

Abstract
Macrophage activation syndrome (MAS), a form of secondary hemophagocytic lymphohistiocytosis, is a frequently fatal complication of a variety of pediatric inflammatory disorders. MAS has been most commonly associated with systemic juvenile idiopathic arthritis (sJIA), as approximately 10% of children with sJIA develop fulminant MAS, with another 30–40% exhibiting a more subclinical form of the disease. Children with other rheumatologic conditions such as systemic lupus erythematosus and Kawasaki disease are also at risk for MAS. Moreover, MAS also complicates various genetic autoinflammatory disorders such as gain of function mutations in the cytosolic inflammasome NLRC4, pediatric hematologic malignancies (e.g., T-cell lymphoma), and primary immunodeficiencies characterized by immune dysregulation. Disease-specific and broadly inclusive diagnostic criteria have been developed to facilitate the diagnosis of MAS. Recently, simple screening tools such as the serum ferritin to erythrocyte sedimentation rate ratio have been proposed. Early diagnosis and rapid initiation of immunosuppression are essential for the effective management of MAS. With a better understanding of the pathophysiology of MAS and the advent of novel therapeutics, a broad immunosuppressive approach to treatment is giving way to targeted anti-cytokine therapies. These treatments include agents that block interleukin-1 (IL-1), IL-6, IL-18, interferon-γ, as well as inhibitors of downstream targets of cytokine signaling (e.g., Janus kinases). Increased early recognition of MAS among pediatric inflammatory disorders combined with the use of effective and less toxic cytokine-targeted therapies should lower the mortality of this frequently fatal disorder.
1. Introduction
The term ‘macrophage activation syndrome’ (MAS) was first employed by Claude Griscelli and collaborators in 1985 to describe seven patients with juvenile idiopathic arthritis (JIA) who developed acute-onset encephalopathy, coagulopathy, and hepatitis [1]. The authors noted activated macrophages on liver biopsy and speculated that “macrophage activation secondary to drug or intercurrent infection” may play a causal role in the clinical presentation [1]. Initially, MAS was considered a complication exclusive to rheumatic diseases, most notably systemic JIA (sJIA) [2].
In recent years, it has become clear that MAS is not a phenomenon uniquely observed in rheumatologic conditions but instead is closely related to hemophagocytic lymphohistiocytosis (HLH) [3–5] (Table 1). Indeed, much of what is known about the pathophysiology of MAS is derived from our understanding of the genetic underpinnings of familial HLH (FHL). FHL is caused by bi-allelic mutations in one of four known FHL genes, perforin 1 (PRF1), unc-13 homolog D (UNC13D), syntaxin11 (STX11), and syntaxin binding protein 2 (STXBP2), all of which are required for cytotoxic granule release by CD8+ T cells and NK cells [6–9]. Defects in FHL genes lead to impaired cytotoxicity, persistence of intracellular infection, and prolonged antigen stimulation that engenders T-cell and macrophage activation as well as massive release of pro-inflammatory cytokines (interleukin [IL]-1β, IL-6, IL-18, tumor necrosis factor [TNF], and interferon gamma [IFNγ]) [10–15]. IFNγ is particularly important in driving disease in FHL. In a mouse model of FHL caused by perforin deficiency, only neutralization of IFNγ rescued the fatal phenotype [10]. In humans, IFNγ blockade appears to ameliorate disease [16]. The clinical features of HLH, including fever, multi-organ failure, cytopenias, coagulopathy, hypertriglyceridemia, hyperferritinemia, hemophagocytosis, elevated acute-phase reactants, and hepatitis, are explained in large part by the cytokine storm that results from defective cytotoxicity [17].
Table 1.
HLH and MAS terminology*
|
Cytokine storm: Encompasses primary and acquired HLH syndromes |
|
|
Primary HLH: HLH caused by a genetic disorder |
Acquired HLH: HLH that develops as a complication of another condition |
| FHL: HLH caused by bi-allelic mutations in one of the known FHL genetic loci (PRF1, UNC13D, STX11, STXBP2) | Infection-associated HLH: HLH that results from an infectious trigger such as EBV |
| HLH associated with genetic disorders: HLH that occurs in the setting of a known genetic disorder, most commonly PIDs (XP1, XLP2, ITK, XMEN, CD27 deficiency, CHS, GS type 2, HPS type 2, CGD) | Rheumatologic HLH/MAS: HLH in the setting of a rheumatologic condition (sJIA, AOSD, SLE, KD, autoinflammatory diseases) |
| Malignancy-associated HLH: HLH that develops in a patient with malignancy | |
| Chemotherapy-associated HLH: HLH that develops as a result of chemotherapy for malignancy |
There is no agreed-upon nomenclature for HLH- and MAS-related diseases.
This table reviews how the given terms are used in this manuscript AOSD adult-onset Still disease, CGD chronic granulomatous disease, CHS Chediak-Higashi syndrome, EBV Epstein-Barr virus, FHL familial hemophagocytic lymphohistiocytosis, GS Griscelli syndrome, HLH hemophagocytic lymphohistiocytosis, HPS Hermansky-Pudlak syndrome, ITK IL-2-inducible T-cell kinase deficiency, KD Kawasaki’s disease, MAS macrophage activation syndrome, PID primary immunodeficiency, sJIA systemic juvenile idiopathic arthritis, SLE systemic lupus erythematosus, XLP1 X-linked lymphoproliferative disease type 1, XLP2 X-linked lymphoproliferative disease type 2, XMEN X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia
Acquired HLH mimics the clinical presentation of FHL and is often triggered by viral infections such as Epstein-Barr virus (EBV) and influenza (infection-associated HLH) or malignancy (malignancy-associated HLH) (Table 1) [18–20]. MAS is now considered a form of acquired HLH in patients with an autoimmune disease (rheumatologic HLH) (Table 1) [4]. In addition to a common clinical phenotype, acquired HLH, MAS, and FHL are all frequently characterized by impaired cytotoxicity and cytokine-driven pathology. NK cell dysfunction has been documented in sJIA patients with active disease and is thought to be due to IL-6-mediated depression of cytotoxic activity, possibly through decreased perforin and granzyme expression [21–23]. A subset of sJIA patients who develop MAS may also possess heterozygous defects in FHL genes (e.g., PRF1, UNC13D) that can also contribute to cytolytic dysfunction via hypomorphic and partial dominant-negative effects [24–26]. Interestingly, a high rate of heterozygous mutations in FHL genes has also been found in patients with fatal influenza infections, indicating a predisposition to aberrant cytolytic killing that is shared with FHL [27, 28]. Infection-associated HLH and MAS have a similar cytokine signature as FHL, including elevated IL-18 and IFNγ [11, 12, 16, 29–34]. In total, FHL, HLH associated with infection and malignancy, and MAS in rheumatologic conditions should be considered a spectrum of the same disease with a commonly shared underlying pathophysiology of impaired cytolytic killing and immune activation leading to a cytokine storm (Fig. 1).
Fig. 1.
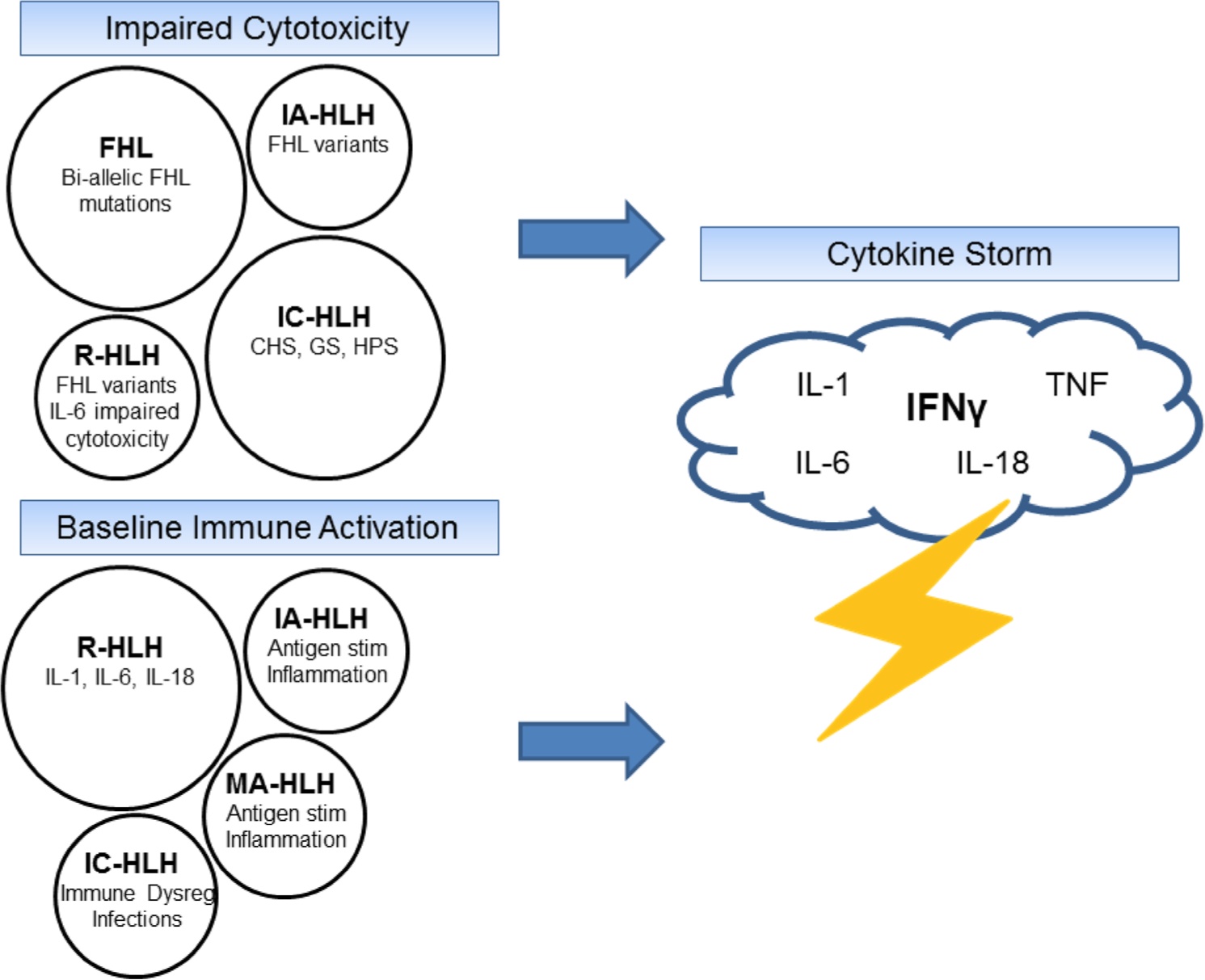
The spectrum of HLH- and MAS-related diseases. The cytokine storm characterized by FHL, acquired HLH, and MAS can be considered a final common pathway that results from impaired cytotoxicity, baseline immune activation, or a combination of both of these factors. CHS Chediak-Higashi syndrome, FHL familial hemophagocytic lymphohistiocytosis, GS Griscelli syndrome, HLH hemophagocytic lymphohistiocytosis, HPS Hermansky-Pudlak syndrome type 2, IA-HLH infection-associated HLH, IC-HLH immune-compromised HLH, IFNγ interferon gamma, IL interleukin, MA-HLH malignancy-associated HLH, MAS macrophage activation syndrome, R-HLH rheumatologic HLH, stim stimulation, TNF tumor necrosis factor
While FHL is rare, acquired HLH and MAS are increasingly recognized in patients with infections and rheumatologic conditions (Fig. 2). Acquired HLH and MAS may result from multiple individual triggers (chronic inflammation, infection, heterozygous defect in cytolysis) that, when combined, breach a threshold level of disease that the immune system is no longer able to counteract (Fig. 1) [35, 36]. Traditionally, it was thought that about 10% of children with sJIA developed MAS; however, it is now understood that many more (30–40%) sJIA patients have subclinical features of the disease [37–39]. Further, MAS has been described in rheumatologic diseases other than sJIA including systemic lupus erythematosus (SLE) and Kawasaki disease (KD) [40–44]. The prevalence of HLH/MAS in critically ill patients with sepsis physiology is almost certainly underdiagnosed by large margins, particularly in patients with cytopenias, coagulopathy, and/or hepatobiliary dysfunction. In a prospective study of intensive care unit (ICU) patients with sepsis and thrombocytopenia, hemophagocytosis was found on bone marrow biopsy in 64% of cases [45]. A similar proportion of patients who died in the ICU were also found to have signs of hemophagocytosis in the bone marrow at autopsy. Hence, acquired HLH and MAS are more common than previously appreciated [46, 47]. The high mortality rates associated with these conditions highlight the importance of early recognition and effective treatment strategies.
