

Abstract
Purpose:
Children with Down syndrome (DS, constitutive trisomy 21) that develop acute lymphoblastic leukemia (DS-ALL) have a 3-fold increased likelihood of treatment-related mortality coupled with a higher cumulative incidence of relapse, compared to other children with B-cell acute lymphoblastic leukemia (B-ALL). This highlights the lack of suitable treatment for DS children with B-ALL.
Experimental design:
To facilitate the translation of new therapeutic agents into clinical trials, we built the first preclinical cohort of patient-derived xenograft (PDX) models of DS-ALL, comprehensively characterized at the genetic and transcriptomic levels, and have proven its suitability for preclinical studies by assessing the efficacy of drug combination between the MEK inhibitor Trametinib and conventional chemotherapy agents.
Results:
Whole exome and RNA-sequencing experiments revealed a high incidence of somatic alterations leading to RAS/MAPK pathway activation in our cohort of DS-ALL, as well as in other pediatric B-ALL presenting somatic gain of the chromosome 21 (B-ALL+21). In murine and human B cell precursors, activated KRASG12D functionally cooperates with trisomy 21 to deregulate transcriptional networks that promote increased proliferation and self-renewal, as well as B-cell differentiation blockade. Moreover, we revealed that inhibition of RAS/MAPK pathway activation using the MEK1/2 inhibitor Trametinib decreased leukemia burden in several PDX models of B-ALL+21, and enhanced survival of DS-ALL PDX in combination with conventional chemotherapy agents such as vincristine.
Conclusions:
Altogether, using novel and suitable PDX models, this study indicates that RAS/MAPK pathway inhibition represents a promising strategy to improve the outcome of DS children with B-cell precursor leukemia.
Keywords: Down syndrome, leukemia, oncogenic cooperation, RAS/MAPK, trametinib
Introduction
B-cell precursor Acute Lymphoblastic Leukemia (B-ALL) is the most common type of childhood malignancy. Children with Down syndrome (DS) face a 27-fold increased risk to develop B-ALL (known as DS-ALL) (1), associated with a worse outcome and a significantly lower Event-free survival (EFS: 64% vs 81%) and overall survival rate (OS: 74% vs 89%) compared to other children (2). Treatment intensification is limited in DS-ALL due to a higher therapy-related morbidity (TRM) (7% vs 2%), ultimately leading to a higher rate of relapses (26% versus 15%) (2, 3). This emphasizes the need to better understand the mechanisms driving leukemia in DS-ALL, so that novel and more targeted treatments can be developed for these children.
At the genetic level, apart from the constitutive trisomy 21, DS-ALL presents a normal karyotype in more than 40% of cases, and a lower incidence of ETV6-RUNX1 or High Hyperdiploid (HeH) karyotypes (2, 4). Half of DS-ALL samples overexpress cytokine receptor-like factor 2 (CRLF2) either through microdeletions on chromosome X (P2RY8-CRLF2 fusion) or translocations to the IGH locus (5–7). JAK2 activating mutations, affecting the Arginine R683 residue located in the pseudokinase domain, are also more frequently found in DS-ALL than in any other subgroups of pediatric B-ALL (8, 9), suggesting an oncogenic cooperation between cytokine signaling activation and constitutive trisomy 21. Other genetic alterations affecting NRAS, KRAS, IKZF1, PAX5 or CDKN2A/B genes are commonly found in DS- and non-DS B-ALL (10, 11).
The development of DS-ALL models, from the transgenic Ts1Rhr mice (trisomic for the Down Syndrome Critical Region (DSCR)) (12), has highlighted the complexity of DS-ALL leukemogenesis in vivo (13). Indeed, four additional alterations affecting CRLF2, JAK2, IKZF1 and PAX5 genes, added to trisomy 21, were required to drive a B-ALL phenotype in this model. Moreover, the lack of cellular models of DS-ALL precludes the development of simple screening assays to identify novel therapeutic approaches.
In non-DS pediatric B-ALL, complete or partial gain of chromosome 21 (+21) is seen in 30-40% of cases. More than 90% of HeH-ALL are tetrasomic for the whole chromosome 21; some HeH cases harbor three additional copies of this chromosome (14). Intrachromosomal amplification of chromosome 21 (a.k.a. iAMP21) is a high-risk cytogenetic abnormality in pediatric B-ALL and is characterized by a pattern of deleted/amplified regions of the chromosome 21, with the aberration on chromosome 21 considered to be the initiating event (15). Trisomy 21 has also been reported in 15% of ETV6-RUNX1 B-ALL (16). Interestingly, the minimal region of amplification on chromosome 21, found in both HeH and iAMP21 B-ALL overlap with the DSCR (14, 15). This strongly suggests that the increased dosage of specific genes located in this DSCR region plays a central role in B cell leukemogenesis, irrespective of whether it results from a constitutive (DS) or somatic (HeH or iAMP21) chromosomal abnormality. Among them, trisomy of Hmgn1 (encoding the High mobility group nucleosomal binding protein 1) has been shown to suppress the Histone H3 tri-methylation of Lys27 (H3K27me3) repressive mark and induce global transcriptional amplification to promote B-cell proliferation and B cell precursor phenotypes (13, 17). Moreover, the DYRK1A (Dual specificity tyrosine-phosphorylation-regulated kinase 1A) regulates B cell proliferation and differentiation (18), and increased dosage of Dyrk1a predisposes to DS-leukemia (19). Together, these results highlight that at least 2 genes within the DSCR promote B cell leukemia. However, the molecular bases of oncogenic cooperation between constitutive trisomy 21 and the somatic alterations found in DS-ALL remain largely unknown.
In this study, we showed that trisomy 21 functionally cooperates with constitutive activation of RAS/MAPK pathway in murine and human B-cell precursors, indicating that disrupting this mechanism may have a therapeutic impact for DS children with B-cell leukemia. We built a comprehensive and preclinical cohort of patient-derived xenograft (PDX) models, and show efficacy of MEK1/2 inhibition on human B-ALL samples harboring NRAS, KRAS, JAK2 and CBL mutations. Altogether, these results indicated that the RAS/MAPK inhibitor Trametinib is therapeutically exploitable to improve the outcome of DS children with B-ALL when combined with conventional chemotherapy agents.
Material and Methods
Human samples and sequencing
Human peripheral blood (PB) and bone marrow (BM) samples were collected from Trousseau, Necker, Tours and Toulouse hospitals (France), and Perth Children’s Hospital (Australia). Nine samples were provided under the MAPPYACTS protocol (clinicaltrial.gov: NCT02613962). All human samples were obtained with the written or signed consent of the patient or parents/guardians. This study conforms to the provisions of the Declaration of Helsinki, was approved by independent ethics committees and complied to institutional, local and national regulations.
Cells were subjected to Ficoll gradient and then either used fresh or frozen in FBS with 10% DMSO. DNA and RNA were extracted using RNA/DNA/Protein Purification Plus kit (Norgen). WES libraries were prepared using SureSelect All Exon V5 Clinical Research Exome V1 or CREV2 kits (Agilent). Sequencing was performed on a HiSeq2000 (Illumina) with a median depth of 100X. Detection of SNVs and small indels were done using Varscan 2.3.7 (20). RNAseq libraries were prepared using SureSelect Automated Strand Specific RNA Library Preparation kit and sequenced to obtain at least 100M reads. Quality control was performed using FastQC (v0.11.7, Babraham Institute) and quantification of transcript expression was performed with Salmon software (v0.11.2)(21). The differential gene expression analysis was performed using R (v3.4.0) and DESeq2 (v1.18.1) (22).
Animal models
Ts1Rhr mice have been previously described (12). 6-8 weeks old Ts1Rhr and wild-type controls bone marrow (BM) cells, after red cells lysis, were cultivated overnight in IMDM medium supplemented with 20% FBS, penicillin (100U/mL)-streptomycin (100μg/mL), 2mM L-Glutamine, 1mM sodium pyruvate, 50nM 2-mercaptoethanol and recombinant murine IL7, SCF and FLT3-L at 10ng/mL. BM cells were spin-infected in culture media containing 5μg/mL polybrene and 7.5mM HEPES buffer as previously described, with retroviral particles produced from the murine stem cell virus (MSCV)-IRES-mCherry or MSCV-IRES-GFP encoding human KRASG12D or NRASG12D respectively. After 24h, 8000 B220+cherry+ sorted cells were plated in M3630 and CFU-preB colonies were scored every week.
