

Abstract
2’-Deoxyguanosin-N1-yl radical (dG(N1-H)•) is the thermodynamically favored one-electron oxidation product of 2’-deoxyguanosine (dG), the most readily oxidized native nucleoside. dG(N1-H)• is produced by formal dehydration of a hydroxyl radical adduct of dG, as well as by deprotonation of the corresponding radical cation. dG(N1-H)•, species that are initially formed as a result of the indirect and direct effects of ionizing radiation, amongst other DNA damaging agents. dG(N1-H)• was generated photochemically (λmax = 350 nm) from an N-aryloxy-naphthalimide precursor (3). The quantum yield for photochemical conversion of 3 is ~0.03, and decreases significantly in the presence O2, suggesting that bond scission occurs from a triplet excited state. dG is formed quantitatively in the presence of excess β-mercaptoethanol. In the absence of a reducing agent, dG(N1-H)• oxidizes 3, decreasing the dG yield to ~50%. Addition of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodGuo) as a sacrificial reductant results in a quantitative yield of dG and two-electron oxidation products of 8-oxodGuo. N-Aryloxy-naphthalimide 3 is an efficient and high yielding photochemical precursor of dG(N1-H)• that will facilitate mechanistic studies on the reactivity of this important reactive intermediate involved in DNA damage.
Graphical Abstract

Introduction
2’-Deoxyguanosine (dG) is the most readily oxidized native nucleoside.1–2 The radical cation (dG•+) resulting directly from one-electron oxidation is critically important for hole transfer in DNA.3–4 In contrast, deprotonation of dG•+ from the N2-amino group or the N1-position to produce dG(N2-H)• and dG(N1-H)•, respectively squelches electron transfer (Scheme 1).5–6 dG(N2-H)• and dG(N1-H)• are also produced via formal dehydration of the C4-hydroxyl radical adduct of dG via an ion pair (C4-OH).7 Of the two neutral radicals, dG(N1-H)• is accepted to be lower in energy.8 It is notable that dG(N1-H)• is produced by both major pathways of γ-radiolysis, the “direct” and “indirect” effects.9 Despite the importance of dG(N1-H)• in DNA oxidation chemistry, there is limited understanding of its reactivity. dG(N1-H)• is a component of mixtures produced from (pulse) radiolysis of dG.10 These studies revealed that it is unreactive with O2.11–12 Photolabile nucleosides are useful tools for generating and studying reactive intermediates involved in nucleic acid oxidation.13–17 dG(N1-H)• was independently generated from 1 and 2, but mass balances were 38%.18–19 We wish to report on a photochemical precursor for dG(N1-H)• that provides the radical in high yield based upon mass balances in photolysates.
Scheme 1.

Formation of dG(N1-H)• and dG(N2-H)•

Although dG(N1-H)• is lower in energy than dG(N2-H)•, the difference in the barriers to their formation from the C4-OH nucleoside is calculated to be < 1 kcal/mol.7 The relationship between dG(N2-H)• and dG(N1-H)• is more complicated in duplex DNA in which the former is the ultimate deprotonation product of dG•+ due to base pairing with dC.6 In addition, contrary to reports that hydroxyl radical preferentially adds to the purine π-bonds, direct formation of dG(N2-H)• by hydrogen atom abstraction has been proposed to be the major pathway for reaction between dG and this species.10, 20–24 dG(N2-H)• is then proposed to isomerize to dG(N1-H)• on the sub-millisecond timescale. High yielding methods independently generating dG(N2-H)• and dG(N1-H)• are desirable to resolve the uncertainties and complexity of their reactivities. dG(N2-H)• and dG(N1-H)• have been produced from photochemical precursors, but the methods are inefficient and/or do not provide suitable mass balances.16, 18–19
Results and Discussion.
Design and preparation of a dG(N1-H)• precursor.
Independent generation of nucleoside radicals and the analogous nucleotide radicals in oligonucleotides has greatly improved our understanding of nucleic acid oxidation.14 However, examples of studies on the heteroatom radicals produced from purine oxidation are more limited.15–16, 25 The N-hydroxypyrid-2(1H)-one (1) and the corresponding thione (2) were designed to produce dG(N1-H)• upon UV photolysis.18–19 The former proved to be a photochemical source of dG(N1-H)• but the mass balance was < 40%. All of the published precursors to heteroatom-centered purine radicals utilize cleavage of weak N-N or N-O bonds.25 Similarly, we pursued the N-aryloxy-1,8-naphthalimide 3 (Scheme 2). Other N-oxy-1,8-naphthalimides undergo efficient N-O bond cleavage under UV excitation.26–27 Furthermore, we postulated that 3 would exhibit strong absorption at wavelengths >300 nm, a desirable property when working with nucleic acids.
