

SUMMARY
Recent studies of bacterial DNA replication have led to a picture of the replisome as an entity that freely exchanges DNA polymerases and displays intermittent coupling between the helicase and polymerase(s). Challenging the textbook model of the polymerase holoenzyme acting as a stable complex coordinating the replisome, these observations suggest a role of the helicase as the central organizing hub. We show here that the molecular origin of this newly-found plasticity lies in the 500-fold increase in strength of the interaction between the polymerase holoenzyme and the replicative helicase upon association of the primase with the replisome. By combining in vitro ensemble-averaged and single-molecule assays, we demonstrate that this conformational switch operates during replication and promotes recruitment of multiple holoenzymes at the fork. Our observations provide a molecular mechanism for polymerase exchange and offer a revised model for the replication reaction that emphasizes its stochasticity.
Keywords: DNA replication, DnaB helicase, DnaG primase, clamp loader–helicase interaction, polymerase turnover, conformational switch
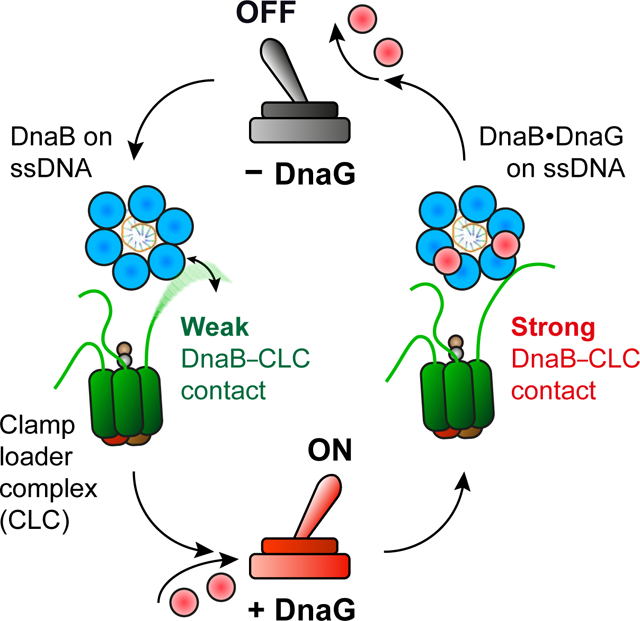
Graphical abstract
INTRODUCTION
The Escherichia coli replisome is composed of at least 12 individual proteins that work together to coordinate leading- and lagging-strand synthesis during the copying of chromosomal DNA (reviewed in Lewis et al., 2016; Figure 1A). Following unwinding of the parental double-stranded (ds) DNA by the DnaB helicase, the DNA polymerase III holoenzyme (Pol III HE) synthesizes DNA on the two daughter strands. The single-stranded (ss) leading strand is displaced by the helicase and duplicated continuously, while the lagging strand is extruded through the DnaB central channel, coated by ssDNA-binding protein (SSB) and converted to dsDNA discontinuously during the production of 1–2 kb Okazaki fragments (OFs) (Kornberg and Baker, 1991). The distinct asymmetry in the mechanism of synthesis of the two strands finds its origin in their opposite polarity, requiring that lagging-strand synthesis take place in a direction opposite that of synthesis of the leading strand. This situation is proposed to result in the formation of a lagging-strand loop (Sinha et al., 1980).
Figure 1. DnaB and Its Binding Partners in the E. coli Replisome.
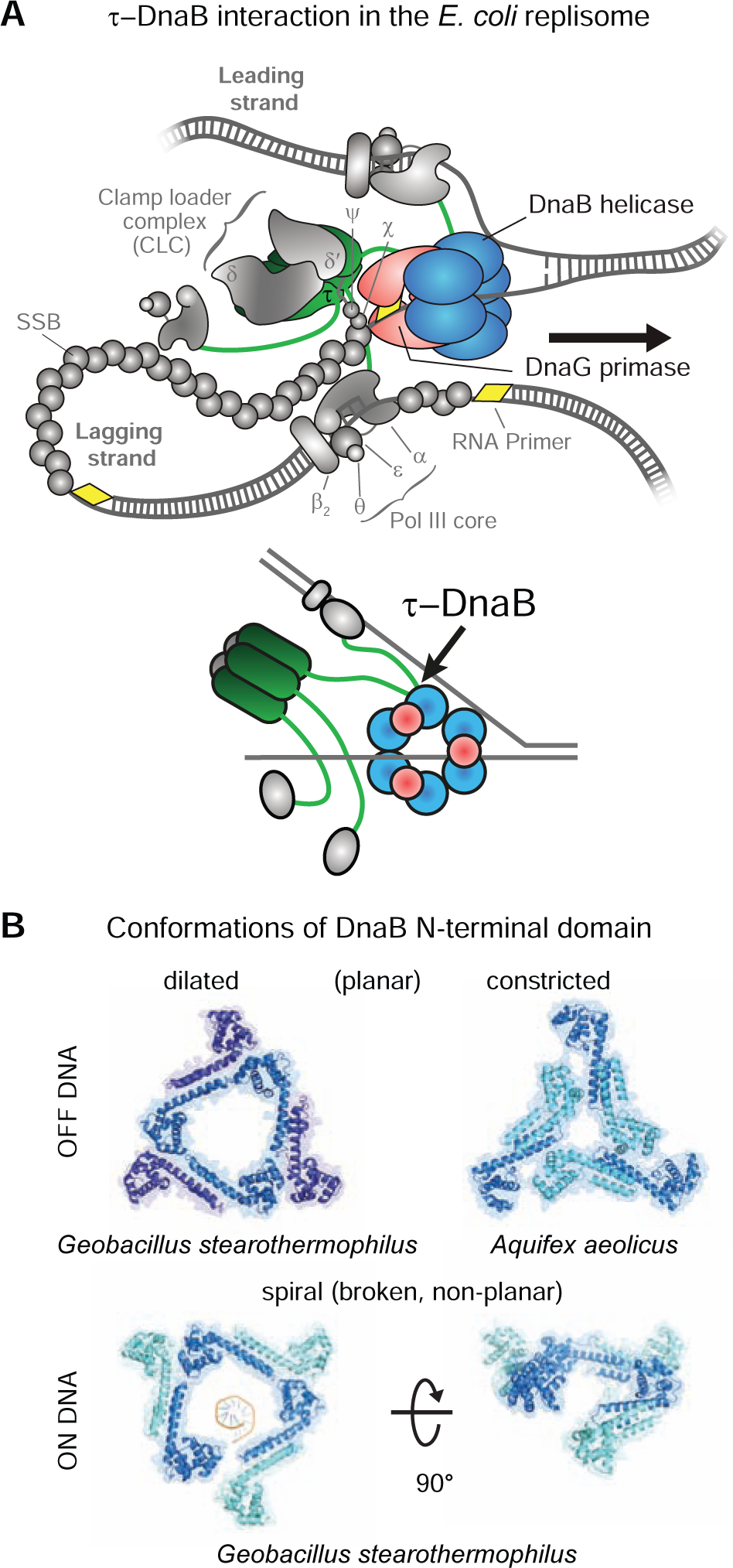
(A) Top: schematic representation of the replisome. DnaB, DnaG, and τ are shown in color; other components of the elongating replisome are in gray. Bottom: Representation of the key interactions between DnaB, DnaG, and τ.
