

Abstract
Nanobodies are special derivatives of antibodies, which consist of single domain fragments. They have become of considerable interest as next-generation biotechnological tools for antigen recognition. They can be easily engineered due to their high stability and compact size. Nanobodies have three complementarity-determining regions, CDRs, which are enlarged to provide a similar binding surface to that of human immunoglobulins. Here, we propose a benchmark testing algorithm that uses 3D structures of already existing protein-nanobody complexes as initial structures followed by successive mutations on the CDR domains. The aim is to find optimum binding amino acids for hypervariable residues of CDRs. We use molecular dynamics simulations to compare the binding energies of the resulting complexes with that of the known complex and accept those that are improved by mutations. We use the MDM4-VH9 complex, (PDB id 2VYR), fructose-bisphosphate aldolase from Trypanosoma congolense (PDB id 5O0W) and human lysozyme (PDB id 4I0C) as benchmark complexes. By using this algorithm, better binding nanobodies can be generated in a short amount of time. We suggest that this method can complement existing immune and synthetic library-based methods, without a need for extensive experimentation or large libraries.
Keywords: Nanobody design, Molecular dynamics, MDM4, Fructose-bisphosphate aldolase, Human lysozyme, CDR, Steered molecular dynamics
Introduction
Antibodies, also called immunoglobulins, are Y-shaped proteins that are the key elements in the adaptive immune system. They recognize unique parts of foreign targets, antigens, in the blood or mucosa and inactivate them. They activate the complementary systems to destroy bacterial cells and facilitate the phagocytosis of foreign substances. Antibodies are composed of two identical copies of heavy (approximately 50 kDa) and light chains (approximately 25 kDa). Disulfide bonds connect heavy chains to each other and to light chains. Each antibody-producing B cell produces a unique type of antibody. There are 5 antibody isotypes: IgG, IgM, IgD, IgE, and IgA that all differ in their heavy chains. Variance in the heavy chains determines their binding to different antigens [1]. Antibody-derived biologics have produced impressive therapeutic results. Advances in antibody engineering have provided improved innovative molecules with impressive achievements in the treatment of several hematological malignancies and tumors. The advantages of them are their enhanced effector function, reduced immunogenicity, prolonged half-life, and reduced side effects. Their large size and hydrophobic binding surfaces pose obstacles to their access to the antigen [2]. The hydrophobic residues of human immunoglobulins required for heavy and light chain interactions are more exposed to the solvent environment. This poses problems in inter- and intramolecular disulfide bridge formations and thus leads to aggregation and poor solubility. Due to these limiting factors, new antibody-formatted treatment options with similar binding specificity and higher stability are required.
Nanobodies are single domain antibody fragments derived from full-length antibodies. They correspond to the variable heavy chain of full-length antibodies, shown in Fig. 1. They hold the functional and structural properties of naturally occurring only heavy chain antibodies derived from members of the Camelid family. Single domain antibody (sdAb) production in the body does not require IgM production, which is the initial response of the body against antigen exposure. Instead, sdAbs can be produced upon immunization [3]. They are highly specific and exert high affinity towards their target. They can be easily expressed in microorganisms. Their toxicity is low, and their tissue penetration is not limited due to their small size. The regions on nanobodies that are responsible for binding and recognition are called complementarity-determining regions (CDR) [4]. The antigen-binding region, paratope, diversity of antibodies is derived from their heavy and light chain combinations. Nanobodies are only composed of a single fragment and compensate this diversification by taking more loop conformations without any structural restrictions in their CDRs. Due to their biochemical functionality and economic benefits, interest in nanobodies has grown in biotechnology and medicine. In order to use nanobodies as a therapeutic reagent, their key properties such as their stability, binding specificity, and affinity to the target antigen must be optimized. One common strategy to improve protein solubility is to replace hydrophobic residues with hydrophilic ones on the binding surface. This modification will contribute both to the stability and solubility of nanobodies [5].
Fig. 1.
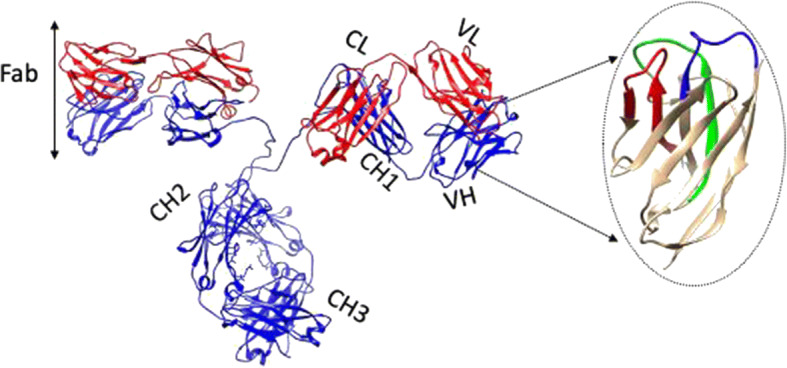
Cartoon representation of an antibody; the heavy chain is blue, and the light chain is red. It has two Fab arms, consisting of two identical heavy (VH-CH1) and light (VL-CL) chain combinations. The constant regions labeled as CH1, CH2, CH3, and CL have the same sequence within the same isotype (IgG, IgM, IgD, IgE, and IgA), and the variable regions labeled as VL and VH determine the antigen specificity of the antibody. The focused VH domain corresponds to the nanobody with 3 antigen-binding regions colored in red, green, and blue
The target specificity of a nanobody is provided by its CDRs. Nanobodies comprise three CDR loops: CDR1, CDR2, and CDR3. CDR3 is the dominating contributor in antigen recognition, while the impact of the first and the second CDR loops are limited. Residue numbers of the CDR amino acids can vary, but a CDR loop can be recognized by the presence of certain amino acid repeats. These repeats are composed of evolutionarily fixed and variable regions, the latter being referred to as hypervariable. The residue position of the hypervariable and conserved residues differs within species but can be determined by using a consensus sequence as a template. A consensus sequence is determined by aligning sequences that share a common feature and determining most commonly expressed or varying amino acids at each position. A previous study showed which CDR residues are conserved and which are hypervariable on llama-derived nanobodies [6]. The increased frequency of some residues in the hypervariable positions supports convergent evolution in those regions.
