

Abstract
SOCS3 is a cytosolic inhibitor of cytokine signaling that suppresses the activation of cytokine receptor-associated JAK kinases. Mechanistically, SOCS3 is recruited to a site in the cytokine receptors known as the SOCS3-interaction motif, and then binds JAK molecules to inhibit their kinase activity. The SOCS3-interaction motif is found in receptors of the gp130 cytokine family but mostly absent from other cytokine receptors, including γc. Thus, SOCS3 has been considered a selective suppressor of gp130 family cytokines, but not γc cytokines. Considering that γc signaling induces SOCS3 expression in T cells, here we revisited the role of SOCS3 on γc signaling. Using SOCS3 transgenic mice, we found that increased abundance of SOCS3 not only suppressed signaling of the gp130 family cytokine IL-6, but also signaling of the γc family cytokine IL-7. Consequently, SOCS3 transgenic mice were impaired in IL-7-dependent T cell development in the thymus and the homeostasis of mature T cells in peripheral tissues. Moreover, enforced SOCS3 expression interfered with the generation of Foxp3+ regulatory T cells which requires signaling by the γc family cytokine IL-2. Collectively, we report an underappreciated role for SOCS3 in suppressing γc cytokine signaling, effectively expanding its scope of target cytokines in T cell immunity.
Keywords: SOCS3, JAK/STAT, γc cytokine, IL-2, Treg cells
Graphical Abstract
SOCS3 requires direct interaction with both the cytokine receptor and JAK to suppress gp130 cytokine signaling. In contrast, when suppressing γc cytokine signaling, SOCS3 does not seem to bind γc or the cytokine proprietary receptor such as IL-2Rβ, but directly interacts with and inhibits JAK1 to suppress IL-2 receptor signaling.

Introduction
While cytokine signaling is critical for T cell development and homeostasis, excessive cytokine signaling is detrimental for T cell survival and function [1, 2]. Multiple layers of negative regulatory mechanisms operate in T cells to prevent uncontrolled cytokine signaling. They include downregulation of cytokine receptor expression, upregulation of phosphatase activities, and induction of inhibitory molecules, such as Protein Inhibitor of Activated STAT (PIAS) or members of the Suppressor Of Cytokine Signaling (SOCS) family [3–5]. Chief among these mechanisms is the role of SOCS family molecules whose significance is illustrated in the embryonic or perinatal lethality of SOCS3- and SOCS1-deficient mice, respectively [6, 7].
Currently, eight SOCS family members have been identified, namely SOCS1–7 and CISH [8]. All SOCS family members share the characteristic of containing a 40-amino acid C-terminal motif that is commonly referred to as the SOCS box. The SOCS box recruits elongin B/C, Cullin 5 and Rbx2 to establish E3 ubiquitin ligase activity, and mediates ubiquitination and proteosomal degradation of SOCS-associated protein substrates [9]. Because all SOCS members also contain an SH2-domain, this mechanism can effectively remove tyrosine phosphorylated signaling molecules that are downstream of cytokine receptors to terminate cytokine signaling. In agreement, genetic deletion or transgenic overexpression of individual SOCS members have unveiled striking effects of SOCS molecules in T cell development and function [8]. Among others, SOCS molecules were found to be critical for correct CD4 versus CD8 T lineage choice in the thymus [10], for anti-viral and anti-tumor immunity [11, 12], and for the homeostatic maintenance of T cells [13].
Interestingly, SOCS box-mediated proteasomal degradation is not the only mechanism how SOCS1 and SOCS3 interferes with cytokine receptor signaling. In fact, the major mechanism that SOCS1 and SOCS3 employ to inhibit cytokine signaling is mediated by a short N-terminal motif known as the Kinase Inhibitory Region (KIR), and which is only found in these two members of the SOCS family [14, 15]. Mechanistically, KIR directly binds to and inhibits receptor-associated JAK molecules, which are cytosolic tyrosine kinases that initiate the proximal signaling downstream of cytokine receptors [16]. Thus, SOCS1 and SOCS3 are unique among SOCS members to block cytokine signaling by suppressing JAK activity. While utilizing the same mechanism for suppression, SOCS1 and SOCS3 do not suppress signaling of the same cytokines. There is a clear distinction between SOCS1- and SOCS3-controlled signaling pathways, which is illustrated in the suppression of IFNγ versus IL-6 receptor signaling. Both cytokine receptors are associated with JAK1, and both SOCS1 and SOCS3 are potent inhibitors of JAK1 activation [17, 18]. Surprisingly, however, SOCS1 and SOCS3 are not equipotent in suppressing IFNγ and IL-6. SOCS1 only inhibits IFNγ and not IL-6 signaling. Conversely, SOCS3 inhibits IL-6 signaling but does not suppress IFNγ or IL-10 signaling [6, 19, 20]. These results revealed that JAK1 suppression by either SOCS1 or SOCS3 is context-dependent, and additional factors are in play to determine the specificity of cytokine suppression.
One of these factors is identified as a SOCS3-binding motif in the intracellular domain of gp130, which is the signal transducing chain of the IL-6 receptor complex [20, 21]. Unlike SOCS1, which can directly bind to JAKs and suppress their activities [17], SOCS3 requires simultaneous binding to both JAK and the cytokine receptor to suppress cytokine signaling [15]. The SOCS3-binding motif is the docking site for SOCS3 on the cytokine receptor, and it is necessary for SOCS3 interaction and suppression of JAK. In agreement, this motif was found in receptors for cytokines whose signaling was suppressed by SOCS3 including LIF, leptin, G-CSF and gp130 family cytokines. Therefore, SOCS3 has been mostly considered as a suppressor of IL-6 and other gp130 family cytokines, but not of cytokines that are targeted by SOCS1, such as interferons, and γc family cytokines. Consequently, SOCS1 and SOCS3 are proposed to have distinct and non-overlapping roles, which was further supported by analyzing mice that lack both SOCS1 and SOCS3 in hematopoietic cells [22]. However, the exact role of SOCS1 and SOCS3 in controlling cytokine signaling remains to be determined.
In the current study, we addressed the substrate specificity of SOCS3 using a newly generated SOCS3 transgenic mouse model. Surprisingly, we found that SOCS3 also inhibited signaling of cytokine receptors that were previously not considered targets of SOCS3 and that lacked clear, distinctive SOCS3-binding motifs. Specifically, increased abundance of SOCS3 interfered with γc cytokine signaling, including IL-7 and IL-2, resulting in impaired thymopoiesis - which depends on IL-7, and diminished generation of Foxp3+CD25+ Treg cells - which requires IL-2, respectively. Thus, our findings expand the spectrum of cytokines that are controlled by SOCS3 and put forward a model where multiple SOCS molecules act in concert to suppress excessive cytokine signaling.
Results
SOCS3 inhibits IL-6 signaling
Cytokine signaling induces the expression of SOCS1 and SOCS3, as illustrated in the increased abundance of both SOCS1 and SOCS3 mRNA in IL-7-stimulated LN CD4+ and CD8+ T cells (Fig. 1A). Upregulating SOCS expression is a critical negative regulatory feedback mechanism in cytokine signaling [8]. However, the exact downstream targets of SOCS1- versus SOCS3-mediated suppression remain unclear, because cytokine stimulation induces expression of both SOCS1 and SOCS3. Thus, to examine the role of SOCS3 independently of SOCS1, we generated genetically engineered mice where a murine Socs3 cDNA is transgenically expressed under the control of a human CD2 mini-cassette. Such SOCS3transgenic (SOCS3Tg) mice were designed to overexpress SOCS3 in all T lineage cells, and we confirmed significantly elevated abundance of SOCS3 transcripts and proteins in SOCS3Tg T cells by semi-quantitative RT-PCR (Fig. 1B) and Western blot analysis (Fig. 1C), respectively. Importantly, SOCS3Tg T cells contained about two- to three-fold more SOCS3 proteins than their WT controls (Fig. 1C), which is similar to the range of SOCS3 protein induction by overnight IL-7 stimulation (Fig. 1D). Thus, the newly generated SOCS3Tg represents a physiological model to assess the role of SOCS3 in modulating cytokine signaling. Because SOCS3 reportedly suppresses signaling of gp130 cytokines [15, 23], next, we examined if the increased abundance of SOCS3 would impair phosphorylation of STAT molecules downstream of gp130 cytokine stimulation. To this end, we assessed intracellular phosphor-STAT3 (pSTAT3) contents in LN T cells of wild-type (WT) and SOCS3Tg mice that were stimulated with IL-6, a major gp130 cytokine [24]. As expected, IL-6-induced STAT3 phosphorylation was severely impaired in SOCS3Tg T cells compared to T cells from WT mice (Fig. 1E and Supporting Information Fig. 1), demonstrating that the overexpressed SOCS3 proteins are functional, and further confirming the detrimental effect of SOCS3 on IL-6 signaling.