Fig. 2.
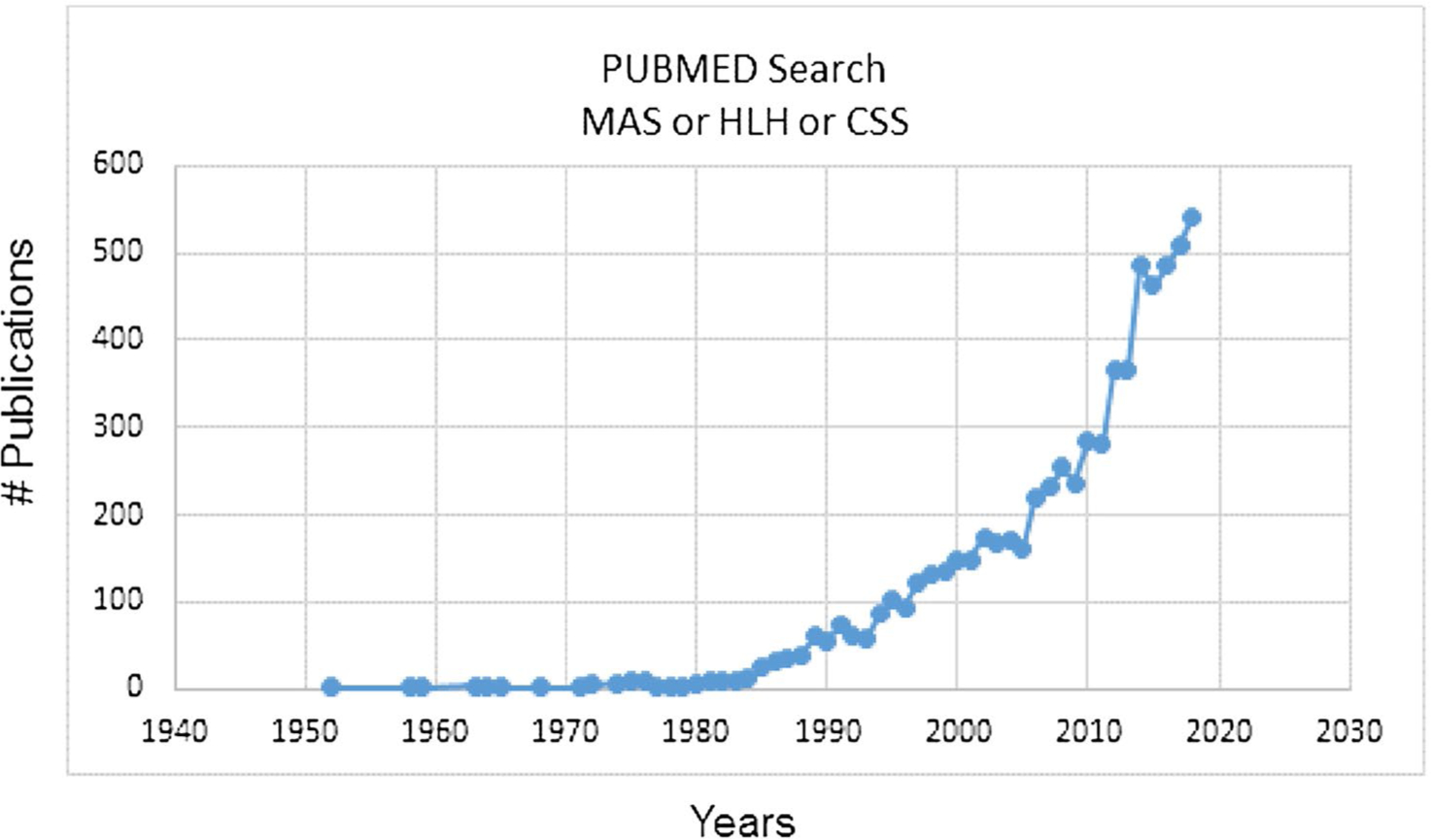
Exponential increase in the number of publications per year cited in PubMed on MAS, HLH, and cytokine storm. CSS cytokine storm syndrome, HLH hemophagocytic lymphohistiocytosis, MAS macrophage activation syndrome
2. Associated Inflammatory Disorders
2.1. Rheumatologic
MAS is a well recognized complication of sJIA and adult-onset Still disease (AOSD) [48, 49]. It is estimated that about 10% of sJIA patients will develop overt MAS, typically with active underlying disease, although infections are also known triggers [38, 48]. In about one-quarter of patients, MAS is noted at sJIA disease onset [38]. Fever is a near universal feature of MAS in sJIA and is characterized by an unremitting pattern that differs from the quotidian fever spikes in sJIA without MAS [38, 48, 49]. As in HLH, hepatosplenomegaly, coagulopathy, encephalopathy, and hepatic dysfunction are common [38, 48, 49]. Elevated d-dimers, transaminitis, and hyperferritinemia are typically noted in > 90% of affected patients while cytopenias, elevated triglycerides, and falling erythrocyte sedimentation rate (ESR) are also frequently observed [38, 48, 49]. Hemophagocytosis on bone marrow biopsy is noted in a majority (60%) but not all sJIA patients with MAS [38]. MAS is a feared complication of sJIA with mortality rates as high as 20% in some case series [48].
While MAS is most often associated with sJIA and AOSD, it is increasingly recognized in other rheumatologic conditions. In a recent retrospective series, close to 10% of childhood-onset SLE patients had MAS, a proportion similar to sJIA [41]. MAS has been reported in KD (1–2% of patients) but less frequently than in sJIA and SLE [42, 44]. MAS in SLE and KD is clinically similar to the presentation in sJIA and is associated with higher mortality rates than observed in patients with SLE and KD alone [40, 42, 43, 50].
Cases of MAS have also been identified in patients with autoinflammatory conditions (disrupted innate immunity) such as hyper-IgD syndrome, familial Mediterranean fever, TNF receptor-associated periodic syndrome, and cryopyrin-associated periodic syndromes [48, 49, 51–54]. Of all the monogenic autoinflammatory disorders, MAS is clearly associated with heterozygous gain of function mutations in NLR-family CARD domain-containing protein 4 (NLRC4) [55, 56]. NLRC4 is a cytosolic inflammasome that is activated in response to bacterial pathogen-associated molecular patterns (PAMPs). Once engaged, NLRC4 converts the precursor form of caspase-1 into a functional enzyme [57, 58]. In turn, caspase-1 cleaves pro-IL-1β and pro-IL-18 and induces pyroptosis or inflammatory cell death that releases these cytokines into the extracellular space [59]. Patients with activating mutations in NLRC4 present with recurrent MAS, early-onset colitis, and markedly elevated levels of IL-18 in a condition called autoinflammation with infantile enterocolitis (AIFEC) [55, 56]. Interestingly, individuals with milder phenotypes including skin predominant features and uveitis have been described [60]. AIFEC patients have normal NK cell function between flares, and the MAS phenotype is thought to be mediated by cytokine excess, particularly IL-1β and IL-18, with IFNγ also playing a role through its induction by IL-18 [55, 56, 61]. Thus, mechanisms other than decreased cytolytic function can also contribute to MAS pathophysiology [62, 63].
The high rates of MAS in chronic inflammatory disorders characterized by IL-1β and IL-18 signatures (sJIA, AOSD, inflammasomopathies) underscore the importance of these cytokines in driving MAS. Yet, MAS is not restricted to IL-1- and IL-18-associated diseases and is observed in conditions such as SLE. This highlights the various pathways that can be engaged in the setting of chronic inflammation that ultimately culminate in MAS.