The structure, consensus sequence, and the hypervariable positions for Lama glama derived nanobodies are given in Fig. 2. The amino acid frequency scheme of the highly variable regions shown with asterisks (*) in Fig. 2b is given as 14%-Y, 12%-G, 10%-S, 9%-D, 7%-T, 6%-R, 6%-A, 5%-L, 5%-V, 4%-N, 3%-F, 3%-E, 3%-I, 3%-W, 2%-Q, 2%-K, 2%-H, 0%-C, and 0%-M. In nature, hypervariable residues in CDRs are mutated naturally to increase the binding affinity and the specificity towards their target antigen. Alignment studies similar to that in Ref [6] can be made for organisms other than Lama glama, and fixed-variable residue templates can be determined.
Fig. 2.

a Cartoon representation of a nanobody. CDR1, 2, and 3 are colored in blue, red, and green, respectively. b Framework of fixed, variable, and highly variable amino acid motifs of each CDR are indicated; amino acids indicated with * are the highly variable ones. Residue positions with more than one type of amino acid which are the variable regions for the Lama glama nanobodies and single residue positions are also shown
Computational screening methods contributed greatly to the optimization and design of antibodies, for which several approaches are available [4, 7]. The common protocol is to start the screening procedure from a known potential binder nanobody retrieved from experimental or computational studies. Experimental strategies employ libraries of phage display, bacterial display, yeast display, ribosome display, and intracellular 2 hybrid selection. Computational approaches utilize docking nanobody structures by using docking servers such as Z-dock, Cluspro, or any other docking software available and enhancing the properties by mutating CDR or non-CDR residues iteratively and measuring the antigen-nanobody affinity values [8–11]. To start the mutation process, the CDR, non-CDR loops, and the locations on the CDR loops, which allow insertions or deletions, must be identified. This can be done by aligning nanobody sequences to known structures belonging to the same species. There are publicly available CDR numbering tools that can be applied to a certain sequence [12]. After the identification of the orientation or the sequence of the nanobody, amino acids to be mutated can be selected to increase the binding affinity for the antigen.
Current computational techniques are based on finding the CDR and non-CDR residues that are in contact with the antigen and performing point mutations to enhance the interaction [13–17]. However, there are no certain criteria to decide which residues to mutate. The optimization strategy proposed in this study is based on finding the hypervariable positions in CDRs by an alignment process and increasing the binding affinity by mutating them iteratively. The residues that are more frequent for a particular species give greater weight in the selection process. This method has been tested on several different proteins: human MDM4, fructose-bisphosphate aldolase from Trypanosoma congolense, and human lysozyme. The PDB codes are 2VYR, 5O0W, and 4I0C, respectively. The three systems are selected because each antigen is bound to a nanobody derived from a different species and the binding affinities are experimentally determined.
Method
The method adopted in this work consists of three parts: (i) determining the hypervariable residues of nanobodies by alignment to a given consensus sequence, (ii) optimizing the binding affinity by successive mutations of hypervariable residues and finding the residue mutation with the minimum interaction energy using molecular dynamics simulations, and (iii) testing the affinity of the nanobody-protein complex using steered molecular dynamics (SMD).
Alignment
Sequences and structures of synthetic and natural nanobodies can be found in databases [18, 19]. Nanobody sequences for specific organisms can be downloaded from the databases and aligned by using several multiple alignment tools available [20–23]. The alignments can be visualized by specific softwares [24, 25]; the consensus sequence and the conserved or hypervariable positions can be determined, and a template can be obtained similar to the llama-derived nanobody framework proposed previously [6]. The amino acid frequency in hypervariable positions, which is denoted as the evolutionary frequency in this study, is analyzed using the alignment results.
Nanobody sequences for Vicugna pacos and Camelus dromedarius are downloaded from the single domain antibody database (sdAb) [19] and aligned separately by using ClustalW, a multiple sequence alignment tool [23]. Alignments are visualized in Unipro U-gene, and hypervariable residues and their evolutionary amino acid frequencies and consensus sequences for CDRs are determined [24]. Residue positions with high gap penalties are not considered, the occurrences of the amino acids in highly variable positions are counted, and the frequency of each amino acid is saved to be used further in the energy update step. A consensus sequence is obtained for each alignment, and hypervariable positions are labeled by asterisks (*) in the templates. Residues corresponding to these positions will be selected for mutation.