Scheme 2.

Generation of dG(N1-H)• from the photolysis of 3
Precursor 3 was readily synthesized using the method developed by Lakshman in which N-hydroxy-1,8-naphthalimide (4) was reacted with 5 that was activated by DABCO (Scheme 3).28 Substitution of the sulfonate in 5 was straightforward when DMF was substituted for the more commonly used DME solvent, due to poor solubility of 4 in the latter. Upon removing the TBMDS protecting groups from 6, precursor 3 was obtained. The UV spectrum of 3 shows absorption maxima at 287 nm (ε = 1.1 × 104 M−1cm−1) which arises from the guanine and another at 346 nm (ε = 1.72 × 104 M−1cm−1), which are attributed to the guanine and 1,8-naphthalimide moieties, respectively (Figure S4).
Scheme 3. Synthesis of dG(N1-H)• precursor 3a.
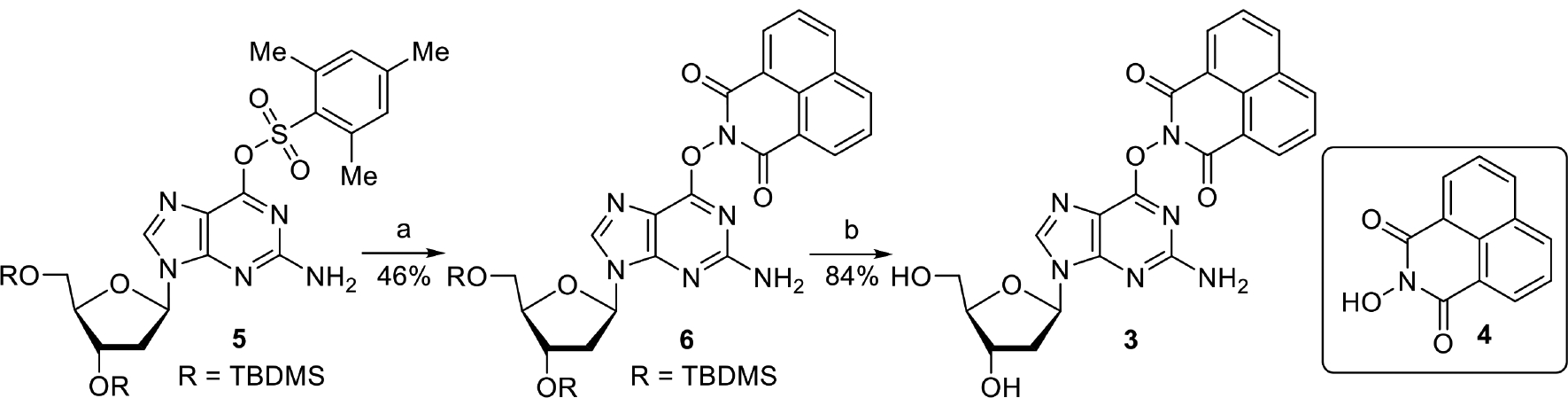
aKey: a) i. DABCO, DMF; ii. 4, DBU b) Et3N•3HF, THF
Photochemical generation of dG(N1-H)• from 3.
When photolyzed in the absence of O2, 3 (0.1 mM) is completely consumed in less than 7 min. The quantum yield of the disappearance of 3 (Φ = 0.03) was determined using N-hydroxy-1,8-naphthalimide triflate as actinometer,26–27 and is 20 times higher than that of the precursor (7) for 2’-deoxyadenosin-N6-yl radical under the same photolysis conditions.15 This increased quantum yield and the high absorbance around 350 nm contribute to the rapid conversion of 3 (Figure S4).