Fig. 1. SOCS3 overexpression suppresses IL-6 signaling in T cells.
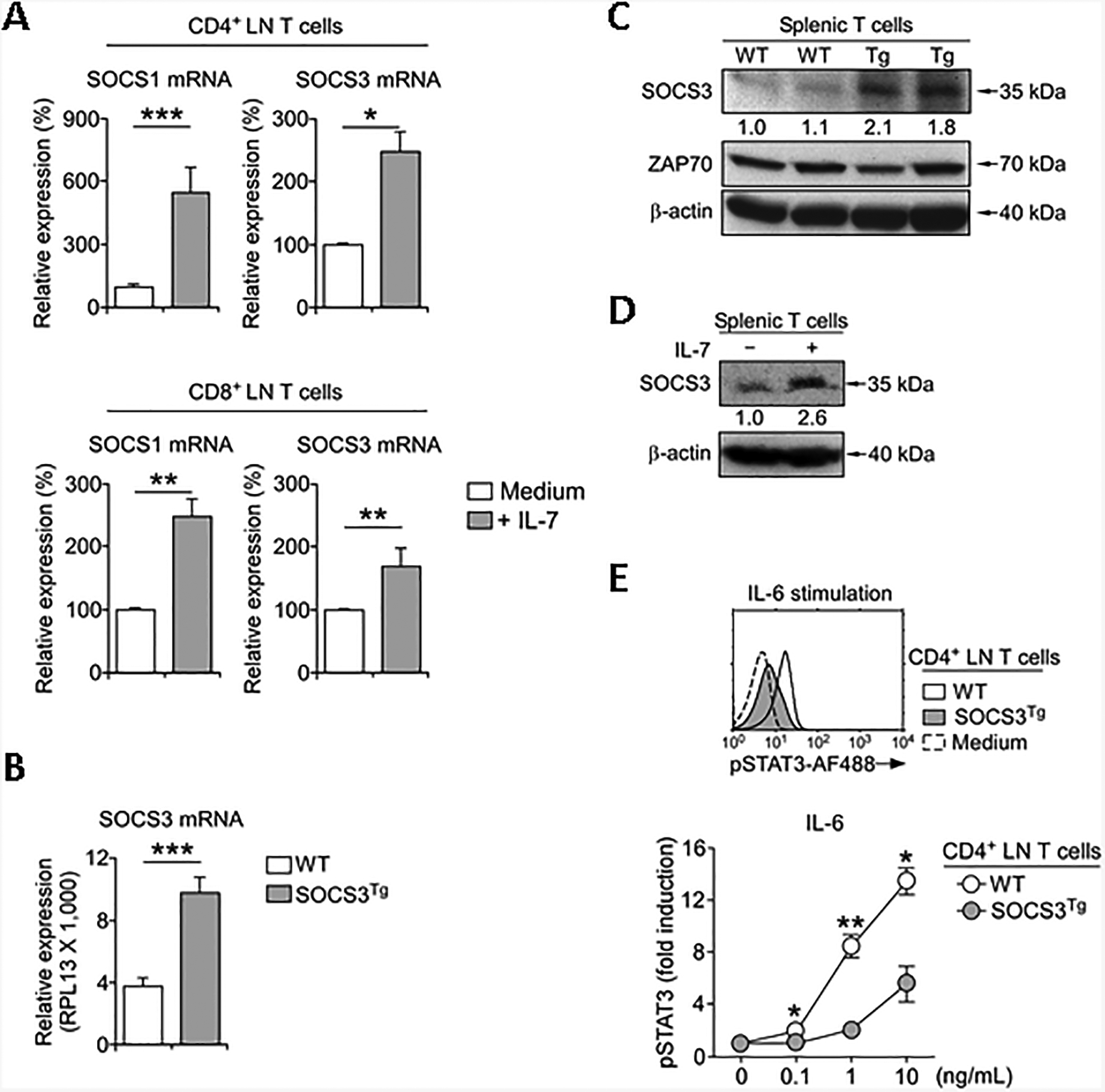
(A) SOCS1 and SOCS3 mRNA contents were assessed by RT-qPCR in purified CD4+ and CD8+ LN T cells that were stimulated overnight with IL-7 (10 ng/ml) or medium alone. Results are summary from each 3 and 5 independent experiments for CD4+ and CD8+ T cells, respectively.
(B) SOCS3 mRNA contents in WT and SOCS3Tg T cells as assessed by RT-qPCR. Results show summary of 3 independent experiments.
(C) SOCS3 protein contents in WT and SOCS3Tg T cells. Total cell lysates from purified WT and SOCS3Tg spleen T cells were assessed by immune blot analysis for SOCS3 protein contents using anti-SOCS3 antibodies. Blots were stripped and re-probed with anti-ZAP-70 and anti–β–actin antibodies as loading controls. Numbers indicate relative SOCS3 protein expression normalized to that in WT T cells, which was set as 1.0, and results are representative of two independent experiments.
(D) Immunoblot analysis of SOCS3 protein expression. Purified splenic T cells were stimulated overnight with IL-7 (10 ng/ml) or medium alone. Next day, cell lysates were prepared and probed for SOCS3 and β-actin. Numbers indicate relative SOCS3 protein expression normalized to medium-treated T cells, which was set as 1.0, and results are representative of two independent experiments.
(E) Intracellular pSTAT3 contents in WT and SOCS3Tg CD4+ LN T cells upon stimulation with increasing amounts of IL-6 for 30 min. Histogram is representative (left, stimulation with 10 ng/mL IL-6), and graph shows summary (right) of 4 independent experiments. Data are shown as mean+/− SEM; *P <0.05, **P<0.01, ***P<0.001.
Increased abundance of SOCS3 impairs T cell development and homeostasis
The IL-6/gp130/STAT3 axis plays a critical role in Th17 cell differentiation [25], but its contribution to thymopoiesis has been considered negligible [26]. Thus, we were surprised to find that the thymus of SOCS3Tg mice was hypotrophic with significantly lower cell numbers, and with a modest but statistically significant loss in CD8 single positive (SP) thymocytes (Fig. 2A). Moreover, frequencies and numbers of peripheral LN T cells in SOCS3Tg mice were also markedly reduced (Fig. 2B), and we further found that SOCS3 overexpression substantially reduced both CD4 and CD8 T cell numbers in the spleen (Supporting Information Fig. 2A). In non-lymphoid tissues, such as the liver, however, the frequencies of conventional αβ T cells as well as invariant NKT (iNKT) cells remained largely unaffected (Supporting Information Fig. 2B). Currently, it is unclear to us whether the transgenic SOCS3 is correctly expressed in these liver-resident T cells or if the survival of liver-resident T cells would be independent of γc signaling. We will address these points in future studies. Among small intestine epithelial lymphocytes, we found that the frequency and number of CD8αβ T cells were substantially decreased, while the CD8αα T cell population, which is mostly dependent on TGF-β [27], was relatively enlarged (Supporting Information Fig. 3A, B). Thus, constitutive SOCS3 expression impairs the survival and homeostasis of CD8αβ T cells in both lymphoid and non-lymphoid tissues.
Fig. 2. Enforced SOCS3 expression impairs T cell development and homeostasis.

(A) CD4 versus CD8 profiles of total (top) and TCRβhi-gated (middle) WT and SOCS3Tg thymocytes. Numbers and frequencies of total and TCRβhi CD8SP thymocytes, respectively (bottom) are summary of 6 independent experiments with total 7 WT and 7 SOCS3Tg mice.
(B) Frequencies (top) and CD4 versus CD8 profile (middle) of WT and SOCS3Tg LN T cells, which were pooled from inguinal, axillary, and mesenteric LNs. Number and frequency of LN T cells (bottom) are summary of 5 independent experiments with total 5 WT and 5 SOCS3Tg mice.
(C) Intracellular pSTAT5 contents in WT and SOCS3Tg CD4+ LN T cells upon stimulation with increasing amounts of IL-7 for 30 min. Histogram is representative (left, stimulation with 1 ng/mL IL-7), and graph shows summary (right) of 4 independent experiments with 5 WT and 6 SOCS3Tg mice.
(D) Cell trace violet dilution of CD45.1+ congenic WT and CD45.2+ SOCS3Tg CD8 donor T cells that were mixed at 1:1 ratio and adoptively transferred into Rag2-deficient lymphopenic host mice by tail vein injection. Donor T cells were recovered 5 days after adoptive transfer. Results are representative of 3 independent experiments with a total of 5 host mice.