Optimization strategy
The strategy developed in our study is to start with a protein-nanobody complex, determine the hypervariable CDR residues according to the given consensus sequences, and then perform 20 independent point mutations on each variable position, starting from alanine up to valine, by using visual molecular dynamics (VMD) [26]. Each mutant structure will be subjected to minimization, annealing, a conventional MD run, and another minimization cycle by using NAMD [27]. In this study, the CHARMM36 force field is used. The interaction energy of the nanobody and the protein for each mutation case is calculated and further updated according to the evolutionary amino acid frequency scheme obtained from the alignment by using an iterative Monte Carlo algorithm (see below), which favors higher evolutionary preferences. The end result provides us with the distribution of possible optimum mutation types on hypervariable positions of CDRs. A broad library of optimal nanobodies can be derived and tested for its affinity and selectivity. The affinity of the new nanobodies can be measured by using SMD [28], by pulling the nanobody away from the protein and measuring the work required to abolish all of the interactions. These SMD simulations provide the dissociation constant KD values, which are useful in comparing the affinity of the optimized and the wild-type nanobodies.
Molecular dynamics min-run cycle
All of the mutant nanobodies in the complex with the protein are solvated in a TIP3P water box, and counter ions are added to neutralize the system. The CHARMM36 force field is used to parametrize the atoms. The time steps of simulations are kept as 2 fs. Initially, the energy of the system is minimized for 8000 steps, and the system gradually heated up to 310 K. The annealing step is followed by an equilibration period at a constant temperature of 310 K and a constant pressure of 1 bar for 10,000 steps. Finally, the energy of the equilibrated system is minimized for another 5000 steps
Interaction energy update by an iterative Monte Carlo algorithm
The interaction energy includes electrostatic, VdW, and non-bonded terms. The majority of the force fields favor the charged residues over the uncharged ones. The interaction energies calculated from the min-run cycle are updated according to the evolutionary probability distributions, (EPD), obtained from the alignment, by using the following algorithm; we assume that the system obeys the Boltzmann statistics and the probabilities are functions of the energy and the temperature of the system. Therefore, the probabilities can be written in the following form:
| 1 |
where E(ii, j) is the interaction energy of each mutant hypervariable position and the antigen obtained from MD min-run cycles, i1 is the hypervariable residue number, j is the mutated residue type (Ala, Arg, Asn, …, Val), and E0 scales with the temperature. Small values of E0 favor low energy residues for each individual. These prior probabilities are further updated according to the EPDs obtained from the alignments. The choice of residues according to the a priori probabilities, Eq. 1, is made as follows: for each position, i1, a random number is generated between 0 and 1, and the jth amino acid mutation for the ith residue is accepted if the random number is between p(i1, j) and p(i1, j + 1). The weights of frequencies on the a priori evolutionary probabilities are introduced as follows: We consider the case where a total of n residues in the nanobody are selected as hypervariable positions. For a given mutated sequence i1, where i1 goes from 1 to n, we represent the calculated distribution from the MD interaction energies by C(i). The EPD is denoted as K(i) where i goes from 1 to n. The divergence, DCD, of the distribution C from the EPD K is calculated according to the divergence equation [29]:
| 2 |
A set of sequences is generated, and the divergence from the EPD is measured according to the flowchart shown in Fig. 3. Initially, the hypervariable positions on the nanobody are detected by comparing the original sequence with the consensus sequence; selected hypervariable positions are mutated to 20 available amino acids and projected to MD min-run cycles. The interaction energies of the single mutant and the antigen are measured by using VMD NAMD Energy plugin. Priori probabilities are calculated from the MD interaction energies, and these energies are further updated by evolutionary probabilities for each amino acid type for a given species. A library of mutant nanobodies that have better binding properties to the antigen is generated as a result of this process. This library is further filtered by measuring the divergence of residue mutations selected for hypervariable positions from the evolutionary probabilities. The sequences that have lower divergence from the EPD are kept, and a mutation library is generated with the proposed probability distributions for each hypervariable position. Mutations are selected from the proposed probability distributions, and the affinities are calculated by the steered molecular dynamics method.
Fig. 3.

Flowchart of the optimization method
Steered molecular dynamics
In SMD, the binding affinity is correlated with the rupture force, where the ligand is totally detached from the protein. It has been shown that non-equilibrium pulling work also correlates well with experimental results [30, 31]. The proposed mutations are applied to nanobodies, and the mutant nanobody is pulled away from the protein by SMD simulations at 310 K [28]. The protein is anchored at a residue close to its center of mass but away from the nanobody binding site; the nanobody is pulled away with constant velocity from a residue away from the binding site. The force vectors required for the pulling are obtained from the simulation log file. The force vectors at each timestep are projected onto the pulling direction, and the projected component is plotted against the pulling distance. The cumulative work is calculated by summing up the area under the force vs distance curve. The distance at which all the hydrogen bonds between the nanobody and the protein diminished, i.e., the rupture distance, is detected from the simulations, and the KD value is calculated from the cumulative work value corresponding to that distance with the following formula:
| 3 |
where the unit of the cumulative work is kj/mol, kT is the product of the Boltzmann constant, k, and the temperature, T. kT is 2.58 kJ/mol at 310 K.
