

Abstract
Pancreatic ductal adenocarcinoma (PDAC) evolves a complex microenvironment comprised of multiple cell types, including pancreatic stellate cells (PSCs). Previous studies have demonstrated that stromal supply of alanine, lipids, and nucleotides supports the metabolism, growth, and therapeutic resistance of PDAC. Here we demonstrate that alanine crosstalk between PSCs and PDAC is orchestrated by the utilization of specific transporters. PSCs utilize SLC1A4 and other transporter(s) to rapidly exchange and maintain environmental alanine concentrations. Moreover, PDAC cells upregulate SLC38A2 to supply their increased alanine demand. Cells lacking SLC38A2 fail to concentrate intracellular alanine and undergo a profound metabolic crisis resulting in markedly impaired tumor growth. Our results demonstrate that stromal-cancer metabolic niches can form through differential transporter expression, creating unique therapeutic opportunities to target metabolic demands of cancer.
Keywords: Pancreatic cancer, metabolism, solute carriers, microenvironment, amino acids
Introduction
Mammalian cells have evolved diverse mechanisms to acquire essential nutrients from the circulation or interstitial fluids to fuel their metabolic demands (1). In cancer, these processes are hijacked to supply the metabolic needs associated with rapid proliferation (2). In pancreatic cancer, endocytotic and lysosomal-dependent scavenging pathways are activated as a catabolic mechanism to supply amino acids essential to sustain proliferation in nutrient austere environments (3-6). In parallel, symbiotic relationships between stromal and cancer cells form through exchange of nutrients and cytokines as a means of relaying pro-growth signals and supplying anabolic precursors, including amino acids and metabolically expensive macromolecules. Metabolic crosstalk is an emerging mechanism of resistance to anti-metabolism therapies (7-11).
Pancreatic ductal adenocarcinoma (PDAC) has a dismal 5-year survival of ~9% with few therapeutic options that lead to prolonged survival. Therapeutic shortcomings can, in part, be attributed to the characteristic stromal (desmoplastic) reaction present in PDAC tumors (12). We previously demonstrated a critical crosstalk of alanine between pancreatic stellate cells (PSCs) and PDAC, highlighting a metabolic support role of the stroma in tumors (11). Nutrient transporters serve as gatekeepers, facilitating metabolic interactions between cells and their surrounding cellular and nutrient environment. A mechanistic understanding of how nutrient sharing occurs within the tumor microenvironment and the metabolic consequences of selectively disrupting crosstalk is required to establish their role in promoting tumor metabolism, growth, and therapeutic resistance. The identification of specific, targetable transporters required for nutrient channeling and tumor metabolism may lead to more effective therapies to restrict nutrient access for deadly cancers, such as PDAC.
In this study, we sought to identify transporters expressed between PSCs and PDAC that are involved in uni-directional channeling of alanine. We applied quantitative stable-isotope tracing and flux analysis to understand the compartmentalization of alanine synthesis, uptake, and utilization in PDAC and to functionally characterize alanine transport mechanisms across a diverse panel of PSC and PDAC cell lines. We identify that PDAC cells selectively express the mitochondrial isoform of glutamic-pyruvic transaminase (GPT2) for de novo synthesis and utilization of alanine. Targeting mitochondrial pyruvate transport, required for de novo synthesis of alanine, resulted in an increased dependence on environmental alanine and revealed heterogeneous absolute demand in a panel of PDAC cell lines. Furthermore, we identify cell type-specific expression of the plasma membrane neutral amino acid transporters SLC1A4 and SLC38A2 and functionally describe their role in facilitating alanine transport in pancreatic stellate and cancer cells. While SLC1A4 was found to cooperate with other transporters in PSCs to maintain environmental alanine levels, SLC38A2 was required for active uptake of alanine from the environment in PDAC. Loss of SLC38A2 in PDAC led to a profound deficiency to internalize and concentrate cytosolic alanine, resulting in increased de novo synthesis and passive efflux to the environment. This increased demand to synthesize alanine caused a profound compartmentalized metabolic crisis, arising from an increased shunting of cytosolic pyruvate and nitrogen sources (e.g. glutamine, BCAA) towards de novo alanine biosynthesis in the mitochondria. From a therapeutic perspective, SLC38A2-deficient cells failed to effectively compensate for loss of SLC38A2 by activating intracellular metabolic pathways or alternative transporter mechanisms. Targeting SLC38A2 significantly impaired the beneficial support of stellate cells in co-injection xenograft models. Utilizing a cDNA withdrawal approach to mimic drugging SLC38A2 we demonstrate that loss of alanine transport leads to significant and durable tumor regression in fully formed tumors, providing rationale that targeting SLC38A2 may be an effective therapeutic strategy for pancreatic cancer.
Results
Characterizing compartmentalization of alanine fate in PDAC and pancreatic stellate cells
We hypothesized that PSC and PDAC cells must exhibit distinct transport mechanisms and nutrient transporters to exchange alanine. To identify cell-specific alanine transport mechanisms, we performed a quantitative analysis of alanine uptake, secretion, and exchange fluxes in a panel of human and primary mouse PDAC and PSC cell lines. Alanine uptake was shared across the majority of PDAC cell lines tested, whereas PSCs primarily efflux alanine (Fig. 1A). Notably, PDAC alanine influx was of similar magnitude to serine and glycine flux, amino acids reported to be critical for cancer cell proliferation (Supplementary Fig. S1A) (13). We observed net alanine secretion in all cell lines cultured in basal media (Fig. 1A). As DMEM does not contain alanine, mass action drives efflux in alanine-limited environments. However, microenvironmental alanine levels measured in both primary human and murine PDAC samples approximate 1 mM and are high relative to that of circulation (plasma alanine ~200-400 μM) (14,15). We hypothesized that stromal cells may supply and maintain tumor environmental alanine concentrations. PSC net alanine efflux was inhibited upon exogenous alanine supplementation (Fig. 1A and 1B, Supplementary Fig. S1B). However, supplementing PSCs with stable-isotope labeled 13C3-alanine revealed that PSCs rapidly exchange extracellular alanine with unlabeled (e.g. synthesized) at a rate ~3x that of the net secretion flux, resulting in substantial turnover of 13C3-alanine in the media over time (Fig. 1B, Supplementary Fig. S1C). Minimal impacts on PSC alanine net secretion or exchange flux were observed in physiological glucose (~5mM) cultures, suggesting that glucose is not limiting for the maintenance of environmental alanine levels (Supplementary Fig. S1D) (14). Notably, PSC exchange flux exceeded PDAC alanine uptake, suggesting that PSCs can meet the alanine demands of PDAC cells.
Fig. 1. Compartmentalized alanine metabolism in PDAC.
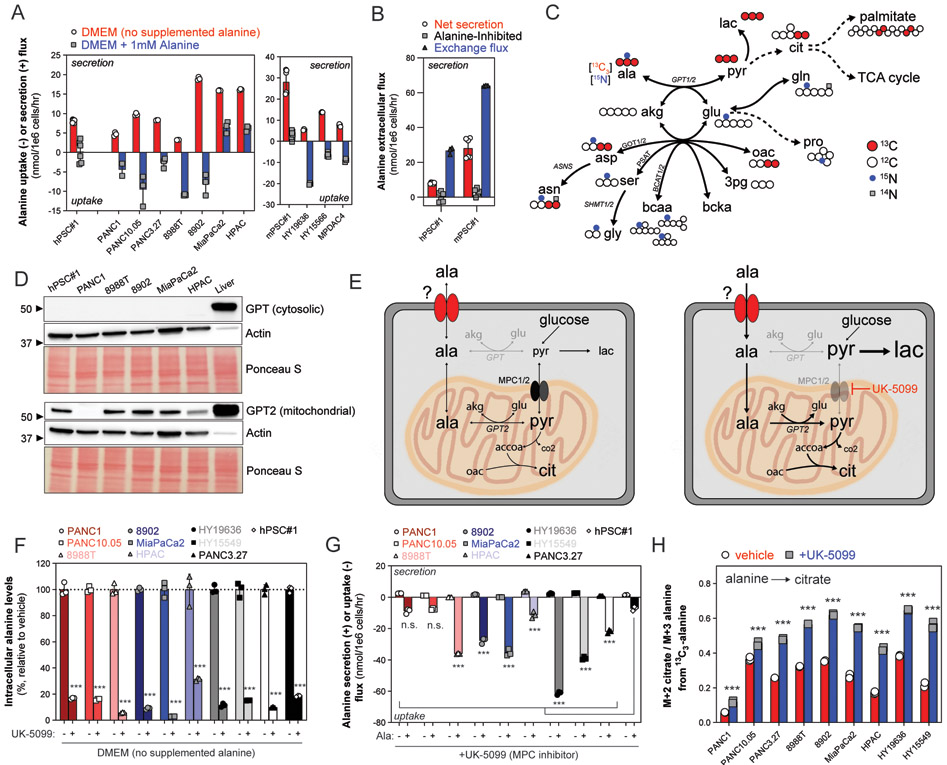
(A) Alanine uptake and secretion flux in a panel of human and mouse PDAC and PSC lines cultured in DMEM or DMEM supplemented with 1mM L-alanine. Extracellular accumulation (+, secretion) or depletion (−, uptake) was measured in conditioned media over 24-72 hours and normalized to the viable cell density over the time course. Error bars depict s.d. of ≥3 technical replicate wells from 1-2 independent experiments normalized to growth of surrogate wells. (B) Alanine exchange flux as compared to net secretion flux and substrate-inhibited flux in human and mouse pancreatic stellate cell lines. 13C3-alanine dilution by unlabeled (M+0) alanine was measured over time, and the molar exchange flux was quantified and normalized by viable cell density over 24 hours. Error bars depict s.d. of ≥3 technical replicate wells from 1-2 independent experiments. Data for alanine net secretion and inhibited flux from same experiment as Fig. 1A. (C) Atom transition map summarizing 13C3-alanine and 15N-alanine contributions to PDAC intracellular metabolism in a diverse panel of human and mouse PDAC cell lines. Large, grey circles depict 13C atom transitions through central carbon metabolism; small, black circles depict 15N atom transitions through the transaminase network. Unlabeled (12C, 14N) atoms depicted as white circles. (D) Immunoblot of cytosolic GPT (top) and GPT2 (bottom) in whole cell lysates (30 μg) extracted from hPSC#1, human PDAC cell lines, or normal murine liver (as positive control for both GPT and GPT2). Immunoblot of actin and Ponceau S staining to indicate equal loading between cell lines. Representative immunoblot from three experiments using two independently collected sets of lysates. (E) Diagram depicting compartmentalized alanine utilization and de novo synthesis pathways. (Left panel) In basal conditions, PDAC cells both de novo synthesize alanine in the mitochondria and import cytosolic alanine into the mitochondria for GPT2-dependent utilization. (Right panel) Inhibiting mitochondrial pyruvate transport, and thus de novo alanine synthesis, increases demand for environmental alanine and reveals mechanism to uncouple MPC activity from pyruvate oxidation. (F) Intracellular alanine levels normalized by cell number in panel of PDAC cell lines and hPSC#1 in response to vehicle (DMSO) or UK-5099 (10 μM for human PDAC, 25μM for murine PDAC) in DMEM. Data are represented as percentage relative to vehicle. Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment. (G) Alanine secretion flux and uptake flux in response to MPC inhibition using UK-5099. Extracellular accumulation (+, secretion) or depletion (−, uptake) was measured in conditioned basal media (DMEM, secretion) or alanine-supplemented media (DMEM + 1mM L-alanine, uptake), respectively, over 24 hours and normalized to the viable cell density over the time course. Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment normalized to growth of surrogate wells. (H) Citrate M+2 labeling normalized to alanine labeling (M+3) in PDAC cells in response to vehicle (DMSO) or MPC inhibition using UK-5099 cultured in DMEM supplemented with 1mM 13C3-alanine. Error bars depict s.d. of three technical replicates. Significance determined with two-way ANOVA using Sidak’s multiple comparisons test in F, G, and H. n.s. p≥0.05, *** p<0.001.
PDAC cells exhibited significant alanine consumption and distinct transport mechanisms relative to PSCs. We hypothesized that alanine was fueling a specific intrinsic demand, and inhibiting either utilization or transport may be an effective strategy to limit cancer cell access to alanine. We performed stable-isotope tracing with 13C3- or 15N- labeled alanine and measured incorporation into biosynthetic and central carbon metabolites or transamination products. Significant labeling from alanine-derived carbon and nitrogen was measured in proteinogenic amino acids, TCA intermediates, de novo synthesized fatty acids, and products of transamination pathways (Fig 1C, Supplementary Fig. S2A-I), suggesting that alanine contributes significantly to bioenergetic and anabolic pathways in PDAC. However, alanine utilization was heterogeneous across the cell panel and failed to reach isotopic steady state (Supplementary Fig. S2C). These data, coupled with alanine secretion in basal DMEM (Fig. 1A), confirm that PDAC cells use a combination of de novo synthesis and uptake from the environment to supply their alanine demands.