2.2. Oncologic
Malignancy-associated HLH occurs in one of two contexts. First, the inflammatory environment created by the neoplasm can trigger HLH (malignancy-triggered HLH). Second, treatment with chemotherapy can result in HLH either from the immunomodulatory effects of the medications or infection from immunosuppression (chemotherapy-associated HLH). HLH in the setting of malignancy is commonly reported in adults and is thought to occur in 1% of adult hematologic malignancies [20, 64]. In pediatric patients with HLH, an occult oncologic process may be missed because children are often presumed to have an underlying genetic defect. Malignancy-associated HLH may be more common in the pediatric population than originally believed. A recent case series by Lehmberg et al. estimated that 8% of pediatric HLH was associated with malignancy [19]. T-cell malignancies, particularly T-cell lymphoma, were most commonly found to trigger HLH in children, while Hodgkin lymphoma and B-cell lymphoma were also reported [19]. Treatment for leukemia and lymphoma were associated with the development of chemotherapy-associated HLH [19, 65]. Immunotherapies such as chimeric antigen receptor T cells have been associated with HLH physiology as well [66]. Unraveling the cause of death in patients with HLH and malignancy is difficult; however, it appears that active HLH increases mortality. Unfortunately, the best treatment for such patients is currently unknown [19].
2.3. Primary Immunodeficiency
HLH has been described in many primary immunodeficiencies (PIDs); however, it is more common in immune defects that impair T cell, vesicle/lysosomal, and macrophage/neutrophil function [67]. Of all the inherited immunodeficiencies, HLH, typically triggered by EBV, is most frequently reported in X-linked lymphoproliferative disease (XLP) types 1 and 2 [68]. The genetic causes of XLP1 (SH2 domain protein 1A, SH2D1A) and XLP2 (BIRC4 gene encoding X-linked inhibitor of apoptosis protein, XIAP) are known; however, the exact mechanism by which mutations in these genes leads to EBV vulnerability and HLH is not entirely understood [69, 70]. Both genes are important in the development and homeostasis of T, NK, and NKT cells, which are also known to be activated in HLH. Interestingly, XIAP deficiency shares some features with AIFEC including inflammatory colitis and markedly elevated levels of IL-18 [68, 71]. Other T-cell PIDs associated with EBV susceptibility and HLH include IL-2-inducible T-cell kinase (ITK) deficiency; X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia (XMEN disease); and CD27 deficiency [72–74].
Chediak-Higashi syndrome (CHS), Griscelli syndrome (GS) type 2, and Hermansky-Pudlak type 2 are a group of diseases characterized by abnormal lysosomal/vesicular biology that results in albinism, primary immunodeficiency, and a predisposition to HLH [67]. CHS is caused by mutations in the lysosomal trafficking regulator (LYST) [75]. GS type 2 is due to mutations in a GTPase encoded by the RAB27A gene that is needed for vesicular transport and fusion with the cell membrane [76]. Hermansky-Pudlak type 2 results from mutations in the AP3B1 gene, which is important for lysosome trafficking [77]. These genetic syndromes all result in decreased perforin-mediated cytolytic activity of lymphocytes as in FHL [63, 78, 79].
Chronic granulomatous disease (CGD) is caused by mutations in the NADPH oxidase complex that is needed by phagocytes to produce reactive oxygen species and kill bacteria and fungi. Affected patients present with recurrent infections and chronic granulomatous inflammation such as colitis. HLH has been reported in CGD in the setting of active infection, particularly Burkholderia cepacia, and often improves once the infection resolves [80–82].
While HLH is reported in several different PIDs, a closer examination reveals a pattern. Certain types of immune defects are more likely to be associated with HLH physiology, such as T-cell immune deficiencies; disorders of vesicle synthesis, transportation and release; and macrophage/neutrophil defects. These conditions share pathways commonly implicated in HLH/MAS, including persistence of infection that results in excessive immune stimulation, T-cell activation, and cytokine excess. The pro-inflammatory cytokine storm that develops is a common thread in most forms of FHL, acquired HLH, and MAS [78, 83].
3. Diagnosis
3.1. HLH-2004 Diagnostic Criteria
The HLH-2004 diagnostic criteria were originally used in the HLH-2004 treatment study evaluating the efficacy of etoposide, dexamethasone, and cyclosporine-based induction therapy before hematopoietic stem-cell transplant (HSCT) (Table 2) [84]. The criteria are often used to diagnose genetic and acquired HLH in the clinical setting. The HLH-2004 criteria reflect some of the common clinical and laboratory features of HLH, including fever, splenomegaly, cytopenias, hypertriglyceridemia, hyperferritinemia, and hemophagocytosis, but also require specialized diagnostic tests such as soluble IL-2 receptor alpha levels (sCD25), NK-cell functional assays, and genetic testing. The HLH-2004 criteria were originally employed in the setting of a clinical trial where a homogenous population of well defined patients was required. The need for genetic and immunologic tests that take days for results may unnecessarily delay the diagnosis and treatment of critically ill patients. Further, some of the HLH-2004 parameters are not sensitive or specific for HLH. Hemophagocytosis is not always noted on bone marrow biopsy in HLH patients, especially in the early stages of the disease [85–87]. Non-nucleated erythrophagocytosis can be seen in many conditions while engulfment of nucleated cells may be more specific to HLH; however, the HLH-2004 criteria do not differentiate between these morphological features [88]. The ferritin cut-off value of 500 μg/L is particularly problematic and lacks specificity for HLH. Ferritin values > 500 μg/L are commonly seen in patients treated with multiple blood transfusions or stem-cell transplant as well as individuals with renal disease, liver disease, malignancy, infection, or hemoglobinopathies [89, 90]. Alternate ferritin cut-off values for HLH such as 2000, 4000, or even 10,000 μg/L have been suggested as more specific for the diagnosis [89, 91, 92]. Finally, the HLH-2004 criteria do not include liver pathology or recently identified biomarkers of HLH including CXCL9 and IL-18 that may be helpful in diagnosis [16, 29–31, 93, 94].
Table 2.
Comparison of diagnostic/classification tools for HLH and MAS
| HLH 2004a | 2016 MAS classification criteriab | MS scorec | Ferritin:ESR ratiod | HScoree | MH scoref |
|---|---|---|---|---|---|
| (1) Molecular diagnosis | (1) Fever in sJIA pt AND |
(1) Calculation ≥ − 2.1 B-coefficient |
(1) Ferritin/ ESR ≥ 21.5 |
(1) Points add to ≥ 169 | (1) Points add to ≥ 60 |
| OR | |||||
| (2) 5/8 Criteria: | (2) Ferritin > 684 ng/ mL AND |
CNS 2.44 Hemorrhagic 1.54 Arthritis − 1.30 |
Immunosuppression 0 (no), 18 (yes) Temp (°C) |
Age at onset (y) 0 (> 1.6), 37 (≤ 1.6) Neutrophils (× 109/L) |
|
| Fever ≥ 38.5 °C Splenomegaly Cytopenias in 2/3 lines Hgb < 9 g/dL Plts < 100 × 103/mL Neutrophils < 1 × 103/ mL TG ≥ 265 mg/dL and/or fibrinogen ≤ 150 mg/dL Hemophagocytosis Low NK cell fxn Ferritin ≥ 500 ng/mL Elevated sIL-2 receptor |
(3) 2/4 Criteria: Plts ≤ 181×103/mL AST > 48 u/L TG > 156 mg/dL Fibrinogen ≤ 360 mg/dL |
Plts (× 109/L) − 0.003 LDH (U/L) 0.001 Fibrinogen (mg/dL) − 0.004 Ferritin (ng/mL) 0.0001 |
0 (< 38.4), 33 (38.4– 39.4), 49 (> 39.4) Organomegaly 0 (no), 23 (H or SM), 38 (HSM) Cytopenias 0 (1 line), 24 (2 lines), 34 (3 lines) Ferritin (ng/mL) 0 (< 2000), 35 (2–6000), 50 (> 6000) TG (mmoles/L) 0 (< 1.5), 44 (1.5–4), 64 (> 4) Fibrinogen (gm/L) 0 (> 2.5) or 30 (≤ 2.5) AST (u/L) 0 (< 30) or 19 (≥ 30) Hemophagocytosis 0 (no) or 35 (yes) |
0 (> 1.4), 37 (≤ 1.4) Fibrinogen (mg/dL) 0 (> 131), 15 (≤ 131) Splenomegaly 0 (no), 12 (yes) Plts (× 109/L) 0 (> 78), 11 (≤ 78) Hgb (g/dL) 0 (> 8.3), 11 (≤ 8.3) |
|
| Sens: N/A | Sens: 73% | Sens: 85% | Sens: 82% | Sens: 93% | Sens: 91% |
| Spec: N/A | Spec: 99% | Spec: 95% | Spec: 78% | Spec: 86% | Spec: 93% |
Sensitivities and specificities in the table refer to the values obtained from the original derivation population in the cited references
AST aspartate aminotransferase, CNS central nervous system involvement, ESR erythrocyte sedimentation rate, fxn function, H hepatomegaly, hemorrhagic hemorrhagic manifestations, Hgb hemoglobin, HLH hemophagocytic lymphohistiocytosis, HSM hepatosplenomegaly, LDH lactic dehydrogenase, MAS macrophage activation syndrome, MH MAS/HLH, MS MAS/sJIA, NK natural killer, Plts platelets, pt patient, s soluble, SM splenomegaly, sens sensitivity, sJIA systemic juvenile idiopathic arthritis, spec specificity, TG triglycerides
Adapted from [84] for the diagnosis of HLH in the HLH-2004 clinical trial
Adapted from [106] to discriminate between sJIA with MAS from active sJIA
MS score = CNS involvement × 2.44 + hemorrhagic manifestations × 1.54 + arthritis × (− 1.3) + Plts × (− 0.003) + LDH × 0.0001 + fibrinogen × (− 0.004) + ferritin × 0.0001
Adapted from [108] to discriminate between sJIA with MAS and active sJIA
Adapted from [109] to assess the risk of acquired HLH in adults
Adapted from [87] to discriminate between primary HLH and MAS
3.2. Classification Criteria for Macrophage Activation Syndrome (MAS) in Systemic Juvenile Idiopathic Arthritis (sJIA)
Identifying evolving MAS in a patient with an underlying inflammatory disorder is difficult. In many respects, the HLH-2004 diagnostic criteria do not adequately discriminate between sJIA patients with MAS/HLH physiology and those with a flare of autoimmune disease [95]. In 2016, a multinational research collaborative of pediatric rheumatologists and hemato-oncologists developed classification criteria for MAS in sJIA based on expert opinion and validated with patient data and a replication cohort (Table 2) [96, 97]. As in the HLH-2004 criteria, fever, hyperferritinemia, thrombocytopenia, hypertriglyceridemia, and hypofibrinogenemia were included with the addition of transaminitis [96, 97]. In the validation cohort, the 2016 MAS classification criteria displayed 73% sensitivity and 99% specificity for MAS in sJIA. It should be noted that this tool was designed as classification criteria for use in clinical trials and was not validated for diagnostic purposes in the clinical setting. These criteria may not capture all patients with MAS and sJIA, particularly those with atypical features.
