

Abstract
Epithelial-derived high-grade serous ovarian cancer (HGSOC) is the deadliest gynecologic malignancy. Roughly 80% of patients are diagnosed with late-stage disease, which is defined by wide-spread cancer dissemination throughout the pelvic and peritoneal cavities. HGSOC dissemination is dependent on tumor cells acquiring the ability to resist anoikis (apoptosis triggered by cell detachment). Epithelial cell detachment from the underlying basement membrane or extracellular matrix leads to cellular stress, including nutrient-deprivation. In this report, we examined the contribution of fatty acid oxidation (FAO) in supporting anoikis resistance. We examined expression Carnitine Palmitoyltransferase 1A (CPT1A) in a panel of HGSOC cell lines cultured in adherent and suspension conditions. With CPT1A knockdown cells, we evaluated anoikis by caspase 3/7 activity, cleaved caspase 3 immunofluorescence, flow cytometry, and colony formation. We assessed CPT1A-dependent mitochondrial activity and tested the effect of exogenous oleic acid on anoikis and mitochondrial activity. In a patient-derived xenograft model, we administered etomoxir, an FAO inhibitor, and/or platinum-based chemotherapy. CPT1A is overexpressed in HGSOC, correlates with poor overall survival, and is upregulated in HGSOC cells cultured in suspension. CPT1A knockdown promoted anoikis and reduced viability of cells cultured in suspension. HGSOC cells in suspension culture are dependent on CPT1A for mitochondrial activity. In a patient-derived xenograft model of HGSOC, etomoxir, significantly inhibited tumor progression.
Keywords: Ovarian cancer, Carnitine Palmitoyltransferase 1A, fatty acid beta oxidation, anoikis resistance
Introduction
Epithelial ovarian cancer (EOC) is the deadliest gynecologic malignancy and accounts for over 220,000 deaths annually worldwide [1]. HGSOC is the most prevalent EOC subtype, carries the highest mortality rate of all gynecologic malignancies, and the overall survival rate at the time of diagnosis is roughly 40% (1,2). This results from most patients presenting with late stage disease (Stage III or IV) after cancer has disseminated throughout the abdomen and pelvis. The current standard of care treatment consists of a combination of surgery and platinum-based chemotherapy (2,3). Unfortunately, most patients develop recurrent, therapy resistant disease (2,4).
HGSOC does not follow classical circulatory and lymphatic dissemination modes of other solid tumors, like breast and endometrial cancers. In contrast, HGSOC originates from precursor lesions called serous intraepithelial carcinoma (STIC) at the distal fimbriated end of the fallopian tube (1,5). STIC dissociate from the extracellular matrix (ECM) and directly disseminate to distant sites within the peritoneal cavity. In normal epithelial cells, loss of attachment to the extracellular matrix (ECM) leads to anoikis (anchorage-independent cell death). Thus, HGSOC dissemination is dependent on tumor cells acquiring the ability to resist anoikis and survive long enough to attach to metastatic sites in the peritoneal cavity or survive in ascites. Metabolic characterization of HGSOC cell lines revealed that cells with a high respiration rate resisted anoikis more effectively (6). HGSOC predominantly migrates to fat-rich distant tissues, such as the omentum. Previous studies report that dissemination to the omentum is partially driven by the nutrient microenvironment (2,7), highlighting a potential shift in the metabolic demands of disseminating HGSOC cells.
Recent research identified fatty acid β-oxidation (FAO) as an essential source of metabolites (e.g., NADH, FADH2, and ATP) fueling cancer growth [reviewed in (8)]. Activation of FAO protects cancer cells against environmental stresses, such as glucose deprivation and hypoxia (9). The rate-limiting step of FAO is the membrane transport of long-chain acyl-CoA from the cytoplasm to the inner mitochondrial matrix across the inner mitochondria by the carnitine palmitoyltransferase system (CPT). The CPT system consists of three components: CPT1, CPT2, and carnitine acylcarnitine translocase (CACT). CPT1 is located on the outer mitochondrial membrane and is responsible for converting acyl-CoA into acylcarnitine. Acylcarnitine is shuttled across the inner mitochondrial membrane by CACT, where it is then converted back to acyl-CoA, ready for β-oxidation and energy substrate generation. Importantly, CPT1 is the rate-limiting step of FAO, and due to its metabolic importance, it is of interest as a possible therapeutic vulnerability. Previous work in HGSOC demonstrated that CPT1A (liver isoform) contributes to anchorage-independent survival, suggesting that pharmacologic CPT1A inhibition could be utilized to inhibit HGSOC progression.
There are several CPT1 inhibitors developed to inhibit FAO. Etomoxir is a specific, irreversible, and orally available CPT1 inhibitor that is highly effective at inhibiting FAO (10,11). Clinical data for etomoxir exists with the majority of early clinical trials examining the potential benefit in chronic heart failure patients. Most recently, a 2007 multicenter randomized control trial was stopped prematurely due to evidence of hepatotoxicity deemed unacceptable for the study population (12). Importantly, however, the reported liver enzyme elevations in this study are reasonable in the cancer setting. Preclinical in vivo models of prostate and colorectal cancers find etomoxir to have anti-neoplastic properties by reduces tumor progression and inhibiting metastasis (13,14). Until now, an in vivo model examining etomoxir in an ovarian cancer model has not been reported.
Ultimately, defining the relationship between anoikis resistance and HGSOC dissemination patterns is important and could provide a strong rationale for therapeutic interventions against HGSOC. Emerging evidence from our lab and others (7,14–16) suggests that elevated FAO is an important adaptation that allows HGSOC to survive ECM-detachment and resist anoikis. Therefore, in this report, we further define the role of FAO and CPT1A in promoting anoikis resistance and demonstrate etomoxir effectively inhibits tumor progression.
METHODS
Cell Culture.
PEO1, OVCAR3, OVCAR4, OVCAR8, OVARY1847 (an OVCAR8 derivative), and OVCAR10 human HGSOC cell lines were authenticated using small tandem repeat (STR) analysis (The University of Arizona Genetics Core) and tested for mycoplasma with MycoLookOut (Sigma, St. Louis, MO). Mycoplasma testing was last performed on the cells Aug. 5th 2019. Cells were cultured for a maximum of 8 weeks. PEO1 cells were purchased from American Type Culture Collection. OVCAR3, OVCAR4, OVCAR8, OVARY1847, and OVCAR10 cells were obtained from the Gynecologic Tumor and Fluid Bank (University of Colorado, Aurora, CO). Cells were cultured in RMPI1640 medium (Gibco Cat #11875–093, 2 mmol/L L-glutamine, 11 mmol/L D-glucose, sodium bicarbonate 2 g/L) supplemented with 1% penicillin-streptomycin (Corning Ca.#30-002-CI) and 10% heat inactivated fetal bovine serum (Access Cell Culture, Cat.# A18003-HI). Charcoal-stripped fetal bovine serum purchased from ThermoFisher (Cat. # 12676029). Cells were maintained in 5% CO2 at 37 °C. Transformed Fallopian Tube Epithelium (tFTE) (17) were obtained from the laboratory of Dr. Ronny Drapkin (University of Pennsylvania, Philadelphia, PA). tFTE cells were maintained in 1:1 DMEM-Ham’s F12 supplemented 1% penicillin-streptomycin and with 2% Ultroser Serum Substitute (CresChem).
Bioinformatic database analysis
Gene Expression Omnibus (GEO) was queried for relevant databases. GSE10971 was examined for relative CPT1A expression. KMplot and The Cancer Genome Atlas (TCGA) Ovarian Serous Cystadenocarcinoma database was accessed via the cBIOPortal (http://www.cbioportal.org/) (18,19). Wheeler et al. 2019 and GSE123290 were examined for gene expression and metabolites (16).
Suspension Culture.
Tissue culture dishes were covered with 6 mg/mL Poly-2-hydroxyethyl methacrylate (Poly-HEMA, Sigma) in 95% ethanol, as previously described (20).
Quantitative reverse transcription polymerase chain reaction (RT-qPCR).
As previously described (21). RNA isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). The Luna Universal One-step RT-qPCR kit (New England BioLabs, Ipswich, MA) was used on a BioRad CFX96 thermocycler with primers for CPT1A (Fwd- CTGAGCCATGAAGCTCTTAGAC; Rev- CCAGTGATGATGCCGTTCTT); 18S RNA was examined as an internal control (Fwd- AACTTTCGATGGTAGTCGCCG; Rev – CCTTGGATGTGGTAGCCGTTT).
Immunofluorescence.