Alanine synthesis and catabolism require activity of compartmentalized glutamic-pyruvic transaminases (GPT, cytosolic; GPT2, mitochondrial), which utilize glutamate as an amino-nitrogen donor and pyruvate (Fig. 1C). To better understand the compartmentalization of alanine metabolism in PDAC, we quantified relative expression of GPT and GPT2 in a panel of PDAC cell lines. Strikingly, PDAC cell lines demonstrated selective transcript and protein expression of the mitochondrial isoform (GPT2) (Fig. 1D, Supplementary Fig. S3A). Thus, de novo alanine synthesis and utilization likely occur simultaneously in the mitochondria of PDAC cells. Indeed, cells expressing low levels of both GPT and GPT2 (e.g. PANC1, HPAC) had significantly lower 13C3-alanine contribution to mitochondrial TCA cycle metabolites (e.g. citrate) relative to cells with high GPT2 expression (Supplementary Fig. S2D). These data suggest that de novo alanine synthesis primarily utilizes mitochondrial pyruvate in PDAC; however, this is surprising given the selective down-regulation or deletion of mitochondrial pyruvate transport in other cancer contexts (e.g. lung, colon, kidney) (16,17). Mitochondrial pyruvate transport is facilitated by the obligate heterodimer consisting of MPC1 and MPC2 (18-20). Down-regulation of MPC1/2 has been reported to be essential for tumor initiation in colon cancer and is thought to promote aerobic glycolysis/Warburg metabolism by decreasing mitochondrial pyruvate oxidation and shifting pyruvate pools to the cytosol (21). Both MPC1 and MPC2 transcripts are expressed in human PDAC cell lines (22), suggesting that PDAC may utilize an alternative mechanism to reduce demand for mitochondrial pyruvate import.
We hypothesized that alanine uptake and utilization by PDAC may act as an alternative means of maintaining compartmentalized pyruvate homeostasis by relieving the need to import pyruvate into the mitochondria for de novo synthesis (Fig. 1E). To explore the dependence on mitochondrial pyruvate transport for alanine synthesis in PDAC, we treated cells with the highly specific MPC2 inhibitor, UK-5099, and quantified impacts on alanine metabolism and transport (Fig. 1E) (23,24). Strikingly, intracellular levels of alanine and net secretion flux significantly decreased by ~70-97% in PDAC cells in response to MPC inhibition (Fig. 1F and 1G). Alanine supplementation rescued the intracellular alanine defect, and all PDAC cells exhibited significantly enhanced alanine uptake in response to MPC inhibition (Fig. 1G, Supplementary Fig. S3B and S3C). Collectively, these data suggest that de novo alanine synthesis is predominantly driven by mitochondrial GPT2 and requires mitochondrial pyruvate transport. Furthermore, inhibition of MPC in contexts where cytosolic alanine synthesis is silenced (e.g. down-regulation of GPT) created a near complete dependence (“pseudo-auxotrophy”) on environmental alanine (Fig. 1F and 1G, Supplementary Fig. S3B). Comparison of pseudo-auxotrophic uptake of alanine across human and mouse PDAC cells revealed heterogeneity in absolute alanine demands that failed to correlate with individual cell preference for alanine uptake or secretion (Fig 1A and 1G, Supplementary Fig. S3C). The divergence between uptake preference and cellular demand for alanine strongly suggests that PDAC meet these requirements through both synthesis and environmental scavenging (Fig. 1E, Supplementary Fig. S3C).
The selective down-regulation of cytosolic GPT argues against alanine utilization in the cytosol. Therefore, we hypothesized that alanine may directly channel into mitochondria for utilization via GPT2 (Fig. 1E). To test this possibility, we cultured cells with 13C3-labeled alanine in the presence of UK-5099 to inhibit glycolytic- and GPT- derived cytosolic pyruvate. Strikingly, we observed a significant increase in alanine anaplerosis in the context of MPC inhibition in all PDAC cell lines tested (Fig. 1H). Furthermore, alanine-derived lactate labeling was significantly suppressed by MPC inhibition, suggesting that MPC inhibition leads to increased shunting of glucose-derived (unlabeled) cytosolic pyruvate towards aerobic glycolysis (Supplementary Fig. S3D). Thus, uptake of alanine from the environment and subsequent mitochondrial import is uncoupled from cytosolic pyruvate and can occur independently of mitochondrial pyruvate transport (Fig. 1E). Together with down-regulation of cytosolic alanine synthesis and utilization, PDAC cells can utilize alanine uptake as a means of simultaneously supplying alanine-derived mitochondrial pyruvate that fuels oxidative TCA cycling whilst sparing glucose-derived cytosolic pyruvate for aerobic glycolysis/Warburg metabolism (Fig. 1E).
Loss of SLC38A2 impairs alanine uptake and intracellular concentration in PDAC
We hypothesized that, in the absence of cytosolic biosynthetic machinery, an important component of this metabolic program in PDAC is a plasma membrane alanine transporter capable of maintaining cytosolic alanine levels. Furthermore, we hypothesized that unique transport mechanisms must exist to facilitate alanine exchange within the PSC-PDAC niche, providing a therapeutic avenue for limiting PDAC access to environmental alanine sources. To identify candidate transporters involved in PSC-PDAC alanine crosstalk, we performed an unbiased analysis of a quantitative proteomics dataset of PDAC and PSC cell lines (25). We initially performed principal component analysis (PCA) of whole proteome or protein subsets involved in metabolic processes or transport (e.g. SLC proteins). Each of these analyses revealed divergent expression profiles in stromal and PDAC cell lines (Supplementary Fig. S4A). We analyzed differentially expressed SLC proteins between PSCs and a commonly used human PDAC line and identified 40 proteins which, excluding mitochondrial (SLC25) and non-amino acid transporters, reduced to five candidates (Fig. 2A and 2B, Supplementary Table S1). Further analysis led to the identification of two plasma membrane-localized neutral amino acid transporters, SLC1A4 and SLC38A2, which were highly expressed in either stromal or PDAC cells, respectively, by both quantitative proteomics and western blot analyses (Fig. 2B, Supplementary Fig. S4B and S4C).
Fig 2. SLC38A2 is the major concentrative alanine transporter in PDAC.

(A) Volcano plot of differentially expressed transporters (‘SLC’ proteins) between hPSC#1 and PANC1 cell lines. Transporters were considered differentially expressed if log2 fold change (FC) ≥ 2 and p-value ≤ 0.05. Transporters involved in amino acid transport are highlighted with colored squares and labeled with protein name. (B) Relative protein expression across panel of non-malignant pancreatic and PDAC cell lines quantified by summing reporter ion counts of peptide-spectral matches for SLC1A4 and SLC38A2. Two tandem mass tag-labeled biological replicates were collected for each cell line. (C & D) Alanine secretion (+) and uptake (−) flux in HY19636, MiaPaCa2, and PANC1 cells cultured in either basal DMEM (C) or DMEM supplemented with 1mM L-alanine (D) for 24 hours. SLC38A2 was suppressed by CRISPR/Cas9 using two sgRNAs targeting SLC38A2 (sgSLC38A2 #1, #3) or a control sgRNA targeting Tomato (sgTom). All experiments were conducted using pools of cells within 1-2 passages after selection. Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment normalized to growth of surrogate wells. (E) Diagram depicting role of SLC38A2 in mediating sodium-dependent, concentrative uptake of alanine in PDAC (top panel). Cells lacking SLC38A2 rely on passive diffusion through other transporter(s) that cannot sustain alanine influx or maintain intracellular concentrations (bottom panel). (F) Intracellular alanine levels normalized by cell number in SLC38A2-deficient (sgSLC38A2 #1) or control (sgTom) HY19636, MiaPaCa2, and PANC1 cells. Data are represented as percentage relative to control (sgTom). Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment. (G) SLC38A2-deficient and control HY19636 cells were supplemented with 1mM L-alanine tert-butyl ester for 24 hours. Esterified alanine internalization, de-esterification, and secretion were measured by quantifying alanine release into conditioned media and normalizing to viable cell density over 24 hours. Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment normalized to growth of surrogate wells. (H) SLC38A2 expression allows PDAC cells to actively re-uptake alanine that is passively exchanged with the environment to allow for intracellular (cytosolic) concentration and enhanced utilization (left panel). SLC38A2-deficient cells do not have another concentrative alanine transport mechanism, and intracellular alanine is passively lost to the environment causing a decrease in intracellular levels and utilization (right panel). Significance determined with two-way ANOVA using Tukey’s multiple comparisons test in C and D; two-way ANOVA using Sidak’s multiple comparisons test in F; t-test in G. * p<0.05, ** p<0.01, *** p<0.001.
The SLC superfamily consists of an estimated 456 genes and pseudogenes classified into 52 subfamilies (26,27). Approximately 25% of SLCs are involved in amino acid transport, including the SLC1 and SLC38 families (28). SLC1A5 has been the focus of several studies in multiple cancer types with broad-spectrum amino acid specificity, including glutamine (29,30). Given the importance of glutamine to PDAC and proliferating cells in vitro (31), we observed similar levels of SLC1A5 and significant glutamine uptake across the cell panel (Supplemental Fig. S1A and S4B). In contrast, SLC1A4/ASCT1 has attracted far less attention beyond its initial characterization as an alanine, serine, cysteine, and threonine transporter and association with inborn errors of metabolism affecting neural development (32,33). SLC38A2/SNAT2 is characterized as an electrogenic sodium-neutral amino acid co-transporter with a preference for alanine and other small neutral amino acids (34). Many transporters are reported to transport alanine, but it remains unclear from biochemical studies alone which transporters function in relevant cellular contexts. Importantly, L-alanine is a reported substrate for both SLC1A4 and SLC38A2 providing rationale that differential transporter expression in PSC and PDAC may facilitate alanine crosstalk (32,34). We assessed expression of SLC38A2 in a panel of human PDAC cell lines to determine whether expression level correlated with alanine uptake preference and/or cellular demands. While we observed SLC38A2 expression in all PDAC cell lines with the majority expressing higher levels compared to stellate cells, SLC38A2 expression level did not correlate with alanine uptake preference or demand (Supplementary Fig. S4D). Furthermore, we observed multiple bands after removal of N-linked glycosylation suggesting differences in post-translational modification and/or isoform selection between cell lines, complicating conclusions of the function of SLC38A2 in PDAC. Therefore, we sought to functionally understand the role of SLC1A4 or SLC38A2 in intact pancreatic stellate and PDAC cells, respectively, through loss-of-function and metabolic flux studies.
Given the lack of a physiologically-relevant stromal and nutrient microenvironment, metabolic studies of cultured cells have limitations. Nonetheless, they have proven indispensable for studying intracellular metabolism and provide a tractable system to dissect the complex machinery involved in nutrient transport. Our data suggests that PDAC cell lines are heterogeneous in their alanine demands and relative preference for uptake versus de novo synthesis (Fig. 1). To functionally characterize the role of SLC38A2 in fulfilling alanine demands in PDAC, we selected representative cell lines that exhibit varying magnitudes of alanine demand (e.g. PANC1, MiaPaCa2, HY19636) and targeted SLC38A2 using CRISPR/Cas9 (Supplementary Fig. S5A and S5B). Upon knockout of SLC38A2, we observed striking alterations in alanine flux supporting a major role for SLC38A2 in the transport of alanine in PDAC cells. In basal conditions, where alanine is predominantly de novo synthesized, knockout of SLC38A2 in PDAC cells significantly enhanced alanine secretion flux (Fig. 2C). Furthermore, alanine uptake was significantly reversed in SLC38A2-deficient cells supplemented with environmental alanine concentrations (Fig. 2D). Irrespective of alanine demand or preference towards uptake versus synthesis, all SLC38A2-deficient PDAC cell lines tested exhibited significantly increased secretion of alanine (Fig. 2D).
We next sought to characterize the functional impacts on intracellular alanine metabolism and transport in SLC38A2-deficient PDAC cells. Loss of SLC38A2 did not completely eliminate alanine transport, suggesting that other transport mechanisms must exist that operate in concert with SLC38A2. We hypothesized that SLC38A2-dependent co-transport of sodium and alanine permits PDAC cells to concentrate cytosolic alanine (Fig. 2E, upper panel). In the absence of a concentrative transport mechanism, regardless of the presence of extracellular alanine, cells would fail to sequester alanine in the cytosol and enhance passive secretion to the environment (Fig. 2E, lower panel). This mechanism is supported by the fact that alanine levels were significantly decreased in SLC38A2-deficient cells relative to control (sgTom) cells regardless of alanine supplementation (Fig. 2F). Importantly, expression of sgRNA-resistant SLC38A2 cDNA fully rescued alanine levels in PDAC, whereas a non-functional mutant with poor sodium affinity failed to rescue (Supplementary Fig. S5C and S5D). SLC38A2-deficient cells failed to maintain similar levels of mitochondrial 13C3-alanine utilization suggesting that passive transport mechanisms were insufficient to sustain mitochondrial alanine anaplerosis in PDAC (Supplementary Fig. S5E). To directly test the hypothesis that SLC38A2 is the major concentrative alanine transporter in PDAC, we quantified the cellular capacity to internalize and concentrate cell-permeable esterified L-alanine, which can diffuse into cells and establish cytosolic levels independent of transport. Strikingly, we observed a significant increase in the secretion of de-esterified alanine in SLC38A2-deficient cells, and a reduced capacity to sequester de-esterified alanine intracellularly (Fig. 2G, Supplementary Fig. S5F). Taken together, these data suggest that de-esterified alanine is passively secreted in the absence of an extracellular concentration gradient (Fig. 2H). In the presence of SLC38A2, cells are capable of actively re-uptaking the passively secreted de-esterified alanine, increasing intracellular levels and counteracting passive efflux (Fig. 2H). Thus, SLC38A2-deficient cells increase passive loss to the environment, decrease alanine utilization, and increase de novo synthesis. Taken together, these data indicate that SLC38A2 is required and the primary driver of concentrative alanine influx in PDAC cells, whereas other mechanisms can only passively equilibrate levels with extracellular concentrations (Fig. 2H).