(E) Intracellular pSTAT6 contents in WT and SOCS3Tg CD4+ LN T cells upon stimulation with increasing amounts of IL-4 for 30 min. Histogram is representative (left, stimulation with 5 ng/mL IL-4), and graph shows (right) summary of 3 independent experiments with 4 WT and 5 SOCS3Tg mice. Data are shown as mean + SEM; *P <0.05, **P<0.01, ***P<0.001.
Defects in thymopoiesis and peripheral T cell homeostasis are more compatible with impaired IL-7 signaling than with diminished IL-6 signaling [28–30]. Consequently, we examined if the increased abundance of SOCS3 would interfere with IL-7 receptor signaling. To this end, we stimulated WT and SOCS3Tg LN T cells with increasing amounts of IL-7 and quantified intracellular pSTAT5 contents in signaled cells. SOCS3Tg T cells showed markedly diminished pSTAT5 contents compared to WT T cells (Fig. 2C and Supporting Information Fig. 4A), while the surface cytokine receptors for IL-7, i.e. IL-7Rα and γc, remained comparable between these cells (Supporting Information Fig. 4B). To further confirm that SOCS3 would suppress IL-7 signaling under in vivo conditions, we employed the lymphopenia-induced homeostatic proliferation model where T cell expansion is strictly dependent on IL-7 signaling [1, 31]. Here, we purified naïve CD8 LN T cells from SOCS3Tg and congenic WT mice, labeled them with cell proliferation indicator dyes, mixed them at 1:1 ratio, and adoptively transferred them into Rag2-deficient (Rag2–/–) lymphopenic host mice. Five days after transfer, we recovered donor T cells from spleen and LN of host mice and examined Cell Trace Violet dilution for each donor population. Notably, we found that SOCS3Tg CD8 T cells were significantly blunted in their IL-7-mediated proliferative response compared to that of WT CD8 donor T cells (Fig. 2D). Altogether, these results document a previously unappreciated role of SOCS3 in suppressing IL-7 receptor signaling, both in vivo and in vitro.
IL-7 is a member of the common γ-chain (γc) family cytokines that shares the γc for ligand binding and signaling [32]. Thus, we wondered if SOCS3 would not only impair IL-7 signaling but also inhibit signaling of other γc cytokines, such as IL-4. To assess this possibility, we stimulated LN T cells from WT and SOCS3Tg mice with increasing dosages of recombinant IL-4. STAT6 phosphorylation is exclusively induced downstream of IL-4 receptor signaling [33, 34], and we found that increased abundance of SOCS3 also blunted IL-4-induced STAT6 activation (Fig. 2E and Supporting Information Fig. 5). Altogether, these results reveal that the substrate specificity of SOCS3 is more promiscuous than previously thought, and that the targets of SOCS3-mediated inhibition encompass other members of the γc cytokine family.
SOCS3 inhibits IL-2 signaling and suppresses Foxp3+CD25+ Treg cell generation
IL-2 is the prototypic member of the γc cytokine family that signals through the hetero-trimeric complex of IL-2 receptor α (IL-2Rα, CD25), IL-2Rβ, and the γc receptor [32, 35]. High dosages of recombinant IL-2 can drive proliferation of CD44loCD62Lhi naïve CD8 T cells in vitro [36], so that we employed this assay to examine a SOCS3 effect on IL-2 signaling. Strikingly, IL-2-driven proliferation was dramatically inhibited in SOCS3Tg CD8 T cells, even as they expressed identical amounts of the IL-2 receptor compared to WT naïve CD8 T cells (Fig. 3A and Supporting Information Fig. 6). Thus, SOCS3 also inhibits IL-2 signaling.
Fig. 3. Increased SOCS3 expression impairs Foxp3+CD25+ Treg cell generation.
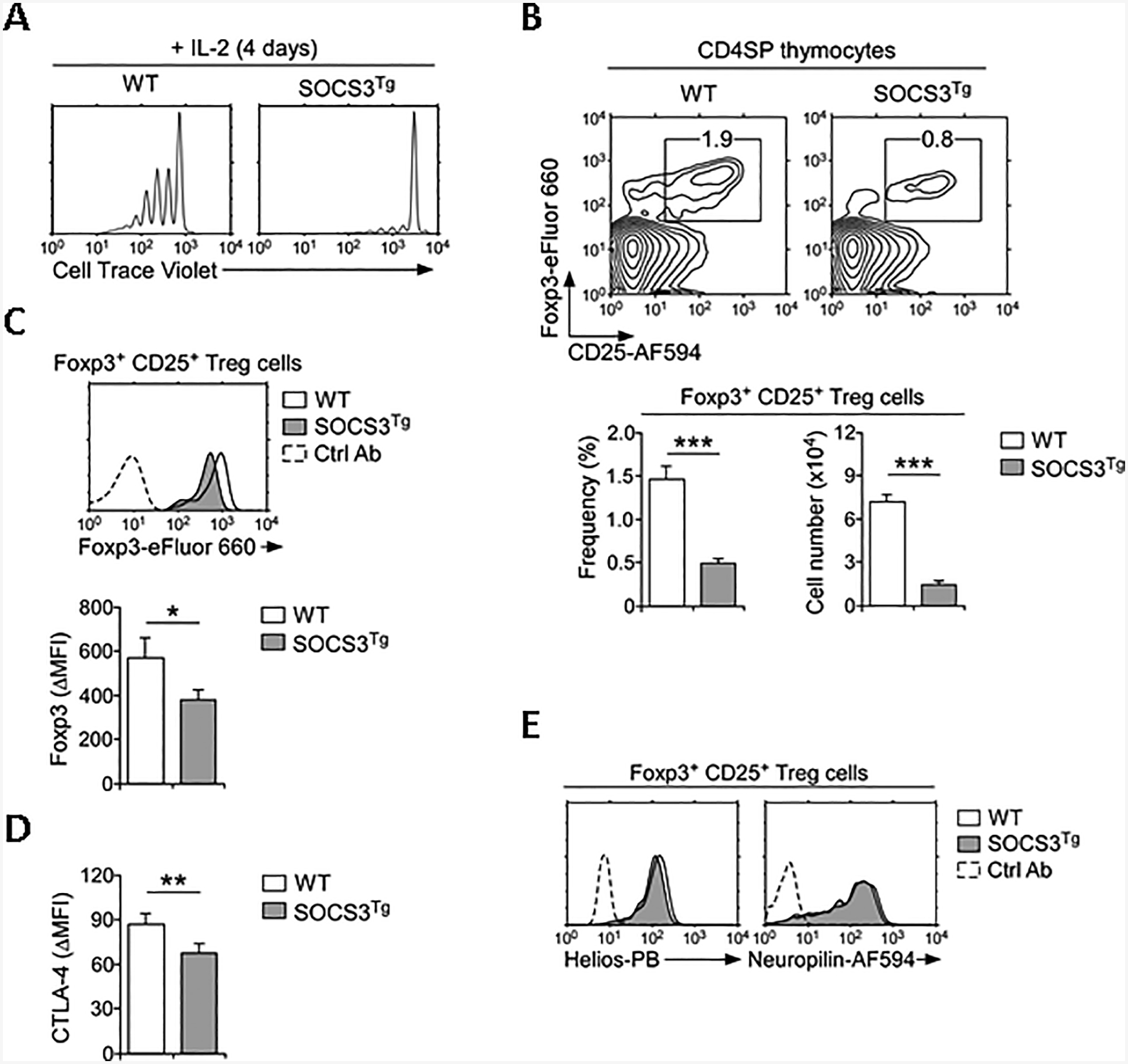
(A) Cell trace violet dilution of sorted CD44loCD62Lhi naïve CD8 T cells from WT and SOCS3Tg mice that were stimulated in vitro with recombinant IL-2 (250 ng/ml) for 4 days. Histograms are representative of 3 independent experiments.
(B) Foxp3+ CD25+ Treg cells were identified among WT and SOCS3Tg CD4SP thymocytes by intracellular staining (top). Frequency and number of thymic Treg cells (bottom) were determined from 3 independent experiments with total 6 WT and 8 SOCS3Tg mice.
(C) Foxp3 protein expression in WT and SOCS3Tg thymic Treg cells. Histogram is representative (left) and bar graph shows quantitation of Foxp3 staining (ΔMFI) of two independent experiment with total 4 WT and 4 SOCS3Tg mice.
(D) CTLA-4 protein expression in WT and SOCS3Tg thymic Treg cells. Bar graph shows quantitation of CTLA-4 staining (ΔMFI) of two independent experiments with total 4 WT and 4 SOCS3Tg mice.