(B) Top: the dilated (left; PDB: 2R6A) and constricted (right; PDB: 4NMN) conformations of DnaB N-tier domains off DNA. Bottom: their open spiral conformation on ssDNA (PDB: 4ESV); ssDNA was removed for clarity in the right panel.
Each OF is initiated by a short RNA primer deposited by the DnaG primase for utilization by Pol III HE. Primase requires interaction with the helicase to stimulate its RNA polymerase activity (Johnson et al., 2000). The DnaB-DnaG contact is established through the interaction between the C-terminal domain of primase, termed DnaGC (Tougu and Marians, 1996; Oakley et al., 2005), and the N-terminal domains of the helicase (Bailey et al., 2007). In E. coli, this interaction is weak and transient, with a KD in the low μM range and fast on/off kinetics (Oakley et al., 2005), whereas in Geobacillus stearothermophilus, DnaB and DnaG form a stable complex that can be isolated and crystallized (Bird et al., 2000; Bailey et al., 2007).
In bacteria, DnaB-family helicases form homohexamers with a distinct two-tiered ring structure. In solution, the C-terminal RecA-like ATPase domains (i.e., the C-tier) that power DNA unwinding have pseudo-six-fold symmetry. In contrast, the N-terminal domains (the N-tier) typically display three-fold symmetry established by a trimer-of-dimers that encircle either a wide open (dilated) central channel that can accommodate dsDNA (Figure 1B, top left; Bujalowski et al., 1994; San Martin et al., 1995; Yu et al., 1996; Bailey et al., 2007; Strycharska et al., 2013), or a narrow (constricted) central pore that is large enough to accommodate only ssDNA (Figure 1B, top right; Strycharska et al., 2013; Arias-Palomo et al., 2019). These contrasting structures suggest that a relatively low energy barrier exists between the dilated and constricted states of DnaB in solution. On DNA however, DnaB-family helicases including those of G. stearothermophilus and phage T7, as well as the complex of the E. coli DnaB helicase with its loader, adopt a spiral “lock-washer” form of both tiers, whereby one of the interfaces in both the hexameric C-tier that coordinates DNA binding and the N-tier is broken (Figure 1B, bottom; Itsathitphaisarn et al., 2012; Gao et al., 2019; Arias-Palomo et al., 2019).
Pol III HE, the replicase that extends RNA primers during most of chromosomal DNA synthesis, interacts with DnaB (Kim et al., 1996; Dallmann et al., 2000; Gao and McHenry, 2001a). Pol III HE is comprised of three functionally distinct subassemblies that can be assembled separately from individual subunits: the Pol III core, the β2 sliding clamp, and the clamp loader complex, CLC (Kelman and O’Donnell, 1995). Pol III cores are heterotrimers of α, ε and θ subunits responsible for DNA synthesis and proofreading (Scheuermann and Echols, 1984; Maki and Kornberg, 1985; Studwell-Vaughan and O’Donnell, 1993; Taft-Benz and Schaaper, 2004). The β2 clamp is a toroidal homodimer (Kong et al., 1992). Once loaded on the DNA by the ATPase activity of the CLC, the clamp stabilizes Pol III on the DNA template and improves processivity through a pair of weak interactions with the α and ε subunits (Naktinis et al., 1996; Dohrmann and McHenry, 2005; Jergic et al., 2013; Fernandez-Leiro et al., 2015). The CLC contains seven proteins and has the composition χψτnγ(3–n)δδ’ (n = 0–3), where physiologically relevant assemblies are thought to have 2 or 3 τ subunits (Reyes-Lamothe et al., 2010; Dohrmann et al., 2016; Lewis et al., 2016). The unique δ and δ’ subunits interact with the three copies of the dnaX gene product, τ and/or γ (Kodaira et al., 1983; Mullin et al., 1983) to assemble a stable ATPase-proficient semi-circular pentamer (Jeruzalmi et al., 2001; Simonetta et al., 2009). Whereas τ is the full-length product of dnaX, γ is a C-terminally truncated version produced as a result of a translational frame-shift (Tsuchihashi and Kornberg, 1990). The accessory subunits χ and ψ form a stable complex that interacts with all three τ/γ subunits of the pentamer via the N-terminal residues in ψ (Gulbis et al., 2004; Simonetta et al., 2009) to assemble the full CLC.
The τ subunit provides physical connectivity between the polymerase and helicase. It has a five-domain structure (Gao and McHenry, 2001b), with the N-terminal domains I–III being identical in γ and responsible for oligomerization and ATP-dependent clamp loading. The C-terminal region that distinguishes τ from γ contains domains IV, which engages DnaB (Gao and McHenry, 2001a), and V, which provides a strong interaction with α (Gao and McHenry, 2001b; Jergic et al., 2007). Consequently, the τ subunit of the CLC plays a key linking role in the replisome: it ensures cohesion of the Pol III–CLC particle (αεθ)n–τnγ(3–n)δδ’–χψ (n = 2–3), termed Pol III*, and links the complex to the helicase (Figure 1A).