To generate the PDX, 0.1-2x106 B-ALL cells from 30 patients (BM or PB) were injected into sub-lethally irradiated (2.5Gy) NOD.Cg-PrkdcscidIl2rgtm1Wjll/SzJ (NSG) mice by intrafemoral (i.f) injection. Subsequent NSG generations were injected with 0.1 to 1×106 cells per animal intravenous (i.v) or i.f. Luciferase expressing models were developed by transduction of B-ALL cells from primografts with lentiviral particles obtained from the FUW-Luc-mCherry-puro vector (a gift from A. L. Kung, Dana Faber Cancer Institute, Boston). Bioluminescence monitoring was performed on mice injected with 150mg/kg of D-Luciferin, imaged with IVIS50 or Ami HT system; Bioluminescence intensity was expressed as photons per second per surface (p/s/cm2 = ROI).
Mice were maintained at the Gustave Roussy or Telethon Kids Institute preclinical facility and all experiments were approved by the French or Australian national animal care and use committee respectively.
Western blotting
To assess the constitutive activation of signalling pathways, B-ALL cells harvested from PDX were starved for 5–6 hours prior to protein extraction, washed and centrifuged at 1200 rpm for 5 min. Cell pellets were lysed in cold RIPA buffer (50mM Tris-HCL pH8, 1mM EDTA, 150mM NaCl, 1% NP40, 0.1% SDS and 0.5% DOC) supplemented with 1X Complete™ (Roche), 50 mM NAF, 2mM PMSF and 1mM Na3VO4. Proteins were separated on a NuPAGE Bis-Tris polyacrylamide gel and transferred onto a nitrocellulose membrane (GE Healthcare). Antibodies were purchased from Cell signaling and SantaCruz (Methods - Table S8). Quantification of band intensities was estimated using ImageJ or ImageQuant softwares.
Drug treatments
In vitro, 0.5-6x105 cells from PDX were plated in 100μL of RPMI 1640 medium supplemented with 10% FBS, penicillin (100U/mL)-streptomycin (100μg/mL), 2mM L-glutamine, and human IL7, SCF and FLT3-L at 10ng/mL. Dexamethasone, Methotrexate, Vincristine sulfate, Selumetinib and Trametinib (all purchased from MedKoo) were diluted in DMSO. After 72h of incubation, MTT assays (absorbance 490nm) was performed by adding 20μL/well of CellTiter 96® AQueous One Solution Reagent (Promega).
In vivo treatments (4 weeks) started when ROI reached 106 p/s/cm2, or when 1% of human blasts where detected in the peripheral blood. NSG mice were then treated with Trametinib (1.5mg/kg, oral gavage 5 times per week) (23), vincristine sulfate (0.5mg/kg, intra-peritoneal injection once a week) (24), or both. Mice were monitored by bioluminescence (IVIS Spectrum and Ami HT) and blood samplings. At sacrifice, flow cytometry analyses were used to determine the leukemic burden in the spleen and BM.
Flow cytometry analyses
For all flow cytometry experiments, cells were stained in PBS 1X supplemented with 2% FBS for 30 minutes at 4°C (Methods – Table S8). Analyses were performed with CANTO-II or CANTO-X instruments (BD), and data were analyzed using FlowJo software (version FlowJo 9.3.2). Murine B220+/mCherry positive cells and human B-ALL blasts were sorted on ARIAIII, Fusion or INFLUX instruments (BD).
Data availability
Whole exome sequencing and RNAseq data from primary samples were deposited into EGA under the accession number EGAS00001003760. RNAseq data from CFU-preB experiments are available on EBI under the accession number E-MTAB-8005.
Statistical analyses
Statistical analyses were performed using GraphPad Prism (version 6, GraphPad), unless otherwise mentioned. The log-rank test was used for survival analyses. For other experiments, statistical significance was evaluated using the Mann-Whitney U test or the two-tailed unpaired Student’s t-test.
Results
Genetic and transcriptomic landscapes of the B-ALL+21 cohort
To investigate the mechanisms of oncogenic cooperation in pediatric B-ALL+21, we collected 8 DS-ALL, 8 iAMP21, 16 HeH and 12 primary patient samples from other subgroups of pediatric B-ALL (referred to as ‘thers’) (Supplementary Table S1), and performed Whole Exome Sequencing (WES) and RNA-sequencing experiments to comprehensively characterize the mutational landscape of this B-ALL cohort. We identified a total of 2067 non-synonymous SNVs or Indels (n=86.1±46 per sample) in coding regions, with an average of 3.6±4.1 acquired predicted driver mutations per sample as determined by Cancer Genome Interpreter (Supplementary Table S2) (25). The most common alterations affected genes encoding signaling effectors (NRAS, KRAS, FLT3, JAK2, CRLF2, SH2B3 and CBL n=27/44 samples), especially in B-ALL+21 as confirmed independently in publicly available pediatric B-ALL cohorts (Supplemental Figure 1A), followed by somatic alterations of transcription factors (n=15), chromatin modifiers (n=12), and cell cycle regulators (n=11) (Figure 1, Supplementary Tables S2 and S3).
Figure 1: Mutational landscape of B-cell Acute Lymphoblastic Leukemia with gain of chromosome 21 (B-ALL+21).
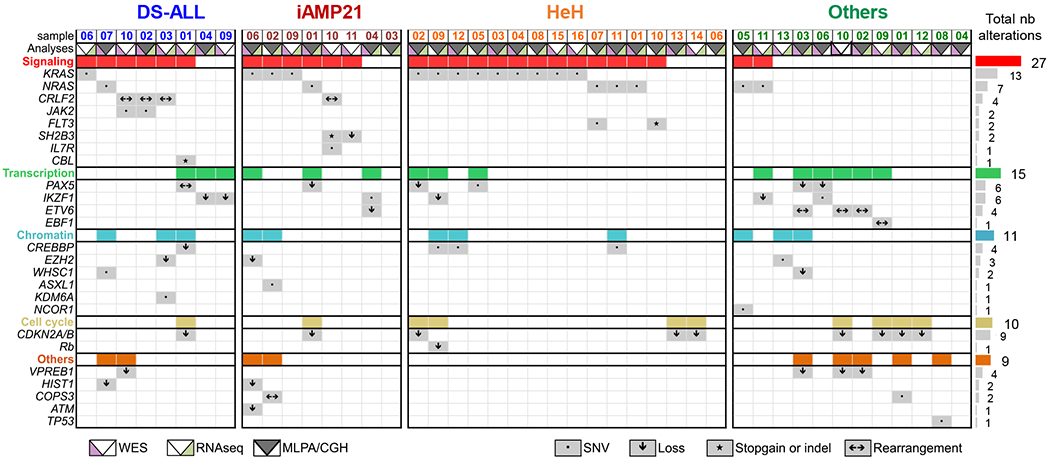
A. Table summarizing selected somatic ‘driver’ alterations obtained from WES, RNAseq and MLPA/CGH assays in our B-ALL cohort: Down syndrome (DS-ALL, n=8), intrachromosomal amplification of the chromosome 21 (iAMP21, n=8), high hyperdiploid (HeH, n=16) and the ‘Others’ subgroup (n=12), see also Supplemental Table 1. Grey boxes represent the presence of a somatic alteration and the symbols specify the type of alteration (SNVs, gains, losses, Indels or rearrangements). The colored boxes (red, green, blue, yellow and orange) integrate all alterations found within the functional subgroups: signaling, transcription, chromatin, cell cycle and others.
