

Abstract
Background
Point mutations and structural variations (SVs) in mitochondrial DNA (mtDNA) contribute to many neurodegenerative diseases. Technical limitations and heteroplasmy, however, have impeded their identification, preventing these changes from being examined in neurons in healthy and disease states.
Methods
We have developed a high-resolution technique—Mitochondrial DNA Structural Variation Sequencing (MitoSV-seq)—that identifies all types of mtDNA SVs and single-nucleotide variations (SNVs) in single neurons and novel variations that have been undetectable with conventional techniques.
Findings
Using MitoSV-seq, we discovered SVs/SNVs in dopaminergic neurons in the Ifnar1−/− murine model of Parkinson disease. Further, MitoSV-seq was found to have broad applicability, delivering high-quality, full-length mtDNA sequences in a species-independent manner from human PBMCs, haematological cancers, and tumour cell lines, regardless of heteroplasmy. We characterised several common SVs in haematological cancers (AML and MDS) that were linked to the same mtDNA region, MT-ND5, using only 10 cells, indicating the power of MitoSV-seq in determining single-cancer-cell ontologies. Notably, the MT-ND5 hotspot, shared between all examined cancers and Ifnar1−/− dopaminergic neurons, suggests that its mutations have clinical value as disease biomarkers.
Interpretation
MitoSV-seq identifies disease-relevant mtDNA mutations in single cells with high resolution, rendering it a potential drug screening platform in neurodegenerative diseases and cancers.
Funding
The Lundbeck Foundation, Danish Council for Independent Research-Medicine, and European Union Horizon 2020 Research and Innovation Programme.
Keywords: Mitochondrial DNA, Neurodegeneration, Single-Cell Sequencing, Disease biomarker, Neuron
RESEARCH IN CONTEXT.
Evidence before this study
Somatic mitochondrial DNA alterations have been linked to neurodegeneration and are found in all types of cancer. However, due to heteroplasmy—the presence of several hundred copies of the circular mitochondrial genome in a cell—it is difficult to identify and analyse disease-causing mutations in mitochondrial DNA. Further, there are no tools with which such mutations can be detected with sufficient accuracy in single cells, especially neurons.
Added value of this study
Our method, Mitochondrial DNA Structural Variation Sequencing (MitoSV-seq), is an advance in single-cell sequencing technology that has allowed us to detect novel mitochondrial DNA mutations in purified dopaminergic neurons in a mouse model of Parkinson disease easily while avoiding contamination. We also confirmed its use in other cell types and cancers in human. Moreover, we found a region of mitochondrial DNA that is prone to mutations in several haematological cancers and across species.
Implications of all the available evidence
MitoSV-seq is a powerful tool for discovering disease-associated mutations in mitochondrial DNA in single cells quickly and specifically. MitoSV-seq has tremendous potential for applications in the development of disease biomarkers and drug screening platforms.
Alt-text: Unlabelled box
1. Introduction
The mitochondrion is an energy-producing organelle that contains its own genome [1]. Although the mitochondrial genome has been believed to be inherited exclusively maternally, a recent study has demonstrated biparental transmission of mtDNA in an autosomal dominant-like pattern [2]. Mitochondrial diseases can arise from defects in nuclear-encoded and mitochondrial-encoded genes [3]. mtDNA diseases have variable clinical features, because mtDNA deficiency affects many tissues, and the involvement of mtDNA genetic mutations has been debated in neurodegenerative disorders, aging, and cancer due to their high energy consumption [3]. The accumulation of mtDNA mutations impairs the electron transport chain, triggers oxidative damage, and generates reactive oxygen species (ROS)—specifically, HO• radicals that form from H2O2 by Fenton reaction—which can contribute to energy failure and lead to neurodegenerative diseases [4,5].
Neurodegenerative disorders, such as Parkinson disease (PD) and Alzheimer disease (AD), are characterized by progressive nervous system dysfunction, but the causal genetic basis of most age-related neurodegenerative diseases is largely unknown. Approximately 5–10% of PD cases arise due to mutations in disease-related genes, but the dysfunction of mitochondria can also accelerate the pathogenesis of PD [6]. In particular, somatic point mutations and larger SVs in mtDNA in postmitotic neurons, especially in dopaminergic (DA) neurons in the substantia nigra (SN), have been speculated to be involved in PD [7], [8], [9], but the inferior sensitivity of mtDNA sequencing techniques has made the identification of mtDNA genetic variations unfeasible [10].
Somatic mitochondrial DNA alterations have been linked to neurodegeneration and are found in all types of cancer [11]. Mitochondria and their genomic instability could be important in several steps of tumour development [12,13], because tumour cells switch from oxidative phosphorylation to glycolysis to meet their energy demands. In the initial stage of tumourigenesis, mtDNA is more likely to be mutated as cancer cells begin adopting aerobic glycolysis. However, in late stages, they shift progressively to glycolytic metabolism, which can promote the selection of cells in which the mutations render them mitochondrial function-independent [14]. Thus, by examining mtDNA-SVs and SNVs in single cells, the ontology of cancer cells and their disease stages can be established.
Recurrent somatic alterations in mtDNA are found in 30% to 100% of tumours, including breast cancer, colorectal carcinomas, and haematological malignancies [12,13,[15], [16], [17], [18], [19]]. Mitochondria in cancerous cells can contain a pool of somatic mtDNA alterations with a level of heteroplasmy that is below current detection limits but has deleterious effects that alter mitochondrial function [20], [21], [22], [23]. Heteroplasmy is the presence of several hundred copies of the circular mitochondrial genome in a cell. Mutations can arise in a small subset of mtDNA molecules, resulting in a heterogeneous pool of mtDNA [24], which nevertheless could have a profound impact. In lung cancer, the rate of mutant heteroplasmy ranges between 0.31% and 97.04% [25]. This aspect becomes a significant issue when studying postmitotic cells, such as neurons, that might have high heteroplasmy that comprises many mutations that are below the 0.5% threshold, because they are not dividing.
The detection of rare heteroplasmic variations in whole mtDNA requires extremely accurate tools to distinguish between a substantial signal and background noise. Previous studies on mtDNA have performed whole-cell sequencing, which typically includes mtDNA, or have applied a combination of long-range polymerase chain reaction (PCR), gel purification, and Sanger sequencing, resulting in the omission of low-frequency heteroplasmic mtDNA deletions [9,[26], [27], [28], [29], [30], [31]]. Deletions are the most common SVs in mtDNA, but some reports indicate the presence of inversions in mtDNA and their correlation with aging and mtDNA instability [32], [33], [34], [35]. Further, most mtDNA deletions reside in the major arc, a region that lies between the origins of replication of the heavy and light strands. The major arc is especially prone to deletions due to its high load of short repeats, causing errors during mtDNA replication or repair [36]. Based on this leading theory, most studies have concentrated on sequencing the major arc rather than the entire mtDNA genome [37,38], necessitating sensitive, whole-mtDNA amplification and sequencing to increase our understanding of mitochondrial genome mutations in relation to diseases. Rolling circle amplification (RCA) is a useful tool for overcoming the difficulty of amplifying circular mtDNA. RCA favours amplification of circular templates (such as circular mtDNA), although linear DNA (genomic DNA and linear mtDNA) can also be amplified with RCA to a lesser extent [39]. RCA was first reported to amplify vector DNA, such as M13, and plasmid DNA from single colonies and plaques [40].
One method of circumventing the technical limitations that are imposed by mtDNA heteroplasmy is single-cell analysis. The past several years have witnessed rapid development of technologies that analyse the genome and transcriptome of single cells in detail [41,42]. Recent evidence from studies of single cells has shown that each cell type has a distinct lineage and function, which determines how they respond to each other and the environment—which has particular relevance in cancer [43]. Cancer scDNAseq (single-cell DNA sequencing) is especially useful for examining the depth of genomic complexity and composite mutations, whereas conventional bulk sequencing is unable to resolve the patterns by which these mutations co-occur in single cells of a tumour [44]. Moreover, amplification of a single-cell genome—also known as a single amplified genome (SAG)—followed by ultra-deep sequencing, provides greater coverage depth of all genomic variations [45]. The evolutionary integration of nuclear mitochondrial DNA segment (NUMT) pseudogenes in the nuclear genome must be considered in mtDNA deep sequencing [46], because they can affect the PCR amplification and read alignment of mtDNA, reducing the coverage of sequencing.
In this study, we describe a method that significantly supports whole-mtDNA sequencing of single cells using sequence-agonist random hexamers (RHs) for amplifying circular mtDNA. We demonstrate that this application has the capacity to detect somatic mtDNA SVs and SNVs in single DA neurons from the murine brain. We have shown that mice that lack Ifnar1 (Ifnar1–/–) develop a PD-like pathology that recapitulates most aspects of the disease in humans, comprising protein aggregation in Lewy body (LB)-like structures, progressive neurodegeneration, and neurological manifestations of PD, including cognitive impairments [47]. In the present study, we used this model as a source of brain DA neurons, giving us access to the affected neurons at an earlier stage of the disease. To perform mtDNA sequencing in a particular type of neurons—in this case, DA neurons—they must be separated from other neurons and other cell types, such as glial cells. The preferred techniques for isolating single neurons have been magnetic cell separation (MACS) [48] and laser capture microdissection (LCM) [9,49], both of which can suffer from nonspecific contamination by other cell types [50].
In this study, as a part of the MitoSV-seq method, single DA neurons were isolated from the SN by fluorescence activated cell sorting (FACS). Phi29 DNA polymerase was used to perform bulk amplification of mtDNA by RCA [30,51,52] (generation of periodic DNA nanotemplates), after which ultra-deep sequencing was applied to detect SNVs and all types of SVs with deep coverage. The RCA, library preparation, and data processing steps included a positive control (PC)—mtDNA that carried a 4.3-kb deletion—allowing us to benchmark and detect known and novel mtDNA variations that are associated with PD pathology in this model that were not identified using conventional techniques. We successfully applied and verified our new MitoSV-seq method and characterised several mutations and variations due to mtDNA instability in various human cancers: breast cancer, colon cancer, and 2 primary cancers of myeloid lineage—acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS). The higher resolution of mtDNA variations that are obtained using MitoSV-seq at the single-cell level demonstrates the power of our technique for single-cell analysis.
