

Abstract
Phylogeography combines ancestry with location and can be translated to intratumor heterogeneity (ITH) to visualize how tumors spread. ITH is common in human tumors, with many genetic and phenotypic differences between regions. The roles of ITH in progression are uncertain because many subclones lack discernable driver mutations. ITH can be visualized by mapping mutations onto microscopic sections, where subclones are directly associated with phenotypes, especially the deeper areas with the more invasive cells that confer worst clinical outcomes. Instead of a stepwise hierarchy where subclones segregate by phenotype with later branching subclones in more invasive areas, multiple subclones share superficial and invasive phenotype and are jigsaw arrayed in vertical columns. Phylogeography shows that both early and late subclones extend from the surface to the invasive front, suggesting that founder cells start with phenotypic plasticity and essentially all the drivers necessary to rapidly grow into large invasive tumors.
Subject Areas: Biological Sciences, Cancer
Graphical Abstract
Biological Sciences; Cancer
Introduction
Multiregional sampling has revealed that many mutations differ between regions of the same human tumor indicating that tumors are subdivided into multiple subclones (McGranahan and Swanton, 2017). This intratumoral heterogeneity (ITH) is very common, but its biological significance is uncertain. The purpose of this review is to outline how direct visualization of ITH with phylogeography can yield better insights into how human tumors grow. Phylogeography is commonly applied to macroscopic populations, such as the worldwide spread of humans, where the locations of individuals give additional insights to the genetic information (Nielsen et al., 2017). Phylogeography combines tumor phylogeny or the ancestral relationships of subclones with their spatial physical locations in the tumor. ITH is currently measured by sampling and sequencing multiple tumor regions and then visualized with ancestral trees and physical maps that document where the samples were obtained. Although this combination of physical location with ancestry is essentially phylogeography, missing is vital visual information on how tumor cells change their phenotypes during tumor growth and subclone formation, which is better documented at the microscopic level. Uncertain are whether subclones have different microscopic phenotypes and how adjacent subclones are physically entangled. Such detailed cell phenotype and topographic information can be obtained by saturation microdissection followed by deep resequencing of individual tumor microscopic sections (Ryser et al., 2020). In this manner, it is possible to infer when subclones arise and their final locations and cellular phenotypes within a single microscopic section.
ITH and Tumor Progression
Precancerous tumors acquire more driver mutations as they progress to larger tumors, as exemplified by the adenoma-cancer sequence, which is the paradigm for tumor progression (Fearon and Vogelstein, 1990). Unclear in such linear progression diagrams are how transitions between stages occur and how to incorporate genetic and cellular phenotypic ITH. Phenotypic ITH reflects that cells in a single tumor do not look alike under the microscope and can be classified as superficial versus invasive, well versus poorly differentiated, and so on. Greater numbers of more sophisticated cellular phenotypes can be quantified with imaging mass cytometry (Jackson et al., 2020). Unclear is how tumors acquire cells with multiple, diverse phenotypes. Microdissection studies of different regions of the same tumor have found that canonical driver mutation ITH is rare (Andreyev et al., 1997; Cross et al., 2018), indicating that these driver mutations are generally acquired early during progression and do not correlate with phenotypic ITH.
Early multiregional sampling found that mutations differ between different parts of the same neoplasm (Maley et al., 2006), but the widespread nature of ITH has become much more evident (Gerlinger et al., 2012) with next-generation sequencing. Essentially every sample from a tumor has a different set of alterations (predominately passenger mutations), indicating most human tumors are subdivided into many distinct subclones. Hence, instead of linear progression, final tumor growth is better represented by branching phylogenies (Figure 1A).
Figure 1.

CRC Phylogeography
(A) Genetic ITH. A typical branching tumor phylogeny inferred from ITH. Final tumor growth starts from the founder cell or MRCA. Clonal mutations are acquired before growth and are present in all cells. Subclonal mutations are acquired during growth, and their subclones (four are illustrated) can be ordered by phylogeny as early and late branching subclones. Note that passenger or driver mutations can be used to distinguish between subclones.
(B) Phenotypic ITH and anatomic barriers to invasion. Histologic landmarks used for clinical staging are illustrated with dotted lines and are the muscularis mucosa (T1), the muscularis propria (T2), and the serosal surface (T3).