(E) Helios and Neuropilin expression in Foxp3+ CD25+ Treg cells of WT and SOCS3Tg CD4SP thymocytes. Histogram is representative of 2 independent experiments with total 4 WT and 4 SOCS3Tg mice. Data are shown as mean +/− SEM; *P <0.05, **P<0.01, ***P<0.001.
IL-2-deficiency results in lethal inflammatory bowel diseases that is primarily attributed to the failure to generate functionally mature Foxp3+CD25+ Treg cells [37–39]. Because SOCS3 suppressed IL-2 signaling, we predicted that enforced SOCS3 expression would also impair the generation of Foxp3+CD25+ Treg cells which requires IL-2 signaling. Analysis of thymic Foxp3+CD25+ Treg cells in SOCS3Tg thymocytes revealed that this was indeed the case. Both frequency and number of Foxp3+CD25+ Treg cells were dramatically reduced among SOCS3Tg CD4 T cells compared to WT cells (Fig. 3B). Moreover, Foxp3 protein amounts in SOCS3Tg Treg cells was substantially diminished, suggesting that SOCS3 interferes with functional maturation of thymic Treg cells (Fig. 3C). Along these lines, we found that expression of CTLA-4, which is an essential regulator of Treg cell function [40], was diminished in SOCS3Tg Foxp3+CD25+ Treg cells (Fig. 3D). Such a decrease, however, was specific to CTLA-4, because the abundance of other thymic Treg-associated molecules such as Helios and Neuropilin remained unaffected (Fig. 3E) [41]. The disparate effects of SOSC3 on CTLA-4 versus Helios expression propose that CTLA-4 expression is potentially controlled by cytokines and can be affected by SOCS3, while Helios expression is induced by TCR signaling and would be independent of SOCS3. Collectively, these results demonstrate that SOCS3 constrains the generation of thymic Foxp3+CD25+ Treg cells and their proper acquisition of an effector phenotype.
SOCS3 impairs IL-2-mediated generation of Foxp3+CD25+ Treg cells
To directly assess if SOCS3 interferes with Foxp3+CD25+ Treg cell generation, next, we purified naïve CD4 LN T cells from WT and SOCS3Tg mice and induced their differentiation into Foxp3+CD25+ Treg cells in the presence of recombinant TGF-β and with increasing amounts of recombinant IL-2. After 4 days of in vitro culture, cells were harvested and assessed for the generation of Foxp3+CD25+ mature Treg cells (Fig. 4A). Here, we found that increased abundance of SOCS3 constrained IL-2-dependent differentiation of Foxp3+CD25+ Treg cells, resulting in diminished frequencies and cell numbers of Foxp3+CD25+ Treg cells (Fig. 4B and Supporting Information Fig. 7). Also, in agreement with the idea that SOCS3 inhibits IL-2 signaling, SOCS3Tg Foxp3+ Treg cells expressed markedly lower amounts of CD25, a classical surface marker that is induced by IL-2 [42] (Fig. 4C). SOCS3, however, did not interfere with TGF-β signaling, because TGF-β-mediated upregulation of Foxp3 did not differ between activated CD4 T cells of WT and SOCS3Tg mice (Supporting Information Fig. 8). Collectively, these results suggest that SOCS3 constrains generation Foxp3+CD25+ Treg cells in vitro, and that it interferes with Foxp3+CD25+ Treg cell generation, presumably by suppressing IL-2 signaling.
Fig. 4. SOCS3 inhibits IL-2 signaling and constrains Foxp3 Treg cell generation.

(A) Generation of Foxp3+ CD25+ Treg cells by in vitro stimulation of naïve CD4 T cells from WT and SOCS3Tg mice, with anti-TCRβ/CD28 in the presence of IL-2 (10 ng/ml) and TGF-β (5 ng/ml).
(B) Frequency and numbers of Foxp3+ CD25+ Treg cells differentiated by IL-2 (0 ~ 25 ng/ml) and TGF-β (5 ng/ml). Graph shows quantitation of 3 independent experiments with total 9 WT and 9 SOCS3Tg mice.
(C) CD25 expression on in vitro generated Foxp3+ CD4+ T cells from WT and SOCS3Tg mice. Histogram is representative of 3 independent experiments with total 9 WT and 9 SOCS3Tg mice. Data are shown as mean +/− SEM; *P <0.05, **P<0.01, ***P<0.001.
SOCS3 impairs IL-2 receptor signaling in Foxp3+CD25+ Treg cells
The idea of SOCS3-mediated suppression of IL-2 signaling was intriguing to us. Thus, we wished to directly examine if increased abundance of SOCS3 would impair IL-2 signaling in Foxp3+CD25+ Treg cells. To this end, we assessed IL-2-induced STAT5 phosphorylation in Foxp3+CD25+ Treg cells of WT and SOCS3Tg mice. We achieved this by stimulating freshly isolated CD4+ T cells from WT and SOCS3Tg mice with increasing amounts of IL-2. Intracellular pSTAT5 contents were then assessed by gating on Foxp3+CD25+ CD4+ Treg cells. Strikingly, IL-2-induced STAT5 phosphorylation was dramatically diminished in SOCS3Tg Foxp3+CD25+ Treg cells, demonstrating a direct inhibitory role of SOCS3 in IL-2 signaling (Fig. 5A).
Fig. 5. SOCS3 impairs IL-2 receptor signaling in Foxp3+ Treg cells.

(A) Intracellular pSTAT5 contents in WT and SOCS3Tg LN Treg cells that were stimulated with IL-2 stimulation (0.3 ng/mL) for 30 min. Histogram is representative (left), and graph shows summary (right) of 2 independent experiments with total 3 WT and 3 SOCS3Tg mice.
(B) Foxp3+ CD25+ Treg cells were identified among WT and SOCS3Tg CD4+ LN T cells by intracellular staining (left). Frequency and number of Treg cells (right) were determined from 2 independent experiments with total 5 WT and 5 SOCS3Tg mice. Data are shown as mean +/− SEM; *P <0.05, **P<0.01, ***P<0.001.
Survival and homeostasis of Foxp3+CD25+ Treg cells mostly depend on IL-2. However, other γc cytokines such as IL-7 also play important roles in these processes [43–46]. Because we found SOCS3 to suppress both IL-2 and IL-7 signaling (Fig. 2C and Fig. 5A), we predicted that Foxp3+CD25+ Treg cell survival would be impaired in SOCS3Tg mice because of a lack in IL-2 and/or IL-7 signaling. This was indeed the case, as both the frequency and number of peripheral Foxp3+CD25+ Treg cells in SOCS3Tg mice were significantly reduced (Fig. 5B). Collectively, these findings illustrate a previously unappreciated role of SOCS3 in suppressing γc cytokine signaling, and they document its detrimental effects on Foxp3+CD25+ Treg cell differentiation and homeostasis.
Increased immune activation and pro-inflammatory cytokine production in SOCS3Tg T cells
Foxp3+ Treg cells are instrumental in maintaining immune tolerance and quiescence [39, 44, 47]. If SOCS3 would indeed interfere with Foxp3+ Treg cell homeostasis, we expected to find aberrant immune activation and inflammation in SOCS3Tg mice. This was indeed the case, because SOCS3Tg CD4 T cells were enriched for CD44hiCD62Llo memory phenotype cells (Fig. 6A) and contained significantly increased frequencies of CD69+ activated cells (Fig. 6B). Such activated phenotype was also observed in CD8 T cells of SOCS3Tg mice (Supporting Information Fig. 9A, B). Moreover, SOCS3Tg T cells produced significantly greater amounts of pro-inflammatory cytokines, such as IL-4 and IFNγ (Fig. 6C and Supporting Information Fig. 9C), suggesting a potential autoimmune phenotype that is triggered by SOCS3 overexpression and presumably mediated by impaired Foxp3+ T cell function.
Fig. 6. SOCS3Tg CD4 T cells are enriched for activated, inflammatory phenotype cells.

(A) Increased frequencies of activated, memory phenotype CD4 T cells in SOCS3Tg mice. Contour plots are representative (left) and bar graph shows summary of 6 independent experiments with total 8 WT and 8 SOCS3Tg mice (right).
(B) CD69+ cell frequency among CD4 LN T cells in WT and SOCS3Tg mice. Dot plots are representative (left) and bar graph shows summary of 4 independent experiments with total 5 WT and 5 SOCS3Tg mice (right).
(C) Intracellular IFNγ and IL-4 contents in PMA and ionomycin stimulated CD4 LN T cells from WT and SOCS3Tg mice. Dot plots are representative (top) and bar graph shows summary of 4 independent experiments with total 7 WT and 7 SOCS3Tg mice (bottom). Data are shown as mean +/− SEM; *P <0.05, **P<0.01, ***P<0.001.