2. Materials and methods
2.1. Animals
Ifnar1–/– mice [53] were backcrossed 20 generations to C57BL/6J mice (IMSR Cat# JAX:000664, RRID:IMSR_JAX:000664) to generate the Ifnar–/– mice that were used in this study [47]. The wild-type animals were Ifnar+/+ littermate mice. Mice were housed in standard facilities. Sex- and weight-matched mice were used in the experiments, which were performed in accordance with the animal use and ethical committee guidelines in Denmark and approved by our institutional review boards (ethical permission #2013−15−2934−00807). Mice were used in triplicate.
2.2. Human samples
Fresh blood samples were obtained from 4 healthy donors (aged 26-46 years, 2 males and 2 females). PBMCs were isolated from fresh blood or buffy coats using the SepMate™ PBMC Isolation kit (85450; Stemcell Technologies). Fresh buffy coats were provided from 3 patients with AML (aged 53-78 years, males) and 3 patients with MDS (aged 60-86 years, females) with multilineage dysplasia, who provided informed consent regarding the collection and use of this material.
Studies of the human samples were approved by the Danish Data Protection Agency (J.nr. 2012-58-0004) and the National Committee on Health Research Ethics (Project ID CVK-1705391).
2.3. Cell culture
Cortical neuron cultures: Primary cortical neuron (CN) cultures were prepared by dissecting the cortex from 1-day-old Ifnar1– /– and Ifnar1+/+ mouse pups and cultured as described [54] for 6 days and 21 days. To obtain pure CNs, cultures were treated with 10 μM FUDR (fluorodeoxyuridine) after 24 hours in culture; then, the culture media was removed and replaced with normal media.
PBMC cultures: The basal culture media comprised RPMI-1640 medium with 1% (v/v) GlutaMAX-I, 1% (v/v) non-essential amino acids, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, 50 U/ml penicillin, 50 μg/ml streptomycin, 1% (v/v) kanamycin, and 5% (v/v) human serum. PBMCs were cultured in a humidified chamber at 37 °C, 5% CO2.
Cancer cell line cultures: The breast cancer cell lines MCF10A (ATCC® CRL-10317™, RRID:CVCL_0598) and MCF7 (ATCC® HTB-22™, RRID:CVCL_0031) and colon cancer cell lines SW480 (ATCC® CCL-228™, RRID:CVCL_0546) and SW620 (ATCC® CCL-227™, RRID:CVCL_0547) were cultured in Dulbecco Modified Eagle's medium (DMEM) with Ham's F12 nutrient supplement and 10% foetal bovine serum (FBS). Cells were cultured in a humidified chamber at 37 °C, 5% CO2.
Induction of mild oxidative stress: Cell lines and PBMCs from healthy donors were treated in triplicate with 100 µM H2O2 for 24 h and washed 3 times with PBS. The cells were then processed similarly as untreated cells for single-cell isolation by cell sorting.
2.4. Single-cell isolation by cell sorting
Dopaminergic neurons: We performed flow cytometry per Guez-Barber et al. [57], with some modifications, to purify single neurons from the SN—ie, DA neurons from dissected brain stems (including the midbrain, pons, and medulla oblongata) from mice. Each BS was placed in a microfuge tube with 1 ml of Low-Fluorescence Hibernate A (HA-LF; Brain Bits) on ice, and the cells were dissociated according to the protocol [55]. Next, 1 ml of Accutase (A1110501; Thermo Scientific™) was added for 15 min at 4 °C to dissociate the cells. Large cell clusters and debris were removed from the cell suspension by serial filtration through 100-μm and 40-μm cell strainers (Falcon 352360 and Falcon 352340; BD Bioscience).
Using Percoll (P1644; Sigma), 3-step density gradient centrifugation (high-density solution: 3426 ml Hibernate A + 824.5 μl Percoll + 97.8 μl 1 M NaCl; medium-density solution: 3600 ml Hibernate A + 650.5 μl Percoll + 76.5 μl 1 M NaCl; low-density solution: 3770 ml Hibernate A + 480.3 μl Percoll + 59.5 μl 1 M NaCl) was then performed to remove small cellular debris [55]. One millilitre of each solution (bottom: high-density; middle: medium-density; top: low-density) was layered carefully in a 15-ml Falcon tube. The filtered cell suspension (from the previous step) was applied to the top of this gradient and centrifuged at 430 × g for 3 min at 4 °C. The cloudy top layer (∼2 ml), containing cell debris, was discarded. The cells in the remaining layers were centrifuged at 550 × g for 5 min at 4 °C. Cell pellets were resuspended in 1 ml of Hibernate A and divided into microfuge tubes for immunolabelling. Cells were fixed in 50% ethanol on ice for 15 min with occasional inversion, pelleted by centrifugation at 600 × g for 2 min, and resuspended in phosphate-buffered saline (PBS). Dopaminergic neurons were co-labelled with antibodies against NeuN (MAB377; Millipore, RRID:AB_2298772) and tyrosine hydroxylase (TH; AB1542; Millipore, RRID:AB_90755), diluted 1:100 in PBS with 1% bovine serum albumin (BSA; A3608, Sigma).
During the incubation, DA neuronal cells were rotated for 30 min at 4 °C. Next, the neurons were washed once by adding 800 μl PBS to each tube and centrifuged at 950 × g for 3 min before being resuspended in PBS. The DA neurons were then incubated with fluorescent labels and rotated for 15 min at 4 °C. TH and NeuN double-positive cells (Fig. 3c) were incubated with Alexa Fluor 488-labelled anti-rabbit (Molecular Probes Cat# A-11070, RRID:AB_142134) and Alexa Fluor 647-labelled anti-mouse (Molecular Probes Cat# A-21236, RRID:AB_141725), diluted 1:1000 in PBS with 1% BSA. The cells were washed again with 800 μl PBS and 1% BSA, centrifuged at 950 × g for 3 min, and resuspended in 1 mL PBS with 1% BSA on ice for the next step.
Fig. 3.

Evaluation of MitoSV-seq method in dopaminergic neurons from a Parkinson disease model, Ifnar1–/– mice. (a) Position of the deletion in the PC. (b) Confirmation of 4.3-kb deletion in the PC. kb, kilobase pair; WT, wild-type. (c) Single DA neuron isolation by FACS. DA, dopaminergic. (d) qPCR using TH primers confirms sorting of DA neurons. TH, tyrosine hydroxylase. (e) RCA of mtDNA from single DA neurons amplifies concatemers that can be separated by enzymatic digestion. (f) Numerical proportions of detected variations in Ifnar1–/– using MitoSV-seq. Pie chart represents total number of SVs and SNVs. (g) Heteroplasmy% distribution for individual SVs detected inIfnar1–/– mice. DEL: deletion; INV: inversion; INS: insertion; TD: tandem duplication. (h) Position of SVs and SNVs detected with MitoSV-seq in Ifnar1–/– mice. Circos plots of mouse mtDNA genome displaying heavy and light origins of replication (OH and LO, respectively), major arc, and D-loop (outer blue circle). mtDNA genes are displayed in the middle circle (grey lines). Depicted are SVs as arches with deletions (blue), tandem duplications (red), inversions (green), and insertions (orange). Thickness of the arches corresponds to SV heteroplasmy. SNVs are marked with short red lines (_). Intensity of red lines corresponds to SNV heteroplasmy. (i) Immunostaining of CN cultures showing their purity. Scale bars equal 10 microns. (j) Flow cytometric analysis, showing 93.6% purity of CN cultures. (k) Comparison of conventional techniques with MitoSV-seq in Ifnar1–/– mice. Each dot represents one variation distributed according to heteroplasmy%. CNs: cortical neurons; D6: DIV=6; D21: DIV=21; W6: 6-week-old brains. (l) Number of reads aligned to the whole mouse reference genome (mm10) obtained from MitoSV-seq and conventional methods. Total number of representative reads from both techniques (app. 3.5 million matching mouse DNA) and the rest of the read not shown in this graph were mapped to mouse mtDNA.
The cells were sorted on a BD FACSAria II (BD Biosciences). A small portion of fixed cells was incubated without antibodies to gate the cells, and another portion was labelled only with secondary antibodies to set the maximum threshold for nonspecific binding. Then, the test samples that were labelled with primary antibodies and fluorescent labels were scanned and sorted at 4 °C. Single DA neurons were sorted into 96-well PCR plates with 2 μl of mild lysis buffer (freshly prepared 0.2% Triton X-100, 93443; Sigma), to be used directly in the DNA amplification step. The plates were kept on ice or in IsoFreeze PCR racks during the sort and stored at −80 °C for longer periods.
Myeloid cell isolation: Single myeloid cells were sorted using CD11b (ab8878; Abcam, RRID:AB_306831) into 96-well PCR plates with 2 μl of mild lysis buffer, and 500,000 cells were sorted into Eppendorf tubes (0030125150; Eppendorf) for bulk experiments.
Single cancer cell isolation: Cancer cells were stained with LIVE/DEAD Fixable Violet (L34955; Invitrogen), and single living cells were sorted into 96-well PCR plates.
2.5. Real-time quantitative PCR
After the purification of DA neuronal cells was confirmed, single cells were treated with DNase. RNA from samples was reverse-transcribed directly into cDNA using the QuantiTect Reverse Transcription kit (205310; Hilden) according to the manufacturer's instructions. cDNA was analysed by real-time qPCR with primers that were designed for TH and two nuclear housekeeping genes (Supplementary Table 22), with each sample repeated in technical triplicate. qPCR was performed using Maxima SYBR Green/ROX qPCR Master Mix (K0222; Thermo Scientific™).