A Japanese group confirmed that the 2016 MAS classification criteria are highly sensitive and specific for full blown MAS in sJIA; however, the tool’s ability to identify early-onset MAS was limited [98]. Monitoring the trend in laboratory studies in an individual patient appears to be essential for the recognition of early MAS [98–100]. sJIA patients typically display elevated white blood cell and platelet counts, ESR, and fibrinogen during active disease. Decreases in these laboratory values in a patient with active inflammation (fever, rash) is concerning, even if the test values remain within normal limits. A further layer of diagnostic complexity is added by the impact of biologic medications on the features of MAS. In particular, tocilizumab treatment (anti-IL-6) masks fever and hyperferritinemia in MAS while lowering platelet, fibrinogen, and AST levels below the values typically seen in patients with sJIA [101]. The result is that the 2016 MAS criteria are less accurate in patients treated with tocilizumab, which is a substantial portion of the sJIA population [101].
The 2016 MAS classification criteria have been applied to other inflammatory conditions that are often complicated by MAS. In SLE and AOSD, the criteria successfully identified a group of febrile patients with high mortality [102, 103]. The classification criteria demonstrated relatively preserved sensitivity in AOSD; however, specificity was decreased due to the poor performance of the triglyceride and ferritin cut-off values in the adult population [102, 104]. Preliminary SLE-specific MAS criteria have also been developed by Parodi and collaborators based on the study of 38 patients with SLE and MAS; however, this study did not have a replication cohort [105]. A recent retrospective review of a different cohort of 403 pediatric SLE patients, including 38 patients with MAS, showed that all SLE patients diagnosed with MAS by the treating physician also met the Parodi MAS criteria [41].
3.3. MAS/sJIA (MS) Score
To address some of the shortcomings of the 2016 classification criteria, the data were reanalyzed with a Bayesian model averaging approach to develop the MAS/sJIA (MS) score (Table 2) [106]. The MS score includes seven parameters and differs from the 2016 classification criteria in that strict cut-off values are not used for laboratory variables [96, 97, 106]. Instead, the patient’s laboratory values are entered into a weighted equation that is used to calculate the final score. In contrast to the 2016 classification criteria, lactic dehydrogenase (LDH) is included while aspartate aminotransferase (AST) and triglycerides are excluded. The platelet count, fibrinogen, and ferritin were retained in the score [96, 97]. Central nervous system (CNS) dysfunction and hemorrhagic manifestations were found to be highly discriminative between active sJIA and sJIA with MAS and were included in the model along with the lack of active arthritis [106]. In the validation cohort, an MS score of −2.1 or higher had 85% sensitivity and 95% specificity [106]. Unlike the 2016 classification criteria, the MS score is validated to aid in the clinical diagnosis of sJIA in MAS.
3.4. Ferritin:ESR Ratio
The ferritin:ESR (ng/mL ÷ mm/h) ratio was also developed in an effort to improve and simplify the 2016 classification criteria (Table 2). Hyperferritinemia is known to rise exponentially as a patient enters HLH/MAS, while the ESR falls due to fibrinogen depletion from the consumptive coagulopathy that is a hallmark of the condition. In 2013, Gorelik and collaborators showed the ferritin:ESR ratio to be superior to ferritin alone in discriminating new-onset sJIA from MAS (n = 28 patients) [107]. Building upon this finding, Eloseily et al. re-evaluated data from the 2016 classification criteria study along with new information from matched febrile patients who were hospitalized for infection [108]. The easy-to-use ferritin:ESR ratio compared favorably to the 2016 classification criteria and was superior to ferritin alone in discriminating between active sJIA and sJIA with MAS (a cut-off of 21.5 showed 82% sensitivity and 78% specificity) [108]. By contrast, ferritin alone outperformed the ferritin:ESR ratio in discerning MAS from children hospitalized with a febrile infection (non-sJIA patients) [108].
3.5. HScore
Diagnosing acquired HLH, which is often triggered by infection or malignancy, poses many of the same problems as identifying MAS in children with active sJIA. The inflammatory environment resulting from the infection or cancer often increases acute-phase reactants, which may render the HLH-2004 laboratory cut-off values less informative. In addition, the HLH-2004 criteria were developed for a pediatric population and may not perform as well in adults where infection- and malignancy-associated HLH is more common [84]. The HScore (reactive hemophagocytic syndrome diagnostic score) was created to accurately identify acquired HLH in adults (Table 2) [109]. It was developed by expert review of clinical cases and the performance of the HScore was verified in a validation cohort [109]. The HScore comprises nine variables, many of which are included in the HLH-2004 criteria (fever, organomegaly, cytopenias, ferritin, triglyceride, fibrinogen, and hemophagocytosis on bone marrow). For each laboratory parameter, a weighted number of points is given based on the degree of abnormality. A score of ≥ 250 is associated with a high probability of acquired HLH (> 99%) [109]. The HScore was not validated in the pediatric population. Since malignancy-associated HLH is more common in adults while infection-associated HLH is more prevalent in children, there is reason to suspect that the HScore may not be generalizable to pediatric patients. Debaugnies and collaborators found that a lower HScore cut-off (> 141) may provide more sensitivity and specificity in children [110].
3.6. MH Score
Given the shared pathophysiology of FHL, acquired HLH, and MAS, these disorders often present in a similar manner. While FHL was once thought to present exclusively in the first year of life, there are patients with autosomal recessive mutations in classic FHL genes who develop their first HLH manifestations at much older ages [111]. Infections such as EBV can trigger a cytokine storm in patients with genetic and non-genetic forms of the disease [18, 86]. Genetic testing can certainly discriminate between FHL and acquired HLH and MAS; however, it can take weeks to obtain results, which is often unhelpful in a critically ill patient. Thus, it can be difficult to determine if an affected patient has an inherited form of HLH. Yet, it is necessary to identify such patients because FHL is associated with high rates of mortality and the standard of care remains chemotherapy and HSCT [86, 112]. The MH (MAS/HLH) score is a diagnostic tool that was validated to differentiate inherited HLH from MAS in sJIA (Table 2) [87]. Surprisingly, ferritin levels did not distinguish between the two study subject groups. Instead, age at onset, neutrophil count, fibrinogen, splenomegaly, platelet count, and hemoglobin were the six variables included, with age < 1.6 years and neutrophil count < 1.4 × 109/L contributing most to the discriminating power of the MH score [87].
In summary, the diagnosis of HLH and MAS remains a challenge. There are no pathognomonic characteristics that define this group of disorders to aid diagnosis. Since HLH and MAS occur most often in individuals with infection, autoimmune diseases, and malignancy, these underlying conditions can easily obscure the evolving cytokine storm. Various tools have been developed to facilitate the diagnosis of HLH and MAS, all of which use clinical symptoms and laboratory abnormalities common to this group of disorders to risk-stratify patients. These diagnostic tools are useful; however, they are not applicable to every clinical circumstance and certainly do not replace the value of a vigilant clinician who is prepared to recognize HLH and MAS in its many forms. In this sense, the ferritin value alone and the ferritin : ESR ratio are simple, inexpensive, timely, and readily available measures that can be used as screening tools for identifying febrile hospitalized HLH and MAS patients and for driving further work-up of these disorders.
4. Treatment
Prompt diagnosis, elimination of inciting triggers, and rapid initiation of immunosuppression are essential for effective management of HLH/MAS. The prognosis for untreated FHL is dismal with close to 100% fatality [113]. Historically, MAS in sJIA was also characterized by mortality rates over 20% [48]. Treatment regimens detailed below have reduced FHL mortality to 40% and MAS to 8% [38, 114]. Patients diagnosed early tend to fare better than those who are identified later in the disease course, highlighting the importance of early diagnosis. Management by a multidisciplinary team of experts including hemato-oncologists, rheumatologists, and intensivists is needed to provide patients with the required supportive care and access to the full complement of treatment options ranging from chemotherapy to cytokine blockade [100].
4.1. Treatment of Triggers
Both genetic and acquired HLH as well as MAS are often triggered by infections, particularly herpesviruses [18, 38, 86]. In patients with EBV-driven HLH, B-cell depletion with rituximab improves clinical parameters of the disease when used in combination with traditional HLH therapies [115]. Other infections should be treated aggressively with antimicrobials and in some cases intravenous immunoglobulin (IVIG). Rarely, patients with acquired HLH and MAS improve with eradication of the infection alone and may not require further immunosuppression [112]. Similarly, patients with MAS induced by active autoimmune disease may respond to increased immunosuppression for the underlying condition.
4.2. Broad Immunosuppression
The first formal treatment protocol for HLH (HLH-94) consisted of 8 weeks of induction therapy with dexamethasone, etoposide (VP-16), and intrathecal methotrexate and continuation therapy with dexamethasone pulses, VP-16, and cyclosporine [116]. Subsequently, patients with FHL or recurrent disease proceeded to HSCT [116]. These medications were selected for their ability to target and kill T cells and also penetrate the CNS. At long-term follow-up, 55% of patients had survived, a marked improvement compared with the near universal mortality in untreated FHL and 10% survival rate of children treated with chemotherapy-based protocols without transplant [86, 113, 116]. In 2004, HLH-94 was slightly modified by starting cyclosporine in the induction phase and adding hydrocortisone intrathecal therapy (HLH-2004), which resulted in decreased time to HSCT but no improvement in survival [84, 114]. Thus, cyclosporine is now frequently avoided, and others have advocated for lowering the VP-16 burden and associated risk of infection [117].
In rheumatologic patients with MAS, glucocorticoid treatment had been recognized as beneficial in the 1980s [1, 2]. Based on the success of HLH-94, cyclosporine in combination with glucocorticoids was used in patients with MAS and several case series reported rapid benefit [1, 49, 118]. Cyclosporine is still used widely in the field of pediatric rheumatology to treat MAS [5]. Refractory cases of MAS are sometimes treated successfully with reduced intensity VP-16 regimens [38]. IVIG is also used frequently based on a small number of studies with reported benefit; however, IVIG monotherapy is rarely successful, and it is typically used in combination with other medications [49, 119–121].
