

SUMMARY
Set2 co-transcriptionally methylates lysine 36 of histone H3 (H3K36), producing mono-, di-, and trimethylation (H3K36me1/2/3). These modifications recruit or repel chromatin effector proteins important for transcriptional fidelity, mRNA splicing, and DNA repair. However, it was not known whether the different methylation states of H3K36 have distinct biological functions. Here, we use engineered forms of Set2 that produce different lysine methylation states to identify unique and shared functions for H3K36 modifications. Although H3K36me1/2 and H3K36me3 are functionally redundant in many SET2 deletion phenotypes, we found that H3K36me3 has a unique function related to Bur1 kinase activity and FACT (facilitates chromatin transcription) complex function. Further, during nutrient stress, either H3K36me1/2 or H3K36me3 represses high levels of histone acetylation and cryptic transcription that arises from within genes. Our findings uncover the potential for the regulation of diverse chromatin functions by different H3K36 methylation states.
Graphical Abstract
In Brief
DiFiore et al. address whether the distinct methyl-lysine states catalyzed by Set2 on nucleosomes have unique or shared functions in transcription and other cellular processes. Their studies show that H3K36me1/2 and H3K36me3 act redundantly in many cellular contexts, including preventing antisense transcription during nutrient stress.
INTRODUCTION
Histone post-translational modifications affect a great variety of DNA-templated processes. Methylation, acetylation, and other modifications are added to histones by chromatin-modifying enzymes (Rothbart and Strahl, 2014; Soshnev et al., 2016). These chemical modifications can alter histone-DNA contacts and often serve as docking sites for effector proteins. In the context of gene expression, the coordinated effort of histone modifications, chromatin-modifying enzymes, and effector proteins helps RNA polymerase II (RNAPII) to gain access to DNA for transcription (Cramer, 2019; Roeder, 2019). A similar coordinated effort is employed to make the genome unreachable by RNAPII at appropriate times to prevent aberrant transcription.
Set2 is a chromatin-modifying enzyme that contributes to the prevention of inappropriate transcription. Set2 methylates histone H3 at lysine 36 (H3K36) (McDaniel and Strahl, 2017; Strahl et al., 2002; Venkatesh and Workman, 2013). In Saccharomyces cerevisiae (hereafter, budding yeast), Set2 is responsible for all forms of H3K36 methylation (H3K36me), mono-, di-, and trimethylation. Set2 binds to the C-terminal domain (CTD) of transcribing RNAPII and catalyzes H3K36me in actively transcribed genes (Kizer et al., 2005; Xiao et al., 2003). H3K36me provides docking sites for several proteins, such as Rpd3S, a histone deacetylase complex, and Isw1b, a nucleosome remodeler. Rpd3S is recruited to chromatin by the plant homeodomain (PHD) fingers in Rco1, and the activity of Rpd3S is stimulated by binding of the Eaf3 chromodomain to H3K36me2 (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005; Li et al., 2007; McDaniel et al., 2016; Ruan et al., 2015). Additionally, Isw1b associates with chromatin by way of the Ioc4 PWWP domain binding to H3K36me3 (Maltby et al., 2012; Smolle et al., 2012). Collectively, these processes ensure transcriptional fidelity by preventing transcription initiation from within gene bodies, a process known as cryptic transcription.
In the absence of Set2 and H3K36me, both sense and antisense cryptic transcription occur across the genome. Cryptic transcription tends to be a consequence of bi-directional transcriptional events at cryptic promoters within gene bodies (Carrozza et al., 2005; Churchman and Weissman, 2011; Joshi and Struhl, 2005; Lickwar et al., 2009; Neil et al., 2009; Xu et al., 2009). Precisely how cryptic sites become accessible in SET2 deletion mutants (set2Δ) is not fully understood; however, a lack of Set2-mediated H3K36me results in increased histone exchange along with increased histone acetylation—both phenomena that may promote the recruitment of RNAPII initiation factors and chromatin remodelers such as RSC that promote chromatin disruption (Li et al., 2007; Pattenden et al., 2010; Smolle et al., 2012; Venkatesh et al., 2012). From a functional standpoint, cryptic transcription in the absence of Set2 does not result in significant growth defects when examined in a nutrient-rich medium, although cell-cycle progression is partially disrupted (Dronamraju et al., 2018; Kim et al., 2016; Lenstra et al., 2011; McDaniel et al., 2017; Venkatesh et al., 2016). In contrast, during nutrient deprivation or carbon source shifting, cryptic antisense transcription can occur at a level sufficient to impair normal transcription and lead to improper gene expression and poor cell growth (Kim et al., 2016; McDaniel et al., 2017).
Although the functions of Set2 and H3K36me have been studied intensely, there are no studies that have thoroughly interrogated which functions of Set2 are directed by the different (mono-, di-, and tri-) H3K36me states. Previous reports suggest that H3K36me3 is dispensable for suppression of cryptic transcription (Hacker et al., 2016; Li et al., 2009; Youdell et al., 2008). In contrast, set2 mutants that harbor only H3K36me1 exhibit cryptic transcription, suggesting that the main functions of H3K36me occur through H3K36me2 (Hacker et al., 2016). However, it is not known whether H3K36me3 function is unique or overlapping with the other H3K36me states.
In this study, we engineered the SET domain of Set2 so that it performed only H3K36me1, H3K36me1/2, or H3K36me3, thus affording a unique opportunity to interrogate the functions of different methylation states. We found that Set2 that generated H3K36me3 and H3K36me1/2 can act redundantly to rescue several canonical phenotypes associated with set2Δ (e.g., caffeine sensitivity and DNA damage response). In contrast, Set2 that generated only low levels of H3K36me1 did not rescue set2Δ phenotypes. However, only H3K36me3 rescued other set2Δ phenotypes (e.g., bur1Δ or spt16–11 bypass). Intriguingly, strains that produced either H3K36me1/2 or H3K36me3 could largely suppress cryptic transcription during nutrient stress. Approximately 60% of the identified cryptic transcript initiation sites contained a degenerate TATA box motif, a fact that suggested how those sites become susceptible to cryptic transcription in the absence of H3K36me. Additionally, H3K36me1/2 or H3K36me3 could function to ensure proper levels of H3K27ac and H3K56ac in genes, pointing to a potential mechanism for preventing inappropriate transcriptional initiation by control of nucleosome remodeling and histone exchange. In sum, our data provide key evidence for the independent and overlapping functions of H3K36me1/2 and H3K36me3 in chromatin biology and in transcriptional regulation.
RESULTS
Phe/Tyr Switch in Set2 Separates H3K36me States in vitro
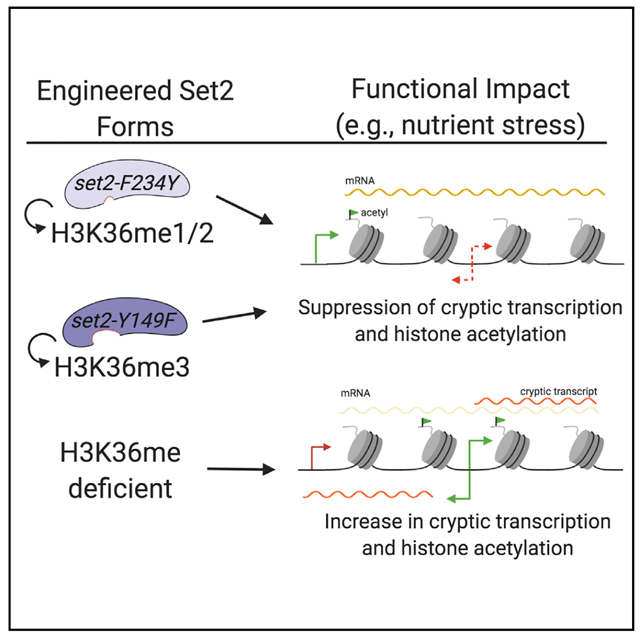
Although previous studies have assessed the effects of losing or limiting H3K36me, no study has comprehensively examined the individual functions of each H3K36me state produced by Set2. To create a system in which we could control the production of H3K36me states, we employed a structural mutagenesis approach for Set2 based on the Phe/Tyr switch phenomenon that alters the SET domain lysine-binding pocket (Cheng et al., 2005; Collins et al., 2005). Briefly, two well-positioned aromatic residues within the SET domain (a phenylalanine [Phe, F] and a tyrosine [Tyr, Y]) can be switched to the opposite aromatic residue to control the size of the catalytic domain. An F to Y mutation creates steric hindrance in the active site with lysine trimethylation, thereby limiting the enzyme to only mono- and dimethylation. However, a Y to F change creates more space and promotes the catalysis of trimethylation. In Set2, Y149 and F234 were predicted to comprise the Phe/Tyr switch (Cheng et al., 2005; Collins et al., 2005) (Figure 1A).
Figure 1. Phe/Tyr Switch in Set2 Separates H3K36 Methylation States in vitro.
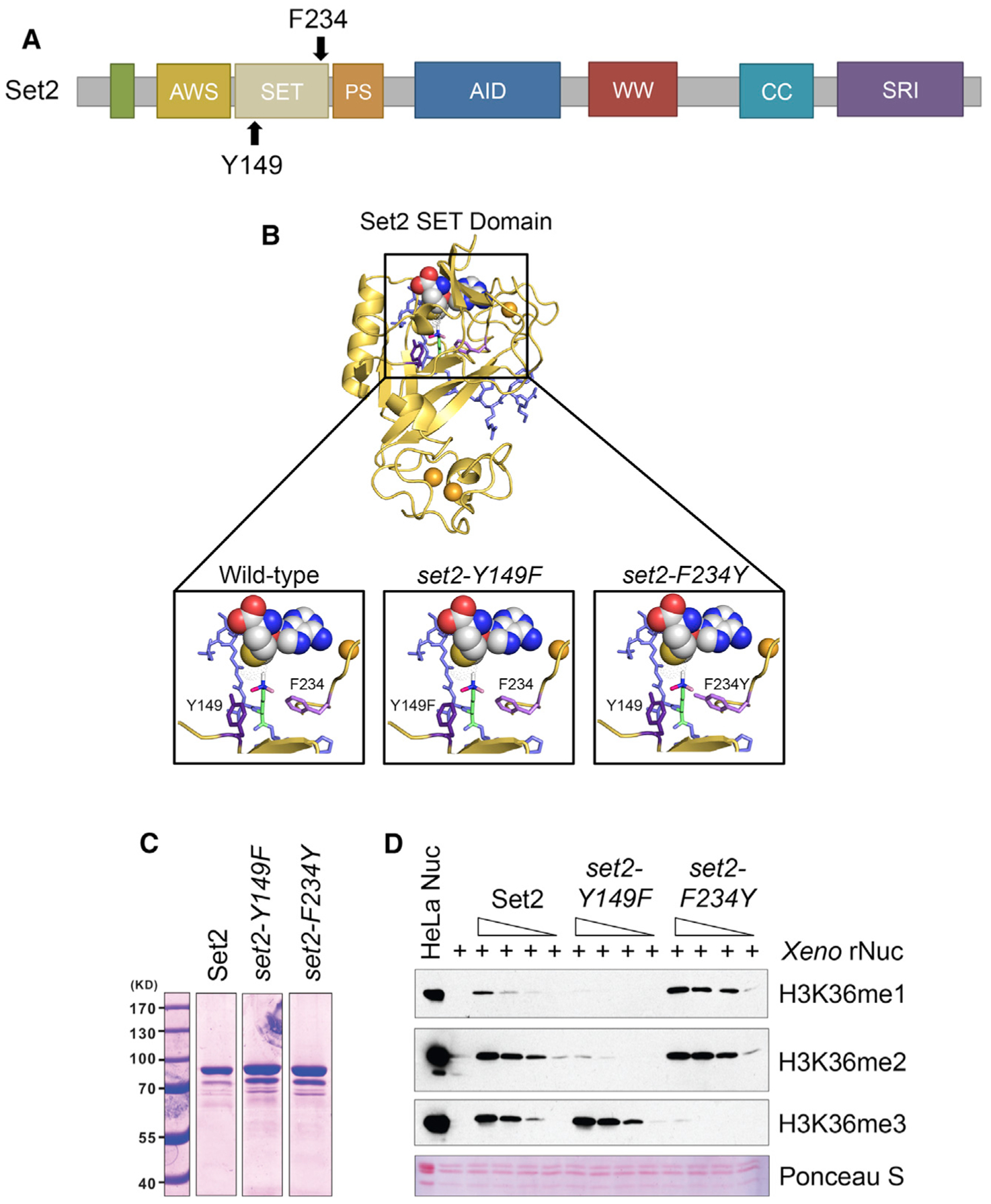
(A) Domain map of Set2 with the Y149 and F234 residues of the Phe/Tyr switch highlighted. The catalytic region of Set2 is composed of the associated with SET (AWS), catalytic SET domain, and post-SET (PS). The autoinhibitory domain (AID) regulates catalytic activity. The C-terminus of Set2 has protein-protein interaction domains, like the coiled-coiled (CC), WW, and Set2-Rpb1 (SRI) protein-protein interaction domain. (B) Model of Set2 SET domain (yellow) bound to H3 peptide (slate) and SAM (yellow, white, blue, and red spheres) with locations of the F234 (light purple) and Y149 (dark purple) that form the Phe/Tyr switch highlighted. H3K36 is green with methylation shown in white, light pink, and dark pink. Orange spheres are zinc ions necessary for catalysis. (C) Coomassie staining of recombinant Set2 and Phe/Tyr switch mutant proteins. Lanes not shown are other purified proteins not used in the following experiments. (D) Western blots of in vitro histone methyltransferase (HMT) assays using the indicated antibodies. In vitro HMT assays were performed with an equal amount of recombinant Set2 and Phe/Tyr switch mutant proteins from insect cells, recombinant Xenopus nucleosomes, and co-factor SAM. HeLa LONs (long oligonucleosomes) were used as a positive control for western blotting.
