

Abstract
Untargeted metabolomics by liquid chromatography–mass spectrometry generates data-rich chromatograms in the form of m/z-retention time features. Managing such datasets is a bottleneck. Many popular data processing tools, including XCMS-online and MZmine2, yield numerous false-positive peak detections. Flagging and removing such false peaks manually is a time-consuming task and prone to human error. We present a web application, Mass Spectral Feature List Optimizer (MS-FLO), to improve the quality of feature lists after initial processing to expedite the process of data curation. The tool utilizes retention time alignments, accurate mass tolerances, Pearson’s correlation analysis, and peak height similarity to identify ion adducts, duplicate peak reports, and isotopic features of the main monoisotopic metabolites. Removing such erroneous peaks reduces the overall number of metabolites in data reports and improves the quality of subsequent statistical investigations. To demonstrate the effectiveness of MS-FLO, we processed 28 biological studies and uploaded raw and results data to the Metabolomics Workbench website (www.metabolomicsworkbench.org), encompassing 1481 chromatograms produced by two different data processing programs used in-house (MZmine2 and later MS-DIAL). Post-processing of datasets with MS-FLO yielded a 7.8% automated reduction of total peak features and flagged an additional 7.9% of features, per dataset, for review by the user. When manually curated, 87% of these additional flagged features were verified false positives. MS-FLO is an open source web application that is freely available for use at http://msflo.fiehnlab.ucdavis.edu.
Graphical Abstract

Processing data effectively and efficiently is a major hurdle in obtaining useable and interpretable data when performing untargeted metabolomics studies via liquid chromatography– tandem mass spectroscopy (LC-MS/MS). There are many data processing programs with powerful peak picking, deconvolution, and alignment algorithms that may be used in untargeted data analysis and peak table creation.1–4 To date, no untargeted data processing program is capable of generating data tables from complex samples without also producing erroneous features, such as duplicate or isotopic peaks. Manually curating an untargeted dataset involves removing duplicate features, isotopic features, chemical noise, and combining multiple ion adducts generated from the same molecule to form a concise feature list and is a time-consuming and error-prone task. There is a need for an automated method of identifying unsolicited features in exported peak tables before proceeding to advanced statistical analysis and identification of unknowns. Incorrect features increase the false discovery rate (FDR) and weaken the statistical power of the study. In addition, false positive peak reports cause users to filter through noise and spend valuable time attempting to identify fabricated metabolites.
Current untargeted data processing programs have numerous settings that can drastically alter the content of the dataset. Ostensibly subtle changes can result in substantial variations in the number of features generated from the same sample set.5 Although a common workflow used by several prevalent software programs follows the same order of processes, from peak detection, deconvolution, and sample alignment, the algorithms and settings employed by the various vendor and open source programs can vary greatly.6 Optimizing these settings is laborious. If parameter thresholds are set too conservatively, truncated data tables may be generated that do not contain a full list of features, essentially underrepresenting the raw data.7 In contrast, under different sets of parameters, data tables may result that contain an overabundance of false features and can be just as detrimental when attempting to extract the representative information from a set of samples. The Mass Spectral Feature List Optimizer (MS-FLO) was designed to examine datasets after initial processing and alert users to erroneous features, thus strengthening the statistical power of the dataset.
Other published tools8–10 have been developed that search datasets for isotopic features and adduct ions by augmenting existing open-source peak-picking programs, such as XCMS or MZmine2. Projects such as CAMERA9 and Allocator8 search for adduct and isotopic features as a means of aiding in metabolite annotation. Ion Fusion10 is a tool that is similar to MS-FLO, which focuses on the removal of isotopes and the fusion of ion adducts; however, the Ion Fusion tool does not account for duplicate or contaminate peaks. The purpose of this paper is not to directly compare the performance of MS-FLO to other available tools, but, instead, to describe the user-friendly features and versatility of MS-FLO. Unlike the other programs in its class, MS-FLO is a stand-alone web-based application that can be used with any commercial or open source data processing software, for recognition and removal of erroneous features in LC-MS datasets. In this paper, we describe the web-based tool MS-FLO and its addition to a generalized untargeted metabolomics workflow. We discuss the performance of this tool and compare data sets post-processed manually to this new automated method of post-processing.