Next, we used RNA-sequencing to identify fusion transcripts in our cohort applying stringent selection criteria in which fusion transcripts must be detected in at least two different algorithms (including deFuse, CRAC, TopHat and TopHat Fusion, Supplementary Table S4). We detected ETV6-RUNX1 fusions in two samples from the ‘Others’ group (Others02 and Others03) and two novel ìn-framè transcripts fusing HM13 (Histocompatibility Minor 13) to REM1 (RRAD and GEM-like GTPase1) in the Others08 sample, and COPS3 to ACAD10 (Acyl-CoA Dehydrogenase Family Member 10) in iAMP02 (Supplementary Figure S1B, C). COPS3, also found to be mutated in Others01, encodes the COP9 signalosome subunit 3, known to mediate the stability of TP53, JUN and SOS1 (26). An òut-of-framè transcript PAX5-CTNND2, associated with the ectopic expression of Catenin Delta 2, was identified in the DS01 sample (Supplementary Figure S1D). RNA-seq experiments also detected the overexpression of CRLF2 in DS02, DS03 and DS010 samples, confirming data obtained by Multiplex ligation-dependent probe amplification (MLPA) and/or flow cytometry analyses.
Together, we detailed the mutational landscape of a cohort of 32 pediatric B-ALL+21 samples, representative of the genetic alterations commonly found in DS-ALL, iAMP21 and HeH, and highlighting a potential oncogenic cooperation between gain of chromosome 21 and RAS/MAPK pathway activation.
Constitutively active KRASG12D cooperates with +21 in murine and human samples
To better understand the molecular bases of this oncogenic cooperation, we first overexpressed KRASG12D in murine bone marrow cells harvested from 6–8 weeks old wild-type (WT) or partially trisomic (Ts1Rhr) mice. In methylcellulose assays, ectopic expression of KRASG12D increased the number and size of CFU-preB colonies in both WT and Ts1Rhr backgrounds (Figures 2A and B). KRASG12D expression in Ts1Rhr B cell precursors also enhanced their self-renewal capacity, as shown by serial replating experiments (Figure 2A). Similar observations were obtained using the mutant NRASG12D (Supplementary Figure S2A). Compared to WT-KRASG12D cells harvested from CFU-pre-B colonies and maintained in liquid cultures, Ts1Rhr-KRASG12D cells have a higher level of constitutive Erk1/2 phosphorylation in starved conditions (Figure 2C). Phenotypic analyses of the CFU colonies obtained at passage 1 (P1) revealed that KRASG12D expression, in wild-type or Ts1Rhr backgrounds, decreased the proportions of pre-pro-B (B220+CD19-Kit+), pre-B (B220+CD19+CD25+) and immature B cells (B220+CD19+IgM+), in favor of pro-B cells (CD43+B220+CD19+) (Supplementary Figure S2C).
Figure 2: Functional cooperation between constitutive trisomy 21 and KRASG12D.
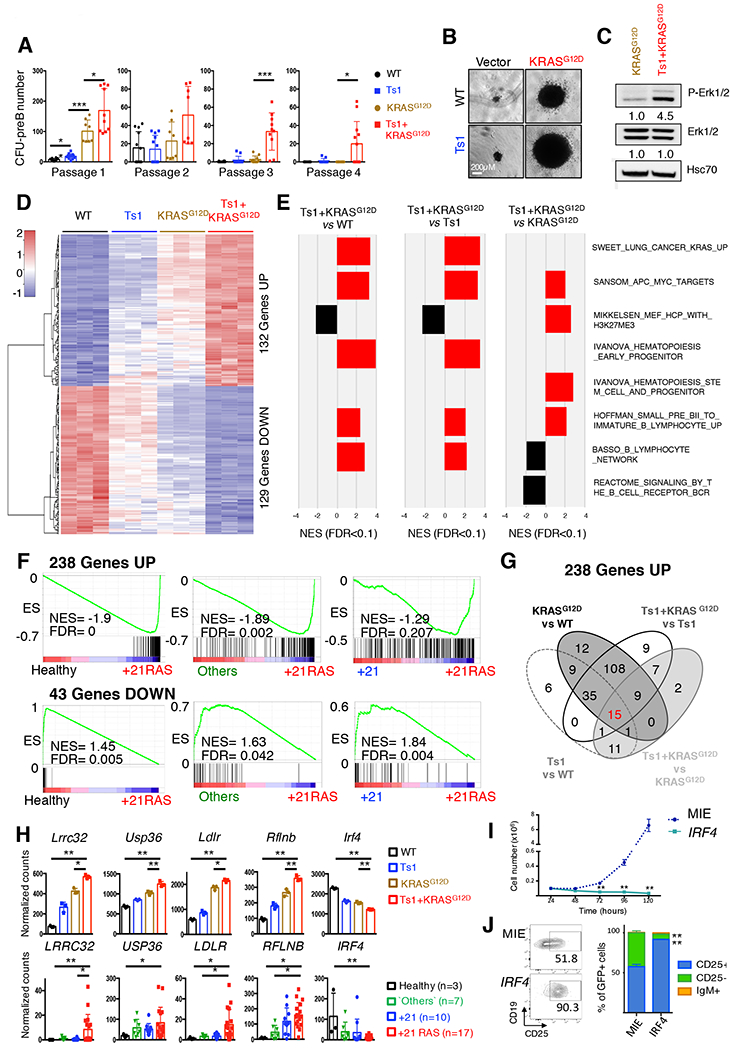
A. Number of CFU-preB colonies obtained per genotype (wild-type: WT and Ts1Rhr: Ts1) transduced with retroviral particles encoding KRASG12D or empty vector across four passages (n=8–10 replicates from 3 independent experiments; *p<0.05, **p<0.01 and ***p<0.001). B. Representative picture of the CFU-preB colonies observed at passage 1 in WT, Ts1, KRASG12D and Ts1-KRASG12D (scale bars, 0.2 mm). C. Western blot assessing the constitutive phosphorylation of Erk1/2 in starved WT-KRASG12D and Ts1Rhr-KRASG12D cells. Quantifications of the band relative intensities are indicated. D. Heatmap representing the expression of the 261 genes commonly deregulated in the WT, Ts1, KRASG12D and Ts1+KRASG12D murine B cell progenitors CFU-preB (132 Upregulated and 129 downregulated genes), n= 3 replicates per condition. E. Gene set enrichment analyses (GSEA) of selected enriched datasets; all FDR < 0.1, see also Supplemental Figure 4 and Table 6. F. GSEA of the 238 upregulated and 43 downregulated genes across subgroups of human B-ALL contained in our cohort: Others, +21 (gain of chromosome 21, no RAS alterations) and +21RAS (gain of chromosome 21 with N/KRAS mutations). G. Venn diagram assessing the association of the 238 upregulated genes among the murine paired comparisons Ts1 vs WT, KRASG12D vs WT, Ts1+KRASG12D vs Ts1 and Ts1+KRASG12D vs KRASG12D. The same analysis was performed with the 43 downregulated genes, leading to the identification of 18 ‘cooperative’ genes (see Supplemental Table 7). H. Normalized expression of five selected ‘cooperative’ genes, commonly deregulated in murine CFU-preB colonies (WT, Ts1, KRASG12D, Ts1+KRASG12D, upper panel) and in human primary B-ALL samples (lower panel). *p<0.05 and **p<0.001. I. Growth of sorted GFP-positive Ts1+KRASG12D ectopically expressing IRF4 compared to empty vector (MIE). **p<0.001. J. Representative dot plots (left panel) and associated bar graphs (right panel, n=3) showing acquisition of CD25 surface marker in IRF4-overexpressing Ts1+KRASG12D cells (at 48h). **p<0.001.
We performed RNA sequencing experiments on WT and Ts1Rhr B220+/CD19+/CD25- B cells harvested at P1, either expressing KRASG12D or not. Compared to WT controls, 2039 genes were significantly deregulated in Ts1Rhr, 8361 genes in KRASG12D, and 8967 genes in Ts1Rhr-KRASG12D (p<0.05) (Supplementary Table S5). Genes encoding surface markers such as Kit (−5.6 fold) and Cd25 (Il2ra, −1.9 fold), or transcription factors known to positively regulate B cell differentiation (i.e: Ikzf1, Pax5, Ebf1) (27) were repressed in KRASG12D and Ts1Rhr-KRASG12D compared to disomic (WT) or trisomic (Ts1Rhr) controls (Supplementary Figure S2C). To identify the transcriptional signatures associated with this oncogenic cooperation, we compared the differentially expressed genes between the paired conditions Ts1Rhr and WT, KRASG12D and WT, Ts1Rhr-KRASG12D and Ts1Rhr, and Ts1Rhr-KRASG12D and KRASG12D (Supplementary Table S5 and Figure S2D). 261 genes were found commonly deregulated in these analyses (132 upregulated and 129 downregulated genes) (Figure 2D), including the upregulation of DSCR genes, trisomic in the Ts1Rhr model (such as Erg, Wrb, Igsf5 and Psmg1), and the repression of several Immunoglobulin kappa (Igk) genes, indicative of a differentiation arrest at the pre-B stage.