To determine whether Set2 Y149 and F234 can function as a Phe/Tyr switch, we created a structural model of the Set2 SET domain by using MODELLER and the crystal structure of SETD2 (PDB: 5JLB) as the template (Webb and Sali, 2016; Yang et al., 2016). The template and alignment were the top hit generated by Iterative Threading ASSEmbly Refinement (I-TASSER) (Zhang, 2008). The model confirmed that Y149 and F234 are part of the lysine-binding pocket (Figure 1B, left panel) and configured to enable Set2 to mono-, di-, and trimethylate H3K36. We then investigated this model for the effect of deploying the Phe/Tyr switch. Modeling showed that the Y149F mutation opened the SET domain binding pocket and eliminated hydrogen bonding between the hydroxyl group of tyrosine and H3K36me (Figure 1B, middle panel). These changes would permit rapid rotation of the target lysine in the presence of S-adenosylmethionine (SAM) and likely increase the potential for higher methylation states on H3K36. Conversely, the F234Y substitution created steric hindrance between the tyrosine hydroxyl group and the target lysine, thus suggesting that this mutant would limit H3K36me3 (Figure 1B, right panel). These findings are consistent with these residues encompassing a working Phe/Tyr switch in Set2.
Lastly, we measured the effect of the Phe/Tyr switch on Set2 enzymatic activity. We performed histone methyltransferase (HMT) activity assays with recombinant Set2 (wild-type [WT], set2-Y149F, and set2-F234Y), SAM, and recombinant Xenopus nucleosomes; methylation states were detected by western blot analysis with antibodies specific to the three modifications (Figures 1C and 1D). We found that set2-Y149F catalyzed predominately H3K36me3, whereas set2-F234Y catalyzed H3K36me1 and H3K36me2 (Figure 1D). Overall, these results demonstrated that the SET domain of Set2 contains a functional Phe/Tyr switch that can be used to produce specific H3K36me states.
Phe/Tyr Switch Mutations in Set2 are Tools to Separate H3K36me States in vivo
To test the effect of the Set2 Phe/Tyr switch on H3K36me in vivo, we generated yeast strains harboring endogenous set2-Y149F or set2-F234Y mutations. For controls, we included a set2Δ strain and a catalytic domain mutant strain (set2-H199L) that was capable of producing only low levels of H3K36me1 in vivo (Hacker et al., 2016; Jha and Strahl, 2014). The set2-Y149F mutation predominately resulted in H3K36me3 and a low level of H3K36me1 (Figure 2A). In contrast, the set2-F234Y mutation limited Set2 methylation to H3K36me1/2 (Figure 2A). As previously described, the absence of Set2 resulted in a complete loss of H3K36me, and the catalytic domain mutant, set2-H199L, showed only a low level of H3K36me1 (Figures 2A and S1A). The SET domain mutations did not alter the amount of Set2 protein (Figure 2B); however, global H3K36me differed from the WT. The set2-Y149F mutant had an average of 56% of the WT H3K36me3 (Figure 2C). set2-F234Y showed an H3K36me2 signal similar to the WT (93%), but a 5-fold increase in H3K36me1 (Figures 2D and 2E). set2-H199L had a reduced H3K36me1 signal compared to the WT. Overall, these results confirmed that the Phe/Tyr switch in Set2 functioned in vivo, and, although global H3K36me levels varied from the WT, we could largely control different methylation states.
Figure 2. Phe/Tyr Switch Mutations in Set2 are Tools to Separate H3K36 Methylation States In Vivo.
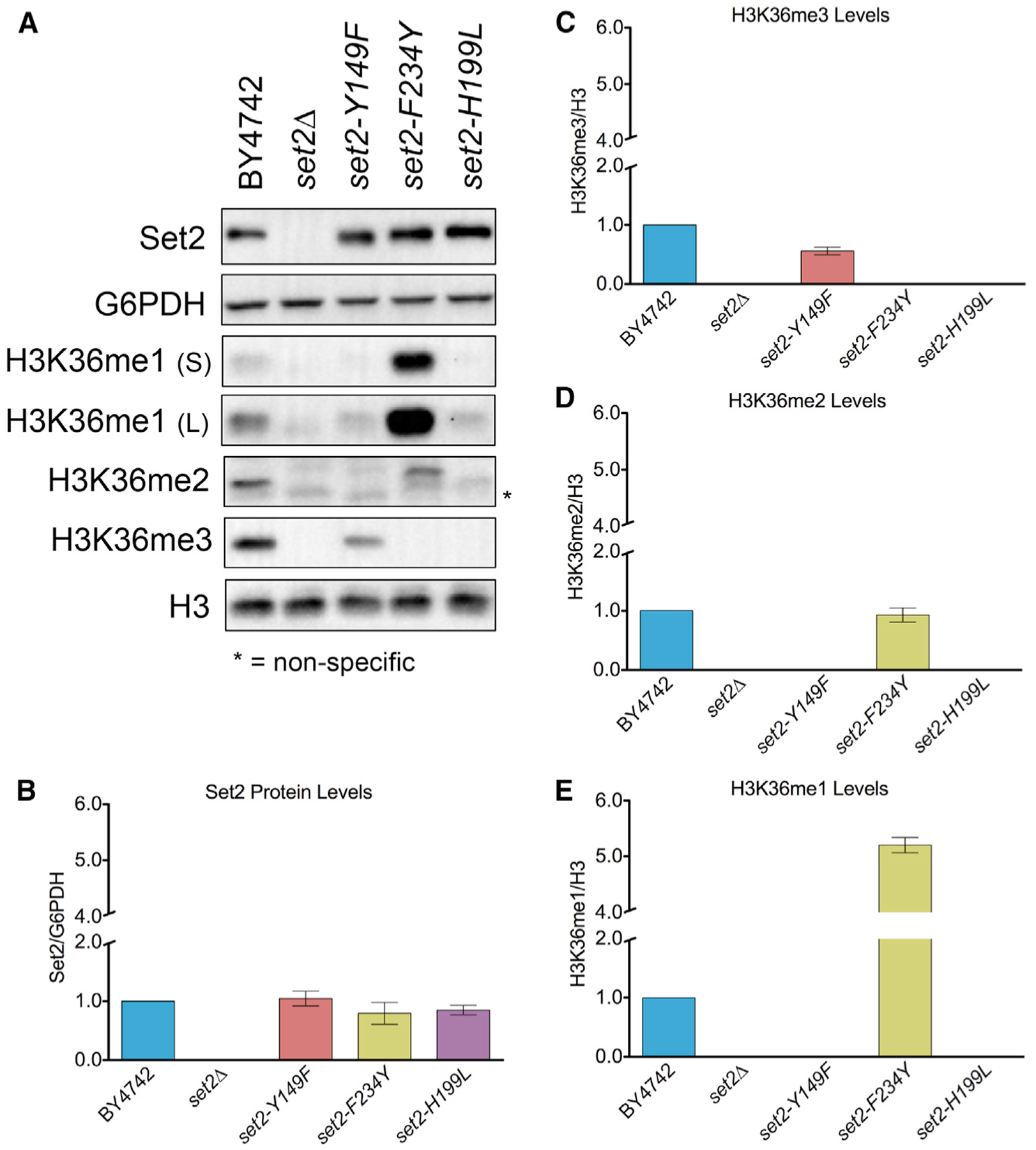
(A) Western blots of yeast strains probed with Set2 and different H3K36me antibodies. S, short exposure; L, long exposure. G6PDH and H3 served as loading controls. (B) Quantification of Set2 normalized to G6PDH in the indicated strains. Data for mutants were normalized relative to BY4742. (C–E) Quantification of H3K36me3 (C), H3K36me2 (D), and H3K36me1 (E) normalized to H3 in the indicated strains. H3K36me1 was quantified using the short exposure western blot. Measurement for all mutants was normalized BY4742. Each bar graph is representative of mean ± SEM of two or more independent biological replicates with a representative replicate shown in (A).
Because some of our downstream genetic analyses would require expression of SET2 from plasmids, we also examined the effect of the set2 mutants in two exogenous expression systems: expression of SET2 and the set2 mutants from their native promoter and overexpression of the same genes from the ADH1 promoter. Western blot analyses for H3K36me in these two systems revealed two important findings. First, the set2-Y149F and set2-F234Y mutants, regardless of expression system, recapitulated the initial in vivo findings that the Phe/Tyr switch could control the methylation states of H3K36 (Figure S1B). Second, and surprisingly, overexpression of SET2 via the ADH1 promoter resulted in robust overexpression of the Set2 protein compared to the expression from its endogenous promoter, yet global H3K36me levels in the WT or set2 mutants were similar to or lower than normal. Thus, although it was possible to greatly overexpress Set2, the amount of methylation that Set2 can produce on chromatin must have a limit, perhaps due to a finite number of Set2 binding sites on the CTD of RNAPII, restricted histone accessibility, limited SAM availability, or a combination of these factors.
Overall, these results were consistent with the in vitro data, and they demonstrated that the Phe/Tyr switch mutants were specific for performing predominately H3K36me3 or H3K36me1/2. These set2 mutants provided tools to examine in vivo the functions of different methylation states of H3K36.
Distinct Methylated Forms of H3K36 are Deposited within or near Transcribed Regions of Genes
We next asked whether the set2 mutants still deposited H3K36me within or near the coding regions of genes in a manner consistent with its known localization and function (Pokholok et al., 2005; Rao et al., 2005; Weiner et al., 2015). We first confirmed by co-immunoprecipitation that the set2 mutants still associated normally with phosphorylated RNAPII (Figure S1C). Consistent with RNAPII association, chromatin immunoprecipitation (ChIP)-qPCR of all H3K36me forms in the set2 mutants confirmed that they were deposited within or near the coding regions of several loci tested (STE11, PMA1, and TDH3; see Figures 3A, 3E, and 3I). Additionally, the H3K36me states detected by ChIP-qPCR were consistent with the aforesaid in vitro HMT and western blot assays (Figures 1C and 2A).