Loss of SLC38A2 results in a metabolic crisis through primary and secondary effects on PDAC metabolism
Alanine supplementation does not stimulate proliferation of PDAC cells cultured in replete nutrient conditions, suggesting that the metabolic costs associated with de novo synthesis do not impact cell fitness in this context (11). However, in nutrient-limited environments similar to those observed in tumors, we hypothesized that scavenging environmental alanine may provide a selective advantage for PDAC cells by offsetting the metabolic costs of de novo synthesis. To mimic nutrient conditions in tumors, we cultured PDAC cells in low amino acid media used in previous studies investigating the metabolic contributions of macropinocytosis in PDAC (15). In this context, alanine supplementation consistently enhanced PDAC cell proliferation by ~15-30%, and loss of SLC38A2 attenuated the growth-promoting effects of alanine in nutrient-limited conditions (Fig. 3A). In addition, loss of SLC38A2 significantly reduced cell proliferation and clonogenic potential in nutrient replete conditions (Fig. 3B and 3C, Supplementary Fig. S6A). Neither alanine nor cell-permeable alanine rescued the proliferation defect in SLC38A2-deficient cells; however, increased passive efflux of alanine occurs in both conditions and SLC38A2 was required for internalization of both analogs (Fig 2G, Supplementary Fig. S5E and S6B). Therefore, we hypothesized that a shift towards decreased alanine utilization and increased passive loss to the environment, arising from deficiencies in alanine internalization, causes a metabolic crisis and decreased proliferative potential in PDAC.
Fig. 3. Loss of SLC38A2 causes a metabolic crisis and suppresses PDAC proliferation.
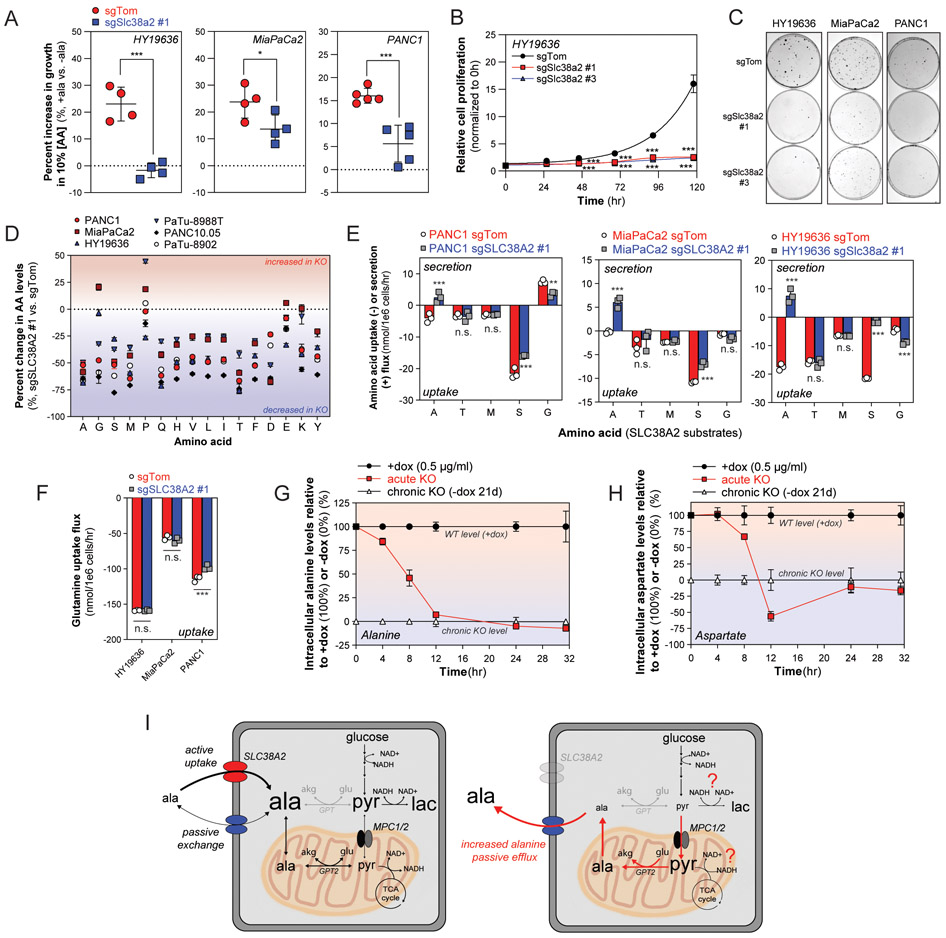
(A) Control (sgTom) or SLC38A2-deficient HY19636, MiaPaCa2, and PANC1 cells cultured in low amino acid DMEM (10% [AA]DMEM) supplemented with or without 1mM L-alanine for 48 hours. Enhanced proliferation with L-alanine supplementation reported as a percent increase relative to growth of cells cultured in basal low amino acid DMEM without L-alanine. Error bars depict s.e.m. of ≥4 independent experiments of 12 technical replicate wells each. (B and C) Cell proliferation (B) of control (sgTom) and SLC38A2-deficient (sgSlc38a2 #1, #3) HY19636 cells cultured in DMEM over five days. Representative clonogenic assay plates in SLC38A2 deficient (sgSlc38a2 #1, #3) or control (sgTom) HY19636, MiaPaCa2, and PANC1 cells cultured in basal DMEM over 7-14 days. Data in B are plotted as cell proliferation relative to day 0 collected after cell attachment. Error bars depict s.d. of four technical replicate wells for each time point; representative data depicted from n≥2 independent experiments. Curves represent fitted logarithmic growth rates. Imaged clonogenic plates representative of three technical replicate plates of ≥2 independent experiments. (D) Increase (+) and decrease (−) in intracellular amino acid levels in SLC38A2-deficient cells (sgSLC38A2 #1) relative to control cells (sgTom). Data are plotted as percent increase or decrease in SLC38A2-deficient cells relative to control cells. All amino acid levels were normalized to cell number (1x106 cells) prior to comparison to control (sgTom). Error bars depict s.d. of ≥3 technical replicate wells per cell line and condition. Dotted line indicates no change and positive and negative values indicate an increase and decrease in amino acid levels in SLC38A2-deficient cells relative to control (sgTom) cells, respectively. (E and F) Alanine and other SLC38A2 substrate (e.g. threonine, methionine, serine, glycine in E; glutamine in F) uptake and secretion fluxes in SLC38A2-deficient (sgSLC38A2 #1) and control (sgTom) PANC1, MiaPaCa2, and HY19636 cells cultured in basal DMEM. Extracellular accumulation (+, secretion) or depletion (−, uptake) was measured in conditioned media over 24 hours and normalized to the viable cell density over the time course. Error bars depict s.d. of ≥3 technical replicate wells normalized to cell growth of surrogate wells. (G and H) Time course of relative intracellular alanine (G) and aspartate (H) levels after acute SLC38A2 loss from doxycycline withdrawal compared to levels measured in SLC38A2-expressing (+dox) or chronic SLC38A2 knockout (-dox chronic) PANC1 pInducer-resSLC38A2WT cells collected over the same time course. Intracellular alanine (G) and aspartate (H) levels in acute SLC38A2 knockout cells (-dox acute) are plotted as a percentage relative to SLC38A2-expressing cells (+dox; set to 100%) and the chronic SLC38A2 knockout levels (chronic −dox; set to 0%). The chronic KO data were treated as a baseline and subtracted from each condition and timepoint; a negative value represents a decrease in amino acid level below that of the chronic SLC38A2 knockout. Error bars depict s.d. of ≥3 technical replicate wells normalized to cell density in surrogate wells per time point. (I) SLC38A2 expressing cells are capable of maintaining compartmentalized concentrations of alanine to support metabolic homeostasis of cytosolic and mitochondrial alanine and pyruvate pools (left panel). Loss of SLC38A2 enhances passive secretion, increasing the demand to synthesize alanine in the mitochondria (right panel). Significance determined with two-way ANOVA using Sidak’s multiple comparison testing in A, E, and F; two-way ANOVA using Dunnett’s multiple comparisons test in B. * p<0.05, ** p<0.01, *** p<0.001.
Alanine utilization contributes to amino acid homeostasis acting as a major amino-nitrogen donor to transaminase networks (Fig. 1C, Supplementary Fig. S2). In addition, SLC38A2 is reported to transport amino acids other than alanine, including glutamine, serine, glycine, methionine, and threonine (34). We hypothesized that defects in other amino acids, due to transport and/or transamination deficiencies, may also contribute to the observed metabolic defect. Strikingly, we observed a significant decrease in total amino acid levels and extensive alterations in the composition of cellular amino acids in SLC38A2-deficient cells (Fig. 3D, Supplementary Fig. S6C and S6D). Despite cell type-specific alterations in individual amino acids, the general dysregulation in amino acid homeostasis was shared across all PDAC cell lines tested, and was fully rescued by a sgRNA-resistant cDNA (Fig. 3D, Supplementary Fig. S6C-E). Quantification of other amino acid extracellular fluxes upon SLC38A2 loss, including SLC38A2 substrates and non-substrates, revealed significant, but relatively minor and cell-line specific effects (Fig. 3E and 3F, Supplementary Fig. S7A and 7B). Cell-line specific changes in uptake or secretion of other SLC38A2 substrates (e.g. serine, glycine), arising from transport limitations and/or metabolic rewiring, may also contribute to the proliferative defect observed in SLC38A2-deficient cells. However, only alanine flux completely reversed in SLC38A2-deficient cells, suggesting that other SLC38A2 substrate fluxes are sustained by redundancy through other transporter systems.
We hypothesized that broad de-regulation of amino acid pools in SLC38A2-deficient cells may be due to secondary metabolic effects arising from alanine transport defects, specifically loss of cytosolic internalization. Alanine may act as an exchange factor for the facilitated diffusion of other amino acids. For example, glutamine and asparagine serve as exchange factors for several amino acids, including BCAAs (35,36). In this model, the sodium gradient produced by the N/K-ATPase provides the free energy for SLC38A2 to concentrate alanine intracellularly against its gradient (Fig. 2E). Subsequently, alanine exchange provides the free energy necessary to facilitate import of other amino acids. To determine whether alanine may serve as an exchange factor, we quantified the impacts of cell-permeable alanine on other intracellular amino acid levels. We hypothesized that esterified alanine would establish intracellular concentrations of alanine following de-esterification in the cytosol. Our initial data demonstrated that esterified alanine was rapidly de-esterified intracellularly and secreted (Fig. 2G), which may be coupled to exchange with other amino acids. However, esterified alanine failed to increase amino acid levels in either control (sgTom) or SLC38A2-deficient cells suggesting that alanine does not function as an exchange factor (Supplementary Fig. S7C). Thus, passive secretion of alanine likely occurs mainly through mass action-driven diffusion.
Many altered amino acids are not reported substrates of SLC38A2 (e.g. glutamate, tyrosine, lysine, BCAAs) or are not transported by PDAC cells (e.g. aspartate) (Fig. 3D, Supplementary Fig. S7A and S7B). To investigate primary and secondary metabolic effects of SLC38A2 loss, we ectopically expressed a dox-inducible sgRNA-resistant cDNA maintained during CRISPR/Cas9-mediated deletion of the endogenous locus and performed metabolomics (Supplementary Fig. S7D). Proteasomal inhibition resulted in accumulation of SLC38A2 in PDAC cells, in-line with previous reports, and withdrawal of doxycycline rapidly depleted SLC38A2 expression (Supplementary Fig. S7E and S7F) (37,38). To allow for secondary metabolic re-wiring, we established chronic SLC38A2-null cells by removing doxycycline for several passages. Upon acute withdrawal of dox, we observed a rapid decrease in SLC38A2 and alanine levels concordant with our chronic knockout data (Supplementary Fig. S7F and S7G). Furthermore, SLC38A2 depletion increased eIF2α phosphorylation but not LC3B lipidation suggesting activation of specific amino acid stress responses (Supplementary Fig. S7F). Acute dox withdrawal resulted in a rapid decrease of intracellular alanine and other SLC38A2 substrate levels to chronic levels (Fig. 3G, Supplementary Fig. S7H). Rapid, delayed decreases in several other amino acids (e.g. aspartate) were also observed upon dox-withdrawal (Fig. 3H). Furthermore, several amino acids decreased significantly below chronic knockout levels (e.g. aspartate, isoleucine, proline) after 12 hours doxycycline withdrawal followed by a compensatory increase to chronic levels after 24 and 36 hours (Fig. 3H, Supplementary Fig. S7H). These data establish that small neutral amino acids, including alanine, serine, and threonine, are transported by SLC38A2 in intact cells. However, only alanine flux was non-redundant, and our data provide evidence of a secondary metabolic crisis impacting both SLC38A2 substrate and other amino acid pools.