4.3. Cytokine-Targeted Therapies
The recombinant human IL-1 receptor antagonist, anakinra, blocks the activity of both IL-1α and IL-1β and was originally developed to reduce inflammation in sepsis [122]. It is commonly used to treat patients with sJIA [123–126]. Starting in 2008, case reports surfaced demonstrating efficacy of anakinra in sJIA patients with MAS, especially at doses > 1–2 mg/kg/day [127–130]. In 2011, Miettunen et al. reported dramatic improvement after anakinra therapy in 12 patients with MAS who were refractory to traditional treatments, including seven patients who met full criteria for HLH [119]. Since that time, IL-1 blockade has been increasingly accepted as a first-line option for MAS associated with sJIA and AOSD [4, 100, 131, 132]. Anakinra treatment in cytokine storm is not limited to rheumatologic patients but is also beneficial in children and adults with infection-associated HLH [133, 134]. Re-analysis of the randomized controlled trial of anakinra in sepsis showed improved survival in patients with signs of MAS treated with anakinra compared with the placebo group (65% vs 35%) [134]. These findings unequivocally demonstrate the safety of high-dose anakinra in patients with active infection. In practice, anakinra is typically used in combination with other medications, particularly glucocorticoids, to treat MAS. There are several recent publications advocating for anakinra in combination with high-dose glucocorticoids, IVIG, and cyclosporine as first-line treatment for acquired HLH instead of the etoposide-based protocols [100, 133, 135, 136]. Anakinra monotherapy is typically appropriate in patients with known sJIA and MAS or in circumstances when glucocorticoids use needs to be delayed for diagnostic purposes (concern for infection or malignancy). Typically, patients respond to anakinra rapidly and lack of improvement within 24–48 h of treatment initiation suggests the need for additional immunosuppression. Interestingly, there is little evidence to support the use of other anti-IL-1 agents such as canakinumab or rilonacept in MAS, which may be due to lack of clinical data, divergent mechanism of action of these drugs, or under-dosing [4].
The efficacy of IL-1 blockade in HLH and MAS has spurred exploration of other cytokine-directed therapies. In one report, recombinant human IL-18 binding protein (rhIL-18BP) ameliorated MAS in a patient with NLRC4 gain of function mutations [61]. Currently, rhIL-18BP is an investigation drug; however, the elevated IL-18 levels in AIFEC, XIAP, HLH, and MAS indicate that it may be a promising therapeutic option in the future [31, 55, 71]. Indeed, results from a phase II open-label study of rhIL-18 PB in 23 AOSD patients showed some indications of efficacy [137].
Based on mouse models and human data, IFNγ is considered a linchpin cytokine in driving HLH and MAS pathophysiology [10–12, 16, 29–34]. Preliminary results from a phase III open-label trial of emapalumab (anti-IFNγ monoclonal antibody) confirm that neutralization of IFNγ is a promising option. Emapalumab was used in combination with dexamethasone and cyclosporine in patients with presumed genetic HLH and demonstrated a 63% response rate (https://www.fda.gov/drugs/fda-approves-emapalumab-hemophagocytic-lymphohistiocytosis). Based on these findings, emapalumab was approved by the Food and Drug Administration (FDA) in 2018 for FHL. There is also an ongoing phase II trial evaluating emapalumab for MAS complicating sJIA. An abstract presented at the 2019 European League Against Rheumatism (EULAR) meeting showed that a complete clinical response was achieved in all six patients enrolled in the study to date [138]. While the final results of these two trials are not yet published, there is a case report in the literature describing successful use of emapalumab in the setting of EBV-associated HLH [139]. In addition, two patients with NLRC4 mutations and early-onset HLH and one patient with neonatal-onset cytopenia with dyshematopoiesis, autoinflammation, rash, and hemophagocytosis (NOCARH syndrome) due to mutations in CDC42 responded to emapalumab treatment (reported in abstracts) [140, 141].
IL-6 blockade has proven beneficial in the specific circumstance of cytokine release syndrome secondary to chimeric antigen receptor T-cell therapy, and from blinatumomab treatment, but there are limited data for its use outside of this setting [142, 143].
An alternate strategy for blocking cytokine effects is to target signaling pathways downstream of cytokines binding their cognate receptors. Many cytokine receptors signal through JAK/STAT pathways. There are four mammalian Janus kinases (JAKs) and seven signal transducers and activators of transcription (STATs) [144]. JAKs dimerize upon cytokine receptor binding and recruit and activate STATs, which ultimately enter the nucleus and induce transcriptional changes [144]. Ruxolitinib is a JAK1/2 inhibitor that blocks signaling of type I interferons, IFNγ, and IL-6, among other cytokines. In mouse models of genetic and acquired HLH, ruxolitinib was effective in ameliorating disease manifestations [145]. There are several case reports in the literature of successful ruxolitinib use as salvage therapy for infection-associated HLH [146–148]. JAK inhibitors possess many appealing characteristics including an ability to block multiple cytokines, a rapid onset of action in patients with HLH/MAS, and existing FDA approval for other conditions. Until more data are available, they are considered mainly salvage medications for HLH/MAS with much promise for future expanded use.
4.4. Other Therapies
Various other therapeutic agents have been used for HLH and MAS spectrum diseases. Anti-thymocyte globulin (ATG) is a polyclonal immunoglobulin that targets T cells. Upfront use of ATG in FHL resulted in significant initial responses rates (73%) but was associated with high rates of infection and relapse [112, 149, 150]. It is currently used as salvage therapy for MAS and HLH with limited information on efficacy [149, 151]. Alemtuzumab or Campath is an anti-CD52 antibody that depletes circulating B and T lymphocytes and other immune cells. In the largest retrospective review to date, alemtuzumab therapy for refractory HLH resulted in a complete response in 0/22 patients and a partial response in 14/22 patients [152]. Plasmapheresis has also been explored anecdotally as therapy for HLH [153–156].
5. Conclusion
Our understanding of HLH and MAS spectrum diseases has expanded, and the available treatment strategies have diversified. Traditionally, glucocorticoids, cyclosporine, and cytotoxic agents were mainstays of therapy. By elucidating the key role of cytokines in driving the pathophysiology of HLH and MAS, entirely new classes of medications have become available. Cytokines can be directly neutralized with biologic agents such as anakinra or emapalumab. Alternately, the action of cytokines can be inhibited by blocking signaling pathways with JAK inhibitors. These cytokine-directed therapies hold much promise in their ability to control inflammation with less toxicity than chemotherapy. Yet, data on how to treat patients in the age of biologics and JAK inhibitors are limited. Rigorous studies must be conducted to directly compare traditional and novel treatment regimens while biomarkers are needed to determine which patients will respond to a given therapy. In the meantime, clinicians are left to rely on expert opinion to guide management (Fig. 3). It is therefore essential that rheumatologists with expertise in cytokine blockade and hemato-oncologists with access to chemotherapy work collaboratively to ensure all patients with HLH and MAS have access to the full spectrum of available therapies.
Fig. 3.
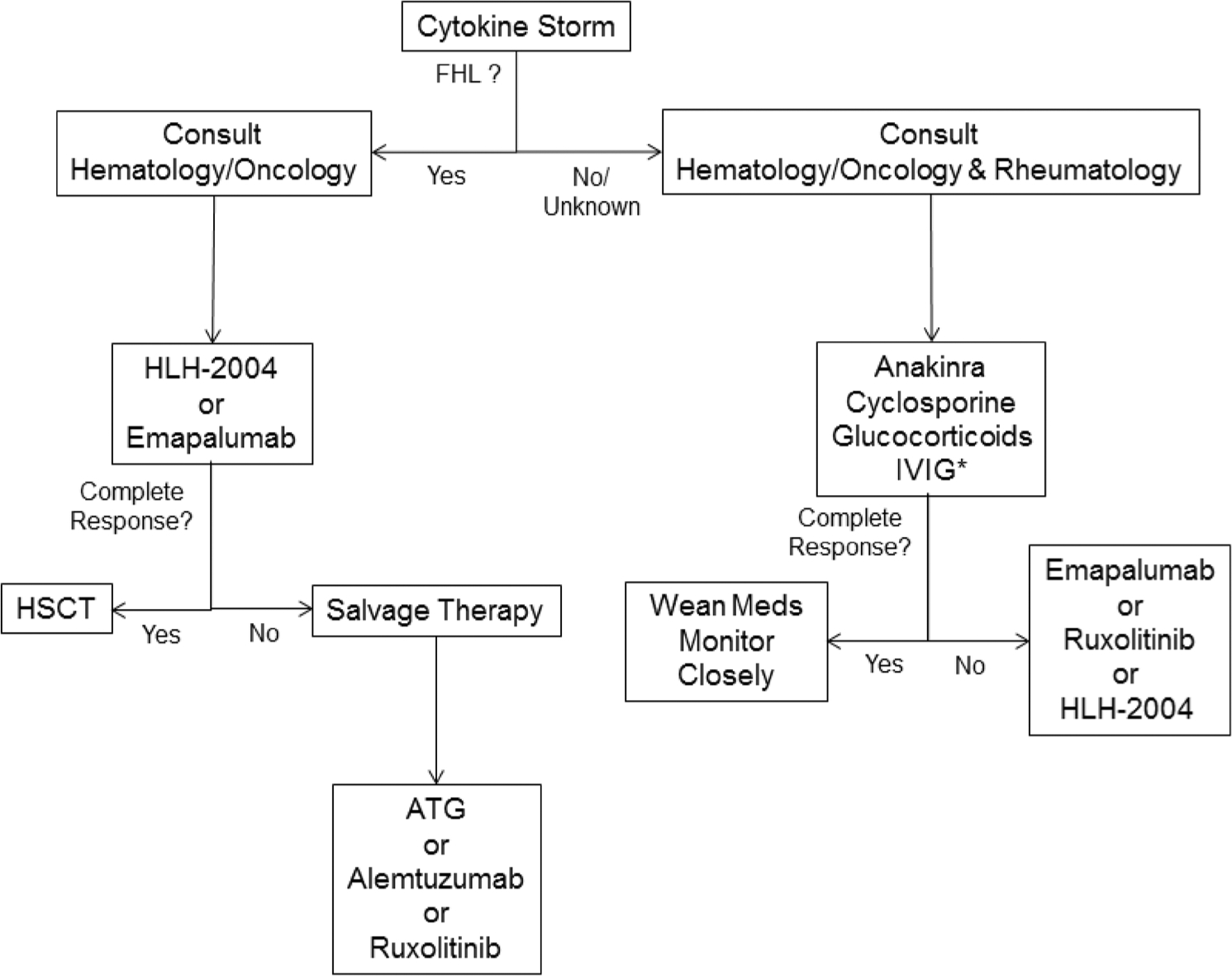
Suggested treatment algorithm for patients with HLH and MAS spectrum diseases. The depicted treatment algorithm is based on expert opinion and currently available data, which are limited. Decisions about treatment are the responsibility of the treating clinician and should always be tailored to individual clinical circumstances. *Anakinra, cyclosporine, glucocorticoids, and IVIG can be used alone or in combination. If anakinra monotherapy is employed, lack of response within 24–48 h suggests the need for additional immunosuppression, particularly high-dose glucocorticoids. ATG anti-thymocyte globulin, FHL familial hemophagocytic lymphohistiocytosis, HLH hemophagocytic lymphohistiocytosis, HSCT hematopoietic stem-cell transplant, IVIG intravenous immunoglobulin, MAS macrophage activation syndrome
Key Points.