Ten replicate SMD simulations were performed for each complex, and the mean cumulative work values are considered in KD calculations.
Results
Alignment results
Vicugna pacos
The consensus sequence for Vicugna pacos-derived nanobodies is obtained and the consensus sequence for each CDR and the frequency of amino acids at each hypervariable position, shown by asterisks (*), are determined.
The consensus sequence templates shown in Fig. 4 are obtained from aligning all Vicugna pacos-derived nanobody sequences available in the single domain antibody database [19]. The positions that are highly variable in the alignments are labeled as the hypervariable positions. Positions that yield high gap scores are not considered in the alignment.
Fig. 4.

Consensus sequence of a CDR1, b CDR2, and c CDR3 loops for Vicugna pacos-derived nanobodies; the asterisks (*) denote the hypervariable positions in the loops
The evolutionary amino acid frequencies given in Table 1 are obtained from Vicugna pacos nanobody sequence alignments. These are the frequency of amino acids in hypervariable positions labeled with an asterisk (*) in Fig. 4.
Table 1.
Amino acid frequencies for hypervariable residue positions shown by asterisks (*) in consensus CDR sequences for Vicugna pacos
| Amino acid letter code | A | R | D | N | C | E | Q | G | H | I | L | K | M | F | P | S | T | W | Y | V |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | 0.090 | 0.054 | 0.043 | 0.040 | 0 | 0.081 | 0 | 0.050 | 0 | 0.062 | 0.062 | 0 | 0 | 0.098 | 0.028 | 0.101 | 0.083 | 0.050 | 0.112 | 0.044 |
Camelus dromedarius
The template for Camelus dromedarius-derived nanobodies is obtained by the same approach. The consensus sequence for each CDR and the frequency of amino acids at each hypervariable position are determined.
The consensus sequence templates shown in Fig. 5 are obtained from aligning all Camelus dromedarius-derived nanobody sequences available in the single domain antibody database. The positions that are highly variable in the alignments are labeled as the hypervariable positions. Positions that yield high gap scores are not considered in the alignment.
Fig. 5.

The consensus sequence of a CDR1, b CDR2, and c CDR3 loops for Camelus dromedarius-derived nanobodies; the asterisks (*) denote the hypervariable positions in the loops
The evolutionary amino acid frequencies given in Table 2 are obtained from Camelus dromedarius nanobody sequence alignments. These are the frequency of amino acids in hypervariable positions labeled with an asterisk (*) in Fig. 5.
Table 2.
Amino acid frequencies for hypervariable residue positions shown by asterisks (*) in consensus CDR sequences for Camelus dromedarius
| Amino acid letter code | A | R | D | N | C | E | Q | G | H | I | L | K | M | F | P | S | T | W | Y | V |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequency | 0.043 | 0.061 | 0.095 | 0.071 | 0 | 0.045 | 0 | 0.110 | 0 | 0.072 | 0 | 0 | 0 | 0.097 | 0.058 | 0.085 | 0.053 | 0 | 0.155 | 0.054 |
Optimization results
Nanobody candidates for human MDM4, with PDB id 2VYR; fructose-bisphosphate aldolase from Trypanosoma congolense, PDB id 5O0W; and human lysozyme, PDB id 4I0C, are proposed, and the hypervariable mutations are compared with a best binder nanobody to studied targets.
MDM4
Human MDM4 is a 490 amino acid, key regulatory protein of tumor suppressor p53. It consists of 3 conserved domains: an N-terminal for binding p53, a zinc-finger domain, and a C-terminal RING domain. Overexpression of MDM4 is associated with tumor growth, suggesting that it could be an important regulator in anticancer strategies. MDM4 binds p53 and decreases its activity and stability. In most cancer studies, p53 is inactivated; thus, reactivating p53 is an attractive strategy for the treatment. As the result of recent studies, MDM proteins, negative regulators of p53, seem to be the druggable and controllable oncogenic targets. Several strategies have been proposed to reactivate p53 by suppressing MDM4. These strategies are based on disrupting the p53-MDM4 interaction by either using a small molecule or a peptidic compound. The small molecule antagonists that disrupt this interaction are nutlin and WK-298 [32, 33]. Although these compounds help to reactivate p53, they do not elicit all the effects of MDM overexpression. Peptidic antagonists have also been developed. Although they have larger interaction surfaces, most of them are unstable in vivo. Another approach is to use mini proteins, nanobodies, to antagonize the interaction between p53 and MDM4. Previously, a successful candidate nanobody consisting of the MDM4-specific single domain was found to inhibit p53-MDM4 interaction with a dissociation constant of 44 nM. This synthetic construct, VH9, is known to be the best binder for MDM4 [34].
Our aim is to use our optimization strategy to detect the wild-type amino acids of the hypervariable positions and propose an amino acid probability distribution for them that will result in a similar binding affinity and selectivity with VH9.