OVCAR3 and OVCAR4 cells were grown in suspension and collected at 48 hours. Cells were fixed in 10% buffered formalin and embedded in paraffin by. The University of Colorado Cancer Center (UCCC) Histology Core. Paraffin sections were deparaffinized, as previously described (21). Mouse anti-CPT1A (1:1000, Abcam, Cat # ab128568), rabbit anti-cleaved caspase (1:1000, Cell Signaling, Cat #9664), rabbit anti- Mitochondrial fission factor (1:1000, Cell Signaling, Cat #84580) were diluted in 1% BSA in TBS, applied to all sections and incubated overnight at 4 °C. The secondary antibodies, anti-mouse Alexa fluor 594 (Thermo Fisher, Ca t#A32731) and anti-rabbit Alexa fluor 488 (Thermo Fisher, Cat #A32731), were applied to the sections and incubated for 60 min. Slides were washed with TBS and mounted with SlowFade Gold antifade reagent with DAPI (ThermoFisher, Cat #S36938). Slides were imaged with a Nikon Eclipse 2000.
Immunohistochemistry.
Paraffin sections were deparaffinized and used for immunostaining, as previously described (21). Rabbit anti-Cleaved Caspase 3 (1:1000, Cell Signaling, Cat #9664) and rabbit anti-KI67 (1:1000, ThermoFisher, Cat #MA5–14520) was diluted to 1:1000 in 1% BSA in TBS, applied to all sections, and incubated overnight at 4 °C. Slides were subsequently washed with TBS, treated with secondary anti-rabbit-HRP (Dako, Cat # K4003), and incubated at room temperature for 60 min. Slides were developed under the Olympus Inverted microscope (Model CKX41) using Liquid 3,3′-diaminobenzidine tetrahydrochloride (DAB) + Substrate Chromagen System (Agilent, Santa Clara, CA Ref#K3468).
Flow Cytometry.
Cells were then fixed with 4% paraformaldehyde for 15 min, centrifuged, and permeabilized with 0.1 % Triton X-100 in 3% Bovine serum albumin(Sigma Cat # A2153) Phosphate Buffered Saline (PBS for 10 min. Cells were washed twice with 1% BSA/PBS, then incubated in 3% BSA/PBS for 10 min. Cells were incubated with 1 μg/mL of the conjugated CPT1A-Alexa Fluor 488 (Abcam, Cat # ab171449) and Cleaved Caspase 3–700 (R&D Systems, Cat # IC835N) in 3% BSA/PBS for 60 min at room temperature. Cells were washed 3X with PBS and resuspended in 500 μl of cold 10% FBS/1% sodium azide/PBS, analyzed by a Beckman Gallios flow cytometer (UCCC Flow Cytometry Core), and FlowJo v9.
Immunoblot.
As previously described (21). Cell lysis was performed using radioimmunoprecipitation assay (RIPA) buffer (150 mmol/L sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS [sodium dodecyl sulfate], 50 mmol/L Tris, pH 8.0) supplemented with complete EDTA-free protease inhibitor cocktail (Roche) and phosphatase inhibitors sodium fluoride (10 mmol/L) and sodium orthovanadate (1 mmol/L). Proteins were transferred to PVDF membrane and immunoblotted with an antibody specific to CPT1A (Cell Signaling Cat. #12252 1:1000) and an antibody specific to β-actin (Abcam, Cat #ab6276). Secondary goat anti-rabbit IRDye 800CW (LI-COR Biosciences, Lincoln, NE, Cat # 926–32211) and goat anti-mouse IRDye 680RD (LI-COR, Cat # 926–68070). Bands were visualized using the LI-COR Odyssey Imaging System. Densitometry was calculated via Image Studio v4 (Licor).
shRNA Knockdown.
Lentiviral packaged as previously described (21). CPT1A-specific shRNAs were obtained from the UC Functional Genomics Facility (CPT1A #1: TRCN0000036279, CPT1A #2: TRCN0000036282, and CPT1A #3 TRCN0000036283) and packaged as previously described (20,21). An empty pLKO.1-puro was utilized as shControl (shCtrl).
Caspase-Glo Assay.
As previously described (22), OVCAR3, OVCAR4, and OVCAR8 cells grown in adherent and suspension conditions were used for a Caspase-Glo 3/7 assay (Promega). Luminescence was measured on a Promega GloMax Multi-detection System.
Clonogenic Assay.
HGSOC cells were cultured for 48 h under adherent or forced-suspension conditions (20,000/well) in 10% FBS/RPMI1640 media. After 48 h, trypsinized adherent cells or suspension cells were plated in a new 24-well plate under adherent conditions. Cells were allowed to attach and grow for 5 days, fixed with 10% acetic acid/10% methanol in PBS, and stained with crystal violet as in (21). Integrated density was calculated with ImageJ (NIH).
Gynecologic Tissue and Fluid Bank (GTFB).
The University of Colorado has an Institutional Review Board approved protocol (COMIRB #07–935) in place to collect tissue from gynecologic patients with both malignant and benign disease processes. All participants are counseled regarding the potential uses of their tissue and sign a consent form approved by the Colorado Multiple Institutional Review Board. The tissues were processed, aliquoted, and stored at −80 °C.
In Vivo Model.
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the University of Colorado IACUC. A developed patient-derived xenograft (PDX) model, PDX-GTFB1016, was utilized to evaluate HGSOC treatment with etomoxir in vivo. PDX-GTFB1016 is derived from a primary tumor collected from a patient diagnosed with Stage IIIC HGSOC. GTFB1016 patient was described to have large volume ascites, peritoneal carcinomatosis, and was chemonaïve at the time of tissue collection. Somatic TP53 and BRCA2 mutations were identified via Novogene PARP CDxi genetic testing. PDX-GTFB1016 cells were implanted by intraperitoneal injection and given 3 weeks to establish tumor before initiation of treatment. Tumor cells were tagged with a GFP/luciferase to facilitate tumor tracking via In Vivo Imaging (IVIS, Perkin Elmer) (21,23). Tumor-bearing mice were randomized based on total flux and divided into four treatment groups: vehicle control (phosphate-buffered saline), cisplatin (0.75 mg/kg, every 3 days via IP injection (24)), etomoxir (13 mg/kg, daily via IP injection) or cisplatin/etomoxir combination for 21 days. Bodyweight was recorded during treatment as a surrogate for toxicity. Each mouse was subjected to necropsy after euthanasia with a CO2 chamber and cervical dislocation. Total tumor weight was measured, and ascites-associated tumor cells were collected.
Seahorse Assay.
Cells were grown in adherent and suspension settings for 24 h. Media was then aspirated, cells washed with PBS, then re-plated with serum-free RPMI for 24 h of forced nutrient deprivation. Media was then aspirated, cells washed with PBS, trypsinized for 5 minutes, and neutralized with serum-free RPMI1640 medium. The cells were pelleted and resuspended in 90 μL/well of Agilent Seahorse XF RPMI medium pH 7.4 (Cat. # 103576–100) and plated on an Agilent Seahorse XF cell culture plate at 4.5×104 cells/per well, with or without etomoxir [40 μmol/L], 8 hours prior to performing the assay. Immediately prior to plating, half of the Agilent Seahorse XF96 cell culture plate (48-wells) was coated with Corning™ Cell-Tak (Cat.# 354240) to allow a monolayer of fixed suspension cells. Cell-Tak plate coating was performed per Agilent recommended protocol. Each well was treated with a [22.4 μg/mL] of Cell-Tak solution for 30 minutes, rinsed with sterile water, and allowed to dry prior to cell plating.
One hour prior to performing the assay, an additional 90 μL/well of Agilent Seahorse XF RPMI medium pH 7.4 enriched with 500 μmol/L carnitine, 50 μmol/L bovine serum albumin, and increasing concentrations of 5, 25, and 50 μmol/L of BSA conjugated oleic acid (Sigma Cat # O1383), with or without 40 μmol/L etomoxir. The Agilent Seahorse XF96 Extracellular Flux Analyzer (Seahorse Biosciences, Billerica, MA) was used to obtain measurements of oxygen consumption rate (OCR). The mitochondrial stress test was performed with the final concentrations of inhibitors per well- 2 μmol/L of oligomycin, 2.5 μmol/L of carbonyl cyanide-p trifluoromethoxyphenylhydrazone (FCCP) and 5 μmol/L of antimycin A with 125 nmol/L Rotenone (25) (Sup. Fig. 3E–G).
Intracellular lipid accumulation.
Intracellular lipid accumulation was measured using the cell-permeable lipophilic dye bodipy (Life Technologies, Cat # D3922). Cells were grown both in adherent and suspension with or without 25 μmol/L oleic acid and fixed with 4% paraformaldehyde/PBS for 10 min. Stained with bodipy (1:500 in PBS/) for 30 min, washed with PBS, and resuspended in 1% BSA/PBS. Cells were examined by a Beckman Gallios flow cytometer and analyzed in FlowJo v9.
Statistical Analysis.