dG was detected (52 ± 2%) in the absence of β-mercaptoethanol (BME, Figure 1a). As in the photochemical studies of 7, the generation of dG in the absence of reducing agents may be attributed to reduction of dG(N1-H)• by the radical precursor (3). In contrast, dG was formed in nearly quantitative yield when the photolysis was carried out in the presence of 10 mM thiol (Figure 1b). Furthermore, the consumption rate of precursor was greater in the presence of thiol. We propose that the accelerated photoconversion is caused by the photoreduction of 3 by BME, and the ensuing fragmentation leads to the formation of dG(N1-H)• and 1,8-naphthalimide (NP, Scheme 2). While the contributions of homocleavage and photoreduction are uncertain, the quantitative generation of dG suggests that dG(N1-H)• is reduced by BME. These observations are in contrast to a previous study in which dG(N1-H)• was generated from 1 and was reported that BME did not react with the radical.19 Suitable reducing agents, albeit not BME, gave rise to similar increases in yields of dA and mass balances from photolyses of 7.15 The product at 16 min was identified as NP by UPLC-MS and HPLC (Figure 2). The lack of formation of N-hydroxy-1,8- naphthalimide (4) was consistent with homolytic N-O bond cleavage and excludes the generation of dG via hydrolysis. The yield of 1,8-naphthalimide (NP) was quantitative, and unlike that of dG was independent of the presence of BME. This is an important observation for interpreting the photochemistry of 3 (vide infra).
Figure 1.

HPLC chromatograms (260 nm detection) of the photolyses of 3 (0.1 mM) under anaerobic conditions in phosphate buffer (10 mM, pH = 7.2). (a) Photolyses of 3 in the absence of BME; (b) photolysis of 3 in the presence of BME (10 mM). The peak at 11 min is attributed to a by-product generated from BME.
Figure 2.

HPLC chromatograms (260 nm detection) of photolysates (5 min) of 3 (0.1 mM) under anaerobic conditions in phosphate buffer (10 mM, pH = 7.2) (a) in the presence of BME (10 mM); (b) in the absence of BME. (c) Solution containing dT (0.1 mM) and NP (0.1 mM). The complete chromatogram is present in Figure S5.
The consumption rate of 3 decreased significantly when the photolyses were carried out in the presence of O2 (Figure 3). The effect of oxygen on the consumption rate of 3 is consistent with the report that the 1,8-naphthalimide triplet excited state is efficiently quenched by O2 to yield 1O2 (Scheme 2).29 This observation indicates that the triplet excited state of 3 undergoes homolytic bond scission to release dG(N1-H)•. Photophysical studies on 1,8-naphthalimide photoacid generators indicate that bond scission occurs from the singlet excited state.27 Although we cannot rule out a contribution of the singlet excited state to the formation of dG(N1-H)•, the involvement of the triplet excited state in this chemistry is different from other 1,8-naphthalimides. In the absence of thiol, 81 ± 1% of the precursor was consumed in 3 h (as opposed to < 7 min under anaerobic conditions, Figure 1a), and the yield of dG was only 14 ± 1%. We propose that the low yield of dG was due to oxidation by 1O2, produced from quenching of triplet exited states of 3 and NP by oxygen.22, 30–33 This proposal was supported by the observation that the yield of dG based on conversion decreased as the photolysis time increased. The yield of dG based on unrecovered 3 decreased from 57 ± 1% at 1 h to 37 ± 4% at 2 h (Figure 3a), indicating that continued photolysis of the mixture of dG and NP in the presence O2 led to destruction of the former. Including BME (10 mM) in aerobic photolyses resulted in a significantly different outcome. dG was formed quantitatively and 3 was completely consumed under these conditions within 1 h (Figure 3b). The accelerated consumption of the precursor is similar to that observed under anaerobic conditions, indicating a possible photoreduction pathway. In addition, BME consumes O2 by reacting with singlet O2, which may contribute to the increased rate of photoconversion as well as higher dG yield.34 Extended photolysis in the presence of O2 eventually led to the reduction of the yield of dG.
Figure 3.

HPLC chromatograms (260 nm detection) of the photolyses of 3 (0.1 mM) under aerobic conditions in phosphate buffer (10 mM, pH = 7.2). (a) Photolyses of 3 in the absence of BME; (b) photolysis of 3 in the presence of BME (10 mM). The peak at 11 min is attributed to a by-product generated from BME.
Secondary redox reactions in the absence of exogenous reducing agent (BME).
To explain the formation of dG at ~50% yield in the absence of BME, we propose that precursor (3) serves as the reducing agent of dG(N1-H)•, but the fate of the portion of the sacrificed precursor was unclear. As previously mentioned, the yield of NP was quantitative under anaerobic conditions regardless of the presence of BME. This observation provided us a hint of the possible products generated from the oxidation of precursor 3 by dG(N1-H)• and naphthalimidyl radical (NP•). Scheme 4 shows a formal representation of the photolysis, in which two products detected by HPLC, dG and NP, result from the single electron reduction by 3. This behavior is consistent with previous reports on the reactivity of purine radicals and similar types of precursors as employed here.15,16 To balance the chemical equation electronically, we postulate that the 50% of 3 that acts as a reducing agent, is ultimately converted into three-electron oxidized forms of dG(N1-H)• (four-electron oxidized forms of dG), such as dSp and dGh (Scheme 4).35 This proposal was corroborated by utilizing Burrow’s previous report on the separation and identification of dSp and dGh.35 As previously reported, the photolysates were initially analyzed by C18-reversed phase HPLC, and the void volume containing the polar four-electron oxidized guanine 2’-deoxynucleosides was collected. The void volume was then analyzed by UPLC-MS equipped with a Hypercarb column, which revealed dSp and dGh (Figure 4).