Figure 3. Distinct Methylated Forms of H3K36 Are Deposited within or near Transcribed Regions of Genes.
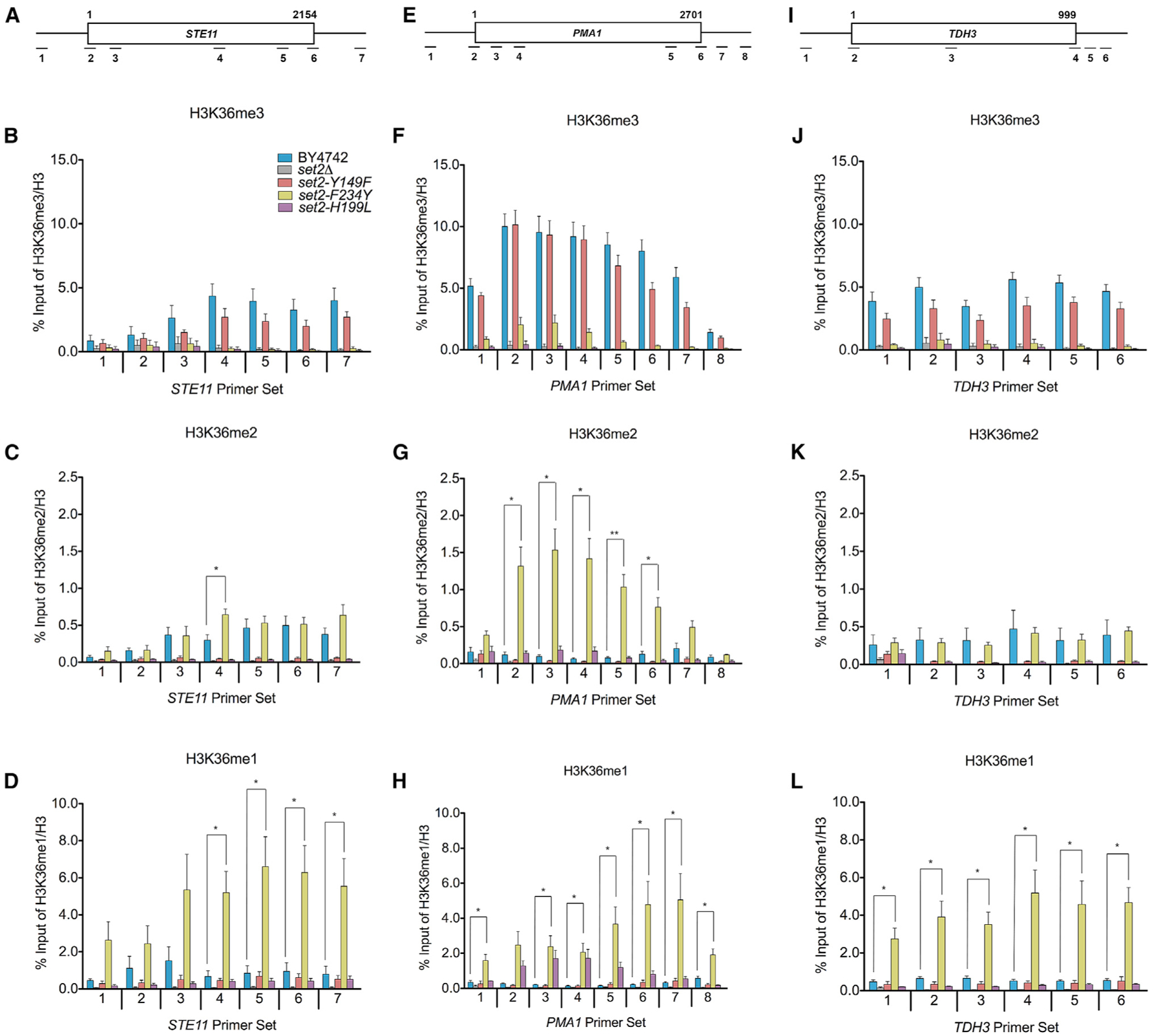
(A) Schematic of STE11 with amplicons indicated below. (B–D) ChIP analysis of H3K36me3, H3K36me2, and H3K36me1 across STE11 in the indicated strains. The legend in (B) is representative for all the data presented in the figure. (E) Schematic of PMA1 with amplicons indicated below. (F–H) ChIP analysis of H3K36me3, H3K36me2, and H3K36me1 across PMA1 in the indicated strains. (I) Schematic of TDH3 with amplicons indicated below. (J–L) ChIP analysis of H3K36me3, H3K36me2, and H3K36me1 across TDH3 in the indicated strains. Data represented as mean ± SEM of three independent biological replicates. Student’s t test was used to obtain p values. Asterisks indicate significance (*p < 0.05; **p < 0.01); non-significant comparisons not shown. All qPCR primers are listed in Table S3.
A closer comparison of the differently methylated H3K36 forms with the WT revealed several key observations. As shown in Figures 3B, 3F, and 3J, there were no significant differences in the H3K36me3 levels between the WT and set2-Y149F at all loci tested. However, for H3K36me2, we observed differences between the WT and set2-F234Y at STE11 and PMA1. A significantly higher H3K36me2 level was detected in the middle of STE11 in set2-F234Y (Figure 3C), whereas at PMA1, a significantly higher H3K36me2 level was detected across the entire gene body (Figure 3G). In contrast, we did not detect any significant difference in H3K36me2 levels between set2-F234Y and the WT at TDH3 (Figure 3K). As expected from the high H3K36me1 western blot signal, set2-F234Y had significantly higher levels of H3K36me1 in the 3′ end of STE11 and across the entirety of PMA1 and TDH3 compared to the WT (Figures 3D, 3H, and 3L). Finally, although global levels of H3K36me1 were lower in set2-H199L, we observed nearly WT levels of H3K36me1 in set2-H199L at the loci examined. At STE11, we did not find significant differences in H3K36me1 between set2-H199L and the WT (Figures 3D and S2B). However, at PMA1, there was significantly more H3K36me1 deposited by set2-H199L in the gene body (Figures 3H and S2D). In contrast, we detected less H3K36me1 at the 5′ end of TDH3 in set2-H199L compared to the WT, although the rest of that locus did not show a significant difference between the mutant and WT. Overall, these results established that the levels of H3K36me in the set2 mutants differed from the WT at certain loci, but the results further confirmed the specificity of the set2 mutants and indicated that differentially methylated forms of H3K36 were localized within or near the bodies of transcribed genes.
H3K36me1/2 and H3K36me3 Function Redundantly in Some Cellular Contexts
To determine whether the different forms of H3K36me are responsible for distinct biological outcomes, we examined the sensitivity of set2 methylation mutants to drugs that affect set2Δ cell growth. We tested caffeine (a TOR1C and MAP kinase inhibitor), rapamycin (a TOR1C inhibitor), phleomycin (a double-strand break-inducing agent), and 6-azauracil (6-AU; a transcriptional elongation inhibitor) (Jha and Strahl, 2014; Kizer et al., 2005; McDaniel et al., 2017). Like previous work, we found that set2Δ and set2-H199L mutant cells were sensitive to caffeine, rapamycin, and phleomycin, and they were resistant to 6-AU (Figures 4A and 4B; Table S1). Intriguingly, similar to the WT, the set2-Y149F and set2-F234Y mutants rescued the drug-induced growth defects from caffeine, rapamycin, and phleomycin (Figure 4A; Table S1). However, only set2-Y149F was as sensitive to 6-AU as the WT, with set2-F234Y having an intermediary phenotype between the WT and set2Δ (Figure 4B). Thus, in some cellular contexts, such as TORC1 signaling and double-strand break repair, H3K36me3 and H3K36me1/2 were functionally redundant, and a low level of H3K36me1 alone could not suffice.
Figure 4. H3K36me1/2 and H3K36me3 Have Unique Phenotypes in Some Cellular Contexts.

(A) Five-fold serial dilutions of BY4742, set2Δ, and set2 mutant strains plated on YPD or YPD containing caffeine (15 mM), rapamycin (25 nM), or phleomycin (10 μg/mL). (B) Five-fold serial dilutions of BY4742, set2Δ, and set2 mutant strains plated on SC-Ura containing DMSO or 200 μg/mL 6-AU. (C) Five-fold serial dilutions of BY4741, bur1Δ, and set2 mutant strains plated on SC-Ura-Leu or SC-Ura-Leu containing 5-fluoorotic (5-FOA). (D) Five-fold serial dilutions of W303, spt16–11, and set2 mutant strains plated on SC-Leu and incubated at 25°C or 34°C. (E) Schematic of FLO8-HIS3 fusion gene reporter to detect cryptic transcription. (F) Schematic of STE11-HIS3 fusion gene reporter to detect cryptic transcription. (G) Five-fold serial dilutions of indicated WT, set2Δ, and set2 mutant strains plated on SC-Ura, SC-Ura-His with 2% galactose, or SC-Ura-His. All spotting assays were repeated three times, and the images shown are representative of the data.
Genetic Interactions of SET2 with BUR1 and SPT16 Reveal Unique Functions for H3K36me1/2 and H3K36me3
In addition to phenotypes of set2Δ cells challenged with drugs, the deletion of SET2 bypasses the lethal or slow growth phenotypes associated with the inactivation or deletion of members of the BUR kinase and FACT (facilitates chromatin transcription) complexes (Biswas et al., 2006; Chu et al., 2006; Keogh et al., 2003). Both complexes regulate aspects of transcription; the BUR kinase complex promotes RNAPII elongation, and the FACT complex is responsible for nucleosome disruption and reassembly. Cells lacking BUR1 (bur1Δ) are not viable, but they survive when SET2 is also deleted (bur1Δ set2Δ). Likewise, cells harboring the FACT mutant allele spt16–11 grow extremely slow at 34°C, and deletion of SET2 bypasses this phenotype. Therefore, we examined our set2 methylation mutants to determine whether the different H3K36me states would bypass the bur1Δ and spt16–11 phenotypes. We confirmed that the presence of set2Δ in the bur1Δ or spt16–11 cells rescued the lethal or slow growth phenotypes, whereas exogenously expressed WT SET2 did not rescue growth (Figures 4C and 4D; Table S1). Surprisingly, cells containing the set2-Y149F allele that catalyzed only H3K36me3 phenocopied WT Set2, whereas the set2-F234Y and set2-H199L alleles that catalyzed H3K36me1/2 or low levels of H3K36me1, respectively, phenocopied the set2Δ cells (Figures 4C and 4D; Table S1). Thus, these findings indicated that H3K36me3 has a function distinct from H3K36me1/2 in relation to Bur1 kinase activity and FACT complex function.