Recent advances have challenged the deterministic view of the replisome as a perfectly orchestrated machine, whereby a single Pol III*, stably bound to the replication fork, replicates DNA in a strictly ordered sequence of events (van Oijen and Dixon, 2015; Monachino et al., 2017; Graham et al., 2017). Instead, frequent turnover of the Pol III* in the replisome has been observed (Yuan et al., 2016; Beattie et al., 2017; Lewis et al., 2017), suggesting that the helicase acts as the central organizing structure as opposed to the polymerase. An explanation for this surprising level of plasticity can be found in the network of weak interactions that enable polymerases from solution to eventually replace those at the fork (Geertsema and van Oijen, 2013; Lewis et al., 2017). However, this explanation seems at odds with the putatively strong and stable interaction between multimeric τ in the CLC (Pritchard et al., 2000; Park et al., 2010) and DnaB (Kim et al., 1996; Gao and McHenry, 2001a).
Here we present an unexpected conformational switch in the DnaB helicase triggered by binding of DnaG primase, that increases the strength of the DnaB–CLC interaction by ~500-fold. We find that in solution DnaB is almost exclusively in the constricted state, as observed in the crystal structure of Aquifex aeolicus DnaB. However, during replication, its N-tier transitions between two or more states in a manner that is controlled by primase interaction. Nevertheless, DnaB remains an active helicase as it samples these separate states. Finally, we use single-molecule visualization of Pol III during replication to show that the primase concentration modulates both the kinetics of polymerase exchange in the replisome and the number of polymerases associated with the replication fork.
Taken together, our observations establish that the interaction between the primase and helicase acts as a switch to control replisomal organization and dynamics. This realization has important ramifications for understanding coordination of leading- and lagging-strand synthesis, the coupling between polymerase and helicase, and the timing of Okazaki-fragment synthesis.
RESULTS
The Interaction Between the Clamp Loader Complex and Helicase is Weak and Transient
Due to its complex stoichiometry, the interaction between the τ subunits in the CLC and DnaB is poorly understood. Previous studies employed surface plasmon resonance (SPR) to identify the region within τ that is responsible for binding to DnaB (Gao and McHenry, 2001a), but the use of immobilized monomeric τ fragments makes it challenging to interpret data in the context of multiple τ subunits within a single CLC interacting with DnaB simultaneously. To study these interactions in a context that closely represents a physiologically relevant system, we isolated (Figure S1A) and immobilized the entire CLC (bioCLC) onto a streptavidin-coated chip surface through biotinylation of the χ subunit (bioχ). We then used the immobilized CLC as a platform to measure interaction with the helicase. Provided ATP was present in the SPR buffer, the CLC remained stably bound on the chip surface, with dissociation t1/2 >10 h (Figure S1B); using ADP instead of ATP reduced t1/2 by ~3-fold.
We then used SPR to test the strength of interaction between wild-type DnaBwt and immobilized τ3CLC (Figure 2A,B). Sensorgrams recorded at a range of concentrations of DnaBwt, [DnaBwt] injected over bioχψτ3δδ’ in a buffer containing ADP (or ATP) revealed unexpectedly fast on-and off-rates (Figure 2B). Responses measured at equilibrium were fit to a 1:1 steady-state affinity (SSA) model (Equation 1, Method Details) to yield a KD value of 1.3 ± 0.2 μM for the bioχψτ3δδ’–DnaB interaction. Similar measurements revealed an almost identical strength and similarly fast kinetics of the bioχψτ1γ2δδ’–DnaB interaction (KD = 4.1 ± 0.3 μM; Figure S1C). Thus, in contrast to expectation based on literature (Kim et al., 1996; Gao and McHenry, 2001a), our data show that the interaction between CLC and DnaBwt is weak and transient, and indicate that it does not depend on the number of τ subunits in the CLC since the difference in KD is not significantly larger as would be expected if multiple τ subunits in a single τ3CLC were contacting a single DnaB hexamer.