Gene Set Enrichment Analyses (GSEA) confirmed that the genes located in the DSCR, were enriched in Ts1Rhr and in Ts1Rhr-KRASG12D compared to their disomic counterparts, as previously described (Supplementary Figure S2E) (13). Using the MSigDB c2 database, GSEA confirmed the enrichment of RAS/MAPK activation datasets, along with APC, MYC, TGFB1 and HDAC targets in wild-type or trisomic cells expressing the mutant KRASG12D; and of PRC2/SUZ12 targets in Ts1Rhr-KRASG12D versus KRASG12D paired comparison in line with previous observations (13). Positive enrichments in transcriptional signatures associated with early hematopoietic stem cells and progenitors were found in both Ts1Rhr and Ts1Rhr-KRASG12D context. Moreover, mature hematopoietic cells and B cell differentiation associated genes sets were negatively enriched in Ts1Rhr-KRASG12D specifically (Figure 2E, Supplementary Figure S3, and Supplementary Table S6), indicative of an earlier B cell differentiation blockade of the Trisomic-KRASG12D B cell precursors. We also identified a Ts1Rhr-KRASG12D specific signature of 1559 deregulated genes (788 down and 771 up) associated with the inhibition of cell cycle and B-cell differentiation, and with activation of stem cell and leukemia transcriptional programs (Supplemental Figure 3, Supplemental Table 5 and 6).
To investigate the relevance of these transcriptional signatures in our datasets of human B-ALL, we first extracted the differentially expressed genes between the B-ALL+21 samples harboring RAS activating mutations and B cell precursors CD34+CD38+CD19+ from healthy donors. 1814 genes were deregulated (p<0.05, Supplemental Table S7), including 238 upregulated and 43 downregulated genes in common with the Ts1Rhr-KRASG12D vs WT comparison. This signature was significantly enriched in B-ALL+21 with RAS mutations compared to other subtypes of B-ALL as shown by GSEA (Figure 2F and Supplementary Figures S4A, B). Among these commonly deregulated genes, 18 genes were enriched in the trisomy 21/RAS mutated contexts in both human and murine sequencing datasets, including upregulation of LRRC32, USP36, LDLR and RFLNB, whose overexpression or function have been correlated with tumor development (Figures 2G, H and Supplementary Table S7) (28–31) (and Human protein atlas available from www.proteinatlas.org). Moreover, a significant downregulation of IRF4, a key regulator of pre-B cell differentiation, was seen in Ts1Rhr-KRASG12D (−1.9 fold) and in B-ALL+21/RAS mutated samples (−11.5 fold). Ectopic expression of IRF4 in Ts1Rhr-KRASG12D cells resulted in growth inhibition and differentiation towards more CD25+ pre-B cells (Figure 2I–J, and supplemental Figure 4C), showing the functional relevance of low IRF4 level in leukemia maintenance.
This combined analysis of murine models and human B-ALL demonstrated that constitutive RAS/MAPK activation functionally cooperates with gain of chromosome 21 to enhance B cell precursors transformation, through the dysregulation of patient-relevant transcriptional signatures that control self-renewal, proliferation and B cell differentiation.
Development of a comprehensive cohort of preclinical models for B-ALL+21
To perform functional studies and assess the efficacy of new pharmacologic agents, we generated patient-derived xenograft (PDX) models. Four out of five DS-ALL samples successfully engrafted, as assessed by flow cytometry analyses from bone marrow and peripheral blood samples (Figures 3A–C and Supplementary Figure S5A). From these primary PDXs, human DS-ALL blasts were transduced with lentiviral constructs encoding mCherry and Luciferase reporter genes, to follow disease progression and response to treatments through non-invasive in vivo imaging. Importantly, based on the expression of the surface markers CD19, CD34, CD38 and TSLPR (Thymic Stromal Lymphopoietin Protein Receptor, a.k.a. CRLF2), these in vivo models conserved the phenotypic architecture of the primary patient (Figures 3A–C and Supplementary Figure S5A). The reliability of these models was confirmed using RNA-sequencing, MLPA and targeted sequencing of the known driver mutations identified in the primary samples (Figure 3D and Supplementary Figures S5B–D). This approach was applied to an extended cohort of 23 B-ALL samples, leading to the successful development of 2 iAMP21, 10 HeH and 5 Others PDX models (Supplemental Table S1). Importantly, the B-ALL+21/RAS mutated PDXs conserved the enrichment of transcriptional signature of 281 genes described previously (Figure 3E), and presented a constitutive phosphorylation of the downstream effectors Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) compared to non-mutated B-ALL PDXs, although the intensity of P-ERK1/2 is variable between individual samples (Figure 3F and Supplementary Figure S5E).
Figure 3: Establishment of a comprehensive cohort of PDX B-ALL+21.
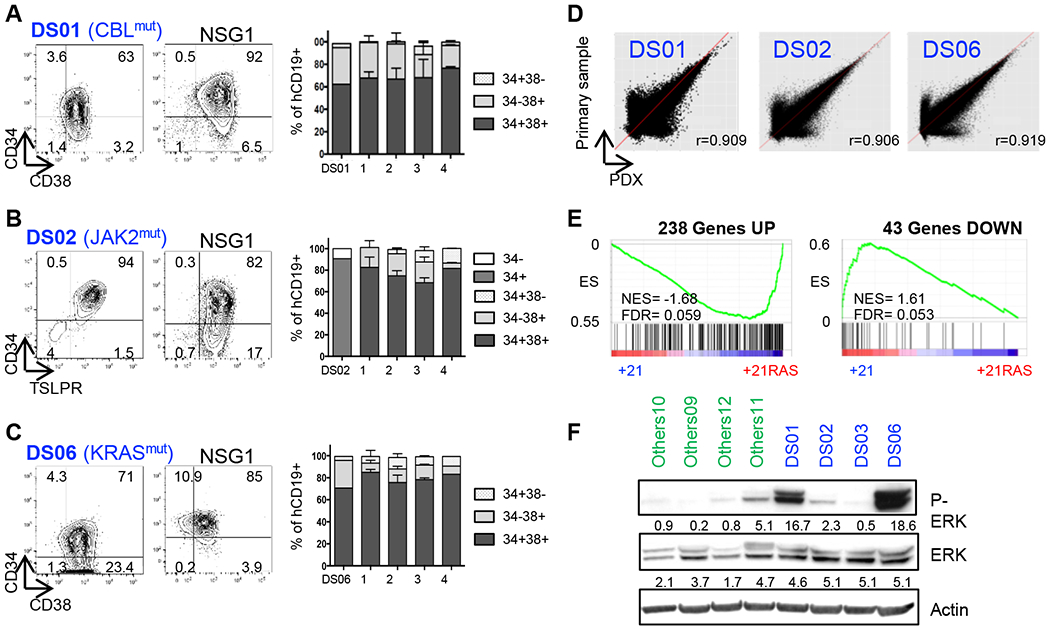
A-C. Left panel Representative flow cytometry analyses of the phenotypes of primary DS-ALL samples (DS01, DS02 and DS06) at diagnosis based on CD34, CD38, TSLPR and CD19 surface markers expression, and corresponding phenotypes from initial to NSG4 (right panel). D. Transcriptome correlation between primary DS01, DS02 and DS06 samples (Y-axis) and their corresponding PDXs (X-axis); r = Pearson correlation coefficient. E. GSEA assessing the enrichment of the 238 upregulated and 43 downregulated genes in B-ALL PDXs. F. Constitutive ERK1/2-phosphorylation assessed by Western blot of freshly harvested B-ALL PDX blasts starved overnight prior to protein extraction.
In summary, we have developed a comprehensive cohort of 21 B-ALL PDX models, including 4 DS-ALL, providing new tools to better understand the mechanisms of oncogenic cooperation in human B-ALL and perform preclinical studies.