Experiments were repeated in triplicate in three independent experiments unless otherwise stated. GraphPad Prism (v8) software was utilized to generate graphs and perform statistical analysis. Quantitative data are expressed as mean ± S.E.M. unless otherwise stated. Analysis of variance (ANOVA) with Fisher’s Least Significant Difference (LSD) was used to identify significances in multiple comparisons. For all other statistical tests, a two-sided unpaired t-test was used unless otherwise noted. The level of significance was set at p < 0.05 for all statistical analyses.
Results
FAO enzymes are enriched in anoikis-resistant high grade serous ovarian cancer cells.
High grade serous ovarian cancer (HGSOC) is known to originate at the distal end of the fallopian tube epithelium, exfoliate from the basement membrane and disseminate throughout the peritoneal cavity in ascites fluid (Figure 1A). Utilizing data from a recently published study (16) we assessed global untargeted metabolomics and RNA-sequencing of HGSOC cells in a dissemination-like environment, forced-suspension. Upon examination of genes and metabolites, we observed enrichment of the Carnitine/Fatty Acid (FA) metabolism pathway (Figure S1A). Metabolomic analysis revealed 109 out of 167 metabolites were differentially enriched in suspension cells in at least one of the two HGSOC cell lines evaluated. Notably, 23% of metabolites were categorized as FA species, and there was a significant increase in acylcarnitine species in both PEO1 and OVARY1847 cells (Figure S1B–C). The RNA-sequencing analysis found 805 differentially regulated genes in HGSOC cells cultured in suspension, with 12.5% of genes associated with FA metabolism. Four (CPT1A, ACAD10, ECI1, and ACADS) of the most differentially expressed genes involved in FAO that were significantly upregulated in suspension cells (Figure 1B) were evaluated in publicly available datasets of normal fallopian tube epithelium or borderline tumors compared to HGSOC tumors. CPT1A was the only gene found to be increased in tumors versus normal fallopian tube epithelium or borderline tissues, suggesting that CPT1A upregulation contributes to disease progression (Figure 1C and Figure S1D). Of the three CPT1 enzymes (CPT1A/B/C), CPT1A has the highest expression in HGSOC tumors (Figure S1E). We also examined the relationship between CPT1A expression and overall survival of patients with HGSOC. Utilizing gene expression from multiple datasets, elevated CPT1A expression in pre-chemotherapy treated patient tumors significantly (Log-rank, p=0.0052) conveyed a worse overall survival compared to tumors with low CPT1A, 50.4 vs. 41.6 months (Figure 1D). CPT1A expression increased in HGSOC, and it correlates to a poor prognosis.
Figure 1. CPT1A expression conveys a poor prognosis in HGSOC and is upregulated in forced suspension culture.
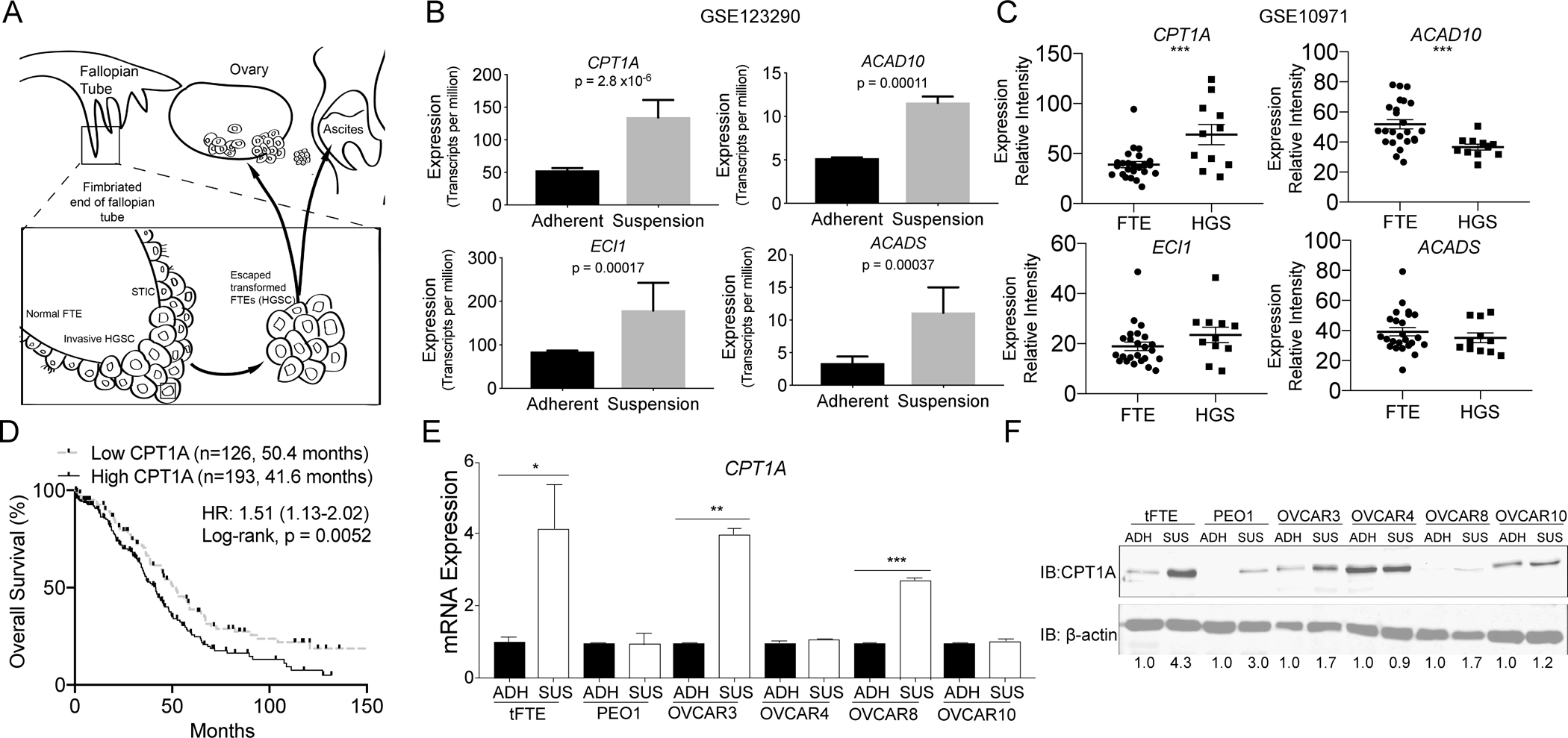
A) HGSOC arises from transformed fallopian tube epithelial cells (tFTE). Cells in serous tubal intraepithelial carcinoma (STIC) lesions within the fallopian tube exfoliate, resist anoikis, and disseminate to the peritoneal cavity. B) Transcriptomic analysis of HGSOC cells cultured in adherent and suspension conditions found increased expression of FA metabolism genes CPT1A, ACAD10, ECI1, and ACADS in suspension cells (GEO: GSE123290). C) Evaluating expression of CPT1A, ACAD10, ECI1, and ACADS in FTE (n=24) compared to high grade serous (HGS) carcinoma (n=11) (GEO: GSE10971) D) Kaplan-Meier curve of pre-treated patients with HGSOC that have high or low CPT1A mRNA expression. High and low expression set by the median. KMplot (51). E) Culture of tFTE and five HGSOC cell lines in adherent and suspension for 48 h. The expression of CPT1A was evaluated via RT-qPCR. Internal control = 18S. F) Same as E, but examined CPT1A protein expression via immunoblot. Loading control = β-actin. Densitometric analysis of CPT1A expression performed with ImageJ. Error bars = S.E.M. Statistical analysis = two-sided t-test. *p<0.05, **p<0.01, ***p<0.001.
CPT1A expression is upregulated in high grade serous ovarian cancer cells upon culture in suspension.
Previous work demonstrated the metabolic reprogramming of HGSOC cells when forced to survive in an anchorage-independent (suspension) condition (16). The data suggested an increase in FAO through elevated CPT1A activity. Therefore, we examined CPT1A mRNA and protein expression in transformed fallopian tube epithelium (FTE) and five HGSOC cell lines – PEO1, OVCAR3, OVCAR4, OVCAR8, and OVCAR10 cells – on adherent tissue culture dishes or in poly-HEMA (suspension) treated tissue culture dishes. Following forced suspension culture for 48 h, CPT1A mRNA expression was elevated in three of six cell lines (Figure 1E). However, forced suspension culture resulted in the upregulation of CPT1A protein expression in four of six cell lines (Figure 1F). In OVCAR10 cells, CPT1A upregulation was modest (up 1.2 fold) compared to the other cell lines. In OVCAR4 cells, CPT1A expression was high in comparison and remained unchanged following forced-suspension cultures. However, upon examination of the protein isolated from OVCAR4 cells, we observed high basal levels of CPT1A protein expression compared to the other cell lines independent of culture conditions. These data highlight that CPT1A is elevated in HGSOC cells cultured in forced-suspension, suggesting a role in anoikis resistance.