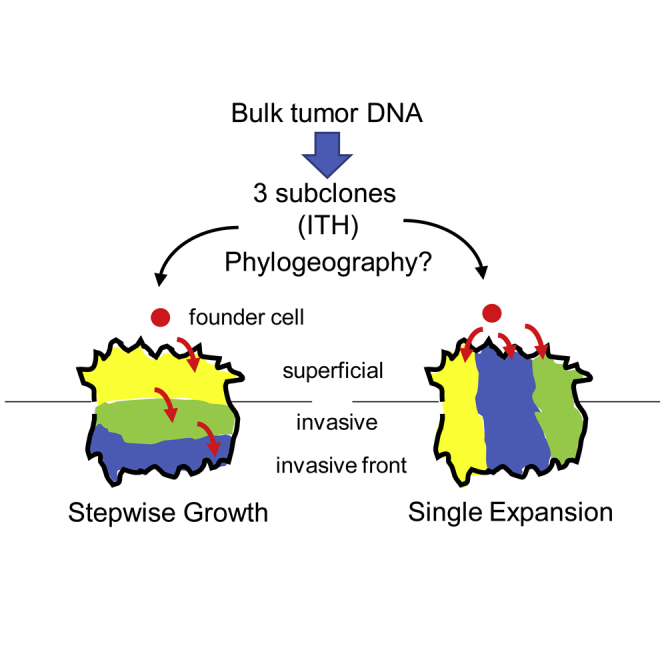
(C) Subclones can be mapped directly onto tissue sections by saturation microdissection and deep resequencing. Although not specified, stepwise progression implies subclone branching (Figure 1A) is due to fitness differences. An early subclone may form a small noninvasive tumor, and each later subclone increases tumor size and invades more deeply. Hence, subclones are layered horizontally by phenotype, with superficial early subclones and later, progressively more invasive subclones. The figure illustrated a stepwise progression phylogeography starting with an early superficial subclone and later deeper invasion of subclones past the T1 and then T2 histologic barriers. Scale bar is 1 cm.
(D) Typical example of CRC phylogeography (Ryser et al., 2020). Consistent with a single expansion by a founder cell with all the drivers and the phenotypic plasticity for rapid growth, subclones are jigsaw arranged in vertical columns that usually span from superficial to invasive regions. There are minimal bottlenecks for invasion and final cell phenotypes depend on their locations or microenvironments. Note that in this scenario, the branching phylogeny in Figure 1A likely represents very early divisions when the tumor was as small as a single gland (Ryser et al., 2018a).
Tumor phylogenies reconstruct the order and timing of genetic alterations during progression. Tumors are clonal, and it is possible to infer the genotype and timing of the final single tumor progenitor cell or the most recent common ancestor (MRCA). This founder cell starts the growth of the final tumor and is the root of the tumor phylogeny (Figure 1A). Mutations that occur before the MRCA will be found in all tumor cells, whereas mutations that arise during growth will be found in subclones that occupy only parts of the final tumor. Hence ITH is generated during the last growth phase. The length and events that occur between this final founder tumor cell and tumor removal are uncertain. One study indicated about 17 years between a large adenoma and its colorectal cancer (CRC) and several years between the final founder cancer cell and metastasis and removal (Jones et al., 2008). A more recent study based on Pan-Cancer Analysis of Whole Genomes data estimated much faster final growth, where a founder cell occurred on average 0.4, 0.3, 0.3, and 0.6 years prior to diagnosis for, respectively, CRC, lung squamous cell carcinoma, ovarian adenocarcinoma, and pancreatic adenocarcinoma (Gerstung et al., 2020).
The creation of multiple subclones during final tumor growth could happen if new driver mutations are acquired during growth, because changes in tumor sizes and phenotypes are correlated with additional driver mutations during early progression (Fearon and Vogelstein, 1990). This scenario of ongoing selection during tumor growth can be modeled, simulating multiple subclones per tumor (Waclaw et al., 2015). Hence, genetic ITH can help explain phenotypic ITH if new subclones with more fit phenotypes progressively arise during growth. Primary CRCs, discussed in this perspective, have both superficial and invasive regions. By the above logic, earlier subclones are superficially located, whereas later branching subclones with more driver mutations should occupy deeper invasive locations.