Scheme 4.

Photolysis product from 3 on the absence of exogenous reductant
Figure 4.

UPLC-MS/MS analysis of the anaerobic photolysis of 3. (a) Extracted-ion chromatogram (EIC) of dGh (calculated (M + H) m/z = 274.1146, observed m/z = 274.1143) and (b) EIC of dSp and Sp (calculated (M + H) m/z = 184.0465, observed m/z = 184.0468).
Our proposed mechanism (Scheme 5) begins with UV-excited 3 undergoing homolytic cleavage of the N-O bond to generate dG(N1-H)• and NP• (Scheme 5a). Precursor 3 reduces dG(N1-H)• or NP• to generate the radical cation 9 and dG or NP, respectively (Scheme 5b, oxidation of 3 by NP• is not shown). Fragmentation of 9 upon reaction with water and subsequent tautomerization results in the formation of 8-oxodGuo and NP• (Scheme 5b). Given the relatively low oxidation potential of 8-oxodGuo (0.74 V/NHE), both dG(N1-H)• (1.29 V/NHE) and NP• (> 2 V/NHE) can be reduced by it, and 8-oxodGuo undergoes two-electron oxidation to generate products such as dGh and dSp (Scheme 5c, oxidation of 8-oxodGuo by dG(N1-H)• is not shown).1, 19, 36–40 It is also possibile that 3 in the excited state oxidizes ground state 3 and 8-oxodGuo, which would lead to the same result. To demostrate whether 8-oxodGuo can serve as the reducing agent, 3 (0.1 mM) was photolyzed in the presence of two equivalents of 8-oxodGuo (Figure 5), a much lower concentration than that of BME (10 mM) used (Figure 1b). The addition of 8-oxodGuo restored the quantitative formation of dG observed under anaerobic conditions in the presence of BME, and one equivalent of 8-oxodGuo was consumed. The yields of dSp and dGh were qualitatively higher for the photolyses carried out in the presence of 8-oxo-dG as expected (Figure 6).
Scheme 5.

Proposed mechanism for secondary redox chemistry
Figure 5.

HPLC chromatograms of the photolyses of 3 (0.1 mM) under anaerobic conditions in phosphate buffer (10 mM, pH = 7.2) in the of presence 8-oxodGuo (0.2 mM) detected at 260 nm.
Figure 6.

UPLC-MS/MS analysis of the anaerobic photolysis of 3 in the presence of 8-oxodGuo (0.2 mM). (a) Extracted-ion chromatogram (EIC) of dGh (calculated (M + H) m/z = 274.1146, observed m/z = 274.1143) and (b) EIC of dSp and Sp (calculated (M + H) m/z = 184.0465, observed m/z = 184.0468).
Conclusions.
We developed a precursor (3) that generates the thermodynamically favored neutral purine radical of 2’-deoxyguanosine (dG(N1-H)•) in high yield. The efficiency of radical formation is also good, particularly under anaerobic conditions. High yields of dG are obtained in the presence of a reducing agent, β-mercaptoethanol. In the absence of exogenous reducing agent, 3 serves as reducing agent, ultimately producing the four-electron dG lesions, dSp and dGh. Dioxygen effects on the photochemistry of 3 indicate that contrary to other 1,8-naphthalimides, cleavage to produce dG(N1-H)• occurs from the excited triplet state. Precursor (3) will be a useful tool for studying the important reactive intermediate, dG(N1-H)•.
Experimental Procedures.
General methods.
Triethylamine, DMF, and DCM were distilled from CaH2 under Ar or under an appropriate vacuum. THF was distilled from Na under Ar. All other reagents were purchased from commercial sources and were used without further purification unless noted otherwise. All reactions were carried out under a positive pressure of argon atmosphere and monitored by TLC on Silica Gel G-25 UV254 (0.25 mm) unless stated otherwise. Spots were detected under UV light and/or by ethanolic solution p-anisaldehyde, aqueous solution of ammonium molybdate, ceric ammonium sulfate, or KMnO4. Column flash chromatography was performed with Silicycle grade 70–230 mesh, 60–200 μm, 60 Å silica. The ratio between silica gel and crude product ranged from 100:1 to 20:1 (w/w). HPLC analyses were carried out using a Waters system equipped with 515 Pumps, 2489 multi-wavelength detector and 2707 auto sampler. NMR data were collected using a Bruker Advance spectrometer. HRMS data were obtained on a Waters Q-TOF mass spectrometer using electrospray ionization.