EXPERIMENTAL SECTION
Software.
Data processing prior to submission to MS-FLO was performed via MZmine2 and MS-DIAL data processing software. To optimize the settings used in these programs, the NIH West Coast Metabolomics Center (WCMC) used validated in-house LC-MS methods to analyze replicate human plasma lipidomics samples spiked with known internal standards. These samples were processed with numerous adjustments to individual parameters in MS-DIAL and MZmine2. The results were then examined for the detection of internal standards and known matrix metabolites. The total number of features detected was also considered, to avoid parameters that yielded datasets with vastly exaggerated feature lists. Although it is beyond the scope of this publication, further details about data processing parameters used in MS-DIAL and MZmine2 can be found in the Supporting Information.
MS-FLO directly accepts files exported from MZmine2, MS-DIAL, and XCMS-online data processing programs. MS-FLO is a freely available, open-source, software written in the Python programming language and utilizes the NumPY and Pandas libraries for data processing and analysis. It is available as a browser-based web service written in AngularJS that can be accessed at http://msflo.fiehnlab.ucdavis.edu. This web tool allows a user to upload files directly from data processing software, specify their desired parameters, run the analysis on our server, and download an archive of the full result set. Demonstration datasets and a user manual, including instructions detailing how to use MS-FLO and how to quickly format a feature list exported from any other data processing program, can also be found at http://msflo.fiehnlab.ucdavis.edu.
Datasets Investigated.
Twenty-eight (28) untargeted LC-QTOF mass spectrometry studies (summing to 1481 chromatograms) were analyzed by the WCMC and processed with MS-FLO to reduce false positive peak reports. Details about samples, studies, mass spectrometry parameters, LC parameters and other metadata are summarized in Table S1 in the Supporting Information. Result tables, raw data, and detailed metadata have been uploaded to the NIH Metabolomics Workbench,11 in order to give the wider metabolomics community the opportunity to benchmark software improvements against publicly available datasets. Feature lists processed with MS-FLO were comprised of both research collaborations and service contract work. Processing of untargeted feature lists was performed via MZmine2 prior to August 2015. Afterward, the laboratory switched to using the newly published MS-DIAL software that can process data from any LC-MS platform for either data-dependent or data-independent MS/MS acquisitions.1
Liquid Chromatography–Mass Spectrometry.
Chromatography conditions varied based on the metabolic questions behind the studies, ranging from charged-surface hybrid reversed-phase LC for lipidomics assessments to hydrophilic interaction chromatography for analysis of biogenic amines. All studies were performed on accurate-mass quadrupole time-of-light mass spectrometers (QTOF) with slight variations in MS1 acquisition rates, positive or negative electrospray ionization voltages, and ion-source parameters. Specific LC and QTOF parameters can be found on the NIH Metabolomics Workbench. Workbench identifiers and brief study summaries are given in Table S1.
RESULTS AND DISCUSSION
Study Design.
The 28 studies were well-distributed between biofluids (11 studies), cell and media studies (9 experiments), and tissue samples (8 studies). Eighty two percent (82%) of the studies were using lipidomics data acquisitions (9 datasets analyzed in negative electrospray ionization, and 14 datasets in positive ESI mode), 11% of the studies investigated biogenic amines by HILIC-QTOF MS, and 7% were regular reversed-phase LC-QTOF MS datasets. The number of samples per study varied greatly, ranging between 3 and 302 samples, with a mean ± standard deviation of 53 ± 75 samples (see Table 1). Because of the wide diversity of methods and matrices, feature counts per dataset in the examined studies varied greatly between 128 and 2806 features (Table S1) after initial data processing.