2.6. MitoSV-seq protocol
2.6.1. Generation of a deleted mtDNA as PC for MitoSV-seq
We generated a 4.3-kb deletion as PC for SV from N2a mt-DNA as follows:
-
a
Mitochondrial fractionation from N2a cells and mtDNA precipitation
N2a cells were scraped into 1X PBS (70011-036, Life Technologies) and pelleted at 300 g for 3 min. The pellet was resuspended in mitochondrial isolation buffer, containing 5 mM HEPES (H3375-100G, Sigma-Aldrich), 210 mM mannitol (63560, Fluka), 70 mM sucrose (84100, Fluka), 0.2 mM EGTA (E3889-100G, Sigma-Aldrich), and 3 mM MgCl2 (M8266-100G, Sigma-Aldrich). Phosphatase inhibitor cocktail (1:500) (P5726-5ML, Sigma-Aldrich) and protease inhibitor cocktail (P8340, Sigma) were also added to the buffer. The cells were lysed in mitochondrial isolation buffer by passing them through a 27G needle (302200, BD Microlance) 10 times, incubating them on ice for 20 min, and passing them through the 27G needle another 10 times. The cells were centrifuged at 3000 rpm for 5 min, and the supernatant was harvested and centrifuged again to clear it of potential nuclear contamination.
The supernatant was then centrifuged at 10,000 g for 10 min to pellet the mitochondrial fraction. The supernatant, containing the cytoplasmic fraction, was harvested and centrifuged again to eliminate any mitochondrial contamination. The 2 mitochondrial pellets were resuspended in mitochondrial isolation buffer, pooled, and centrifuged at 10,000 g for 10 min and resuspended in RIPA (89900, Thermo Scientific™) to form the mitochondrial fraction. All centrifugation steps were performed at 4 °C.
To precipitate the mtDNA from fractionated mitochondria, one-tenth of the sample volume of proteinase K (1017738, QIAGEN) was added and incubated at 56 °C for 10 min. One-tenth of the sample volume of sodium acetate (TA942068, Merck) was added after the incubation, followed by the addition of 2.5 times the sample volume of absolute ethanol (1680766, CCS Healthcare). After being mixed thoroughly, the samples were left at -20°C overnight and then centrifuged at full speed for 20 min at 4 °C. The DNA pellets were washed with 70% ethanol (1680766, CCS Healthcare) and centrifuged at full speed for 15 mins at 4 °C. The pellets were left to dry before being dissolved in nuclease-free water.
-
b
Introduction of 4.3-kb deletion in mouse mtDNA
A restriction map of mouse mtDNA was predicted using Gene Runner (v6.5.51) to identify restriction enzymes with two cleavage sites in mtDNA. NheI (R0131S, BioLabs) was used to digest the mtDNA at 5930 bp and 10,395 bp, yielding two mtDNA fragments of 4345 bp and 11,950 bp. The digestion was performed at 37 °C for 3 hours.
The 11,950-bp fragment was purified from an agarose gel using the GenepHlow™ Gel/PCR Kit (DFH100, Geneaid) (Fig. 3a-b) and ligated using T4 DNA Ligase (EL0014, Thermo Scientific™) for 1 hour at room temperature before enzyme inactivation. The deletion in the PC was confirmed by PCR (Supplementary Table 1).
2.6.2. mtDNA amplification by conventional methods
Total DNA from CNs and four types of malignant cells was purified using the DNeasy Blood & Tissue Kits (69504, QIAGEN). Circular mtDNA was amplified using two pairs of site-specific primers (MTL primers) that were designed for human mtDNA (Supplementary Table 1) and KOD Hot Start Master Mix (71842, Merck) according to the manufacturer's protocol. Amplified PCR fragments were purified from agarose gels and processed for sequencing. The quantity and size distribution of the mitochondrial amplicons (MTL‐F1 + MTL‐R1 and MTL‐F2 + MTL‐R2) were determined using Agilent DNA 12000 chips (5067-1508; Agilent Technologies) on an Agilent Technologies 2100 Bioanalyzer (G2939BA: Agilent Technologies).
2.6.3. MitoSV-seq mtDNA amplification
In MitoSV-seq, RCA of the circular mtDNA was performed using Phi29 DNA Polymerase (Templiphi; GE Healthcare 25-6400-10). Two microlitres of the cell lysate from 20 single DA neurons (10 individual Ifnar1+/+ and Ifnar1–/– cells each), 10 single myeloid cells from each patient with AML and MDS and healthy donors, and 10 single cells from each cell line (SW480, SW620, MCF10A, and MCF7) were used as the template to be amplified with Phi29 DNA polymerase and RHs according to the manufacturer's protocol. For bulk amplification, the entire genome was isolated using the DNeasy Blood & Tissue Kits (69506; QIAGEN), and 2 ul (10 ng) was used for RCA.
Amplification was performed for 2 hours at 30 °C; longer amplification times favoured genomic DNA amplification over mtDNA. The PCR products were loaded onto an agarose gel (A9539; Sigma) to assess the amplification of concatemers. TempliPhi-amplified DNA was quantified by PicoGreen® dsDNA quantitation assay (P7581; Invitrogen). To confirm the RCA of the mtDNA and to increase the efficiency of the library preparation for NGS, concatemers were digested using XhoI (FD0694; Thermo Scientific™), which has only one restriction site in mouse mtDNA, into linear 16.3-kb fragments (Fig. 3e). The resulting concatemers were processed directly for sequencing.
To verify the propensity for circular DNA to be amplified, RCA was performed as described above using purified mtDNA (circular) and XhoI-linearized mtDNA, followed by purification (GenePhlow, Geneaid).
2.6.4. Next-generation sequencing (NGS) and data processing
The digested concatemers from RCA and amplicons from the site-specific PCR were processed for library preparation using the Nextera XT DNA Library Preparation Kit (FC-131-1096, San Diego) according to the manufacturer's instructions. AMPPure XP beads were used to purify the DNA library and provide a size selection step to remove short library fragments. Bioanalyzer-based normalization was conducted using the Agilent High-Sensitivity DNA kit (5067-4626, Agilent Technologies) to determine the molarities of the libraries. The libraries were pooled at equimolar amounts and run at 1.4 picomolar (spiked with 1.4 picomolar PhiX) in paired-end 300-cycle high-mode on an Illumina NextSeq 500 (Illumina Inc.), which produced ∼400 million reads.
FASTQ files were transferred automatically to BaseSpace and quality-trimmed using Trimmomatic v0.36 [56], and adaptors were removed using cutadapt v1.16. Reads were aligned using Bowtie2 [57] with default parameters against the mouse mtDNA reference sequence NC_005089 and the mtDNA reference sequence NC_012920.1. Considering that mtDNA has a circular genome, alignment tools use a linear form as the reference to align sequence reads. As a result, the reads that cover the artificial breakpoint in the circular genome are not aligned properly and are discarded. We used two frames of the mtDNA—one with a breakpoint in the replication control region to “start” at position 1 and “end” at position 16,299 for mouse (16,569 for human) and a shifted mtDNA sequence with a breakpoint in the internal region of the genome at position 3710. Thus, the “shifted” reference sequence begins at original position 3710 and ends at original position 3709. Two frames were also used for the human mtDNA sequence.
Mpileup was used to call SNVs with BAM files (SAM tools v0.1.19). Variant calling was performed using VarScan2 (version 2.3.6) using default parameters for SNVs and INDELs. VarScan Somatic was used to compare the variations between wild-type cells (as germlines) and knockout cells (as tumours). To maximize the specificity, reads were not considered if their mapping quality was below 30 and if their average per-base sequencing quality fell below 30. Pindel v0.2.5 [58] was used to detect breakpoints of large structural variations, including deletions, insertions, duplications, and inversions. Detected SVs were discarded if fewer than 20 unique reads with an upstream anchor and fewer than 20 unique reads with downstream anchors spanned the variant area to maximize specificity by setting stricter filtering criteria. Heteroplasmy% was calculated by dividing the total number of mutations at a certain nucleotide position by the total number of genomes that was represented at the position, multiplied by 100.
To analyse contamination by NUMTs, trimming and alignment were performed using BWA (as described above), but instead of being aligned to mouse mtDNA, it was mapped to mm10 or the entire mouse genome, and reads that were lost due to NUMTs were quantified.
3. Results
3.1. Overview of MitoSV-seq method
To examine the function and association of mtDNA mutations in human disorders, a sensitive method is required to scan all parts of mtDNA and characterise all types of mtDNA mutations that confer their selective advantages in progressive human diseases. To this end, we developed the MitoSV-seq method to detail all types of mtDNA genetic damage with various frequencies from a broad range of cell types in a species-independent manner. MitoSV-seq generates a clear pattern of mtDNA alterations that might affect mitochondrial function in relation to neurodegeneration and cancers.
The MitoSV-seq workflow typically consists of 3 steps: (i) preparation of single cells, (ii) preparation of a positive control (PC), and (iii) RCA and ultra-deep sequencing (Fig. 1). For the first step, single cells can be obtained from solid tissues, such as brain (in this study, brain stem but equally applicable to other parts of the brain); fluids, including peripheral blood; and cell cultures. Single cells are sorted using cell specific markers directly in lysis buffer. A PC for SV was established by engineering a 4.3-kb deletion in the mtDNA to be used as a premise for the experiment. RCA is performed on the PC and cell lysate using RHs, dNTPs (deoxyribonucleotide triphosphates), and Phi29 DNA polymerase to produce high-molecular-weight concatemers. To increase the efficiency of the library preparation step, the concatemers were digested to 16.3-kb mtDNA fragments.
Fig. 1.

Overview of MitoSV-seq method. Samples were collected from mouse brain, cancer cell lines, and human buffy coats. Single cells were isolated by FACS using cell-specific markers. PC preparation is optional, and although it is not necessary, it is highly recommended for mtDNA sequencing. PCs and single-cell lysates were amplified by RCA. High-molecular-weight concatemers were digested using XhoI. Tagmentation and indexing were performed using the Nextera XT DNA Library Preparation Kit. Libraries were pooled and run for paired-end 300-cycle high-mode sequencing on a NextSeq 500. NGS data were analysed using standard DNA-seq methods. Pindel [58] was applied for SV calling. N2a, Neuro-2a; mtDNA, mitochondrial DNA; PC, positive control; RCA, rolling circle amplification; RHs, random hexamers; dNTP, deoxyribonucleotide triphosphate; DA, dopaminergic.
