

Abstract
Modulating signaling pathways including Wnt and Hippo can induce cardiomyocyte proliferation in vivo. Applying these signaling modulators to human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) in vitro can expand CMs modestly (< 5-fold). Here, we demonstrate massive expansion of hiPSC-CMs in vitro (i.e. 100 to 250-fold) by glycogen synthase kinase-3β (GSK-3β) inhibition using CHIR99021 and concurrent removal of cell-cell contact. We show GSK-3β inhibition suppresses CM maturation while contact removal prevents CMs from cell cycle exit. Remarkably, contact removal enabled 10- to 25-times greater expansion beyond GSK-3β inhibition alone. Mechanistically, persistent CM proliferation required both LEF/TCF activity and AKT phosphorylation but was independent from Yes associated protein (YAP) signaling. Engineered heart tissues from expanded hiPSC-CMs showed comparable contractility to those from unexpanded hiPSC-CMs, demonstrating uncompromised cellular functionality after expansion. In summary, we uncovered a molecular interplay that enables massive hiPSC-CMs expansion for large-scale drug screening and tissue engineering applications.
Keywords: cardiomyocytes, proliferation, expansion, maturation, induced pluripotent stem cells, Wnt signaling, GSK3
Graphical Abstract
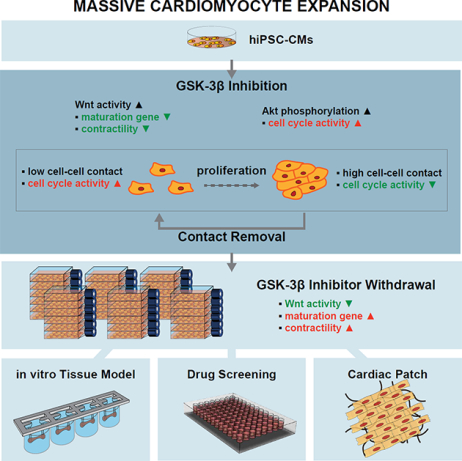
eTOC:
Deriving large number of hiPSC-cardiomyocytes would be beneficial for large-scale tissue engineering and drug screening applications. Buikema et al. show that GSK3β inhibition combined with removal of cell-cell contact enables massive expansion of hiPSC-cardiomyocytes with comparable function to non-expanded cells.
Introduction
Adult mammalian heart has limited capacity for regeneration. Cardiac injuries such as myocardial infarction lead to significant cardiomyocyte (CM) loss and subsequent heart failure with significant morbidity and mortality. Cell-based therapeutic approaches such as injection of stem cell-derived cells or transplantation of engineered cardiac tissue patches have shown promise to re-muscularize the damaged myocardium and enhance cardiac functions (Chong et al., 2014; Grego-Bessa et al., 2007; Liu et al., 2018; Ogle et al., 2016; Senyo et al., 2014; Shiba et al., 2016). Since the myocardium is densely packed with CMs, cell-based therapy requires introductions of billions of human CMs (Chong et al., 2014; Liu et al., 2018). In principle, pluripotent stem cells provide an unlimited supply of human CMs, however, in practice the generation of therapeutically relevant number of human CMs still remains an extremely labor-and time-intensive process, despite robust and highly efficient directed differentiation protocols.
To overcome this hurdle, several studies have focused on identifying molecules for promoting proliferation of differentiated CMs from pluripotent stem cells (PSCs) (Mills et al., 2019; Sharma et al., 2018; Titmarsh et al., 2016; Uosaki et al., 2013). However, when these molecules were applied to induce expansion of human PSC-derived CMs in vitro, the extent of proliferation has generally been modest (3- to 5-fold), which limits the use of these cells for cell-based therapeutic applications.
This modest extent of proliferation in vitro may result from contact inhibition of proliferation, which is a natural regulatory process of normal tissue morphogenesis, homeostasis, and regeneration. Contact inhibition of proliferation has been well-described in many cell types other than cardiomyocytes including epithelial cells and endothelial cells (Grazia Lampugnani et al., 2003; Kim and Asthagiri, 2011; Puliafito et al., 2012), where cell proliferation is cell-density dependent. When human induced pluripotent stem cells (hiPSCs) undergo directed cardiac specification and differentiation in vitro, the differentiated CMs become fully confluent in a dish. It is in this densely-packed cardiomyocytes condition that screening of mitogen candidates have been performed (Mills et al., 2017, 2019; Sharma et al., 2015; Titmarsh et al., 2016). Hence, we asked whether the removal of contact inhibition would further promote hiPSC-CM proliferation beyond that achieved in the presence of mitogen. In fact, our previous studiy on compensation of experimentally-ablated CM loss in the fetal heart by residual unablated CMs and another study on targeted disruption of cell-cell contacts in the mouse heart show evidence of cell cycle re-entry and enhanced CM proliferation in vivo (Li et al., 2015; Sturzu et al., 2015). Therefore, we hypothesize that combined inhibition of cell-cell contacts and treatment with CM mitogen would enable continuous CM proliferation and expansion.
Here, we demonstrate the ability to massively expand functional hiPSC-CMs by continuous treatment of GSK-3ȕ inhibitor CHIR-99021and concomitant removal of cell-cell contact. We chose CHIR-99021 (CHIR) as the hiPSC-CM mitogen because several studies have previously demonstrated that it was the most potent CM mitogen in vitro (Mills et al., 2017, 2019; Sharma et al., 2018; Titmarsh et al., 2016). We show that hiPSC-CMs exhibit contact-mediated inhibition of proliferation, indicating that the removal of cell-cell contacts is essential to enable the maintenance of their proliferative capacity through continuous passaging. We show that the withdrawal of CHIR or treatment with C59, a small molecule Wnt antagonist, result in rapid cell-cycle exit and restoration of mature contractile, electrophysiological, and cellular characteristics. Further mechanistic studies identified the role of canonical Wnt signal via LEF/TCF to delay hiPSC-CM maturation, and the stimulatory role of AKT T308 and HSP27 phosphorylation in enhancing hiPSC-CM proliferation by increasing cell cycle activity. Lastly, we demonstrate that the expanded hiPSC-CMs exhibit fully differentiated and contractile phenotype as the unexpanded hiPSC-CMs and are therefore an excellent cell source for the mass production of engineered heart tissues. Together, these studies reveal a robust method, the mechanistic explanation, and a proof-of-principle demonstration of a translational approach to achieve massive hiPSC-CM expansion (>1 billion) needed for future cardiac regenerative strategies in patients with cardiomyopathy or congenital heart defects.
Results
Glycogen synthase kinase-3β (GSK-3β) inhibition promotes hiPSC-CM proliferation in a cell-density dependent manner through direct cell-cell contact
Previous studies have shown CHIR-mediated GSK-3β inhibition leads to cardiomyocyte proliferation in mouse fetal heart, embryonic stem cell-derived cardiomyocytes (Buikema et al., 2013a, 2013b) and hiPSC-CM (Mills et al., 2017; Sharma et al., 2018). Despite the known role of GSK-3β inhibitor as a mitogen, the extent of cardiomyocyte proliferation has been reported to be modest (< 3- to 5-fold increase in total cell number) (Mills et al., 2017; Sharma et al., 2018) and were all performed in densely-plated cell cultures. We investigated whether disruption of cell-cell contact by low-density passaging would further promote hiPSC-CM proliferation. iPSC lines from healthy individuals were differentiated into cardiomyocytes using small molecule-based Wnt-modulated protocol (Figure 1A) (Lian et al., 2012, 2013). The differentiating cells displayed stage-specific gene expression for pluripotent stem cells (NANOG), mesoderm (MESP1) and cardiac progenitor cells (Isl1) before committing to the cardiomyogenic lineage (Figure S1A–B). When hiPSC-CMs were passaged at low-density, the total cell number increased by 8-, 8-, and 4-folds at each passage, resulting overall in a 241-fold increase in hiPSC-CM number (Figure 1B). In contrast, the fold-change in cell number in densely passaged group and w/o passaging group remained comparable, and at the end of three passages or 3 weeks, the number increase was ~10-fold (Figure 1B). The CM number increase with low-density passage was supported by their increase in cell cycle activity (Figure 1C–F) compared with densely passaged or un-passaged CMs assessed by cell cycle marker Ki67 (Figure 1C–D). Moreover, fraction of mitotic CMs was at 1% without passaging and remained at 1% upon passaging at high cell density, but it was only when CMs were passaged at low-density that led to significant increase in mitotic CM fraction up to 10% (Figure 1E–F). This indicates that the maintenance of low cell density is necessary to sustain active cell cycle activity in CMs. Taken together, we demonstrate that massive expansion of hiPSC-CMs can be achieved through sparse passaging in the presence of CHIR.
Figure 1:
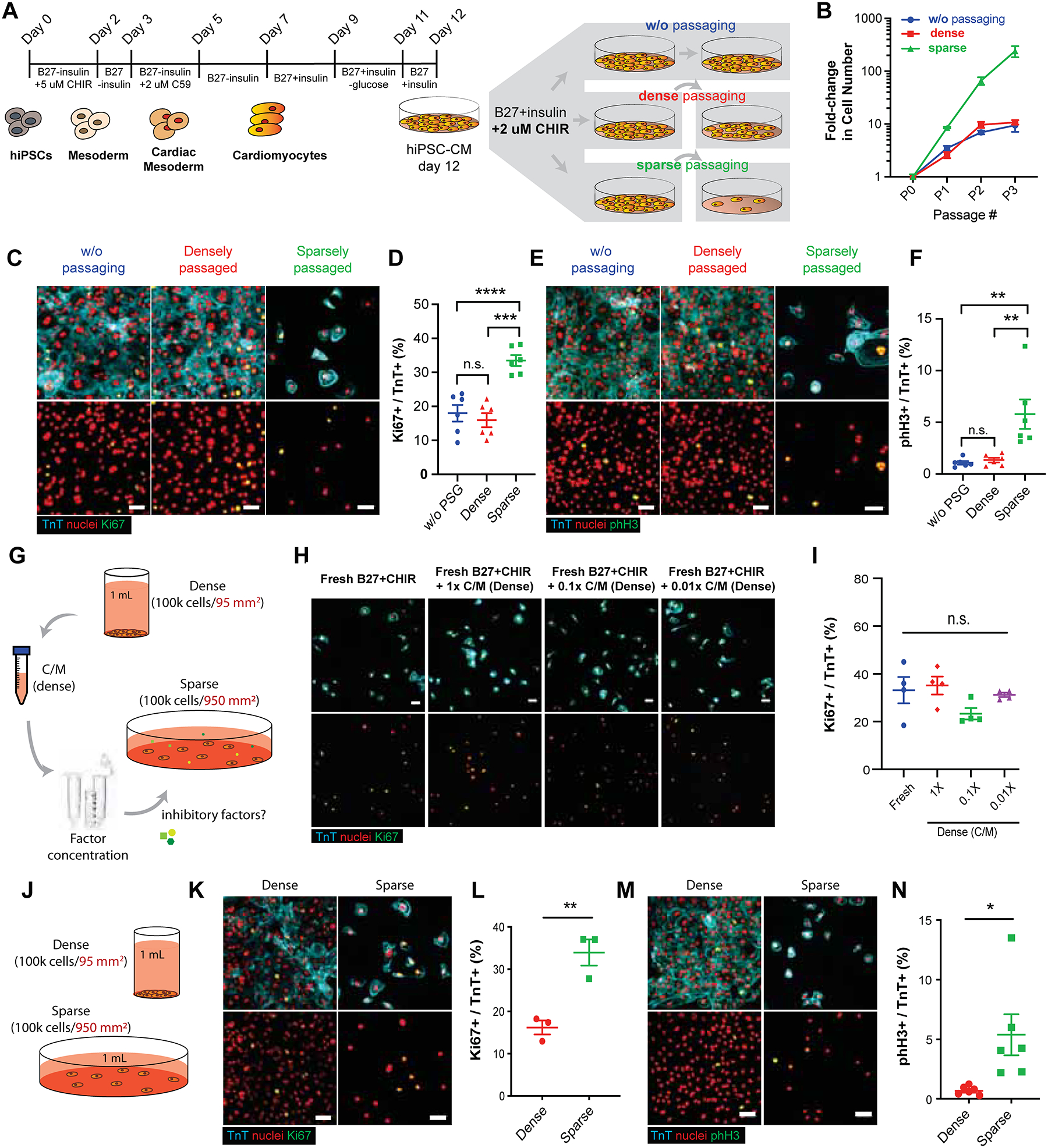
GSK-3β inhibition promotes hiPSC-CM proliferation in a cell-density dependent manner. (A) Schematic of Wnt-modulated directed cardiac differentiation and subsequent expansion. Day 12, hiPSC-CMs were plated densely (~100,000 cells/cm2), sparsely (~10,000 cells/cm2) for serial passaging or cultured without passaging (~500,000–1M cells/cm2) in the presence of CHIR99021 (CHIR) (2.0 μM). (B) Expansion of hiPSC-CMs represented as fold increase over day 12 for each passage (P) number. (C) Immunofluorescence images and (D) the quantification of proliferation marker Ki67 (green), cardiac troponin T (TnT) (cyan) and nuclei (red) in hiPSC-CMs. (E) Immunofluorescence images and (F) the quantification of mitotic cardiomyocytes assessed by phospho-histone H3 (pHH3) (green), TnT (cyan) and nuclei (red). (B, D, F) Data are means ± SEM. *p<0.05, ****p<0.001, n.s. p>0.05 by One-way ANOVA with Tukey’s post hoc multiple comparisons test. (G) Illustration of conditioned media collection of densely cultured cells, subsequent factor concentration and application to sparsely cultured hiPSC-CMs. (H) Immunofluorescence images of Ki67 (green), TnT (cyan), and nuclei (red). (I) Quantification of the percentages of Ki67+/TnT+ cells in H. Data are means ± SEM. n.s. p>0.05 by One-way ANOVA Dunnett’s post hoc multiple comparisons to control “no C/M” group (blue circle). (J) Schematic of culturing the fixed number of hiPSC-CMs in dense or sparse condition. (K, M) Immunofluorescence and (L, N) the quantification of the percent (K, L) proliferative cardiomyocytes (Ki67+/TnT+) or (M, N) mitotic cardiomyocytes (pHH3+/TnT+). Data are means ± SEM. *p<0.05, **p<0.01 by unpaired t-test. Supplementary Table 1 specifies the replicates per experiment.