Figure 2. The Dilated Form of DnaB Interacts Tightly with bioχψτ3δδ’.
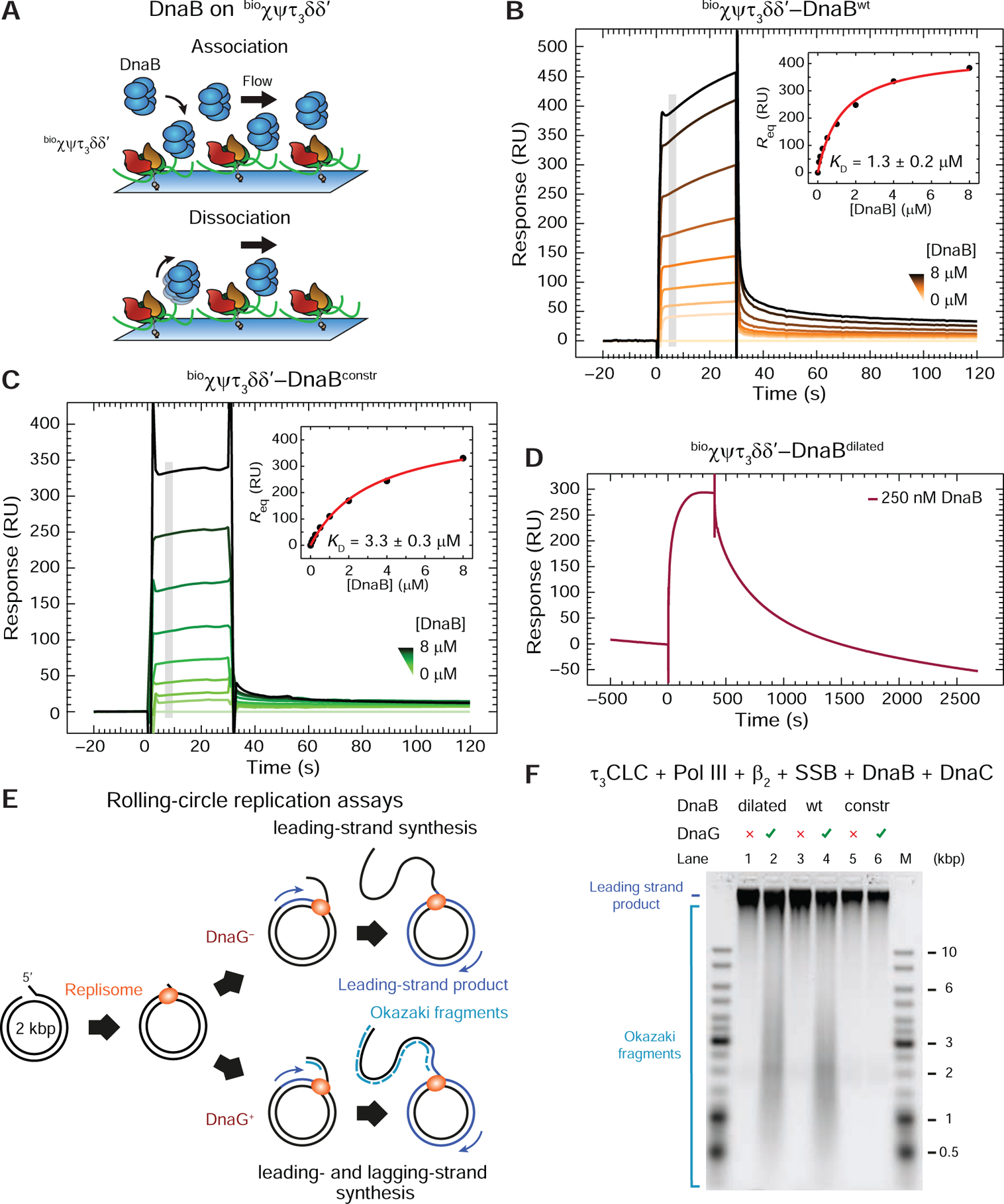
(A) Cartoon representation of the SPR experiment used to measure the binding of DnaB versions in solution to immobilized τ3CLC.
(B) and (C) SPR sensorgrams show association (30 s) and dissociation phases of serially-diluted 0.0625–8 μM DnaBwt (B) and DnaBconstr (C) on bioχψτ3δδ’, including 0 μM control. Responses at equilibrium, determined by averaging values in the gray region, were fit (insets) using a 1:1 steady state affinity (SSA) model to derive values of KD (as indicated) and Rmax = 440 ± 20 RU (B), 460 ± 20 RU (C). Errors are S.E. of the fit.
(D) DnaBdilated (250 nM) is injected for 400 s and its dissociation monitored over 2000 s.
(E) Representation of bulk replication assays with rolling-circle substrates in the absence or presence of primase to enable lagging-strand synthesis.
(F) Alkaline agarose gel showing resolved leading- (all lanes) and lagging-strand (DnaG+ lanes) products generated by replisomes in the absence of primase (odd lanes) or its presence (even lanes), using DnaBdilated, DnaBwt, and DnaBconstr (as indicated).
See also Figure S1.
There are Two Modes of Clamp Loader Complex–Helicase Interaction
Recent studies indicated that the τ subunit could be interacting differently with the dilated and constricted states of DnaB (Strycharska et al., 2013). This conclusion was based on DNA-unwinding assays with mutant versions of the helicase that were stabilized in either the dilated (DnaBdilated) or constricted (DnaBconstr) states (Figure 1B, top), and a τ peptide. We tested both mutants for their interaction with immobilized τ3CLC by SPR. Whereas DnaBconstr exhibited binding kinetics and strengths similar to that of DnaBwt (KD = 3.3 ± 0.3 μM; Figure 2C), injection of 250 nM DnaBdilated resulted in markedly slower dissociation from bioχψτ3δδ’ (Figure 2D). The similarity of the kinetics of DnaBwt and DnaBconstr interacting with τ3CLC and the stark difference between those of DnaBwt and DnaBdilated suggest that E. coli DnaBwt is mostly not in the dilated state in solution.
In a crystal structure of the G. stearothermophilus DnaB6•(DnaGC)3 complex, the three C-terminal domains of primase each bind to two adjacent subunits of the dilated N-tier of DnaB (Figure S2A). However, in the constricted A. aeolicus DnaB (Figure 1B, top right), one of the two asymmetric DnaGC contact points is buried and unavailable for DnaG binding (Strycharska et al., 2013). In agreement with the structures, it was further reported that DnaBdilated is able to interact with DnaG and support priming activity whereas DnaBconstr could not sustain priming at all. We compared the activities of DnaBwt and the two mutants in a leading- and lagging-strand replication assay (Figure 2E, DnaG+ path) and found that, unlike DnaBwt and DnaBdilated, DnaBconstr was incapable of sustaining OF synthesis upon addition of DnaG (Figure 2F; lanes 6 cf. 2 and 4). In contrast, all three helicases, including DnaBconstr were proficient in leading-strand synthesis (Figure 2E, DnaG− path, and 2F, lanes 1, 3 and 5). The observation that conformationally-fixed DnaBdilated and DnaBconstr are both active helicases implies that the principal state of the DnaB N-tier on DNA is either unchanged or neither dilated nor constricted. The latter possibility is in agreement with the crystal structure of DnaB on DNA showing a spiral rather than planar conformation of the N-tier domains (Figure 1B, bottom). The observation that DnaBwt supports the production of OFs indicates that the helicase is able to explore dilated-like states on DNA due either to primase or ssDNA binding, or both.