The crystal structure for MDM4-VH9 with PDB id 2VYR is used. VH9 is a synthetic construct; thus, to determine which consensus sequence to utilize in the interaction energy update step, the VH9 sequence is aligned with all the consensus sequences for Lama glama, Vicugna pacos, and Camelus dromedarius, and the sequence similarity is measured to understand the homology of the VH9 nanobody with other species. Percent identity score, which refers to a quantitative homology measure, is calculated by multiplying the number of matching amino acids by 100 and dividing it by the length of the aligned sequence. Closely related sequences are expected to yield a higher percent similarity. The percent identity matrix of consensus sequences from other species and VH9 is shown in Table 3. VH9 and Lama glama alignment gives a higher percent similarity score; therefore, VH9 is more homologous to Lama glama, and its consensus sequence can be used to update MD interaction energies of VH9. The template shown in Fig. 2b for Lama glama, obtained from a previous study, is used to update the MD energies [6]. The hypervariable positions were detected according to the Lama glama template, and the optimization procedure is applied. According to the template, the highly variable residue positions for each CDR are residue numbers 30, 31, 32, and 33 for CDR1; 53 for CDR2; and 94, 95, 96, 97, 98, 99, 100, 101, 102, and 104 for CDR3.
Table 3.
Sequence identity matrix of nanobody consensus sequences and VH9, measured by Clustal12.1
| VH9 | Vicugna pacos | Lama glama | Camelus dromedarius | |
|---|---|---|---|---|
| VH9 | 100.0 | 62.3 | 69.3 | 66.9 |
| Vicugna pacos | 62.3 | 100.0 | 70.8 | 66.4 |
| Lama glama | 69.3 | 70.8 | 100.0 | 69.3 |
| Camelus dromedarius | 66.9 | 66.4 | 69.3 | 100.0 |
The optimum residue mutation types for selected hypervariable positions on VH9 in the complex with MDM4 are obtained by the optimization method described in the flowchart in Fig. 3. The mutations on the sequence with the least divergence from the evolutionary frequency are compared with the wild-type amino acids in Table 4. The affinity of the mutant nanobody and the wild-type VH9 is compared with SMD. A constant pulling speed of 30 Å/ns is applied.
Table 4.
Residue types at hypervariable positions for WT VH9 and mutant VH9 optimized for MDM4
| 30 | 31 | 32 | 33 | 53 | 94 | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 | 104 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | GLU | GLU | TYR | ALA | ALA | TYR | TYR | CYS | ALA | LYS | PRO | TRP | TYR | PRO | MET |
| Mutant | GLU | TYR | THR | ARG | HIS | TYR | SER | GLY | GLY | TYR | ARG | TYR | ARG | GLY | ASP |
Affinities are calculated from the SMD plots as shown in Fig. 6. The rupture distance is 10.34 Å for WT VH9 and 11.83 Å for mutant VH9. Force values are obtained from the simulation log files, and the cumulative work values are measured by calculating the area under the force vs distance plots. The KD values are calculated from the cumulative work values corresponding to rupture distances. Calculated KD values are 18.12 nM and 9.24 nM for WT and mutant VH9, respectively.
Fig. 6.

Mean force (pN) vs distance (Å) and mean cumulative work (kJ/mol) vs distance (Å) plots for a WT VH9 and b mutant VH9
Fructose-bisphosphate aldolase from Trypanosoma congolense
Trypanosoma genus belongs to a diverse group of parasites, which cause various diseases in humans and livestock. Infections of this genus lead to human African trypanosomiasis (HAT) and animal African trypanosomiasis (AAT). Specifically, Trypanosoma congolense is responsible for infections and leads to major economic losses. A proper diagnostic tool is required to detect the infection in animals and proceed with a selective treatment. Recent studies have shown that nanobodies can be used as a diagnostic tool to recognize Trypanosoma congolense fructose-1,6-bisphosphate aldolase (TcoALD). It is a well-conserved glycolytic enzyme among the Trypanosoma genus. The affinity of the nanobody, Nb474, determined for enzyme detection has a dissociation affinity constant of 73.83 pM [35].
The aim is to use this optimization strategy to propose similar amino acids of the WT Nb474 hypervariable positions, which will yield in a similar binding affinity and selectivity with Nb474.
The crystal structure for TcoALD-Nb474 with PDB ID 5O0W is used. The template for Vicugna pacos is used to update the MD energies since it is an alpaca-derived Nb. The hypervariable positions are detected according to the template, and the optimization procedure is applied. According to the template, the highly variable residue positions for each CDR are residue numbers 28, 31, and 32 for CDR1; 53 for CDR2; and 103, 104, 105, 106, 108, 110, 115, 125, and 126 for CDR3.
The optimum residue mutation types for selected hypervariable positions on Nb474 in the complex with TcAldolase are obtained by the optimization method described in the flowchart in Fig. 3. The mutations on the sequence with the least divergence from the evolutionary frequency are compared with the wild-type amino acids in Table 5. The affinity of the mutant nanobody and the wild-type Nb474 is compared with SMD. A constant pulling speed of 30 Å/ns is applied.
Table 5.