However, several observations indicate subclone branching may not correlate with phenotypic progression. Although cancers have hundreds of mutations, most appear to be neutral passenger mutations, with very few bona fide selective driver mutations (Vogelstein et al., 2013). Most driver mutations are clonal and arise before the start of growth (Ryser et al., 2018a; Reiter et al., 2018). The paucity of subclonal driver mutations suggests branching may instead reflect subclones distinguished by random passenger mutations that accumulate during tumor growth (Williams et al., 2018). Such subclones would have similar fitness and occupy different tumor regions, but the sizes of the subclones would more depend on how early they arose during growth. However, the definition of driver mutations is controversial, and potentially more subtle copy number, mutational, or epigenetic differences could lead to selective advantages and subclonal outgrowth. Phylogeography can resolve whether later arising subclones do in fact localize with more invasive phenotypes, which would give credence to subclone fitness differences.
Another observation that is difficult to explain with stepwise progression is the common finding that metastases often branch early on tumor trees (Reiter et al., 2018). If metastases arise stepwise after growth and invasion, its subclones should branch much later on its ancestral tree. Early branching suggests this phenotype is present very early, and indeed a quantitative analysis (Hu et al., 2019) comparing metastatic and primary mutations indicates that metastatic subclones often absconded before their primary tumors were visible (<0.01 cm3).
Visualizing ITH
Multiregional sampling of bulk tumor regions can reveal ITH between gross phenotypes, such as differences between primary cancers and their metastases (Reiter et al., 2018; Hu et al., 2019). One way to better understand branching tumor phylogenies (Figure 1A) with respect to more subtle phenotypic changes is to physically map subclones onto tumor sections on microscope slides. Phylogeography visualizes for each subclone when they arise and their final locations and phenotypes. Because depth of invasion is a critical determinate of patient survival, human tumors are oriented on microscopic sections as cross sections that span superficial and deeper regions. Just like genetic ITH, there is considerable visual phenotypic heterogeneity between cells in the same tumor, and potentially this phenotypic ITH corresponds to genetic ITH.
Most multiregional studies sample the lateral spread of cells across the surface of a tumor. With microscopic sections it is possible to specifically examine with microdissection the critical downward invasive growth of tumor cells (Figure 1B). Downward growth is a critical dimension because the depth of invasion largely determines clinical outcomes. Progressively deeper histologic landmarks are the muscularis mucosa (in situ versus T1 invasion), the muscularis propria (T2), and the serosal surface (T3). Histologic examination routinely subclassifies or stages CRCs based on the depth of invasion.
Although not specified, the logic of stepwise progression implies that the clinical transitions from T1 to T2 to T3 invasion occur in discrete steps via sequential clonal evolution, where new driver mutations allow progressively deeper invasion past each physical barrier. A T1 tumor will be stalled at the muscularis propria barrier until a new subclonal driver mutation confers the ability to invade this physical barrier. By this model of sequential stepwise bottlenecks, subclones are layered horizontally. Earlier, less invasive subclones should be superficial, whereas later branching subclones with “more advanced” phenotypes should be found in more deeply invasive regions (Figure 1C).
An alternative and simpler model is when a founder tumor cell starts with all the driver mutations needed for growth (Figure 1D). In this “Big Bang” scenario (Sottoriva et al., 2015; Williams et al., 2018), most subclonal mutations are passenger mutations and ITH represents mutations acquired during the growth of subclones with equivalent malignant potentials (i.e., neutral evolution). There is still selection for growth, but this selection is conferred by the clonal drivers present in all the cells. In this scenario, the phenotype of a cell is not subclone specific but rather depends on where it ends up in the final tumor. Hence, progeny of the first founder cell grows quickly in all directions (laterally and deeply) and subclones are organized in radiant columns that span superficial and invasive regions (Figure 1D). Because the malignant potential is conferred by common clonal driver mutations, there are no phenotypic bottlenecks and multiple subclones can share invasive phenotypes.