Preparation of 6.
Compound 528 (135 mg, 0.2 mmol), and DABCO (22 mg, 0.4 mmol) were dissolved in DMF (2 mL), and stirred for 30 min. DBU (45 mg, 0.3 mmol) and N-1,8-hydroxynaphthalimide (4, 213 mg, 1 mmol) were added, and the reaction was kept at room temperature overnight. The reaction was quenched by water and extracted by DCM (3 × 10 mL). The combined organic phase was dried over Na2SO4, filtered, and the mixture was concentrated under vacuum. The product was purified by flash chromatography on a silica column. Elution with 50% EtOAc in hexanes gave 6 as a yellow solid (64 mg, 46%). 1H NMR (400 MHz, CDCl3) δ 8.63 (d, J = 7.3 Hz, 2H), 8.27 (d, J = 8.2 Hz, 2H), 7.95 (s, 1H), 7.78 (t, J = 7.8 Hz, 2H), 6.26 (t, J = 6.2 Hz, 1H), 4.55 (dd, J = 9.2, 4.3 Hz, 1H), 3.99 – 3.89 (m, 1H), 3.80 (dd, J = 11.2, 3.0 Hz, 1H), 3.73 (dd, J = 11.2, 3.0 Hz, 1H), 2.50 (dt, J = 12.4, 6.1 Hz, 1H), 2.41 – 2.25 (m, 1H), 0.89 (s, 9H), 0.85 (s, 9H), 0.08 (s, 6H), 0.04 (s,3H), 0.03 (s, 3H). 13C {1H} NMR (101 MHz, CDCl3) δ 159.9, 158.7 155.18, 139.2, 134.7, 131.9, 131.8, 127.7, 127.0, 122.7, 113.8, 87.5, 83.8, 71.4, 62.5, 41.1, 25.9, 25.7, 18.3, 17.9, −4.6, −4.8, −5.4, −5.5. HRMS (ESI-TOF) m/z calcd for C34H47N6O6Si2 (M + H)+ = 691.3096, found m/z = 691.3097.
Preparation of 3.
Compound 6 (64 mg, 0.092 mmol) was dissolved in THF (1 mL). While stirring, Et3N•3HF (161 mg, 1 mmol) was slowly added. The reaction was stirred at 25 °C overnight. The reaction was concentrated under vacuum and purified by flash chromatography on a silica column. Elution with 5% MeOH in DCM gave 3 as a yellow foam (35.7 mg, 84%). 1H NMR (400 MHz, MeOH-d4) δ 8.64 (d, J = 6.8 Hz, 2H), 8.48 (d, J = 7.8 Hz, 2H), 8.20 (s, 1H), 7.90 (s, 2H), 6.37 (s, 1H), 4.57 (s, 1H), 4.03 (s, 1H), 3.83 (d, J = 10.1 Hz, 1H), 3.74 (d, J = 10.1 Hz, 1H), 2.80 (s, 1H), 2.41 (s, 1H). 13C {1H} NMR (101 MHz, DMSO) δ 160.1, 159.8, 140.1, 135.9, 132.1, 132.0, 127.9, 127.4, 122.5, 112.0, 88.1, 83.3, 71.1, 62.0. HRMS (ESI-TOF) m/z calcd for C22H19N6O6 (M + H)+ = 463.1366, found m/z = 463.1369.
Photolysis of the precursor and subsequent HPLC analysis.