Mechanistic insights into SOCS3-mediated suppression of γc signaling
SOCS molecules can inhibit cytokine signaling by multiple pathways, such as by direct suppression of JAK kinase activities but also by ubiquitylation and proteasomal degradation of cytokine receptors and associated kinases [48]. However, inhibition of proteasomal degradation did not alter γc expression of SOCS3Tg T cells compared to WT T cells when treated with MG-132 in vitro (Supporting Information Fig. 10A, B). These results suggest that it is unlikely that SOCS3 mediates suppression by direct degradation of the γc cytokine receptor. Instead, we consider it more likely that SOCS3 would directly inhibit the enzymatic activity of receptor-associated JAKs. Both SOCS1 and SOCS3 proteins contain a short motif called Kinase Inhibitory Region (KIR) which is inserted into the catalytic cleft of JAKs to suppress their kinase activity [14]. Notably, SOCS3 is distinct from SOCS1 as it cannot directly bind JAKs but needs to anchor itself to the cytokine receptor to bind JAKs. This requirement limits the substrate specificity of SOCS3 [15]. When inhibiting gp130 cytokine receptor signaling, SOCS3 binds a phosphorylated tyrosine motif (pTyr757) on gp130 that serves also the docking site for the phosphatase SHP-2 [21, 49]. The same motif is also found around pTyr729 of the G-CSF receptor [50]. Because SOCS3 inhibited γc cytokine signaling, we examined if such a SOCS3 motif would be present in the intracellular domain of γc. The cytosolic tail of γc contains four conserved tyrosine residues [32], and mutagenesis studies demonstrated the first 52 residues of this domain being essential for JAK3 binding and signaling function [51]. When examining the γc sequence by motif alignment and protein homology analysis, however, we failed to identify a clear consensus SOCS3-binding motif (Supporting Information Fig. 11). Consequently, SOCS3 might bind to the γc receptor using an unconventional motif or might inhibit γc signaling without binding to γc and rather associating with JAK molecules. Alternatively, and not mutually exclusively, SOCS3 might also inhibit γc cytokine signaling through binding to γc cytokine proprietary receptors, e.g. IL-2Rβ, and IL-7Rα, etc. Addressing these questions will be critical in understanding the substrate specificity and role of SOCS3 in regulating T cell immunity.
Discussion
SOCS3 is a potent suppressor of cytokine receptor signaling that has been primarily associated with inhibiting the signaling of IL-6 and other members of the gp130 family cytokines [18, 52]. In contrast, here we show that SOCS3-mediated suppression is not limited to gp130 family cytokines but that SOCS3 also targets γc cytokines. In agreement, enforced SOCS3 expression in T lineage cells impacted thymopoiesis and dysregulated T cell homeostasis, two major events that require signaling by the γc cytokine IL-7. Importantly, increased abundance of SOCS3 also interfered with IL-2 signaling, which resulted in impaired Foxp3+ Treg cell generation, both in vitro and in vivo. Collectively, these results put forward SOCS3 as an inhibitor of γc cytokine signaling, and they provide new insights into the suppressive mechanism of SOCS3-mediated inhibition of JAK.
SOCS3 is thought to suppress a rather limited spectrum of cytokines, which is proposed to be imposed by a unique mechanism that SOCS3 utilizes to inhibit cytokine receptor signaling. Instead of directly binding to receptor-associated JAKs, as is the case for SOCS1 [17], SOCS3 requires binding to both JAK and the cytokine receptor at the same time [15, 23]. Thus, the cytokine specificity of SOCS3 originates from its distinct requirement to bind specific pTyr motifs on the cytoplasmic domain of the cytokine receptor before it can associate with JAKs and suppress their activation. A consensus binding motif for SOCS3 has been previously identified as pY-(S/A/V/Y/F)-hydrophobic-(V/I/L)-hydrophobic (H/V/I/Y), and this motif was found to be necessary for SOCS3 recruitment to cytokine receptor intracellular domains [53]. In agreement with its requirement for SOCS3-binding, the SOCS3-interaction motif was present in cytokine receptors whose signaling is suppressed by SOCS3, and they include gp130 as well as receptors for LIF, G-CSF and leptin [15, 23]. Cytokines of the γc family, on the other hand, have been considered being refractory to SOCS3-mediated suppression. Such notion is supported by structural studies where SOCS3 was found being unable to repress the kinase activity of JAK3, which is the JAK family kinase that specifically associates with γc [23, 32]. Along these lines, we analyzed and searched for SOCS3 motifs within the γc receptor amino acid sequence but failed to identify canonical SOCS3 binding sites. These results were obtained using web-based search engines, such as PATTINPROT (PRABI-Lyon-Gerland), and we further confirmed them by MOTIF and Motif Scan analysis (ExPASy, SIB, Swiss). Thus, we conclude that SOCS3 is unlikely to bind the cytosolic domain of γc, and, even if bound, it would not be able to suppress JAK3 activity [23].
Consequently, we were surprised to find that the increased abundance of SOCS3 proteins markedly inhibited signaling of γc cytokines, including that of IL-7, IL-4, and IL-2. As a potential resolution, we considered the possibility that SOCS3 would bind to the cytokine proprietary receptors, such as IL-7Rα, IL-4Rα or IL-2Rβ, to suppress signaling. All proprietary receptors of the γc family are associated with JAK1, but not with JAK3. Accordingly, SOCS3 could suppress cytokine signaling by interfering with JAK1 and not JAK3 activation. On the other hand, such a scenario posits that the cytokine proprietary receptors would contain pTyr motifs that can serve as docking sites for SOCS3. Analyzing the amino acid sequences of the proprietary receptors for potential SOCS3 motifs, however, did not reveal any canonical SOCS3-binding sites. In particular, we did not find canonical SOCS3 motifs in the intracellular domain of the IL-7Rα or IL-2Rβ receptor (data not shown). These data suggest that SOCS3-mediated suppression of γc cytokines could utilize mechanisms distinct from those for suppressing gp130 cytokines. Alternatively, SOCS3 could bind to unconventional pTyr motifs of IL-7Rα or IL-2Rβ cytosolic domains. In fact, human SOCS3 binding to JAK1 and JAK2 could be further enhanced by the presence of IL-2Rβ [54]. Thus, these results suggested that IL-2Rβ can serve as a scaffold for SOCS3 and JAK1/2 interaction. Notably, the same study also showed that SOCS3 transfection suppressed IL-2 signaling in BaF3 cells, which agrees with our results that identify SOCS3 as a suppressor of IL-2 signaling [54]. Nonetheless, we cannot exclude the possibility that SOCS3 inhibition of γc signaling would be independent of the Kinase Inhibitory Region (KIR) [14], and that it rather employs a mechanism used by most other SOCS family members, which is to compete with STAT molecules and block their recruitment to cytokine receptors [48].
The identification of SOCS3 as a suppressor of γc cytokines has wide-ranging implications for understanding its negative regulatory effect on cytokine receptor signaling. Among others, γc cytokines contribute to T helper cell differentiation, so that IL-2 signaling promotes Th2 cell differentiation while IL-21 signaling facilitates Th17 cell differentiation [55]. Consequently, the suppression of γc cytokine signaling would interfere with T helper lineage differentiation. Along these lines, overexpression of SOCS3 was found to impair interleukin-17 production, but it has been unclear why this would be the case [56]. Because SOCS3 inhibits γc cytokine signaling, we consider it likely that SOCS3 suppresses IL-21 signaling to constrain the differentiation of Th17 cells. Also, it was previously reported that SOCS3 is highly expressed in Th2, but not in Th1 cells [57]. Because overexpression of a myc-tagged SOCS3 transgene increased the generation of IL-4-producing Th2 cells [57], SOCS3 possibly suppresses the signaling of cytokines that inhibits Th2 cell differentiation. The precise molecular mechanism underlying this observation, however, remains unclear.
In addition to CD4 T helper cells, SOCS3 also affects Foxp3+ Treg cells. Under steady-state conditions, Treg cells do not express discernible amounts of SOCS3 proteins, and in vitro activation of Treg cells does not induce SOCS3 protein expression either [58]. However, retroviral expression of SOCS3 constrained the effector function and expansion of Treg cells, demonstrating an inhibitory effect of SOCS3 on Treg cells [59]. Thus, SOCS3 interferes with Treg cell differentiation and function, but it was unclear why this would be the case. Our current finding, that SOCS3 suppresses IL-2 signaling in Treg cells suggest that SOCS3 must be excluded from Treg cells to ensure effective IL-2 signaling. Foxp3 is pro-apoptotic and induces cell death by repressing anti-apoptotic Bcl-2 expression and promoting phosphorylation of Bim and JNK [47]. Unless counteracted by pro-survival cytokine signals, such as by IL-2, Foxp3 proteins are lethal to CD4 T cells. Thus, maximizing IL-2 signaling in Treg cells is crucial for effective Treg cell generation and maintenance. Avoiding SOCS3 expression would be important to do so, and mechanistically this might involve the transcription factor Id3. Foxp3+ Treg cells express large amounts of Id3 which is an inhibitor of E proteins and which suppresses their DNA-binding activities [60]. The E protein family transcription factor E47 upregulates expression of SOCS3, so that Id3-deficient Treg cells were found to contain large amounts of SOCS3, which in turn resulted in the loss of Foxp3 expression [61]. Our study provides additional insights into this observation as it demonstrates that SOCS3 controls the amount of Foxp3 using a cytokine-mediated circuit by suppressing IL-2 signaling.