H3K36me1/2 and H3K36me3 Have Unique and Shared Functions in Repressing Cryptic Transcription at Reporter Loci
An extensively documented phenotype of set2Δ cells is the appearance of sense and antisense cryptic transcripts that arise from genic regions (McDaniel and Strahl, 2017). Although our studies and others have revealed that H3K36me3 is dispensable for preventing cryptic transcription (Hacker et al., 2016; Li et al., 2009; Youdell et al., 2008), it was not known whether H3K36me3 alone can support the suppression of cryptic transcription in the absence of H3K36me1/2. To answer this question, we used two reporter genes, FLO8 and STE11, that each have internal cryptic initiation sites (CISs) and the HIS3 gene integrated out-of-frame and downstream of the CIS. When the FLO8 and STE11 loci are transcribed from canonical promoters, HIS3 is not transcribed in-frame, and the cells cannot survive on media lacking histidine (Silva et al., 2012; Wang et al., 2015). However, when cryptic transcription occurs at the CIS, HIS3 is transcribed in-frame, and cells survive without histidine (Figures 4E and 4F). Thus, we first confirmed that cells with WT SET2 suppressed cryptic transcription and did not grow on media lacking histidine, whereas set2Δ cells did not suppress cryptic transcription at either locus (Figures 4G and 4H; Table S1). At the FLO8 locus, which was under the GAL promoter and constitutively expressed in cells growing on galactose-containing media, both H3K36me3 and H3K36me1/2 were sufficient to suppress cryptic transcription similar to WT SET2 (Figure 4G; Table S1). Surprisingly, at the STE11 locus, expressed from its native promoter and at a lower level than FLO8, only H3K36me3 prevented cryptic transcription, whereas cells with H3K36me1/2 or a low level of H3K36me1 did not suppress (Figure 4H; Table S1). Thus, at STE11, H3K36me3 alone is sufficient to suppress cryptic transcription. Additionally, these results suggested that H3K36me3 and H3K36me1/2 have different, but sometimes overlapping, functions in preventing cryptic transcription. In particular, highly and lowly expressed genes may have different requirements for H3K36me.
During Nutrient Deprivation, H3K36me1/2 or H3K36me3 Prevents Antisense Transcription
Having established that different forms of H3K36me are necessary to repress cryptic transcription at specific loci, we examined this fact globally. Previous work from our lab demonstrated that robust bidirectional cryptic transcription occurs at 439 genes in set2Δ cells during nutrient deprivation (McDaniel et al., 2017). We used this system as a tool to learn the genome-wide requirement of the different H3K36me states to prevent cryptic transcription. We performed stranded RNA sequencing (RNA-seq) at 0 and 60 min following nutrient deprivation in WT, set2Δ, set2-Y149F, set2-F234Y, and set2-H199L cells. We examined sense and antisense transcriptional signals in the genomic regions that surround the high- and intermediate-confidence cryptic initiation sites of the aforesaid 439 genes. After 60 min of nutrient deprivation, we observed a global increase in sense and antisense transcription surrounding the CIS in set2Δ and set2-H199L cells (Figure 5A). Interestingly, cryptic transcription was not completely repressed in set2-Y149F or set2-F234Y; both mutants showed a slight increase in antisense transcription upstream of the CIS compared to the WT (Figure 5A). When we separated the CIS-containing genes based on gene expression levels, we observed that set2-Y149F or set2-F234Y repressed sense and antisense cryptic transcription similar to the WT in highly and lowly expressed genes (Figure S3A). In contrast, set2Δ and set2-H199L did not repress any form of cryptic transcription in highly or lowly expressed genes (Figure S3A). Importantly, we verified that nutrient deprivation did not alter the existence of the H3K36me states catalyzed by the set2 mutants (Figure 6A). These results revealed that H3K36me1/2 or H3K36me3 can function to prevent cryptic transcription genome-wide, and low levels of H3K36me1 do not have a large part in repressing cryptic transcription.
Figure 5. Function of H3K36 Methylation States in Cryptic Transcription Regulation.
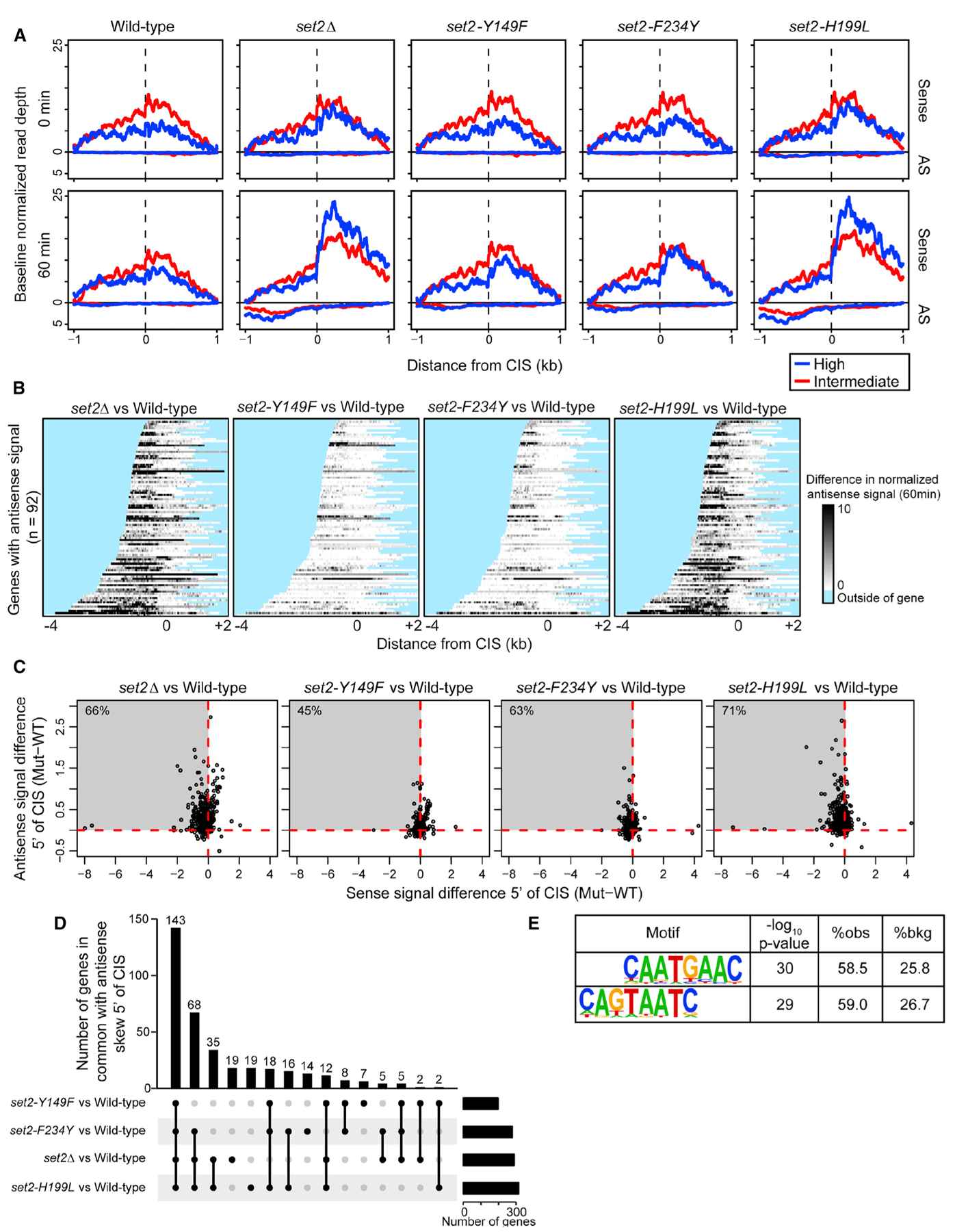
(A) Sense and antisense normalized transcriptional signal (reads per million mapped) across 439 high- (blue) and intermediate- (red) confidence cryptic initiation sites (CISs) defined previously. Signal was averaged across three independent biological replicates and plotted for 0 min (top) and 60 min (bottom) following nutrient deprivation for each genetic model. The minimum value for each line was adjusted to 0 (y axis) to adjust for subtle differences in baseline expression and to focus on the position and range in magnitude of signal (see McDaniel et al., 2017). (B) Heatmap of antisense transcription, plotted as the difference in antisense signal between set2 mutant and WT (mutant-WT) at 60 min following nutrient deprivation. Normalized signal is plotted for 92 genes shown previously to have antisense transcription between the CIS and transcription start site. Darker gray indicates more antisense signal in mutant than in WT. Regions outside of the gene body are masked blue. (C) Scatterplot of sense and antisense signal differences (mutant-WT) in the mean per-base coverage over the gene regions between the CIS and transcription start site, for the 439 high- and intermediate-confidence CISs. Each point represents the CIS of a given gene; points that extend into the upper-left quadrant indicate a decrease in sense transcription (relative to WT) and a concomitant increase in antisense transcription. The percentage of all 439 genes falling in this quadrant is supplied in the top left of each panel. (D) UpSet plot showing overlaps for genes in the upper-left quadrants of (C). In total, 143 genes exhibited this antisense skew relative to WT in all four set2 mutants. (E) Significantly enriched sequence motifs discovered in the 100 bp surrounding the 439 CISs, requiring at least 2-fold enrichment relative to local background sequence.
Figure 6. Alteration of Global H3K27ac and H3K56ac in set2 Methylation Mutants.
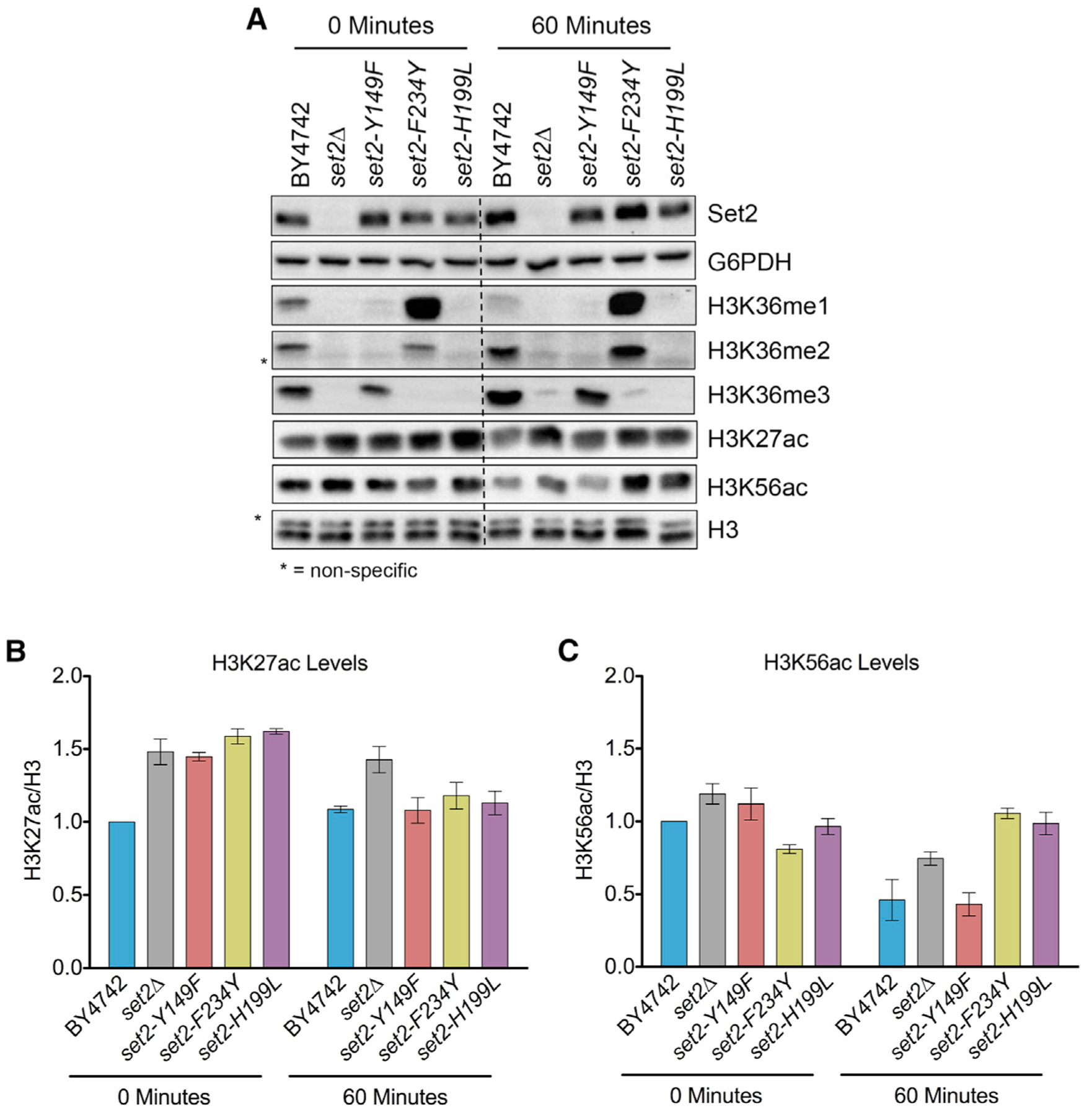
(A) Western blots of indicated strains probed with Set2, H3K36me1, H3K36me2, H3K36me3, H3K27ac, and H3K56ac antibodies. G6PDH and H3 served as loading controls. (B and C) Quantification of H3K27ac (B) and H3K56ac (C) normalized to H3. All measurements are normalized relative to BY4742 at 0 min. Each bar graph is representative of mean ± SEM of two or more independent biological replicates with a representative replicate shown in (A).