Saturation microdissection and targeted deep resequencing (Ryser et al., 2020) reveal that CRC subclones are jigsaw arrayed in millimeter-wide columns that share phenotypes rather than being layered horizontally by phenotype (Figure 1D). For a typical section, there was a median of three subclones per slide. Although exact subclone boundaries could not be determined, in situ studies indicate both smooth and infiltrative borders between subclones (Baker et al., 2017). The subdivision of adenocarcinomas into distinct glands may help physically restrict cell migration. Most (76%) large subclones shared both invasive and superficial phenotypes. Moreover, when the subclones were organized by ancestry, subclones with invasive phenotypes arose from both early and late phylogenetic branches. Early and late branching subclones also had similar sizes. These CRC phylogeographies are more consistent with single expansions by founder cells with all the driver mutations needed for growth rather than a stepwise mechanism where invasion occurs later during growth by a minority of more advanced subclones.
The phylogeography from one microscope slide (Figure 1D) visualizes only a very small slice of the total tumor expansion, which can extend many centimeters (Figure 2). Sampling more regions provides opportunities to see whether any model of tumorigenesis is reproducible even when the same tumor is viewed from a slightly different spatial perspective. Single neutral expansions produce exponentially more subclones that grow apart during growth (Figure 2), and therefore more subclones should be found with more sampling of the same tumor. Still consistent with single expansions, the phylogeographies of other microscope slides from the same tumors had multiple related but distinct subclones that were also oriented in vertical columns with shared superficial and invasive phenotypes (Ryser et al., 2020). The trend for increasingly more subclones with more samples from the same tumor indicates that any differences between the subclones are increasingly moot.
Figure 2.

Growth in All Directions by Progeny of a Founder Cell with all the Necessary Driver Mutations and Phenotypic Plasticity Can Explain CRC Phylogeography
(A) A single expansion with growth in all directions can explain the spherical macroscopic shapes typical of CRCs. Growth in all directions and phenotypic plasticity can help explain the jigsaw arranged subclone columns with both invasive and superficial phenotypes (Figure 1D).
(B) Early branching metastases could reflect that the first few downward-growing tumor cells have greater opportunities to invade deeply and reach the vasculature. Physical access of upward-growing superficial cells to the invasive front becomes increasingly more difficult as the tumor becomes larger.
Colocalizing subclones and their phylogenies onto tissue sections confers additional information on when the branching occurs. For example, the phylogeography (Figure 1D) is inconsistent with initial superficial growth followed months to years later by a new single more fit subclone with an invasive phenotype. Because subclonal mutations detectable by conventional exome sequencing (i.e., variant allele frequencies >5%) arise early during growth (Ryser et al., 2018a), detectable subclones are created early during growth and grow in all directions at the same time. Additional smaller subclones per microscope slide could be detectable with even deeper sequencing.
Mechanisms for Changing Tumor Cell Phenotype
CRCs appear to grow from single founder cells into their final tumors in less than a year (Gerstung et al., 2020). A single cell can grow into a visible tumor (∼a billion cells or ∼1 cm3 tumor) in about 30 divisions, but how these cells rapidly acquire multiple cellular phenotypes is uncertain. Phylogeography does not quantify the time intervals between branchpoints but indicates many CRC subclones acquire and share multiple phenotypes. Here we examine three broad mechanisms that can change cell phenotypes. The first mechanism is mutational, exemplified by the adenoma-cancer sequence. This stepwise mutation accumulation is relatively slow as decades are needed to acquire a small number of driver mutations. This mechanism does not readily explain how multiple subclones can share multiple phenotypes because subclonal driver mutations appear rare (Ryser et al., 2018a; Reiter et al., 2018).
Epigenetic mechanisms, which can be subdivided into epigenetic remodeling and phenotypic plasticity, more readily allow a single genome to have multiple phenotypes because epigenetic configurations dictate expression. Epigenetic remodeling can be rapid and is exemplified by normal hematopoiesis, where progeny from a stem cell can differentiate into multiple mature cell types. This differentiation occurs over several weeks, and different hematopoietic cell types are distinguished by epigenetic difference at genes critical for their cell phenotypes (Farlik et al., 2016).