Finally, we found that SOCS3Tg T cells were hyperactivated and expressed increased amounts of pro-inflammatory cytokines which agree with having a defect in immunosuppressive Foxp3+ Treg cells. Unlike Foxp3-deficient scurfy mice, however, SOCS3Tg mice survived long-term and did not display a disease phenotype that is usually associated with complete Foxp3+ Treg cell deficiency [38, 39, 44]. Consequently, either the few remaining Foxp3+ Treg cells in SOCS3Tg mice suffice to maintain peripheral immune tolerance or the effector functions of SOCS3Tg T cells are impaired and fail to trigger autoimmunity. It would be necessary to assess and characterize infiltration of pro-inflammatory T cells and myeloid cells in peripheral tissues to discern these possibilities, and we aim to address this question in future studies.
Altogether, here we identified SOCS3 as a broad-spectrum inhibitor of cytokine signaling, including the cytokines of the γc family. Because SOCS3 is unlikely to directly bind γc, the inhibitory mechanism employed by SOCS3 to suppress γc signaling is presumably distinct from the inhibitory mechanism employed by SOCS3 to suppress gp130 cytokine signaling. Understanding this mechanism and deciphering the precise role of SOCS3 in γc cytokine-dependent events are critical questions that remain to be addressed.
Experimental Procedures
Mice
C57BL/6 (B6) mice of both sexes were obtained from the Charles River Laboratories (Frederick, MD), and analyzed between 6–12 weeks of age. To generate SOCS3Tg transgenic mice, a cDNA construct of SOCS3 was cloned into a human CD2 enhancer-promoter-based vector and injected into fertilized B6 oocytes. Transgene expression was identified by genomic DNA PCR screening using the following primers; Forward 5’- ATGGTCACCCACAGCAAGTTTCCC-3’; Reverse: 5’-CTGCCAGCCCTCTTC CATC-3’. The PCR condition was as follows: one cycle of denaturation for 3 min at 95 °C, followed by 34 cycles of 30 sec at 95 °C, 30 sec at 60 °C, 30 sec at 72 °C, followed by a final extension cycle of 5 min at 72 °C. Animal experiments were approved by the NCI Animal Care and Use Committee. All mice were cared for in accordance with NIH guidelines.
Flow cytometry
Flow cytometry data were acquired on FACS Calibur II, LSR Fortessa or LSR II flow cytometers (BD Biosciences, San Jose, CA) and analyzed using software designed by the Division of Computer Research and Technology, NCI. Use of flow cytometry and data analyses strictly adhered to the guidelines as recently proposed [62]. Single cell suspensions for FACS-staining were prepared from the indicated organs as previously described [63]. LN cells were pooled from inguinal, axillary and mesenteric LN. Live cells were gated using forward scatter exclusion of dead cells stained with Ghost Dye™ Violet 510 (Tonbo Bioscience, San Diego, CA) or propidium iodide. Fixation and permeabilization were performed with Foxp3 Transcription Factor Staining Buffer kit according to the manufacturer’s instructions (eBioscience Thermo Fisher, Waltham, MA). Antibodies with the following specificities were used for staining: Foxp3 (FJK-16s), CD45 (30-F11), IL-2Rβ (TM-β1) were from Invitrogen Thermo Fisher (Waltham, MA); TCRβ (H57–597), CD25 (PC61.5), IL-7Rα (A7R34), CD44 (IM7) were from eBioscience; CD8α (53–6.7), CD62L (MEL-14), CD69 (H1.2F3), IL-4Rα (mIL4R-M1), IL-6Rα (D7715A7), human CD3 (Leu4), CD25 (7D4), CD132 (4G3), IgG1κ, pSTAT3 (pY705), pSTAT5 (pY694) and pSTAT6 (pY641) were from BD Biosciences; CD4 (GK 1.5), CTLA-4 (UC10–4F10–11) were from Tonbo Biosciences; Helios (22F6), CD8β (53–5.8), gp130 (4H1B35) were from Biolegend (San Diego, CA); Neuropilin was from R&D systems (Minneapolis, MN). Fluorochrome-conjugated CD1d tetramers loaded with PBS-57 and unloaded controls were obtained from the NIH tetramer facility (Emory University, Atlanta, GA). Gating strategies for both cell surface and intracellular staining are indicated in Supporting Information Fig. 12, 13, 14 and 15.
Intracellular staining for pSTAT and Foxp3
To assess cytokine-induced STAT phosphorylation, freshly isolated LN T cells from WT and SOCS3Tg mice were stimulated with recombinant cytokines for 30 min, and then fixed with 2% paraformaldehyde in PBS, followed by permeabilization with MeOH or MeOH/acetone and anti-pSTAT antibodies, as previously described [64]. To determine Foxp3 expression, cells were fixed on ice using a Foxp3 intracellular staining kit (eBioscience). Cells were washed with permeabilization buffer (eBioscience) and stained with Alexa Fluor 660-conjugated anti-Foxp3 antibodies (FJK-16s, Invitrogen) for 30 min at room temperature. Cells were washed, re-fixed using 2% paraformaldehyde on ice, and permeabilized using 90% methanol. Cells were then stained with anti-pSTAT5 (Clone 47, BD Biosciences) at room temperature for 60 min, followed by staining with anti-CD4 (GK1.5, Tonbo Biosciences) and anti-CD25 (PC61.5, eBioscience) antibodies.
Intracellular staining for cytokine production
Freshly isolated LN cells from WT or SOCS3Tg mice were stimulated with PMA (50 ng/ml) and ionomycin (1 μM) (both from Sigma) for 4 hours in the presence of brefeldin A (eBioscience). Dead cells were excluded by counterstaining with Aqua Live/Dead (Invitrogen). Surface staining was performed prior to cell fixation and permeabilization using the Foxp3 Transcription Factor Staining Buffer kit (eBioscience). Intracellular IL-4 and IFNγ contents were assessed using anti-IL-4 (clone 11B11, eBioscience) and anti-IFNγ (clone XMG1.2, BioLegend) antibodies.
IL-2-induced naive CD8 T cell proliferation
CD44lo naïve CD8+ T cells were FACS-sorted from WT and SOCS3Tg mice and labeled with 1 μM Cell Trace Violet (Life Technologies Thermo Fisher, Waltham, MA) as previously described [31]. Labeled cells were resuspended into cell culture media (5 × 106 cells/ml) with recombinant IL-2 (250 ng/ml), and incubated for 4 days at 37 °C and 7.5% CO2 before analysis.
Adoptive T cell transfer
Naïve CD8 donor T cells were FACS-sorted from lymph node cells of CD45.2+ SOCS3Tg and CD45.1+ congenic WT mice. Purified donor T cells were then loaded with Cell Trace Violet (Invitrogen) as previously described [31], and then mixed at 1:1 ratio for adoptive transfer. Total 10 × 106 cells were tail-vein injected into each Rag2–/– lymphopenic mice, and donor T cells were recovered after 5 days from spleen and lymph nodes of host mice. Cell Trace Violet dilutions were assessed by flow cytometry in WT and SOCS3Tg-origin T cells that were identified based on their congenic markers.
In vitro regulatory T cell differentiation
Naïve CD4 T cells were induced to differentiate into Foxp3+CD25+ Treg cells in vitro as previously described [64]. In brief, total splenocyte and LN T cells were FACS-sorted using surface markers for CD4, TCRβ and CD44. Enriched cells were stimulated with 1 μg/ml plate bound anti-CD3/CD28 for 4 days in the presence of 5 ng/ml TGF-β and increasing amounts (0–25 ng/ml) of recombinant IL-2 (Peprotech). After differentiation, viable cells were counted, and expression of CD25 and intracellular Foxp3 was determined by flow cytometry.