For the alignment and data analysis, we used two frames of mtDNA to maintain coverage around the artificial breakpoint in the circular genome. The average sequencing depth across the genome was 30 ± 2.30 for single cells. To circumvent the contamination of NUMTs in our analytical pipeline [59], we aligned the sequencing reads to the mtDNA genome rather than the entire genome (mm10 and hg19), preventing the loss of reads due to alignment to NUMTs. The analysis included visualisation of SVs and SNVs using Circos plots—the recommended schematic representation for mtDNA-SVs—which can be created for all types of SVs and SNVs using Circlize package 0.4.7. The script for autogenerating circos plots for mtDNA-SVs and SNVs is provided as part of this study. The lists of SVs and SNVs must be prepared in standardised browser extensible data (BED) format.
3.2. Advantage of MitoSV-seq over existing approaches
There are many commercial protocols for sequencing mtDNA that use Sanger sequencing or NGS from various suppliers that support the identification of mtDNA mutations and structural variations. These approaches are inadequately robust to amplify and sequence all types of SVs and SNVs in mtDNA. Some of them are insufficiently sensitive to detect minor heteroplasmic variations, although these low-frequency variations can still affect mitochondrial function in regulating synaptic transmission, brain function, and cognition in aging [60]. Similarly, certain mtDNA mutations, even those with a low frequency, could confer a growth advantage to a small population of treatment-resistant cancer cells, which in turn could potentiate recurrence or generate metastatic potential. Despite attempts to sequence mtDNA in single cells, most studies have merely analysed a small region of mtDNA [primarily the D-loop, the noncoding displacement (D) loop] [61,62], which might underestimate the importance of all mitochondrially encoded genes in human disorders.
The MitoSV-seq method is easy to set up for many species (tested in mouse and human) in a high-throughput manner due to the application of RHs rather than site-specific primers, which can hybridise to DNA from bulk cells and that from a single cell. Bench implementation confirmed the use of MitoSV-seq in the identification of all types of mtDNA genetic variations in various disease-specific cell types. MitoSV-seq also assesses mtDNA damage at the single-cell level with better sensitivity and output compared with bulk samples due to the greater sequencing depth that can be obtained with the mtDNA copies of a single cell. Bulk sequencing reduces the depth of sequencing for each copy of mtDNA, diluting the detected variations.
Fig. 2 presents a step-by-step comparison of mtDNA sequencing between conventional methods and our newly developed high-resolution MitoSV-seq technique. Conventional methods have focused primarily on brain homogenates or bulk cortical neuron (CN) cultures to identify mtDNA variations in neurodegenerative conditions. Because DA neurons are more sensitive to mitochondrial deficiency and accumulate mtDNA damage more frequently than other cell types in the brain, using brain homogenates might dilute the number of variations and their heteroplasmy.
Fig. 2.

Step-by-step schematic between MitoSV-seq and conventional methods. Conventional methods have focused primarily on brain homogenate, bulk cortical neurons (CNs), or single DA neurons isolated by MACS or LCM, followed by PCR amplification of mtDNA using site-specific primers designed mainly for the D-loop or major arc. Amplified PCR fragments are usually purified from agarose gels and processed for NGS library preparation and sequencing. MitoSV-seq benefits from FACS in characterising and isolating single cells from a heterogeneous pool of cells using cell-specific markers. Single cells are sorted and used directly for the amplification step. Concatemers from circular mtDNA are generated using RHs and phi29 DNA polymerase in a rolling circle manner. Addition of our custom PC allows to us to first set up the analytical pipeline to find a known PC deletion. High-molecular-weight concatemers are digested and used for NGS library preparation and ultra-deep sequencing. MACS, magnetic cell separation; LCM, laser capture microdissection; PCR; polymerase chain reaction; FACS; fluorescence-activated cell sorting; RHs, random hexamers; RCA, rolling circle amplification; PC, positive control.
Several studies have attempted to determine the distribution of mtDNA variations in single DA neurons. The most commonly used and recommended techniques for isolating single neurons have been magnetic cell separation (MACS) [48] and laser capture microdissection (LCM) [9,49], which have a higher probability of nonspecific contamination by other cell types. Nonspecific contamination during MACS can arise from the adsorption of background cells to the capture device. Further, immunomagnetic techniques can only separate cells into positive and negative populations, which is not useful for double or multiple selection of cells using specific antibodies [63]. Also, the separation of a minute population of cells or single cells from a heterogeneous population using LCM can be inaccurate and is more prone to contamination from neighbouring cells, compounded by the limitations of being combined with multiple antibody stains. The sample preparation for LCM can affect the integrity of mtDNA, and the quality of microdissected cells might fall short of the quality that is required for further analysis [50].
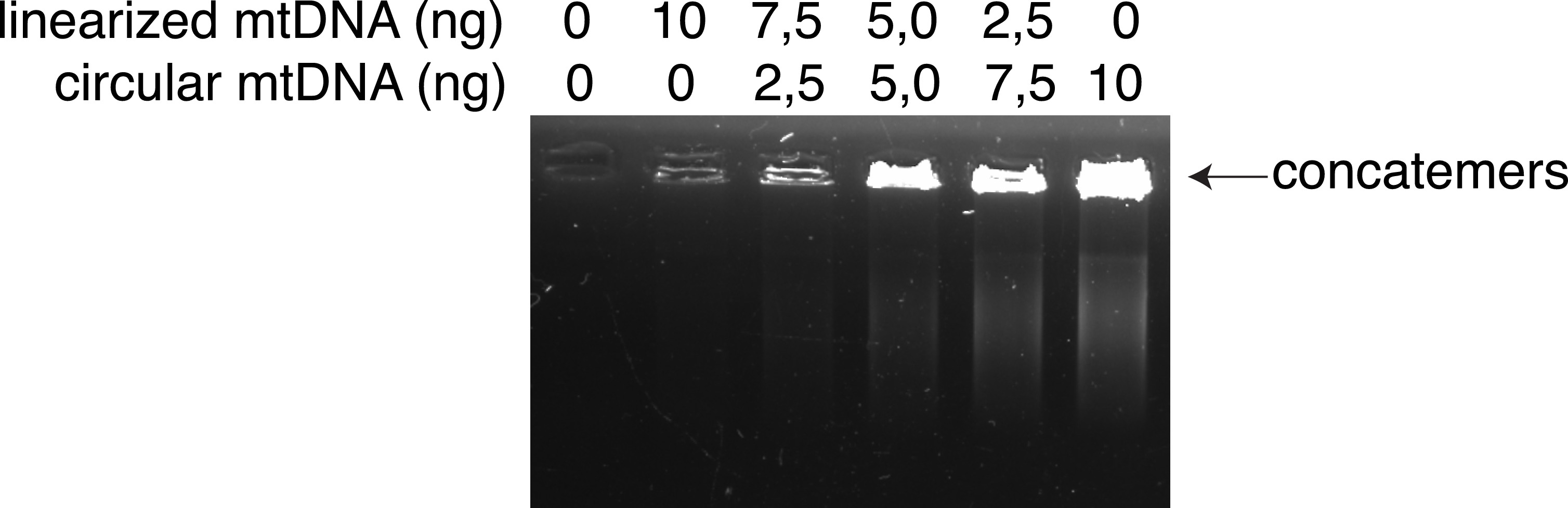
In this study, we confirmed that FACS is an efficient and feasible technique for characterising and sorting various cell types in a heterogeneous cell population, based on the exclusion of duplets, cell size, granularity, and combination of specific cell markers that are stained with fluorescently labelled antibodies. Also, FACS ensures the selection of live cells on live/dead cell gating. To prevent the degradation of mtDNA, times for performing the steps should be considered. For isolating single DA neurons, the time from dissection of the brain to fixation should be under 30 min. Buffy coats and resuspended cell cultures should be kept on ice before fixation. Another issue regarding the conventional methods is the amplification of mtDNA by primer-specific PCR and gel purification. The combination of these techniques can lead to the loss of infrequent SVs, which cannot be observed on an agarose gel for excision and purification. MitoSV-seq applies RCA using RHs, with a greater tendency to amplify circular DNA over linearized DNA (Supp. Fig. 1), promoting the amplification of circular mtDNA over linearized DNA. Further, RCA amplifies DNA from various sections, which will not be affected by any SV or point mutation in the mtDNA.
3.3. Evaluation of MitoSV-seq in single dopaminergic neurons from a PD model: Ifnar1–/– mice
The efficacy of the method was evaluated in Ifnar1–/– mice, a mouse model of PD. Neurons in this model experience oxidative phosphorylation damage to mtDNA, which induces neuronal accumulation of senescent or damaged mitochondria [47]. Initially, we struggled to determine the genetic basis of the mitochondrial damage in Ifnar1– /– mice using conventional techniques, because only two SNVs and no SVs were seen in days in vitro (DIV)=6 CN cultures, versus 27 SNVs and no SVs in DIV=21 CN cultures (Fig. 3). To overcome this deficiency, we designed the MitoSV-seq method and compared its efficacy with conventional techniques.
One advantage of using animal models to study neurodegeneration is that we can focus on earlier stages of the disease, when dopaminergic neurons in the SN are still alive. We chose 6-week-old mice, the time at which brain development has completed [64] but before most PD pathological features, such as LB and motor dysfunction, are observed [47]. Based on reports that have challenged the ability of NGS to find deletions and other structural variations in mtDNA [9,[26], [27], [28], [29], [30], [31]], we designed an internal PC for MitoSV-seq by introducing a 4.3-kb deletion from 5930 bp to 10,395 bp into mouse mtDNA to benchmark our method (Fig. 3a-b). The addition of this PC allowed us to establish our analytical pipeline to detect a known PC deletion, after which we applied our method to other samples.
To purify single DA neurons from the SN, we performed flow cytometry to select double-positive neurons for NeuN (neuronal nuclei) and tyrosine hydroxylase (TH) in Ifnar1+/+ and Ifnar1–/– mice (Fig. 3c). We validated the purity of this neuronal sorting by qPCR, which demonstrated the expression of TH in isolated single cells (Fig. 3d). The mtDNA that was released from each single cell and the PC was amplified with Phi29 DNA polymerase and RHs. Concatemers were digested to create linear mtDNA fragments and increase the efficiency of the library preparation for NGS (Fig. 3e). Ultra-deep sequencing (UDS) of mtDNA yielded ∼1 Gbp per individual neuron and detected 127 SVs and 143 SNVs in 7 of 10 neurons (70%) in Ifnar1–/– mice (Fig. 3f) compared with WT samples.