Macrophage activation syndrome (MAS) is a well known, frequently fatal complication of inflammatory pediatric rheumatic diseases (e.g., systemic juvenile idiopathic arthritis), genetic autoinflammatory disorders (e.g., NLRC4 mutation), and hematologic malignancies (e.g., T-cell lymphoma).
Diagnostic criteria have been developed to aid in the identification of MAS in the setting of specific diseases (e.g., systemic juvenile idiopathic arthritis) and more broadly (e.g., HScore).
Novel therapies targeting pro-inflammatory cytokines (e.g., anti-interferon-γ) are being explored for less toxic, yet effective, approaches for treating children with MAS.
Funding and Conflicts of Interest
This work was partially supported by the Rheumatology Research Foundation’s Investigator Award (L.A.H.), National Institute of Arthritis and Musculoskeletal and Skin Diseases, P30 AR070253-01 and K08 AR073339-01 (L.A.H.). L.A.H. has received speaking fees from Swedish Orphan Biovitrum (Sobi) (< US$1000). R.Q.C.’s work was supported in part by grants from the Histiocytosis Association, the Center for Genomic Medicine at the University of Alabama at Birmingham & HudsonAlpha Institute for Bio-technology, and an investigator-initiated clinical trial funded by Sobi.
References
- 1.Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–6. 10.1016/s0022-3476(85)80072-x. [DOI] [PubMed] [Google Scholar]
- 2.Silverman ED, Miller JJ 3rd, Bernstein B, Shafai T. Consumption coagulopathy associated with systemic juvenile rheumatoid arthritis. J Pediatr. 1983;103(6):872–6. 10.1016/s0022-3476(83)80704-5. [DOI] [PubMed] [Google Scholar]
- 3.Bracaglia C, Prencipe G, De Benedetti F. Macrophage activation syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J. 2017;15(1):5 10.1186/s12969-016-0130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–68. 10.1038/nrrheum.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravelli A, Davi S, Minoia F, Martini A, Cron RQ. Macrophage activation syndrome. Hematol Oncol Clin N Am. 2015;29(5):927–41. 10.1016/j.hoc.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–9. [DOI] [PubMed] [Google Scholar]
- 7.Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–73. [DOI] [PubMed] [Google Scholar]
- 8.zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter JI, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–34. 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 9.Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18–2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–92. 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8 + T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–43. 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 11.Takada H, Takahata Y, Nomura A, Ohga S, Mizuno Y, Hara T. Increased serum levels of interferon-gamma-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. 2003;133(3):448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–90. 10.1016/j.jpeds.2011.11.046. [DOI] [PubMed] [Google Scholar]
- 13.Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–22. [PubMed] [Google Scholar]
- 14.Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. 2015;212(3):307–17. 10.1084/jem.20140964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang M, Bracaglia C, Prencipe G, Bemrich-Stolz CJ, Beukelman T, Dimmitt RA, et al. A heterozygous RAB27A mutation associated with delayed cytolytic granule polarization and hemophagocytic lymphohistiocytosis. J Immunol. 2016;196(6):2492–503. 10.4049/jimmunol.1501284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jordan MB, Locatelli F, Allen C, De Benedetti F, Grom AA, Ballabio M, Ferlin G, Nl-0501–04 Study Group, De Min C. A novel targeted approach to the treatment of hemophagocytic lymphohistiocytosis (HLH) with an anti-interferon gamma (IFNγ) monoclonal antibody (mAb), NI-0501: first results from a pilot phase 2 study in children with primary HLH. Blood. 2015;126(23):LBA–3. [Google Scholar]
- 17.Brisse E, Wouters CH, Matthys P. Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 2015;26(3):263–80. 10.1016/j.cytogfr.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Brisse E, Wouters CH, Andrei G, Matthys P. How viruses contribute to the pathogenesis of hemophagocytic lymphohistiocytosis. Front Immunol. 2017;8:1102 10.3389/fimmu.2017.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Muller I, Suttorp M, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170(4):539–49. 10.1111/bjh.13462. [DOI] [PubMed] [Google Scholar]
- 20.Machaczka M, Vaktnas J, Klimkowska M, Hagglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–9. 10.3109/10428194.2010.551153. [DOI] [PubMed] [Google Scholar]
- 21.Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142(3):292–6. 10.1067/mpd.2003.110. [DOI] [PubMed] [Google Scholar]
- 22.Cifaldi L, Prencipe G, Caiello I, Bracaglia C, Locatelli F, De Benedetti F, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 2015;67(11):3037–46. 10.1002/art.39295. [DOI] [PubMed] [Google Scholar]
- 23.Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005;7(1):R30–7. 10.1186/ar1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hazen MM, Woodward AL, Hofmann I, Degar BA, Grom A, Filipovich AH, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(2):567–70. 10.1002/art.23199. [DOI] [PubMed] [Google Scholar]
- 25.Kaufman KM, Linghu B, Szustakowski JD, Husami A, Yang F, Zhang K, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014;66(12):3486–95. 10.1002/art.38793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vastert SJ, van Wijk R, D’Urbano LE, de Vooght KM, de Jager W, Ravelli A, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology. 2010;49(3):441–9. 10.1093/rheumatology/kep418. [DOI] [PubMed] [Google Scholar]
- 27.Schulert GS, Zhang M, Fall N, Husami A, Kissell D, Hanosh A, et al. Whole-exome sequencing reveals mutations in genes linked to hemophagocytic lymphohistiocytosis and macrophage activation syndrome in fatal cases of H1N1 influenza. J Infect Dis. 2016;213(7):1180–8. 10.1093/infdis/jiv550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bracaglia CSE, Da Ros M, De Fusco C, Micalizzi M, Cetica V, Ciambotti B, Coniglio ML, Insalaco A, De Benedetti F, Arico M. Mutations of familial hemophagocytic lymphohistiocytosis (FHL) related genes and abnormalities of cytotoxicity function tests in patients with macrophage activation syndrome (MAS) occuring in systemic juvenile idiopathic arthritis (sJIA). Pediatr Rheumatol Online J. 2014;12(Supp1):53.25540605 [Google Scholar]
- 29.Bracaglia C, de Graaf K, Pires Marafon D, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon-gamma and interferon-gamma-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–72. 10.1136/annrheumdis-2015-209020. [DOI] [PubMed] [Google Scholar]
- 30.Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-gamma. Rheumatology. 2015;54(8):1507–17. 10.1093/rheumatology/keu524. [DOI] [PubMed] [Google Scholar]
- 31.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018;131(13):1442–55. 10.1182/blood-2017-12-820852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Investig. 2011;121(6):2264–77. 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prencipe G, Caiello I, Pascarella A, Grom AA, Bracaglia C, Chatel L, et al. Neutralization of IFN-gamma reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol. 2018;141(4):1439–49. 10.1016/j.jaci.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 34.Strippoli R, Carvello F, Scianaro R, De Pasquale L, Vivarelli M, Petrini S, et al. Amplification of the response to Toll-like receptor ligands by prolonged exposure to interleukin-6 in mice: implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum. 2012;64(5):1680–8. 10.1002/art.33496. [DOI] [PubMed] [Google Scholar]
- 35.Strippoli R, Caiello I, De Benedetti F. Reaching the threshold: a multilayer pathogenesis of macrophage activation syndrome. J Rheumatol. 2013;40(6):761–7. 10.3899/jrheum.121233. [DOI] [PubMed] [Google Scholar]
- 36.Brisse E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and secondary haemophagocytic lymphohistiocytosis: differences and similarities. Br J Haematol. 2016;174(2):203–17. 10.1111/bjh.14147. [DOI] [PubMed] [Google Scholar]
- 37.Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–8. [PubMed] [Google Scholar]
- 38.Minoia F, Davi S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160–9. 10.1002/art.38802. [DOI] [PubMed] [Google Scholar]
- 39.Bleesing J, Prada A, Siegel DM, Villanueva J, Olson J, Ilowite NT, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–71. 10.1002/art.22416. [DOI] [PubMed] [Google Scholar]
- 40.Gavand PE, Serio I, Arnaud L, Costedoat-Chalumeau N, Carvelli J, Dossier A, et al. Clinical spectrum and therapeutic management of systemic lupus erythematosus-associated macrophage activation syndrome: a study of 103 episodes in 89 adult patients. Autoimmun Rev. 2017;16(7):743–9. 10.1016/j.autrev.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 41.Borgia RE, Gerstein M, Levy DM, Silverman ED, Hiraki LT. Features, treatment, and outcomes of macrophage activation syndrome in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(4):616–24. 10.1002/art.40417. [DOI] [PubMed] [Google Scholar]
- 42.Latino GA, Manlhiot C, Yeung RS, Chahal N, McCrindle BW. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol Oncol. 2010;32(7):527–31. 10.1097/MPH.0b013e3181dccbf4. [DOI] [PubMed] [Google Scholar]
- 43.Bennett TD, Fluchel M, Hersh AO, Hayward KN, Hersh AL, Brogan TV, et al. Macrophage activation syndrome in children with systemic lupus erythematosus and children with juvenile idiopathic arthritis. Arthritis Rheum. 2012;64(12):4135–42. 10.1002/art.34661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Gong F, Zhu W, Fu S, Zhang Q. Macrophage activation syndrome in Kawasaki disease: more common than we thought? Semin Arthritis Rheum. 2015;44(4):405–10. 10.1016/j.semarthrit.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Francois B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med. 1997;103(2):114–20. 10.1016/s0002-9343(97)00136-8. [DOI] [PubMed] [Google Scholar]
- 46.Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14(4):307–8. 10.1038/ni.2554. [DOI] [PubMed] [Google Scholar]
- 47.Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients—a postmortem clinicopathologic analysis. Crit Care Med. 2004;32(6):1316–21. 10.1097/01.ccm.0000127779.24232.15. [DOI] [PubMed] [Google Scholar]
- 48.Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421–6. 10.1136/adc.85.5.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephan JL, Kone-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285–92. [DOI] [PubMed] [Google Scholar]