Photolyses were carried out in Pyrex tubes using a Rayonet photochemical reactor (Southern New England Ultraviolet) equipped with a merry-go-round apparatus and 16 lamps having a maximum output at 350 nm. Reaction mixtures (50 μL each) containing 3 (100 μM), thymidine (100 μM) in buffer (10 mM phosphate, pH 7.2), and BME (10 mM) when appropriate, were photolyzed at room temperature under aerobic or anaerobic conditions. Samples for anaerobic reactions were degassed by three freeze-pump-thaw cycles at 2 mTorr and flame sealed under vacuum. The reaction mixtures (including unphotolyzed controls) were analyzed by reversed-phase HPLC while being monitored at 260 nm. HPLC was performed on an Phenomenex Luna C-18 column (250 × 4.6 mm) using water and acetonitrile as eluents (1 mL/min) from t = 0 to 1 min holding 3% ACN, from t = 1 to 10 min, from 3% - 28% ACN linearly, and then from t = 15 to 20 min, from 28% - 97% ACN. The column was equilibrated for 20 min following a rapid return to the starting conditions. Peaks corresponding to 2’-deoxyguanosine, thymidine, 3 and NP eluted at 7.4, 8.3, 15.5 and 16.0 min, respectively. The peaks were integrated and quantified against the internal standard thymidine. After collecting the void volume of the above injections carried out on the C18-reverse phase column, dSP and dGh analyses were carried out on UPLC-MS system equipped with a Thermo Scientific Hypercarb column (130 Å, 5 μm, 2.1 × 100 mm), The gradient (A, 0.1% formic acid; B, acetonitrile, 0.3 mL/min) was 0% B at t = 0 min, from 0–90% B linearly from t = 0 to 10 min and holding at 90% B from t = 10 to 12 min. The column was equilibrated for 3 min following a rapid return to the starting conditions.
Determination of the quantum yield for photoconversion of 3 in acetonitrile under continuous photolysis.
The light flux of the photoreactor was determined by N-1,8-hydroxynaphthalimide triflate actinometry in acetonitrile. N-Hydroxy-1,8-naphthalimide triflate has a 0.23 quantum yield for N-O bond cleavage.26–27 Solutions containing the actinometer (50 μL, 50 mM) were degassed using 3 freeze-pump-thaw cycles and sealed under high vacuum prior to photolysis. The extent of reaction of the actinometer was measured by UV absorbance at 360 nm. The conversion of the actinometer was 12.4% in 30 s. The % transmittance of the actinometer solution was less than 0.1% after the photolysis. Therefore, the change of transmittance was negligible during photolysis. Using eqn. 8, the light flux was calculated to be (2.5 ± 0.1) × 10−7 Einstein•min−1. Solutions containing 3 (50 μL, 100 μM) had an OD350 at 1.72. The samples were degassed and photolyzed for 1 min using the calibrated photoreactor. The extent of reaction of 3 was determined by HPLC using thymidine as internal standard. The conversion of 3 was 30.7 ± 0.9 %. The quantum yield for conversion of precursor 3 was calculated using eqn. 9, and determined to be 0.03.
| eqn. 1 |
| eqn. 2 |
Supplementary Material
Acknowledgement.
We are grateful for support of this research from the National Institute of General Medical Sciences (GM-054996, GM-131736).
Footnotes
Supporting Information. NMR spectra of new compounds, UV-VIS spectrum of 3 and complete chromatograms of photolysis of 3. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Steenken S; Jovanovic SV, How Easily Oxidizable is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc 1997, 119, 617–618. [Google Scholar]
- 2.Thapa B; Schlegel HB, Calculations of pKa’s and Redox Potentials of Nucleobases with Explicit Waters and Polarizable Continuum Solvation. J. Phys. Chem. A 2015, 119, 5134–5144. [DOI] [PubMed] [Google Scholar]
- 3.Genereux JC; Barton JK, Mechanisms for DNA Charge Transport. Chem. Rev 2010, 110, 1642–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanvah S; Joseph J; Schuster GB; Barnett RN; Cleveland CL; Landman U, Oxidation of DNA: Damage to nucleobases. Acc. Chem. Res 2010, 43, 280–287. [DOI] [PubMed] [Google Scholar]