Residue types at the hypervariable positions for WT Nb474 and mutant Nb474 optimized for TcoALD
| 28 | 31 | 32 | 53 | 103 | 104 | 105 | 106 | 108 | 110 | 115 | 125 | 126 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | ALA | TYR | TYR | ARG | ASP | THR | THR | ASP | TYR | SER | TYR | ASP | TYR |
| Mutant | ARG | GLU | ALA | HIS | ASN | ASP | LEU | GLN | LEU | LYS | TYR | ASP | GLN |
Affinities are calculated from the SMD plots given in Fig. 7. The rupture distance is 11.09 Å for WT Nb474 and 12.07 Å for mutant Nb474. Force values are obtained from the simulation log files, and the cumulative work values are measured by calculating the area under the force vs distance plots. The KD values are calculated from the cumulative work values corresponding to rupture distances. The calculated KD values are 31.22 pM and 634.5 pM for WT and mutant Nb474, respectively.
Fig. 7.

Mean force (pN) vs distance (Å) and mean cumulative work (kJ/mol) vs distance (Å) plots for a WT Nb474 and b mutant Nb474
Human lysozyme
Human lysozyme (HuL) belongs to the c-type class of lysozymes. It is a 130 amino acid protein that is capable of both hydrolysis and trans-glycosylation and plays a vital role in host defense. Several variants of this protein were found to be related with familial systemic non-neuropathic amyloidosis. It has been revealed that several nanobodies are available that prevent the formation of pathogenic aggregates and inhibit fibril formation by the amyloidogenic variants. These nanobodies, cAbHuL5 and cAbHuL5G, can be used to treat protein misfolding diseases. cAbHuL5 and cAbHuL5G display an affinity of 460 nM and 310 nM for the wild-type HuL [36].
The aim is to use this method to propose similar amino acids of the WT cAbHuL5 hypervariable positions, which will yield a similar binding affinity and selectivity for cAbHuL5.
The crystal structure for HuL-cAbHuL5 with PDB ID 4I0C is used. The template for Camelus dromedarius is used to update the MD energies. The hypervariable positions were detected according to the template, and the optimization procedure is applied. According to the template, the highly variable residue positions for each CDR are residue numbers 27, 28, 29, 30, and 31 for CDR1; 55 and 56 for CDR2; and 97, 98, 101, 110, 111, 112, 113, and 115 for CDR3.
The optimum residue mutation types for selected hypervariable positions on cAbHuL5 in the complex with HuL are obtained by the optimization method described in the flowchart in Fig. 3. The mutations on the sequence with the least divergence from the evolutionary frequency are compared with the wild-type amino acids in Table 6. The affinity of the mutant nanobody and the wild-type cAbHuL5 is compared with SMD. A constant pulling speed of 30 Å/ns is applied.
Table 6.
Residue types at the hypervariable positions for WT cAbHuL5 and mutant cAbHuL5 optimized for HuL
| 27 | 28 | 29 | 30 | 31 | 55 | 56 | 97 | 98 | 101 | 110 | 111 | 112 | 113 | 115 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | LEU | SER | THR | THR | VAL | PHE | PRO | LYS | THR | PHE | SER | ARG | ALA | TYR | HIS |
| Mutant | LEU | ASN | ASP | GLU | GLN | ASP | GLN | ALA | LEU | LEU | TRP | SER | LYS | TRP | VAL |
Affinities are calculated from the SMD plots given in Fig. 8. The rupture distance is 11.01 Å for WT cAbHuL5 and 12.11 Å for mutant cAbHuL5. Force values are obtained from the simulation log files, and the cumulative work values are measured by calculating the area under the force vs distance plots. The KD values are calculated from the cumulative work values corresponding to rupture distances. The calculated KD values are 0.15 nM and 1.48 pM for WT and mutant cAbHuL5, respectively.
Fig. 8.

Mean force (pN) vs distance (Å) and mean cumulative work (kJ/mol) vs distance (Å) plots for a WT cAbHuL5 and b mutant cAbHuL5
Discussion
In this article, we present a method to improve the binding of nanobodies to their known protein targets. Binding of known protein-nanobody complexes is used as a benchmark study. Evolutionarily variant residues on nanobodies are determined, and nanobodies with similar affinities to the best binders are generated. The KD values for wild-type nanobodies are replicated with 10 replicates of steered molecular dynamics simulations, and the affinity of mutated nanobodies is compared with the results of experimentally detected values.
VH9 is the best binding nanobody to the human MDM4 N-terminal domain synthesized so far. The experimentally determined dissociation constant of VH9 bound to the human MDM4 N-terminal domain is 44 nM [34]. The dissociation constant we detected in our SMD simulations is 18.12 nM, which is in the same nM range as the experimentally determined value. The dissociation constant of the mutant VH9 was measured as 9.24 nM.
The contact residues shown in Fig. 9 indicate that this optimization strategy is useful for increasing the contact points of the nanobody. The contact points between the antigen and the mutant nanobody are increased upon mutation. The measured KD values for the mutant nanobody show that the optimization method is promising and nanobodies with similar affinities to experimentally determined nanobodies can be detected. The interaction of non-CDR residues and MDM4 in mutant VH9 is improved. The binding surface area, calculated by using Cocomaps, of the nanobody with MDM4 is increased from 792.2 to 893.55 Å2 in the mutant structure [37]. The affinities of both nanobodies for MDM4 are in the nM range. Pairwise interacting residues of the WT and mutant nanobody are compared in Fig. 9. Increased interactions within the complex cause an increase in the binding affinity.