Clamp Loader Complex–Helicase Affinity Increases ~500-Fold upon DnaGC Binding
To test the possibility that DnaG binding to DnaB is sufficient to trigger the conformational transition in the helicase and induce strong interaction with the CLC, we used DnaGC (Loscha et al., 2004), a C-terminal domain of primase that has all the determinants for DnaB binding. Both DnaG and DnaGC interact similarly weakly with DnaBwt with KD values of 2.8 and 4.9 μM in 150 mM NaCl, respectively (Oakley et al., 2005). Injection of 0.5 μM DnaBwt together with 5 μM DnaGC and 1 mM ATP (50 mM NaCl) over immobilized τ3CLC elicited a much higher response compared to DnaBwt alone (Figure S2B). This stronger binding was not ATP specific, since the use of ADP resulted in a similar response (Figure S2C). Injection of 5 μM DnaGC in the absence of DnaB did not yield a response, showing that the signal is not caused by direct binding of DnaGC to τ3CLC (Figure S2B). Critically, fast off kinetics were observed when DnaGC was present during the association of DnaB with the CLC, but omitted during the dissociation phase (Figures 2B cf. S2B). Considering that DnaGC appears to dramatically increase the affinity of DnaB for CLC, the fast dissociation detected appeared to violate the thermodynamic principle that each ligand (CLC or DnaGC) must increase the affinity of DnaB for the other. To further investigate this behavior, we measured the binding of DnaBwt to immobilized τ3CLC by fixing the solution [DnaBwt] to 125 nM while titrating DnaGC over a range of 0.5–8 μM (Figure S2D). Despite the presence of three binding sites on DnaB for DnaGC, the data were readily fit by a simple SSA model of one-to-one binding, as if only one of the three sites on DnaB had been bound by DnaGC. The measured KD of 1.74 ± 0.09 μM and the fast kinetics were similar to the reported binding of DnaBwt to individual immobilized DnaG molecules (Oakley et al., 2005). These observations can be reconciled by a model that describes a cooperative transition in DnaB, with the weak binding of the first DnaGC initiating a conformational transition in the DnaB hexamer towards a dilated state that has at least a 10-fold higher affinity for a second (and probably third) DnaGC.
The strength of the interaction between τ3CLC and the DnaBwt•DnaGC complex could now be determined accurately by measuring responses at equilibrium for various [DnaBwt] in the presence of 5 μM DnaGC and fitting against the calculated [DnaBwt•DnaGC] (see Equation 2, Method Details) using an SSA model (KD = 2.6 ± 0.3 nM; Figure 3A). The increase in the strength of DnaBwt‒CLC interaction by up to 500-fold in the presence of DnaGC (Figure 3A cf. 2B) indicates that the binding of primase triggers a conformational switch in DnaB towards a dilated state, stabilizing its binding to the CLC. As expected, injection of DnaBconstr in the presence of DnaGC resulted in a much weaker response, consistent with the inability of this conformation to stably interact with primase (Figure S2E).
Figure 3. DnaGC–DnaB Association Strengthens the bioχψτ3δδ’–DnaB Interaction.

(A) Top: representation of the phases of an SPR experiment used to measure the binding of DnaBwt to immobilized τ3CLC in the presence of DnaGC during association only. Bottom: SPR sensorgrams showing association (150 s) and dissociation of DnaB•DnaGC to and from bioχψτ3δδ’ over a 0.5–32 nM range of serially-diluted DnaBwt (including 0 nM control) together with 5 μM DnaGC. Equilibrium responses, determined by averaging values in the gray regions of the sensorgrams, were fit (inset) against the calculated DnaBwt•DnaGC concentrations (0–23.7 nM, see Method Details) to obtain KD = 2.6 ± 0.3 nM and Rmax = 460 ± 10 RU. Errors are S.E. of the fit.
(B) Top: representation of DnaBwt association (with DnaGC, step I) and dissociation (with, step II and without DnaGC, step III) during an SPR experiment. Bottom: DnaGC slows the dissociation of DnaBwt. Sensorgram shows the association of DnaBwt•DnaGC during a 60 s injection of 32 nM DnaBwt with 5 μM DnaGC (step I), followed by dissociation in presence of 5 μM DnaGC (step II) and second, rapid dissociation phase without DnaGC (step III).
See also Figure S2.
The presence of DnaGC (5 μM) in the dissociation phase slows down τ3CLC•DnaB dissociation to a lifetime of several hundreds of seconds (Figure 3B). Hence the DnaB conformation is regulated by primase–helicase interaction, thereby controlling the affinity of DnaB for the CLC. On the other hand, the CLC cannot lock DnaB in its dilated state. We thus identified two functional forms of the helicase–CLC interaction: one with weak and the other with strong affinity that depends exclusively on cooperative primase–helicase interactions.
A Single τ Subunit in the Clamp Loader Complex Binds the Helicase
Our data indicated that in the absence of DnaGC a single copy of τ in τ3CLC contacts a single DnaB, so we investigated whether the tighter binding in the presence of DnaGC is due to DnaB interacting with multiple copies of τ in the same CLC. We immobilized comparable amounts of τ1CLC and τ2CLC cf. τ3CLC onto distinct flow cells (Figure S2F) and tested interaction with DnaBwt•DnaGC. As before, binding of DnaBwt to τ1 and τ2CLC at 5 μM DnaGC was similarly strong (KDs of 10 ± 1 and 3.0 ± 0.3 nM, respectively; Figure S2G, H) to that of τ3CLC (2.6 ± 0.3 nM; Figure 3A), again arguing that a single τ subunit within the τ3CLC is responsible for binding a single helicase. To prove that the observed difference in KD is insignificant, we compared fit Rmax values (Table 1), the saturating response when all immobilized CLC on the surface have been bound to DnaBwt•DnaGC, normalized to the number of immobilized τ1CLC molecules (Method Details). The results show that 2.3 ± 0.2 and 3.0 ± 0.2-fold more DnaBwt•DnaGC binds to τ2CLC and τ3CLC at saturation, respectively, compared to τ1CLC (Table 1), confirming that a single copy of τ in the CLC binds DnaB.
Table 1.