- 5.Rokhlenko Y; Geacintov NE; Shafirovich V, Lifetimes and Reaction Pathways of Guanine Radical Cations and Neutral Guanine Radicals in an Oligonucleotide in Aqueous Solutions. J. Am. Chem. Soc 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rokhlenko Y; Cadet J; Geacintov NE; Shafirovich V, Mechanistic Aspects of Hydration of Guanine Radical Cations in DNA. J. Am. Chem. Soc 2014, 136, 5956–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A; Pottiboyina V; Sevilla MD, Hydroxyl Radical (OH•) Reaction with Guanine in an Aqueous Environment: A DFT Study. J. Phys. Chem. B 2011, 115, 15129–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adhikary A; Kumar A; Becker D; Sevilla MD, The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Sonntag C, Free-Radical-Induced DNA Damage and Its Repair Springer-Verlag: Berlin, 2006. [Google Scholar]
- 10.Candeias LP; Steenken S, Structure and Acid-Base Properties of One-Electron-Oxidized Deoxyguanosine, Guanosine, and 1 -Methylguanosine. J. Am. Chem. Soc 1989, 111, 1094–1099. [Google Scholar]
- 11.Al-Sheikhly M, The reactivity of adenyl and guanyl radicals towards oxygen. Radiat. Phys. Chem 1994, 44, 297–301. [Google Scholar]
- 12.Candeias LP; Steenken S, Reaction of HO· with guanine derivatives in aqueous solution: formation of two different redox-active OH-adduct radicals and their unimolecular transformation reactions. Properties of G(−H)·. Chem. - Eur. J 2000, 6, 475–484. [DOI] [PubMed] [Google Scholar]
- 13.Giese B; Beyrich-Graf X; Erdmann P; Giraud L; Imwindelried P; Müller SN; Schwitter U, Cleavage of Single-Stranded 4’-Oligonucleotide Radicals in the Presence of O2. J. Am. Chem. Soc 1995, 117, 6146–6147. [Google Scholar]
- 14.Greenberg MM, Reactivity of Nucleic Acid Radicals. Adv. Phys. Org. Chem 2016, 50, 119–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng L; Griesser M; Pratt DA; Greenberg MM, Aminyl Radical Generation via Tandem Norrish Type I Photocleavage, β-Fragmentation: Independent Generation and Reactivity of the 2’-Deoxyadenosin-N6-yl Radical. J. Org. Chem 2017, 82, 3571–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L; Lin L; Qu K; Adhikary A; Sevilla MD; Greenberg MM, Independent Photochemical Generation and Reactivity of Nitrogen-Centered Purine Nucleoside Radicals from Hydrazines. Org. Lett 2017, 19, 6444–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manetto A; Georganakis D; Leondiadis L; Gimisis T; Mayer P; Carell T; Chatgilialoglu C, Independent Generation of C5’-Nucleosidyl Radicals in Thymidine and 2’-Deoxyguanosine. J. Org. Chem 2007, 72, 3659–3666. [DOI] [PubMed] [Google Scholar]
- 18.Vrantza D; Kaloudis P; Leondiadis L; Gimisis T; Vougioukalakis GC; Orfanopoulos M; Gasparutto D; Cadet J; Encinas S; Paris C; Miranda MA, Modification of Guanine with Photolabile N-Hydroxypyridine-2(1H)-thione: Monomer Synthesis, Oligonucleotide Elaboration, and Photochemical Studies. Helv. Chim. Acta 2006, 89, 2371–2386. [Google Scholar]
- 19.Kaloudis P; Paris C; Vrantza D; Encinas S; Perez-Ruiz R; Miranda MA; Gimisis T, Photolabile N-hydroxypyrid-2(1H)-one derivatives of guanine nucleosides: a new method for independent guanine radical generation. Org. & Biomol. Chem 2009, 7, 4965–4972. [DOI] [PubMed] [Google Scholar]
- 20.Cadet J; Wagner JR, Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys 2014, 557, 47–54. [DOI] [PubMed] [Google Scholar]
- 21.Cadet J; Wagner JR, DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harbor Perspect. Biol 2013, 5, A012559/1-A012559/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cadet J; Douki T; Ravanat J-L, Oxidatively Generated Damage to the Guanine Moiety of DNA: Mechanistic Aspects and Formation in Cells. Acc. Chem. Res 2008, 41, 1075–1083. [DOI] [PubMed] [Google Scholar]
- 23.Chatgilialoglu C; D‚ Angelantonio, M.; Kciuk, G.; Bobrowski, K., New Insights into the Reaction Paths of Hydroxyl Radicals with 2’-Deoxyguanosine. Chem. Res. Toxicol 2011, 24, 2200–2206. [DOI] [PubMed] [Google Scholar]