- 50.Hiraki LT, Silverman ED. Genomics of systemic lupus erythematosus: insights gained by studying monogenic young-onset systemic lupus erythematosus. Rheum Dis Clin N Am. 2017;43(3):415–34. 10.1016/j.rdc.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Bader-Meunier B, Florkin B, Sibilia J, Acquaviva C, Hachulla E, Grateau G, et al. Mevalonate kinase deficiency: a survey of 50 patients. Pediatrics. 2011;128(1):e152–9. 10.1542/peds.2010-3639. [DOI] [PubMed] [Google Scholar]
- 52.Rossi-Semerano L, Hermeziu B, Fabre M, Kone-Paut I. Macrophage activation syndrome revealing familial Mediterranean fever. Arthritis Care Res. 2011;63(5):780–3. 10.1002/acr.20418. [DOI] [PubMed] [Google Scholar]
- 53.Horneff G, Rhouma A, Weber C, Lohse P. Macrophage activation syndrome as the initial manifestation of tumour necrosis factor receptor 1-associated periodic syndrome (TRAPS). Clin Exp Rheumatol. 2013;31(3 Suppl 77):99–102. [PubMed] [Google Scholar]
- 54.Rigante D, Emmi G, Fastiggi M, Silvestri E, Cantarini L. Macrophage activation syndrome in the course of monogenic autoinflammatory disorders. Clin Rheumatol. 2015;34(8):1333–9. 10.1007/s10067-015-2923-0. [DOI] [PubMed] [Google Scholar]
- 55.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–6. 10.1038/ng.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet. 2014;46(10):1135–9. 10.1038/ng.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7(6):569–75. 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 58.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204(13):3235–45. 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356(6372):768–74. 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 60.Volker-Touw CM, de Koning HD, Giltay JC, de Kovel CG, van Kempen TS, Oberndorff KM, et al. Erythematous nodes, urticarial rash and arthralgias in a large pedigree with NLRC4-related autoinflammatory disease, expansion of the phenotype. Br J Dermatol. 2017;176(1):244–8. 10.1111/bjd.14757. [DOI] [PubMed] [Google Scholar]
- 61.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139(5):1698–701. 10.1016/j.jaci.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Canna SW, Behrens EM. Not all hemophagocytes are created equally: appreciating the heterogeneity of the hemophagocytic syndromes. Curr Opin Rheumatol. 2012;24(1):113–8. 10.1097/BOR.0b013e32834dd37e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tesi B, Bryceson YT. HLH: genomics illuminates pathophysiological diversity. Blood. 2018;132(1):5–7. 10.1182/blood-2018-05-845818. [DOI] [PubMed] [Google Scholar]
- 64.Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–16. 10.1016/S0140-6736(13)61048-X. [DOI] [PubMed] [Google Scholar]
- 65.Strenger V, Merth G, Lackner H, Aberle SW, Kessler HH, Seidel MG, et al. Malignancy and chemotherapy induced haemophagocytic lymphohistiocytosis in children and adolescents-a single centre experience of 20 years. Ann Hematol. 2018;97(6):989–98. 10.1007/s00277-018-3254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–22. 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Faitelson Y, Grunebaum E. Hemophagocytic lymphohistiocytosis and primary immune deficiency disorders. Clin Immunol. 2014;155(1):118–25. 10.1016/j.clim.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 68.Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011;117(5):1522–9. 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 69.Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature. 1998;395(6701):462–9. 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- 70.Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–4. 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 71.Wada T, Kanegane H, Ohta K, Katoh F, Imamura T, Nakazawa Y, et al. Sustained elevation of serum interleukin-18 and its association with hemophagocytic lymphohistiocytosis in XIAP deficiency. Cytokine. 2014;65(1):74–8. 10.1016/j.cyto.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 72.Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Investig. 2009;119(5):1350–8. 10.1172/jci37901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. 2015;136(3):703–12. 10.1016/j.jaci.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 74.Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2 + regulation of immunity against Epstein–Barr virus. Blood. 2014;123(14):2148–52. 10.1182/blood-2013-11-538686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagle DL, Karim MA, Woolf EA, Holmgren L, Bork P, Misumi DJ, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14(3):307–11. 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- 76.Menasche G, Pastural E, Feldmann J, Certain S, Ersoy F, Dupuis S, et al. Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet. 2000;25(2):173–6. 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- 77.Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat Immunol. 2003;4(11):1111–20. 10.1038/ni1000. [DOI] [PubMed] [Google Scholar]
- 78.Crayne CB, Albeituni S, Nichols KE, Cron RQ. The immunology of macrophage activation syndrome. Front Immunol. 2019;10:119 10.3389/fimmu.2019.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Filipovich AH, Chandrakasan S. Pathogenesis of hemophagocytic lymphohistiocytosis. Hematol Oncol Clin N Am. 2015;29(5):895–902. 10.1016/j.hoc.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 80.Parekh C, Hofstra T, Church JA, Coates TD. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer. 2011;56(3):460–2. 10.1002/pbc.22830. [DOI] [PubMed] [Google Scholar]
- 81.Valentine G, Thomas TA, Nguyen T, Lai YC. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: a case report. Pediatrics. 2014;134(6):e1727–30. 10.1542/peds.2014-2175. [DOI] [PubMed] [Google Scholar]
- 82.Alvarez-Cardona A, Rodriguez-Lozano AL, Blancas-Galicia L, Rivas-Larrauri FE, Yamazaki-Nakashimada MA. Intravenous immunoglobulin treatment for macrophage activation syndrome complicating chronic granulomatous disease. J Clin Immunol. 2012;32(2):207–11. 10.1007/s10875-011-9616-5. [DOI] [PubMed] [Google Scholar]
- 83.Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin N Am. 2012;59(2):329–44. 10.1016/j.pcl.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 85.Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014;141(1):62–71. 10.1309/AJCPMD5TJEFOOVBW. [DOI] [PubMed] [Google Scholar]
- 86.Arico M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197–203. [PubMed] [Google Scholar]
- 87.Minoia F, Bovis F, Davi S, Insalaco A, Lehmberg K, Shenoi S, et al. Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr. 2017;189(72–8):e3 10.1016/j.jpeds.2017.06.005. [DOI] [PubMed] [Google Scholar]
- 88.Gars E, Purington N, Scott G, Chisholm K, Gratzinger D, Martin BA, et al. Bone marrow histomorphological criteria can accurately diagnose hemophagocytic lymphohistiocytosis. Haematologica. 2018;103(10):1635–41. 10.3324/haematol.2017.186627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1227–35. 10.1002/pbc.21423. [DOI] [PubMed] [Google Scholar]
- 90.Schram AM, Campigotto F, Mullally A, Fogerty A, Massarotti E, Neuberg D, et al. Marked hyperferritinemia does not predict for HLH in the adult population. Blood. 2015;125(10):1548–52. 10.1182/blood-2014-10-602607. [DOI] [PubMed] [Google Scholar]
- 91.Lehmberg K, McClain KL, Janka GE, Allen CE. Determination of an appropriate cut-off value for ferritin in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2014;61(11):2101–3. 10.1002/pbc.25058. [DOI] [PubMed] [Google Scholar]
- 92.Saeed H, Woods RR, Lester J, Herzig R, Gul Z, Monohan G. Evaluating the optimal serum ferritin level to identify hemophagocytic lymphohistiocytosis in the critical care setting. Int J Hematol. 2015;102(2):195–9. 10.1007/s12185-015-1813-1. [DOI] [PubMed] [Google Scholar]
- 93.Takada H, Ohga S, Mizuno Y, Suminoe A, Matsuzaki A, Ihara K, et al. Oversecretion of IL-18 in haemophagocytic lymphohistiocytosis: a novel marker of disease activity. Br J Haematol. 1999;106(1):182–9. [DOI] [PubMed] [Google Scholar]
- 94.Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. 2010;49(9):1645–53. 10.1093/rheumatology/keq133. [DOI] [PubMed] [Google Scholar]
- 95.Davi S, Minoia F, Pistorio A, Horne A, Consolaro A, Rosina S, et al. Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66(10):2871–80. 10.1002/art.38769. [DOI] [PubMed] [Google Scholar]
- 96.Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016;68(3):566–76. 10.1002/art.39332. [DOI] [PubMed] [Google Scholar]
- 97.Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016;75(3):481–9. 10.1136/annrheumdis-2015-208982. [DOI] [PubMed] [Google Scholar]
- 98.Shimizu M, Mizuta M, Yasumi T, Iwata N, Okura Y, Kinjo N, et al. Validation of classification criteria of macrophage activation syndrome in Japanese patients with systemic juvenile idiopathic arthritis. Arthritis Care Res. 2018;70(9):1412–5. 10.1002/acr.23482. [DOI] [PubMed] [Google Scholar]
- 99.Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. Expert consensus on dynamics of laboratory tests for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. RMD Open. 2016;2(1):e000161 10.1136/rmdopen-2015-000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Halyabar O, Chang MH, Schoettler ML, Schwartz MA, Baris EH, Benson LA, et al. Calm in the midst of cytokine storm: a collaborative approach to the diagnosis and treatment of hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Pediatr Rheumatol Online J. 2019;17(1):7 10.1186/s12969-019-0309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schulert GS, Minoia F, Bohnsack J, Cron RQ, Hashad S, Kon EPI, et al. Effect of biologic therapy on clinical and laboratory features of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. Arthritis Care Res. 2018;70(3):409–19. 10.1002/acr.23277. [DOI] [PubMed] [Google Scholar]
- 102.Ahn SS, Yoo BW, Jung SM, Lee SW, Park YB, Song JJ. Application of the 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome in patients with adult-onset still disease. J Rheumatol. 2017;44(7):996–1003. 10.3899/jrheum.161286. [DOI] [PubMed] [Google Scholar]
- 103.Ahn SS, Yoo BW, Jung SM, Lee SW, Park YB, Song JJ. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Semin Arthritis Rheum. 2017;47(2):216–21. 10.1016/j.semarthrit.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 104.Tada Y, Inokuchi S, Maruyama A, Suematsu R, Sakai M, Sadanaga Y, et al. Are the 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome applicable to patients with adult-onset Still’s disease? Rheumatol Int. 2019;39(1):97–104. 10.1007/s00296-018-4114-1. [DOI] [PubMed] [Google Scholar]