Fig. 9.

2D plot of MDM4-CDR residue interactions for a WT VH9 and b mutant VH9. Nanobody CDR residues are shown in stick representation
A diagnostic assay, employing the smallest antigen-binding Nb474, was prepared to detect T. congolense infections in animals. This nanobody specifically recognizes T. congolense fructose-1,6-bisphosphate aldolase. The affinity of this specific interaction is experimentally found to be 73.83 pM, indicating very tight binding. In our SMD simulations, we calculated this interaction as 31.22 pM [35]. The mutant Nb474 is generated by mutating its hypervariable residues according to the optimization scheme introduced in Fig. 3. The affinity of mutant Nb474 for TcoAld is measured as 634.5 pM. Both WT and the mutant Nb474 have affinities in the same magnitude of order.
The contacting residues in Fig. 10 and the SMD plots in Fig. 7 show that similar binding patterns and affinities can be achieved by using this optimization strategy. The contacting residues on TcAldolase with WT and mutant nanobodies shown in Fig. 10 are similar. The TcAldolase-nanobody interface area is calculated by using Cocomaps [37], 820.75 Å2 and 915.55 Å2 for WT and mutant, respectively. Both affinities of mutant and WT nanobodies are in the pM range, and the interactions of WT Nb474 are preserved in the mutant case.
Fig. 10.

2D plot of TcAldolase-CDR residue interactions for a WT Nb474 and b mutant Nb474. Nanobody CDR residues are shown in stick representation
The reported nanobody for human lysozyme inhibits fibril formation by preventing the unfolding and structural reorganization of the α-domain. The studies provide evidence that nanobodies can be used as therapeutic reagents for protein misfolding diseases. The suggested nanobody has an affinity of 460 nM towards HuL, which is obtained as 0.15 nM in our SMD simulations [36]. The proposed mutant according to the optimization method shows an affinity in the low pM range, 1.48 pM.
The nanobody generated by mutating its hypervariable CDR residues has higher affinity towards HuL. Figure 11 shows that the mutant nanobody has more contact points with HuL. The binding surface is increased from 662.95 to 691.6 Å2 upon mutation. We can conclude that tighter binding nanobodies can be proposed by this optimization method.
Fig. 11.

2D plot of HuL-CDR residue interactions for a WT cAbHuL5 and b mutant cAbHuL5. Nanobody CDR residues are shown in stick representation
Nanobodies are currently emerging, powerful biologics used for treatment and diagnostic purposes. Their small size, increased stability and solubility, and large binding regions make nanobodies great therapeutic reagent candidates. Experimentally screening the best binder by phage display-based methods is time-consuming; therefore, computational tools, such as MD and SMD, can be used instead to design a nanobody for a specific target and characterize its binding properties.
In this study, an optimization method is proposed by selecting the evolutionarily variable residues to design conformationally selective nanobodies. Nanobody-protein interaction is improved by applying point mutations on hypervariable residues and selecting energetically, evolutionarily favorable mutants. Affinities of the mutant and WT nanobodies can be compared by SMD. The unbinding direction can be optimized by comparing the SMD results with the actual experimental results.
This study reveals that mutations on hypervariable CDR residues improve the binding affinity of nanobodies and can detect experimentally proven best binders. The currently proposed method can be extended for wider applications, and many tighter binding nanobodies can be designed for specific targets within the nM-pM affinity range. We conclude that optimizing antigen-nanobody binding properties with our proposed computational method will generate a reliable source of nanobody candidates for specific targets, and it should be useful in a vast variety of immunotherapeutic applications.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P.: B cells and antibodies. In: Molecular biology of the cell. 4th edition. Garland Science, (2002)
- 2.Kremer L, Garcia-Sanz JA. Is the recent burst of therapeutic anti-tumor antibodies the tip of an iceberg? Front. Immunol. 2018;9:442. doi: 10.3389/fimmu.2018.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drabek D, Janssens R, de Boer E, Rademaker R, Kloess J, Skehel J, Grosveld F. Expression cloning and production of human heavy-chain-only antibodies from murine transgenic plasma cells. Front. Immunol. 2016;7:619. doi: 10.3389/fimmu.2016.00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiller KE, Tessier PM. Advances in antibody design. Annu. Rev. Biomed. Eng. 2015;17:191–216. doi: 10.1146/annurev-bioeng-071114-040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trevino SR, Scholtz JM, Pace CN. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007;366(2):449–460. doi: 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon C, Baier AS, Pascolutti R, Wegrecki M, Zheng S, Ong JX, Erlandson SC, Hilger D, Rasmussen SGF, Ring AM, Manglik A, Kruse AC. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018;25(3):289–296. doi: 10.1038/s41594-018-0028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norman, R.A., Ambrosetti, F., Bonvin, A.M., Colwell, L.J., Kelm, S., Kumar, S., Krawczyk, K.: Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief. Bioinform. (2019) [DOI] [PMC free article] [PubMed]