Pharmacologic inhibition of MEK1/2 decreases leukemia burden in B-ALL+21
To assess the suitability of our murine and PDX models we developed, we tested the efficacy of MEK (Mitogen-Activated Protein Kinase (MAPK) kinases) inhibitors, which are essential downstream mediators of the RAS/MAPK pathway. First, the efficacy of the MEK1 inhibitor Selumetinib (AZD6244) (32) and the MEK1/2 inhibitor Trametinib (GSK1120212) (33) was tested on the Ts1Rhr-KRASG12D cells and on four human B-ALL+21 samples freshly harvested from PDXs (Figures 4A, B and Supplementary Figures S6A–C). Both inhibitors significantly decreased cell viability and were associated with a dose-dependent decreased phosphorylation of the ERK1/2 in several models, with Trametinib treatment being up to 60-fold more cytotoxic than Selumetinib in human samples. Next, we investigated the efficacy of Trametinib on 9 additional B-ALL+21 (2 iAMP21 and 7 HeH) and 5 ‘Others’ samples (Figure 4C and Supplementary Figure S6C). All B-ALL subtypes responded to Trametinib with a significantly lower IC50 in DS-ALL (IC50= 0.04 μM) and HeH (IC50= 0.06 μM) samples compared to ‘Others’ (IC50= 3.9 μM), and was associated with an increased proportion of annexinV-positive cells (Figures 4C and 4D, and Supplementary Figure S6D). In vivo, after 4 weeks of treatment with Trametinib (1.5mg/kg/day, oral gavage, 5 days a week), a decreased proportion of human CD45+ cells was observed in the bone marrow and spleen of treated animals (DS02) compared to vehicle controls, associated with lower levels of ERK1/2 phosphorylation in sorted CD45+/CD19+ splenic cells (Figure 4E, F).
Figure 4: RAS/MAPK pathway inhibition decreases viability of B-ALL cells in vitro and leukemia burden in vivo.
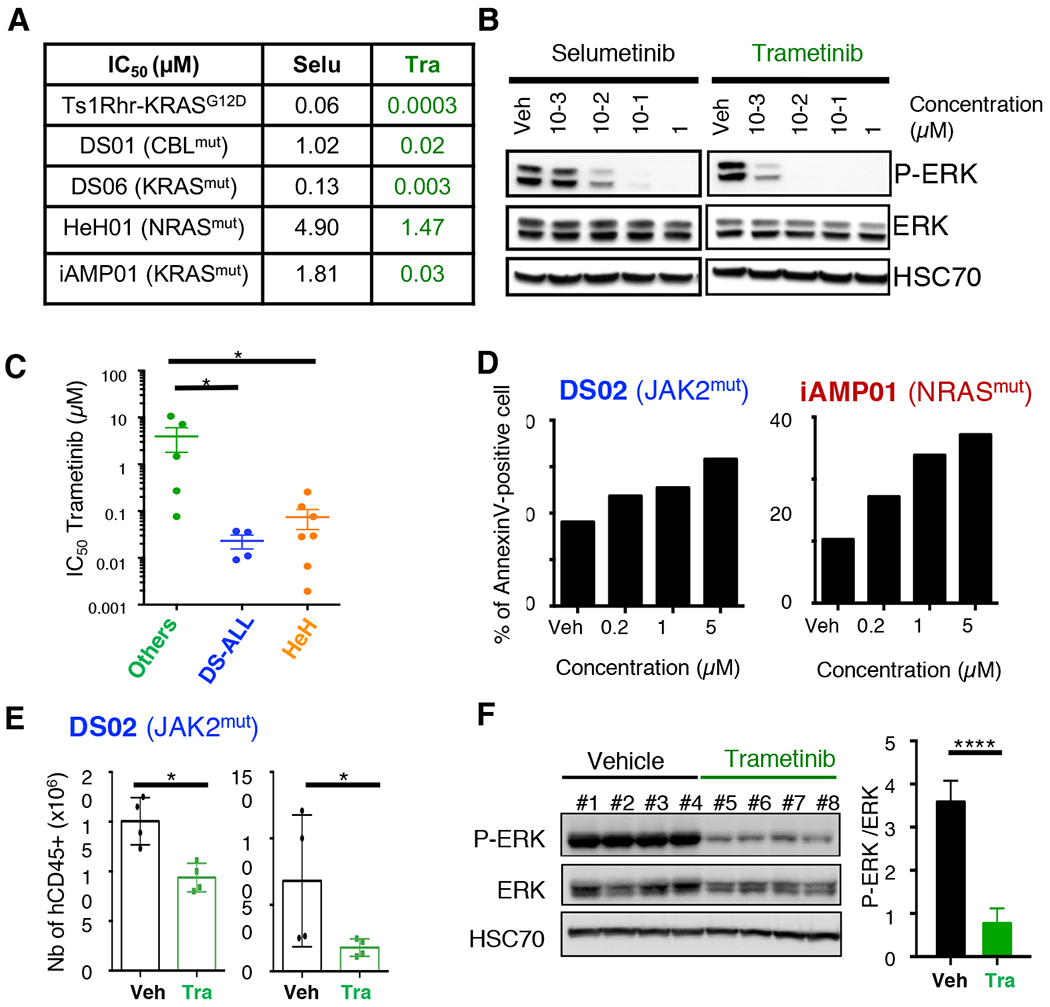
A. Table comparing the IC50 values (μM) of Selumetinib (Selu) and Trametinib (Tra) in Ts1Rhr-KRASG12D cells and in four different B-ALL+21 PDX cells with constitutive RAS/MAPK pathway activation. B. Representative western blots assessing the efficacy of Selumetinib and Trametinib on ERK1/2 phosphorylation (P-ERK) on HeH02 PDX cells after 6h of treatment in vitro. C. Averaged IC50 values obtained in Others (n=5), DS-ALL (n=4) and HeH (n=7) PDX samples. *p=0.05. Regression curves used to calculate these IC50 values from several NSG recipients are represented in Supplemental Figure 6D. D. Histogram plots representing the percentage of Annexin-V positive DS02 and iAMP01 cells at 48h. E. Absolute number of human CD45-positive cells detected by flow cytometry in the bone marrow (left panel) and spleen (right panel) of DS02 recipient mice, at the end of a 4 weeks treatment with Trametinib (Tra, 1.5mg/kg, oral gavage). *p=0.05. F. Effect of Trametinib on ERK1/2 phosphorylation assessed by Western blot in flow sorted CD45/CD19 human DS02 cells at the end of the 4-weeks in vivo treatment (left panel); Intensities were normalized using HSC70 and ERK total protein (right panel). ****p<0.0001.
Next, we assessed the efficacy of Trametinib on the survival of two B-ALL+21 models carrying RAS activating mutations: iAMP01 (NRASG12V) and HeH09 (KRASA146T). Four weeks treatments with Trametinib decreased leukemia burden, associated with an increased survival compared to vehicles (Figures 5A–F). Importantly, similar results were observed in two DS-ALL PDX models that harbor mutations in genes encoding upstream effectors of the RAS/MAPK pathway (Figures 5G–J).
Figure 5: Trametinib treatment decreases leukemia progression in vivo.