The identified SVs were distributed among 4 types with various percentages of heteroplasmy: 41 deletions (DELs), 24 inversions (INVs), 38 insertions (INSs), and 24 tandem duplications (TDs) (Fig. 3g). One INV and 1 TD reached 100% and 67%, respectively—ie, greater than the 50% heteroplasmy that had been suggested as the threshold for heteroplasmy that was necessary for pathogenicity. The SVs resided in mitochondrial tRNA genes, rRNA-encoding genes (mt-Rnr1), and the protein-encoding genes mt-Nd2, mt-Co1, mt-Atp6, mt-Co3, mt-Nd4, mt-Nd5, and mt-Nd6 (Fig. 3h; Supplementary Table 1). Mutations in mt-ND4, encoding mitochondrial NADH dehydrogenase 4, have been associated with Leber optic atrophy [65] and MELAS syndrome [66]. Mutations in mt-ND5, the gene for mitochondrial NADH dehydrogenase 5, have been reported in Parkinson disease and LEIGH syndrome [67]. The genes that encode mitochondrial tRNAs are highly susceptible to point mutations and deletions, which are a primary cause of mitochondrial dysfunction and are linked to a wide range of pathologies, including cardiomyopathy, pigmentary retinopathy, sensorineural deafness, cataract, cerebellar ataxia, and diabetes mellitus [68].
MitoSV-seq identified SNVs in Ifnar1–/– neurons at higher resolution, which were distributed throughout the mtDNA (Fig. 3h; Supplementary Table 2). Among 7 cells (of 10) in Ifnar-/- mice, 6 shared SNVs (Supplementary Table 3). To compare the output of MitoSV-seq with conventional techniques, we used cortical neuronal cultures at DIV=6 and DIV=21. The purity of the primary neuronal cultures (∼93%) was confirmed by immunofluorescence and flow cytometry (Fig. 3i and 3j, respectively). mtDNA was amplified according to the conventional techniques and sequenced, generating ∼1 Gbp per sample. Two SNVs and no SVs were detected in DIV=6 CN cultures, and 27 SNVs were seen in the 21-day-old cultures (Fig. 3k). Notably, MitoSV-seq detected SVs and SNVs in various regions of mtDNA with optimal sensitivity, which was not provided by the conventional method (Fig. 3). The SNVs in Ifnar1+/+ single DA neurons were considered to be germline and were filtered out in Ifnar1–/– animals. We also compared these methods with regard to contamination by NUMTs (Fig. 3L). We observed an approximately 20-fold decrease in NUMTs following the MitoSV-seq approach. These findings establish MitoSV-seq as a superior method over conventional approaches, identifying more mutations and deletions with greater heteroplasmy, especially from single cells. These data also emphasize the need to analyse the entire mtDNA sequence specifically in age-related disorders, rather than merely focusing on the major arc or D-loop.
3.4. MitoSV-seq applications
-
A
Validation of MitoSV-seq in breast cancer cell lines
We validated the MitoSV-seq method by examining the underlying mtDNA mutations in breast cancer. We used two breast cancer cell lines—MCF7, a tumourigenic cell line, and the nontumourigenic epithelial line MCF10A—to determine the function of mtDNA instability in human diseases in single cells (30 single cells, purified from each line, Fig. 4a) and bulk samples (2.5 million cells from each line). Next, we measured the effects of H2O2, an mtDNA oxidative damage-inducing agent, as a positive condition and thus validated the capacity of MitoSV-seq to study mtDNA integrity.
Fig. 4.

Validation of MitoSV-seq in breast cancer cell lines. (a) Single-cell isolation from MCF10A and MCF7 cell lines. Cell were stained with LIVE/DEAD Fixable Violet, and single living cells were sorted. (b) Evaluation of MitoSV-seq in bulk vs single cells.
Biological samples were used in triplicate. B: bulk; SC: single cell. (c) Schematic of distribution of human mtDNA detected in bulk and single cells by MitoSV-seq. Total numbers of detected variations in each condition and their overlapping variations are depicted in the Venn diagrams in the middle. Circos plots of human mtDNA genome displaying heavy and light origins of replication (OH and LO, respectively), major arc, and D-loop (outer blue circle). mtDNA genes are displayed in the middle circle (grey lines). Shown are SVs as arches with deletions (blue), tandem duplications (red), inversions (green), and insertions (orange). Thickness of the arches corresponds to SV heteroplasmy. SNVs are marked with short red lines (_) according to their positions in mouse mtDNA. Intensity of red lines corresponds to SNV heteroplasmy. (d) Mild oxidative stress with H2O2 in MCF10A and MCF7 cell lines introduces SVs and SNVs in mtDNA. T: treated. (e) Comparison of conventional technique with MitoSV-seq to identify SVs in bulk sample. C, conventional technique; N, new (MitoSV-seq).
MitoSV-seq detected more variations in single cells compared with bulk samples for each line. We did not identify any SVs or SNVs in bulk or individual MCF10A cells, as expected (Fig. 4b). More SVs and SNVs were detected in single MCF7 cells (108 variations in total) compared with bulk MCF7 populations (19) (Fig. 4c). The distribution of variations in single versus bulk cells is shown in Fig. 4c. In single-cell sequencing, SVs were observed only in the major arc. No variations were common between single and bulk cells. Notably, in only single-cell sequencing did we find the previously reported rs193302991 for familial breast cancer (OMIM:114480).
One possible technical explanation for the lack of common variations is that we collected 2.5 million cells for bulk sequencing and 30 single cells per line; thus, using bulk samples will reduce the efficiency of the technique due to there being more copies of mtDNA that need to be amplified and sequenced. Consequently, bulk sequencing dilutes the number of detectable variations, whereas single-cell sequencing detects variations more precisely. Biologically, this dispersion in variations might be attributed to the random mutagenic effect of oxidative stress in damaged mitochondria. Mild oxidative stress treatment effected more damage in MCF10A and MCF7 cells, indicating the direct mutagenic effects of ROS on mtDNA (Fig. 4d). These data confirm the contribution of ROS to mtDNA damage and demonstrate the impressive ability of MitoSV-seq to identify structural variations in mtDNA.
The MitoSV-seq method (N/New) was also compared with conventional techniques (C) using bulk samples, because mtDNA from a single cell is insufficient for amplification using the two pairs of site-specific primers that are required in the conventional methods. Total DNA from ∼2.5 million cells was extracted from each biological replicate, amplified using site-specific primers, and prepared for sequencing. Notably, MitoSV-seq identified more variations in bulk samples (9 SVs) than conventional techniques (4 SVs), with 1 SV overlapping between methods (Fig. 4e).
-
B
Validation of MitoSV-seq in colon cancer cell lines
To confirm the broader application of MitoSV-seq, we compared the colon cancer cell line SW480 with SW620, a colon cancer cell line that has been isolated from a metastatic site. Ten single living cells from each replicate were prepared for sequencing (Fig. 5a). Single-cell and bulk sequencing was performed using MitoSV-seq.
Fig. 5.

Validation of MitoSV-seq in colon cancer cell lines. (a) Single living cell isolation from SW480 and SW620 cell lines. (b) Evaluation of MitoSV-seq in bulk cells vs single cells in SW480 and SW620 cell lines. B: bulk; SC: single cell. (c) Distribution of variations in bulk and single SW620 cells with MitoSV-seq. Venn diagram represents total number of detected variations in each condition and their overlap. Circos plots of human mtDNA genome displaying heavy and light origins of replication (OH and LO, respectively), major arc, and D-loop (outer blue circle). mtDNA genes are displayed in the middle circle (grey lines). Also shown are SVs as arches with deletions (blue), tandem duplications (red), inversions (green, and insertions (orange). Thickness of the arches corresponds to SV heteroplasmy. SNVs are marked with short red lines (_) according to their positions in mouse mtDNA. Intensity of red lines corresponds to SNV heteroplasmy. (d) Identification of SVs and SNVs on treatment of SW480 and SW620 cell lines after mild oxidative stress induction. T: treated. (e) Comparison of conventional technique with MitoSV-seq in bulk sample. C, conventional technique; N, new (MitoSV-seq).
We found more variations in single SW480 cells compared with the bulk samples (Fig. 5b). More SVs and SNVs were also noted at the single-cell level in SW620 cells. However, the level of heteroplasmy of the variations was nearly the same as in the bulk sequencing, perhaps due to the nature of SW620 cells, which have more copies of mtDNA (∼2 times more) compared with SW480 cells [69], which will dilute the result. Overall, we found 117 variations in single SW620 cells (10 single cells per replicate) and 13 variations in bulk SW620 cells (∼2.5 million cells per replicate), with no overlap. MitoSV-seq detected larger SVs even in single cells: 2 large TDs (8.7 kb and 7.1 kb) were seen using MitoSV-seq in single SW620 cells (Fig. 5c). In addition to identifying novel variations only in single SW620 cells, MitoSV-seq detected 2 reported variations for colorectal cancer, COSM6716769 [70] and COSM6716773 [71] (Supplementary Table 8-11). Further, mild oxidative stress resulted in a higher load of mtDNA damage in SW480 and SW620 cells (Fig. 5d). Compared with conventional techniques, the same variations were detected in SW480 cells, but MitoSV-seq identified more SVs in the SW620 line (Fig. 5e).
-
C
Validation of MitoSV-seq in peripheral blood mononuclear cells from healthy donors and AML patients
PBMCs were isolated from four healthy donors and cultured with or without oxidative stress, and mtDNA was prepared and sequenced by MitoSV-seq. We characterized 15 SVs in untreated PBMCs and 19 SVs after treatment (Fig. 6a). Although 15 mutations were detected in healthy PBMCs, their mean heteroplasmy was ∼8%; thus, they might not be deleterious. In support of our findings, low-frequency, potentially high-pathogenicity mtDNA mutations have been reported in healthy humans [72]. At low frequencies, these harmful mtDNA mutations are benign, but at higher frequency, they are likely to cause mitochondrial dysfunction. Further, mild H2O2 treatment increased the number of SVs to 19, with a mean heteroplasmy of ∼13% (Fig. 6b).