Table 1.
Details from MS-FLO Log Files of 28 Datasets Processed by MS-FLO
| mean ± standard deviation | |
|---|---|
| number of samples | 3–302 |
| duplicates | |
| flaggeda | 16 ± 26 |
| auto removedb | 5 ± 10 |
| adducts pairs | |
| flaggedc | 32 ± 40 |
| auto joinedd | 71 ± 71 |
| isotopes flaggedd | 31 ± 34 |
| sum all flagged features | 79 ± 101 |
| initial features | 996 ± 657 |
| final features | 918 ± 605 |
| features auto removed | |
| mean ± standard deviation | 78 ± 71 |
| percentage | 7.8% |
| total features flagged and removed | 15.7% |
Similarity <85%.
Similarity >85%.
0.5 < R2 <0.8.
R2 ≥ 0.8.
Developing and Implementing MS-FLO.
The mass spectral feature list optimizer removes erroneous peak reports and flags ambiguous mass-to-charge-retention time features (m/z-RT, later: “features”) that should be manually checked by users. Figure 1 shows how to integrate MS-FLO into a generalized workflow for the generation and processing of untargeted LC-MS-based metabolomics data. Four categories of erroneous peaks were defined: (1) actual duplicate feature reports based on the analytical techniques used, specifically for mass accuracy and expected chromatographic peak widths; (2) isotopes of the main monoisotopic features that most software programs claim to efficiently remove; (3) different charged variants of chemical adducts of the neutral molecules that together represent all ion species of a molecule; and (4) consistent contaminant ions that may stem from solvent noise, instrument artifacts or infusion ions, column degradation, or other repetitive features.
Figure 1.
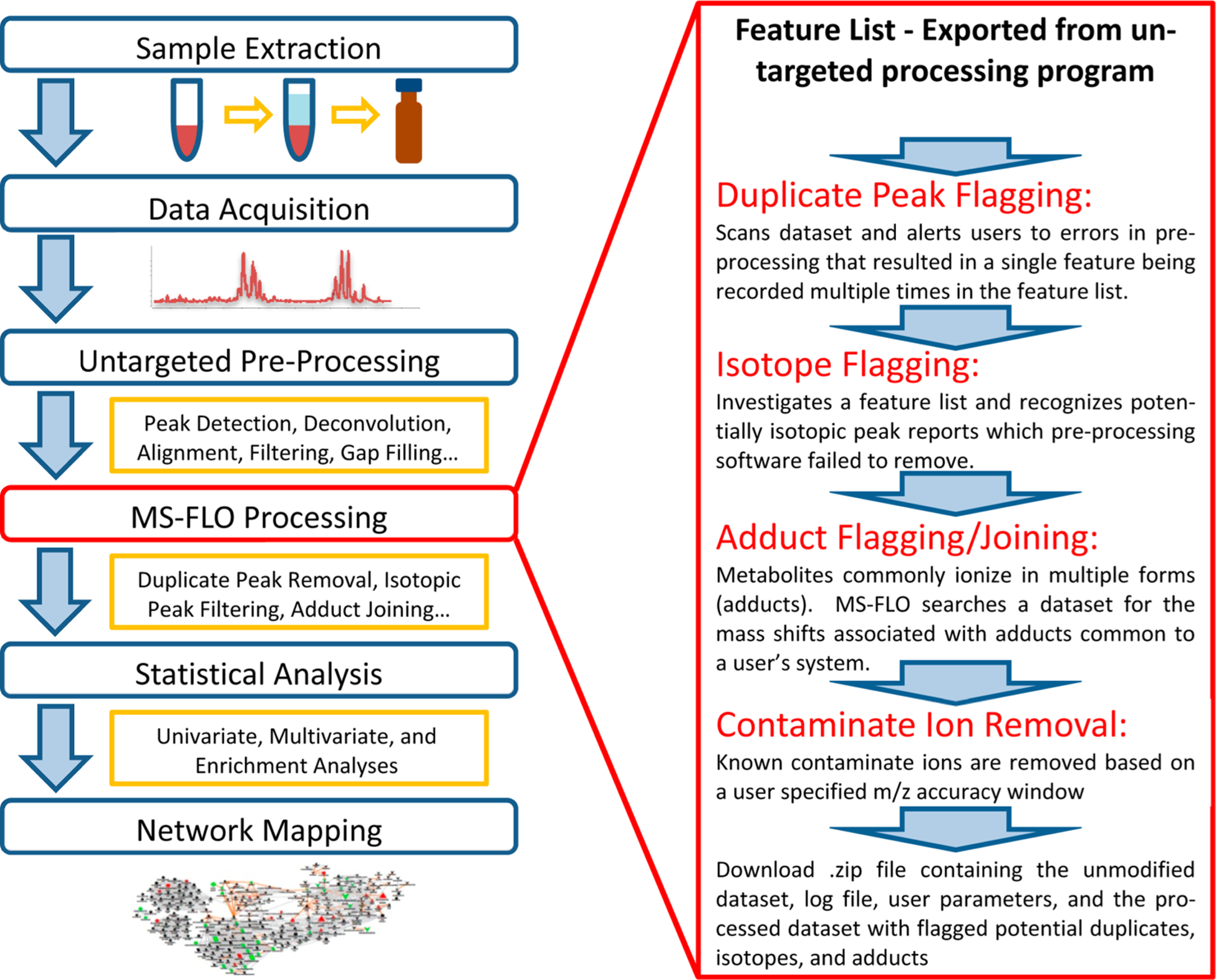
Workflow for the generation and processing of untargeted LC-MS-based metabased metabolomics data. Four categoriesbolomics data, and the incorporation of MS-FLO post-processing into said workflow.
Duplicate Peak Removal.
Duplicate peak removal involves scanning the dataset to identify peaks that have been incorrectly aligned as separate peaks by data processing software. MS-FLO’s algorithm utilizes user-defined settings for peak similarity based on retention time variance, m/z precision, and similarity of peak heights found in a defined percentage of samples. Both features must