Real-time PCR
CD4 and CD8 LN T cells were isolated using rat anti-mouse CD4 or CD8 antibodies and anti-rat IgG-conjugated BioMag beads (Qiagen, Germantown, MD). Cells were cultured overnight at 37°C with or without IL-7 (10 ng/mL). Total RNA from thymocytes and purified cells was isolated by TriZol (Invitrogen Thermo Fisher) or with the RNeasy Mini kit (Qiagen). RNA was reverse transcribed into cDNA by oligo(dT) priming with the QuantiTect Reverse transcription kit (Qiagen). Quantitative RT-PCR (qRT-PCR) was performed with a QuantStudio7 Flex System and the QuantiTect SYBR Green detection system (Qiagen) or TaqMan Probe detection system (Thermo Fisher, Waltham, MA). SYBR green primer sequences are as follows. Socs1 (Forward: 5’-CCGCTCCCACTCCGATTA-3’; Reverse: 5’-GCACCAAGAAGGTGCCCA-3’), Socs3 (Forward: 5’-TTTCGCTTCGGGACTAGCTC-3’; Reverse: 5’-TTGCTGTGGGTGACCATGG-3’), Hprt (Forward: 5’-GCGATGATGAACCAGGTTATGA-3’; Reverse: 5’-ACAATGTGATGGCCTCCCAT 3’). The following TaqMan assays were used: Socs1 Rn01643811_s1, Socs3 Mm01249143_g1 and Rpl13a Mm01612986 gH (Thermo Fisher).
Western blot analysis
Cell lysates were prepared from WT or SOCS3Tg spleen T cells by isolation with anti-mouse IgG-conjugated BioMag beads (Qiagen). Purified T cells were washed in ice-cold PBS and lysed in standard lysis buffer (1% Nonidet P-40, 10 mM Tris (pH 7.5), 150 mM NaCl, 2 mM EGTA, 50 mM β-glycerophosphate, 2 mM Na3VO4, 10 mM NaF), and protease inhibitors (Roche Bioscience, Palo Alto, CA). Immunoblot analysis were performed as described [65]. Briefly, samples were run on SDS-PAGE and transferred to PVDF membranes by electro blot. The membrane was blocked with buffer containing 5% skim milk for 1 hour at room temperature. Membranes were incubated with the indicated antibodies for 4 hours followed by HRP-conjugated anti-mouse IgG. The immunoblots were developed by enhanced chemiluminescence (SuperSignal™ West Pico Chemiluminescent Substrate, Thermo Fisher Scientific, Waltham, MA). Following antibodies were used for immune blot: anti-SOCS3 (Clone 516919, R&D systems), anti-β-actin (C4, Santa Cruz Biotechnology, Dallas, TX) and anti-ZAP70 (1E7.E, Santa Cruz Biotechnology).
Motif alignment and protein homology analysis
The murine cytoplasmic domains of gp130 and the γc cytokine receptors were taken from UniProtKB database. Different sets of sequence were aligned using default options using the Align tool within the UniProKB server. Cytoplasmic sequences were manually inspected to try to identify SOCS3 motif sites around the tyrosine residues. Furthermore, the sequences were fed into web-based search engines of PATTINPROT (PRABI-Lyon-Gerland) MOTIF, and Motif Scan analysis (ExPASy, SIB, Swiss) for location of SOCS3 motif sites. Analysis of the human cytoplasmic domains of the cytokine receptors were also analyzed but did not reveal any defining motif patterns.
Statistical analysis
Statistical differences were analyzed by unpaired or paired two-tailed Student t-test. P values of less than 0.05 were considered significant. *P <0.05, **P<0.01, ***P<0.001. All statistical analyses were performed using GraphPad Prism 6.
Supplementary Material
Acknowledgments
We thank Drs. A. Singer (NCI), X. Tai (NCI), and C. Hong (Pusan National University) for their critical review of this manuscript. This work was supported by the Intramural Research Program of the US National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Conflict of Interest
The authors declare no commercial or financial conflict of interest.
REFERENCES
- 1.Kimura MY, Pobezinsky LA, Guinter TI, Thomas J, Adams A, Park JH, Tai X and Singer A, IL-7 signaling must be intermittent, not continuous, during CD8(+) T cell homeostasis to promote cell survival instead of cell death. Nat Immunol 2013. 14: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunig T, The rise and fall of the CD28 superagonist TGN1412 and its return as TAB08: a personal account. Febs j 2016. 283: 3325–3334. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL and Singer A, Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity 2004. 21: 289–302. [DOI] [PubMed] [Google Scholar]
- 4.Shuai K, Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 2006. 16: 196–202. [DOI] [PubMed] [Google Scholar]
- 5.Linossi EM, Babon JJ, Hilton DJ and Nicholson SE, Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev 2013. 24: 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D and Hilton DJ, SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999. 98: 597–608. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AW, Robb L, Rakar S, Hartley L, Cluse L, Nicola NA, Metcalf D, Hilton DJ and Alexander WS, Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci U S A 2001. 98: 9324–9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer DC and Restifo NP, Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol 2009. 30: 592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linossi EM and Nicholson SE, The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 2012. 64: 316–323. [DOI] [PubMed] [Google Scholar]
- 10.Luckey MA, Kimura MY, Waickman AT, Feigenbaum L, Singer A and Park JH, The transcription factor ThPOK suppresses Runx3 and imposes CD4(+) lineage fate by inducing the SOCS suppressors of cytokine signaling. Nat Immunol 2014. 15: 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inagaki-Ohara K, Kondo T, Ito M and Yoshimura A, SOCS, inflammation, and cancer. Jakstat 2013. 2: e24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kedzierski L, Linossi EM, Kolesnik TB, Day EB, Bird NL, Kile BT, Belz GT, Metcalf D, Nicola NA, Kedzierska K and Nicholson SE, Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog 2014. 10: e1004134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davey GM, Starr R, Cornish AL, Burghardt JT, Alexander WS, Carbone FR, Surh CD and Heath WR, SOCS-1 regulates IL-15-driven homeostatic proliferation of antigen-naive CD8 T cells, limiting their autoimmune potential. J Exp Med 2005. 202: 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN and Yoshimura A, The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J 1999. 18: 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA and Babon JJ, SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 2013. 20: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darnell JE Jr., Kerr IM and Stark GR, Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994. 264: 1415–1421. [DOI] [PubMed] [Google Scholar]
- 17.Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, Callaghan K, Nicola NA, Kershaw NJ and Babon JJ, The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun 2018. 9: 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babon JJ, Varghese LN and Nicola NA, Inhibition of IL-6 family cytokines by SOCS3. Semin Immunol 2014. 26: 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW and Alexander WS, SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 2003. 4: 540–545. [DOI] [PubMed] [Google Scholar]
- 20.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R and Murray PJ, SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol 2003. 4: 546–550. [DOI] [PubMed] [Google Scholar]
- 21.Schmitz J, Weissenbach M, Haan S, Heinrich PC and Schaper F, SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J Biol Chem 2000. 275: 12848–12856. [DOI] [PubMed] [Google Scholar]
- 22.Ushiki T, Huntington ND, Glaser SP, Kiu H, Georgiou A, Zhang JG, Metcalf D, Nicola NA, Roberts AW and Alexander WS, Rapid Inflammation in Mice Lacking Both SOCS1 and SOCS3 in Hematopoietic Cells. PLoS One 2016. 11: e0162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS and Nicola NA, Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 2012. 36: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose-John S, Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol 2018. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, Aono H, Ishihara K, Huseby E, Betz UA, Murakami M and Hirano T, IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol 2007. 19: 695–702. [DOI] [PubMed] [Google Scholar]
- 26.Hong C, Nam AS, Keller HR, Ligons DL, Park JY, Yoon HW, Park JJ, Luckey MA and Park JH, Interleukin-6 expands homeostatic space for peripheral T cells. Cytokine 2013. 64: 532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivares-Villagomez D and Van Kaer L, Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol 2018. 39: 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCaughtry TM, Etzensperger R, Alag A, Tai X, Kurtulus S, Park JH, Grinberg A, Love P, Feigenbaum L, Erman B and Singer A, Conditional deletion of cytokine receptor chains reveals that IL-7 and IL-15 specify CD8 cytotoxic lineage fate in the thymus. J Exp Med 2012. 209: 2263–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong C, Luckey MA and Park JH, Intrathymic IL-7: the where, when, and why of IL-7 signaling during T cell development. Semin Immunol 2012. 24: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrette F and Surh CD, IL-7 signaling and CD127 receptor regulation in the control of T cell homeostasis. Semin Immunol 2012. 24: 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim HK, Chung H, Kwon J, Castro E, Johns C, Hawk NV, Hwang S, Park JH and Gress RE, Differential Cytokine Utilization and Tissue Tropism Results in Distinct Repopulation Kinetics of Naive vs. Memory T Cells in Mice. Front Immunol 2019. 10: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waickman AT, Park JY and Park JH, The common gamma-chain cytokine receptor: tricks-and-treats for T cells. Cell Mol Life Sci 2016. 73: 253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M and McKnight SL, An interleukin-4-induced transcription factor: IL-4 Stat. Science 1994. 265: 1701–1706. [DOI] [PubMed] [Google Scholar]