Fig. 6.

Validation of MitoSV-seq in healthy donors and AML patients. (a) Effect of mild oxidative stress on mtDNA genetic variations in blood samples. MtDNA variations were identified in bulk cells using MitoSV-seq. (b) Heteroplasmy rates of detected SVs in PBMCs without and with treatment. (c) Single myeloid cell isolation. AML, acute myeloid leukaemia. (d) Comparison of MitoSV-seq efficiency on bulk and single cells. B: bulk; SC: single cell. (e) Distribution of variations in bulk and single myeloid cells with MitoSV-seq. Venn diagram represents total number of detected variations in each condition and their overlap. Circos plots of human mtDNA genome displaying heavy and light origins of replication (OH and LO, respectively), major arc, and D-loop (outer blue circle). mtDNA genes are displayed in the middle circle (grey lines). Also shown are SVs as arches with deletions (blue), tandem duplications (red), inversions (green), and insertions (orange). Thickness of the arches corresponds to SV heteroplasmy. SNVs are marked with short red lines (_) according to their positions in mouse mtDNA. Intensity of red lines corresponds to SNV heteroplasmy. (f) Performance of conventional and the newly developed technique in myeloid cells healthy donors and AML patients. C, conventional technique; N, new (MitoSV-seq).
Ten single CD11b-positive myeloid cells were then sorted from AML patients and healthy donors (in total, 30 single cells for each condition) (Fig. 6c). For bulk sequencing, 200,000 myeloid cells were sorted. By single-cell sequencing, more SVs and SNVs appeared in healthy myeloid cells and AML patients (Fig. 6d). Bulk sequencing of AML myeloid cells by MitoSV-seq generated 229 variations, whereas at the single-cell level, 231 variations were observed, with no overlap (Fig. 6e; Supplementary Tables 14-17). At the single-cell level, we identified SVs in the major arc and throughout the D-loop and other regions of mtDNA (Fig. 6e).
MitoSV-seq of bulk samples detected 9 SVs in AML, whereas the conventional method identified 6 SVs, 3 of which were shared between techniques (Fig. 6f).
-
D
Validation of MitoSV-seq in MDS
Thirty single CD11b-positive myeloid cells were sorted from MDS patients and healthy donors (Fig. 7a), and 200,000 myeloid cells were sorted from each individual for bulk sequencing. By MitoSV-seq, more variations were detected in single MDS cells (254 variations) compared with the bulk samples (143), 2 of which were shared (Fig. 7b and 7c; Supplementary Table 18-21). More variations were also noted in single myeloid cells from healthy individuals (Fig. 7b). Although the distribution of SVs and SNVs at the single-cell and bulk levels was similar in MDS patients, single-cell sequencing provided better resolution of the variations. At the single-cell level, in addition to our novel variations, we identified 2 SNVs that have been reported in haematopoietic and lymphoid tissue tumours: COSM56566133 and COSM5656134 [73]. Using conventional techniques for bulk MDS samples, we identified 2 SVs, compared with 34 SVs by MitoSV-seq (Fig. 7d).
Fig. 7.

Validation of MitoSV-seq in MDS patients. (a) Single myeloid cell isolation. MDS, myelodysplastic syndrome. (b) Evaluation of MitoSV-seq method in bulk and single myeloid cells from MDS patients. B, bulk; SC, single cell. (c) Allocation of variations detected by MitoSV-seq in bulk and single myeloid cells from MDS patients. Venn diagram represents total number of detected variations in each condition and their overlap, showing two detected variations at both levels. Circos plots of human mtDNA genome displaying heavy and light origins of replication (OH and LO, respectively), major arc, and D-loop (outer blue circle). mtDNA genes are displayed in the middle circle (grey lines). Also shown are SVs as arches with deletions (blue), tandem duplications (red), inversions (green), and insertions (orange). Thickness of the arches corresponds to SV heteroplasmy. SNVs are marked with short red lines (_) according to their positions in mouse mtDNA. Intensity of red lines corresponds to SNV heteroplasmy. (d) Comparison of MitoSV-seq with conventional techniques in bulk malignant myeloid cells from MDS patients. C, conventional technique; N, new (MitoSV-seq).
3.5. Extended somatic mtDNA mutations are associated with tumour progression
Based on our results, MitoSV-seq provided a universal map of the mtDNA genome in four human malignancies at the single-cell level. By multiple comparison of the tumours, several mutations were shared between groups of cancers; AML and MDS had the most variations in common (9) (Fig. 8a). Notably, genomic variations were seen in metabolism-related genes for all four malignancies (Fig. 8b), and a specific SV was shared by all four cancers, NADH dehydrogenase 5 (MT-ND5), indicating the importance of its function and likely its disruption in cancers. Supporting our findings, 65% of somatic truncating mutations have been reported to occur in NADH dehydrogenase 5 (MT-ND5) in colon adenocarcinoma, rectal adenocarcinoma, acute myeloid leukaemia, ovarian serous carcinoma, and cystadenocarcinoma [74].
Fig. 8.

Universal mtDNA genome comparison of four malignancies at single-cell level. (a) Spectrum of overlapping mutations in four tumours. Venn diagram depicts common variations in multiple comparisons of four types of tumours. Common variations and their relative genes are shown under various conditions. The most common variations exist between AML and MDS. (b) Alignment of all SVs found in the four malignancies. SVs are shown as arches (MCF7, red; SW620, green; AML, pink; MDS, blue). The only shared damaged mtDNA region in all of the tumours is highlighted. (c) Distribution of variations in AML and MDS single myeloid cells, showing nearly the same pattern of mtDNA damage. SVs are shown as arches with AML SVs in pink and MDS SVs in blue. (d) Segregation of eight common mutations in AML and MDS patients according to their heteroplasmy%.
The patterns of mtDNA damage in single AML and MDS cells were similar, although in the latter, the damage expanded to more regions (Fig. 8c). This finding is consistent with the nature of these myeloid lineage cancers—ie, on average, 30% of patients with MDS develop from AML during disease progression [75,76]. Of the eight overlapping variations that we found between AML and MDS, seven had slightly higher heteroplasmy in MDS, whereas the m.3524C>G mutation reached 100% heteroplasmy in AML patients (Fig. 8d). Using our novel MitoSV-seq method, these data expand the evidence that mtDNA damage that is related to defective metabolism is associated with tumour progression.
4. Discussion
Examining mutations that are associated with mitochondrial disorders remains a challenge due to phenotypic variability, genetic heterogeneity, and technical limitations .[9,26,27,[29], [30], [31],77,78] The precise identification of all mtDNA mutations in mitochondrial disorders is essential to understand the clinical variability, proper diagnosis, and prevention of many diseases [79]. Somatic mutations accumulate in mtDNA with age and are hypothesised to function in age-related neurodegenerative diseases, such as PD, but large increases in mtDNA mutations have not been seen in PD [49].
One limitation of studies on PD-associated mtDNA mutations is that due to the nature of the test, most of them have been performed on postmortem patient brains and thus at a late stage of the disease [28,77,78]. At this time, dopaminergic neurons in the substantia nigra that have accumulated high levels of mutations have most likely undergone cell death and are thus absent from the analysis. Although it is not feasible in patients, neurons can be accessed from the brain at an early stage of the disease in animal models. Thus, we used a mouse model of PD, Ifnar1–/–, to purify DA neurons, allowing us to better understand the impact of mtDNA alterations during early progression of the disease.
Mitochondrial dysfunction and the subsequent alterations in oxidative phosphorylation have also been hypothesized to be involved in tumourigenesis [80]. According to the Warburg hypothesis, cancer originates from a non-neoplastic cell that adopts anaerobic metabolism to survive after injury to its respiratory system, prompting a model in which tumours are initiated by constant damage to mitochondria [14]. In support of this model, somatic mtDNA mutations have been increasingly observed in human cancers, although their nature and diagnostic and therapeutic implications are poorly understood [21].
One major obstacle in the diagnosis of mitochondrial diseases is the variability of the symptoms, perhaps caused by the heteroplasmy of mtDNA damage, meaning that different versions of the mitochondrial genome can coexist in each cell, disrupting many cellular functions. Each cell contains thousands of copies of mtDNA; thus, the proportion of mutants in any one cell changes over time [42,81]. Having several copies of mtDNA allows each mitochondrion to function normally, even when some copies are mutated, allowing cells to carry high loads of mutant mitochondrial DNA before their function is affected. Consequently, disease symptoms emerge only when the mutant load exceeds a specific threshold [82]. This threshold is lower for certain cell types, such as postmitotic neurons, which depend highly on mitochondria due to their high-energy metabolism [83] and lack the capacity to proliferate, as a means of mitigating the damage that accumulates. Determining the proportion of mutations in mtDNA at the single-cell level will allow us to increase our understanding of mitochondrial diseases.
There are many mtDNA sequencing methods, but they are insufficiently sensitive and specific to detect mtDNA SVs and their relative heteroplasmy, because the signal that is generated by low-heteroplasmy variations cannot be distinguished easily from background noise, even with the latest NGS technology [84]. Although we observed mutation hotspots in tissues and species, there were few common mutations between cells within replicates, indicating that it is not possible to assess high levels of heteroplasmy in postmitotic neurons that harbour damaged mitochondria or in various cancers by bulk sequencing analysis. The development and application of MitoSV-seq helped us identify mtDNA variations in single neurons from an animal model of PD and single malignant human cells in solid and fluid tissues. This new method has the potential to be applied to a broad range of diseases, cell and tissue types, samples, and species in the study of mtDNA mutations and dysfunction. Another significant advantage of MitoSV-seq is that it requires few cells, using as low as 10 single cells from a biological sample to generate a comprehensive profile of mtDNA variations.