Next, we wanted to further characterize the antisense transcription that occurred in the set2 mutants. We previously showed that in set2Δ, 92 of 121 high-confidence CISs exhibited robust antisense transcription that extended to the transcription start site. Here, we found that relative to the WT, set2Δ and set2- H199L cells exhibited increased antisense signal that extended to the transcription start site, irrespective of gene length, whereas this was rarely observed in set2-Y149F and set2-F234Y cells or when comparing WT replicates (Figures 5B and S3B). Previously, our lab and others had suggested that by extending into gene promoter regions, this antisense transcription interfered with and reduced sense transcription (Huber et al., 2016; McDaniel et al., 2017). To examine this idea for the set2 mutants, we compared the sense and antisense transcriptional signals between the WT and each of the set2 mutants, focusing specifically on the regions between the CIS and transcription start sites. Any signal in these regions should reflect normal sense transcription and cryptic antisense transcription, but not cryptic sense transcription, because cryptic sense transcription would occur downstream of the CIS. Indeed, we frequently observed decreases in sense transcription and concomitant increases in antisense transcription, with the strongest effects on both strands occurring in set2Δ and set2-H199L cells (Figure 5C). The effects observed here were more pronounced than the effects we observed between two WT replicates compared as a control (Figure S3C). Moreover, many of the same genes exhibited similar effects for all the set2 mutants, particularly set2Δ, set2-H199L, and set2-F234Y cells (Figure 5D; Table S5). Together, these data suggested that during nutrient deprivation, cryptic antisense transcription downregulates sense transcription from certain CIS-containing genes in the absence of H3K36me1/2 or H3K36me3.
Lastly, we wondered whether the sequences surrounding the CIS harbored any particular sequence motifs that may explain why cryptic transcription initiates there and not else-where. We searched the sequences ± 100 bp of the 439 high- and intermediate-confidence CISs and found a significant enrichment of a (T/C)AAT motif, which may represent a degenerate TATA box element (Figure 5E) (Basehoar et al., 2004; Lubliner et al., 2013). We did not observe this motif for a random sampling of 439 low-confidence CISs (Figure S3D). Therefore, these data suggested that cryptic transcription may initiate from TATA-like elements, but their suppression requires H3K36me1/2 or H3K36me3. Mechanistically, H3K36me1/2 and H3K36me3 may be capable of recruiting histone deacetylase complexes (HDACs) and preventing histone exchange to maintain a repressive chromatin environment near these TATA-like elements.
Alteration of Global H3K27ac and H3K56ac in set2 Methylation Mutants
Given the ability of H3K36me1/2 and H3K36me3 to suppress antisense transcription during nutrient stress, we next sought to determine whether these methylation states would also suppress the co-transcriptional histone acetylation associated with the appearance of cryptic transcripts (Carrozza et al., 2005; Du and Briggs, 2010; Joshi and Struhl, 2005). H3K36me limits co-transcriptional histone acetylation in two main ways: by recruitment and activation of the Rpd3S deacetylation complex that removes H3 and H4 N-terminal tail acetylation (e.g., H3K27ac); and by the ability of H3K36me to repel the histone exchange factor Asf1, which helps deposit the histone exchange-associated mark H3K56ac (Merker et al., 2008; Venkatesh et al., 2012; Williams et al., 2008). Before evaluating the effects of the set2 mutants on histone acetylation, we first examined whether nutrient stress affected the amounts of Set2 protein or degrees of H3K36me. The set2 mutants showed only a slight reduction in Set2 protein levels after nutrient stress, but they maintained their H3K36me specificity (Figures 6A and S4A–S4D). Interestingly, global H3K36me1 levels were about equal under normal growth conditions and nutrient stress, whereas H3K36me2 and H3K36me3 increased after nutrient stress in set2-F234Y and set2-Y149F, respectively.
Examination of global H3K27ac levels in the WT and our set2 mutants revealed several interesting observations. Under normal growth conditions, H3K27ac levels were higher in set2Δ and the set2 mutants compared to the WT (Figures 6A and 6B). Global H3K27ac levels remained steady in WT cells after nutrient stress and H3K27ac level in set2-Y149F, set2-F234Y, and set2-H199L decreased to that of the WT. In contrast, H3K27ac levels remained high in set2Δ strains after nutrient depletion. These results were consistent with the known function of Set2-dependent H3K36me in recruiting and stimulating the activity of the Rpd3S histone deacetylase complex.
When looking at H3K56ac, most strains had similar levels in normal growth conditions, with set2-F234Y showing a slightly lower level (Figures 6A and 6C). Intriguingly, the WT and set2-Y149F showed close to a 50% reduction in H3K56ac after nutrient stress. However, set2Δ, set2-F234Y, and set2-H199L did not show a similar reduction in H3K56ac. Thus, the data suggest that during nutrient stress, H3K36me3 functions to reduce global H3K56ac levels because only strains containing H3K36me3 had reduced H3K56ac.
H3K36me1/2 and H3K36me3 Are Important for Proper H3K27ac and H3K56ac Localization
After investigating the global effects on H3K27ac and H3K56ac levels during nutrient stress, we next sought to determine how the localization of these histone acetylation events would be affected in the WT and our set2 mutants. Using ChIP-qPCR, we examined H3K27ac and H3K56ac at STE11 and SPB4 in WT, set2Δ, set2-Y149F, set2-F234Y, and set2-H199L strains before and after nutrient stress. Both loci showed nutrient stress-induced cryptic transcription in our RNA-seq dataset, and both genes produced cryptic transcripts as assayed by northern blot (Cheung et al., 2008; McDaniel et al., 2017). On the basis of where the CIS was detected in the RNA-seq data, we designed qPCR primers near the predicted CIS (for STE11, primer set 4; for SPB4, primer set 5) and other locations across the genes (Figures 7A, 7F, S5A, and S5F).
Figure 7. H3K36me1/2 and H3K36me3 Are Important for Proper H3K27ac Localization.
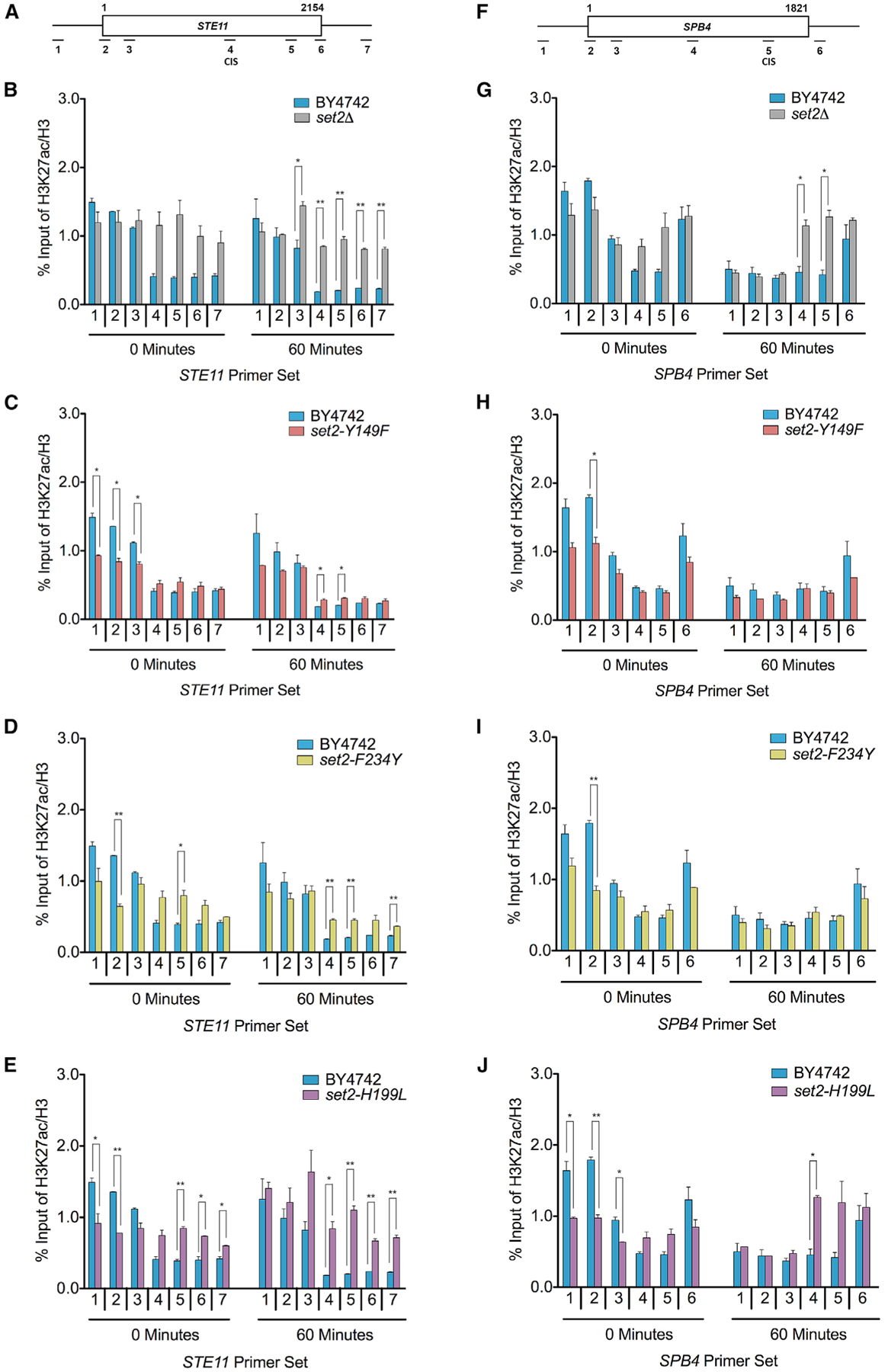
(A) Schematic of STE11 with amplicons indicated below. Predicted CIS located within primer set 4. (B–E) ChIP analysis of H3K27ac across STE11 in the indicated strains and time points. (F) Schematic of SPB4 with amplicons indicated below. Predicted CIS located within primer set 5. (G–J) ChIP analysis of H3K27ac across SPB4 in the indicated strains and time points. Data represented as mean ± SEM of two independent biological replicates. Student’s t test was used to obtain p values. Asterisks indicate significance (*p < 0.05; **p < 0.01); non-significant comparisons are not shown. All qPCR primers are listed in Table S3.