We then asked whether hiPSC-CMs at high cell density inhibit CM proliferation and induce cell cycle exit by their secretion of anti-proliferative paracrine factors (Figure 1G). We did not find any significant changes in the proportion of Ki67-expressing hiPSC-CMs between different conditioned media treatment groups (Figure 1H–I). Next, we examined whether the presence of cell-cell contact induces cell cycle exit in hiPSC-CMs (Figure 1J). Interestingly, densely plated hiPSC-CMs significantly down-regulated Ki67 and pHH3, suggesting that cell-cell contact inhibits cell cycle activity in hiPSC-CMs (Figure 1K–N). Taken together, we demonstrate here that cell-cell contact is a major barrier to hiPSC-CM proliferation and that prevention of cell-cell contact is critical to maintain CHIR-induced cardiomyocyte proliferation.
Glycogen synthase kinase-3β inhibition and low-density passaging result in massive expansion of beating hiPSC-CMs
To determine the maximum extent of hiPSC-CMs expansion through combined GSK-3β inhibition and low-density passaging, we treated hiPSC-CMs continuously with or without 2.0 μM CHIR (CHIR vs. CTR), and serially passaged at low-density (Figure 2A). Remarkably, in the presence of CHIR, the beating hiPSC-CMs continue to divide without notable cell death (Figure S1C) and can be passaged up to 4–5 times over 60+ days (Figure 2B–D). Control dimethyl-sulfoxide (DMSO)-treated hiPSC-CMs cease to proliferate after the first or second passage (Figure 2B–D). Interestingly, despite the capacity for continuous cell division in the presence of CHIR, these hiPSC-CMs remain capable of spontaneous beating (Supplementary Movie 1). Typically, ~2 million hiPSC-CMs (e.g. 1 well in a 6-well plate) at day 12 of differentiation generate 300–900 million hiPSC-CMs (Figure 2E). In addition, the expansion capacity was reproducible in CMs from 4 different iPSC lines (CVI-111, CVI 113, CVI 202 and CVI 273) (Figure 2F). Moreover, no significant difference in expansion capacity was observed among the same batch of day 12 CVI-111 hiPSC-CMs that were either cryopreserved or used directly (Figure S1D). In total, continuous treatment of day 12 hiPSC-CMs with CHIR for >30 days resulted in 100 to 250-fold increase in total CM number when compared to CTR hiPSC-CMs at similar time points (Figure 2G–H). Furthermore, CHIR-treatment appeared to maintain, if not slightly enrich, CM purity as opposed to control DMSO-treated hiPSC-CMs which decreased CM purity with each passage (Figure 2I–J), most likely due to over-growth of non-myocytes. To examine the progression of cell cycle activity of CHIR-treated hiPSC-CMs over multiple passaging, the percentage of troponin T (TnT) positive hiPSC-CMs that express the cell cycle marker Ki67 was assessed at each passage after treatment with CHIR or DMSO (CTR) (Figure S1E). Interestingly, while CTR hiPSC-CMs showed a rapid decline in their expression of Ki67 with each passage where only a small number of proliferative cells can be observed after passage 2 (P2), CHIR-treatment with low-density passaging resulted in significant (P = 4.42E-05) extension (up to 5 passages) of the presence of proliferating cells. Moreover, we found that CHIR-treated hiPSC-CMs indeed proceed through each stage of the cell cycle, undergo cytokinesis and remain more mono-nucleated (Figure 2K–M and S1F–I). Furthermore, our immunostaining data suggests that hiPSC-CMs disassemble their aligned sarcomeres during active mitosis, suggesting the diminution or absence of active hiPSC-CM beating during the mitotic phase of cell division (Figure 2K–M). Collectively, these data support the ability of GSK-3ȕ inhibition and contact removal via serial passaging to maintain hiPSC-CM proliferation.
Figure 2:
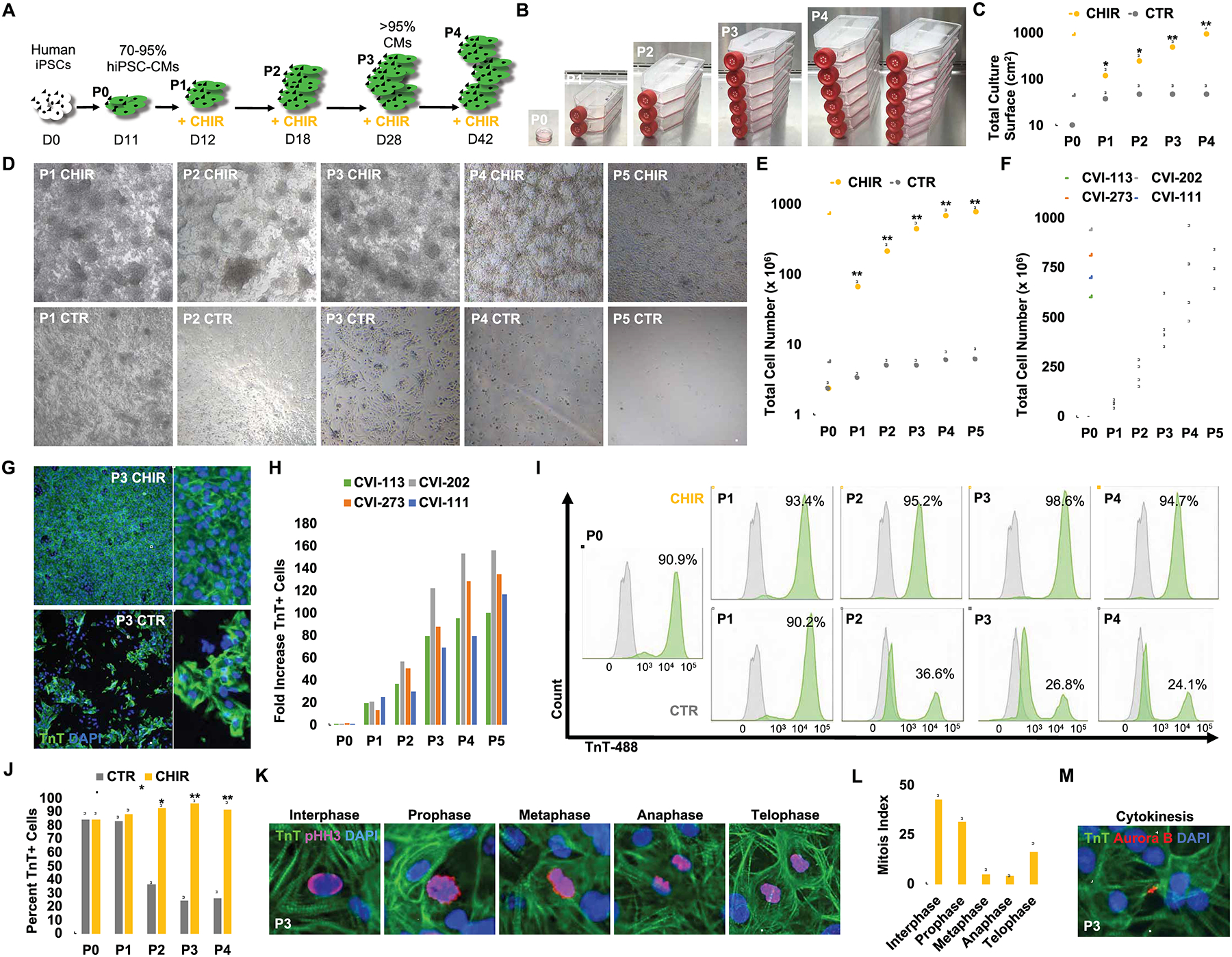
GSK-3β inhibition and long-term low-density passaging results in massive expansion of beating hiPSC-CMs. (A) Schematic timeline of hiPSC-CM expansion and passaging. (B) Representative images of hiPSC-CM flasks from initial 10 cm2 confluent dish at passage 0 (P0) to multiple T-175 cm2 cell culture flasks at end of passage 4. (C) Total surface area (cm2) coverage by hiPSC-CMs at each passage. (D) Representative bright-field images of hiPSC-CMs treated with CHIR (2.0 μM) or DMSO (CTR) prior to being passaged. Note that CHIR-treated cells reach confluence again prior to next passaging while DMSO-treated cells become progressively more sparse. Same dilution factor was applied to both treatment conditions. (E) Quantification of total cell numbers from P0 to P5. (F) Graph displaying the expansion for multiple passages (P) of low-density cultured and CHIR treated CMs derived from four different hiPSC lines. (G) Immunofluorescence analysis for TnT expression at P3 for hiPSC-CMs treated with DMSO (CTR) or CHIR. (H) Fold increase in TnT+ cells at each passage in CHIR-treated over DMSO treated cells for 4 indicated hiPSC lines. (I) Representative flow cytometry plots of TnT expression (green) and unstained controls (grey) in DMSO (CTR) and CHIR-treated cells over multiple passaging. (J) Average percentages of TnT+ cells from flow cytometric analysis in (I). (K) Representative confocal microscopy images of P3 (> day 28) CHIR-treated hiPSC-CMs at different phases of mitosis. (L) Quantification of proliferating hiPSC-CMs at distinct mitotic phases from (K). (M) Immunofluorescence image of Aurora B kinase expression in TnT+ cells undergoing cytokinesis. Scale bars represent 100μm, Bar charts represent mean±SD. *p<0.05 and **p<0.005 by unpaired t-test. Supplementary Table 1 specifies the replicates per experiment.