With this approach, we found an unexpectedly high frequency of heteroplasmic mtDNA variations in 70% of single DA neurons. In all four tumours that we studied, we observed more variations at the single-cell level, demonstrating the relevance of somatic mutational heterogeneity in oncology and the superiority of single-cell over bulk sequencing. Notably, although MitoSV-seq produces high-resolution data even with a limited number of cells, this technique has the potential for upscaling into a high-throughput platform. Single-cell MitoSV-seq provides higher coverage of circular mtDNA, because all of the reads are from mtDNA copies of a single cell, as shown in other studies [85]. This greater coverage could thus also prevent relevant mtDNA variations from being masked, due to other variations predominating in a smaller proportion of cells when using bulk cells or total tissue.
Nevertheless, MitoSV-seq can be applied to bulk cells, providing greater coverage of mtDNA mutations compared with conventional methods. In this study, we confirmed that FACS is an efficient and feasible technique for characterising dopaminergic and other neurons, various brain cell types, and malignant myeloid cells. FACS has advantages in purifying specific cells, based on single or multiple cell-specific markers, over previously recommended techniques for isolating single neurons, including MACS [48] and LCM [9,49], because both are prone to nonspecific contamination [50]. Moreover, RCA favours the amplification of circular templates. Using RHs in our method effected uniform amplification of circular mtDNA, regardless of sequence changes due to SVs and point mutations in a specific region of mtDNA. MitoSV-seq is sequence-agnostic and can be used to sequence mtDNA from human, mouse, and other organisms, because circular mtDNA can be amplified using RHs rather than site-specific primers. The PC that we have developed is a major advantage in the quality control of the MitoSV-seq method and can be applied to validate other mtDNA sequencing methods. The RCA approach that is implemented in MitoSV-seq is easy to set up in a high-throughput manner compared with classical PCR amplification, providing greater coverage of single-cell mtDNA amplification and sequencing and being universally applicable to a variety of biological samples, such as tissue and bulk cell extracts.
Notably, using the MitoSV-seq method specifically with our designed PC enabled us to scan the entire mtDNA, yielding many SV types, including deletions, inversions, short insertions, and tandem duplications. MitoSV-seq identified many mtDNA mutations that were otherwise unidentifiable using conventional methods and linked them to the potential mitochondrial basis of DA neuronal death in a PD model. The broad applicability of MitoSV-seq was confirmed in various cancers, expanding the evidence for altered metabolism as a critical hallmark of colon, breast, and haematopoietic cancers (AML and MDS), supporting defective oxidative phosphorylation in tumourigenesis.
Our comparison of defective mtDNA regions in these tumours highlighted MT-ND5 (NADH dehydrogenase 5) as a genetic hotspot for mutations in cancers. NADH dehydrogenase 5 is a mitochondrial respiratory complex I subunit and the largest and most complicated component of the respiratory chain [86]. In cancer cells, defective complex I potentiates tumour formation, resistance to cell death stimuli, and metastasis by increasing ROS levels [87]. Further, mutations in MT-ND5 have been linked to MDS [88], AML [89], colon adenocarcinoma [74], and breast cancer [90]. The accumulation of mutations in MT-ND5 has been suggested to inhibit oxidative phosphorylation, causing the malfunction of mitochondria and thus conferring selective growth advantages early in oncogenesis [87], also implicating the clinical use of MT-ND5 mutations as tumour biomarkers.
MitoSV-seq could significantly improve the identification of disease-associated mitochondrial DNA mutations, the lack of which has impeded research in this field [9,26,27,[29], [30], [31],77,78], and increase our understanding of the differences in mitochondrial biology in a cell-specific manner. MitoSV-seq has the potential to assist in examining the mtDNA damage that is associated with cancer cell ontology, progression, and metastatic changes in increasing numbers of purified single cells. MitoSV-seq has widespread applicability in mtDNA sequencing, including in disease studies, conservation genetics, and forensic identification, in which mtDNA sequence data are often gathered. Our newly developed MitoSV-seq method for single cells has significantly greater sensitivity and high-throughput capacity compared with previous approaches, and its broad use might encourage its application in the identification of cancer-relevant mtDNA mutations and mtDNA that are associated with metastasis and as a high-throughput platform to examine the potential and outcomes of cancer therapies.
5. Funding
This project received funding for S.I-N. from the Lundbeck Foundation (R223-2016-849) and Danish Council for Independent Research-Medicine (DFF-6110-00658), for E.J. from a Lundbeck Foundation postdoctoral fellowship (R288-2018-1095), and for E.T. from a Lundbeck Foundation postdoctoral fellowship (R219-2016-536) and the European Union's Horizon 2020 Research and Innovation Programme under Marie Skłodowska-Curie grant agreement No. 703217. None of the funding sources had any involvement in the study design; the collection, analysis, or interpretation of data; the writing of the report; or the decision to submit this paper for publication.
6. Declaration of Competing Interest
The authors declare no competing interests.
7. Author contributions
E.T. and S.I-N. initiated the project. All co-authors contributed to the development of the protocols and techniques. E.J. developed and conducted all experiments, performed the statistical analysis of the data, and wrote the first version of the manuscript. J.W. contributed to the new technical progression, assisted with processing the NGS data, and prepared the script to autogenerate circos plots for mtDNA-SVs and SNVs. K.G. provided patient material and clinical data. All authors read and contributed to the final manuscript.
The sequences that are reported here have been assigned SRA accession ID# PRJNA636990.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ebiom.2020.102868.
Appendix. Supplementary materials
Supplementary Fig. 1. Rolling circle amplification (RCA) preferentially amplifies circular mtDNA template over linearized mtDNA.
RCA of purified mtDNA template containing increasing proportions of circular mtDNA over linearized mtDNA. The arrow indicates concatemers of amplified mtDNA. The amounts of each type of mtDNA template are indicated in ng/reaction.
{kind=link}
References
- 1.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luo S, Valencia CA, Zhang J. Biparental Inheritance of Mitochondrial DNA in Humans. Proc Natl Acad Sci U S A. 2018;115(51):13039–13044. doi: 10.1073/pnas.1810946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6(5):389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cha MY, Kim DK, Mook-Jung I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med. 2015;47:e150. doi: 10.1038/emm.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy PH. Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease. CNS Spectr. 2009;14(8 Suppl 7):8–13. doi: 10.1017/s1092852900024901. discussion 6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7(3):207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 7.Dolle C, Flones I, Nido GS. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. 2016;7:13548. doi: 10.1038/ncomms13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coxhead J, Kurzawa-Akanbi M, Hussain R, Pyle A, Chinnery P, Hudson G. Somatic mtDNA variation is an important component of Parkinson's disease. Neurobiol Aging. 2016;38 doi: 10.1016/j.neurobiolaging.2015.10.036. 217 e1–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantuti-Castelvetri I, Lin MT, Zheng K. Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol Aging. 2005;26(10):1343–1355. doi: 10.1016/j.neurobiolaging.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Wong LJ. Challenges of bringing next generation sequencing technologies to clinical molecular diagnostic laboratories. Neurotherapeutics. 2013;10(2):262–272. doi: 10.1007/s13311-012-0170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y, Bai RK, Trieu R, Wong LJ. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim Biophys Acta. 2010;1797(1):29–37. doi: 10.1016/j.bbabio.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fliss MS, Usadel H, Caballero OL. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287(5460):2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 13.Bai RK, Wong LJ. Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over-replication in patients with mitochondrial disease. J Mol Diagn. 2005;7(5):613–622. doi: 10.1016/S1525-1578(10)60595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Sharma LK, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009;19(7):802–815. doi: 10.1038/cr.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumimoto H, Yamane Y, Nishimoto Y. Frequent somatic mutations of mitochondrial DNA in esophageal squamous cell carcinoma. Int J Cancer. 2004;108(2):228–231. doi: 10.1002/ijc.11564. [DOI] [PubMed] [Google Scholar]
- 16.Liu VW, Shi HH, Cheung AN. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61(16):5998–6001. [PubMed] [Google Scholar]
- 17.Tan DJ, Bai RK, Wong LJ. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002;62(4):972–976. [PubMed] [Google Scholar]
- 18.Habano W, Nakamura S, Sugai T. Microsatellite instability in the mitochondrial DNA of colorectal carcinomas: evidence for mismatch repair systems in mitochondrial genome. Oncogene. 1998;17(15):1931–1937. doi: 10.1038/sj.onc.1202112. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Akhtari M, Alachkar H. Characterization of mutations in the mitochondrial encoded electron transport chain complexes in acute myeloid leukemia. Sci Rep. 2018;8(1):13301. doi: 10.1038/s41598-018-31489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linnartz B, Anglmayer R, Zanssen S. Comprehensive scanning of somatic mitochondrial DNA alterations in acute leukemia developing from myelodysplastic syndromes. Cancer Res. 2004;64(6):1966–1971. doi: 10.1158/0008-5472.can-03-2956. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006;25(34):4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 22.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 23.Bonora E, Porcelli AM, Gasparre G. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66(12):6087–6096. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 24.Jazin EE, Cavelier L, Eriksson I, Oreland L, Gyllensten U. Human brain contains high levels of heteroplasmy in the noncoding regions of mitochondrial DNA. Proc Natl Acad Sci U S A. 1996;93(22):12382–12387. doi: 10.1073/pnas.93.22.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi Y, Wei Y, Wang Q. Heteroplasmy of mutant mitochondrial DNA A10398G and analysis of its prognostic value in non-small cell lung cancer. Oncol Lett. 2016;12(5):3081–3088. doi: 10.3892/ol.2016.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bender A, Krishnan KJ, Morris CM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 27.Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann Ny Acad Sci. 2008;1147:21–29. doi: 10.1196/annals.1427.016. [DOI] [PubMed] [Google Scholar]