- 105.Parodi A, Davi S, Pringe AB, Pistorio A, Ruperto N, Magni-Manzoni S, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 2009;60(11):3388–99. 10.1002/art.24883. [DOI] [PubMed] [Google Scholar]
- 106.Minoia F, Bovis F, Davi S, Horne A, Fischbach M, Frosch M, et al. Development and initial validation of the MS score for diagnosis of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2019. 10.1136/annrheumdis-2019-215211. [DOI] [PubMed] [Google Scholar]
- 107.Gorelik M, Fall N, Altaye M, Barnes MG, Thompson SD, Grom AA, et al. Follistatin-like protein 1 and the ferritin/erythrocyte sedimentation rate ratio are potential biomarkers for dysregulated gene expression and macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2013;40(7):1191–9. 10.3899/jrheum.121131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eloseily EMA, Minoia F, Crayne CB, Beukelman T, Ravelli A, Cron RQ. Ferritin to erythrocytoe sedimentation rate ratio: simple measure to identify macrophage activation syndrome in systemic juvenile idiopathic arthritis. ACR Open Rheumatol. 2019. 10.1002/acr2.11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–20. 10.1002/art.38690. [DOI] [PubMed] [Google Scholar]
- 110.Debaugnies F, Mahadeb B, Ferster A, Meuleman N, Rozen L, Demulder A, et al. Performances of the H-Score for diagnosis of hemophagocytic lymphohistiocytosis in adult and pediatric patients. Am J Clin Pathol. 2016;145(6):862–70. 10.1093/ajcp/aqw076. [DOI] [PubMed] [Google Scholar]
- 111.Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13–4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794–8. 10.1182/blood-2011-07-370148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041–52. 10.1182/blood-2011-03-278127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221–30. [DOI] [PubMed] [Google Scholar]
- 114.Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728–38. 10.1182/blood-2017-06-788349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemoimmunotherapeutic regimens. Br J Haematol. 2013;162(3):376–82. 10.1111/bjh.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henter JI, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–73. 10.1182/blood-2002-01-0172. [DOI] [PubMed] [Google Scholar]
- 117.Ehl S, Astigarraga I, von Bahr Greenwood T, Hines M, Horne A, Ishii E, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH steering committee of the histiocyte society. J Allergy Clin Immunol Pract. 2018;6(5):1508–17. 10.1016/j.jaip.2018.05.031. [DOI] [PubMed] [Google Scholar]
- 118.Ravelli A, De Benedetti F, Viola S, Martini A. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr. 1996;128(2):275–8. [DOI] [PubMed] [Google Scholar]
- 119.Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology. 2011;50(2):417–9. 10.1093/rheumatology/keq218. [DOI] [PubMed] [Google Scholar]
- 120.Chen RL, Lin KH, Lin DT, Su IJ, Huang LM, Lee PI, et al. Immunomodulation treatment for childhood virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. 1995;89(2):282–90. 10.1111/j.1365-2141.1995.tb03302.x. [DOI] [PubMed] [Google Scholar]
- 121.Emmenegger U, Frey U, Reimers A, Fux C, Semela D, Cottagnoud P, et al. Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol. 2001;68(1):4–10. [DOI] [PubMed] [Google Scholar]
- 122.Fisher CJ Jr., Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271(23):1836–43. [PubMed] [Google Scholar]
- 123.DeWitt EM, Kimura Y, Beukelman T, Nigrovic PA, Onel K, Prahalad S, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. 2012;64(7):1001–10. 10.1002/acr.21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–54. 10.1136/ard.2010.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–55. 10.1002/art.30128. [DOI] [PubMed] [Google Scholar]
- 126.Ter Haar NM, van Dijkhuizen EHP, Swart JF, van Royen-Kerkhof A, El Idrissi A, Leek AP, et al. Treatment to target using recombinant interleukin-1 receptor antagonist as first-line monotherapy in new-onset systemic juvenile idiopathic arthritis: results from a five-year follow-up study. Arthritis Rheumatol. 2019;71(7):1163–73. 10.1002/art.40865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Durand M, Troyanov Y, Laflamme P, Gregoire G. Macrophage activation syndrome treated with anakinra. J Rheumatol. 2010;37(4):879–80. 10.3899/jrheum.091046. [DOI] [PubMed] [Google Scholar]
- 128.Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis. 2011;17(1):23–7. 10.1097/RHU.0b013e318205092d. [DOI] [PubMed] [Google Scholar]
- 129.Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008;4(11):615–20. 10.1038/ncprheum0919. [DOI] [PubMed] [Google Scholar]
- 130.Kahn PJ, Cron RQ. Higher-dose Anakinra is effective in a case of medically refractory macrophage activation syndrome. J Rheumatol. 2013;40(5):743–4. 10.3899/jrheum.121098. [DOI] [PubMed] [Google Scholar]
- 131.Lenert A, Yao Q. Macrophage activation syndrome complicating adult onset Still’s disease: a single center case series and comparison with literature. Semin Arthritis Rheum. 2016;45(6):711–6. 10.1016/j.semarthrit.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 132.Neel A, Wahbi A, Tessoulin B, Boileau J, Carpentier D, Decaux O, et al. Diagnostic and management of life-threatening Adult-Onset Still Disease: a French nationwide multicenter study and systematic literature review. Crit Care. 2018;22(1):88 10.1186/s13054-018-2012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med J Soc Crit Care Med World Fed Pediatr Intensive Crit Care Soc. 2014;15(5):401–8. 10.1097/PCC.0000000000000078. [DOI] [PubMed] [Google Scholar]
- 134.Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–81. 10.1097/CCM.0000000000001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wohlfarth P, Agis H, Gualdoni GA, Weber J, Staudinger T, Schellongowski P, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically ill adult patients with hemophagocytic lymphohistiocytosis. J Intensive Care Med. 2017. 10.1177/0885066617711386. [DOI] [PubMed] [Google Scholar]
- 136.Kumar B, Aleem S, Saleh H, Petts J, Ballas ZK. A personalized diagnostic and treatment approach for macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in adults. J Clin Immunol. 2017;37(7):638–43. 10.1007/s10875-017-0439-x. [DOI] [PubMed] [Google Scholar]
- 137.Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Kotter I, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77(6):840–7. 10.1136/annrheumdis-2017-212608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.De Benedetti F, Borgan P, Grom A, Quartier P, Schneider R, De Graaf K, et al. OP0204 Emapalumab, an interferon gamma-blocking monoclonal antibody, in patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis [abstract]. Ann Rheum Dis. 2019;78:178. [Google Scholar]
- 139.Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. 2019;3(1):47–50. 10.1182/bloodadvances.2018025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bracaglia C, Prencipe G, Insalaco A, Caiello I, Marucci G, Pecoraro R, et al. Emapalumab, an anti-interferon gamma monoclonal antibody in two patients with NLRC4-related disease and severe hemophagocytic lymphohistiocytosis (HLH) [abstract]. Arthritis Rheumatol. 2018;70(suppl 9):1547. [Google Scholar]
- 141.Lam MT, Coppola S, Krumbach OH, Prencipe G, Insalaco A, Cifaldi C, et al. FRI0540 A novel autoinflammatory disease characterized by neonatal-onset cytopenia with autoinflammation, rash, and hemophagocytosis (NOCARH) due to aberrant CDC42 function [abstract]. Ann Rheum Dis. 2019;78(suppl 2):964. [Google Scholar]
- 142.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154–7. 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18(4):374–84. 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127(13):1666–75. 10.1182/blood-2015-12-684399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zandvakili I, Conboy CB, Ayed AO, Cathcart-Rake EJ, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a second experience. Am J Hematol. 2018;93(5):E123–5. 10.1002/ajh.25063. [DOI] [PubMed] [Google Scholar]
- 147.Slostad J, Hoversten P, Haddox CL, Cisak K, Paludo J, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a single patient experience. Am J Hematol. 2018;93(2):E47–9. 10.1002/ajh.24971. [DOI] [PubMed] [Google Scholar]
- 148.Broglie L, Pommert L, Rao S, Thakar M, Phelan R, Margolis D, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. 2017;1(19):1533–6. 10.1182/bloodadvances.2017007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, Neven B, Picard C, Blanche S, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–8. 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]
- 150.Marsh RA, Jordan MB, Talano JA, Nichols KE, Kumar A, Naqvi A, et al. Salvage therapy for refractory hemophagocytic lymphohistiocytosis: a review of the published experience. Pediatr Blood Cancer. 2017. 10.1002/pbc.26308. [DOI] [PubMed] [Google Scholar]
- 151.Coca A, Bundy KW, Marston B, Huggins J, Looney RJ. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol. 2009;132(1):10–8. 10.1016/j.clim.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 152.Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–9. 10.1002/pbc.24188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nusshag C, Morath C, Zeier M, Weigand MA, Merle U, Brenner T. Hemophagocytic lymphohistiocytosis in an adult kidney transplant recipient successfully treated by plasmapheresis: a case report and review of the literature. Medicine. 2017;96(50):e9283 10.1097/MD.0000000000009283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kinjo N, Hamada K, Hirayama C, Shimizu M. Role of plasma exchange, leukocytapheresis, and plasma diafiltration in management of refractory macrophage activation syndrome. J Clin Apheresis. 2018;33(1):117–20. 10.1002/jca.21570. [DOI] [PubMed] [Google Scholar]
- 155.Simon DW, Aneja R, Carcillo JA, Halstead ES. Plasma exchange, methylprednisolone, IV immune globulin, and now anakinra support continued PICU equipoise in management of hyper-ferritinemia-associated sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome/secondary hemophagocytic lymphohistiocytosis syndrome*. Pediatr Crit Care Med J Soc Crit Care Med World Fed Pediatr Intensive Crit Care Soc. 2014;15(5):486–8. 10.1097/PCC.0000000000000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Demirkol D, Yildizdas D, Bayrakci B, Karapinar B, Kendirli T, Koroglu TF, et al. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care. 2012;16(2):R52 10.1186/cc11256. [DOI] [PMC free article] [PubMed] [Google Scholar]