Single-cell phenotypic assessment of hiPSC-CMs following CHIR treatment
CM maturation is accompanied by formation of highly structured sarcomere, bi- or multi-nucleation, and cell cycle exit (Bassat et al., 2017; Bersell et al., 2009; Senyo et al., 2014; Uygur and Lee, 2016). The observed massive expansion of beating hiPSC-CMs following GSK-3β inhibition and low-density plating (Figure 2) raises the possibility that besides cell-cell contact inhibition (Figure 1), prevention of maturation may also in part explain their retained capacity to proliferate. Patterned CHIR-treated single CM that is undergoing mitotic cell division marked by pHH3, exhibited complete sarcomere disassembly (Figure 3A). Moreover, control (CTR) hiPSC-CMs demonstrated highly organized and aligned sarcomeres, while CHIR-treated hiPSC-CMs on micropatterns exhibited markedly reduced alignment and organization of sarcomeres (Figure 3A–B). Quantification of z-disc-registered sarcomeric α-actinin confirmed the observed sarcomere disorganization in CHIR-treated hiPSC-CMs (Figure 3C–D). Interestingly, when the Wnt inhibitor C59 was added to cells that had previously expanded with CHIR treatment (CHIR>C59), these cells improved their sarcomeric organization (Figure 3B–D). This supports the reversibility of GSK-3β inhibition signaling effects on hiPSC-CMs. Imaging-based assessment of contractile properties of CTR and CHIR-treated hiPSC-CMs demonstrated a decrease in force generation with CHIR treatment (Figure 3E and Supplementary Movie 2–4) that recovered upon replacing CHIR with C59 or after the withdrawal of CHIR (data not shown). Additional single-cell calcium studies of age-matched CTR- and CHIR-treated and CHIR withdrawn hiPSC-CMs showed no difference in their spontaneous action potentials, calcium transients, beating frequency nor amplitude (Figure 3F–H).
Figure 3:
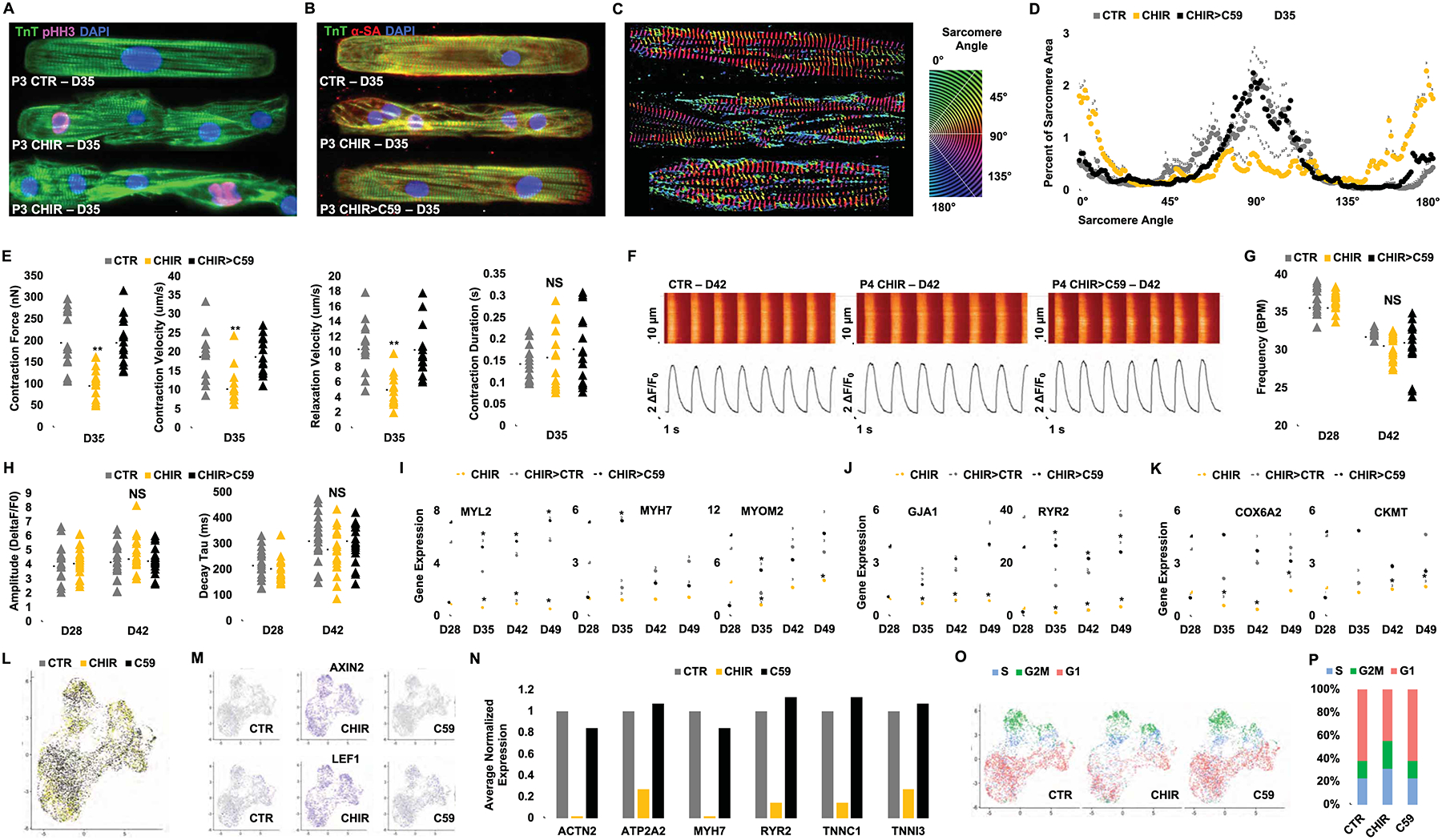
Phenotypic assessment of hiPSC-CMs following GSK-3β inhibition and removal of contact inhibition (A, B) Confocal microscopy images of P3 (D35) hiPSC-CMs treated with either DMSO (1:5000) (CTR) or CHIR (2.0 μM) since P0 and (for CHIR-treated cells in B) subsequently treated for 6 days with either continuation of CHIR (2.0 μM) or replacement of CHIR with C59 (2.0 μM) (CHIR>C59) and immunostained on micropatterned surfaces for the expression of troponin T (TnT) and sarcomeric α-actinin (α-SA) or phospho Histone H3 (pHH3). Treated hiPSC-CMs were micropatterned as single cells to adopt rectangular morphology with 7:1 aspect ratio. (C-D) Representative images of distribution of sarcomere alignment. Color codes represent angle of α-SA with respect to cellular long axis (e.g. Z-disc-registered α-SA angle = 90°) (n=10) (E) Contractility measurements in cells treated in (B). (F) Ca2+ transients (Fluo-4AM) fluorescence expressed relative to baseline [F/F0] in hiPSC-CMs at day (D) 42 in hiPSC-CMs treated with DMSO (CTR), CHIR (CHIR) or CHIR followed by C59 (CHIR>C59). Plots displaying the Ca2+ transient (G) Frequency, (H) Amplitude and Decay for each group. Fold increases in the expression of sarcomere (I), electrophysiological (J), and metabolic genes (K) at the indicated days in hiPSC-CMs treated with CHIR from D12 to D28 followed by either continued treatment with CHIR, CHIR withdraw (CHIR>CTR) or replacement of CHIR with C59 (CHIR>C59). (L) UMAP plots of day 12 hiPSC-CMs treated with DMSO (1:2500) (grey), CHIR (4.0 μM) (yellow), or C59 (4.0 μM) (black) for 24 hrs. (M) FeaturePlot of Wnt target genes. (N) Average expression of the selected mature cardiac genes normalized to CTR. (O) UMAP plots for cell cycle analysis, S-phase (S) (blue), G2/M-phase (G2M) (green) and G1-phase (red). (P) Bar graph of the percentage of cells in different cell cycle phases. Scale bars represent 100μm. Dot plots represent biological replicates and average. Bar charts represent mean. Graphs represent average±SD. *p<0.05 and **p<0.005 by unpaired t-test. NS=not significant. Supplementary Table 1 specifies the replicates per experiment.
Real-time quantitative PCR analysis revealed CHIR-treated hiPSC-CMs downregulate markers associated with cardiomyocyte maturation (MYL2, TNNI3, MYOM2), excitation (GJA1), contractility (RYR2) and metabolism (COX6A2, CKMT) compared to those treated with DMSO or C59, supporting maturation arrest phenotype at transcriptional level (Figure 3I–K).
To further characterize global transcriptional changes in individual hiPSC-CMs in response to GSK-3β inhibition, we performed single-cell RNA sequencing in day 12 hiPSC-CMs treated for 24 hours with either DMSO (CTR), CHIR (4.0 μM), or C59 (4.0 μM) (Figure 3L). We captured a total of 8,381 cells and performed 93,552 mean reads per cell resulting in a median of 1,297 genes analyzed per cell. Unsupervised analysis revealed 5 cell populations characterized as non-proliferative and proliferative ventricular CMs, non-proliferative and proliferative atrial CMs and fibroblast, present in all 3 treatment samples (Figure S2A–D). We found that Wnt target genes such as AXIN2 and LEF1 were upregulated in almost all cells treated with CHIR (Figure 3M). Furthermore, mature cardiac genes were dramatically downregulated in CHIR-treated hiPSC-CMs and conversely correlated with the up-regulation of Wnt signaling target genes (Figure 3N). Interestingly, CHIR treatment resulted in a larger subset of proliferative and immature atrial and ventricular-like hiPSC-CMs (Figure 3O–P and Figure S2E). Importantly, GSK-3β inhibition did not result in increased expression of the cardiac progenitor markers such as CKIT, ISL1 or MESP1/2 (Figure S2F), suggesting that expansion of functional hiPSC-CM with CHIR does not lead to expansion of a rare population of residual cardiac mesoderm or second-heart-field progenitor cells (Wu et al., 2008). Furthermore, expression of key cardiac transcription factors such as GATA4, TBX5, MEF2C and NKX2.5 (Ieda et al., 2010), did not alter significantly in hiPSC-CMs treated with CHIR versus CTR and C59 (Figure S2G). Overall, these data strongly support the inhibition of hiPSC-CM maturation by CHIR/GSK-3β inhibition, a phenomenon that has also been observed in second heart field-derived cardiac progenitor cells and other cell types (Sato et al., 2004; Yin et al., 2014).
YAP activity is uncoupled from cell density-dependent CHIR-induced hiPSC-CM proliferation.
The Hippo signaling pathway has been shown to regulate heart growth and Wnt signaling-mediated cardiomyocyte proliferation (Heallen et al., 2011). Hippo pathway consists of a core kinase cascade that modulates activity of transcriptional co-activator, yes-associated protein (YAP), which is one of the major nuclear effectors of Hippo pathway. Previous studies have shown that overexpression of active YAP is sufficient to induce cardiomyocyte proliferation (Bassat et al., 2017; von Gise et al., 2012; Lin et al., 2015). We examined whether YAP is responsible for density-dependent proliferation of CHIR-treated hiPSC-CMs. Immunostaining of YAP revealed that ratio of nuclear to cytoplasmic YAP increases when hiPSC-CMs are sparsely plated (Figure 4A–B). Given the positive correlation between nuclear YAP translocation and cardiomyocyte proliferation (Figure 1K–N, 4A–B), we next asked whether this increase in YAP nuclear translocation results from mechanosensing of stiffness. Since densely-packed hiPSC-CMs would sense their microenvironment to be softer than sparsely-plated hiPSC-CMs that are cultured directly over tissue culture plastic, we addressed whether substrate stiffness could alter the proliferation of hiPSC-CMs via YAP activity (Figure 4C). Interestingly, we found no difference in the fraction of Ki67+ CMs with changing substrate stiffness (Figure 4D–E), despite a significant increase in nuclear translocation of YAP when hiPSC-CMs were cultured on stiff hydrogel (60 kPa) or rigid plastic (Figure 4F–G). In line with this finding, reduced YAP nuclear translocation via treatment of verteporfin (Figure 4H–I), an inhibitor of YAP nuclear translocation, did not lead to a reduction in hiPSC-CM proliferation (Figure 4J–K). In summary, these data demonstrate an uncoupling of CHIR and cell-cell contact regulated hiPSC-CM proliferation from nuclear YAP activity.