When examining H3K27ac localization, we found several differences between the set2 mutants. In set2Δ, there were higher H3K27ac levels compared to the WT near the CIS of STE11 and SPB4 and in the 3′ regions of STE11, particularly after nutrient stress (Figures 7B and 7G). These data were consistent with previous reports that in the absence of H3K36me, Rpd3S was unable to deacetylate nucleosomes (Drouin et al., 2010; Govind et al., 2010). In contrast, for set2-Y149F and set2-F234Y, changes in H3K27ac were different at STE11 and SPB4. At STE11 after nutrient stress, both set2-Y149F and set2-F234Y had significantly higher H3K27ac compared to the WT near the CIS and immediately 3′ (Figures 7C and 7D). However, at SPB4, H3K27ac levels in both mutants were nearly identical to the WT before and after nutrient stress (Figures 7H and 7I). One commonality in set2-Y149F and set2-F234Y was the significantly lower H3K27ac levels compared to the WT at the promoter before nutrient stress. Similarly, set2-H199L had significantly reduced H3K27ac at the promoter regions of STE11 and SPB4 before nutrient stress (Figures 7E and 7J). However, the changes in H3K27ac near the promoter may be due to Rpd3L and Set3C recruitment, as those complexes localize to 5′ ends of genes (Kim et al., 2012; Shi et al., 2007; Wang et al., 2011). Nonetheless, in set2-H199L, H3K27ac levels were higher near the CIS of STE11 and 3′ regions of both genes, particularly in STE11. These results demonstrated that in certain contexts, H3K36me1/2 or H3K36me3 can ensure proper H3K27ac levels in gene bodies to repress cryptic transcription.
The set2 mutants also showed clear differences in H3K56ac localization before and after nutrient stress. In set2Δ, there were significantly higher H3K56ac levels compared to the WT near the CIS and in the 3′ regions of STE11 and SPB4, particularly after nutrient stress (Figures S5B and S5G). Interestingly, we also found higher levels of H3K56ac near the CIS of STE11 under normal growth conditions. This observation suggested that in the absence of H3K36me, sites near TATA-like elements are prone to histone exchange, and this exchange is exacerbated during nutrient stress. In contrast, set2-Y149F and set2-F234Y generally showed similar levels of H3K56ac compared to the WT near the CIS and in the 3′ regions of STE11 and SPB4 (Figures S5C, S5D, S5H, and S5I). However, there were significant differences in each mutant in the upstream noncoding, promoter, and promoter proximal regions; both set2-Y149F and set2-F234Y had decreased H3K56ac signals compared to the WT. Although set2-H199L showed robust cryptic antisense transcription, there were no significant differences in H3K56ac compared to the WT near the CIS or in the 3′ coding regions, suggesting that alteration of something other than histone exchange was responsible for the appearance of cryptic antisense transcription at these loci during nutrient stress (Figures S5E, S5J, and S6). Together, these data supported a model in which H3K36me1/2 or H3K36me3 can ensure proper H3K56ac levels in gene bodies and repress histone exchange.
DISCUSSION
During transcriptional elongation, Set2 catalyzes H3K36me1, H3K36me2, and H3K36me3 and suppresses cryptic transcription (Kim et al., 2016; McDaniel and Strahl, 2017; Venkatesh et al., 2016). However, it was unclear whether the three methylation states of H3K36 have distinct biological functions and which methyl state(s) is important for the suppression of cryptic transcription. We have demonstrated that H3K36me1/2 and H3K36me3 have unique and shared functions, particularly regarding the suppression of antisense transcription. By taking advantage of a functional Phe/Tyr switch in the SET domain of Set2, we controlled the methylation products generated by this enzyme and established the in vivo functions of different H3K36me states. We observed that in certain cellular contexts, such as rescuing the set2Δ phenotype caused by the absence of the Bur1 kinase or Spt16 of the FACT complex, H3K36me1/2 and H3K36me3 had distinct activities. However, in other contexts—such as sensitivity to caffeine, rapamycin, and phleomycin—H3K36me1/2 and H3K36me3 were functionally equivalent. Similarly, in nutrient stress, H3K36me1/2 or H3K36me3 suppressed antisense cryptic transcription from within gene bodies at 439 genes with previously identified CISs. Without H3K36me1/2 or H3K36me3, we observed antisense cryptic transcription across the genome, and many loci also had decreases in sense transcription, in agreement with previous findings (McDaniel et al., 2017). We also found evidence of a degenerate TATA box motif in loci with CISs, suggesting that DNA sequence contributes to the sensitivity of these sites to cryptic transcription. Mechanistically, H3K36me1/2 and H3K36me3 have an important function in ensuring proper levels of H3K27ac and H3K56ac in genes, particularly during nutrient stress. By maintaining proper histone acetylation levels within genes, cells prevent inappropriate nucleosome remodeling and histone exchange that likely governs cryptic initiation events (Pattenden et al., 2010). Overall, these findings reveal different functions of the H3K36me states in a diverse set of transcriptional programs.
Our study shows that in certain cellular contexts, H3K36me1/2 and H3K36me3 are interchangeable, thus providing flexibility to dynamic processes. This equivalency may exist partly because Eaf3 and Ioc4 bind to both H3K36me2 and H3K36me3, as previously shown by in vitro assays (Carrozza et al., 2005; Joshi and Struhl, 2005; Li et al., 2009; Maltby et al., 2012). Additionally, both H3K36me2 and H3K36me3 peptides are refractory to Asf1 binding (Venkatesh et al., 2012). The recruitment or repelling of these effector proteins by sites with either H3K36me2 or H3K36me3 maintains a proper transcriptional response in certain contexts and at certain genes, even in the absence of one of those marks. Future work examining other cellular contexts and stresses will provide more insight into the roles of H3K36me1/2 and H3K36me3. Although previous data supported a model wherein H3K36me3 was dispensable for certain cellular processes, particularly suppression of cryptic transcription, it was counterintuitive that the cell would use energy to catalyze an unnecessary mark (Hacker et al., 2016; Li et al., 2009; Youdell et al., 2008). Our findings suggest that instead of H3K36me3 being dispensable, it often has a redundant function with H3K36me1/2, thus affording the cell flexibility, particularly in response to stress.
However, our study provides evidence that in certain contexts, H3K36me3 has a unique function for which the other H3K36me forms cannot substitute. Particularly, H3K36me1/2, but not H3K36me3, genetically interacted with Bur1 and Spt16. With Bur1 acting upstream of Set2 during transcription, it is unclear how Bur1 activity relates to specific H3K36me states (Dronamraju and Strahl, 2014). For the FACT complex, in vitro results indicated that Spt16 can bind H3K36me2, but not H3K36me3, peptides (Venkatesh et al., 2012). Perhaps nucleosomes with H3K36me3 are refractory to FACT binding, which may have protected WT and set2-Y149F cells from FACT-dependent nucleosome remodeling. Further work to ascertain how H3K36me3 functions in the Bur1 pathway and in FACT complex-mediated nucleosome exchange will further highlight how specific histone modifications facilitate distinct processes, as proposed by the histone code hypothesis (Strahl and Allis, 2000).
We were unable to ascribe a function for H3K36me1 in any of the Set2 phenotypes that we examined. This lack of functionality may have been because the low levels of H3K36me1 observed in set2-H199L were not enough to elicit an activity, or there simply was no function for H3K36me1 in these phenotypes; we cannot distinguish between these possibilities. Notably, all known reader domains of H3K36me interact with H3K36me2 and H3K36me3, not H3K36me1 (McDaniel and Strahl, 2017). Pryde et al. (2009) suggested that H3K36me1 recruits the replication factor Cdc45 to origins. We did not interrogate this particular activity; thus, we cannot rule out that H3K36me1 may function in replication. H3K36me1 may be targeted by a yet-to-be-identified reader that functions in origin activity.
Interestingly, we identified an enriched sequence motif that resembles a degenerate TATA box, within 100 bp of almost 60% of high- and intermediate-scoring CISs. Although these sites were identified previously, it was not known what predisposed them to cryptic initiation events, as many of the loci are neither linked directly to nor highly transcribed during the nutrient stress response (McDaniel et al., 2017). Possibly, the degenerate TATA sequence motif, which is similar to A/T-rich promoter sequences, contributes to the initiation of transcription at these loci. In addition to the DNA sequence, our work suggests that the chromatin architecture determines whether cryptic transcription can initiate at these sites. We observed that H3K36me1/2 and H3K36me3 are important in maintaining proper H3K27ac and H3K56ac levels at two sites of cryptic transcription. These results connect initiation of cryptic transcription to known Set2 functions—Rpd3S recruitment and activation and repelling Asf1—and provide mechanistic understanding of how Set2-mediated H3K36me prevents cryptic transcription.
There is robust evidence that different methylation states at other histone lysine residues—such as H3K4, H3K9, and H3K27—have unique functions, but this information was lacking for H3K36 (Hyun et al., 2017). We have investigated the functions of different H3K36me states in vivo, particularly H3K36me3. Many studies have examined the consequences of the complete absence of H3K36me or the absence of one or more methylation states; however, there has been no comprehensive analysis of the function of each H3K36me state (Gopalakrishnan et al., 2019; Leung et al., 2019; McDaniel and Strahl, 2017). The distinct activities of H3K36me in certain contexts and interchangeable functions in others presented in this study advances our understanding of how H3K36me contributes to transcriptional control, particularly in repressing cryptic transcription during the nutrient stress response. Our findings establish a foundation for elucidating mechanisms for how the H3K36me states contribute to a variety of cellular processes conserved across eukaryotes that are important for gene expression, development, and disease.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Strahl (brian_strahl@med.unc.edu).
Materials Availability
All reagents generated in this study are available from the Lead Contact without restriction.
Data Code Availability
The RNA-seq data generated during this study are available at GEO: GSE125684 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). Original data for Western blots and spotting assays have been deposited to Mendey Data: https://doi.org/10.17632/25fpx9dzsk.1 (https://data.mendeley.com/datasets/25fpx9dzsk/1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains and Plasmids
Yeast strains were created using Delitto Perfetto (Stuckey and Storici, 2013) or the PCR Toolbox (Janke et al., 2004). Yeast strains used in this study are listed in Table S2 and were created using the primers in Table S3. To construct the FLAG-tagged wild-type Set2 plasmid for the baculovirus system, the SET2 ORF was generated by standard PCR reaction from genomic DNA and subcloned into XhoI/NotI digested pBL532 (a derivative of pBacPAK8 that contains an N-terminal HIS Tag and FLAG tag separated by a TEV protease digestion site). All other plasmids were created using site-directed mutagenesis (Agilent) with the primers in Table S3. Plasmids used in this study are listed in Table S4.
METHOD DETAILS
Alignment and Molecular Modeling
The SETD2 crystal structure (Yang et al., 2016) was isolated from the Protein Data Bank (PDB: 5JLB) and served as a template for I-TASSER modeling of Set2 (Zhang, 2008). Set2 was visualized in PyMOL, and mutations were modeled with MODELLER (Webb and Sali, 2016).