Compartmentalized redox crisis and compensatory amino acid catabolism driven by increased passive alanine efflux
Our previous data suggests that PDAC cells down-regulate cytosolic alanine biosynthetic machinery and rely on SLC38A2 to maintain cytosolic concentrations of alanine. While our data supports multiple amino acids are transported by SLC38A2, only alanine uptake was completely suppressed in SLC38A2-deficient cells (Fig. 3E and 3F, Supplementary Fig. S7A and S7B). In SLC38A2-deficient conditions, cells increase passive efflux of alanine to the environment, both in basal and alanine-supplemented conditions, increasing the demand to synthesize alanine (Fig. 3I). We hypothesized that increased mitochondrial alanine synthesis demand would lead to compartmentalized impacts on pyruvate metabolism (Fig. 3I). To explore this hypothesis, we generated isogenic control (sgTom) and Slc38a2-knockout (sgSlc38a2 #1) clonal knockouts ectopically expressing sgRNA-resistant SLC38A2 cDNA (Fig. 4A). Similar to pooled knockouts, Slc38a2-deficient cells had significantly suppressed proliferation and clonogenic potential (Fig. 4B and 4C). Pyruvate is required for NAD+ regeneration in the cytosol (via LDH activity) and supplies mitochondrial NADH (via PDH activity) for electron transport chain (ETC) function (Fig. 3I). We hypothesized that the increased cytosolic pyruvate import into the mitochondria would lead to a misbalancing of cytosolic and mitochondrial NAD(H). Strikingly, both ECAR and OCAR significantly decreased by ~35% in Slc38a2-knockout but not control (sgTom) cells after SLC38A2 depletion, suggesting profound decreases in both glycolytic and respiratory potential (Fig. 4D, Supplementary Fig. S8A).
Fig. 4. SLC38A2 deficiency causes compartmentalized redox crisis and switch to catabolic metabolism.
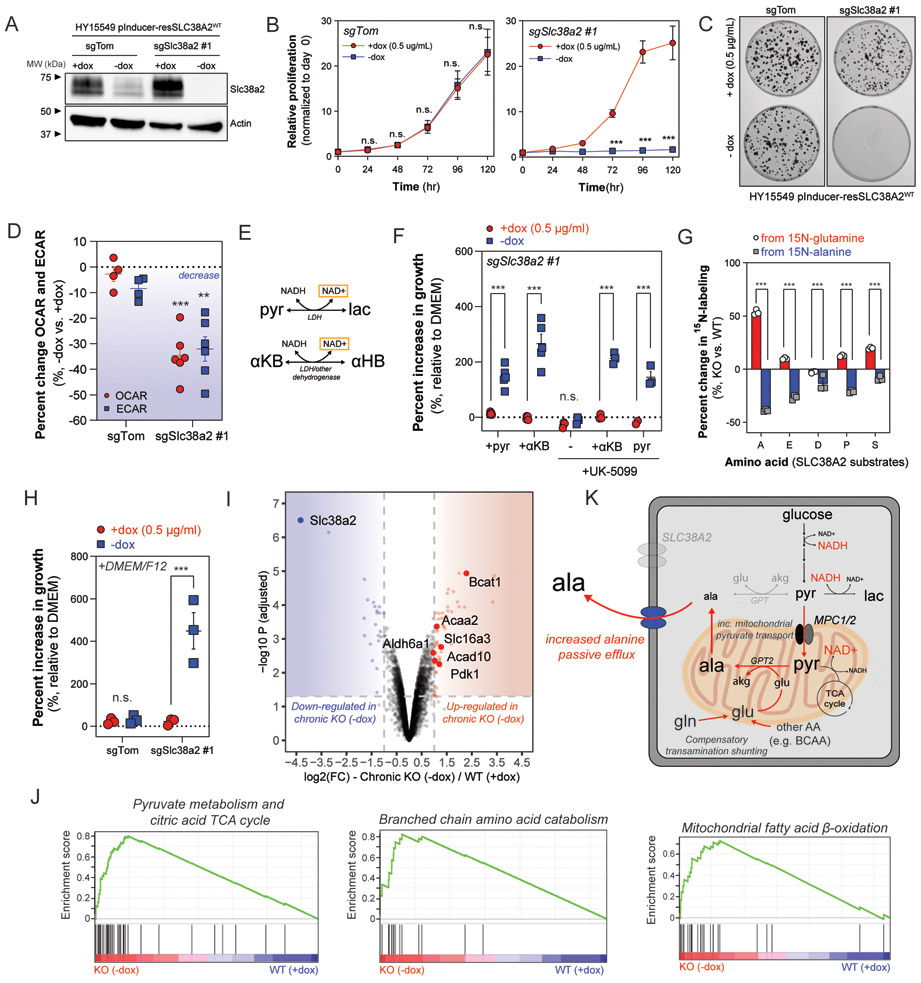
(A) Immunoblot of Slc38a2 and Actin in whole cell lysates (30 μg) collected from clonally derived HY15549 control (sgTom) and Slc38a2-deficient (sgSlc38a2 #1) cells ectopically expressing sgRNA-resistant SLC38A2 cDNA under control of doxycycline-inducible promoter. Cells were cultured in the presence of dox (0.5 μg/ml) or after washout of dox for 24 hours prior to lysis. Withdrawal of doxycycline leads to rapid decrease in SLC38A2 expression in Slc38a2-deficient HY15549 cells. Representative immunoblot of two independent experiments. (B and C) Cell proliferation (B) and representative plates from clonogenic assay (C) of clonally derived control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) HY15549 cells cultured in DMEM +dox or −dox over five days (B) or 7-10 days (C). Data in B are plotted as cell proliferation relative to day 0 collected after cell attachment. Error bars in B depict s.e.m. of ≥3 independent experiments of four technical replicate wells for each time point. Representative clonogenic plates in C selected from ≥3 independent experiments of three technical replicate plates for each condition. (D) Decrease in extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in clonally derived HY15549 control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) cell lines after withdrawal of doxycycline for 24 hours. Data are plotted as a percent increase or decrease of −dox compared to +dox (0.5 μg/ml). Error bars depict s.e.m. of ≥4 independent experiments of ≥10 technical replicate wells for each condition. (E) Pyruvate (pyr) is reduced by NADH by LDH to produce lactate (lac) and regenerate NAD+. α-ketobutyrate (αKB) can be reduced by LDH or other dehydrogenases to produce α-hydroxybutyrate (αHB) and regenerate NAD+. (F) Percent increase in proliferation of HY15549 Slc38a2-deficient cells in DMEM supplemented with doxycycline (+dox 0.5 μg/ml) or without doxycycline (-dox). Cells were supplemented with control (blank DMEM), pyruvate (1mM), α-ketobutyrate (1mM) and either vehicle (DMSO) or UK-5099 (25 μM) and allowed to grow for 96 hours. Data are plotted as a percent increase compared to control DMEM. Error bars depict s.e.m. of ≥3 independent experiments of four technical replicate wells for each condition. (G) Stable isotope labeling from either 15N-alanine or α-15N-glutamine in intracellular alanine (A), glutamate (E), aspartate (D), proline (P), or serine (S) after 24 hours incubation with either tracer. Data are plotted as a percent increase or decrease in HY15549 Slc38a2-deficient cells cultured without doxycycline (KO, -dox) or with doxycycline (WT, +dox 0.5 μg/ml). Error bars depict s.d. of ≥3 technical replicate wells from one independent experiment per tracer. (H) Percent increase in proliferation of HY15549 Slc38a2-deficient cells in DMEM supplemented with doxycycline (+dox 0.5 μg/ml) or without doxycycline (-dox). Cells were supplemented with DMEM or 50:50 DMEM/F12 and allowed to grow for 96 hours. Data are plotted as a percent increase compared to control DMEM. Error bars depict s.e.m. of ≥3 independent experiments of four technical replicate wells for each condition. (I) Volcano plot of differentially expressed proteins between chronic Slc38a2 knockout (chronic KO −dox) and Slc38a2-expressing (WT +dox 0.5 μg/ml) HY15549 cells. Proteins were considered differentially expressed if log2 fold change (FC) ≥ 1 or ≤ −1 and adjusted p-value ≤ 0.05. Metabolic enzymes involved in BCAA catabolism (Bcat1, Aldh6a1), mitochondrial fatty acid β-oxidation (Acaa2, Acad10), and pyruvate metabolism (Slc16a3, Pdk1) that are highly expressed in chronic knockout cells are highlighted with red squares and labeled with protein name. (J) Gene set enrichment analysis (GSEA) of differentially expressed proteins between chronic Slc38a2 knockout (KO −dox) and Slc38a2-expressing (WT +dox). (K) Diagram depicting compartmentalized redox crisis occurring as a result of increased passive efflux of alanine in SLC38A2-deficient cells. Increased mitochondrial pyruvate transport results in compartmentalized NAD(H) crisis reducing both glycolytic and respiratory potential. Decreased utilization of alanine and increased synthesis shifts other amino-nitrogen donors (e.g. glutamine, BCAA) towards mitochondrial de novo alanine synthesis. Significance determined with two-way ANOVA using Sidak’s multiple comparison testing in B, D, F, G, and H. * p<0.05, ** p<0.01, *** p<0.001.
Mitochondrial respiration supports aspartate biosynthesis in cells (39). Indeed, acute depletion of SLC38A2 significantly reduced respiratory potential and led to a reduction in intracellular aspartate levels by ~90% (Fig. 4D, Supplementary Fig. S8B). Pyruvate or alternative electron acceptors (e.g. α-ketobutyrate) can rescue proliferation of ETC-deficient cells by regenerating NAD+ (Fig. 4E) (39). Furthermore, excess pyruvate would relieve redox pressures associated with increased mitochondrial pyruvate transport. We hypothesized that a compartmentalized redox imbalance was driving the metabolic crisis in SLC38A2-deficient cells. Pyruvate or α-ketobutyrate (αKB) supplementation significantly rescued proliferation and clonogenic potential of SLC38A2-deficient cells (Fig. 4F, Supplementary Fig. S8C). The proliferative rescue by either electron acceptor in SLC38A2-deficient cells was independent of mitochondrial pyruvate transport, suggesting that SLC38A2-deficient cells have defective cytosolic NAD+ regeneration consistent with an increased mitochondrial shunting of pyruvate for alanine synthesis (Fig. 4F).
We cultured SLC38A2 knockout cells with 13C3-alanine or 15N-alanine to quantify alterations in alanine utilization after acute SLC38A2 depletion. Strikingly, contribution to mitochondrial TCA cycle metabolites (e.g. citrate) and contribution to transaminase products (e.g. glutamate, aspartate, proline, serine) were significantly decreased after acute SLC38A2 depletion (Fig. 4G, Supplementary Fig. S8D). We hypothesized that other amino-nitrogen donors may compensate for the decreased capacity to utilize alanine. We cultured cells with α-15N-glutamine and found that utilization of glutamine and other (e.g. unlabeled) nitrogen donor(s) compensated for the decreased alanine contribution in SLC38A2-deficient cells (Fig. 4G, Supplementary Fig. S8E). Strikingly, no compensatory increase in aspartate was observed, suggesting deficient aspartate production and an incomplete metabolic rewiring in response to SLC38A2 loss (Fig. 4G). These data suggest that glutamine utilization is increased and confirms that its transport through SLC38A2 is not limiting, in-line with the minimal impacts on glutamine uptake in SLC38A2-deficient cells (Fig. 3F, Supplementary Fig. S7A). To relieve the need to synthesize non-essential amino acids, we supplemented SLC38A2-deficient cells with a rich medium containing all 20 amino acids (e.g. DMEM/F12) and observed a near complete rescue in clonogenic potential and a significant rescue of proliferation (Fig. 4H, Supplementary Fig. S8C). Taken together, these data suggest that SLC38A2 drives a major metabolic crisis associated with deficiencies in compartmentalized alanine metabolism that results in an ineffective metabolic compensation.
Intracellular metabolic networks are intrinsically plastic, and targeting individual enzymes often leads to significant metabolic re-wiring to compensate for loss of activity of a particular metabolic node (40,41). In parallel, transporters often have multiple substrates and multiple transporters for a given metabolite, providing reason to believe that redundancy may limit their efficacy as drug targets in cancer. However, it is important to study the context-specific requirements for specific transporters to understand the durability in targeting a single transporter and potential compensatory mechanisms that may exist. To evaluate potential redundant alanine transport mechanisms in PDAC, we generated a chronic knockout cell line after long-term culture in the absence of doxycycline and conducted quantitative proteomics comparing SLC38A2-expressing cells (+dox), acute SLC38A2-deficient cells (-dox acute), and chronic SLC38A2 knockout cells (-dox chronic). Surprisingly, chronic knockout cells retained the growth defect observed in acute deletion cells suggesting that cells ineffectively adapt to loss of SLC38A2 (Supplementary Fig. S8F). We observed several SLC proteins that were significantly up-regulated in chronic knockout cells; however, none were associated with alanine transport (Supplementary Fig. S8G). Interestingly, the lactate transporter Slc16a3 (Mct4) was significantly up-regulated in SLC38A2 knockout cells, suggesting that cells attempt to adapt to a decreased glycolytic capacity by enhancing lactate secretion (Fig. 4I, Supplementary Fig. S8G). We conducted gene set enrichment analysis (GSEA) to understand how SLC38A2-deficient cells attempt to metabolically compensate for loss of SLC38A2. In-line with our stable-isotope tracing results, we identify that proteins involved in pyruvate and TCA cycle metabolism are significantly up-regulated (e.g. Slc16a3, Pdk1) suggesting a diversion of mitochondrial pyruvate away from oxidative TCA cycling, via Pdk1 phosphorylation and inactivation of pyruvate dehydrogenase complex required for mitochondrial pyruvate oxidation (Fig. 4I and 4J, Supplementary Fig. S8H). Furthermore, we observe significant enrichment in proteins involved in mitochondrial fatty acid β-oxidation (e.g. Acaa2, Acad10) and branched chain amino acid (BCAA) catabolism (e.g. Bcat1, Aldh6a1), suggesting that cells increase alternative supply pathways for mitochondrial NADH and acetyl-CoA (β-oxidation) and amino-nitrogen (BCAA catabolism) (Fig. 4I and 4J, Supplementary Fig. S8H). These results provide evidence that non-SLC38A2 substrate (e.g. BCAA) levels may decrease as SLC38A2-deficient cells switch from anabolic metabolism to a more catabolic state. Collectively, these results suggest that cytosolic alanine transport defects drive a specific compartmentalized metabolic crisis, and that cells lacking SLC38A2 undergo an ineffective metabolic compensation (Fig. 4K).