Figure 4:
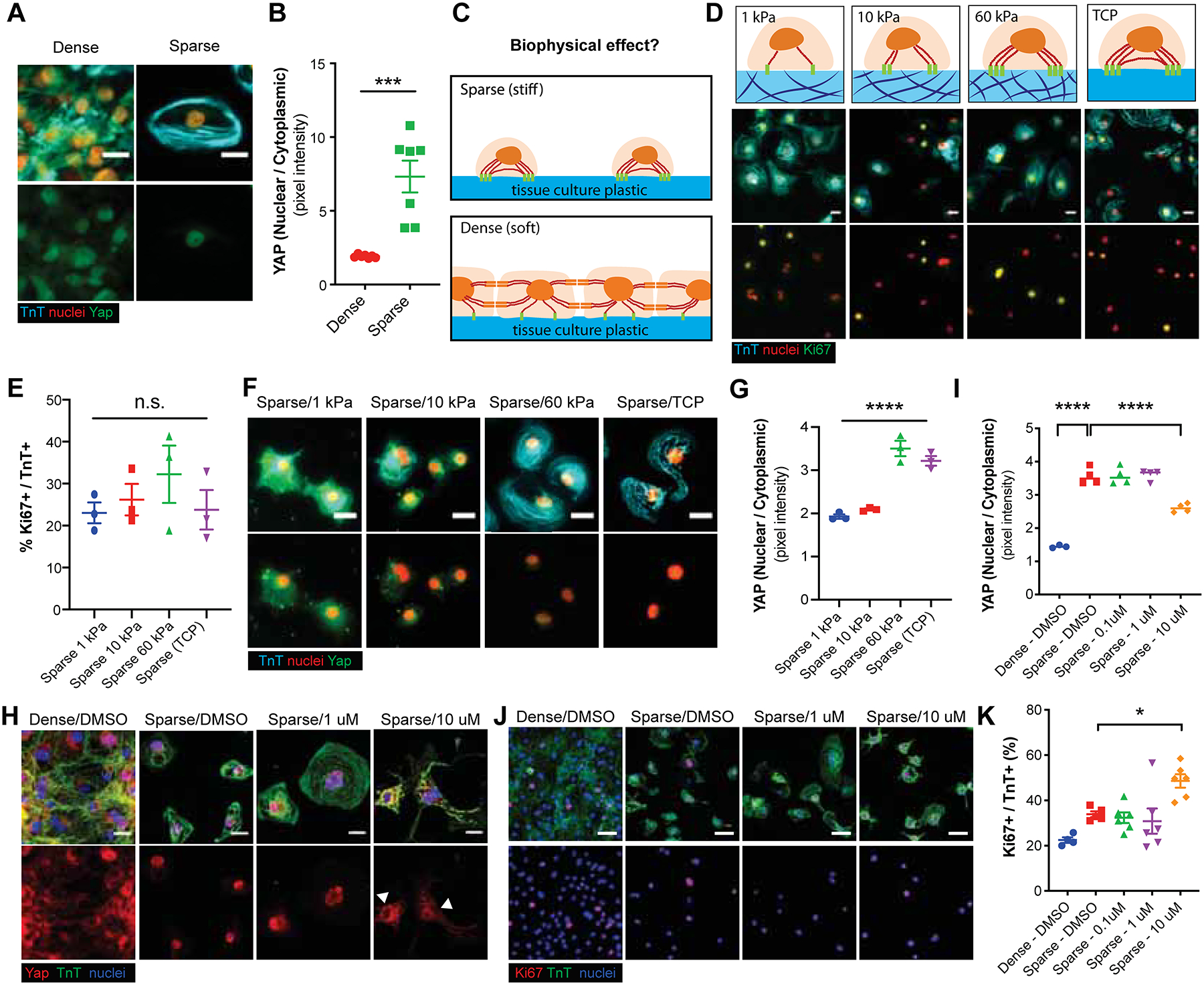
Density-dependent CHIR-induced hiPSC-CM proliferation is uncoupled from YAP activity. (A) Representative immunofluorescence images for yes-associated protein (YAP) (green), cardiac troponin T (TnT) (cyan) and nuclei (red) in dense or sparse culture conditions. (B) Quantification of nuclear to cytoplasmic YAP ratios. Data are means ± SEM. ****p<0.0001 by unpaired t test. (C) Schematic displaying potential biophysical effects on sparse and dense cell culture. (D) Immunofluorescence for Ki67 (green), TnT (cyan) and nuclei (red) in hiPSC-CMs cultured on substrates with varying stiffness (kPa) or on tissue culture plastic (TCP) (~GPa). (E) Quantification of the percent Ki67+/TnT+ cells in D. (F, G) Effect of substrate stiffness on YAP localization. (F) Representative immunofluorescence images and (G) the quantification of nuclear/cytoplasmic ratios of YAP in hiPSC-CMs. (I, H) YAP localization in hiPSC-CMs cultured in sparse or dense culture conditions with or without YAP inhibitor Verteporfin (1.0 or 10 μM). (J, K) Representative immunofluorescence images and the quantification of Ki67+ (red), TnT+ (green) cells (blue) for the same conditions as in H. Data are means ± SEM. *p<0.05. ****p<0.0001 by One-way ANOVA Dunnett’s post hoc multiple comparisons to control “Sparse-DMSO” group. Supplementary Table 1 specifies the replicates per experiment.
Mechanistic analysis of GSK-3β inhibition and low-density culture on hiPSC-CM proliferation reveals an interplay between canonical Wnt and AKT signaling
To further elucidate the underlying molecular mechanisms, we first examined whether CHIR acts downstream of Wnt ligand binding to its receptor Frizzled and co-receptor low-density lipoprotein receptor 5/6 (LRP 5/6) (Nusse and Clevers, 2017). We found that a panel of Wnt ligands (i.e. canonical Wnt agonist Wnt3A and a previously described Wnt surrogate ligand scFv-DKK1c (Janda et al., 2017; Yan et al., 2017) in combination with R-Spondin (RSPO)) robustly mediated immature hiPSC-CM expansion comparable to CHIR-treatment, suggesting CHIR acts downstream of Wnt ligand activity (Figure S3A–G).
Since GSK-3β is known to be involved in multiple cellular processes beyond canonical Wnt signaling modulation (Beurel et al., 2015) and we show here that cell contact is a potent inhibitor of hiPSC-CM proliferation, we sought to examine whether a β-catenin-independent mechanism is involved in hiPSC-CM proliferation. Interestingly, we found that while CHIR-induced activation of the TCF/LEF luciferase reporter was completely abolished in the presence of PNU74654, a specific TCF/LEF β-catenin signaling blocker (Trosset et al., 2006) (Figure 5A), the CHIR-mediated hiPSC-CM proliferation decreased only by ~50% (Figure 5B). This suggests that a β-catenin-independent mechanism contributes to roughly half of the proliferative activity observed with CHIR treatment. We confirmed, by real time quantitative PCR, the induction of Wnt signaling target gene expression (e.g. Axin2, LEF, and CCND2) following CHIR treatment in hiPSC-CMs and the ability of PNU74654 treatment to abolish this increase (Figure 5C). We also show that the expression of markers of hiPSC-CM maturation (MYL2, MYH7, MYOM1) were down-regulated by CHIR treatment and recovered fully upon the co-treatment with PNU746554 (Figure 5C). These results demonstrate that β-catenin-mediated canonical Wnt signaling prevents hiPSC-CM maturation and is responsible for roughly 50% of the observed CHIR-induced expansion of hiPSC-CMs.
Figure 5:
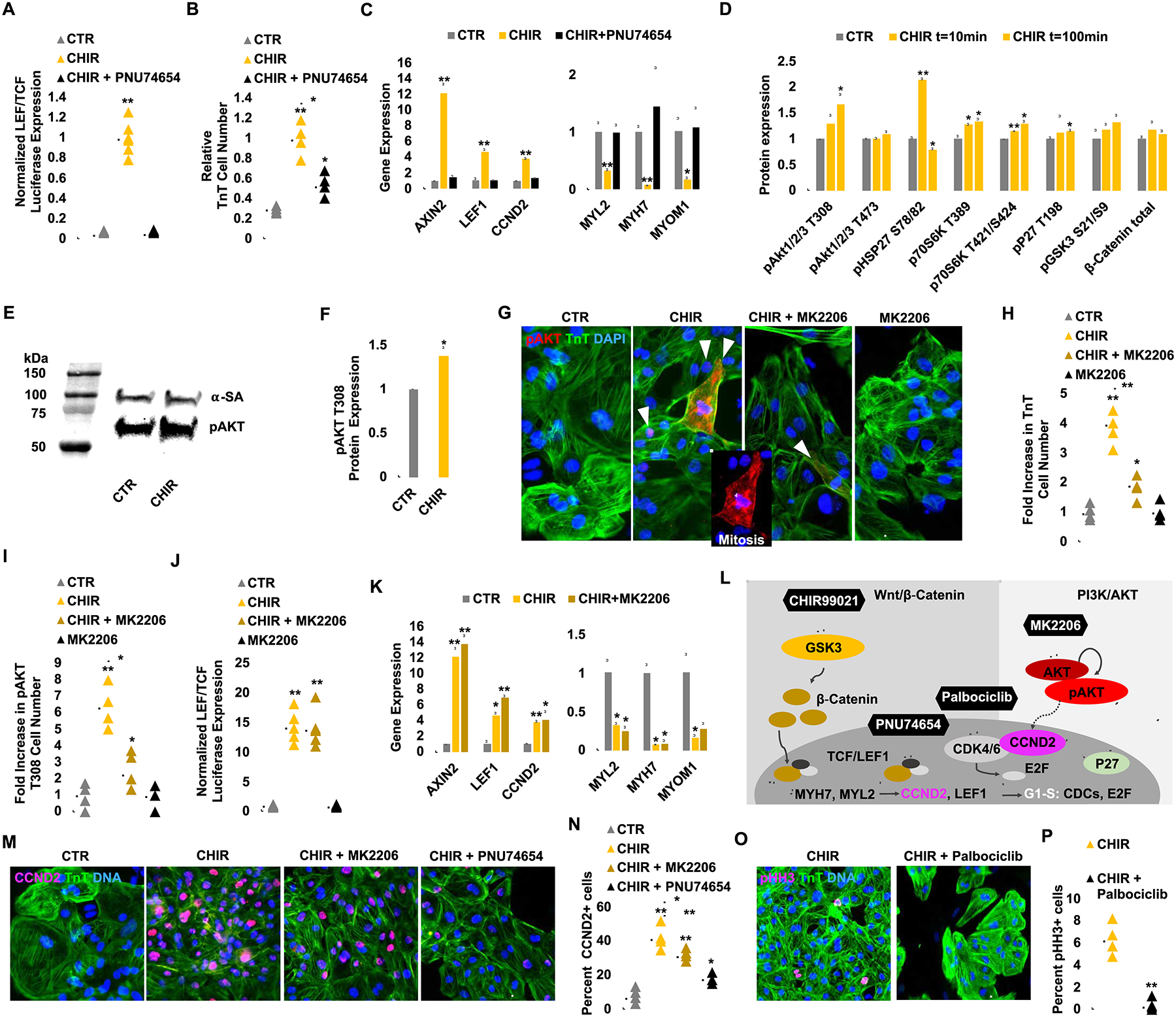
CHIR and low-density plating activate β-catenin and AKT signaling to enhance hiPSC-CM proliferation via Cyclin D2-dependent kinases and prevent maturation via repression of sarcomere gene expression. (A) TOPFlash (TCF/LEF) luciferase analysis of hiPSC-CMs treated with CTR or CHIR with or without PNU74654 for 24 hrs. (B) Fold increase in TnT+ cell number after DMSO (CTR) or CHIR (2.0 μM) treatment with or without PNU74654 (32 μM). (C) Normalized gene expression of Wnt target genes and maturation markers. Note the complete recovery of maturation gene expression when canonical Wnt signaling is abolished by PNU74654 treatment. (D) A mini-screen of 43 kinase targets after CHIR treatment demonstrated an increase in phosphorylation (p) levels at AKT, HSP27, and others. (E) Confirmation of AKT T308 phosphorylation by western blot. Control lanes were removed. (F) Quantification of pAKT protein expression level after CHIR treatment. (G) Immunofluorescence analysis for pAKT T308 expression in TnT+ (green) day 20 hiPSC-CMs cultured for 6 days with the indicated treatment. (H, I) Quantification of the number of TnT+ (H) and pAKT T308+ (I) cells in G. (J) TOPFlash luciferase analysis of hiPSC-CMs after treatment of CHIR for 24 hrs with or without MK2206 (1.0 μM). (K) Expression of Wnt target genes and maturation markers after treatment with the indicated compounds. Note that AKT signaling is not changing the Wnt dependent maturation related gene expression. (L) A schematic diagram of the inhibitory relationship between GSK3 and downstream canonical Wnt signaling and their de-repression with CHIR. The role of PNU74654 to inhibit β-catenin-TCF/LEF activity is highlighted. Dashed lines indicate a correlation between AKT-CCND2. (M) Immunofluorescence analysis for Cyclin D2 (CCND2) (pink) expression in TnT+ (green) day 20 hiPSC-CMs cultured for 6 days with the indicated compounds. Quantification of the number of CCND2+/TnT+ cells per treatment group (N). (O) Immunofluorescence analysis for phospho Histone H3 (pHH3) (pink) and TnT (green) expression at day 20 hiPSC-CMs cultured for 6 days with the indicated compounds. Quantification of the number of pHH3+/TnT+ cells per treatment group (P). Scale bars represent 100μm, Dot plots represent biological replicates and average. Bar charts represent mean±SD. *p<0.05 and **p<0.005 by unpaired t-test. Supplementary Table 1 specifies the replicates per experiment.