fall within the user-defined RT and m/z windows to be considered a potential duplicate. Potential duplicates are then compared based on the percentage of samples that have comparable peak intensity for both features, with the user defining the minimum percent similarity required to automatically remove one of the features. MS-FLO retains the feature that first occurs in the data table when two features are found to have a similarity percentage above the value defined by the user. Potential duplicates that do not meet the user-defined minimum percent similarity are flagged for manual review. Users also have the option to set a peak height threshold; this threshold dictates the peak height difference at which MS-FLO will consider peak heights to be equal. Figure S1 shows an excerpt of a .csv file that has been exported from MS-FLO and contains examples of true and false flagged duplicates.
Isotope Matching.
Isotope matching searches all features in a dataset to identify potential isotopic relationships. The default value is 1.003355 Da, which is the difference between a 13C isotope and naturally occurring 12C; however, the user can insert any mass difference relevant to their dataset. The user defines windows of mass accuracy and RT tolerance that MS-FLO uses to identify features as potential isotopes. Once MS-FLO identifies potential isotopes, it conducts a Pearson’s correlation analysis and generates coefficients of determination (R2) across all samples for each potential isotope pair(s). The user may choose to define a minimum R2 value that must be achieved for potential isotopic relationships to be flagged for manual inspection. The exported .csv file will contain a new column with information about the flagged isotope pair(s) including the R2 value, a delta RT value detailing the RT proximity of the two features, and a peak height ratio, which represents the intensity of the potential isotopic peak to its parent averaged across all samples (see Figure S2 in the Supporting Information). The peak height ratio determines the ratio of a potential isotopic peak to its parent ion, giving the user valuable information in determining if the flagged peak is a true isotope or a coeluting ion.