Western Blotting
All strains were grown to saturation in YPD before being diluted to an OD600 of 0.2 and grown to OD600 ~1 in YPD at 30°C. For nutrient deprivation experiments, cells were isolated, washed with water, and resuspended in SD media (2% glucose, 0.15% yeast nitrogen base, 0.5% ammonium sulfate) (Cheung et al., 2008). Ten ODs of cells were collected at each time point and western blotting was performed after extraction of proteins by TCA lysis (Keogh et al., 2006a, 2006b). Briefly, 10 ODs of cells were resuspended in 200 μL of TCA Lysis Buffer (10 mM Tris pH 8.0, 10% TCA, 25 nM NH4OAc, 1 mM Na2EDTA) and incubated on ice for 10 min before centrifugation at max speed for 5 min. The supernatant was aspirated, the pellet resuspended in 100 μL of Resuspension Buffer (0.1 M Tris pH 11 and 3% SDS) and heated at 95C for 5 min. Samples were then centrifuged at max speed for 1 min, 90 ul of supernatant was moved to a fresh tube and quantified before diluting 2x with loading buffer and stored at 4C. Lysates were separated by SDS-PAGE, transferred to PVDF membrane, and probed overnight at 4°C with Set2 (in-house), G6PDH (Sigma A9521–1VL), H3K36me1 (abcam 9048), H2K36me2 (in-house), H3K36me3 (abcam 9050), H3K27ac (Active Motif, 39133), H3K56ac (Active Motif, 39281), H3 C-terminal (EpiCypher 13–0001), or RNAPII CTD phospho Ser2 (Active Motif 61083) primary antibodies (see Key Resources Table). Membranes were washed in TBS (Tris-buffered saline)-Tween (50 mM Tris-HCl, 150 mM NaCl, and 0.5% Tween 20). Membranes were incubated with HRP-conjugated anti-rabbit (GE Healthcare NA934V; 1:10,000) or HRP-conjugated anti-rat (Jackson ImmunoResearch 312–036-045; 1:10,000) antibody and probed with ECL Prime (GE Healthcare).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Set2 | In House | N/A |
| Anti-G6PDH antibody, polyclonal | Sigma | Cat#A9521–1VL |
| Anti-H3K36me1 | Abcam | ab9048 |
| Anti-H3K36me2 | In House | N/A |
| Anti-H3K36me2 | Active Motif | Cata#39255 |
| Anti-H3K36me2 | Abcam | ab9049 |
| Anti-Histone H3K36me3 | Abcam | ab9050 |
| Anti-H3K27ac | Active Motif | Cat#39133 |
| Anti-H3K56ac | Active Motif | Cat#39281 |
| Anti-histone H3 antibody, polyclonal | EpiCypher | 13–0001 |
| Normal Rabbit IgG | Cell Signaling Technology | 2729 |
| Anti-RNAPII CTD phospho Ser2, clone 3E10 | Active Motif | Cat#61083 |
| Anti-HA Tag | 3ethyl | Cat#A190–108A |
| HRP-conjugated anti-rabbit | GE Healthcare | Cat#NA934V |
| HRP-conjugated anti-mouse | GE Healthcare | Cat#NA931V |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Caffeine | Sigma-Aldrich | Cat#C0750–100G |
| Rapamycin | SIGMA (Roche) | Cat#R8781 |
| Phleomycin | Sigma-Aldrich | Cat#P9546–5MG |
| 6-Azauracil | SIGMA (Roche) | Cat#A1757 |
| 5-FOA | RPI | F10501–10.0 |
| Acid Phenol Chloroform | Ambion/Thermo Fisher Scientific | Cat#AM9722 |
| iTaq Universal SYBR ® Green Supermix | 3iorad | Cat#172–5124 |
| Protein A Agarose | SIGMA (Roche) | Cat #11134515001 |
| FLAG_M2 Agarose beads | Sigma-Aldrich | A2220 |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat#10009D |
| SPRI Beads (Agencourt AMPure XP) | Beckman Coulter | Cat#A63881 |
| ECL Prime | GE Healthcare | Cat#45002401 |
| DNase | Promega | M610A |
| SAM | Sigma-Aldrich | Cat#A7007–25MG |
| Set2 | This Study | N/A |
| set2-Y149F | This study | N/A |
| set2-F234Y | This study | N/A |
| Xenopus nucleosomes | This study | N/A |
| Critical Commercial Assays | ||
| TruSeq Stranded Total RNA with RiboZero Gold Library Prep Kit | Illumina | Cat#RS-122–2301 |
| RNeasy Mini Kit | QIAGEN | Cat#74106 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | 200523 |
| ChIP DNA Clean and Concentrator | Zymo | Cat#D5205 |
| Deposited Data | ||
| RNA-seq Data | This study | GEO: GSE125684 |
| Raw data for western blots and spotting assays | This study | Mendeley Data: https://doi.org/10.17632/25fpx9dzsk.1 (https://data.mendeley.com/datasets/25fpx9dzsk/1) |
| Experimental Models: S. cerevisiae Strains | ||
| Yeast Strains | This study and other sources | Table S2 |
| Oligonucleotides | ||
| Oligos | This study and other sources | Table S3 |
| Recombinant DNA | ||
| Plasmids | This study and other sources | Table S4 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| I-TASSER | Zhang, 2008 | https://zhanglab.ccmb.med.umich.edu/I-TASSER/ |
| PyMOL | N/A | https://pymol.org/2/ |
| MODELLER | Webb and Sali, 2016 | https://salilab.org/modeller/ |
| STAR | Dobin et al., 2013 | N/A |
| BEDtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/motif/ |
Recombinant Protein Purification from Baculovirus Expression System
Ten mL of wild-type Set2, set2-Y149F, or set2-F234Y P2 virus was inoculated into 100 mL freshly conditioned Sf21 cells (1×106 cells/ mL), supplemented with 10% FBS, 1% P/S, and cultured at 27°C for 48 h. Cells were collected by centrifugation at 3,000 rpm for 5 min in a table-top centrifuge and washed with 10 mL 1x PBS buffer. Cell pellets were lysed in 10 mL BV-lysis buffer (50 mM HEPES pH 7.9, 500 mM NaCl, 10% glycerol, 2mM MgCl2 and 0.2% Triton) on ice for 30 min. The lysates were clarified by ultracentrifugation (Beckman 50.2 Ti 40,000 rpm) for 30 min. Flag beads (200 μl, Sigma) were added to the supernatant and incubated at 4°C for at least 2 h. After incubation, beads were washed three times with 10 mL BV-lysis buffer and eluted with 600 μL of FLAG-elution buffer (50 mM HEPES pH 7.9, 100 mM NaCl, 2 mM MgCl2, 0.02% NP40; 10% glycerol) containing 500 μg/mL of 3xFlag peptide. Elutions were concentrated with a 10-kD cut-off concentrator (Amicon).
Histone Methyltransferase Assay
Standard HMT reactions were performed in a 40 μl system containing 1x HMT buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM MgCl2, 2 mM DTT, 5% glycerol), 4 μg or 3-fold serial dilution of recombinant Set2 protein, 4 μg of Xenopus recombinant nucleosomes supplemented with 10 μM unlabeled S-adenosylmethionine (Sigma) (Li et al., 2003). Reactions were incubated at 30°C for 1 h. Immunoblots were performed with antibodies against H3K36me3 (abcam 9050), H3K36me2 (abcam 9049), and H3K36me1 (abcam 9048) to detect specific histone methylation states.
Co-immunoprecipitation
All strains were grown to saturation in SC-Leu before being diluted to an OD600 of 0.4 and grown to OD600 ~2.0 in 100 mL of SC-Leu at 30°C. The cells were pelleted and washed with 30 mL of distilled H2O. The pellets were resuspended in 500 μL of Extraction Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 1 mM Mg(C2H3O2)2, 1 mM imidazole, 0.1% NP-40, 0.5 mM EDTA, and 10% glycerol) (Kizer et al., 2005) and split equally into two tubes. Glass beads (BioSpec Products) were added to bring the total volume to 1 mL and samples were vortexed for 10 min at 4°C. Lysates were collected into fresh tubes via centrifugation, and the lysates were cleared at max speed for 15 min at 4°C. Protein concentration was measured via Bradford assay (Bio-Rad). A 25 μL aliquot was used for input and 1 mg/mL of protein was incubated overnight in 600 μL of Extraction Buffer at 4°C with 5 μL of HA antibody (Bethyl). Antibody was conjugated to 40 μL Protein A Agarose beads (Roche) for 2 h at 4°C before being washed with Extraction Buffer and eluted with 50 μL of 1x SDS buffer. Samples were heated at 95°C for 5 min and loaded onto an 8% SDS-PAGE gel. Data are representative of two independent biological replicates.
Chromatin Immunoprecipitation and Real-Time PCR
Yeast strains were grown to saturation in YPD before being diluted to an OD600 of 0.2 and grown to an OD600 of ~1 at 30°C. For nutrient deprivation experiments, cells were isolated, washed with water, and resuspended in SD media (2% glucose, 0.15% yeast nitrogen base, 0.5% ammonium sulfate) (Cheung et al., 2008). Fifty ODs of cells were collected at each time point and ChIP was performed as described with modifications (Ahn et al., 2009). For precipitation of proteins, 2 ul of H3K36me1 (abcam 9048), H2K36me2 (Active Motif 39255), H3K36me3 (abcam 9050), H3K27ac (Active Motif, 39133), H3K56ac (Active Motif, 39281), or H3 C-terminal (EpiCypher, 13–0001) was used and IgG (Cell Signaling 2729) was the negative control. DNA was eluted in 30 μL of elution buffer (Zymo) and diluted 1:20. Two μl of the diluted DNA was subjected to qPCR using SYBR Green (Bio-Rad) and primers described in Table S3.
Spotting Assays
Yeast strains were grown at 30°C to saturation and diluted to an OD600 of 0.5 prior to spotting 5-fold serial dilutions on plates at 30°C or 34°C, as indicated, for 2–4 days. Plates contained the indicated concentrations of caffeine (Sigma-Aldrich), rapamycin (Roche), phleomycin (Sigma-Aldrich), 6-azauracil (6AU) (Roche), or 5-fluoorotic (5-FOA) (RPI). For the Bur1 growth assay, BUR1 deletion shuffle strains were grown on media lacking uracil to maintain the wild-type BUR1 plasmid before plating on media containing the 5-FOA. All experiments were performed at least three times.
RNA-seq Library Preparation and Sequencing
Yeast strains were grown to saturation in YPD before being diluted to an OD600 of 0.2 and grown to an OD600 of ~1 at 30°C. Cells were isolated, washed with water, and resuspended in SD media (2% glucose, 0.15% yeast nitrogen base, 0.5% ammonium sulfate) (Cheung et al., 2008). Ten ODs of cells were collected at each time point and RNA was isolated by acid phenol (Ambion/Thermo Fisher Scientific) extraction. Five μg RNA was treated with DNase (Promega) and purified (RNeasy column, QIAGEN). Next, 2.5 μg of the purified RNA was processed with yeast-specific rRNA depletion beads (Illumina). Strand-specific bar-coded sequencing libraries were then prepared (TruSeq Stranded Total RNA Library Preparation Kit, Illumina and SPRI beads, Beckman Coulter). Libraries were pooled and sequenced across two lanes (Hi-Seq 4000, Illumina). Data are representative of three independent biological replicates.
Sequencing Alignment
Reads were trimmed and filtered of adaptor sequences using cutadapt, and required to have at least 90% of bases with quality scores exceeding 20. Reads were aligned to the reference yeast genome (sacCer3) using STAR. Sense and anti-sense reads were analyzed separately (Dobin et al., 2013). Coverage across genomic features was computed using BEDtools (Quinlan and Hall, 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
Western Blotting
Western blots were quantified with ImageJ software for two or more independent biological replicates. Quantification of Set2 was normalized to G6PDH. Data for mutants were normalized relative to BY4742. Quantification of H3 PTMs was normalized to H3. Measurement for all mutants were normalized BY4742 and in nutrient deprivation experiments, measurements were normalized to BY4742 at 0 min. Each bar graph is representative of mean ± SEM of two or more independent biological replicates.
Chromatin Immunoprecipitation and Real-Time PCR
Data represented as mean ± SEM of two independent biological replicates. Student’s t test was used to obtain p-values and asterisks indicate significance (* p < 0.05; ** p < 0.01).
Sequencing Analysis
Significantly enriched motifs were discovered using HOMER, with CIS ± 100bp as foreground and the 100 bp flanking sequence on either side as background (Heinz et al., 2010). As a control, we randomly selected 439 low-confidence CIS and repeated the motif analysis. Signal was normalized to total sequencing depth and biological replicates were averaged where appropriate.
Supplementary Material
Highlights.