An even more rapid way to change cell phenotype is called phenotypic plasticity (Easwaran et al., 2014). Phenotypic plasticity allows a single genome and epigenome to confer different cell phenotypes and is exemplified by the rapid differentiation of cells within intestinal crypts. The major crypt cell types (absorptive and secretory) have markedly different visual phenotypes and arise from the same stem cells. The microenvironment dictates cell phenotype because differentiation occurs within days when a cell moves upward out of the crypt base and depends on the phenotypes of neighboring cells (lateral inhibition). Unlike the epigenetic remodeling of hematopoiesis, differentiated secretory and absorptive crypt cells have nearly identical epigenomes. The underlying mechanism is open or broadly permissive chromatin (Kim et al., 2014) where the genome is configured or poised to express either adsorptive or secretory specific genes, and cell phenotype is determined by transcription factors induced by the microenvironment.
An epigenetic mechanism is more likely to explain how multiple CRC subclones can share multiple phenotypes. Perhaps not surprisingly, the epigenetic mechanism hijacked by CRC appears to be the phenotypic plasticity of normal colon crypts rather than the epigenetic remodeling of hematopoiesis. There are many common recurrent epigenetic changes in CRCs such as aberrant CRCs enhancers (Cohen et al., 2017), and multiregional sampling of the same tumor can address whether epigenomes are stable during tumor growth. DNA methylation was nearly identical between opposite sides of 16 colorectal tumors (Ryser et al., 2018b). Observed differences between tumor regions were more consistent with random drift rather than specific epigenetic remodeling because the methylation of gene-associated CpG sites was preferentially conserved, with greater changes outside of coding regions or in non-expressed genes. Although only superficial regions were sampled, the relative stability of gene-associated DNA methylation during growth is more consistent with phenotypic plasticity rather than epigenetic remodeling.
Phenotypic plasticity allows a subclone to express both superficial and invasive phenotypes because microenvironments or locations determine tumor cell phenotype. Phenotypic plasticity would favor rapid growth in less than a year because once tumor cells start to proliferate, their progeny could adapt to their current microenvironments and could continuously migrate and colonize other microenvironments. Stepwise sequential evolution may be too slow for rapid growth, even for cells with several driver mutations. Recent studies document the mismatch between numbers of driver mutations and tumorigenesis. Many somatic mutations commonly accumulate in normal tissues, with some normal cells harboring multiple driver mutations (Martincorena, 2019; Lee-Six et al., 2019). By contrast, even after whole-genome sequencing, some cancers lack discernable driver mutations (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020). Tumors that require additional subclonal driver mutations or epigenetic remodeling for stepwise growth may not often become clinically detectable. Instead, once tumor cells start to proliferate, phenotypic plasticity mediated by permissive chromatin or epigenetic plasticity (Flavahan et al., 2017) may be critical to catalyze widespread growth. The features that confer phenotypic plasticity are potentially additional therapeutic targets. Single-cell sequencing studies have also demonstrated that breast tumor subclones also have multiple phenotypes (Casasent et al., 2018).
Future Directions: Tissue Sections as Data Scaffolds
Phylogeography, such as the spread of humans across the world, reconstructs “what” happened but does not explain “how.” However, knowing what happened can help frame what questions to ask and how to ask them. Tumors grow in many ways (Williams et al., 2019) and mechanisms likely vary between tumors that grow stepwise (Figure 1C) versus as single expansions (Figure 1D). Overlaying additional data types upon the same microscopic sections can enhance integration. For example, although it is difficult to reliably measure chromatin in very small microdissected samples, the topography of epigenomes might be very different in stepwise versus single tumor expansions. Relatively uniform epigenomes in superficial and invasive tumor regions would favor phenotypic plasticity, whereas regional differences could identify how subclones independently acquire invasive properties.
Phylogeography could help organize and visualize the many other data types that can be generated by modern technologies. Tissue sections can be used as patient-specific spatial scaffolds to visualize and organize other commonly measured informational molecules (chromatin, RNA, protein) with modern immunohistochemical and in situ hybridization methods. For example, imaging mass cytometry can identify multiple tumor communities with different phenotypes even within very small (<1 mm2) regions of breast cancers (Jackson et al., 2020). Such very detailed phenotypic spatial maps can be integrated with the phylogeography of the same microscopic sections. RNA in situ methods such as BaseScope can localize subclones directly on microscope slides (Baker et al., 2017). Newer tissue clearing methods (Tanaka et al., 2017) could potentially allow phylogeography reconstructions in three dimensions. Such spatially organized data integrated with ancestral information can help visualize how individual tumors grow. A potential goal for data scientists is to organize ever increasing volumes of data onto tissue sections, allowing more integrated clinical descriptions of human tumors.