Relationship between CPT1A upregulation and anoikis
To further identify if HGSOC cells with elevated CPT1A expression demonstrated increased resistance to anoikis, we evaluated OVCAR3 and OVCAR4 cells grown in suspension for 48 h using immunofluorescence (IF, Figure 2A–B). Cells were co-stained for cleaved caspase 3 (CC3) and CPT1A. Consistent with CPT1A known localization, we observed a cytosolic accumulation of CPT1A consistent with mitochondrial localization, which was confirmed with a mitochondrial marker, Mitochondrial Fission Factor (MFF) (Figure S2A). We confirmed a low background IF signal (Figure S2B). In OVCAR3 cells, we observed variable expression of CPT1A, but visually observed cells with low CPT1A expression were CC3-positive (Figure 2A, arrow). In OVCAR4 cells, which have a high basal CPT1A expression, CPT1A expression was uniform across cells (Figure 2B). Using flow cytometry, we measured the CPT1A/CC3 mutual exclusivity on OVCAR3 and OVCAR4 cells grown in suspension for 48 h. The sensitivity of the flow cytometry assay is superior to IF. The CPT1A antibody clone was knockout validated (Abcam), and we further confirmed the specificity of the CPT1A antibody was using CPT1A knockdown cells (Figure S2C). In OVCAR3 and OVCAR4 cells after 48 h in suspension culture, 40% and 80% of cells were positive for CPT1A expression, and 6% and 2% were CC3 positive, respectively (Figure 2C–D). However, in OVCAR3 and OVCAR4 cells, 0.7% and 0.3% were double CPT1A/CC3 positive, respectively. In OVCAR3, OVCAR4, and OVCAR8 cells, we assessed cell viability of cells following being cultured in suspension for 48 h with a crystal violet clonogenic assays. The loss of viability was consistent with CPT1A expression (Figure 1F, 2E–F). These data suggest that elevated CPT1A is correlated to the attenuation of suspension-induced apoptosis.
Figure 2. CPT1A upregulation in suspension cells protects against anoikis.
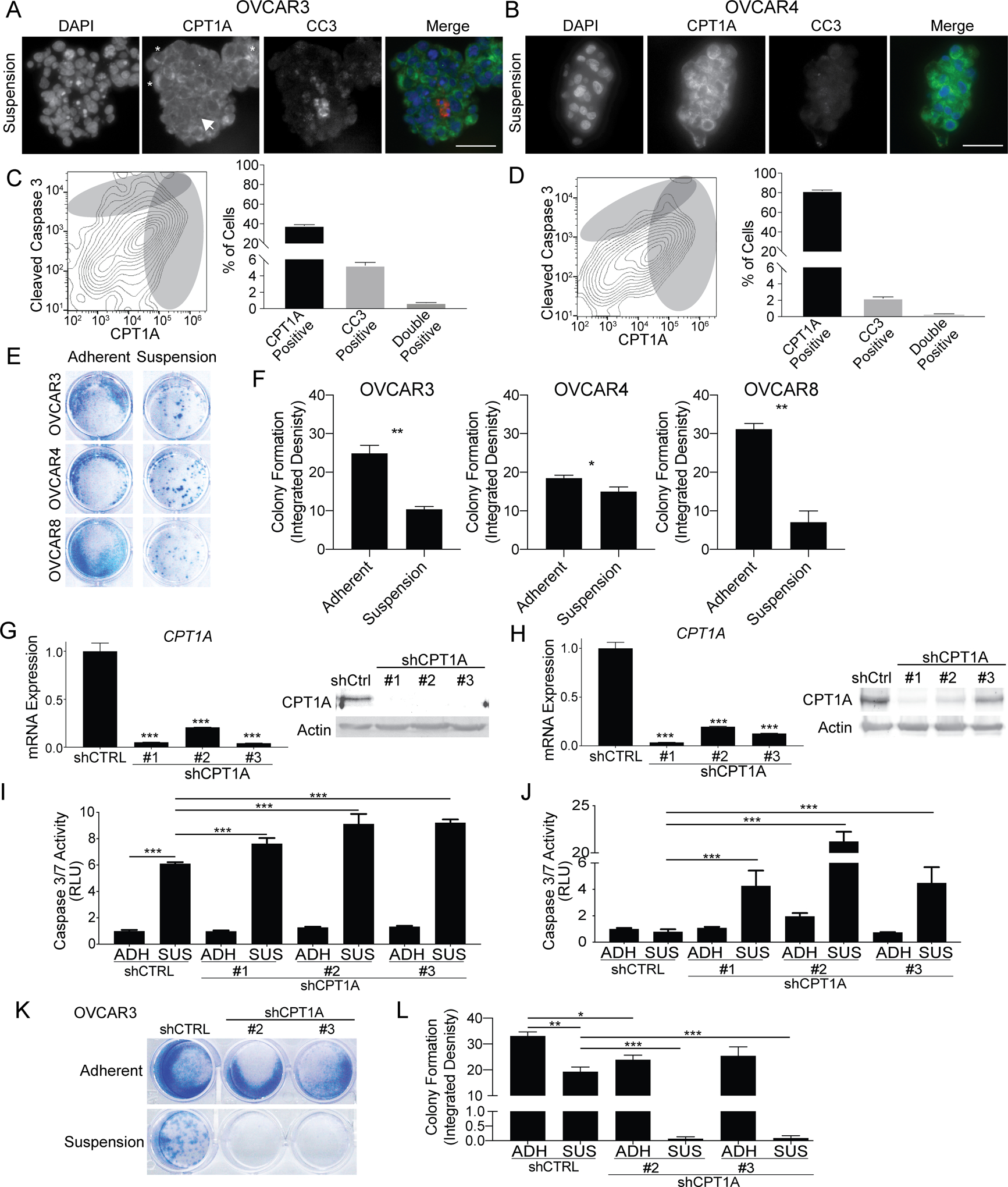
A) OVCAR3 cells cultured in suspension for 48 h were embedded in histogel and utilized for immunofluorescence against CPT1A (green) and cleaved caspase 3 (CC3, red). White arrow = low CPT1A and high CC3 Asterisk = high CPT1A cell. Scale bar = 50 microns. B) Same as A, but with OVCAR4 cells. C) OVCAR3 cells cultured in suspension for 48 h fixed and stained for CPT1A-488 and CC3–700. Cells analyzed via flow cytometry. The frequency of CPT1A and CC3 positive cells (gray circles) were calculated. D) Same as C, but with OVCAR4 cells. E) OVCAR3, OVCAR4, and OVCAR8 cells cultured in suspension for 48 h and were plated in adherent condition. After 24 h, cells were fixed and used for a crystal violet colony formation assay. F) The integrated density of crystal violet measured. G) OVCAR3 cells were transduced with shControl (shCtrl) and three independent shCPT1A (#1–3) constructs. CPT1A knockdown confirmed via RT-qPCR and immunoblot. H) Same as E, but with OVCAR4 cells. I) OVCAR3 shCtrl and shCPT1A cells were cultured in adherent (ADH) or suspension (SUS) for 48 h. Caspase 3/7 activities were measured via CaspaseGlo assay. J) Same as G, but with OVCAR4 cells. K) OVCAR3 shCtrl and shCPT1A cultured in adherent (ADH) or suspension (SUS) for 48 h, plated into adherent conditions, and incubated for 5 days. Cells were stained with crystal violet. L) Same as K, integrated density measured (ImageJ). Error bars = S.E.M. Statistical analysis = ANOVA. *p<0.05, **p<0.01, ***p<0.001.
To elucidate the direct role of CPT1A-mediated HGSOC anoikis resistance, we evaluated the impact of CPT1A knockdown in HGSOC cell lines grown in both adherent and suspension conditions. Three independent small hairpin RNAs (shRNA) specific for CPT1A or a control (shControl) were transduced into OVCAR3 or OVCAR4 cells. We observed a 70–90% knockdown of CPT1A and confirmed CPT1A protein knockdown (Figure 2G–H). OVCAR3 (CPT1A-low) or OVCAR4 (CPT1A-high) shControl and shCPT1A cells were cultured for 48 h in adherent or forced suspension. As a readout of apoptosis, caspase 3/7 activities were measured using a luminescence-based assay, which gives a broader indication of caspase activity compared to CC3 flow cytometry. In CPT1A-low OVCAR3 shControl cells, forced suspension resulted in a significant increase in apoptosis compared to adherent cells (Figure 2I). Moreover, shCPT1A OVCAR3 cells cultured in suspension, apoptosis was further induced compared to suspension culture shControl cells. In contrast, in CPT1A-high OVCAR4 shControl cells, forced suspension failed to induce caspase 3/7 activity and measurable apoptosis, but caspase 3/7 activity was significantly elevated in shCPT1A OVCAR4 cells cultured in suspension (Figure 2J). In OVCAR3 and OVCAR4 cells, CPT1A knockdown did not significantly increase caspase 3/7 activities in adherent conditions. Subsequently, cell viability was assessed in OVCAR3 shControl and shCPT1A cells following 48 h in forced suspension culture. Cell viability was significantly reduced in OVCAR3 shCPT1A compared to shControl cells (Figure 2K–L). These data highlight that knockdown of CPT1A potentiated apoptosis and reduced cell viability of cells cultured in suspension.