SLC38A2 is highly expressed in human and murine PDAC and localized to the plasma membrane
The PDAC microenvironment is thought to be nutrient and oxygen austere owing to the intense fibrotic stroma, increased interstitial pressure, and leaky vasculature (14,42). Thus, nutrients within the PDAC microenvironment are likely locally supplied and shared between stromal cell populations. We hypothesized that differential expression of nutrient transporters allows for the formation of metabolic niches within the TME, including PSC-PDAC alanine crosstalk. To determine whether PDAC-specific SLC38A2 expression is also observed in vivo, we stained tumors derived from the well-established pancreatic cancer ‘KPC’ (LSL-KrasG12D; Trp53lox/+; p48Cre+) mouse model for SLC38A2. Strikingly, we observed strong and specific staining in PDAC cells and low or no staining in the surrounding stroma (Fig. 5A). As SLC38A2 is thought to be ubiquitously expressed in normal tissues (43), we stained normal murine pancreas and liver and observed punctate intracellular staining suggesting that SLC38A2 in normal tissues may be predominantly non-plasma membrane localized (Fig. 5B and 5C). Furthermore, we performed immunohistochemical analysis for SLC38A2 expression in human PDAC tissue microarrays consisting of 136 patient tumor specimens (Fig. 5D). SLC38A2 was expressed in PDAC cells in majority of human PDAC samples (114/136 tumors; ~84%). In contrast, SLC38A2 expression in stromal cells was observed only in ~7% of human PDAC samples (9/136) (Fig. 5D and 5E). Thus, PDAC cells may intrinsically activate expression of SLC38A2 to facilitate alanine crosstalk during tumorigenesis. To confirm the localization of SLC38A2, we expressed SLC38A2-GFP in PDAC cells and non-malignant human pancreatic ductal epithelial (HPDE) cells, mPSC#1, canine kidney epithelial (MDCK) cells and determined localization patterns by confocal microscopy. In-line with our previous results we observed robust plasma membrane localization of SLC38A2-GFP in PDAC cells, whereas non-malignant cells localize SLC38A2 predominantly to intracellular endomembranes (Fig. 5F, Supplementary Fig. S9A). While ubiquitously expressed, SLC38A2 is not considered a common essential gene in large-scale CRISPR knockout studies (22). Furthermore, SLC38A2 is expressed in non-malignant cells in vitro, albeit at lower levels (Fig. 2B, Supplementary Fig. S4D). We hypothesized that SLC38A2 may be specifically required by PDAC cells, independent of localization, to supply their increased alanine demand compared to non-malignant cells (Fig. 1G). Indeed, knocking out SLC38A2 in non-malignant cell lines (e.g. hPSC#1, mPSC#1, IMR-90, HPDE) did not illicit a significant growth defect (Fig. 5G, Supplementary Fig. S9B). Taken together, the striking selective expression of SLC38A2 in human PDAC tumors relative to the stroma and the PDAC-specific dependency may provide a therapeutic window for limiting cancer cell access to stromal-supplied and environmental alanine.
Fig. 5. Differential SLC38A2 expression in vivo, localization, and dependence in PDAC and non-malignant cells.
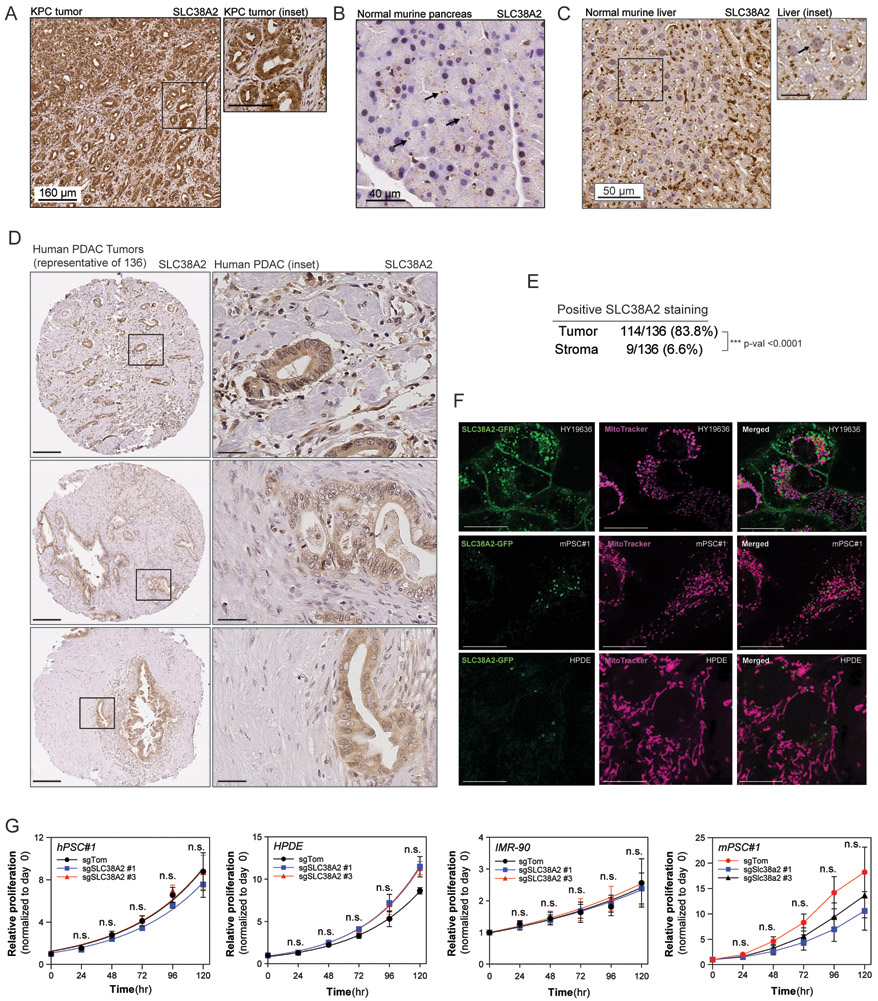
(A) (A-C) Immunohistochemical analysis of Slc38a2 in (A) pancreatic tumors derived from the KPC (LSL-KrasG12D; Trp53lox/+; p48Cre+) mouse model, (B) normal murine pancreas, and (C) normal murine liver. Representative fields from KPC (4x), normal pancreas (10x), and normal liver (4x) derived from two mice. Scale bars indicated in panels; scale bars for (A) inset is 90 μm and for (C) inset is 20 μm. Arrows indicate instances of punctate SLC38A2 in normal pancreas and liver. (D) Representative tumors from immunohistochemical analysis of SLC38A2 in tumor microarray of 136 human PDAC tumor specimens. Scale bars indicated in panels; 200 μm and inset is 40 μm. (E) Positive SLC38A2 staining observed in 114/136 tumors, 9/136 of corresponding stroma. (F) Representative live cell images of HY19636 (top panel), mPSC#1 (center panel), and HPDE (bottom panel) cells transiently transfected with 3 μg of SLC38A2-GFP (green) overnight. Live cells were imaged using MitoTracker. Representative fields containing 1-2 cells from ≥5 fields from ≥3 independent experiments. Scale bars are indicated in each panel; 5 μm. (G) Cell proliferation of non-malignant control (sgTom) or SLC38A2-deficient (sgSLC38A2 #1, #3) hPSC#1, HPDE, IMR-90, and mPSC#1 cells cultured in DMEM over five days. Data in are plotted as cell proliferation relative to day 0 collected after cell attachment. Error bars depict s.e.m. of ≥3 independent experiments of four technical replicate wells for each time point. Significance determined with two-tailed Fisher’s exact test in E; two-way ANOVA using Dunnett’s multiple comparison testing in G. * p<0.05, ** p<0.01, *** p<0.001.
Multiple passive transporters, including SLC1A4, are required for PSC alanine secretion and exchange
We next set out to understand how alanine is exported from stellate cells. Given the specific expression of SLC1A4 in PSCs (Fig. 2A and 2B), we utilized RNAi to suppress its expression and quantified impacts on alanine secretion and exchange fluxes (Supplementary Fig. S10A). Knockdown of SLC1A4 in PSCs resulted in a significant reduction in net alanine secretion flux and reduced kinetics of extracellular 13C3-alanine dilution arising from exchange (Supplementary Fig. S10B and S10C). Interestingly, we observed no significant impact on other reported substrate fluxes, such as serine and threonine, supporting the concept that transporter specificity is influenced by cellular context (Supplementary Fig. S10D). Cysteine, although a reported substrate of SLC1A4, is rapidly oxidized to cystine in vitro and was not detectable at appreciable levels in conditioned media. Notably, we did not observe a significant impact on PSC proliferation upon knockdown of SLC1A4 (Supplementary Fig. S10E). The magnitude of inhibition on alanine secretion, while significant, was not complete suggesting that SLC1A4 operates in concert with other diffusive transporters to facilitate alanine secretion by PSCs. Co-targeting multiple diffusive neutral amino acid transporters may be an effective strategy to abolish PSC alanine secretion. However, we hypothesize that targeting SLC38A2 may be a more robust strategy for inhibiting the alanine demands of pancreatic cancer, as SLC38A2 was indispensable for alanine uptake and inhibition would limit access to other environmental alanine sources that may exist.
Disrupting SLC38A2-dependent alanine uptake affects tumor initiation and maintenance in subcutaneous and orthotopic models of PDAC
To evaluate whether targeting SLC1A4- or SLC38A2- mediated alanine crosstalk is sufficient to inhibit PSC-PDAC alanine crosstalk in vivo, we utilized a co-injection strategy similar to previous studies (7,11). PSC-PDAC co-injection significantly promotes tumor initiation and growth (7,11); therefore, we performed limiting dilution subcutaneous xenografts by injecting a sub-optimal number of control or SLC38A2 knockdown PDAC cells alone or co-injected with PSCs (Supplementary Fig. S11A). In agreement with previous studies, we observed enhanced tumor initiation when PDAC cells were co-injected with PSCs (Supplementary Fig. S11B-D). Notably, the enhanced tumor initiation was completely abolished by knockdown of SLC38A2 (Fig. 6A). Knockdown of SLC1A4 in PSCs failed to illicit a similar reduction in tumor initiation in this co-injection model, consistent with the fact that SLC1A4 knockdown was insufficient to completely inhibit PSC alanine transport (Supplementary Fig. S11E). To further demonstrate the role of SLC38A2-mediated alanine crosstalk in tumor initiation and growth using a more physiologically relevant model, we performed orthotopic xenograft and syngeneic allograft studies using SLC38A2 knockdown or knockout cells. Consistent with subcutaneous xenograft results, SLC38A2-deficient cells exhibited significantly delayed tumor initiation as well as decreased tumor burden in both orthotopic xenografts and syngeneic allografts (Fig. 6B, Supplementary Fig. S11F). While orthotopic tumor burden was significantly reduced using pooled CRISPR/Cas9 SLC38A2-knockout cells, we observed evidence of escape of SLC38A2-expressing cells in large tumors arising from silent repair of the CRISPR/Cas9-mediated double strand breaks (Supplementary Fig. S11F and S11G). To determine if apoptosis was contributing to the decreased tumor burden, we stained tumors for cleaved caspase 3; however, we did not observe a significant increase in SLC38A2-deficient tumors suggesting that increased cell death was not contributing to the tumor burden defect (Supplementary Fig. S11H).
Fig. 6. SLC38A2 is vital for PDAC tumor initiation and growth.

(A) Tumor initiation was significantly suppressed by SLC38A2 knockdown (shSLC38A2 #1, #4) compared to control (shGFP) PANC1 cells (2x105) co-injected with hPSC#1 cells (1x106) in a subcutaneous xenograft model. Subcutaneous tumors were monitored bi-weekly by caliper measurement and considered formed if length and width were measured to be ≥1mm each. Each injected flank was considered a possible tumor when calculating percent tumor-free. (B) Tumor initiation was significantly suppressed by SLC38A2 knockdown (shSLC38A2 #1, #4) compared to control (shGFP) PANC1 cells (5x105) in an orthotopic xenograft model. Orthotopic xenografts were monitored by 3-D ultrasound bi-weekly and considered tumors if volume ≥1mm3. (C) Subcutaneous (SubQ) and orthotopic tumor maintenance strategy for deconvoluting tumor initiation versus maintenance effects with acute SLC38A2 deletion. Mice were pre-treated with chow containing doxycycline (625ppm) for three days prior to injection and for five (orthotopic) or seven (subcutaneous) days after injection to allow for SLC38A2 expression during tumor initiation period. Following tumor initiation, mice were switched to new cages with regular chow (no doxycycline). (D) Tumor initiation was rescued by doxycycline-induced SLC38A2 expression in Slc38a2-deficient (sgSlc38a2 #1) HY15549 cells. No significant difference was observed in tumor volume, measured by caliper measurement at day 7 after injection, between control (sgTom) and Slc38a2-deficient (sgSlc38a2 #1) tumors. (E) Tumor volume of subcutaneous control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) tumors following dox withdrawal after seven day initiation period. Significant tumor regression in Slc38a2-deficient tumors was observed following withdrawal of SLC38A2. (F) Final tumor weight of subcutaneous (left panel) and orthotopic (right panel) control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) tumors. Subcutaneous tumors were resected and weighed at 20 days (sgTom) or 64 days (sgSlc38a2 #1) after dox-withdrawal. Orthotopic tumors were resected and weighed after 21 days dox-withdrawal. Significance determined with log-rank Mantel-Cox test in A-B; two-tailed t-test in D and F; two-way ANOVA using Sidak’s multiple comparisons test in E. * p<0.05, ** p<0.01, *** p<0.001.