Adduct Joining.
Adduct joining screens all features in the dataset for candidate molecular ions and adduct ions to be joined or flagged based on user-defined parameters. Joining refers to the summing of the peak intensities of at least two features. Given that the type of adducts present are dependent on the LC-MS system used, the adduct types and m/z difference between the respective adduct ions and molecular ions must be defined. The user may select common adducts from a drop-down menu. Users also have the option to manually add complementary adducts that might be unique to their system (e.g., dimers, doubly charged molecules, lithium adducts). Subsequently, the user defines m/z and RT tolerance windows, in addition to a R2 cutoff threshold similar to the schema used for duplicate features and isotopes to search for adducts. The software suggests a default peak height correlation of R2 > 0.8 across all samples to automatically join features, but no default peak height correlation for flagging features to alert users for manual investigations of potential adducts. MS-FLO queries each feature against all other features in the dataset. If any two features meet the aforementioned criteria, then they will either be joined automatically or flagged for manual review. For instance, if the feature list contains two candidate features within the user-defined m/z and RT tolerance windows, but exhibit an R2 value of <0.8, then these features will be flagged as potential adducts. For R2 ≥ 0.8, such features will be automatically joined. If more than two features are matched, they will be flagged but not joined, regardless of the R2 value. Figure S3 in the Supporting Information shows an excerpt of a .csv file exported from MS-FLO and contains examples of automatically matched adducts as well as true and false flagged adducts.
Removal of Contaminant Ions.
Contaminant background ions from solvents, system ghost ions, or reference masses can momentarily spike during data acquisition and be characterized as peaks by the data processing software. Contaminant Ion Removal has no limit to the number of ions that can be entered into the system and can be easily used as a tool to remove features also found in extraction blanks from a dataset. Users can remove such features by entering the accurate mass(es) in MS-FLO’s Contaminant Ion Removal module. The software will then use the prespecified m/z tolerance and remove any feature that falls in the m/z range of that ion. Figure S4 in the Supporting Information shows the MS-FLO user interface with recommended default settings.
Once erroneous features have been identified or flagged by MS-FLO, a new feature report list is generated. MS-FLO performs the following post-processing steps for report generation. MS-FLO reformats the feature list to a user-friendly design to increase usability. First, the software creates a unique identifier for each feature by shortening and combining the retention time and m/z values into a new column in the exported data set. For instance, a feature with a retention time of 3.851 min and an m/z of 812.6129 would be given the unique identifier 3.85_812.61. Next, the post-processor removes decimals from peak height or area values, rounding up or down to the nearest integer, thus creating a leaner data table. Once data are submitted to MS-FLO, the user observes a live log of the software actions. When post-processing is complete, a .zip file will automatically download. Contained in the .zip file is the original unmodified feature list, a summary of the parameters defined by the user (.ini file), a log of all the actions performed by MS-FLO, and the processed feature list. The .ini file can later be uploaded directly to MS-FLO to save the user time when processing future datasets.
Evaluating the Performance of MS-FLO.
For evaluating the number of erroneous features from open access untargeted metabolomics software, MZmine2 and later MS-DIAL were used. Data processed using XCMS-online can also be uploaded directly to MS-FLO; however, the WCMC has found it harder to optimize XCMS to yield a minimal number of false positives. MZmine2 results were investigated for 18 studies (683 chromatograms) and MS-DIAL results were investigated for 10 studies (798 chromatograms). Summary statistics, including the numbers of removed or flagged features were compiled from MS-FLO log files. MS-FLO parameters are given in Table 2.
Table 2.