Metabolic reprogramming of HGSOC related to anoikis resistance
Based on previously published RNA-seq and metabolomics data (16), we hypothesized that elevated CPT1A expression promotes FAO to generate ATP as HGSOC cells are disseminating and resisting anoikis. To further evaluate suspension-induced metabolic reprogramming and the role of FAO, we evaluated lipid accumulation via a fluorescently tagged lipid stain, bodipy of OVCAR3, and OVCAR8 cells cultured in adherent and suspension conditions. Forcing cells into suspension culture significantly reduced bodipy fluorescence signal (Figure 3A–B and Figure S3A–B), suggesting that cells in suspension utilize lipid stores. Consistently, Oil Red O accumulation was also diminished in OVCAR3 cells cultured in forced suspension (Figure S3C–D). During dissemination, HGSOC cells have a predilection for colonizing the fat-rich omentum; thus, we wanted to modulate the microenvironment to recapitulate the omental niche. Previous metabolic analysis of the FA composition omentum identified the monounsaturated FA (MUFA) account for 33.5% of omental-associated adipocyte FA species (26). More specifically, oleic acid (18:1) accounts for 27.2%. We, therefore, wanted to determine if the addition of increasing concentrations of exogenous oleic acid could rescue suspension-induced lipid depletion. Exogenous oleic acid prevented lipid depletion of OVCAR3 and OVCAR8 cells cultured in suspension (Figure 3A–B and Figure S3A–B). Possible FA utilization is through CPT1A and FAO; thus, we next wanted to assess mitochondrial function.
Figure 3. HGSOC cells in forced-suspension demonstrate a dependency of FA metabolism to promote anoikis resistance.
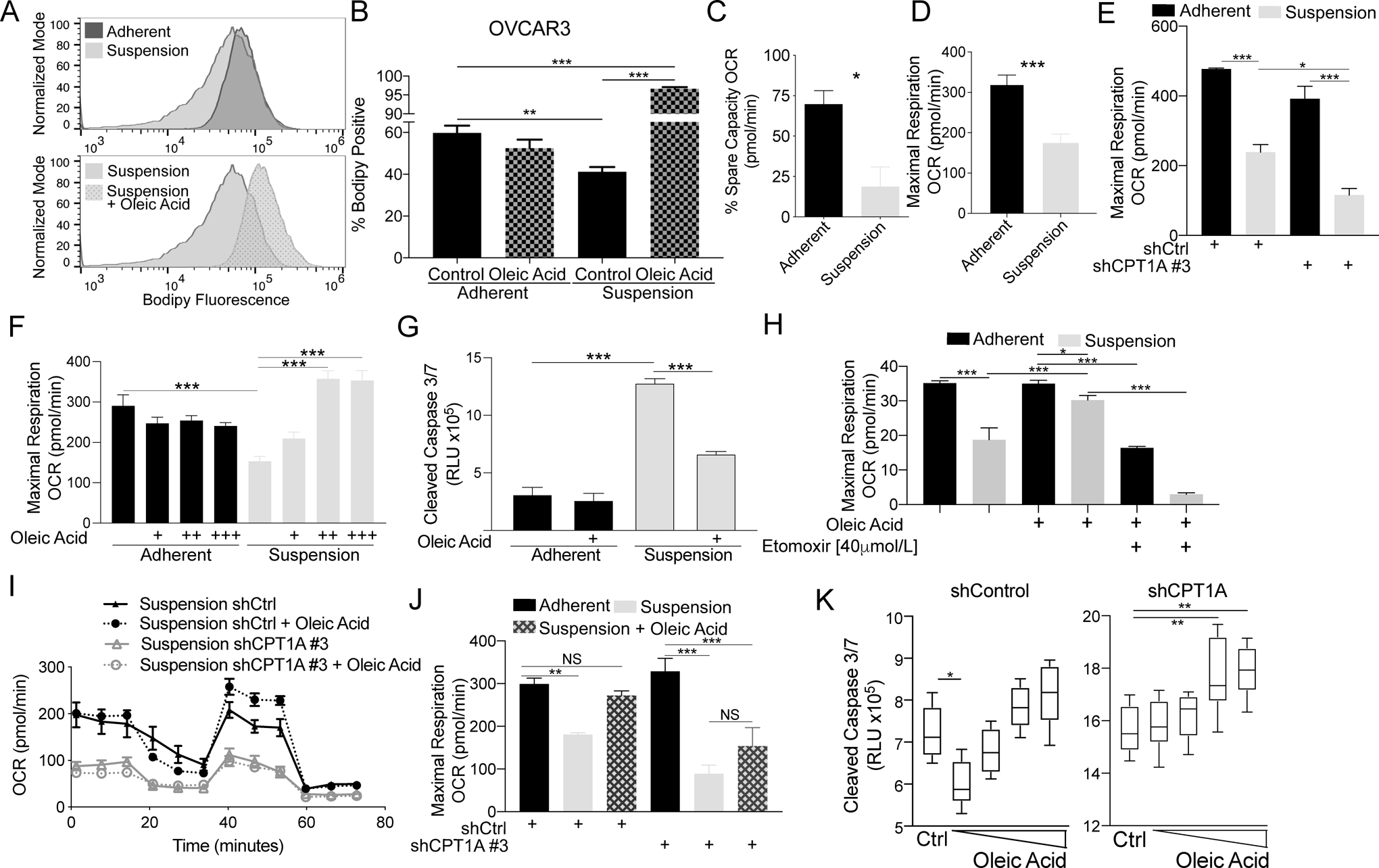
A) OVCAR3 cells cultured with or without BSA-conjugated oleic acid in adherent or suspension for 48 h were incubated with fluorescent bodipy, and cells were analyzed via flow cytometry. B) Quantification of bodipy positive cells. C) OVCAR3 cells cultured in adherent and suspension for 48 h were utilized for mitochondrial stress test analysis with an Agilent Seahorse XF96 Analyzer. Percentage of spare capacity oxygen consumption rate (OCR) calculated with the Wave program. D) Same as C, but examined maximal respiration. E) OVCAR3 shCtrl and shCPT1A #3 cells were cultured in adherent and suspension for 48 h and utilized for mitochondrial stress test analysis with an Agilent Seahorse XF96 Analyzer. Maximal respiration is graphed. F) OVCAR3 cells cultured in adherent and suspension for 48 h with increasing doses of oleic acid (+ 5, ++ 25, and +++ 50 μmol/L). Maximal respiration was measured. G) OVCAR3 cells were cultured in adherent or suspension for 48 h with oleic acid. Caspase 3/7 activities were measured via CaspaseGlo. H) OVCAR3 cells cultured in adherent and suspension for 48 h. The mitochondrial stress test analysis was performed with oleic acid and/or etomoxir. Maximal respiration was measured. I) OCR measured in OVCAR3 shControl (shCtrl) and shCPT1A with oleic acid (25 μmol/L). J) Maximal respiration of OVCAR3 shControl and shCPT1A cultured in adherent and suspension-cultured supplemented with or without oleic acid. K) Caspase 3/7 activities measured in OVCAR3 shControl (shCtrl) and shCPT1A with increasing concentrations of oleic acid (5 – 100 μmol/L). Error bars = S.E.M. Statistical analysis = ANOVA. *p<0.05, **p<0.01, ***p<0.001.
To directly evaluate the mitochondrial activity, the Seahorse Extracellular Flux Analyzer was used to evaluate the oxygen consumption rate (OCR) through the mitochondrial stress test assay. Both the percentage spare capacity and maximal respiration were significantly reduced in OVCAR3 cells cultured in suspension for 48 h (Figure 3C–D and Figure S3E). Loss of CPT1A significantly exacerbated the reduced maximal respiration compared to shControl cultured in suspension (Figure 3E), suggesting that although respiration is reduced in suspension conditions, CPT1A contributes to maintaining some degree of respiration. The addition of oleic acid significantly rescued maximal respiration in a dose-dependent manner (Figure 3F). Exogenous oleic acid rescued lipid depletion and respiration rate, so next assessed apoptosis. Exogenous oleic acid significantly reduced caspase 3/7 activity in suspension-cultured OVCAR3 and OVCAR8 cells (Figure 3G and Figure S3F). These data demonstrate HGSOC cells in suspension partially maintain mitochondrial respiration through CPT1A, utilize oleic acid for respiration and to resist apoptosis.