Engineered forms of Set2 can control Set2 methylation products
H3K36me1/2 and H3K36me3 act redundantly in many cellular contexts
H3K36me1/2 or H3K36me3 can prevent cryptic transcription during nutrient stress
Cryptic transcription in the absence of H3K36me is linked to increased H3 acetylation
ACKNOWLEDGMENTS
We thank Brenda Temple of the R. L. Juliano Bioinformatics Core Facility for assistance with protein modeling and Howard Fried for editorial suggestions. B.L. acknowledges support from the National Key R&D Program of China (2018YFC1004500) and the National Natural Science Foundation of China (31872817). J.V.D. was supported in part by a grant from the National Institute of General Medical Sciences under award FT32 GM007092. B.D.S. acknowledges support from National Institutes of Health (NIH) grant R35 GM126900. T.S.P. and J.M.S. were supported by The Eunice Kennedy Shriver National Institute of Child Health and Human Development (U54HD079124) and the National Institute of Neurological Disorders (NINDS) (P30NS045892).
Footnotes
DECLARATION OF INTERESTS
B.D.S. acknowledges he is a co-founder and scientific advisory board member of EpiCypher, Inc.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107751.
SUPPORTING CITATIONS
The following reference appears in the Supplemental Information: Du et al., 2008; Grzechnik et al., 2015; Mumberg et al., 1995; Nourani et al., 2006; Voth et al., 2014.
REFERENCES
- Ahn SH, Keogh MC, and Buratowski S (2009). Ctk1 promotes dissociation of basal transcription factors from elongating RNA polymerase II. EMBO J. 28, 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, and Pugh BF (2004). Identification and distinct regulation of yeast TATA box-containing genes. Cell 116, 699–709. [DOI] [PubMed] [Google Scholar]
- Biswas D, Dutta-Biswas R, Mitra D, Shibata Y, Strahl BD, Formosa T, and Stillman DJ (2006). Opposing roles for Set2 and yFACT in regulating TBP binding at promoters. EMBO J. 25, 4479–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, and Workman JL (2005). Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592. [DOI] [PubMed] [Google Scholar]
- Cheng X, Collins RE, and Zhang X (2005). Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct 34, 267–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung V, Chua G, Batada NN, Landry CR, Michnick SW, Hughes TR, and Winston F (2008). Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol. 6, e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Sutton A, Sternglanz R, and Prelich G (2006). The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol. Cell. Biol 26, 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, Selker EU, Shinkai Y, and Cheng X (2005). In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyl-transferases. J. Biol. Chem 280, 5563–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P (2019). Organization and regulation of gene transcription. Nature 573, 45–54. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronamraju R, and Strahl BD (2014). A feed forward circuit comprising Spt6, Ctk1 and PAF regulates Pol II CTD phosphorylation and transcription elongation. Nucleic Acids Res. 42, 870–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronamraju R, Jha DK, Eser U, Adams AT, Dominguez D, Choudhury R, Chiang YC, Rathmell WK, Emanuele MJ, Churchman LS, and Strahl BD (2018). Set2 methyltransferase facilitates cell cycle progression by maintaining transcriptional fidelity. Nucleic Acids Res. 46, 1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin S, Laramée L, Jacques PÉ, Forest A, Bergeron M, and Robert F (2010). DSIF and RNA polymerase II CTD phosphorylation coordinate the recruitment of Rpd3S to actively transcribed genes. PLoS Genet. 6, e1001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du HN, and Briggs SD (2010). A nucleosome surface formed by histone H4, H2A, and H3 residues is needed for proper histone H3 Lys36 methylation, histone acetylation, and repression of cryptic transcription. J. Biol. Chem 285, 11704–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du HN, Fingerman IM, and Briggs SD (2008). Histone H3 K36 methylation is mediated by a trans-histone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 22, 2786–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan R, Marr SK, Kingston RE, and Winston F (2019). A conserved genetic interaction between Spt6 and Set2 regulates H3K36 methylation. Nucleic Acids Res. 47, 3888–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, Hu C, Swaminathan V, Workman JL, Li B, and Hinnebusch AG (2010). Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell 39, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzechnik P, Gdula MR, and Proudfoot NJ (2015). Pcf11 orchestrates transcription termination pathways in yeast. Genes Dev. 29, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker KE, Fahey CC, Shinsky SA, Chiang Y-CJ, DiFiore JV, Jha DK, Vo AH, Shavit JA, Davis IJ, Strahl BD, et al. (2016). Structure/ Function Analysis of Recurrent Mutations in SETD2 Reveals a Critical and Conserved Role for a SET Domain Residue in Maintaining Protein Stability and H3K36 Trimethylation. J. Biol. Chem 291, jbc.M116.739375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber F, Bunina D, Gupta I, Khmelinskii A, Meurer M, Theer P, Steinmetz LM, and Knop M (2016). Protein Abundance Control by Non-coding Antisense Transcription. Cell Rep. 15, 2625–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, and Kim J (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, and Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Jha DK, and Strahl BD (2014). An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun 5, 3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, and Struhl K (2005). Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell 20, 971–978. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Podolny V, and Buratowski S (2003). Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Mol. Cell. Biol 23, 7005–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. (2005). Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, et al. (2006a). A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 439, 497–501. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Mennella TA, Sawa C, Berthelet S, Krogan NJ, Wolek A, Podolny V, Carpenter LR, Greenblatt JF, Baetz K, and Buratowski S (2006b). The Saccharomyces cerevisiae histone H2A variant Htz1 is acetylated by NuA4. Genes Dev. 20, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Xu Z, Clauder-Münster S, Steinmetz LM, and Buratowski S (2012). Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell 150, 1158–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee BB, Oh YM, Zhu C, Steinmetz LM, Lee Y, Kim WK, Lee SB, Buratowski S, and Kim T (2016). Modulation of mRNA and lncRNA expression dynamics by the Set2-Rpd3S pathway. Nat. Commun 7, 13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, and Strahl BD (2005). A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol 25, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NACH, Margaritis T, van de Pasch LAL, van Heesch SAAC, Brok MO, Groot Koerkamp MJA, et al. (2011). The specificity and topology of chromatin interaction pathways in yeast. Mol. Cell 42, 536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CS, Douglass SM, Morselli M, Obusan MB, Pavlyukov MS, Pellegrini M, and Johnson TL (2019). H3K36 Methylation and the Chromodomain Protein Eaf3 Are Required for Proper Cotranscriptional Spliceosome Assembly. Cell Rep. 27, 3760–3769.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Howe L, Anderson S, Yates JR III, and Workman JL (2003). The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem 278, 8897–8903. [DOI] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Lee D, Seidel C, and Workman JL (2007). Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science 316, 1050–1054. [DOI] [PubMed] [Google Scholar]
- Li B, Jackson J, Simon MD, Fleharty B, Gogol M, Seidel C, Workman JL, and Shilatifard A (2009). Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem 284, 7970–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Rao B, Shabalin AA, Nobel AB, Strahl BD, and Lieb JD (2009). The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS ONE 4, e4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubliner S, Keren L, and Segal E (2013). Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 41, 5569–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltby VE, Martin BJE, Schulze JM, Johnson I, Hentrich T, Sharma A, Kobor MS, and Howe L (2012). Histone H3 lysine 36 methylation targets the Isw1b remodeling complex to chromatin. Mol. Cell. Biol 32, 3479–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SL, and Strahl BD (2017). Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci 74, 3317–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SL, Fligor JE, Ruan C, Cui H, Bridgers JB, DiFiore JV, Guo AH, Li B, and Strahl BD (2016). Combinatorial histone readout by the dual plant homeodomain (PHD) fingers of Rco1 mediates Rpd3S chromatin recruitment and the maintenance of transcriptional fidelity. J. Biol. Chem 291, 14796–14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SL, Hepperla AJ, Huang J, Dronamraju R, Adams AT, Kulkarni VG, Davis IJ, and Strahl BD (2017). H3K36 Methylation Regulates Nutrient Stress Response in Saccharomyces cerevisiae by Enforcing Transcriptional Fidelity. Cell Rep. 19, 2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker JD, Dominska M, Greenwell PW, Rinella E, Bouck DC, Shibata Y, Strahl BD, Mieczkowski P, and Petes TD (2008). The histone methylase Set2p and the histone deacetylase Rpd3p repress meiotic recombination at the HIS4 meiotic recombination hotspot in Saccharomyces cerevisiae. DNA Repair (Amst.) 7, 1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Müller R, and Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, and Jacquier A (2009). Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457, 1038–1042. [DOI] [PubMed] [Google Scholar]
- Nourani A, Robert F, and Winston F (2006). Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol. Cell. Biol 26, 1496–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattenden SG, Gogol MM, and Workman JL (2010). Features of cryptic promoters and their varied reliance on bromodomain-containing factors. PLoS ONE 5, e12927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. (2005). Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527. [DOI] [PubMed] [Google Scholar]
- Pryde F, Jain D, Kerr A, Curley R, Mariotti FR, and Vogelauer M (2009). H3 k36 methylation helps determine the timing of cdc45 association with replication origins. PLoS ONE 4, e5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B, Shibata Y, Strahl BD, and Lieb JD (2005). Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell. Biol 25, 9447–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder RG (2019). 50+ years of eukaryotic transcription: an expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol 26, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, and Strahl BD (2014). Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan C, Lee CH, Cui H, Li S, and Li B (2015). Nucleosome contact triggers conformational changes of Rpd3S driving high-affinity H3K36me nucleosome engagement. Cell Rep. 10, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Kachirskaia I, Walter KL, Kuo J-HA, Lake A, Davrazou F, Chan SM, Martin DGE, Fingerman IM, Briggs SD, et al. (2007). Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem 282, 2450–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AC, Xu X, Kim HS, Fillingham J, Kislinger T, Mennella TA, and Keogh MC (2012). The replication-independent histone H3-H4 chaperones HIR, ASF1, and RTT106 co-operate to maintain promoter fidelity. J. Biol. Chem 287, 1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, and Workman JL (2012). Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat. Struct. Mol. Biol 19, 884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soshnev AA, Josefowicz SZ, and Allis CD (2016). Greater Than the Sum of Parts: Complexity of the Dynamic Epigenome. Mol. Cell 62, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, and Allis CD (2000). The language of covalent histone modifications. Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, and Allis CD (2002). Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol 22, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckey S, and Storici F (2013). Gene knockouts, in vivo site-directed mutagenesis and other modifications using the delitto perfetto system in Saccharomyces cerevisiae. Methods Enzymol. 533, 103–131. [DOI] [PubMed] [Google Scholar]
- Venkatesh S, and Workman JL (2013). Set2 mediated H3 lysine 36 methylation: regulation of transcription elongation and implications in organismal development. Wiley Interdiscip. Rev. Dev. Biol 2, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, and Workman JL (2012). Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 489, 452–455. [DOI] [PubMed] [Google Scholar]
- Venkatesh S, Li H, Gogol MM, and Workman JL (2016). Selective suppression of antisense transcription by Set2-mediated H3K36 methylation. Nat. Commun 7, 13610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Takahata S, Nishikawa JL, Metcalfe BM, Näär AM, and Stillman DJ (2014). A role for FACT in repopulation of nucleosomes at inducible genes. PLoS ONE 9, e84092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Zhou BO, and Zhou JQ (2011). Histone H3 lysine 4 hypermethylation prevents aberrant nucleosome remodeling at the PHO5 promoter. Mol. Cell. Biol 31, 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Niu Y, and Li B (2015). Balancing acts of SRI and an auto-inhibitory domain specify Set2 function at transcribed chromatin. Nucleic Acids Res. 43, 4881–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, and Sali A (2016). Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics 54, 1–55, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hsieh T-HS, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, and Friedman N (2015). High-resolution chromatin dynamics during a yeast stress response. Mol. Cell 58, 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SK, Truong D, and Tyler JK (2008). Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc. Natl. Acad. Sci. USA 105, 9000–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, and Strahl BD (2003). Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 17, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, and Steinmetz LM (2009). Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zheng X, Lu C, Li GM, Allis CD, and Li H (2016). Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 30, 1611–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdell ML, Kizer KO, Kisseleva-Romanova E, Fuchs SM, Duro E, Strahl BD, and Mellor J (2008). Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol. Cell. Biol 28, 4915–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.