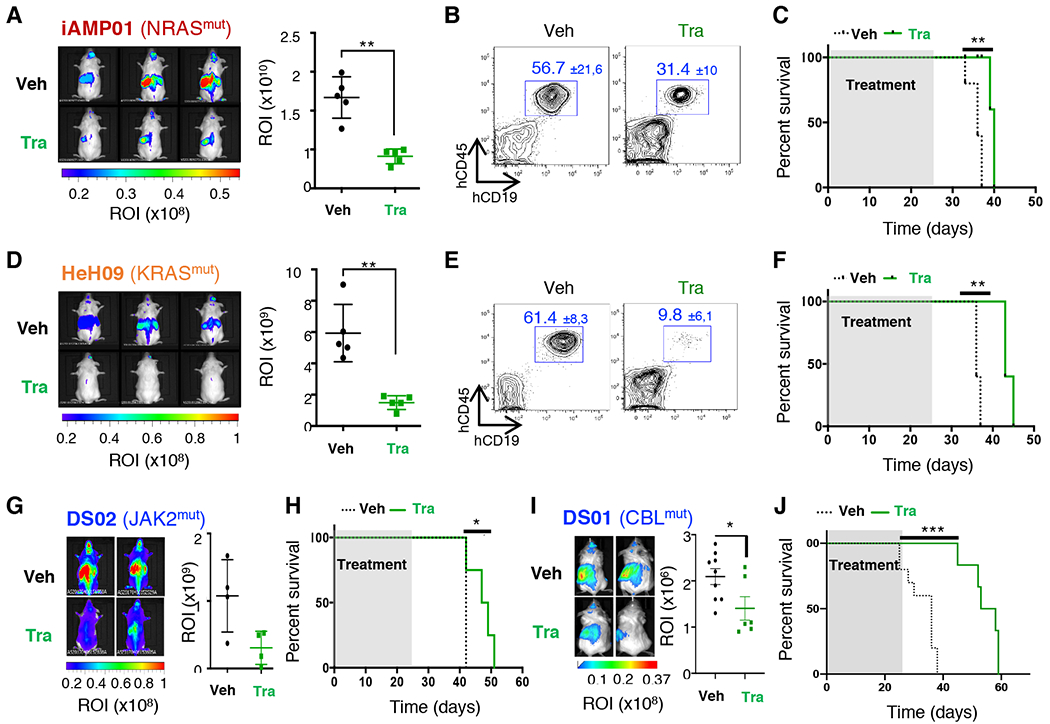
A. Left panel: Whole-body bioluminescence images after 4 weeks of treatment of iAMP01 PDX model. Right Panel: Absolute quantification of ROI (photons/second/surface [p/s/cm2]) between both groups (n=5 mice per group). **p=0.008. B. Proportion of human CD45+CD19+ cells in the peripheral blood at the end of treatment (average percentages ± SD are indicated). C. Survival curves of the iAMP01 PDX model treated with vehicle (black) or Trametinib (1.5 mg/kg, green), (n=5 mice per group), **p=0.003. D-F. Efficacy of Trametinib in the HeH09 PDX model (n=5 mice per group), **p=0.0008 (in D), **p=0.002 (in F). G-H. Efficacy of Trametinib in the DS02 model (n=4 mice per group), *p=0.04. (in H). I-J. Efficacy of Trametinib in the DS01 model (n=6–9 mice per group), *p=0.04 (in I), ***p=0.0003 (in J).
Together, these results indicated that pharmacological inhibition of MEK1/2 delayed disease development in murine models of B-ALL+21 presenting a constitutive activation of the RAS/MAPK pathway, irrespective of whether they carry genetic alterations affecting NRAS, KRAS, JAK2, and CBL genes.
Trametinib synergizes with conventional chemotherapy in DS-ALL models
To investigate the benefice of combining Trametinib with standard chemotherapy, dose-response profile of B-ALL PDX blasts to Dexamethasone, Vincristine and Methotrexate treatments were defined in vitro. Most of our samples responded to Vincristine and Dexamethasone (Figure 6A and Supplementary Figures S7A, B). Next, we performed drug combinations studies using Dexamethasone, Vincristine, Methotrexate or Idarubicin in combination with Trametinib, and evaluated B-ALL blast viability by live cell imaging. Combination index calculation unveiled a synergy between Trametinib and Vincristine in both DS-ALL and HeH-ALL samples (Figure 6A, B and Supplementary Figures S7C) (34). A synergistic effect was also seen between Trametinib and Dexamethasone in all four DS-ALL models.
Figure 6: Trametinib synergizes with Vincristine to increase survival of DS-ALL PDX.
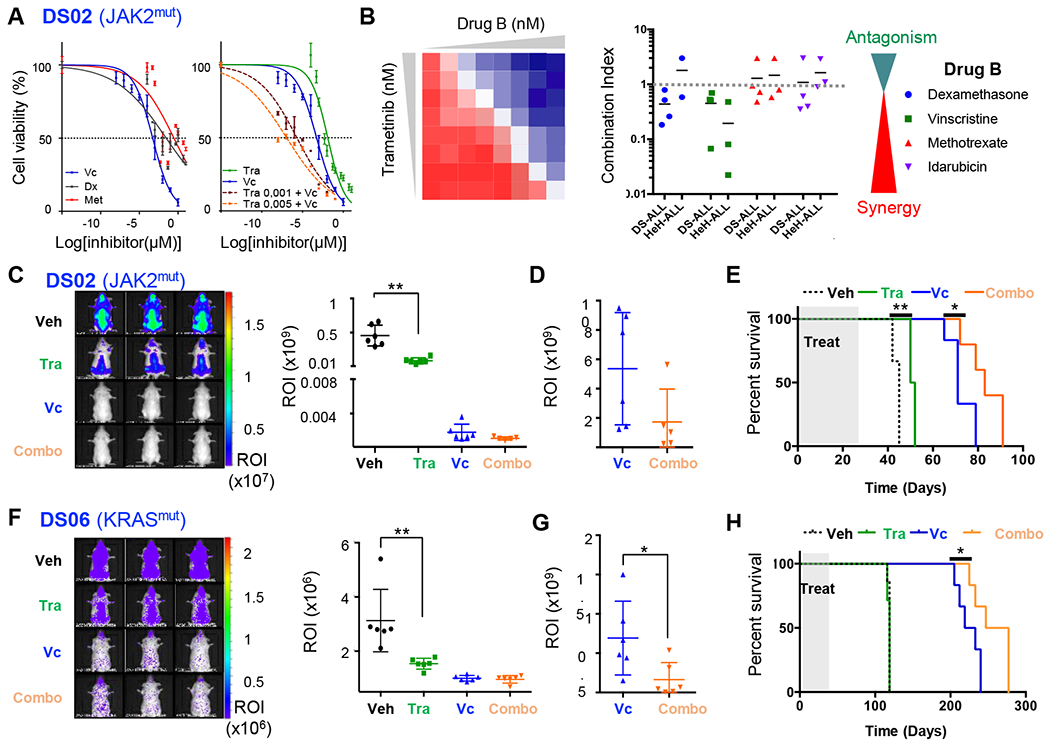
A. Left panel: Dose-response curves of single agent treatment for Vincristine (Vc, blue), Dexamethasone (Dx, purple) and Methotrexate (Met, red) on DS02 PDX cells for 72h; Right panel: Dose-response curves assessing the combination of Vincristine with Trametinib (Tra) at 2 doses: 1 nM (brown) and 5 nM (orange), compared to Vc and Tra alone. B. Drug combination studies of Trametinib with Dexamethasone, Vincristine, Methotrexate and Idarubicin performed on DS-ALL (n=3–4 PDX) and HeH (n=2–3 PDX). Combination indices (CI) were calculated according to (34) and represented in the right panel with CI<1 = synergy and CI>1 = antagonism. C and F. Left panels: Representative whole-body bioluminescence images of luciferase-positive DS02 and DS06 PDXs at the end of the treatment with Trametinib (1.5 mg/kg) and/or Vincristine (0.5mg/kg). Right panels: Absolute quantification of bioluminescence in ROI (photons/second/surface [p/s/cm2]) during the last week of treatment (Day 24), *p<0.05; **p<0.01. D and G. End point measurement of ROI in DS02 (D, day 64) and DS06 (G, day 205). E and H. Survival curves obtained for DS02 (E) and DS06 (G) PDXs (n=5 to 7 mice per arm). *p<0.05; **p<0.01.
Based on the synergy observed between Trametinib and Vincristine in vitro, efficacy of this combination was evaluated in two luciferase-expressing DS-ALL PDX models: DS02 (CRLF2/JAK2 positive) and DS06 (KRAS positive). Treatment was initiated after the detection of bona fide engraftment (recipients presenting >106 p/s/cm2 in total flux (ROI)), using 1.5 mg/kg/day of Trametinib, 0.5mg/kg of Vincristine (once per week), or both for 4 weeks. Trametinib alone significantly decreased leukemia burden at day 24 of treatment in both models (Figures 6C, F), confirming our previous observations. In survival analyses, we observed a decreased intensity in ROI between Vincristine alone and combo (Trametinib + Vincristine) group at the endpoint measurement for DS02 (day 64, Figure 6D), and DS06 (day 205, Figure 6G) models, which was also associated with an increase in survival for both DS-ALL PDXs (Figures 6E, H).