Binding Parameters for bioCLC‒DnaBwt•DnaGC Interactions
| Interaction with DnaBwt•DnaGC | Immobilized CLC (RU) | MW (kDa) | KD (nM) | Normalized Rmax (RU) | Rmax / Rmax, τ1CLC |
|---|---|---|---|---|---|
| bioτ1CLC | 1630 | 277 | 10 ± 1 | 163 ± 8 | 1 |
| bioτ2CLC | 1955 | 301 | 3.0 ± 0.3 | 370 ± 10 | 2.3 ± 0.2 |
| bioτ3CLC | 1830 | 325 | 2.6 ± 0.3 | 490 ± 10 | 3.0 ± 0.2 |
Equilibrium constant (KD) and stoichiometry of binding of DnaBwt•DnaGC at saturation (from normalized Rmax) for interactions with immobilized bioτ1CLC, bioτ2CLC and bioτ3CLC, determined by fitting of equilibrium responses in Figures 3A and S2GH to Equation 1 (see Method Details). The errors are S.E. of the fits.
Strong Helicase–Clamp Loader Complex Interaction Stimulates the Activity of a Destabilized Replisome
The primase-induced tightening of DnaB–CLC binding and the strongly altered kinetics of the interaction is expected to affect replisome organization and dynamics. To demonstrate the importance of the strong helicase–clamp loader interaction in a functional context, we first used a rolling-circle leading-strand replication assay (Figure 2E, DnaG− path). In this assay (Mok and Marians, 1987), the β2–Pol IIIlead–CLC–DnaB connectivity stimulates the simultaneous unwinding of dsDNA by DnaB and primer-extension DNA synthesis by a leading-strand Pol III. With the aim to preferentially destabilize Pol III* on primer-template (p/t) DNA to highlight the contribution of other interactions within the replisome, a strategy we have used before (Jergic et al., 2013), and to ensure that DNA synthesis is entirely a result of coupled Pol III*–DnaB synthesis, we modified the assay such that the stability of Pol III* on DNA is compromised by leaving out β2, SSB and χψ (Figure 4A). While the omission of β2 is known to result in distributive DNA synthesis, all omitted components comprise elements of the p/t DNA–β2–Pol IIIlead–τ–ψ-χ–SSB–ssDNAin trans link that enables DnaB-independent strand-displacement (SD) synthesis by Pol III HE (Yuan and McHenry, 2009), a possibility we intended to prevent. The omission of SSB additionally prevents uncoupled χψ-less (−χψ)Pol III*–DnaB synthesis given that this situation favors re-zipping of non-replicated dsDNA behind the helicase, and stalling of the destabilized SD-incompetent polymerase (for uncoupled synthesis, see Dallmann et al., 2000; Graham et al., 2017). Hence our conditions artificially elevate the importance of the remaining DnaB–τ3δδ’ link and allow us to visualize its functional dependence on [DnaGC].
Figure 4. DnaGC-Induced CLC–DnaB Interaction Stimulates Leading-Strand Synthesis by Destabilized Replisomes.
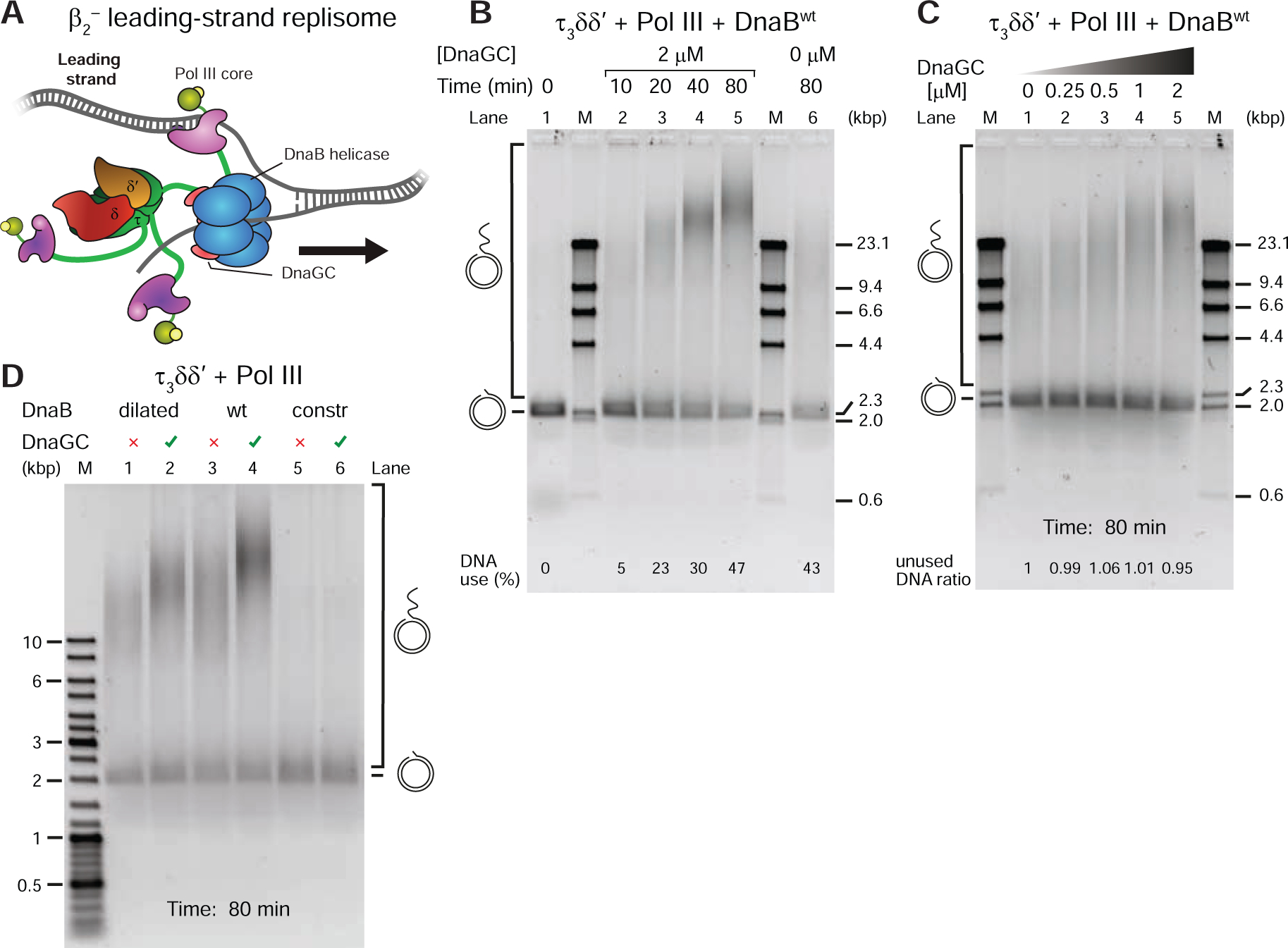
(A) Destabilized replisome that synthesizes only the leading strand in the absence of β2, SSB and χψ, used in (B–D).
