH, Kuns RD, Locke KR, et al. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J Exp Med 2015;212(8):1303–21 doi 10.1084/jem.20150329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, et al. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J Neuroinflammation 2015;12:247 doi 10.1186/s12974-015-0464-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss L, Lubin I, Factorowich I, Lapidot Z, Reich S, Reisner Y, et al. Effective graft-versus-leukemia effects independent of graft-versus-host disease after T cell-depleted allogeneic bone marrow transplantation in a murine model of B cell leukemia/lymphoma. Role of cell therapy and recombinant IL-2. J Immunol 1994;153(6):2562–7. [PubMed] [Google Scholar]
- 32.Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, et al. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest 2001;107(12):1581–9 doi 10.1172/JCI12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garred P, Hetland G, Mollnes TE, Stoervold G. Synthesis of C3, C5, C6, C7, C8, and C9 by human fibroblasts. Scand J Immunol 1990;32(5):555–60. [DOI] [PubMed] [Google Scholar]
- 34.Fischetti F, Tedesco F. Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 2006;39(5):417–28 doi 10.1080/08916930600739712. [DOI] [PubMed] [Google Scholar]
- 35.Andoh A, Kinoshita K, Rosenberg I, Podolsky DK. Intestinal trefoil factor induces decay-accelerating factor expression and enhances the protective activities against complement activation in intestinal epithelial cells. J Immunol 2001;167(7):3887–93. [DOI] [PubMed] [Google Scholar]
- 36.Barchet W, Price JD, Cella M, Colonna M, MacMillan SK, Cobb JP, et al. Complement-induced regulatory T cells suppress T-cell responses but allow for dendritic-cell maturation. Blood 2006;107(4):1497–504 doi 10.1182/blood-2005-07-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Groot AS, Ross TM, Levitz L, Messitt TJ, Tassone R, Boyle CM, et al. C3d adjuvant effects are mediated through the activation of C3d-specific autoreactive T cells. Immunol Cell Biol 2015;93(2):189–97 doi 10.1038/icb.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med 2017;377(22):2167–79 doi 10.1056/NEJMra1609337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raedler H, Heeger PS. Complement regulation of T-cell alloimmunity. Curr Opin Organ Transplant 2011;16(1):54–60 doi 10.1097/MOT.0b013e3283425419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RA, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a-C3aR interaction. Blood 2008;111(4):2452–61 doi blood-2007-06-095018 [pii] 10.1182/blood-2007-06-095018. [DOI] [PubMed] [Google Scholar]
- 41.Hayashida JN, Nakamura S, Toyoshima T, Moriyama M, Sasaki M, Kawamura E, et al. Possible involvement of cytokines, chemokines and chemokine receptors in the initiation and progression of chronic GVHD. Bone Marrow Transplant 2013;48(1):115–23 doi 10.1038/bmt.2012.100. [DOI] [PubMed] [Google Scholar]
- 42.Gerard C Inflammatory chemokines: tuned in, turned on, dropped out. Nat Immunol 2005;6(4):366–8 doi ni0405-366 [pii] 10.1038/ni0405-366. [DOI] [PubMed] [Google Scholar]
- 43.Castor MG, Pinho V, Teixeira MM. The role of chemokines in mediating graft versus host disease: opportunities for novel therapeutics. Front Pharmacol 2012;3:23 doi 10.3389/fphar.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolb HJ. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 2008;112(12):4371–83 doi 10.1182/blood-2008-03-077974. [DOI] [PubMed] [Google Scholar]
- 45.Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat Rev Cancer 2004;4(5):371–80 doi 10.1038/nrc1365. [DOI] [PubMed] [Google Scholar]
- 46.Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood 2011;118(17):4705–13 doi 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.