To evaluate the viability of SLC38A2 as a therapeutic target for PDAC tumor maintenance, we utilized a dox-inducible cDNA withdrawal approach to deconvolute effects on tumor initiation and tumor maintenance in both syngeneic subcutaneous and orthotopic tumor models. Mice were fed doxycycline-containing chow prior to injection and during tumor initiation, to express sgRNA-resistant SLC38A2, which fully rescued tumor initiation (Fig. 6C and 6D). Withdrawal of doxycycline to mimic drugging SLC38A2 in fully formed tumors resulted in a rapid and significant regression of Slc38a2-knockout but not control (sgTom) subcutaneous tumors (Fig. 6E and 6F). At endpoint, no tumors were detected in the pancreata of mice injected with Slc38a2-knockout cells, suggesting either failed initiation or more likely tumor regression (Fig. 6F). Furthermore, the regression of subcutaneous Slc38a2-knockout tumors was durable over 60 days, consistent with our previous data suggesting an ineffective metabolic adaptation to loss of SLC38A2 (Fig. 6E). Collectively, our results suggest that SLC38A2 is vital to the initiation and maintenance of pancreatic tumors, and targeting SLC38A2 may be a viable approach to treat this deadly disease.
Discussion
Our work demonstrates that pancreatic cancer and stellate cells form a niche to exchange alanine through differential expression of SLC38A2 and SLC1A4. While SLC1A4 was not essential for PSC alanine secretion, SLC38A2 was required for the uptake and cytosolic concentration of alanine in PDAC. Metabolic niches play an important physiological role in tissue function. In intestinal crypts, Paneth cells supply lactate to intestinal stem cells as an anaplerotic substrate to fuel oxidative mitochondrial metabolism (44). Our data illustrates that specific alanine transporters are required to exchange alanine within the PDAC tumor niche. For intestinal and tumor lactate exchange, this has been demonstrated through differential expression of monocarboxylate and glucose transporters to channel lactate from hypoxic to normoxic regions of the tumor (45,46). Glucose uptake and utilization is mediated by both transporter and hexokinase activity to prevent exchange by phosphorylating intracellular glucose and decrease intracellular glucose concentrations to drive increased transport (47). In contrast, alanine and other amino acids are not enzymatically modified for intracellular sequestration. Alternatively, we demonstrate that cells have evolved a complex network of transporters; involving both active and passive transport mechanisms critical for concentration and equilibration, respectively; that together facilitate flux and sustain metabolic homeostasis. Not surprisingly, cancer cells hijack specific transporters in this network to enhance transport of nutrients required to fuel their increased metabolic demands for specific substrates.
Our data demonstrate that PDAC have evolved high demands for alanine relative to stellate cells (Fig. 1). To fulfill these demands, PDAC cells engage a compartmentalized metabolic program consisting of selective expression of mitochondrial alanine utilization/synthesis machinery and plasma membrane SLC38A2 expression. In addition to alanine, PDAC cells have significant demands for the amino acid glutamine that supplies carbon, nitrogen, and reducing equivalents (e.g. NADPH) through a non-canonical pathway involving shuttling of mitochondrial and cytosolic aspartate (31). Efforts to target the glutamine requirements of PDAC, through inhibition of glutaminase activity required for glutamine anaplerosis, led to complete compensation and failed to illicit an impact on PDAC tumor growth in vivo (40). Indeed, activation of other amino-nitrogen and anaplerotic pathways, mainly mitochondrial GPT2, was sufficient to compensate for glutaminase inhibition in other contexts (48). Directly inhibiting transport of these nutrients may be a more efficacious approach to targeting the metabolic demands of PDAC and other cancers. Several reports have demonstrated that targeting glutamine transport through inhibition of SLC1A5/ASCT2 has profound impacts on tumor growth in gastric and breast cancer (49,50). In addition, targeting other amino acid transporters, including SLC6A14 and SLC7A11/xCT, have also been reported to have significant impacts on tumor growth in PDAC (51,52). Collectively, these studies and ours suggest that metabolite transport is less plastic than previously thought, and targeting key transporters may be a viable therapeutic strategy for cancer.
In addition to transporting nutrients from the microenvironment, PDAC activate macropinocytosis and autophagy to scavenge interstitial and/or intracellular protein. Activation of these pathways confers a survival advantage in nutrient-limiting environments and contributes to therapeutic resistance (3-6). Surprisingly, scavenging pathways were unable to rescue the proliferation defect in SLC38A2-deficient PDAC cells either in vitro or in vivo suggesting that transporter utilization may be required for retention of amino acids derived from catabolic processes. It is well appreciated that PDAC cells activate scavenging pathways in response to nutrient-limiting conditions to sustain growth (3). The converse also occurs whereby autophagy-deficient cells have been reported to upregulate amino acid transport to survive in glutamine-limited conditions (53). Importantly, transporters are required to release amino acids derived from lysosomal-dependent catabolic processes, such as autophagy and macropinocytosis, and are critical components of lysosomal amino acid-sensing pathways (54-57). Understanding the relationship between catabolic and anabolic pathway utilization and transporter engagement is critical for identifying effective therapeutic combinations to target the metabolic requirements of cancer cells.
Collectively, our results provide a new perspective for understanding the complex metabolic relationship between cancer cells and the surrounding stroma. As novel metabolite crosstalks are discovered in cancer, understanding the specific transporters involved may suggest potential therapeutic targets for limiting cell access to nutrients that promote growth and/or therapeutic resistance. Previous reports have suggested that transcriptional activation of SLC38A2 in response to environmental stimuli (e.g. amino acid deficiency, hypertonicity, hypoxia) involves ATF4, hypoxia inducible factors, and members of the MAP kinase family (ERK and JNK) (38,58). Our data suggests that plasma membrane localization may also be a key regulatory requirement for SLC38A2 activity. It is unclear what trafficking processes direct SLC38A2 to the plasma membrane; however, several possible mechanisms exist that parallel other amino acid transporters, including through post-translational modifications (e.g. phosphorylation, glycosylation) and/or endosomal trafficking (59-62). Thus, SLC38A2 activity and dependence in PDAC is likely driven by multi-faceted regulation of expression and localization. However, our data demonstrates that expression of SLC38A2 was insufficient to predict the magnitude of dependence on its activity or the alanine preference of individual cell lines. Therefore, it may be more important to understand the mechanistic underpinnings driving the increased demand to predict alanine requirements and dependence on SLC38A2 in heterogeneous diseases such as cancer. Given the potent anti-tumor effects of SLC38A2 loss in fully formed PDAC tumors and the fact that it appears to be dispensable in non-transformed cells, we believe that targeting alanine uptake and utilization through inhibition of SLC38A2 is a promising therapeutic approach in this deadly disease.
Materials and Methods
Cell culture
The cell lines PANC1, PANC3.27, HPAC, MiaPaCa2, 8988T, PANC10.05, IMR-90, MDCK, and 8902 were obtained from ATCC or the DSMZ. hPSC#1, mPSC#1, and HPDE cells were obtained as previously described (11). Primary murine PDAC cell lines (HY19636, MPDAC4, and HY15549) were isolated from KPC tumors using established protocols (63). All cell lines were maintained in a central lab cell bank. Cultures were verified to be negative for mycoplasma by PCR at least once prior to all experiments and before freezing of cell stocks. All cell lines were authenticated by STR DNA fingerprinting every 1-2 years and by visual inspection prior to experiments and freezing of cell line stocks in central cell bank.
Proliferation assays
Cell proliferation was performed at variable densities depending on growth rate in 24 well plates for each time point (HY19636 and HY15549 at 1-3,000 cells/well, PANC1 and MiaPaCa2 at 5,000 cells/well, hPSC#1 and mPSC#1 at 6,000 cells/well, IMR-90 and HPDE at 10,000 cells/well). Clonogenic assays were performed for 14 days (PANC1) or 7-10 days (MiaPaCa2, HY9636, HY15549) at 1,000 cells/plate in 6 cm dishes. For clonogenic rescue, cells were plated into 6 cm dishes containing doxycycline (0.5 μg/ml), pyruvate (1mM), or DMEM/F12 (50:50) as indicated in each figure. Proliferation rescue was performed for four days at 1,000 cells/well. The following day, media containing 2x concentrations of rescue metabolite or 100% F12 (for 50:50 DMEM/F12 experiments) was added to wells.
Low amino acid growth assays were performed by seeding cells in low amino acid DMEM (25mM glucose, 10% [AA]DMEM, 10% FBS) in clear 96-well plates (Corning). Custom DMEM without amino acids was obtained from US Biological (D9800), and 10% [AA]DMEM media was prepared by mixing custom and basal DMEM at 9:1 ratio. Wells contained media supplemented with 2x concentrations of alanine to reach final experimental condition after dilution with cell suspension. Cell growth after 48 hours was assessed by CellTiter-Glo 2 (Promega) and normalized to an initial measurement made after cells attached overnight.
Stable-isotope tracing, metabolite profiling, and flux analysis
For stable-isotope tracing experiments, cells were plated into 6-well plates at 0.25-0.5x106 cells/well and cultured for 24 hours in DMEM supplemented with 1mM labeled alanine and 10% dialyzed FBS (Gibco). For MPC inhibition tracing experiments, cells were plated at 0.25-0.5x106 cells/well and washed once with PBS before addition of 1mM 13C3-alanine or L-alanine and either vehicle (DMSO) or UK-5099 for 24 hours. Conditioned media was collected and briefly centrifuged at 1,000xg for 10 minutes to remove cell debris prior to extraction. For alanine exchange experiments, PSCs were plated at 0.1x106 cells/well into 6-well plates and cultured with 1mM 13C3-alanine and 10% dialyzed FBS. Conditioned media and intracellular metabolites were collected every 24 hours from independent plates per timepoint.
For metabolite extraction, cells were washed with cold 0.9% NaCl followed by addition of 500 μl methanol (−20°C) and 200 μl HPLC grade water (4°C). Cells were scraped and transferred into vials containing 500 μl chloroform (4°C) and vortexed at 4°C for 10-15 minutes. Aqueous and inorganic layers were separated by cold centrifugation for 15 minutes. 300 μl of the aqueous layer containing polar metabolites was transferred to sample vials (Agilent) and evaporated by SpeedVac (Savant Thermo) prior to derivatization and GC-MS analysis. Fatty acid labeling from 13C3-alanine was measured by transferring 400 μl of the inorganic layer to a glass vial and evaporating under nitrogen flow in a needle evaporator prior to transesterification and GC-MS analysis. To measure proteogenic amino acids, the insoluble interphase containing proteins was washed three times with HPLC-grade acetone, to remove free metabolites, and allowed to dry overnight with gentle nitrogen flow. The resulting pellet was hydrolyzed in 2N HCl at 95°C for two hours with occasional vortexing and dried overnight under nitrogen flow prior to derivatization and GC-MS analysis. For amino acid and metabolite profiling, approximately 0.5-1x106 cells were extracted using methanol:water:chloroform containing 2.5 nmol of uniformly (13Cx,15Nx)-labeled amino acids including L-alanine, L-lysine, L-hisitidine, L-arginine, L-tyrosine, L-phenylalanine, L-methionine, L-glutamic acid, L-aspartic acid, L-leucine, L-isoleucine, L-valine, L-threonine, L-proline, L-serine, and glycine; 4 nmol of 13C5-labeled L-glutamine; 0.5 nmol 13C3-labeled pyruvate; 5 nmol 13C3-labeled lactate; 1.25 nmol 13C6,15N2-labeled cystine, and 1μg of norvaline. For media extraction, 5 μl of initial or conditioned media was extracted in 250 μl of 80% methanol solution containing the above isotope-labeled internal standards. The abundance of each metabolite was quantified by the following equation:
Where Xstandard is the molar amount for each added standard (e.g. 2.5 nmol for alanine, 5 nmol for lactate) and %M0X is the relative abundance of unlabeled (M+0) species ‘X’ corrected for natural isotope abundance. Multiple surrogate wells were pooled and counted to normalize metabolite abundances by cell number.
For cDNA withdrawal experiments, cells were plated into 6-well plates at 0.1-0.25x106 cells/well in DMEM containing doxycycline (0.5 μg/ml). For time course cDNA withdrawal, cells were washed twice with PBS to remove residual doxycycline and cultured in DMEM with or without doxycycline to initiate time course. Metabolites were extracted at indicated times and surrogate wells were counted for normalization. Kinetic changes in relative amino acid levels with acute SLC38A2 cDNA withdrawal were calculated by subtracting a baseline value (chronic SLC38A2 KO; set to “0%”) from the control (+dox) and experimental conditions (acute KO) followed by normalization to positive control level (+dox; set to “100%”).