- 24.Chatgilialoglu C; D’Angelantonio M; Guerra M; Kaloudis P; Mulazzani QG, A Reevaluation of the Ambident Reactivity of the Guanine Moiety Towards Hydroxyl Radicals. Angew. Chem. Int. Ed 2009, 48, 2214–2217. [DOI] [PubMed] [Google Scholar]
- 25.Kuttappan-Nair V; Samson-Thibault F; Wagner JR, Generation of 2’-Deoxyadenosine N6-Aminyl Radicals from the Photolysis of Phenylhydrazone Derivatives. Chem. Res. Toxicol 2010, 23, 48–54. [DOI] [PubMed] [Google Scholar]
- 26.Malval J-P; Morlet-Savary F; Allonas X; Fouassier J-P; Suzuki S; Takahara S; Yamaoka T, On the cleavage process of the N-trifluoromethylsulfonyloxy-1,8-naphthalimide photoacid generator. Chemical Physics Letters 2007, 443, 323–327. [Google Scholar]
- 27.Malval J-P; Suzuki S; Morlet-Savary F; Allonas X; Fouassier J-P; Takahara S; Yamaoka T, Photochemistry of Naphthalimide Photoacid Generators. The Journal of Physical Chemistry A 2008, 112, 3879–3885. [DOI] [PubMed] [Google Scholar]
- 28.Lakshman MK; Ngassa FN; Keeler JC; Dinh YQV; Hilmer JH; Russon LM, Facile Synthesis of O6-Alkyl-, O6-Aryl-, and Diaminopurine Nucleosides from 2’-Deoxyguanosine. Org. Lett 2000, 2, 927–930. [DOI] [PubMed] [Google Scholar]
- 29.Aveline BM; Matsugo S; Redmond RW, Photochemical Mechanisms Responsible for the Versatile Application of Naphthalimides and Naphthaldiimides in Biological Systems. J. Am. Chem. Soc 1997, 119, 11785–11795. [Google Scholar]
- 30.Ravanat J-L; Cadet J, Reaction of Singlet Oxygen with 2’-Deoxyguanosine and DNA. Isolation and Characterization of the Main Oxidation Products. Chem. Res. Toxicol 1995, 8, 379–388. [DOI] [PubMed] [Google Scholar]
- 31.Hickerson RP; Prat F; Muller JG; Foote CS; Burrows CJ, Sequence and Stacking Dependence of 8-Oxoguanine Oxidation: Comparison of One-Electron vs Singlet Oxygen Mechanisms. J. Am. Chem. Soc 1999, 121, 9423–9428. [Google Scholar]
- 32.Sheu C; Kang P; Khan S; Foote CS, Low-Temperature Photosensitized Oxidation of a Guanosine Derivative and Formation of an Imidazole Ring-Opened Product. J. Am. Chem. Soc 2002, 124, 3905–3913. [DOI] [PubMed] [Google Scholar]
- 33.Ye J; Muller JG; Luo W; Mayne CL; Shallop AJ, Formation of 13C-, 15N-, and 18O- labeled Guanidinohydantoin From Guanosine Oxidation with Singlet Oxygen. Implications for Structure and Mechanism. J. Am. Chem. Soc 2003, 125, 13926–13927. [DOI] [PubMed] [Google Scholar]
- 34.Devasagayam TPA; Sundquist AR; Di Mascio P; Kaiser S; Sies H, Activity of thiols as singlet molecular oxygen quenchers. J. Photochem. Photobiol. B: Biology 1991, 9, 105–116. [DOI] [PubMed] [Google Scholar]
- 35.Alshykhly OR; Fleming AM; Burrows CJ, 5-Carboxamido-5-formamido-2-iminohydantoin, in Addition to 8-oxo-7,8-Dihydroguanine, Is the Major Product of the Iron-Fenton or X-ray Radiation-Induced Oxidation of Guanine under Aerobic Reducing Conditions in Nucleoside and DNA Contexts. J. Org. Chem 2015, 80, 6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munk BH; Burrows CJ; Schlegel HB, An Exploration of Mechanisms for the Transformation of 8-Oxoguanine to Guanidinohydantoin and Spiroiminodihydantoin by Density Functional Theory. J. Am. Chem. Soc 2008, 130, 5245–5256. [DOI] [PubMed] [Google Scholar]
- 37.Luo W; Muller JG; Rachlin EM; Burrows CJ, Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem. Res. Toxicol 2001, 14, 927–938. [DOI] [PubMed] [Google Scholar]
- 38.Luo W; Muller JG; Rachlin EM; Burrows CJ, Characterization of Spiroiminodihydantoin as a Product of One-Electron Oxidation of 8-Oxo-7,8-dihydroguanosine. Org. Lett 2000, 2, 613–616. [DOI] [PubMed] [Google Scholar]
- 39.Steenken S; Jovanovic SV; Bietti M; Bernhard K, The Trap Depth (in DNA) of 8-Oxo-7,8-dihydro-2’deoxyguanosine as Derived from Electron-Transfer Equilibria in Aqueous Solution. J. Am. Chem. Soc 2000, 122, 2373–2374. [Google Scholar]
- 40.Kucheryavy P; Khatmullin R; Mirzakulova E; Zhou D; Glusac KD, Photoinduced Electron Transfer in Naphthalimide-Pyridine Systems: Effect of Proton Transfer on Charge Recombination Efficiencies. J. Phys. Chem. A 2011, 115, 11606–11614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.