To identify the β-catenin-independent process that accounts for the low density plus CHIR-mediated hiPSC-CM proliferation, we screened a library of 43 kinases with known functions (Figure 5D and S4A) and found a significant upregulation of AKT1/2/3 phosphorylation at residue T308 (Figure 5D), a residue that is required for AKT activation and cell division (Liu et al., 2014). In addition, we found an increased phosphorylation of HSP27, an AKT binding protein, as well as p70S6K, a downstream AKT target (Figure 5D) (Conejo et al., 2002; Song et al., 2005). Using an antibody directed against the T308 residues of phosphorylated AKT (pAKT), we confirmed by western blotting that the active (i.e. phosphorylated) form of AKT is rapidly increased following CHIR treatment at low cell density (Figure 5E–F). Immunostaining of day 12 hiPSC-CMs revealed that T308 pAKT was abundantly expressed in the cytoplasm of mitotic hiPSC-CMs in the CHIR-treated group (Figure 5G and S4B).
Next, we assessed the contribution of T308 pAKT in hiPSC-CM proliferation by using a previously described highly selective AKT phosphorylation inhibitor MK2206 (Lindsley et al., 2007). When co-treated with MK2206, CHIR and low density-induced hiPSC-CM proliferation decreased by ~50% compared with CHIR treatment alone and pAKT expression reduced (Figure 5G–I). Furthermore, we found while MK2206 treatment inhibits AKT T308 phosphorylation (Figure 5I), it does not affect downstream ȕ-catenin signaling, as assessed by the LEF/TCF luciferase reporter (Figure 5J), nor does it alter the expression of genes associated with CM maturation or Wnt signaling (Figure 5K). These data reveal the presence of β-catenin-independent signaling that target downstream effects of GSK-3ȕ inhibition and low cell density via activation of AKT signaling to induce hiPSC-CM proliferation (Figure 5L). (PI3K)-AKT pathway is a well-known regulator of Cyclin D2 protein stabilization and cell-cycle progression (Mirzaa et al., 2014). Interestingly, the addition of MK2206 (AKT inhibitor) or PNU74654 (LEF/TCF inhibitor) reduced amounts of Cyclin D2 positive hiPSC-CMs (Figure 5MN), indicating that Cyclin D2 activity integrates the contribution by β-catenin signaling and AKT phosphorylation to overall hiPSC-CM proliferation. Previously, Type D cyclins were reported to assemble with Cyclin-dependent-kinases 4/6 (CDK4/6) and form enzymatically active holoenzyme complexes to propel cell cycle progression (Sherr et al., 2016). To address whether CDK4/6 also play an integrative role in CHIR and low cell density-mediate proliferation, CHIR-treated, sparse hiPSC-CMs were treated with palbociclib, a potent small molecular inhibitor of CDK4/6, and shown mitosis was nearly completely abrogated (Figure 5O–P). Taken together, these data demonstrate that GSK-3β inhibition and concomitant cell contact removal induce a molecular interplay to enhance LEF/TCF activity via canonical β-catenin signaling and activate AKT phosphorylation at T308 to enhance Cyclin D2 activity and promote hiPSC-CM proliferation.
Long-term GSK-3β inhibition does not preclude terminal differentiation and maturation of expanded hiPSC-CMs and their capacity to form functional cardiac tissue
Given the diverse role for GSK-3β signaling to regulate cell lineage commitment and differentiation, a prolonged exposure to CHIR raises concerns regarding possibilities of hiPSCCM phenotype conversion or oncogenic transformation that prevent them from appropriate maturation following expansion, a finding that would significantly hamper their use for tissue engineering applications or regenerative approaches. To address this, we expanded hiPSC-CMs for 4 serial passages in continuous presence of CHIR followed by its withdrawal from the culture media for several weeks. When compared to age-matched controls, previously long-term expanded cells had phenotypically recovered to normal as assessed by immunohistochemistry for α-sarcomeric actinin and real-time PCR for several mature sarcomeric and ion channel genes (Figure 6A–B). Next, as a functional proof-of-principle, we created engineered heart tissues from P4 CHIR expanded hiPSC-CMs and age-matched non-expanded CMs (Figure 6C). Mass-produced (Figure 6D) engineered heart tissues from expanded hiPSC-CMs exhibited normal sarcomere alignment and fully functional integration when applied in engineered heart tissues (Figure 6E). Moreover, mass produced engineered heart tissues from multiple hiPSC lines (CVI-111 and CVI-273) have comparable or slightly enhanced functional properties when compared to those generated from age-matched unexpanded control cells (Figure 6F and Supplementary Movie 5 and 6).
Figure 6:
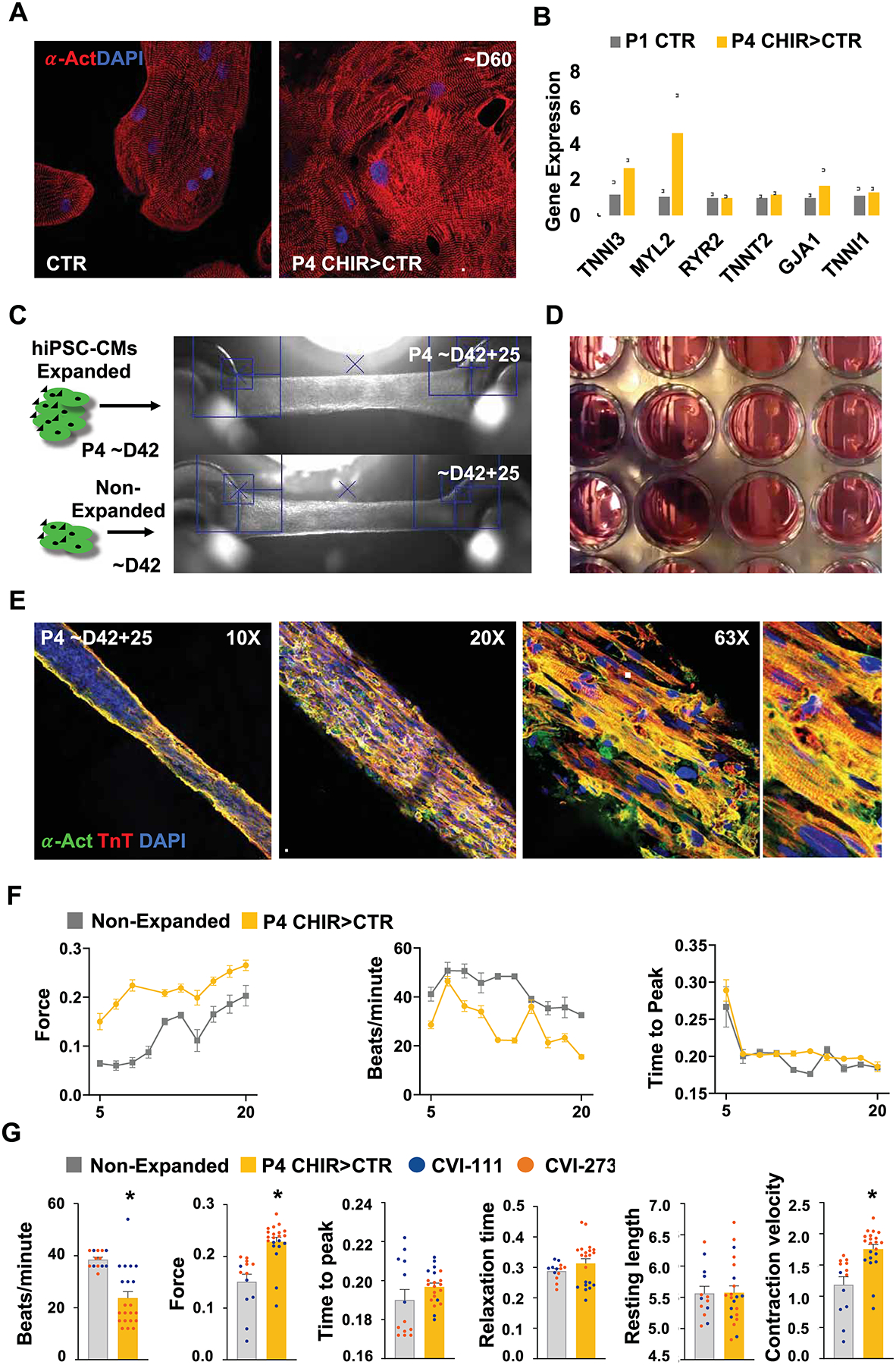
Long-term GSK3β inhibition with low-density plating does not preclude hiPSC CMs terminal differentiation and maturation in functional cardiac tissue. (A) Immunofluorescence image of alpha-actinin (α-Act) and (B) qPCR analysis of sarcomeric gene expression in passage 4 (P4) expanded vs non-expanded hiPSC-CMs at day 42 (~D42) of differentiation. (C) Brightfield images of engineered heart tissues produced from non-expanded or P4 expanded hiPSC-CMs at ~D42 and further cultured for 25-days. (D) Brightfield image of engineered heart tissues in a 24-well plate format. (E) Immunofluorescence microscopy images of alpha-actinin (α-Act) and Troponin T (TnT) in cardiac tissue generated from previously expanded hiPSC-CMs. (F) Graphs displaying development of contraction force (mN), beating rate and time to peak (T1) in heart tissues generated from previously expanded (P4 CHIR>CTR) (yellow) or non-expanded (grey) hiPSC-CMs measured at time points between day (D) 5 tot 20. (G) Plots for contraction analysis (contraction force (mN), beating rate, time to peak (T1), relaxation time (s), resting length (mm), contraction velocity (mN/s) in engineered heart tissue 14 days after formation for the indicated treatment and hiPSC lines. Scale bars represent 100μm. Bar charts represent mean±SD. Dot plots represents biological replicates and mean±SEM. *p<0.05 by unpaired t-test. Supplementary Table 1 specifies the replicates per experiment.
The proliferative response of hiPSC-CMs to GSK-3β inhibition raises the possibility that CHIR may act to stimulation cell division in proliferation-competent CMs when applied the late embryonic or postnatal heart in vivo. To investigate this, pregnant mice at E16.5 was treated with a once daily injection of CHIR or DMSO carrier (CTR) for 4 consecutive days, followed by harvesting. Interestingly, at E20.5 CHIR-treated embryos contained 1.8-fold increase in mitotic CMs when compared to the embryos treated with DMSO control (Figure S5A–C). Analysis of individual cell size revealed no difference in the size of single CMs in hearts from CHIR-administered mice compared to those treated with DMSO (Figure S5D–E). These data suggest the potential for residual hyperplasia of CMs during late gestation with GSK-3β inhibition, despite substantially attenuated proliferative effect compared to in vitro CHIR-treated hiPSC-CMs. Furthermore, GSK-3ȕ inhibition with daily CHIR injections for 6 consecutive days in postnatal mice (P10) did not result in increased myocardial growth when compared to controls (Figure S5F–G). These data support the progressive decline in proliferative capacity of embryonic CMs in vivo and the likely inhibitory effects of cell-cell contact that prevents significant CM expansion upon in vivo GSK-3β inhibition.