- 34.Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM, Cleveland JL, Pierce JH, Keegan AD, Nelms K and et al. , Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol 1995. 15: 3336–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spolski R, Li P and Leonard WJ, Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol 2018. 18: 648–659. [DOI] [PubMed] [Google Scholar]
- 36.Cho JH, Kim HO, Surh CD and Sprent J, T cell receptor-dependent regulation of lipid rafts controls naive CD8+ T cell homeostasis. Immunity 2010. 32: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC and Horak I, Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993. 75: 253–261. [DOI] [PubMed] [Google Scholar]
- 38.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N and Lafaille JJ, Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med 2002. 196: 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontenot JD, Rasmussen JP, Gavin MA and Rudensky AY, A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 2005. 6: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 40.Walker LS, Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun 2013. 45: 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh K, Hjort M, Thorvaldson L and Sandler S, Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naive mice. Sci Rep 2015. 5: 7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim HP, Kelly J and Leonard WJ, The basis for IL-2-induced IL-2 receptor alpha chain gene regulation: importance of two widely separated IL-2 response elements. Immunity 2001. 15: 159–172. [DOI] [PubMed] [Google Scholar]
- 43.Fan MY, Low JS, Tanimine N, Finn KK, Priyadharshini B, Germana SK, Kaech SM and Turka LA, Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep 2018. 25: 1204–1213 e1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, Gasteiger G, Feng Y, Fontenot JD and Rudensky AY, An essential role for the IL-2 receptor in Treg cell function. Nat Immunol 2016. 17: 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim GY, Ligons DL, Hong C, Luckey MA, Keller HR, Tai X, Lucas PJ, Gress RE and Park JH, An in vivo IL-7 requirement for peripheral Foxp3+ regulatory T cell homeostasis. J Immunol 2012. 188: 5859–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bayer AL, Lee JY, de la Barrera A, Surh CD and Malek TR, A function for IL-7R for CD4+CD25+Foxp3+ T regulatory cells. J Immunol 2008. 181: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tai X, Erman B, Alag A, Mu J, Kimura M, Katz G, Guinter T, McCaughtry T, Etzensperger R, Feigenbaum L, Singer DS and Singer A, Foxp3 transcription factor is proapoptotic and lethal to developing regulatory T cells unless counterbalanced by cytokine survival signals. Immunity 2013. 38: 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshimura A, Naka T and Kubo M, SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007. 7: 454–465. [DOI] [PubMed] [Google Scholar]
- 49.Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, Metcalf D, Hilton DJ, Nicola NA and Baca M, Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A 2000. 97: 6493–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuang D, Qiu Y, Haque SJ and Dong F, Tyrosine 729 of the G-CSF receptor controls the duration of receptor signaling: involvement of SOCS3 and SOCS1. J Leukoc Biol 2005. 78: 1008–1015. [DOI] [PubMed] [Google Scholar]
- 51.Tsujino S, Di Santo JP, Takaoka A, McKernan TL, Noguchi S, Taya C, Yonekawa H, Saito T, Taniguchi T and Fujii H, Differential requirement of the cytoplasmic subregions of gamma c chain in T cell development and function. Proc Natl Acad Sci U S A 2000. 97: 1051410519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR and Yoshimura A, IL-6 induces an antiinflammatory response in the absence of SOCS3 in macrophages. Nat Immunol 2003. 4: 551–556. [DOI] [PubMed] [Google Scholar]
- 53.De Souza D, Fabri LJ, Nash A, Hilton DJ, Nicola NA and Baca M, SH2 domains from suppressor of cytokine signaling-3 and protein tyrosine phosphatase SHP-2 have similar binding specificities. Biochemistry 2002. 41: 9229–9236. [DOI] [PubMed] [Google Scholar]
- 54.Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS and Johnston JA, SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol 1999. 19: 4980–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin JX and Leonard WJ, The Common Cytokine Receptor gamma Chain Family of Cytokines. Cold Spring Harb Perspect Biol 2018. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romain M, Taleb S, Dalloz M, Ponnuswamy P, Esposito B, Perez N, Wang Y, Yoshimura A, Tedgui A and Mallat Z, Overexpression of SOCS3 in T lymphocytes leads to impaired interleukin-17 production and severe aortic aneurysm formation in mice--brief report. Arterioscler Thromb Vasc Biol 2013. 33: 581–584. [DOI] [PubMed] [Google Scholar]
- 57.Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, Komine O, Hamano S, Himeno K, Inagaki-Ohara K, Cacalano N, O’Garra A, Oshida T, Saito H, Johnston JA, Yoshimura A and Kubo M, SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med 2003. 9: 1047–1054. [DOI] [PubMed] [Google Scholar]
- 58.Pillemer BB, Xu H, Oriss TB, Qi Z and Ray A, Deficient SOCS3 expression in CD4+CD25+FoxP3+ regulatory T cells and SOCS3-mediated suppression of Treg function. Eur J Immunol 2007. 37: 2082–2089. [DOI] [PubMed] [Google Scholar]
- 59.Yu CR, Kim SH, Mahdi RM and Egwuagu CE, SOCS3 deletion in T lymphocytes suppresses development of chronic ocular inflammation via upregulation of CTLA-4 and expansion of regulatory T cells. J Immunol 2013. 191: 5036–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyazaki M, Miyazaki K, Chen S, Itoi M, Miller M, Lu LF, Varki N, Chang AN, Broide DH and Murre C, Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat Immunol 2014. 15: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rauch KS, Hils M, Lupar E, Minguet S, Sigvardsson M, Rottenberg ME, Izcue A, Schachtrup C and Schachtrup K, Id3 Maintains Foxp3 Expression in Regulatory T Cells by Controlling a Transcriptional Network of E47, Spi-B, and SOCS3. Cell Rep 2016. 17: 2827–2836. [DOI] [PubMed] [Google Scholar]
- 62.Cossarizza A, Chang HD, Radbruch A, Acs A, Adam D, Adam-Klages S, Agace WW, Aghaeepour N, Akdis M, Allez M, Almeida LN, Alvisi G, Anderson G, Andra I, Annunziato F, Anselmo A, Bacher P, Baldari CT, Bari S, Barnaba V, Barros-Martins J, Battistini L, Bauer W, Baumgart S, Baumgarth N, Baumjohann D, Baying B, Bebawy M, Becher B, Beisker W, Benes V, Beyaert R, Blanco A, Boardman DA, Bogdan C, Borger JG, Borsellino G, Boulais PE, Bradford JA, Brenner D, Brinkman RR, Brooks AES, Busch DH, Buscher M, Bushnell TP, Calzetti F, Cameron G, Cammarata I, Cao X, Cardell SL, Casola S, Cassatella MA, Cavani A, Celada A, Chatenoud L, Chattopadhyay PK, Chow S, Christakou E, Cicin-Sain L, Clerici M, Colombo FS, Cook L, Cooke A, Cooper AM, Corbett AJ, Cosma A, Cosmi L, Coulie PG, Cumano A, Cvetkovic L, Dang VD, Dang-Heine C, Davey MS, Davies D, De Biasi S, Del Zotto G, Dela Cruz GV, Delacher M, Della Bella S, Dellabona P, Deniz G, Dessing M, Di Santo JP, Diefenbach A, Dieli F, Dolf A, Dorner T, Dress RJ, Dudziak D, Dustin M, Dutertre CA, Ebner F, Eckle SBG, Edinger M, Eede P, Ehrhardt GRA, Eich M, Engel P, Engelhardt B, Erdei A, Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park JY, DiPalma DT, Kwon J, Fink J and Park JH, Quantitative Difference in PLZF Protein Expression Determines iNKT Lineage Fate and Controls Innate CD8 T Cell Generation. Cell Rep 2019. 27: 2548–2557 e2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Waickman AT, Ligons DL, Hwang S, Park JY, Lazarevic V, Sato N, Hong C and Park JH, CD4 effector T cell differentiation is controlled by IL-15 that is expressed and presented in trans. Cytokine 2017. 99: 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zvezdova E, Mikolajczak J, Garreau A, Marcellin M, Rigal L, Lee J, Choi S, Blaize G, Argenty J, Familiades J, Li L, Gonzalez de Peredo A, Burlet-Schiltz O, Love PE and Lesourne R, Themis1 enhances T cell receptor signaling during thymocyte development by promoting Vav1 activity and Grb2 stability. Sci Signal 2016. 9: ra51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.