(B) Time course of rolling-circle DNA synthesis by the destabilized replisome, at indicated times, in the presence of 2 μM DnaGC (lanes 1–5) and in its absence (lane 6), resolved on an agarose gel.
(C) Serially-diluted DnaGC (0.25–2 μM, including zero) were supplemented into individual rolling-circle reactions and the replication products visualized on a gel.
(D) DNA replication products generated by destabilized replisomes ± DnaGC, using DnaBdilated, DnaBwt, and DnaBconstr (as indicated).
See also Figure S3.
A time-course assay at constant [DnaGC] (Figure 4B, lanes 2–5) showed progressively longer products synthesized and more DNA templates consumed in time. Moreover, in the absence of DnaGC, replication was significantly less efficient and equivalent to the level in the DnaGC-dependent reaction at early time points (Figure 4B, lanes 6 cf. 2 and 3). The narrow distribution of product sizes points to the distributive nature of DNA synthesis, as expected considering the absence of β2. Using progressively increasing [DnaGC] at a fixed time point confirms dependence of the synthesis efficiency on DnaGC (Figure 4C).
Consistent with distributive DNA synthesis in the absence of β2, increasing [Pol III*] led to an increase in product lengths and their abundance, both with and without DnaGC, while the pattern of template utilization was similar (Figure S3A; left cf. right). This result aligns with the observation that increasing [DnaGC] does not itself affect template utilization (Figure 4C). However, the product lengths increased more sharply in the presence of DnaGC in the lower range of [Pol III*] (Figure S3A; left cf. right). These data argue that the rate limiting step in these reactions is (re)loading of Pol III* and its stability on the p/t DNA, rather than the rate of DNA unwinding by DnaB, i.e. DnaB is capable of supporting much higher DNA unwinding rates even in the absence of primase. Considering that (a) DnaGC has no enzymatic activity, nor known interacting partners other than DnaB and SSB; (b) the physical coupling between the helicase and Pol III*, which greatly stimulates leading-strand replication (Kornberg and Baker, 1991; Kim et al., 1996) was found to be essential in this assay since there was no DNA synthesis with γ3CLC cf. τ3CLC (Figure S3B); (c) helicase-independent Pol III SD synthesis cannot occur in the absence of β2, SSB or χψ (Yuan and McHenry, 2009; Jergic et al., 2013); (d) the efficiency of replication increases in the range of [DnaGC] that titrates DnaB (Figure S2D) and stabilizes the DnaB–CLC interaction (Figure 3); (e) helicase loading was unaffected by increasing [DnaGC] (as observed by constant template utilization in Figure 4C); (f) significantly longer products of synthesis by Pol III* were observed in presence of DnaGC, i.e. without DnaGC, 8 nM Pol III* was necessary to achieve similar product lengths to 1 nM Pol III* with DnaGC, in spite of higher template utilization and more efficient Pol III* reloading at 8 nM (Figure S3A); and (g) DnaB is reported to be stable on ssDNA for very long periods (~60 min; Pomerantz and O’Donnell, 2010), we conclude that the progressively higher efficiencies in DNA synthesis with increasing [DnaGC] (Figure 4C) are due to DnaGC-induced strengthening of the Pol III*–DnaB interaction.
DnaBdilated Displays the Two Populations of States of the N-Tier on DNA
Our expectation was that DnaBdilated should be insensitive to the addition of DnaGC, had the stabilized dilated conformation of its N-tier and strong interaction with CLC been preserved on DNA. Unexpectedly, the covalently-crosslinked DnaBdilated responded to the addition of DnaGC much like DnaBwt, resulting in more efficient DNA synthesis (Figure 4D, lanes 1–4). These data suggest that, once on DNA and active, DnaBdilated may no longer interact strongly with the CLC as it does in the absence of DNA. This indicates an altered organization of the N-tier domains that abrogates the strong CLC–DnaB interaction on DNA in the absence of DnaGC, which also cannot be constricted since crosslinks would not allow this organization. That DnaGC was able to stimulate replication using DnaBdilated (and DnaBwt) confirms that the energy of primase–helicase interaction is sufficient to overcome the barrier between the two distinct populations of states of the N-tier of DnaB on DNA, switching the CLC–DnaB binding mode from weak to strong (i.e., the energy barrier must be much lower off DNA given that crosslinking was sufficient to promote strong CLC–DnaB interaction even in the absence of DnaG).
Interestingly, DnaBconstr did not provide a platform for detectable DNA synthesis in the absence of DnaGC, showing a defect in helicase loading (Figure 4D, lane 5). This particular defect was not so apparent in the context of processive leading-strand replication with the full replisome, but it could have contributed to lower efficiency (much less final product was produced; Figure 2F, lanes 5 cf. 1 and 3). As expected, DnaGC did not relieve the inhibition (Figure 4D, lane 6) since DnaBconst cannot provide a platform for interaction with DnaGC (Figure 2F, lane 6). That DnaBdilated, whose N-tier cannot resume the constricted state, is proficient in DNA synthesis yet is still stimulated by DnaGC and that DnaBconstr is very inefficient in loading indicate that the conformation of the DnaBwt N-tier on DNA is neither strictly dilated nor constricted. The simplest explanation consistent with structural data (Figure 1B) is that it is no longer planar, but still becomes more dilated-like on interaction with DnaG, strengthening the interaction with the CLC.