- 8.Uchański T, Zögg T, Yin J, Yuan D, Wohlkönig A, Fischer B, Rosenbaum DM, Kobilka BK, Pardon E, Steyaert J. An improved yeast surface display platform for the screening of nanobody immune libraries. Sci. Rep. 2019;9(1):382. doi: 10.1038/s41598-018-37212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hassanzadeh-Ghassabeh G, Devoogdt N, De Pauw P, Vincke C, Muyldermans S. Nanobodies and their potential applications. Nanomedicine. 2013;8(6):1013–1026. doi: 10.2217/nnm.13.86. [DOI] [PubMed] [Google Scholar]
- 10.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017;12(2):255. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefranc, M.P.: IMGT unique numbering for the variable (V), constant (C), and groove (G) domains of IG, TR, MH, IgSF, and MhSF. Cold Spring Harb. Protoc. 2011(6), 633–642 (2011). 10.1101/pdb.ip85 [DOI] [PubMed]
- 13.Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front. Immunol. 2017;8:1603. doi: 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muyldermans S. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 15.Li T, Pantazes RJ, Maranas CD. OptMAVEn–a new framework for the de novo design of antibody variable region models targeting specific antigen epitopes. PLoS One. 2014;9(8):e105954. doi: 10.1371/journal.pone.0105954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barderas, R., Desmet, J., Timmerman, P., Meloen, R., Casal, J.I.: Affinity maturation of antibodies assisted by in silico modeling. Proc. Natl. Acad. Sci. U.S.A. 105(26), 9029–9034 (2008) [DOI] [PMC free article] [PubMed]
- 17.Mahajan SP, Meksiriporn B, Waraho-Zhmayev D, Weyant KB, Kocer I, Butler DC, Messer A, Escobedo FA, DeLisa MP. Computational affinity maturation of camelid single-domain intrabodies against the nonamyloid component of alpha-synuclein. Sci. Rep. 2018;8(1):17611. doi: 10.1038/s41598-018-35464-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuo J, Li J, Zhang R, Xu L, Chen H, Jia X, Su Z, Zhao L, Huang X, Xie W. Institute collection and analysis of nanobodies (iCAN): a comprehensive database and analysis platform for nanobodies. BMC Genomics. 2017;18(1):797. doi: 10.1186/s12864-017-4204-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilton, E.E., Opyr, M.P., Kailasam, S., Kothe, R.F., Wieden, H.-J.: sdAb-DB: the single domain antibody database. ACS Synth. Biol. 7(11), 2480–2484 (2018). 10.1021/acssynbio.8b00407 [DOI] [PubMed]
- 20.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassmann T, Frings O, Sonnhammer EL. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 2008;37(3):858–865. doi: 10.1093/nar/gkn1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 23.Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J.: Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7(1) (2011). [DOI] [PMC free article] [PubMed]
- 24.Okonechnikov K, Golosova O, Fursov M, Team U. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28(8):1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- 25.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 27.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr Opin Struc Biol. 2001;11(2):224–230. doi: 10.1016/S0959-440X(00)00194-9. [DOI] [PubMed] [Google Scholar]
- 29.Kullback S, Leibler RA. On information and sufficiency. Ann. Math. Statist. 1951;22(1):79–86. doi: 10.1214/aoms/1177729694. [DOI] [Google Scholar]
- 30.Vuong QV, Nguyen TT, Li MS. A new method for navigating optimal direction for pulling ligand from binding pocket: application to ranking binding affinity by steered molecular dynamics. J. Chem. Inf. Model. 2015;55(12):2731–2738. doi: 10.1021/acs.jcim.5b00386. [DOI] [PubMed] [Google Scholar]
- 31.Truong DT, Li MS. Probing the binding affinity by Jarzynski’s nonequilibrium binding free energy and rupture time. J. Phys. Chem. B. 2018;122(17):4693–4699. doi: 10.1021/acs.jpcb.8b02137. [DOI] [PubMed] [Google Scholar]
- 32.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer. 2013;13(2):83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 2016;11:617–644. doi: 10.1146/annurev-pathol-012414-040349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu GW, Vaysburd M, Allen MD, Settanni G, Fersht AR. Structure of human MDM4 N-terminal domain bound to a single-domain antibody. J. Mol. Biol. 2009;385(5):1578–1589. doi: 10.1016/j.jmb.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 35.Pinto, J., Odongo, S., Lee, F., Gaspariunaite, V., Muyldermans, S., Magez, S., Sterckx, Y.G.-J.: Structural basis for the high specificity of a Trypanosoma congolense immunoassay targeting glycosomal aldolase. PLoS Negl. Trop. Dis. 11(9), e0005932 (2017) [DOI] [PMC free article] [PubMed]
- 36.De Genst E, Chan P-H, Pardon E, Hsu S-TD, Kumita JR, Christodoulou J, Menzer L, Chirgadze DY, Robinson CV, Muyldermans S. A nanobody binding to non-amyloidogenic regions of the protein human lysozyme enhances partial unfolding but inhibits amyloid fibril formation. J. Phys. Chem. B. 2013;117(42):13245–13258. doi: 10.1021/jp403425z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R. COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics. 2011;27(20):2915–2916. doi: 10.1093/bioinformatics/btr484. [DOI] [PubMed] [Google Scholar]