Progression is heterogeneous with evidence of both ongoing selection and neutral evolution during tumor growth (Sun et al., 2017; Williams et al., 2018). One way forward is to first identify and study tumors with the reduced complexity of neutral evolution where growth is more homogeneous. Most CRCs appear to have phylogeographies more compatible with single “Big Bang” expansions (Sottoriva et al., 2015; Williams et al., 2018), where their founder cells started with all the driver mutations and the phenotypic plasticity to rapidly grow into large tumors. The final tumor expansion is the culmination of decades-long mutation accumulation and selection, and the founder cell likely starts already with very high fitness levels (Barber et al., 2015). Such a “born to be bad” scenario (Ryser et al., 2018a) could help spatially explain the common finding of early branching metastases, because during early growth in all directions, the initially downward-growing cells would invade first and have the best physical access to the vasculature (Figure 2). For such simple single expansions, it should be possible with multiregional sampling and phylogeny to go back in time to visualize the genome and epigenome of the founder cell, to better understand how some human tumors start to grow and keep growing. Better information on how human tumors start to grow may help strategies for cancer prevention and early detection.
Acknowledgments
Supported by a grant from the National Cancer Institute (NCI), USA (grant no. U54CA217376).
Author Contributions
The author wrote the review.
References
- Andreyev H.J., Tilsed J.V., Cunningham D., Sampson S.A., Norman A.R., Schneider H.J., Clarke P.A. K-ras mutations in patients with early colorectal cancers. Gut. 1997;41:323–329. doi: 10.1136/gut.41.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A.M., Huang W., Wang X.M., Jansen M., Ma X.J., Kim J., Anderson C.M., Wu X., Pan L., Su N. Robust RNA-based in situ mutation detection delineates colorectal cancer subclonal evolution. Nat. Commun. 2017;8:1998. doi: 10.1038/s41467-017-02295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber L.J., Davies M.N., Gerlinger M. Dissecting cancer evolution at the macro-heterogeneity and micro-heterogeneity scale. Curr. Opin. Genet. Dev. 2015;30:1–6. doi: 10.1016/j.gde.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasent A.K., Schalck A., Gao R., Sei E., Long A., Pangburn W., Casasent T., Meric-Bernstam F., Edgerton M.E., Navin N.E. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018;172:205–217. doi: 10.1016/j.cell.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A.J., Saiakhova A., Corradin O., Luppino J.M., Lovrenert K., Bartels C.F., Morrow J.J., Mack S.C., Dhillon G., Beard L. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 2017;8:14400. doi: 10.1038/ncomms14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross W., Kovac M., Mustonen V., Temko D., Davis H., Baker A.M., Biswas S., Arnold R., Chegwidden L., Gatenbee C. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018;2:1661–1672. doi: 10.1038/s41559-018-0642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H., Tsai H.C., Baylin S.B. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell. 2014;54:716–727. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M., Halbritter F., Müller F., Choudry F.A., Ebert P., Klughammer J., Farrow S., Santoro A., Ciaurro V., Mathur A. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell. 2016;19:808–822. doi: 10.1016/j.stem.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon E.R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A.J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M., Jolly C., Leshchiner I., Dentro S.C., Gonzalez S., Rosebrock D., Mitchell T.J., Rubanova Y., Anur P., Yu K. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–128. doi: 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Ding J., Ma Z., Sun R., Seoane J.A., Scott Shaffer J., Suarez C.J., Berghoff A.S., Cremolini C., Falcone A. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019;51:1113–1122. doi: 10.1038/s41588-019-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson H.W., Fischer J.R., Zanotelli V.R.T., Ali H.R., Mechera R., Soysal S.D., Moch H., Muenst S., Varga Z., Weber W.P., Bodenmiller B. The single-cell pathology landscape of breast cancer. Nature. 