Discussion
A major goal of cardiac regenerative medicine is the restoration of functional cardiac tissue in damaged or defected hearts. Identification of a strategy to stimulate proliferation of preexisting CMs would allow for repairing failing hearts induced by CM loss and generating patient-specific, engineered heart tissue for regenerative therapy or drug screening applications. Here, we achieved massive expansion of functional hiPSC-CMs in vitro through concomitant GSK-3β inhibition and removal of cell-cell contact. While we and others have previously demonstrated the ability of canonical Wnt signaling activation or direct GSK-3β inhibition to transiently expand mouse and human PSC-CMs in vitro, the extent remained modest (<10-fold) (Buikema et al., 2013b; Mills et al., 2019; Sharma et al., 2018; Titmarsh et al., 2016; Uosaki et al., 2013). In this study, we show that direct cell-cell contact is a major inhibitor for continuous CM proliferation, and the modest expansion of hiPSC-CMs by GSK-3β inhibition alone can be overcome by removal of cell-cell contact following multiple passages, which can cumulatively result in a 250-fold expansion (Figure 1 and 2). Mechanistically, we demonstrate that GSK-3β inhibition suppresses CM maturation via LEF/TCF activity, while concomitant removal of cell-cell contact stimulates the cell cycle activation by AKT T308 phosphorylation (Figure 3 and 5). The combined CHIR treatment with low density hiPSC-CM culture allowed for continuous cell cycle activation and CM proliferation, independent from upregulation of YAP activity (Figure 1 and 4). These findings support the role of Wnt/ β-catenin signaling in suppressing hiPSC-CM maturation and uncover a role of cell-cell contact in inhibiting CM proliferation. As a proof-of-concept, we show the feasibility of using hiPSC-CMs expanded by the presented method for mass production of engineered heart tissues (Figure 6). We believe the ability to generate hiPSC-CMs at this scale will greatly facilitate in vitro disease modeling, high throughput drug screening and in vivo tissue engineering applications. In summary, we demonstrate massive expansion of human cardiomyocytes in vitro by simultaneously “removing brakes and pushing accelerators” (He and Zhou, 2017).
Our finding here complements the recent studies demonstrating the generation of a large number of cardiac cells for regenerative applications with noted differences. First, this study reveals an important finding that mitotic cardiomyocytes quickly lose proliferation capacity via juxtacrine signaling (Figure 1), highlighting the importance of removal of cell-cell contact. Contact inhibition of cell proliferation is an essential regulatory process to control tissue growth during embryonic development and tissue homeostasis, and is known to be dysregulated in uncontrolled tumor growth (Kim et al., 2011). Historically for continuous cell growth upon reaching high cell confluence, reducing cell density and removing contact inhibition have been standard in vitro cell culture procedure, also known as passaging. However, passaging of hiPSC-CMs has not been possible because passaging alone leads to expanding only nonmyocytes (Figure 2F and 2H). We show a remarkable role of CHIR in delaying hiPSC-CM maturation and widening their mitotic window to enable continuous CM passaging and expansion (Figure 2 and 3). In addition, this study represents a unique strategy to generate a massive amount of CMs from differentiated CMs. Earlier studies have shown to produce cardiovascular progenitor cells using growth factor combinations during hiPSC differentiation or small molecules plus overexpression of reprogramming factor for direct lineage conversion into cardiovascular progenitors (Birket et al., 2015; Zhang et al., 2016). Given the mix population of the differentiated progeny of multipotent cardiovascular progenitors, this strategy to generate a massive amount of pure hiPSC-CMs provides a unique opportunity. For example, ~10 billion hiPSC-CMs (the total estimated number of CMs in an adult heart) with >95% purity can be prepared from two 6-wells plate of day 12 hiPSC-CMs. Mechanistically, none of previous studies that demonstrated the ability of GSK-3β inhibition and Wnt signaling to induce CM division (Buikema et al., 2013b; Heallen et al., 2011; Kerkela et al., 2008; Titmarsh et al., 2016; Tseng et al., 2006; Uosaki et al., 2013), has reported that canonical Wnt signaling activation via GSK-3β inhibition prevents CM maturation and extends the proliferative window of immature CMs (Figure 2 and 5). Furthermore, we report CHIR/low density-induced hiPSC-CM proliferation is independent from YAP activity (Figure 4), which was previously shown to be sufficient for promoting cardiomyocyte proliferation mediated by β-catenin (Heallen et al., 2011; Mills et al., 2017). Since β-catenin was a required mediator of Hippo signaling to induce cardiomyocyte proliferation and cardiomegaly (Heallen et al., 2011), CHIR treatment may have overwritten the effect of YAP modulation, thereby revealing a YAP-independent effect of contact inhibition. Further study is being actively conducted to further elucidate the underlying molecular mechanisms.
One concern associated with massively expanding CMs is the possibility of their oncogenic transformation. We demonstrate that after the withdrawal of CHIR, hiPSC-CMs can mature normally and display a similar degree of sarcomere organization, electrophysiological response, and force generation as control hiPSC-CMs that were withheld from GSK-3β inhibition (Figure 3 and Figure 6). Gene expression analysis showed an increase in most sarcomere and ion-channel gene expression upon withdrawal of GSK-3β inhibition (Figure 3). Furthermore, we observed that proliferating hiPSC-CMs are mostly mononucleated (Figure 2 and 3) and that after withdrawal of CHIR, the number of binucleated CMs increased while the fraction of cell-cycle-entered hiPSC-CMs decreased (Figure S1H–I). Our single-cell RNA sequencing data also supports the maintenance of the immature state of hiPSC-CMs by Wnt signaling pathway activation as a mean to extend their proliferative window, leading to a moderate level of increase in the expression of cell cycle proteins compared with untreated hiPSC-CMs (Figure 3 and 5 and S2). Taken together, this confirms that GSK-3β inhibition in immature hiPSC-CMs serves to increase proliferation by arresting these cells from their normal maturation process and not by driving these beating CMs into an alternative fate that is more proliferative, as with overexpression of cell cycle genes, which should minimize the potential for oncogenic transformation.
Finally, one of the most promising therapeutic strategies to treat ischemic heart failure is to generate new CMs in an injured myocardium. Our in vivo studies indicate CHIR treatment during late gestation results in significantly more mitotic CMs, while this effect was not found 10 days after birth (Figure S5). This indicates that GSK-3β inhibition alone is not sufficient for cardiac regeneration in the postnatal and adult phases. Nevertheless, these findings point to the potential for residual hyperplasia of CMs during late gestation with Wnt stimulation as a therapy for certain forms of congenital heart disease such as hypoplastic left heart syndrome since a recent in-depth study indicates dysregulation of Wnt signaling as one of the contributors to impaired growth (Liu et al., 2017).
Study Limitations
Some limitations should be considered when interpreting the results of this study. First, while we observed a strong expansion capacity of day 12 hiPSC-CMs, this capacity is not unlimited. This suggests that additional factors contribute to the proliferative decline of hiPSCCM with maturation. In line with this finding we found that late gestational but not postnatal administration of CHIR is able to increase in vivo cardiomyocyte proliferation. Second, our mechanics studies focused on the inhibition of GSK-3β signaling to stimulate hiPSC-CM proliferation. While we identified the powerful negative effect of cell-cell contact on hiPSC-CM proliferation, the precise molecular mechanism of this effect remains unknown. Whether the engagement of cell surface molecules between two adjacent cells leads to a change in downstream signaling event that suppresses hiPSC-CM proliferation will require further investigation.
Summary
We demonstrated a strategy for the massive expansion of beating hiPSC-CMs with simultaneous GSK-3β inhibition and low cell density culture that ultimately generated a 250-fold increase in the number of hiPSC-CMs, which is suitable for multiple translational/regenerative applications. Furthermore, the in vivo control of late gestational CM proliferation may lead to promising regenerative strategies for treating patients with congenital heart defects.
STAR*METHODS
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sean M. Wu, smwu@stanford.edu.
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The single cell RNA sequencing datasets that were generated for this manuscript are available under the GEO accession number: GSE148586.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Four previously established hiPSC lines (SCVI-111, Sendai virus reprogrammed, peripheral blood mononuclear cells (PBMCs), male; SCVI-113, Sendai virus reprogrammed, PBMCs, male; SCVI-202 Sendai virus reprogrammed, PBMCs, female; SCVI-273, Sendai virus reprogrammed, PBMCs, female (Matsa et al., 2016)) were maintained in DMEM/F12 (ThermoFisher) supplemented with the essential eight (E8) (Thermo Fisher) growth factors in a Matrigel (Corning) coated (1:400 for 24h) polystyrene 2D culture system. Upon 80–90% confluency, cells were dissociated in PBS with 0.5mM EDTA for 5–10 minutes at 37 °C. Dissociation was performed with gentle trituration to obtain small aggregates of undifferentiated hiPSCs. Passaging was performed in 1:15–20 splitting ratios or 10,000 cells per cm2 to achieve low density and reach full confluency within 4–5 days. For the first 24h after replating, 10βM of ROCK inhibitor Y-27632 was included in the hiPSC maintenance media. hiPSC-CM differentiation was performed using the previously described canonical Wnt stimulation and inhibition protocol (Lian et al., 2013) in RPMI 1640-based differentiation media supplemented with B27 minus insulin (Invitrogen). Between day 0–2, a gradient of CHIR99021 (Seleckchem) concentrations (3.0, 4.0, 5.0, 6.0, 7.0, 8.0 μM) was used. Between day 3–5, Wnt-C59 (2.0 μM) (Selleckchem) was added to the differentiation media. At day 7, B27 with insulin was added to the differentiation media. At day 11, wells that contain more than 80% beating cells were treated with TrypLE Select Enzyme 10X (Invitrogen) at 37 °C for 10–40 minutes. Gentle rocking was performed every 10 minutes. Preparations of single dissociated cells were generated with very gentle trituration and transferred to a 15 mL conical tube containing a wash buffer (PBS with 20% FBS). Cells were gently centrifuged at 1000 RPM for 3 minutes and immediately replated in 1:10 to 1:15 split ratios in RPMI 1640 + B27 1× with 10% Knock Out Serum Replacement (Gibco) and Thiazovivin 1.0 μM (Selleckchem). At day 12, hiPSC-CMs were further expanded for downstream assays in RPMI 1640 + B27 1× differentiation media supplemented with 2.0 to 4.0 μM CHIR99021 (Selleckchem). For the first 24h after passaging, 10% Knock Out Replacement Serum and Thiozovivin 1.0 μM were added to the differentiation media.
Generation of eGFP-anillin+ expressing hiPSCs
The previously described eGFP-anillin expression cassette (Hesse et al., 2012) was inserted into the AAV-CAGGS vector (Addgene #22212). hiPSCs (iLB-C1–30m-r12), kindly provided by Dr. O. Brüstle (Univ. of Bonn), were co-transfected (NEONTransfection, Invitrogen) with the AAV-CAGGS-eGFP-anillin vector, the hAAVS1 1L TALEN vector (Addgene #35431) and the hAAVS1 1R TALEN vector (Addgene, #35432) for targeted integration into the AAVS1 locus and several independent eGFP-anillin expressing hiPSC lines were generated. Targeted integration into the AAVS1 locus was proven by a PCR strategy, copy numbers were determined by qPCR. The eGFP-anillin+ expressing hiPSC line used was shown to have one specific AAVS1 integration and one random integration site without known functional consequence.
METHOD DETAILS
Contact inhibition study
For high cell-cell contact condition (i.e. dense), hiPSC-CMs were either left unpassaged or passaged without splitting, while for low cell-cell contact condition (i.e. sparse), the cells were passaged at 1:10 – 1:15 splitting ratio, depending on cell confluency of the original well. To examine paracrine effect, the conditioned media was collected from densely plated hiPSC-CMs, concentrated using Pierce™ protein concentrator (3K MWCO; Thermoscientific 88512). The concentrated factors were diluted with fresh expansion media (B27 + CHIR) at different ratios (1×, 0.1×, 0.01×), then given to sparsely plated hiPSC-CMs. To examine direct cell-cell contact effect, 100,000 hiPSC-CMs were plated in small surface area (i.e. 48-well-plate) to induce dense culture, while the same number of cells were plated in large surface (i.e. 6-well-plate) for sparse condition. For immunostaining, cells were fixed at 4% paraformaldehyde for 15 min at room temperature, washed three times with washing buffer (0.1% Tween-20/PBS), permeabilized and blocked with blocking buffer (3% bovine serum albumin/2% goat serum/0.01% saponin/PBS for 30 min at room temperature. Corresponding primary antibodies were diluted at desired ratio in blocking buffer (TnT: 1:250, Ki67: 1:200, phH3: 1:400) and were incubated with cells overnight at 4 °C on shaker. Next day, the cells were washed three times with washing buffer, each time for 5 min at room temperature on shaker. Then, the appropriate secondary antibodies were diluted in blocking buffer at 1:300 dilution ratio and were incubated with cells for 1 hr at room temperature on shaker. After the incubation, the samples were washed three times to remove any unbound secondary antibodies. Nuclei were counterstained using Dapi.