Through pharmacologic and genetic approaches, we assessed whether CPT1A is required for the oleic acid-dependent rescue of anoikis resistance and maximal respiration. Etomoxir is an irreversible pharmacologic inhibitor of CPT1A (27). In suspension cells, etomoxir abrogated the oleic acid-dependent rescue of maximal respiration (Figure 3H). Also, etomoxir further reduced viability in a dose-dependent fashion in cells cultured in forced-suspension (Figure S3I). In shCPT1A cells, we examined whether oleic acid could rescue the maximal respiration. Consistent with etomoxir treatment, the addition of exogenous oleic acid failed to rescue maximal respiration OCR in shCPT1A cells, highlighting the dependency of CPT1A to utilize MUFA (Figure 3I–J). In shControl and shCPT1A cells cultured in charcoal-stripped media in suspension, we added increasing concentrations of exogenous oleic acid and evaluated caspase 3/7 activity. In shControl cells, we observed that lower concentrations of oleic acid significantly reduced caspase 3/7 activities (Figure 3K); however, consistent with previous reports, higher concentrations of free fatty acid promoted apoptosis (28,29). In contrast, oleic acid at any concentration failed to decrease caspase 3/7 activities in shCPT1A cells cultured in suspension (Figure 3K). Taken together, in suspension-cultured HGSOC cells, CPT1A utilizes MUFAs to maintain mitochondrial respiration and to promote anoikis resistance.
Pharmacological inhibition of CPT1A reduced tumor growth rate, ascites production, and altered dissemination patterns.
Prior translational studies found the CPT1A pharmacologic inhibitor, etomoxir, attenuates tumor growth in colorectal and prostate cancer animal models (13,14). We observed a significant increase in CPT1A expression between adherent and suspension HGSOC cells in vitro, which suggests a potential therapeutic opportunity. Platinum-based chemotherapy (i.e., carboplatin/cisplatin) in combination with paclitaxel is standard-of-care for HGSOC; therefore, we also wanted to assess whether targeting CPT1A was comparable to cisplatin.
To determine the effects of CPT1A inhibition in vivo, we established an HGSOC patient-derived xenograft (PDX) model from sample GTFB 1016 (PDX-GTFB1016). This patient was diagnosed with stage IIIC HGSOC with associated large volume ascites, somatic TP53, and BRCA2 mutations, and was chemonaïve. GTFB1016 tumor cells were isolated from patient ascites, and tumors were propagated through NOD scid gamma (NSG) mice. Passaged PDX-GTFB1016 cells were tagged with GFP/luciferase (21). PDX-GTFB1016 progresses similarly to clinical manifestations of HGSOC evidenced by both solid tumors and ascites, making it a suitable model to examine CPT1A inhibition (Figure S4B).
We inoculated luciferase-expressing PDX-GTFB1016 cells into NSG mice via intraperitoneal (IP) injection and allowed 3 weeks for tumor establishment. Mice were randomized into four treatment groups based on luminescence intensity and were treated with vehicle control (n=7), cisplatin-only (n=7), etomoxir-only (n=7), or cisplatin/etomoxir combination (n=7) (Figure S4A). Representative images of control and treated mice on Day 0 and Day 21 are shown in Figure 4A. Measured by the percent change in total flux (photons/second), we observed all treatments resulted in a significant reduction in tumor growth at the end of the study compared to control (Figure 4B). Notably, we observed a significant reduction in tumor growth in the etomoxir-only treatment growth, although to a lesser extent than the cisplatin-treated groups. No significant change was observed between the cisplatin-only and cisplatin/etomoxir combination group.
Figure 4. Inhibition of CPT1A inhibits tumor progression.
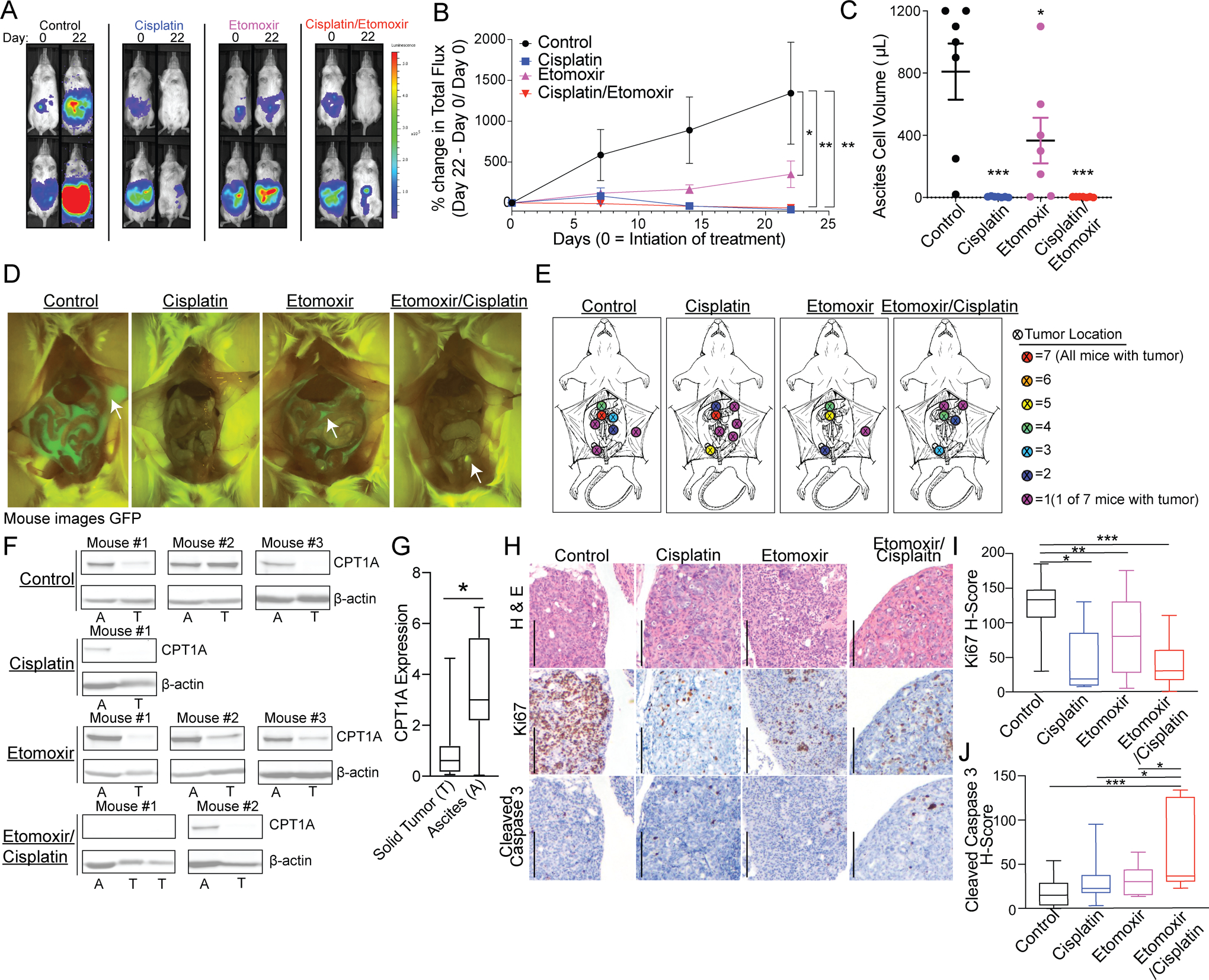
A) PDX-GTFB1016 cells tagged with GFP/luciferase were implanted in NSG mice. Tumor-bearing mice were treated for 21 days with vehicle control (n=7), cisplatin (n=7), etomoxir (n=7), or cisplatin/etomoxir combination (n=7). Representative luminescence images of mice at day 0 of treatment and after cessation of treatment, day 22. B) Percent change in total flux over the course of treatment. C) The volume of ascites-associated tumor cells on day 22. D) Representative images of GFP signal on day 22. E) Compilation of tumor dissemination in all of the groups. Color represents the absolute number of tumors observed at the indicated location. F) Expression of CPT1A in ascites-associated tumor cells (A) versus matched solid tumors (T). G) Same as F, densitometric analysis of CPT1A expression. H) Immunohistochemistry of H&E, Ki67, and cleaved caspase 3. Scale bar = 200 microns. I) Histological score of Ki67 expression blindly calculated based on the percentage and intensity of the positive stain. J) Histological score of cleaved caspase 3 expression blindly calculated based on the percentage and intensity of the positive stain. Error bars = S.E.M. Statistical analysis = ANOVA. *p<0.05, **p<0.01, ***p<0.001.