Taken together, these preclinical studies showed that Trametinib combined with conventional chemotherapy, improved outcomes of DS-ALL PDX models presenting a constitutive of the RAS/MAPK pathway.
Discussion
Children with Down Syndrome are predisposed to develop B-ALL and show an inferior prognosis compared to patients without DS due to a high risk of treatment-related morbidity and an increased rate of relapse (2, 3). In this study, we investigated the genetic landscape in DS-ALL and revealed that the oncogenic cooperation between constitutive trisomy 21 and RAS/MAPK pathway activation promotes cell proliferation, self-renewal and B-cell differentiation blockade associated with transcriptional alterations that can be modeled in murine cells. We also developed clinically-relevant models of human DS-ALL, and other pediatric B-ALL+21 samples, to show that targeting the RAS/MAPK pathway, through MEK1/2 inhibition, in combination with currently used chemotherapeutic agents is a promising strategy to improve the outcome for these children.
Constitutive RAS/MAPK pathway activation is a hallmark of cancer cells which in B-ALL, can result either directly through somatic mutations affecting NRAS and KRAS genes or indirectly, through the alterations of upstream regulators, such as IL7R/CRLF2/JAK2 cytokine signaling or the FLT3 receptor tyrosine kinase axis (35). These results are in line with recent studies in DS-ALL (10, 11), and in other B-ALL+21 samples (36–38). Interestingly, somatic gain of chromosome 21 is frequently seen in CRLF2-rearranged and BCR-ABL B-ALL (39–41), further highlighting the strong association between constitutive or acquired +21 and aberrant activation of signaling effectors. At the transcriptional level, we confirmed the low incidence of recurrent fusion transcripts leading to chimeric proteins in DS-ALL samples (11). Altogether, our cohort is representative of the genetic, genomic and transcriptomic landscape of childhood B-ALL+21, thus highlighting the suitability of our PDX models to better understand the mechanisms driving B cell leukemogenesis and test new pharmacological compounds.
At the molecular level, Lane et al. showed that trisomy of the Hmgn1 gene, located in the DSCR, reactivates the expression of genes regulated by H3K27me3 to promotes B cell leukemogenesis; a phenotype conserved in human primary B-ALL+21 specimens (13). Here, our transcriptomic analyses suggested that KRASG12D B cell precursors are negatively enriched in datasets regulated by the PRC2 complex, thus antagonizing the effect of Hmgn1 trisomy, and emphasizing a molecular interplay between RAS/MAPK activation and H3K27 modifications, as seen in other tumor types (42, 43). Of importance, other genes of the DSCR may participate to leukemia development and/or maintenance in B-ALL+21, such as Dyrk1A shown to regulate B-cell proliferation and differentiation (18). To date, the chromosome 21 genes that functionally cooperates with RAS/MAPK pathway activation in human B cell leukemia remain elusive and will require further investigations.
Here, we identified a transcriptional signature reflecting the cooperation between gain of chromosome 21 and RAS/MAPK pathway activation that is conserved in primary B-ALL, and including overexpression LRRC32 and of USP36. LRRC32 encodes the pro-tumorigenic Glycoprotein A Repetitions Predominant (GARP), known as critical regulator of latent TGFß activation and overexpressed in B cell malignancies (28, 29). The Ubiquitin Specific Peptidase USP36 has been shown to interact with FBW7 to regulate MYC stability (30) and enhance ERK and AKT signaling pathways (31). IRF4, a key regulator of B cell differentiation, was also significantly downregulated in Ts1Rhr-KRASG12D B cell precursors and in primary B-ALL+21 samples with constitutive RAS activation compared to healthy controls. In murine hematopoiesis, double knock-out Irf4−/−8−/− B cells display a hyper-proliferative phenotype and are arrested at the pre-B stage (44). With regards to leukemogenesis, loss of Irf4 was also shown to facilitate BCR-ABL or Myc-induced B cell leukemia (45, 46), and we showed that decreased expression of IRF4 may also be implicated in DS-ALL samples with constitutive RAS/MAPK activation. These differential expressions in LRCC32, USP36 and IRF4 correlated with GSEA showing positive enrichments of TGFß signaling and MYC targets, and negative enrichments of late pre-B cells and BCR receptor signaling signatures. Thus, the Ts1Rhr-KRASG12D model represents a useful tool to further characterize the underlying molecular mechanisms of oncogenic cooperation in DS-ALL.
Given that DS children with B-ALL have a poor prognosis, targeting the molecular mechanisms underlying the proliferation of leukemic blasts could improve their outcome. Although CRLF2/JAK2 signaling is enhanced in half of DS-ALL and recent observations suggested that only high doses of the JAK1/JAK2 inhibitor, Ruxolitinib, decrease survival of leukemia cells (11), Jak2 was reported as dispensable for B-cell leukemia maintenance (47). The DS-ALL PDX models we developed in this study will allow us to assess the efficacy of Ruxolitinib treatment in future preclinical assays to establish the therapeutic benefit for DS-ALL harboring JAK2 activating mutations . Studies have recently shown that RAS mutations are associated with inferior prognosis in non-DS B-ALL (48), that several subtypes of B cell leukemia carrying RAS mutations were sensitive to MEK inhibition (49–51), and that Selumetinib synergized with dexamethasone to decrease viability of B-ALL cells (52). Here, we revealed that DS-ALL are sensitive to Trametinib and further decrease leukemia burden and prolong survival in several PDX models when combined with Vincristine, regardless of the genetic alterations leading to RAS/MAPK pathway activation (RAS, JAK2 and CBL mutations). Overall, these results support the potential benefice of integrating Trametinib to conventional chemotherapies that include both Dexamethasone and Vincristine, to improve treatment efficacy of Down syndrome children with B-ALL.
Supplementary Material
Relevance.
B-cell precursor Acute Lymphoblastic Leukemia (B-ALL) is the most common type of childhood malignancies, accounting for more than 20% of all pediatric cancers worldwide. Although survival rates have significantly improved, many children with B-ALL continue to have a poor prognosis, and suffer from treatment related toxicity and relapse; clinical features that are exemplified in Down syndrome children with acute lymphoblastic leukemia (known as DS-ALL). Among the most frequent somatic alterations seen in childhood B-ALL, gain of chromosome 21 (+21) and RAS/MAPK alterations are found in 30–40% and 50%, respectively. In this study, using DS-ALL as a paradigm, we showed that both events functionally cooperate and that treatment with the MEK inhibitor Trametinib improved the survival in patient-derived xenograft models of childhood B-ALL+21 with RAS activating mutations. The comprehensive repository of preclinical models developed here is of general relevance to investigate new targeted therapies in childhood leukemia.
Acknowledgements
We thank M. Deloger, G. Meurice, T. Dayris from the bioinformatic facility of Gustave Roussy Institute (Villejuif, France), L. Chia (Telethon Kids Institute, Australia) and F. Boudia for their support in analysis of next generation sequencing data; P. Rameau (PFIC, Gustave Roussy, France) for flow cytometry analyses; C. Thiollier and C. K. Lopez (INSERM U1170, Gustave Roussy, France); G. Chua and S. Singh (Telethon Kids Institute, Australia) for processing some PDX models; V. Frimantas (Children’s Research Centre, Switzerland) for establishing drug combination experiments; S. Lagarde (CRCT Toulouse, France); T. Adam-de-Beaumais (Gustave Roussy, France) for the access to the genetic data of primary samples. This work was supported by the Leukemia Research Foundation (SM, 2012), the European Hematology Association (SM, Research fellowship 2013), Fondation ARC, GEFLUC, Fondation Lejeune (#1485 and #1806), Channel 7 Telethon 2017 - Lexus Ball for Cancer, and the Children’s Leukaemia and Cancer Research Foundation (CLCRF, Australia), by the Genomic Core Facility of Gustave Roussy (TA2014-CAIG to CI, TA2016-ANLA to APL), MAPPYACTS (INCa PHRC-K14–175 and Imagine for Margo), Fondation ARC grant MAPY201501241, Imagine for Margo), PDX development (Société Française de Lutte contre les Cancers et les Leucémies de l’Enfant et l’Adolescent (SFCE), Fondation Enfants et Santé, Fondation AREMIG, Association Thibault BRIET). RSK is supported by a Fellowship from the National Health and Medical Research Council of Australia (NHMRC APP1142627). JDC is supported in part by the NIH (CA101774). TM and ED are supported by PAIR-Pédiatrie/CONECT-AML (COllaborative Network for Children and Teenagers with Acute Myeloblastic Leukemia: INCa-ARC-LIGUE_11905 and Association Laurette Fugain). APL was supported by Cancéropôle Ile-de-France and Fondation pour la Recherche Médicale. JPB was supported by the Swiss National Research Foundation and the Clinical Research Priority Program of the University of Zurich. SM is supported by a Fellowship from Cancer Council Western Australia (CCWA).