- 28.Simon DK, Matott JC, Espinosa J, Abraham NA. Mitochondrial DNA mutations in Parkinson's disease brain. Acta Neuropathol Commun. 2017;5(1):33. doi: 10.1186/s40478-017-0433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang T. Next generation sequencing to characterize mitochondrial genomic DNA heteroplasmy. Curr Protoc Hum Genet. 2011 doi: 10.1002/0471142905.hg1908s71. Chapter 19: Unit19 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marquis J, Lefebvre G, Kourmpetis YAI. MitoRS, a method for high throughput, sensitive, and accurate detection of mitochondrial DNA heteroplasmy. BMC Genomics. 2017;18(1):326. doi: 10.1186/s12864-017-3695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris J, Na YJ, Zhu H. Pervasive within-Mitochondrion Single-Nucleotide Variant Heteroplasmy as Revealed by Single-Mitochondrion Sequencing. Cell Rep. 2017;21(10):2706–2713. doi: 10.1016/j.celrep.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blakely EL, Rennie KJ, Jones L. Sporadic intragenic inversion of the mitochondrial DNA MTND1 gene causing fatal infantile lactic acidosis. Pediatr Res. 2006;59(3):440–444. doi: 10.1203/01.pdr.0000198771.78290.c4. [DOI] [PubMed] [Google Scholar]
- 33.Musumeci O, Andreu AL, Shanske S. Intragenic inversion of mtDNA: a new type of pathogenic mutation in a patient with mitochondrial myopathy. Am J Hum Genet. 2000;66(6):1900–1904. doi: 10.1086/302927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang JN, Seluanov A, Gorbunova V. Mitochondrial inverted repeats strongly correlate with lifespan: mtDNA inversions and aging. Plos One. 2013;8(9):e73318. doi: 10.1371/journal.pone.0073318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9(9) doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoffner JM, Lott MT, Voljavec AS, Soueidan SA, Costigan DA, Wallace DC. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: a slip-replication model and metabolic therapy. Proc Natl Acad Sci U S A. 1989;86(20):7952–7956. doi: 10.1073/pnas.86.20.7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nido GS, Dolle C, Flones I. Ultradeep mapping of neuronal mitochondrial deletions in Parkinson's disease. Neurobiol Aging. 2018;63:120–127. doi: 10.1016/j.neurobiolaging.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 38.Yang JN, Seluanov A, Gorbunova V. Mitochondrial Inverted Repeats Strongly Correlate with Lifespan: mtDNA Inversions and Aging. Plos One. 2013;8(9) doi: 10.1371/journal.pone.0073318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dean FB, Hosono S, Fang L. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99(8):5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–1099. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tim Stuart RS. Integrative single- cell analysis. Nature Reviews Geneticsvolume. 2019;20:257–272. doi: 10.1038/s41576-019-0093-7. [DOI] [PubMed] [Google Scholar]
- 42.Duan M, Tu J, Lu Z. Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations. Molecules. 2018;23(2) doi: 10.3390/molecules23020323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodzic E. Single-cell analysis: Advances and future perspectives. Bosn J Basic Med Sci. 2016;16:313–314. doi: 10.17305/bjbms.2016.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sierant MC, Choi J. Single-cell ssequencing in cancer: recent applications to immunogenomics and multi-omics tools. Genomics Inform. 2018;16(4):e17. doi: 10.5808/GI.2018.16.4.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lasken RS. Single-cell genomic sequencing using multiple displacement amplification. Curr Opin Microbiol. 2007;10(5):510–516. doi: 10.1016/j.mib.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 46.Lopez JV, Yuhki N, Masuda R, Modi W, O'Brien SJ. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J Mol Evol. 1994;39(2):174–190. doi: 10.1007/BF00163806. [DOI] [PubMed] [Google Scholar]
- 47.Ejlerskov P, Hultberg JG, Wang J. Lack of neuronal IFN-beta-IFNAR causes lewy body- and parkinson's disease-like dementia. Cell. 2015;163(2):324–339. doi: 10.1016/j.cell.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467(1):60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- 49.Lin MT, Cantuti-Castelvetri I, Zheng K. Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Ann Neurol. 2012;71(6):850–854. doi: 10.1002/ana.23568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang P, Liang M, Zhang C. Biochemical evidence for a mitochondrial genetic modifier in the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum Mol Genet. 2016;25(16):3613–3625. doi: 10.1093/hmg/ddw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ni T, Wei G, Shen T. MitoRCA-seq reveals unbalanced cytocine to thymine transition in Polg mutant mice. Sci Rep. 2015;5:12049. doi: 10.1038/srep12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gregory MT, Bertout JA, Ericson NG. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic Acids Res. 2016;44(3):e22. doi: 10.1093/nar/gkv915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erlandsson L, Blumenthal R, Eloranta ML. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr Biol. 1998;8(4):223–226. doi: 10.1016/s0960-9822(98)70086-7. [DOI] [PubMed] [Google Scholar]
- 54.Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2(6):1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- 55.Guez-Barber D, Fanous S, Harvey BK. FACS purification of immunolabeled cell types from adult rat brain. J Neurosci Methods. 2012;203(1):10–18. doi: 10.1016/j.jneumeth.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25(21):2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santibanez-Koref M, Griffin H, Turnbull DM, Chinnery PF, Herbert M, Hudson G. Assessing mitochondrial heteroplasmy using next generation sequencing: A note of caution. Mitochondrion. 2019;46:302–306. doi: 10.1016/j.mito.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Picard M, McEwen BS. Mitochondria impact brain function and cognition. Proc Natl Acad Sci U S A. 2014;111(1):7–8. doi: 10.1073/pnas.1321881111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao YG, Ogasawara Y, Kajigaya S. Mitochondrial DNA sequence variation in single cells from leukemia patients. Blood. 2007;109(2):756–762. doi: 10.1182/blood-2006-01-011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma H, Singh A, Sharma C, Jain SK, Singh N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005;5:34. doi: 10.1186/1475-2867-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Q, Wang G. Mitochondrial dysfunction in Parkinson's disease. Transl Neurodegener. 2016;5:14. doi: 10.1186/s40035-016-0060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106-107:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallace DC, Singh G, Lott MT. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242(4884):1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 66.Lertrit P, Noer AS, Jean-Francois MJ. A new disease-related mutation for mitochondrial encephalopathy lactic acidosis and strokelike episodes (MELAS) syndrome affects the ND4 subunit of the respiratory complex I. Am J Hum Genet. 1992;51(3):457–468. [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor RW, Morris AA, Hutchinson M, Turnbull DM. Leigh disease associated with a novel mitochondrial DNA ND5 mutation. Eur J Hum Genet. 2002;10(2):141–144. doi: 10.1038/sj.ejhg.5200773. [DOI] [PubMed] [Google Scholar]
- 68.Yarham JW, Elson JL, Blakely EL, McFarland R, Taylor RW. Mitochondrial tRNA mutations and disease. Wiley Interdiscip Rev RNA. 2010;1(2):304–324. doi: 10.1002/wrna.27. [DOI] [PubMed] [Google Scholar]
- 69.Lin CS, Liu LT, Ou LH, Pan SC, Lin CI, Wei YH. Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol Rep. 2018;39(1):316–330. doi: 10.3892/or.2017.6087. [DOI] [PubMed] [Google Scholar]
- 70.Giannakis M, Mu XJ, Shukla SA. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;17(4):1206. doi: 10.1016/j.celrep.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giannakis M, Mu XJ, Shukla SA. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15(4):857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A. 2014;111(29):10654–10659. doi: 10.1073/pnas.1403521111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morin RD, Assouline S, Alcaide M. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin Cancer Res. 2016;22(9):2290–2300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 74.Larman TC, DePalma SR, Hadjipanayis AG. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A. 2012;109(35):14087–14091. doi: 10.1073/pnas.1211502109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Porwit A. Role of flow cytometry in diagnostics of myelodysplastic syndromes–beyond the WHO 2008 classification. Semin Diagn Pathol. 2011;28(4):273–282. doi: 10.1053/j.semdp.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 76.Chen J, Kao YR, Sun D. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. 2019;25(1):103–110. doi: 10.1038/s41591-018-0267-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simon DK, Lin MT, Zheng L. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson's disease. Neurobiol Aging. 2004;25(1):71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 78.Wei W, Keogh MJ, Wilson I. Mitochondrial DNA point mutations and relative copy number in 1363 disease and control human brains. Acta Neuropathol Commun. 2017;5(1):13. doi: 10.1186/s40478-016-0404-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5(11) doi: 10.1101/cshperspect.a021220. a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Urra FA, Munoz F, Lovy A, Cardenas C. The Mitochondrial Complex(I)ty of Cancer. Front Oncol. 2017;7:118. doi: 10.3389/fonc.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naue J, Horer S, Sanger T. Evidence for frequent and tissue-specific sequence heteroplasmy in human mitochondrial DNA. Mitochondrion. 2015;20:82–94. doi: 10.1016/j.mito.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 82.Stewart JB, Larsson NG. Keeping mtDNA in shape between generations. PLoS Genet. 2014;10(10) doi: 10.1371/journal.pgen.1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carelli V, Chan DC. Mitochondrial DNA: impacting central and peripheral nervous systems. Neuron. 2014;84(6):1126–1142. doi: 10.1016/j.neuron.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Casoli T, Spazzafumo L, Di Stefano G, Conti F. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer's disease neurodegeneration. Front Aging Neurosci. 2015;7:142. doi: 10.3389/fnagi.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106(1):18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- 86.Cardenas C, Miller RA, Smith I. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142(2):270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garcia-Heredia JM, Carnero A. Decoding Warburg's hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget. 2015;6(39):41582–41599. doi: 10.18632/oncotarget.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gupta M, Madkaikar M, Rao VB. Mitochondrial DNA variations in myelodysplastic syndrome. Ann Haematol. 2013;92(7):871–876. doi: 10.1007/s00277-013-1706-4. [DOI] [PubMed] [Google Scholar]
- 89.Triska P, Kaneva K, Merkurjev D. Landscape of Germline and somatic mitochondrial dna mutations in pediatric malignancies. Cancer Res. 2019;79(7):1318–1330. doi: 10.1158/0008-5472.CAN-18-2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hertweck KL, Dasgupta S. The landscape of mtDNA modifications in cancer: a tale of two cities. Front Oncol. 2017;7:262. doi: 10.3389/fonc.2017.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Rolling circle amplification (RCA) preferentially amplifies circular mtDNA template over linearized mtDNA.
RCA of purified mtDNA template containing increasing proportions of circular mtDNA over linearized mtDNA. The arrow indicates concatemers of amplified mtDNA. The amounts of each type of mtDNA template are indicated in ng/reaction.