To quantify extracellular uptake and secretion fluxes, the molar change in extracellular metabolites was normalized to the cell density over the time course of the experiment using the following equation:
Where AAt and AAo are the final and initial molar amounts of each amino acid, respectively, quantified using labeled internal standards, Xo is the initial cell density, k is the exponential growth rate (hr−1), and t is the media conditioning time. To quantify alanine exchange flux, the percent dilution of exogenous 13C3-alanine was multiplied by the molar quantity of alanine in the media over the conditioning period normalized to cell density over time.
Oxygen consumption rate and extracellular acidification rate
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were simultaneously measured using a Seahorse XFe96 analyzer (Agilent). HY15549 cells were seeded into Seahorse XFe96 plates at 10,000 cells/well in either basal DMEM (-dox) or doxycycline-containing DMEM (0.5 μg/ml) 24h prior to assay. Prior to the assay, DMEM (25mM glucose, 2mM glutamine, no sodium bicarbonate) was exchanged and incubated for ~30 min at 37°C in a non-CO2 incubator (Thermo). OCR and ECAR were measured in response to sequential injections of oligomycin (1 μM), FCCP (0.5 μM), and rotenone/antimycin (1 μM/1 μM). Cells were immediately lysed using 30 μL/well Reagent A (Bio-Rad) immediately following each assay, and protein was quantified using the DC protein assay kit (Bio-Rad). OCR and ECAR were normalized to milligrams total protein, as quantified by a standard curve.
Chemicals
13C3-labeled alanine (CLM-2184-H), 15N-labeled alanine (NLM-454), 13C5-labeled glutamine (CLM-1822-H), 13C3-labeled lactate (CLM-1579), 13C3-labeled pyruvate (CLM-2440), and 13Cx,15Nx-labeled amino acid standard mix (MSK-A2) were acquired from Cambridge Isotope Laboratories. D-glucose (Sigma), DMSO (Sigma), doxycycline hyclate (Sigma), oligomycin (Cayman Chemicals), sodium pyruvate (Sigma), 2-ketobutyric acid sodium salt hydrate (Sigma), rotenone (Cayman Chemicals), FCCP (Cayman Chemicals), antimycin A (Cayman Chemicals), L-alanine (Sigma), L-alanine tert-butyl ester (Alfa Aesar), UK-5099 (Cayman Chemicals), methoxyamine hydrochloride (Sigma), and MTBSTA + 1% TBDMSCl (Sigma).
Antibodies and western blot
Proteins were extracted using RIPA buffer containing fresh protease (Roche) and phosphatase (Roche) inhibitor cocktails on ice for 30 minutes. Where indicated, lysates were deglycosylated using PNGaseF (removes only N-linked glycosyl groups, New England Biolabs) according to modified manufacturer protocol with all steps conducted at 37°C. Lysates were not boiled prior to separation on SDS-PAGE gels as heating above 50°C causes complete loss of SLC38A2 signal. Note that loss of signal following heating was specific to SDS-PAGE, and not immunohistochemical staining, as heating of hydrophobic transmembrane proteins led to aggregation and insolubility in aqueous buffers irrespective of presence of detergent. Membranes were blocked in either 5% nonfat milk or bovine serum albumin dissolved in TBS-t according to antibody manufacturer recommendations. Primary antibodies were incubated overnight at 4°C with gentle agitation using the following antibodies and dilutions: anti-SLC38A2 (1:500; MBL, BMP081), anti-SLC1A4 (1:1000; Cell Signaling Technologies, 8442), anti-cleaved caspase-3 (1:1000, Cell Signaling Technologies, 9664S), anti-N/K-ATPase (1:5000; Abcam, ab76020), anti-Actin (1:10,000; Sigma, A4700), anti-pS51-eIF2α (1:1000; Cell Signaling Technologies, 3398S), anti-total eIF2α (1:1000; Cell Signaling Technologies, 2103S), anti-GPT (1:1000; Proteintech, 16897-1-AP), anti-GPT2 (1:1000; Proteintech, 16757-1-AP), and anti-LC3B (1:1000; Novus, NB100-2220). After TBS-t washing, membranes were incubated with peroxidase-conjugated secondary antibodies, anti-rabbit (1:1000-1:5000; Cell Signaling Technologies, 7045S) or anti-mouse (1:2500-10,000; Cell Signaling Technologies, 7076S), for 1 hour at room temperature and imaged by chemiluminescence (Bio-rad 1705061) using a ChemiDoc (Bio-Rad).
Lentiviral shRNA and sgRNA targets
All shRNA vectors were obtained from the Sigma MISSION TRC library in glycerol stock form. The target sequence and TRC number for each shRNA are as follows: shGFP: GCAAGCTGACCCTGAAGTTCAT; shSLC38A2 #1: GGAGAAGATACTGTGGCAA (TRCN0000020243); shSLC38A2 #4: GAATACCAAGAGTTGTTTCTA (TRCN0000020241), shSLC1A4 #4: TGTACACCAGGGATCTGTTTG (TRCN0000418203), shSlc1a4 #3: CTTCACCAATTTGCTCGTCAT (TRCN0000326081). These shRNAs were selected after screening 5-7 shRNAs by qPCR and western blot. All sgRNAs were designed using the Broad sgRNA Designer (Broad Institute), cloned into pLentiCRISPRv2 (Addgene, plasmid #52961), and sequence verified prior to transfection. The target sequences are as follows: sgTom: GCCACGAGTTCGAGATCGA, sgSLC38A2/sgSlc38a2 #1: TAATCTGAGCAATGCGATTG, sgSLC38A2/sgSlc38a2 #3: TCTTATGCCATGGCTAATAC. Both sgSLC38A2 #1 and #3 target a homologous sequence present in mouse Slc38a2 and when used in mouse cell lines are indicated as sgSlc38a2 #1 and #3. Lentivirus were produced by transfecting 293T cells with pLKO or pLentiCRISPRv2 constructs, pMD2.G (Addgene, plasmid #12259), and psPAX2 (Addgene, plasmid #12260) using standard Lipofectamine 3000 (ThermoFisher) protocol. All experiments using shRNA and sgRNA were conducted using pools of cells after selection. Clonally derived control (sgTom) and Slc38a2-deficient (sgSlc38a2 #1) HY15549 cells expressing pInducer-resSLC38A2WT were derived by plating single cells in 96-well plates. Cells were cloned in DMEM containing doxycycline (0.5 μg/ml), refreshed every five days, to express SLC38A2 and limit selection pressure that may arise during establishment and outgrowth of clones. Each individual clone was validated for both knockout and expression of inducible sgRNA-resistant SLC38A2 by western blot. Six individual control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) clones were pooled after individual validation. Chronic Slc38a2 knockout HY15549 cell lines were generated by maintenance in DMEM (-dox) for a total of ten passages. Cells were split 1:8 every six days for a total of 60 days.
Immunohistochemistry
Sections were deparaffinized and rehydrated, and antigen retrieval was performed in a steamer for 20 minutes in 10 mM pH 6.0 citrate buffer containing 0.05% Tween-20. Slides were incubated in 3% hydrogen peroxide and 50% methanol for 30 minutes and blocked in 5% goat serum and 1% bovine serum albumin in TBS-t for 30 minutes at room temperature. Primary antibody (anti-SLC38A2, 1:1000, MBL BMP081; anti-cleaved caspase-3, 1:1000, Cell Signaling Technologies, 9664S) diluted in blocking buffer was added to the sections and incubated overnight at 4°C. Sections were washed with TBS-t and incubated with biotin-conjugated secondary antibody (anti-rabbit 1:200; Vector Labs BA-1000) for 30 minutes at room temperature and Avidin-Biotin Complexes (Vector Labs PK-4000) for 30 minutes. Slides were developed by 3,3-diaminobenzidine (Vector Labs SK-4100) for 3-5 minutes and quenched with water followed by hematoxylin staining for 3-5 minutes. Sections were dehydrated and mounted in permount mounting medium (Fisher Scientific). Cleaved caspase-3 staining was quantified by DAB color deconvolution and quantification of optical density using Image J.
Quantitative proteomics and bioinformatics analysis
Quantitative proteomics data (Fig. 2, Supplementary Fig. S4) were generously supplied by Joseph D. Mancias (25). Principal Component Analysis (PCA) and differential expression analysis were conducted on log2 fold changes using Matlab (MathWorks). Metabolism proteins were queried from a published list of approximately 3,000 proteins (64). Analysis of transporter proteins included all gene products with ‘SLC’ in the gene symbol and excluded ABC transporters.
Quantitative proteomics of isogenic HY15549 cell lines was performed as previously described (40). In brief, roughly 5 million cells were harvested and washed twice with cold PBS. Cell pellets were resuspended in 500 μl lysis buffer (8 M urea in 200 mM EPPS buffer, pH 8.5) and sonicated at lower power for 2 min (10 seconds on plus 10 seconds off). Lysates were quantified by Pierce BCA Assay Kit (Thermo Fisher Scientific) and adjusted to 1 μg/μl using 200 mM EPPS buffer. 100 μg protein from each sample was reduced by 10 μl 5 mM dithiothreitol in dark for 30 min followed by alkylation with 10 μl 15 mM iodoacetamide solution in dark for 30 min. Protein was precipitated via chloroform-methanol by mixing 100 μl reduced and alkylated lysate with 300 μl methanol, 100 μl chloroform, and 200 μl water and centrifuging at 10,000xg for 5 min. Pellets were washed with 300 μl methanol, resuspended in 100 μl EPPS buffer, and digested with MS-grade trypsin (1:50, protein-to-enzyme ratio) and LysC (1:50) at 37 °C overnight. 10 μl tandem mass tag (TMT, Thermo Fisher Scientific) reagent (20 μg/μl) and 30 μl anhydrous acetonitrile were added to each digested sample and incubated at room temperature for 1 hour. TMT-labeled samples were eventually pooled and subjected to C18 SPE (Sep-Pak, Waters). Eluted sample was fractioned into 12 parts via a pH reversed-phage HPLC. Each fraction was injected to an Orbitrap Fusion Lumos mass spectrometry (Thermo Fisher Scientific) with a Proxeon Easy-nLC 1200 LC pump.
Raw mass spectra were analyzed via a Sequest-based in-house pipeline. Quantitative proteomics was normalized to total intensity of each TMT channel. Pairwise comparisons were performed using LIMMA software package (3.40.2). False discovery rate (FDR) was calculated by adjusting p value by Benjamini-Hochberg method. Each comparison was subject to gene set enrichment analysis (GSEA) via Broad GSEA software (4.0) using Reactome Gene Sets (c2.cp.reactome.v7.0.symbols.gmt) from MsigDB. GSEA results were further analyzed in Cytoscape (3.7.1) platform, and pathway enrichment maps were generated. All statistical analysis and plots relevant to quantitative proteomics were performed on R (3.6.0) platform.
Microscopy imaging
Live cell imaging of SLC38A2-GFP was performed on cells transiently transfected using lipofectamine 3000 in 35mm plates that incorporate a No. 1.5 cover-slip-covered well (Mattek Corp) with an inverted Zeiss 800 laser scanning confocal microscope (Oberkochen). MitoTracker™ Red CMXRos was used to stain mitochondria and indicate live cells. Cells were incubated in media containing MitoTracker for 15 minutes, after which the media was aspirated and replaced with fresh media.
In vivo xenograft and allograft experiments and ultrasound tumor monitoring
For subcutaneous co-injection tumor initiation studies, PANC1 cells were infected with lentiviral shRNAs targeting SLC38A2 or GFP (control) and selected with puromycin (2 μg/mL) for three days. 0.2x106 PANC1 shGFP or shSLC38A2 cells were resuspended in 100 μl HBSS with or without 1x106 hPSC#1 cells and subcutaneously injected into bilateral lower flanks of 7-8 week old NCr nude mice (Taconic). A similar protocol was followed for studies using hPSC#1 shGFP and shSLC1A4 cells co-injected with PANC1 cells. Tumor initiation was monitored 2-3 times per week by caliper measurement and was considered if length and width were measured to be ≥1mm each.
For orthotopic xenograft experiments, PANC1 or HY19636 cells were infected with lentiviral shRNAs or sgRNAs targeting SLC38A2 or GFP/Tomato (control) and selected with puromycin (2 μg/mL) for three days. 0.5x106 PANC1 cells or 0.1x105 HY19636 cells were resuspended in 10 μl HBSS and 10 μl growth factor-reduced matrigel (Corning 356231) per injection and kept on ice. Female 7-8 week old NCr nude or C57BL/6J mice were used for xenograft and allograft experiments, respectively. An incision was made near the spleen which was gently removed from the peritoneal space to expose the pancreas. The 20 μl cell:matrigel suspension was slowly injected into the tail of the exposed pancreas using either a Hamilton or insulin syringe (BD324702). After injection, the needle was held in place by tweezers briefly to allow the matrigel to polymerize before gently removing the needle and re-introducing the spleen and pancreas into the animal. The peritoneum was sutured and the wound was closed with surgical staples. Buprenorphine was administered by intraperitoneal injection immediately after surgery and every 12 hours for 48 hours. After injection, all mice were allowed to recover from surgery for five days before screening for tumor initiation by non-invasive 3-D ultrasound imaging (VisualSonics Vevo 770) twice a week under anesthesia using 1-3% isoflurane via nose cone. Ultrasound image collection was conducted un-blinded; tumor volume quantification was conducted blinded. Tumor initiation was considered if volume ≥1mm3 and sustained or increased in volume over the course of the experiment. Euthanasia and tumor resection was performed at the conclusion of the experiment and tumors were confirmed by histology.