Definition of MS-FLO Parameters Used for Both the Archived and the Manually Investigated Studies
| parameter | value |
|---|---|
| m/z tolerance | 0.01 Da |
| RT tolerance | |
| for duplicates | 0.2 min |
| for adducts | 0.02 min |
| peak height tolerance | 500 |
| minimum peak match ratio | 85% |
| minimum R2 for isotopes | 0.5 (isotopes described had an R2≥ 0.8) |
| reference ions | Pos: 922.00982, 121.0508; Neg: 119.0363, 966.0007 |
| adducts positive mode (adduct1, adduct2, mass difference, lower R2 value, upper R2 value) | ([M+NH4]+, [M+Na]+, 4.956, 0.0, 0.8), ([M+H]+, [M+Na]+, 21.9787, 0.0, 0.8), ([M+H]+, [M+NH4]+, 17.0265, 0.0, 0.8) |
| adducts negative mode (adduct1, adduct2, mass difference, lower R2 value, upper R2 value) | ([M-H]−, [M+Cl]−, 35.9761, 0.0, 0.8); ([M-H]−, [M+HCOO]−, 46.0049, 0.0, 0.8); ([M+Cl]−, [M+HCOO]−, 24.0445, 0.0, 0.8) |
We first analyzed how many features were automatically removed, joined, or flagged in the 28 studies uploaded to the Metabolomics Workbench (www.metabolomicsworkbench.org). On average, these studies were comprised of 996 features, 77.5 of which were automatically removed, including 4.7 duplicates, 71.2 adducts, and 1.6 contaminate ions. An additional 15.8 duplicates, 32.3 adducts, and 37 isotopes were flagged for manual user inspection (Table S1). In total, 15.7% of peaks reported in the 28 datasets were either automatically removed or flagged for user review.
To validate MS-FLO’s selection of features removed or flagged for potential removal, five independent studies were manually investigated (Studies 3, 10, 16, 17, and 28 from Table S1). These studies were diverse in sample matrix as well as in the type of data acquisition methods used. On average, MZmine2 and MS-DIAL reported 726 features, of which 102 were joined or removed (including validation of flagged features). On average, 87% of the flagged features were joined or removed after manual inspection (Table 3), showing that the flagging parameters were set very conservatively. Indeed, when inspecting features that were automatically removed, none were found to be incorrectly selected. Flagged features included flagged adducts, flagged isotopes, and flagged duplicates. There were no systematic differences found between these categories, with respect to the validity of the flagging and eventual removal of redundant features, or differences between sample matrices or data acquisition methods. This result showed that the software performed as expected. Approximately 10% of the flagged features were not confirmed as erroneous features, since we did not have lower limits of correlations in peak intensities.
Table 3.
Details from Five Datasets Processed to Completion To Investigate Functionality of Flagging Features
| post-processor parameter | Study 17 | Study 10 | Study 28 | Study 03 | Study 16 | mean | removed, joined |
|---|---|---|---|---|---|---|---|
| number of samples | 23 | 100 | 22 | 302 | 17 | 93 | |
| adducts automatcheda | 19 | 45 | 33 | 119 | 8 | 45 | |
| flagged adductsb | 24 | 63 | 6 | 21 | 2 | 23 | |
| flagged adducts joinedb | 20 | 57 | 5 | 20 | 1 | 21 | 89% |
| total adducts joined | 39 | 102 | 38 | 139 | 9 | 65 | 96% |
| duplicates autoremovedc | 0 | 34 | 2 | 2 | 0 | 8 | |
| duplicate peaks flagged | 1 | 19 | 1 | 17 | 4 | 8 | |
| flagged duplicates removed | 0 | 17 | 1 | 17 | 2 | 7 | 88% |
| duplicate peaks removed | 0 | 51 | 3 | 19 | 2 | 15 | 94% |
| isotopes flaggeda | 8 | 57 | 28 | 29 | 1 | 25 | |
| total isotopes removed | 7 | 54 | 25 | 18 | 1 | 21 | 85% |
| total flagged features | 33 | 139 | 35 | 67 | 7 | 56 | |
| total flagged removed features | 27 | 128 | 31 | 55 | 4 | 49 | 87% |
| features before MS-FLO | 744 | 1364 | 498 | 841 | 181 | 726 | |
| features after MS-FLO | 698 | 1156 | 432 | 665 | 169 | 624 | |
| total rows removed | 46 | 208d | 66 | 176 | 12 | 102 | 14% |
Cutoff: R2 ≥ 0.8.