Footnotes
Conflict of Interest
JDC receives research support from Forma Therapeutics and Scholar Rock, is a consultant for Sierra Oncology, and the Scientific Advisor of the MPN Research Foundation. The other authors declare no conflict of interest.
References
- 1.Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with Down syndrome. Genet Med. 2016;18(11):1151–7. [DOI] [PubMed] [Google Scholar]
- 2.Buitenkamp TD, Izraeli S, Zimmermann M, Forestier E, Heerema NA, van den Heuvel-Eibrink MM, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123(1):70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ceppi F, Stephens D, den Hollander BS, Krueger J, Whitlock J, Sung L, et al. Clinical presentation and risk factors of serious infections in children with Down syndrome treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2016;63(11):1949–53. [DOI] [PubMed] [Google Scholar]
- 4.Forestier E, Izraeli S, Beverloo B, Haas O, Pession A, Michalova K, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111(3):1575–83. [DOI] [PubMed] [Google Scholar]
- 5.Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–98. [DOI] [PubMed] [Google Scholar]
- 6.Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood. 2010;115(5):1006–17. [DOI] [PubMed] [Google Scholar]
- 8.Malinge S, Ben-Abdelali R, Settegrana C, Radford-Weiss I, Debre M, Beldjord K, et al. Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood. 2007;109(5):2202–4. [DOI] [PubMed] [Google Scholar]
- 9.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484–92. [DOI] [PubMed] [Google Scholar]
- 10.Nikolaev SI, Garieri M, Santoni F, Falconnet E, Ribaux P, Guipponi M, et al. Frequent cases of RAS-mutated Down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun. 2014;5:4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzman O, Savino AM, Gombert M, Palmi C, Cario G, Schrappe M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A. 2017;114(20):E4030–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson LE, Richtsmeier JT, Leszl J, Reeves RH. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science. 2004;306(5696):687–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46(6):618–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulsson K, Forestier E, Lilljebjorn H, Heldrup J, Behrendtz M, Young BD, et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010;107(50):21719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loncarevic IF, Roitzheim B, Ritterbach J, Viehmann S, Borkhardt A, Lampert F, et al. Trisomy 21 is a recurrent secondary aberration in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion. Genes Chromosomes Cancer. 1999;24(3):272–7. [PubMed] [Google Scholar]
- 17.Mowery CT, Reyes JM, Cabal-Hierro L, Higby KJ, Karlin KL, Wang JH, et al. Trisomy of a Down Syndrome Critical Region Globally Amplifies Transcription via HMGN1 Overexpression. Cell Rep. 2018;25(7):1898–911 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson BJ, Bhansali R, Diebold L, Cook DE, Stolzenburg L, Casagrande AS, et al. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J Exp Med. 2015;212(6):953–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122(3):948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koboldt DC, Larson DE, Chen K, Ding L, Wilson RK. Massively parallel sequencing approaches for characterization of structural variation. Methods Mol Biol. 2012;838:369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, et al. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013;15(2):143–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuels AL, Beesley AH, Yadav BD, Papa RA, Sutton R, Anderson D, et al. A pre-clinical model of resistance to induction therapy in pediatric acute lymphoblastic leukemia. Blood Cancer J. 2014;4:e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018;10(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zarich N, Anta B, Fernandez-Medarde A, Ballester A, de Lucas MP, Camara AB, et al. The CSN3 subunit of the COP9 signalosome interacts with the HD region of Sos1 regulating stability of this GEF protein. Oncogenesis 2019;8(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Yoshida T, Georgopoulos K. Transcriptional circuits in B cell transformation. Curr Opin Hematol. 2017;24(4):345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metelli A, Wu BX, Fugle CW, Rachidi S, Sun S, Zhang Y, et al. Surface Expression of TGFbeta Docking Receptor GARP Promotes Oncogenesis and Immune Tolerance in Breast Cancer. Cancer Res. 2016;76(24):7106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arribas AJ, Rinaldi A, Chiodin G, Kwee I, Mensah AA, Cascione L, et al. Genome-wide promoter methylation of hairy cell leukemia. Blood Adv. 2019;3(3):384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc Natl Acad Sci U S A. 2015;112(12):3734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SY, Choi J, Lee DH, Park JH, Hwang YJ, Baek KH. PME-1 is regulated by USP36 in ERK and Akt signaling pathways. FEBS Lett. 2018;592(9):1575–88. [DOI] [PubMed] [Google Scholar]
- 32.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–83. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31. [DOI] [PubMed] [Google Scholar]
- 34.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 35.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinclair PB, Ryan S, Bashton M, Hollern S, Hanna R, Case M, et al. SH2B3 inactivation through CN-LOH 12q is uniquely associated with B-cell precursor ALL with iAMP21 or other chromosome 21 gain. Leukemia. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47(6):672–6. [DOI] [PubMed] [Google Scholar]
- 38.Malinowska-Ozdowy K, Frech C, Schonegger A, Eckert C, Cazzaniga G, Stanulla M, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015;29(8):1656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell LJ, Jones L, Enshaei A, Tonin S, Ryan SL, Eswaran J, et al. Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2017;56(5):363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vesely C, Frech C, Eckert C, Cario G, Mecklenbrauker A, Zur Stadt U, et al. Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia. 2017;31(7):1491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wetzler M, Dodge RK, Mrozek K, Stewart CC, Carroll AJ, Tantravahi R, et al. Additional cytogenetic abnormalities in adults with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a study of the Cancer and Leukaemia Group B. Br J Haematol. 2004;124(3):275–88. [DOI] [PubMed] [Google Scholar]
- 42.Agger K, Cloos PA, Rudkjaer L, Williams K, Andersen G, Christensen J, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23(10):1171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514(7521):247–51. [DOI] [PubMed] [Google Scholar]
- 44.Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003;17(14):1703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acquaviva J, Chen X, Ren R. IRF-4 functions as a tumor suppressor in early B-cell development. Blood. 2008;112(9):3798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pathak S, Ma S, Trinh L, Eudy J, Wagner KU, Joshi SS, et al. IRF4 is a suppressor of c-Myc induced B cell leukemia. PLoS One. 2011;6(7):e22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim SK, Knight DA, Jones LR, Vervoort S, Ng AP, Seymour JF, et al. JAK2 is dispensable for maintenance of JAK2 mutant B-cell acute lymphoblastic leukemias. Genes Dev. 2018;32(11–12):849–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jerchel IS, Hoogkamer AQ, Aries IM, Steeghs EMP, Boer JM, Besselink NJM, et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia. 2018;32(4):931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124(23):3420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryan SL, Matheson E, Grossmann V, Sinclair P, Bashton M, Schwab C, et al. The role of the RAS pathway in iAMP21-ALL. Leukemia. 2016;30(9):1824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerstjens M, Pinhancos SS, Castro PG, Schneider P, Wander P, Pieters R, et al. Trametinib inhibits RAS-mutant MLL-rearranged acute lymphoblastic leukemia at specific niche sites and reduces ERK phosphorylation in vivo. Haematologica. 2018;103(4):e147–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polak A, Kiliszek P, Sewastianik T, Szydlowski M, Jablonska E, Bialopiotrowicz E, et al. MEK Inhibition Sensitizes Precursor B-Cell Acute Lymphoblastic Leukemia (B-ALL) Cells to Dexamethasone through Modulation of mTOR Activity and Stimulation of Autophagy. PLoS One. 2016;11(5):e0155893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole exome sequencing and RNAseq data from primary samples were deposited into EGA under the accession number EGAS00001003760. RNAseq data from CFU-preB experiments are available on EBI under the accession number E-MTAB-8005.