For tumor maintenance studies (Fig. 6C), 0.25x105 pInducer-resSLC38A2WT HY15549 control (sgTom) or Slc38a2-deficient (sgSlc38a2 #1) cells were prepared and injected subcutaneously (uni-laterally; as detailed above) or orthotopically (as detailed above). Mice were fed a doxycycline-containing chow (625 ppm, Evigo) for three days prior to injection and for 5 days (orthotopic) or 7 days (subcutaneous) following injection to allow for sgRNA-resistant SLC38A2 cDNA expression during tumor initiation. Following initiation, mice were fed regular chow (-dox) and tumor size was monitored 2-3 times per week by caliper measurement for subcutaneous experiments.
All mouse experiments with human and mouse PDAC cell lines were approved by the NYU Institutional Animal Care and Use Committee (IACUC) under protocol numbers IA16-00507 and IA16-01331. Human PDAC tumor microarrays were approved by the University of Texas M.D. Anderson Cancer Center Institutional Review Board (IRB) under protocol number LAB05-0854.
Molecular cloning
To clone sgRNA-resistant SLC38A2-WT and SLC38A2-N82A cDNA, codons recognized by the target sequence were silently mutated to prevent sgRNA recognition by fragment PCR of SLC38A2 cDNA (Dharmacon MGC, clone ID 3874551). Fragments containing sgRNA-resistant sequence and AAC→GCT mutation (N82A) were isolated by PCR and assembled by Gibson assembly (New England Biolabs). The assembled fragments were PCR amplified and recombined into pDONR221 using Gateway assembly (Invitrogen). Inserts were recombined into either pLentiCMVBlast (Addgene, plasmid #17451) or pInducer20 (Addgene, plasmid #44012) using Gateway assembly (Invitrogen). To clone SLC38A2-GFP, SLC38A2 cDNA was amplified from pDONR221-SLC38A2-WT by PCR to include restriction sites for XhoI (5’) and EcoRI (3’). The PCR product for SLC38A2 and the pEGFP-N1 vector were digested with XhoI and EcoRI and ligated into pEGFP-N1 (Clonetech) using the Rapid DNA Dephos and Ligation Kit (Roche). Colonies were sequenced to confirm correct insert sequence and, if necessary, if tetO repeats were fully intact.
Statistical analysis
Statistical analysis was performed using Prism (GraphPad). When comparing multiple groups to a common control (for example, experiments performed using two sgRNAs targeting SLC38A2 compared to control sgRNA), a one- or two- way ANOVA test was performed and followed up by the Dunnett’s test to compare multiple groups. When comparing multiple groups to each other, a one- or two- way ANOVA test was performed and followed up by Sidak’s multiple comparisons test. Unpaired, 2-tailed Student’s t-tests were performed when comparing two groups to each other. Log-rank Mantel-Cox tests were performed when conducting survival curve statistical analysis. Fisher’s exact test was performed when comparing tumor versus stroma SLC38A2 staining in human PDAC tumor microarray. Statistical analysis was performed on the means of technical replicates from each independent experiment. Technical sampling of a single biological sample was not performed. For all significance analyses, groups were considered significantly different if p-value <0.05. Detailed statistical information, including statistical test used and exact p-values, are included in Supplemental Information if not included in figure.
Supplementary Material
Statement of Significance: This work identifies critical neutral amino acid transporters involved in channeling alanine between pancreatic stellate and PDAC cells. Targeting PDAC-specific alanine uptake results in a metabolic crisis impairing metabolism, proliferation, and tumor growth. PDAC cells specifically activate and require SLC38A2 to fuel their alanine demands that may be exploited therapeutically.
Acknowledgments
The authors would like to thank Haoqiang Ying for providing the HY19636 and HY15549 cell lines. The authors would like to thank all members of the Kimmelman and Pacold labs for critical discussions and guidance. Specifically, the authors would like to thank Michael Pacold for feedback on the manuscript. The authors would like to thank Caitlyn Bowman for helpful discussions and suggestions surrounding mitochondrial pyruvate transport and alanine. The authors would like to acknowledge the Experimental Pathology Research Laboratory for their expertise and assistance with processing, embedding, sectioning tissue sections, and imaging of slides. This work was primarily supported by NIH/NCI grants R01CA157490, R01CA188048, P01CA117969, R35CA232124; ACS Research Scholar Grant RSG-13-298-01-TBG; NIH grant R01GM095567; P30CA016087; and by a Stand Up To Cancer-Lustgarten Foundation Pancreatic Cancer Interception Translational Cancer Research Grant (Grant Number: SU2C-AACR-DT26-17) to A. Kimmelman. Financial support was provided to J. Paulo by NIH/NIGMS grant R01GM132129 and to H. Wang by NIH/NCI grant R01CA196941. D. Biancur was supported by a NIH/NCI Predoctoral to Postdoctoral Fellow Transition Award (F99CA245822). K. Yamamoto was supported by a Uehara Memorial Foundation Research Fellowship. S. Parker was supported by an American Cancer Society – New York Cancer Research Fund grant (132942-PF-18-215-01-TBG). Stand Up To Cancer is a division of the Entertainment Industry Foundation. The above mentioned research grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C.
Financial Support: This work was supported by National Cancer Institute Grants R01CA157490, R01CA188048, P01CA117969, R35CA232124; ACS Research Scholar Grant RSG-13-298-01-TBG; NIH grant R01GM095567; P30CA016087; and by a Stand Up To Cancer-Lustgarten Foundation Pancreatic Cancer Interception Translational Cancer Research Grant (Grant Number: SU2C-AACR-DT26-17) to A.C.K.; R01GM132129 to J.A.P.; R01CA196941 to H.W.; D.E.B. was supported by a NCI Predoctoral to Postdoctoral Fellow Transition Award F99CA245822; K.Y. was supported by a Uehara Memorial Foundation Research Fellowship; S.J.P. was supported by American Cancer Society grant 132942-PF-18-215-01-TBG. Stand Up To Cancer is a division of the Entertainment Industry Foundation. The above mentioned research grant is administered by the American Association for Cancer Research, the Scientific Partner of SU2C.
Footnotes
Conflict of Interest: A.C.K. has financial interests in Vescor Therapeutics, LLC. A.C.K. is an inventor on patents pertaining to KRAS regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the SAB of Rafael/Cornerstone Pharmaceuticals. A.C.K. is a consultant for Deciphera Pharmaceuticals.
References
- 1.Palm W, Thompson CB. Nutrient acquisition strategies of mammalian cells. Nature 2017;546(7657):234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011;27:441–64. [DOI] [PubMed] [Google Scholar]
- 3.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497(7451):633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nature communications 2017;8:16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer discovery 2014;4(8):905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer discovery 2018;8(3):276–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer discovery 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell metabolism 2019;29(6):1390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duong MN, Geneste A, Fallone F, Li X, Dumontet C, Muller C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017;8(34):57622–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017;152(6):1492–506 e24. [DOI] [PubMed] [Google Scholar]
- 11.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016;536(7617):479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013;62(1):112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012;336(6084):1040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer research 2015;75(3):544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schell JC, Olson KA, Jiang L, Hawkins AJ, Van Vranken JG, Xie J, et al. A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Molecular cell 2014;56(3):400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rauckhorst AJ, Taylor EB. Mitochondrial pyruvate carrier function and cancer metabolism. Curr Opin Genet Dev 2016;38:102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012;337(6090):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012;337(6090):93–6. [DOI] [PubMed] [Google Scholar]
- 20.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Molecular cell 2014;56(3):414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bensard CL, Wisidagama DR, Olson KA, Berg JA, Krah NM, Schell JC, et al. Regulation of Tumor Initiation by the Mitochondrial Pyruvate Carrier. Cell metabolism 2020;31(2):284–300 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170(3):564–76 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halestrap AP. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. The Biochemical journal 1975;148(1):85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Molecular cell 2014;56(3):425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulo JA, Mancias JD, Gygi SP. Proteome-Wide Protein Expression Profiling Across Five Pancreatic Cell Lines. Pancreas 2017;46(5):690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cesar-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, et al. A Call for Systematic Research on Solute Carriers. Cell 2015;162(3):478–87. [DOI] [PubMed] [Google Scholar]
- 27.Perland E, Fredriksson R. Classification Systems of Secondary Active Transporters. Trends in pharmacological sciences 2017;38(3):305–15. [DOI] [PubMed] [Google Scholar]
- 28.Schioth HB, Roshanbin S, Hagglund MG, Fredriksson R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Molecular aspects of medicine 2013;34(2–3):571–85. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu K, Kaira K, Tomizawa Y, Sunaga N, Kawashima O, Oriuchi N, et al. ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. British journal of cancer 2014;110(8):2030–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P, Ge M, Hu J, Li X, Che L, Sun K, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017;66(1):167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013;496(7443):101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zerangue N, Kavanaugh MP. ASCT-1 is a neutral amino acid exchanger with chloride channel activity. The Journal of biological chemistry 1996;271(45):27991–4. [DOI] [PubMed] [Google Scholar]
- 33.Srour M, Hamdan FF, Gan-Or Z, Labuda D, Nassif C, Oskoui M, et al. A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clinical genetics 2015;88(1):e1–4. [DOI] [PubMed] [Google Scholar]
- 34.Yao D, Mackenzie B, Ming H, Varoqui H, Zhu H, Hediger MA, et al. A novel system A isoform mediating Na+/neutral amino acid cotransport. The Journal of biological chemistry 2000;275(30):22790–7. [DOI] [PubMed] [Google Scholar]
- 35.Krall AS, Xu S, Graeber TG, Braas D, Christofk HR. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nature communications 2016;7:11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009;136(3):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann TM, Cwiklinski E, Shah DS, Stretton C, Hyde R, Taylor PM, et al. Effects of Sodium and Amino Acid Substrate Availability upon the Expression and Stability of the SNAT2 (SLC38A2) Amino Acid Transporter. Frontiers in pharmacology 2018;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nardi F, Hoffmann TM, Stretton C, Cwiklinski E, Taylor PM, Hundal HS. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. The Journal of biological chemistry 2015;290(13):8173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015;162(3):552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature communications 2017;8:15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biancur DE, Kimmelman AC. The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochimica et biophysica acta Reviews on cancer 2018;1870(1):67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyssiotis CA, Kimmelman AC. Metabolic Interactions in the Tumor Microenvironment. Trends in cell biology 2017;27(11):863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatanaka T, Huang W, Wang H, Sugawara M, Prasad PD, Leibach FH, et al. Primary structure, functional characteristics and tissue expression pattern of human ATA2, a subtype of amino acid transport system A. Biochimica et biophysica acta 2000;1467(1):1–6. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017;543(7645):424–7. [DOI] [PubMed] [Google Scholar]
- 45.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. The Journal of clinical investigation 2008;118(12):3930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America 2013;110(10):3919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Irwin DM, Tan H. Evolution of glucose utilization: glucokinase and glucokinase regulator protein. Molecular phylogenetics and evolution 2014;70:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim M, Gwak J, Hwang S, Yang S, Jeong SM. Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 2019;38(24):4729–38. [DOI] [PubMed] [Google Scholar]
- 49.Lu J, Chen M, Tao Z, Gao S, Li Y, Cao Y, et al. Effects of targeting SLC1A5 on inhibiting gastric cancer growth and tumor development in vitro and in vivo. Oncotarget 2017;8(44):76458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016;35(24):3201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daher B, Parks SK, Durivault J, Cormerais Y, Baidarjad H, Tambutte E, et al. Genetic Ablation of the Cystine Transporter xCT in PDAC Cells Inhibits mTORC1, Growth, Survival, and Tumor Formation via Nutrient and Oxidative Stresses. Cancer research 2019;79(15):3877–90. [DOI] [PubMed] [Google Scholar]
- 52.Coothankandaswamy V, Cao S, Xu Y, Prasad PD, Singh PK, Reynolds CP, et al. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br J Pharmacol 2016;173(23):3292–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang N, Yang X, Yuan F, Zhang L, Wang Y, Wang L, et al. Increased Amino Acid Uptake Supports Autophagy-Deficient Cell Survival upon Glutamine Deprivation. Cell reports 2018;23(10):3006–20. [DOI] [PubMed] [Google Scholar]
- 54.Verdon Q, Boonen M, Ribes C, Jadot M, Gasnier B, Sagne C. SNAT7 is the primary lysosomal glutamine exporter required for extracellular protein-dependent growth of cancer cells. Proceedings of the National Academy of Sciences of the United States of America 2017;114(18):E3602–E11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017;171(3):642–54 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015;519(7544):477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015;347(6218):188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morotti M, Bridges E, Valli A, Choudhry H, Sheldon H, Wigfield S, et al. Hypoxia-induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proceedings of the National Academy of Sciences of the United States of America 2019;116(25):12452–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olivares L, Aragon C, Gimenez C, Zafra F. The role of N-glycosylation in the targeting and activity of the GLYT1 glycine transporter. The Journal of biological chemistry 1995;270(16):9437–42. [DOI] [PubMed] [Google Scholar]
- 60.Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Molecular cell 2017;67(1):128–38 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McNally KE, Faulkner R, Steinberg F, Gallon M, Ghai R, Pim D, et al. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nature cell biology 2017;19(10):1214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burd C, Cullen PJ. Retromer: a master conductor of endosome sorting. Cold Spring Harb Perspect Biol 2014;6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 2003;17(24):3112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Minton DR, Nam M, McLaughlin DJ, Shin J, Bayraktar EC, Alvarez SW, et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Molecular cell 2018;69(4):610–21 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.