With respect to toxicity, the cisplatin only treated mice had a significant decrease in weight over the course of treatment (Figure S4B). In contrast, etomoxir-only and combination-treated mice did not have a significant weight loss. These data highlight that while cisplatin was superior at reducing tumor burden, etomoxir managed to reduce to tumor burden without inducing weight loss. Furthermore, while not significant (p = 0.131), combining etomoxir with cisplatin partially rescued the observed weight loss (Figure S4B). Future studies will examine if the combination would allow a dose reduction in cisplatin, while still giving an equivalent reduction in tumor burden.
At the time of necropsy, visible GFP-positive ascites was collected, followed by a collection of abdominal washings. Ascites cell volume was measured, and we observed a significant reduction in all treatment groups compared to vehicle control (Figure 4C). Ascites and tumor collection were accomplished using a GFP luminescence filter at the time of necropsy (Figure 4D). We noted that the tumor distribution was different across all treatment groups. In both the vehicle control and cisplatin mice, 100% of mice within these groups were positive for omental tumors (Figure 4E). In contrast, we observed that 71% (5 of 7) of etomoxir and 57% (4 of 7) of etomoxir/cisplatin-treated mice were positive for omental tumors (Figure 4E). Notably, while cisplatin-treated mice had reduced tumor burden, the tumors that did grow were more likely to grow on the pelvic fat pad (yellow dot), suggesting that cisplatin could potentially be driving dissemination to a fat-rich niche.
Since the in vitro data found an increase in CPT1A expression in cells grown in suspension compared to adherent cells, we compared CPT1A protein expression in matched tumor cells collected in ascites (A) versus solid tumor (ST) within the same animal. It should be noted that not all mice had both GFP-positive ascites-associated cells and solid tumors. In mice that had both ascites and solid tumors, CPT1A protein expression was compared via immunoblot. Consistent with our in vitro findings, we observed an increase in CPT1A protein in ascites-associated tumor cells (Figure 4F–G). We did note that CPT1A in ascites-associated tumor cells from etomoxir treated mice was elevated compared to the other treatment groups suggesting a compensatory upregulation in tumor cells that persist following treatment (Figure 4F), which was observed in other etomoxir-treated models (30).
Conversely, treatment with cisplatin-only or cisplatin/etomoxir resulted in reduced CPT1A expression (Figure 4F). We next utilized immunohistochemistry (IHC) against Ki67 (a marker of proliferation) and cleaved caspase 3 (a marker of apoptosis) on solid tumors (Figure 4H). IHC tumor staining was blindly scored, and a histology score (H-score) was calculated for each tumor section by assigning tissue staining percentage and staining intensity (3). Compared to vehicle control-treated mice, all of the treatment groups had a significant decrease in Ki67 H-score, with the combination group having the smallest average H-score (Figure 4I). Concerning apoptosis, we observed that the single agents (cisplatin or etomoxir) failed to induce a significant increase in cleaved caspase 3. Only the combination-treated tumors compared to the vehicle, etomoxir alone, or cisplatin alone had significantly increased cleaved caspase 3 (Figure 4J). Notably, the low observed cleaved caspase 3 signal, even in the cisplatin only treatment, highlights the limitation of only performing IHC analysis at the study endpoint. Overall, etomoxir effectively inhibited HGSOC tumor progression in vivo.
Discussion
In this study, we observed, CPT1A is highly expressed in models of HGSOC cell dissemination. Also, ECM-detachment and anoikis resistance drives a metabolic reprogramming towards a predilection for FAO potentially to maintain cellular energy demands and drive tumor redistribution to fat-rich tissues within the abdominopelvic cavity. We observed that CPT1A contributes to anoikis resistance, and targeting CPT1A significantly inhibited HGSOC tumor progression.
Altered energy metabolism is a hallmark of cancer, and there is compelling evidence for both alterations of glucose metabolism and lipid metabolism (8,31). A recent study suggests that mitochondrial FAO is an important metabolic adaptation of cancer cells in response to energy-deprived environments and may be a required adaptation for cancer cell proliferation and survival (3,9,15,32–34). HGSOC cells frequently disseminate and colonize the adipocyte-enriched omentum. Notably, the co-culture of adipocytes and HGSOC cells promotes the upregulation of CPT1A, anoikis resistance, and the transfer of free fatty acids from adipocytes to HGSOC cells via fatty acid-binding protein 4 (7). Our data found that the addition of an FA enriched in the omentum, oleic acid, promoted CPT1A-dependent anoikis resistance and attenuated metabolic stress. In contrast to cells in suspension, cells cultured in suspension could potentially be utilizing the oleic acid differently, such lipid droplet formation. Notably, high concentrations of oleic acid promoted increased apoptosis, highlighting an equilibrium of oleic acid’s pro-tumorigenic and anti-tumorigenic properties (35–37). Furthermore, prolonged utilization of FAO leads to elevated levels of reactive oxygen species (ROS), which in the case of disseminating HGSOC, could further promote disease progression and mutagenesis [reviewed in (38)]. Future work will assess the interplay between oleic acid, CPT1A, and ROS production in ovarian cancer progression.
In the context of HGSOC, there is still a significant need to describe both transcriptional and post-translational regulation of CPT1A to understand the underlying metabolic adaptation of ECM-detachment further. CPT1 is reported to be overexpressed in several different solid tumor types, including HGSOC (15,33). Shao et al. found that CPT1A overexpression contributed to cell cycle deregulation and poor survival of ovarian cancer patients. While the primary cell lines used in this study, OVCAR3, OVCAR4, and OVCAR8, are all considered HGSOC, each cell line has a unique mutational profile (39), which could contribute to differential CPT1A regulation. Little is known regarding the post-translational regulation of CPT1A, but one study found that phosphorylation of CPT1A led to increased catalytic activity (40). Interestingly, we observed cell lines without increased CPT1A mRNA had an upregulation at the protein level suggesting post-translational regulation such as ubiquitination or phosphorylation.
CPT1A transcription has been established to be regulated through several factors, including androgen receptor (41), peroxisome proliferator-activated receptor (PPAR, (42)), PPAR gamma coactivator (PGC-1a, (43)), and CCAAT/enhancer-binding protein beta (44). CPT1A mRNA expression is also regulated via epigenetic mechanisms, such as promoter methylation and histone modifications (45,46). In our study, etomoxir also increased CPT1A expression, which is likely a compensatory mechanism in response to the block in enzymatic activity (30). The possibility that an inactive CPT1A in the mitochondria promotes apoptosis by sequestering BCL2 is intriguing (47). The robust growth inhibition observed with the etomoxir + cisplatin combination could be explained by the intersection of orthogonal pathways that, when optimized, could more effectively signal apoptosis. Caveats to this work are the potential confounding factor of etomoxir-induced ROS, which has been shown to promote oncogenic signaling (48). Also, independent of CPT1A and FAO, excess etomoxir can promote macrophage polarization and inhibit T-cell differentiation (49,50). Etomoxir’s off-target effects related to anti-tumor immunity, while important, did not likely contribute to the anti-tumor response observed in the immune-compromised mice, but future studies will assess the intersections of CPT1A inhibition and anti-tumor immunity.
With respect to the relationship between FAO/CPT1A and anoikis resistance, several recent studies in colorectal, prostate, and ovarian cancers have demonstrated the need for metabolic reprogramming to promote metastasis and survival in anchorage-independent environments (13–15). Our findings corroborate these studies and provide further rationale for targeting CPT1A and FAO in multiple tumor types. Consistently, we found in an HGSOC PDX model that etomoxir significantly reduced tumor growth rate and promoted a differential dissemination pattern. Notably, this only represents a single PDX model, and future pre-clinical work will evaluate a panel of PDX models, with an emphasis on carboplatin-resistant models. Etomoxir was well tolerated with minimal weight loss over the course of treatment. Also, combining etomoxir with platinum-based chemotherapy did not antagonize therapeutic response, suggesting a combinatorial approach is possible and may provide a more durable anti-tumor response. Etomoxir has been investigated in clinical trials but not in the oncology setting, where its anti-tumor properties potentially outweigh its off-target cytotoxic effects. Overall, FAO and CPT1A contribute to HGOSC anoikis resistance, suggesting that future clinical trials evaluating CPT1A inhibition in patients with HGSOC are warranted.
Supplementary Material
Implications:
Targeting FAO in HGSOC to promote anoikis and attenuate dissemination is a potential approach to promote a more durable anti-tumor response and improve patient outcomes.