Cardiac tissue generation
Engineered Heart Tissues were with small modifications generated as described previously (Hansen et al., 2010). Briefly, induced pluripotent stem cell-derived cardiomyocytes were detached and resuspended in culture medium containing bovine fibrinogen (5 mg/ml), aprotinin (2.5 μg/ml) and 10% Matrigel. EHTs of 1×106 cardiomyocytes each were casted by mixing the reconstitution mix (97 μl/EHT) with thrombin (3 μl/EHT, 100 U/ml) and pipetting it into the casting molds. Fibrin polymerization (37 °C, 7% CO2, 98% RH, 2 h) led to the formation of a muscle strip. The EHTs were transferred to culture medium (Dulbecco’s modified Eagle’s medium) containing 10% horse serum, 1% penicillin/streptomycin, 10 μg/ml insulin and 33 μg/ml aprotinin and maintained at 21% oxygen, 7% CO2 and 37 °C in a humidified cell culture incubator for 25 days. Contractility measurements were performed in culture medium as described previously (Hansen et al., 2010).
Small molecules / growth factors
PNU74654, MK2206, CHIR99021, C59 and Palbociclib were obtained commercially. CHIR99021 and C59 were used depending on the assay at 2.0 or 4.0 μM final concentration and PNU74654, MK2206 and Palbociclib were used at 32, 1.0 and 1.0 μM, respectively. For in vitro studies, recombinant scFv-DKK1c and RSPO were expressed and purified as previously described (Janda et al., 2017) and used in final concentrations of 200 and 25 nM, respectively. Recombinant Wnt3A protein was purchased commercially and added in 100 ng/mL final concentration.
Protein expression analysis
Immunocytochemistry was performed with overnight incubation of primary antibodies (listed below) followed by 2-hour incubation with Alexa-conjugated fluorescent secondary antibodies. Immunostaining images were captured using confocal (Zeiss LSM 710) or epifluorescence (Leica DM IL) microscopy.
Kinase phosphorylation screening assays were performed using the Proteome Profiler Human Phospho-Kinase Array Kit containing 43 human kinases. Western blotting was performed to validate the kinase screen results. Total protein expression was measured with a gel imager and quantified based on pixel intensity (BioRad). Only lanes from the same gel were used for displaying and quantification.
Luciferase assays
Day 12 hiPSC-CMs were transfected for 48 hours with a TCF reporter plasmid or the mutant reporter plasmid using Lipofectamine. 72 h after transfection cells were treated with various small molecules for an additional 24 h and then lysed and mixed with luciferase substrate. The expression of firefly luciferase was finally measured using a 96-well micro plate reader.
Real-time PCR expression analysis
For quantitative analysis of gene expression, RNA was extracted with a lysis buffer from hiPSC-CMs and stored at −80 °C. Total RNA from each sample was purified from cell lysate using a RNA purification kit. cDNA was made using a cDNA synthesis kit. Quantitative PCR was performed using a 96-well thermocycler system (Biorad) with SYBR Green substrate for 40 cycles. All primers sequences were obtained from the PrimerBank (Massachusetts General Hospital / Harvard Medical School) online database. Oligos were synthesized at Stanford University.
Single cell gene expression analysis
For single cell gene expression analysis, day 12 hiPSC-CMs were treated with DMSO, CHIR99021 or C59 for 24 hours. At the end of treatment hiPSC-CM aggregates were dispersed with TrypLE Select Enzyme as described above and dissociated with gentle trituration. Single cells were washed and re-suspended in ice cold RPMI 1640 with B27 supplement and then captured using the 10x Genomics platform. RNA amplification and generation of cDNA expression libraries were performed prior to sequencing using HiSeq-4000 (Illumina). The three data files (CTR, CHIR and C59) (GEO accession number: GSE148586) were merged and subsequent analysis was performed using Seurat software. Cells with poor quality and doublets/multiplets were filtered out based on aberrant gene numbers, total transcript counts, percentage of reads to mitochondrial genes, and housekeeping gene counts (GAPDH, SOD1, BLVRB, PSMB3). To remove batch effect, Seurat integration tool was employed (Stuart et al., 2019).
Flow cytometry
Freshly isolated hiPSC-CMs were fixed in PFA 4% for 5 min and incubated with primary antibody against TnT for 1 hr. After multiple washings, an Alexa488-conjugated mouse secondary antibody was added for 30 min of incubation followed by washings to remove unbound antibodies. Samples were then analyzed using the FACSCalibur® flow cytometer (BD Biosciences) and data processed using the FlowJo® software (TreeStar).
Electrophysiological studies
hiPSC-CM treated with DMSO, CHIR, or CHIR>C59 were passaged 3 times in RPMI 1640 + B27 Supplement for an equivalent number of days and seeded sparsely on Matrigel© coated 8 mm coverslips. Cells on coverslips were immersed in extracellular solution containing 140 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 10 mM glucose, at pH 7.4. Patch electrodes were filled with an intracellular solution containing 140 mM potassium gluconate, 10 mM NaCl, 2 mM MgCl2, 10 mM HEPES, 1 mM EGTA, 4 mM Mg-ATP, and 0.3 mM Na-GTP, at pH 7.3, hence, giving resistances of ~2–5 M’Ω. Spontaneous CM action potentials were recorded at room temperature using a sharp current clamp mode. For calcium transient recording, iPSC-CMs were loaded with 5 μM Fluo-4 AM for 5–10 mins at 37 °C and were washed 3 times afterward. Spontaneous calcium transients were observed at 37 °C with a Carl Zeiss LSM 510 confocal microscopy (Göttingen, Germany) and a 63× objective (Plan-Apochromat 63×/1.40 Oil DIC M27). Calcium signalings were recorded using line-scanning mode, and the Imaging data were analyzed with a custom-made Mat lab algorithm. Transient amplitude was expressed as ΔF/F0.
Single cell patterning and contractility
For single cell contractile assays, hiPSC-CMs were plated on polyacrylamide hydrogels of 10 kPa stiffness with micro-patterned Matrigel© to achieve a final hiPSC-CM aspect ratio of 7:1 (Lee et al., 2020). After 3–6 days of culture in the presence of DMSO, CHIR, or CHIR>C59, contractility of hiPSC-CMs were measured by recording contractile motion of at least 10 individual cells per group using high speed microscope camera (frame rate: 75 fps, shutter speed: 1/150 sec) and motion velocity analysis was performed using Sony Cell Motion Imaging System (SI8000) (Hayakawa et al., 2014). Briefly, motion velocity analysis calculates contraction/relaxation velocity and deformation distance by computationally measuring movement of pixels in the consecutive video images. Since cells are patterned into known surface area (1800 μm2) and attached to hydrogel substrate of known stiffness (10 kPa), maximum contractile force was calculated using Young’s modulus equation.
After recordings, cells were fixed immunostained with antibodies to cTnT and α-actinin, counterstained with DAPI, and imaged using Zeiss LSM710 confocal microscope. Quantification of sarcomere alignment was performed using FIJI software with an additional analysis package (Orientation J) to determine the angle of z-disc-registered sarcomeric α-actinin. For example, 90 ° angle of z-disc-registered sarcomeric α-actinin represents myofibril alignment along cellular long axis.
Biophysical effect examination using hydrogel substrate
To modulate substrate stiffness, polyacrylamide hydrogels were prepared as previously described (Lee et al., 2019, 2020). Briefly, 40% of acrylamide and 2% bisacrylamide stock solution was diluted with deionized water to make polyacrylamide hydrogel precursor solution with the final concentration of acrylamide at 8% and the final concentration of bisacrylamide at 0.06%, 0.15%, and 0.6%, to achieve hydrogel stiffness of 1, 10, and 60 kPa, respectively. The hydrogel precursor solution was sandwiched between two coverslips and photo-crosslinked under light (365 nm, 4 mW/cm2) for 5 min. After photocrosslinking the hydrogel, one coverslip was peeled off to reveal a hydrogel substrate. Day 24 hiPSC-CMs were plated on the hydrogel substrate and cultured for 5 days in expansion media (B27+CHIR 2μM) for 5 days before fixation. Immunostaining was done as described above. Rabbit-derived Yap antibody was diluted at 1:50.
YAP inhibition study
To pharmacologically inhibit YAP activity, a well-known inhibitor, verteporfin (SML0534, Sigma) was administered to day 22 hiPSC-CMs at different concentrations (0.1, 1, 10, 50, 100 μM) for 24 hours. 50, 100 μM verteporfin treatment led to cell death. Next day, the cells were fixed and stained for YAP, TnT, and Ki67 as described above.
In vivo studies in mice
Pregnant mice at gestational day 16.5 and 4-day-old CD1 mice (Jackson Laboratory, Bar Harbor, ME) were given 4–6 consecutive daily intraperitoneally injections of CHIR at a dose of 50mg/kg. After 4 days (embryonic studies) or 6 days (neonatal studies) post-injection, euthanasia was performed by first sedating the mice via isoflurane (inhalant, 2% in 100% oxygen, neonate placed on a warm pad), followed by a secondary cervical dislocation. Death was verified after euthanasia and prior to disposal. All animal experiments were approved by the animal care and use committee (APLAC) at Stanford University. All experiments were performed in accordance with relevant guidelines and regulations of Stanford University. Body and heart weights were measured by a blinded observer. Freshly isolated adult hearts were dissected from mouse chest cavity and washed in PBS to remove excess blood. The postnatal hearts were incubated in 30% sucrose in phosphate buffered solution (PBS) overnight followed by stepwise incubation with a graded concentration of OCT in PBS for cryo-sectioning. Following cryopreservation, hearts were cut into 10 μm sections and lightly fixed in 4% paraformaldehyde in PBS prior to immunostaining. All quantitative analyses of the histological sections were performed on numerically coded animals in an observer-blinded fashion to prevent subjective bias in data analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell counts were performed with Luna-FL fluorescence cell counter, BD flow cytometry system, Image J software or manually. Numerical data are presented individually or as mean ± standard deviation (SD), standard error (SE) or standard error of the mean (SEM). Statistical significance was performed using Excel or Prism software. Values of p<0.05 were considered statistically significant.
Supplementary Material
SUPPLEMENTARY VIDEOS
Movie 1. CHIR-treated hiPSC-CMs one day after passaging (Passage 2). Related to Figure 2.
Movie 2. Single-cell patterned hiPSC-CMs (DMSO treated). Related to Figure 3.
Movie 3. Single-cell patterned hiPSC-CMs (CHIR treated). Related to Figure 3.
Movie 4. Single-cell patterned hiPSC-CMs (CHIR treatment, followed by C59). Related to Figure 3.
Movie 5. Engineered heart tissue created from non-expanded hiPSC-CMs. Related to Figure 6.