2020;578:615–620. doi: 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- Jones S., Chen W.D., Parmigiani G., Diehl F., Beerenwinkel N., Antal T., Traulsen A., Nowak M.A., Siegel C., Velculescu V.E. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. U S A. 2008;105:4283–4288. doi: 10.1073/pnas.0712345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.H., Li F., Ferreiro-Neira I., Ho L.L., Luyten A., Nalapareddy K., Long H., Verzi M., Shivdasani R.A. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature. 2014;506:511–515. doi: 10.1038/nature12903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H., Olafsson S., Ellis P., Osborne R.J., Sanders M.A., Moore L., Georgakopoulos N., Torrente F., Noorani A., Goddard M. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–537. doi: 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- Maley C.C., Galipeau P.C., Finley J.C., Wongsurawat V.J., Li X., Sanchez C.A., Paulson T.G., Blount P.L., Risques R.A., Rabinovitch P.S., Reid B.J. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006;38:468–473. doi: 10.1038/ng1768. [DOI] [PubMed] [Google Scholar]
- Martincorena I. Somatic mutation and clonal expansions in human tissues. Genome Med. 2019;11:35. doi: 10.1186/s13073-019-0648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N., Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Nielsen R., Akey J.M., Jakobsson M., Pritchard J.K., Tishkoff S., Willerslev E. Tracing the peopling of the world through genomics. Nature. 2017;541:302–310. doi: 10.1038/nature21347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J.G., Makohon-Moore A.P., Gerold J.M., Heyde A., Attiyeh M.A., Kohutek Z.A., Tokheim C.J., Brown A., DeBlasio R.M., Niyazov J. Minimal functional driver gene heterogeneity among untreated metastases. Science. 2018;361:1033–1037. doi: 10.1126/science.aat7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser M.D., Min B.H., Siegmund K.D., Shibata D. Spatial mutation patterns as markers of early colorectal tumor cell mobility. Proc. Natl. Acad. Sci. U S A. 2018;115:5774–5779. doi: 10.1073/pnas.1716552115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser M.D., Yu M., Grady W., Siegmund K., Shibata D. Epigenetic heterogeneity in human colorectal tumors reveals preferential conservation and evidence of immune surveillance. Sci. Rep. 2018;8:17292. doi: 10.1038/s41598-018-35621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser M.D., Mallo D., Hall A., Hardman T., King L.M., Tatishchev S., Sorribes I.C., Maley C.C., Marks J.R., Hwang E.S., Shibata D. Minimal barriers to invasion during human colorectal tumor growth. Nat. Commun. 2020;11:1280. doi: 10.1038/s41467-020-14908-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A., Kang H., Ma Z., Graham T.A., Salomon M.P., Zhao J., Marjoram P., Siegmund K., Press M.F., Shibata D., Curtis C. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R., Hu Z., Sottoriva A., Graham T.A., Harpak A., Ma Z., Fischer J.M., Shibata D., Curtis C. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nat. Genet. 2017;49:1015–1024. doi: 10.1038/ng.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N., Kanatani S., Tomer R., Sahlgren C., Kronqvist P., Kaczynska D., Louhivuori L., Kis L., Lindh C., Mitura P. Whole-tissue biopsy phenotyping of three-dimensional tumours reveals patterns of cancer heterogeneity. Nat. Biomed. Eng. 2017;1:796–806. doi: 10.1038/s41551-017-0139-0. [DOI] [PubMed] [Google Scholar]
- Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waclaw B., Bozic I., Pittman M.E., Hruban R.H., Vogelstein B., Nowak M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525:261–264. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.J., Werner B., Heide T., Curtis C., Barnes C.P., Sottoriva A., Graham T.A. Quantification of subclonal selection in cancer from bulk sequencing data. Nat. Genet. 2018;50:895–903. doi: 10.1038/s41588-018-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.J., Sottoriva A., Graham T.A. Measuring clonal evolution in cancer with genomics. Annu. Rev. Genomics Hum. Genet. 2019;20:309–329. doi: 10.1146/annurev-genom-083117-021712. [DOI] [PubMed] [Google Scholar]