R2 < 0.8.
Similarity ≥85%.
One contaminate ion feature removed.
Next, we compared five additional archived studies that were manually investigated before MS-FLO was developed. We reanalyzed the final reports from those studies and compared how many more false positive features were now detected by MS-FLO that were missed by human inspection alone. Results are given in Table 4. Overall, the manually inspected archived studies had 681 features reported on average, of which an average of 71.4 were subsequently removed by MS-FLO. Among these erroneous features were 3 duplicates, and a mean of 28.6 adducts and 39.8 isotope pairs. We found for both validation sets that ghost ions (contamination ion series features) were already effectively removed through MZmine2 and MS-DIAL software results by subtracting peaks found in method blank injections from those reported in the actual samples. For these five studies, data were not uploaded to the metabolomics workbench (www.metabolomicsworkbench.org), because biological study design information was too sparse to be entered into the public domain.
Table 4.
Details from Five Archived Datasets Processed Manually before Development of MS-FLO, Then Processed with MS-FLO
| post processor parameter | plasma (+) lipids | meat and cheese (−) lipids | meat and cheese (+) lipids | mouse liver, intestine, serum (+) HILIC | wine (+) reverse phase | mean | joined, removed |
|---|---|---|---|---|---|---|---|
| number of samples | 90 | 38 | 38 | 53 | 70 | 58 | |
| adducts automatcheda | 40 | 0 | 17 | 31 | 10 | 20 | |
| flagged adductsb | 8 | 0 | 10 | 38 | 12 | 14 | |
| flagged adducts joinedb | 8 | 0 | 6 | 22 | 9 | 9 | 66% |
| total adducts joined | 48 | 0 | 23 | 53 | 19 | 29 | 86% |
| duplicates autoremovedc | 0 | 0 | 0 | 5 | 0 | 1 | |
| duplicate peaks flagged | 4 | 1 | 4 | 4 | 2 | 3 | |
| flagged duplicates removed | 1 | 1 | 4 | 4 | 0 | 2 | 67% |
| duplicate peaks removed | 1 | 1 | 4 | 9 | 0 | 3 | 75% |
| isotopes flaggeda | 11 | 21 | 134 | 51 | 5 | 44 | |
| total isotopes removed | 11 | 19 | 132 | 34 | 3 | 40 | 90% |
| total flagged features | 23 | 22 | 148 | 93 | 19 | 61 | |
| total flagged removed features | 20 | 20 | 142 | 60 | 12 | 51 | 83% |
| features before MS-FLO | 415 | 320 | 628 | 1096 | 948 | 681 | |
| features after MS-FLO | 355 | 300 | 469 | 1000 | 926 | 610 | |
| total feature rows | 60 | 20 | 159 | 96 | 22 | 71 | 10% |
Cutoff R2 ≥u0.8.
R2 < 0.8.
Similarity ≥85%.
Why Does MS-FLO Remove Isotopes but Summarize Adducts?
While isotopic features are highly correlated, adduct features often do not strongly correlate. It has been observed that the ion adduct with maximum peak height can be formed from various adducts (i.e., [M+Na]+ or [M+NH4]+) across a large set of samples. Depending on peak intensity, the ratio of ion adducts can vary greatly (Figure S5 in the Supporting Information). This makes adduct aggregation more complicated. There is a great discrepancy in the literature in how to handle features that form multiple ion adducts. In difference to MS-FLO, previously published attempts8–10 were designed to be integrated within data-processing software, specifically XCMS, instead of being deployed as stand-alone, posthoc data curation tool.