Movie 6. Engineered heart tissue created from expanded hiPSC-CMs. Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cardiac Troponin T | Fisher | MS-295 |
| KI67 | Abcam | Ab15580 |
| Phospho-Histone 3 | Cell Signaling | 9701 |
| alpha sarcomeric actinin | Sigma-Aldrich | A7811 |
| MLC2V | Abcam | ab48003 |
| phospo AKT T308 | Cell Signaling | #9275 |
| Cyclin D2 | Santa Cruz | SC-452 |
| YAP | Santa Cruz | SC15407 |
| Proteome Profiler Human Phospho-Kinase Array Kit | R&D systems | ARY003B |
| Biological Samples | ||
| TCF reporter plasmid | Addgene | M50 |
| Mutant TCF reporter plasmid | Addgene | M51 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CHIR99021 | Selleckchem | S2924 / S1263 |
| C59 | Selleckchem | S737 |
| Verteporfin | Sigma | SML0534 |
| PNU74654 | Selleckchem | S8429 |
| MK2206 | Selleckchem | S1078 |
| Palbociclib | Selleckchem | S1116 |
| Wnt3A | R&D systems | 5036-WN-010 |
| Lipofectamine | Invitrogen | L3000001 |
| luciferase substrate | Promega | E1910 |
| Critical Commercial Assays | ||
| Bovine fibrinogen | Sigma | F8630 |
| Aprotin | Sigma | A1153 |
| Matrigel | Corning | 35234 |
| Thrombin | Sigma | T7513 |
| Insulin | Sigma | I9278 |
| ROCK inhibitor Y-27632 | Selleckchem | S1049 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| HotStart-IT Sybr | Invitrogen | 75762 |
| Superscript cDNA kit | Invitrogen | 11904018 |
| RPMI 1640 | Invitrogen | 12633012 |
| B27 supplement | Invitrogen | 17504044 |
| Deposited Data | ||
| Single cell RNA sequencing | 10x | GSE148586 |
| Experimental Models: Cell Lines | ||
| hiPSC line | Stanford Biobank | SCVI111 |
| hiPSC line | Stanford Biobank | SCVI113 |
| hiPSC line | Stanford Biobank | SCVI202 |
| hiPSC line | Stanford Biobank | SCVI273 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mice | Jackson Laboratory | 000664 |
| Software and Algorithms | ||
| Prism 8 | Graphpad | |
| Excel 2016 | Microsoft | |
| Seurat | Satijalab.org | |
| FlowJo® | TreeStar | |
Highlights:
GSK3β inhibition-mediated hiPSC-cardiomyocyte proliferation is cell density dependent
GSK3β inhibition with reduced cell-cell contact massively expands hiPSC-cardiomyocytes
LEF/TCF activity inhibits hiPSC-cardiomyocyte maturation without promoting cell cycling
Long-term expansion does not alter cardiomyocyte contractile function
Acknowledgements
This work was supported by a UMC Utrecht Clinical Fellowship and Netherlands Heart Institute Fellowship (to J.B.); Stanford Child Health Research Institute Postdoctoral Fellowship and NIH NRSA Postdoctoral Fellowship 5F32HL142205 (to S.L.); NIH Pathway to Independence Award 1K99HL127295–01A1 (to V.S.); The Richard and Helen DeVos Foundation (to S.J.); Netherlands Heart Foundation (CVON-Dosis 2014–40), and Netherlands Organization for Sciences (NWO)-ZonMW (VICI 91818602) (to J.V.); R01 HL145676, R01 HL146690 and P01 HL141084 (to J.C.W); NIH (OD004411, HL099776, LM012179), and the Endowed Faculty Scholar Award of Lucile Packard Foundation for Children and Child Health Research Institute at Stanford (to S.M.W.). The sequencing data was generated on an HiSeq purchased with funds from NIH under award number S10OD018220. We thank members of the Sean Wu lab for manuscript critique and Andrew Olson at the Stanford Neuroscience Imaging Core Facility for assistance with confocal imaging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Conflict of Interest
A provisional patent has been filed with the US Patent and Trademark Office regarding the effect of bioactive lipids plus Wnt signaling activation on hiPSC-CM proliferation/expansion.
REFERENCES
- Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Umansky KB, Yifa O, Kain D, Rajchman D, Leach J, Bassat DR, et al. (2017). The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 547, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K, Arab S, Haring B, and Kühn B (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270. [DOI] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, and Jope RS (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther 148, 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birket MJ, Ribeiro MC, Verkerk AO, Ward D, Leitoguinho AR, den Hartogh SC, Orlova VV, Devalla HD, Schwach V, Bellin M, et al. (2015). Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat. Biotechnol 33, 970–979. [DOI] [PubMed] [Google Scholar]
- Buikema JW, Zwetsloot P-PM, Doevendans PA, Sluijter JPG, and Domian IJ (2013a). Expanding Mouse Ventricular Cardiomyocytes through GSK-3 Inhibition. Curr Protoc Cell Biol 61, 23.9.1–23.9.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buikema JW, Mady AS, Mittal NV, Atmanli A, Caron L, Doevendans PA, Sluijter JPG, and Domian IJ (2013b). Wnt/ȕ-catenin signaling directs the regional expansion of first and second heart field-derived ventricular cardiomyocytes. Development 140, 4165–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JJH, Yang X, Don CW, Minami E, Liu Y-W, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, et al. (2014). Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejo R, de Alvaro C, Benito M, Cuadrado A, and Lorenzo M (2002). Insulin restores differentiation of Ras-transformed C2C12 myoblasts by inducing NF-kappaB through an AKT/P70S6K/p38-MAPK pathway. Oncogene 21, 3739–3753. [DOI] [PubMed] [Google Scholar]
- von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, et al. (2012). YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. U.S.A 109, 2394–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, et al. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol 161, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grego-Bessa J, Luna-Zurita L, del Monte G, Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama Y, Chen H, et al. (2007). Notch Signaling is Essential for Ventricular Chamber Development. Dev Cell 12, 415–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, Eder A, Bönstrup M, Flato M, Mewe M, Schaaf S, Aksehirlioglu B, Schwoerer AP, Schwörer A, Uebeler J, et al. (2010). Development of a drug screening platform based on engineered heart tissue. Circ. Res 107, 35–44. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Kunihiro T, Ando T, Kobayashi S, Matsui E, Yada H, Kanda Y, Kurokawa J, and Furukawa T (2014). Image-based evaluation of contraction-relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: Correlation and complementarity with extracellular electrophysiology. J. Mol. Cell. Cardiol 77, 178–191. [DOI] [PubMed] [Google Scholar]
- He L, and Zhou B (2017). Cardiomyocyte proliferation: remove brakes and push accelerators. Cell Research 27, 959–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, and Martin JF (2011). Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 332, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M, Raulf A, Pilz G-A, Haberlandt C, Klein AM, Jabs R, Zaehres H, Fügemann CJ, Zimmermann K, Trebicka J, et al. (2012). Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nature Communications 3, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda CY, Dang LT, You C, Chang J, de Lau W, Zhong ZA, Yan KS, Marecic O, Siepe D, Li X, et al. (2017). Surrogate Wnt agonists that phenocopy canonical Wnt and β-catenin signalling. Nature 545, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkela R, Kockeritz L, MacAulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR, et al. (2008). Deletion of GSK-3β in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118, 3609–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-H, and Asthagiri AR (2011). Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J Cell Sci 124, 1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N-G, Koh E, Chen X, and Gumbiner BM (2011). E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. U.S.A 108, 11930–11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Stanton AE, Tong X, and Yang F (2019). Hydrogels with enhanced protein conjugation efficiency reveal stiffness-induced YAP localization in stem cells depends on biochemical cues. Biomaterials 202, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yang H, Chen C, Venkatraman S, Darsha A, Wu SM, Wu JC, and Seeger T (2020). Simple Lithography-Free Single Cell Micropatterning using Laser-Cut Stencils. JoVE e60888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Gao E, Vite A, Yi R, Gomez L, Goossens S, van Roy F, and Radice GL (2015). Alpha-catenins control cardiomyocyte proliferation by regulating Yap activity. Circ. Res 116, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, and Palecek SP (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U.S.A 109, E1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, and Palecek SP (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc 8, 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, Guo H, van Gorp PRR, Wang D-Z, and Pu WT (2015). Pi3kcb links Hippo-YAP and PI3K-AKT signaling pathways to promote cardiomyocyte proliferation and survival. Circ. Res 116, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Barnett SF, Yaroschak M, Bilodeau MT, and Layton ME (2007). Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Top Med Chem 7, 1349–1363. [DOI] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yagi H, Saeed S, Bais AS, Gabriel GC, Chen Z, Peterson KA, Li Y, Schwartz MC, Reynolds WT, et al. (2017). The complex genetics of hypoplastic left heart syndrome. Nat. Genet 49, 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-W, Chen B, Yang X, Fugate JA, Kalucki FA, Futakuchi-Tsuchida A, Couture L, Vogel KW, Astley CA, Baldessari A, et al. (2018). Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol 36, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E, Burridge PW, Yu K-H, Ahrens JH, Termglinchan V, Wu H, Liu C, Shukla P, Sayed N, Churko JM, et al. (2016). Transcriptome Profiling of Patient-Specific Human iPSC-Cardiomyocytes Predicts Individual Drug Safety and Efficacy Responses In Vitro. Cell Stem Cell 19, 311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, et al. (2017). Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci USA 114, E8372–E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RJ, Parker BL, Quaife-Ryan GA, Voges HK, Needham EJ, Bornot A, Ding M, Andersson H, Polla M, Elliott DA, et al. (2019). Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 24, 895–907.e6. [DOI] [PubMed] [Google Scholar]
- Mirzaa G, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, et al. (2014). De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat. Genet 46, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Ogle BM, Bursac N, Domian I, Huang NF, Menasché P, Murry CE, Pruitt B, Radisic M, Wu JC, Wu SM, et al. (2016). Distilling complexity to advance cardiac tissue engineering. Sci Transl Med 8, 342ps13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puliafito A, Hufnagel L, Neveu P, Streichan S, Sigal A, Fygenson DK, and Shraiman BI (2012). Collective and single cell behavior in epithelial contact inhibition. PNAS 109, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, and Brivanlou AH (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med 10, 55–63. [DOI] [PubMed] [Google Scholar]
- Senyo SE, Lee RT, and Kühn B (2014). Cardiac regeneration based on mechanisms of cardiomyocyte proliferation and differentiation. Stem Cell Res 13, 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Zhang Y, Buikema JW, Serpooshan V, Chirikian O, Kosaric N, Churko JM, Dzilic E, Shieh A, Burridge PW, et al. (2018). Stage-specific Effects of Bioactive Lipids on Human iPSC Cardiac Differentiation and Cardiomyocyte Proliferation. Scientific Reports 8, 6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Abbasi C, Lazic S, Teng ACT, Wang D, Dubois N, Ignatchenko V, Wong V, Liu J, Araki T, et al. (2015). Evolutionarily conserved intercalated disc protein Tmem65 regulates cardiac conduction and connexin 43 function. Nature Communications 6, 8391. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Beach D, and Shapiro GI (2016). Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 6, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba Y, Gomibuchi T, Seto T, Wada Y, Ichimura H, Tanaka Y, Ogasawara T, Okada K, Shiba N, Sakamoto K, et al. (2016). Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538, 388–391. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, and Bao S (2005). The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med 9, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, Sharma A, Xu AF, Engels MC, Feistritzer R, et al. (2015). The Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation 132, 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titmarsh DM, Glass NR, Mills RJ, Hidalgo A, Wolvetang EJ, Porrello ER, Hudson JE, and Cooper-White JJ (2016). Induction of Human iPSC-Derived Cardiomyocyte Proliferation Revealed by Combinatorial Screening in High Density Microbioreactor Arrays. Scientific Reports 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosset J-Y, Dalvit C, Knapp S, Fasolini M, Veronesi M, Mantegani S, Gianellini LM, Catana C, Sundström M, Stouten PFW, et al. (2006). Inhibition of protein-protein interactions: the discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins 64, 60–67. [DOI] [PubMed] [Google Scholar]
- Tseng A-S, Engel FB, and Keating MT (2006). The GSK-3 Inhibitor BIO Promotes Proliferation in Mammalian Cardiomyocytes. Chemistry & Biology 13, 957–963. [DOI] [PubMed] [Google Scholar]
- Uosaki H, Magadum A, Seo K, Fukushima H, Takeuchi A, Nakagawa Y, Moyes KW, Narazaki G, Kuwahara K, Laflamme M, et al. (2013). Identification of chemicals inducing cardiomyocyte proliferation in developmental stage-specific manner with pluripotent stem cells. Circ Cardiovasc Genet 6, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uygur A, and Lee RT (2016). Mechanisms of Cardiac Regeneration. Dev Cell 36, 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM, Chien KR, and Mummery C (2008). Origins and fates of cardiovascular progenitor cells. Cell 132, 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. (2017). Non-equivalence of Wnt and R-spondin ligands during Lgr5 + intestinal stem-cell self-renewal. Nature 545, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Farin HF, van Es JH, Clevers H, Langer R, and Karp JM (2014). Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods 11, 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cao N, Huang Y, Spencer CI, Fu J-D, Yu C, Liu K, Nie B, Xu T, Li K, et al. (2016). Expandable Cardiovascular Progenitor Cells Reprogrammed from Fibroblasts. Cell Stem Cell 18, 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY VIDEOS
Movie 1. CHIR-treated hiPSC-CMs one day after passaging (Passage 2). Related to Figure 2.
Movie 2. Single-cell patterned hiPSC-CMs (DMSO treated). Related to Figure 3.
Movie 3. Single-cell patterned hiPSC-CMs (CHIR treated). Related to Figure 3.
Movie 4. Single-cell patterned hiPSC-CMs (CHIR treatment, followed by C59). Related to Figure 3.
Movie 5. Engineered heart tissue created from non-expanded hiPSC-CMs. Related to Figure 6.
Movie 6. Engineered heart tissue created from expanded hiPSC-CMs. Related to Figure 6.
Data Availability Statement
The single cell RNA sequencing datasets that were generated for this manuscript are available under the GEO accession number: GSE148586.