ACKNOWLEDGEMENTS
We would like to acknowledge Drs. Katherine M. Aird, Theresa Powell, and Andrew P. Bradford for critical reading of this manuscript. We acknowledge Dr. Philip Owens for the use of his Nikon microscope. This work was supported by the National Institutes of Health (Bitler, R00CA194318), The Department of Defense OCRP (Bitler, OC170228), The American Cancer Society (Sawyer ACS-IRG, 16-184-56, Schlaepfer RSG-16-256-01-TBE, Bitler RSG-19-129-01-DDC), The Cancer League of Colorado (Schlaepfer AWD-193286) and the University of Colorado Cancer Center Support Grant (P30CA046934).
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Lee Y, Miron A, Drapkin R, Nucci MR, Medeiros F, Saleemuddin A, et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J Pathol 2007;211:26–35 [DOI] [PubMed] [Google Scholar]
- 2.Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet 2014;384:1376–88 [DOI] [PubMed] [Google Scholar]
- 3.Parmar MK, Ledermann JA, Colombo N, du Bois A, Delaloye JF, Kristensen GB, et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON4/AGO-OVAR-2.2 trial. Lancet 2003;361:2099–106 [DOI] [PubMed] [Google Scholar]
- 4.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015;15:668–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Steen S, Bulten J, Van de Vijver KK, van Kuppevelt TH, Massuger L. Changes in the Extracellular Matrix Are Associated With the Development of Serous Tubal Intraepithelial Carcinoma Into High-Grade Serous Carcinoma. Int J Gynecol Cancer 2017;27:1072–81 [DOI] [PubMed] [Google Scholar]
- 6.Dier U, Shin DH, Hemachandra LP, Uusitalo LM, Hempel N. Bioenergetic analysis of ovarian cancer cell lines: profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS One 2014;9:e98479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011;17:1498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013;13:227–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deep G, Schlaepfer IR. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. Int J Mol Sci 2016;17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull DM, Bartlett K, Younan SI, Sherratt HS. The effects of 2[5(4-chlorophenyl)pentyl]oxirane-2-carbonyl-Co-A on mitochondrial oxidations. Biochem Pharmacol 1984;33:475–81 [DOI] [PubMed] [Google Scholar]
- 11.Wolf HP, Engel DW. Decrease of fatty acid oxidation, ketogenesis and gluconeogenesis in isolated perfused rat liver by phenylalkyl oxirane carboxylate (B 807–27) due to inhibition of CPT I (EC 2.3.1.21). Eur J Biochem 1985;146:359–63 [DOI] [PubMed] [Google Scholar]
- 12.Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) 2007;113:205–12 [DOI] [PubMed] [Google Scholar]
- 13.Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther 2014;13:2361–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang YN, Zeng ZL, Lu J, Wang Y, Liu ZX, He MM, et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018 [DOI] [PubMed] [Google Scholar]
- 15.Shao H, Mohamed EM, Xu GG, Waters M, Jing K, Ma Y, et al. Carnitine palmitoyltransferase 1A functions to repress FoxO transcription factors to allow cell cycle progression in ovarian cancer. Oncotarget 2016;7:3832–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheeler LJ, Watson ZL, Qamar L, Yamamoto TM, Sawyer BT, Sullivan KD, et al. Multi-Omic Approaches Identify Metabolic and Autophagy Regulators Important in Ovarian Cancer Dissemination. iScience 2019;19:474–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karst AM, Drapkin R. Primary culture and immortalization of human fallopian tube secretory epithelial cells. Nat Protoc 2012;7:1755–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler LJ, Watson ZL, Qamar L, Yamamoto TM, Post MD, Berning AA, et al. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis 2018;7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto TM, McMellen A, Watson ZL, Aguilera J, Ferguson R, Nurmemmedov E, et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol Carcinog 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon MA, Babbs B, Cochrane DR, Bitler BG, Richer JK. The long non-coding RNA MALAT1 promotes ovarian cancer progression by regulating RBFOX2-mediated alternative splicing. Mol Carcinog 2018 [DOI] [PubMed] [Google Scholar]
- 23.Bitler BG, Nicodemus JP, Li H, Cai Q, Wu H, Hua X, et al. Wnt5a Suppresses Epithelial Ovarian Cancer by Promoting Cellular Senescence. Cancer Res 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res 2016;76:6320–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Windt GJ, Chang CH, Pearce EL. Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr Protoc Immunol 2016;113:3 16B 1–3 B 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garaulet M, Perez-Llamas F, Perez-Ayala M, Martinez P, de Medina FS, Tebar FJ, et al. Site-specific differences in the fatty acid composition of abdominal adipose tissue in an obese population from a Mediterranean area: relation with dietary fatty acids, plasma lipid profile, serum insulin, and central obesity. Am J Clin Nutr 2001;74:585–91 [DOI] [PubMed] [Google Scholar]
- 27.Kruszynska YT, Sherratt HS. Glucose kinetics during acute and chronic treatment of rats with 2[6(4-chloro-phenoxy)hexyl]oxirane-2-carboxylate, etomoxir. Biochem Pharmacol 1987;36:3917–21 [DOI] [PubMed] [Google Scholar]
- 28.Jiang L, Wang W, He Q, Wu Y, Lu Z, Sun J, et al. Oleic acid induces apoptosis and autophagy in the treatment of Tongue Squamous cell carcinomas. Sci Rep 2017;7:11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Schwarz S, Ahlemeyer B, Grzeschik S, Klumpp S, Krieglstein J. Oleic acid causes apoptosis and dephosphorylates Bad. Neurochem Int 2005;46:127–35 [DOI] [PubMed] [Google Scholar]
- 30.Schlaepfer IR, Glode LM, Hitz CA, Pac CT, Boyle KE, Maroni P, et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[(18)F]Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol Imaging Biol 2015;17:529–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 32.Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017;541:41–5 [DOI] [PubMed] [Google Scholar]
- 33.Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis 2016;7:e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 2015;126:1925–9 [DOI] [PubMed] [Google Scholar]
- 35.Moon HS, Batirel S, Mantzoros CS. Alpha linolenic acid and oleic acid additively down-regulate malignant potential and positively cross-regulate AMPK/S6 axis in OE19 and OE33 esophageal cancer cells. Metabolism 2014;63:1447–54 [DOI] [PubMed] [Google Scholar]
- 36.Navarro-Tito N, Soto-Guzman A, Castro-Sanchez L, Martinez-Orozco R, Salazar EP. Oleic acid promotes migration on MDA-MB-231 breast cancer cells through an arachidonic acid-dependent pathway. Int J Biochem Cell Biol 2010;42:306–17 [DOI] [PubMed] [Google Scholar]
- 37.Vinciguerra M, Carrozzino F, Peyrou M, Carlone S, Montesano R, Benelli R, et al. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J Hepatol 2009;50:1132–41 [DOI] [PubMed] [Google Scholar]
- 38.Saed GM, Diamond MP, Fletcher NM. Updates of the role of oxidative stress in the pathogenesis of ovarian cancer. Gynecol Oncol 2017;145:595–602 [DOI] [PubMed] [Google Scholar]
- 39.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerner J, Distler AM, Minkler P, Parland W, Peterman SM, Hoppel CL. Phosphorylation of rat liver mitochondrial carnitine palmitoyltransferase-I: effect on the kinetic properties of the enzyme. J Biol Chem 2004;279:41104–13 [DOI] [PubMed] [Google Scholar]
- 41.Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijon MA, et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017;8:56051–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popeijus HE, van Otterdijk SD, van der Krieken SE, Konings M, Serbonij K, Plat J, et al. Fatty acid chain length and saturation influences PPARalpha transcriptional activation and repression in HepG2 cells. Mol Nutr Food Res 2014;58:2342–9 [DOI] [PubMed] [Google Scholar]
- 43.Song S, Attia RR, Connaughton S, Niesen MI, Ness GC, Elam MB, et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol 2010;325:54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charos AE, Reed BD, Raha D, Szekely AM, Weissman SM, Snyder M. A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells. Genome Res 2012;22:1668–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frazier-Wood AC, Aslibekyan S, Absher DM, Hopkins PN, Sha J, Tsai MY, et al. Methylation at CPT1A locus is associated with lipoprotein subfraction profiles. J Lipid Res 2014;55:1324–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moody L, Xu GB, Chen H, Pan YX. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim Biophys Acta Gene Regul Mech 2019;1862:141–52 [DOI] [PubMed] [Google Scholar]
- 47.Chowdhury PS, Chamoto K, Kumar A, Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8(+) T Cells and Facilitates Anti-PD-1 Therapy. Cancer Immunol Res 2018;6:1375–87 [DOI] [PubMed] [Google Scholar]
- 48.O’Connor RS, Guo L, Ghassemi S, Snyder NW, Worth AJ, Weng L, et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci Rep 2018;8:6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab 2018;28:490–503 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 2018;28:504–15 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer 2012;19:197–208 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.