Allocator8 and CAMERA9 use adduct and isotope information to aid in spectral annotation, instead of focusing on quantitative aspects, as in MS-FLO. MS-FLO is applied after feature annotation with the single purpose of removing false positives to decrease the severity of FDR corrections in statistical analyses. Given the variable nature of relative adduct ion intensities across all samples in a single analysis, MS-FLO presents the best way to reduce the total number of features while still accurately representing the information in the samples by combining all adducts of a given metabolite.
CONCLUSIONS
MS-FLO presents a versatile user-friendly post-processing tool specifically designed to (1) improve data quality and decrease false positive peaks, by removing features that are not biologically relevant and combining features that represent the same compound and (2) reduce time spent on manual curation of datasets. The duplicate removal, isotope matching, adduct joining, and removal of contaminant ions in MS-FLO can be applied individually or simultaneously to best serve the user’s specific needs. MS-FLO is a useful tool to add to any untargeted LC-MS-based metabolomics workflow.
Supplementary Material
ACKNOWLEDGMENTS
Michael Sa contributed by uploading many of the studies investigated to the NIH Metabolomics Workbench.11 This work was funded by NIH No. U24 DK097154 and NIH No. R01 DK104351.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b04372.
Details regarding the parameter settings in the data processing programs to create the datasets submitted to MS-FLO; detailed information about the formatting of any dataset for submission to MS-FLO; supplemental figures giving examples of duplicate peaks, isotopic peaks, and multiple adduct peaks and examples of MS-FLO exports related to each of the above-mentioned features; supplemental figures depicting the MS-FLO user interface; supplemental table giving detailed information about studies, including analytical techniques used, sample information, and a summary of the MS-FLO log files from the post-processing of those studies (PDF)
The authors declare no competing financial interest.
An early version of MS-FLO was presented as a poster at the 2015 Metabolomics Society Conference in San Francisco, CA, USA. MS-FLO is also currently live and publicly available at http://msflo.fiehnlab.ucdavis.edu/.
REFERENCES
- (1).Tsugawa H; Cajka T; Kind T; Ma Y; Higgins B; Ikeda K; Kanazawa M; VanderGheynst J; Fiehn O; Arita M Nat. Methods 2015, 12 (6), 523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G Anal. Chem 2006, 78 (3), 779–87. [DOI] [PubMed] [Google Scholar]
- (3).Pluskal T; Castillo S; Villar-Briones A; Oresic M BMC Bioinf. 2010, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Castillo S; Gopalacharyulu P; Yetukuri L; Orešič M Chemom. Intell. Lab. Syst 2011, 108 (1), 23–32. [Google Scholar]
- (5).Zheng H; Clausen MR; Dalsgaard TK; Mortensen G; Bertram HC Anal. Chem 2013, 85 (15), 7109–16. [DOI] [PubMed] [Google Scholar]
- (6).Cajka T; Fiehn O Anal. Chem 2016, 88 (1), 524–545. [DOI] [PubMed] [Google Scholar]
- (7).Eliasson M; Rännar S; Madsen R; Donten MA; MarsdenEdwards E; Moritz T; Shockcor JP; Johansson E; Trygg J Anal. Chem 2012, 84 (15), 6869–6876. [DOI] [PubMed] [Google Scholar]
- (8).Kessler N; Walter F; Persicke M; Albaum SP; Kalinowski J; Goesmann A; Niehaus K; Nattkemper TW PLoS One 2014, 9 (11), e113909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kuhl C; Tautenhahn R; Bottcher C; Larson TR; Neumann S Anal. Chem 2012, 84 (1), 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zeng Z; Liu X; Dai W; Yin P; Zhou L; Huang Q; Lin X; Xu G Anal. Chem 2014, 86 (8), 3793–3800. [DOI] [PubMed] [Google Scholar]
- (11).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS Nucleic Acids Res. 2016, 44, D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.