Binding of Primase Does Not Inhibit DnaB Helicase Activity
It is unknown whether the helicase stalls during OF priming in E. coli. It is also not known how long primase is bound to the helicase during each OF cycle. It is therefore important to know whether the helicase–primase interaction per se is sufficient to stall the helicase. Graham et al. (2017) found no evidence for DnaG-dependent pausing of leading-strand replication, and our data indicate that the DnaGC–DnaB interaction stimulates leading-strand synthesis through strengthening of the Pol III*–DnaB link (Figure 4). However, major experimental challenges in characterizing the DNA-unwinding activity of an E. coli helicase–primase complex include the multivalency and transience of the interactions in conjunction with the likelihood of different helicase conformations on DNA. To overcome the stoichiometric heterogeneity of DnaB•(DnaGC)n (n = 0–3 in solution) and to ensure that the helicase remains continuously in the dilated-like state on DNA with strong affinity for the CLC, we used a disulfide-linked construct termed DnaB~GC, in which facile crosslinking of three DnaGCR568C/C492L molecules with DnaB6F102C was achieved through one of the two asymmetric contact points in each DnaGC. The DnaBF102C mutant interacts 200-fold less efficiently with DnaGC, while DnaGCR568C/C492L interacts 30-fold more weakly with DnaBwt than wild-type. Nevertheless, the disulfide crosslinking efficiency of these mutants is nearly 100%, ensuring that for each hexamer of DnaB, all three primase sites are occupied by DnaGC (Z.-Q.X. and A.T.Y.L., unpublished).
To characterize DnaB~GC, we used SPR to monitor its binding to immobilized τ3CLC. This experiment resulted in characteristically slow dissociation (Figure 5A) as observed with DnaBdilated (Figure 2D) or DnaBwt in the presence of DnaGC (Figure 3B). Moreover, similarly to DnaBwt and DnaBconstr, DnaBF102C alone exhibited a much lower response at equilibrium, accompanied by fast kinetics (Figure 5A). These data indicate that DnaB~GC is indeed mostly in a dilated state in solution and interacts strongly with the CLC.
Figure 5. A Cross-Linked DnaB~GC Complex is an Active Helicase.
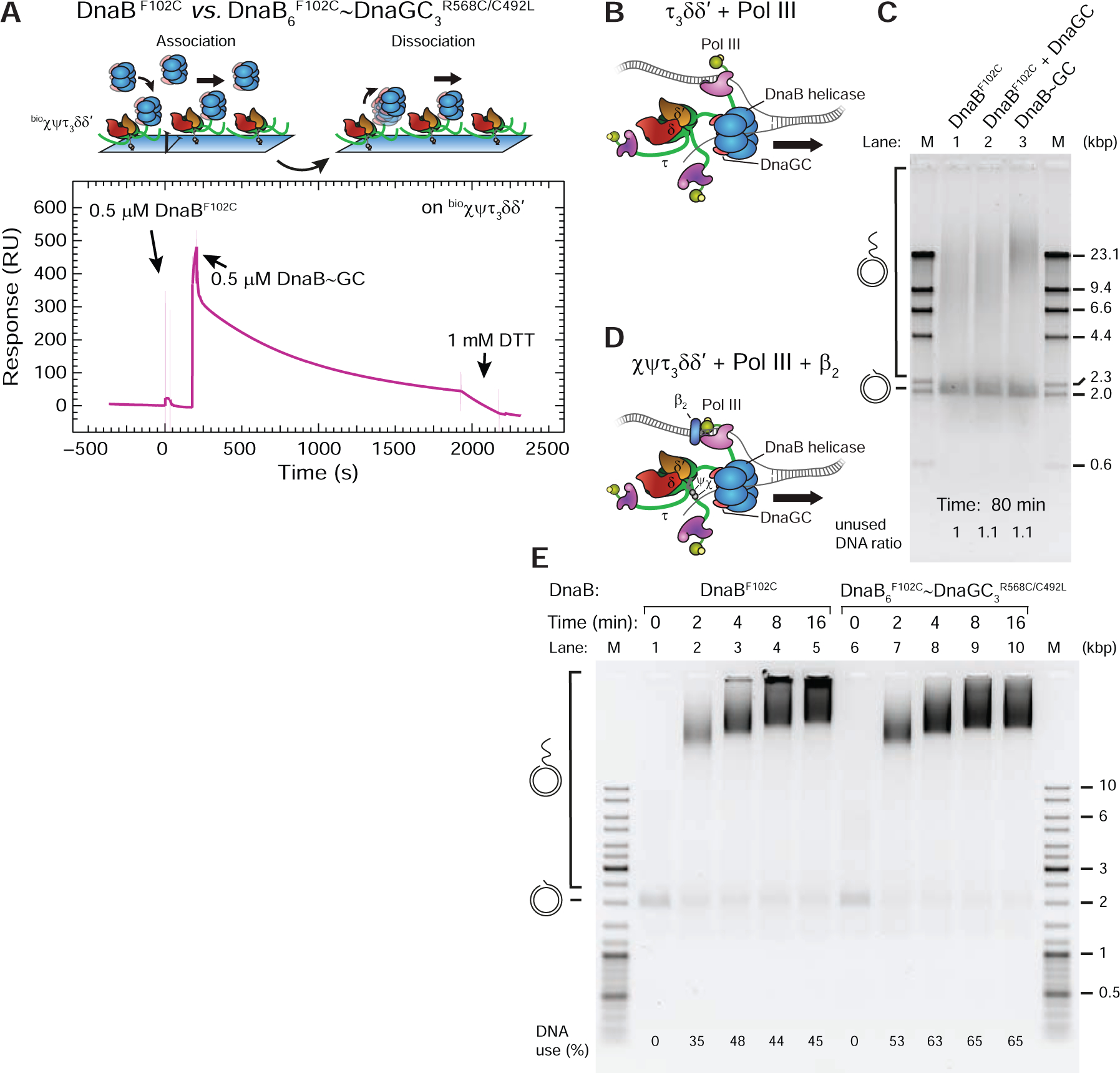
(A) Top: association and dissociation phases of an SPR experiment to visualize binding of cross-linked DnaB~GC to immobilized bioχψτ3δδ’. Bottom: sensorgrams showing consecutive injections of DnaBF102C alone and cross-linked DnaB6F102C~DnaGC3R568C/C492L (DnaB~GC). During DnaB~GC dissociation, dithiothreitol injected at ~1800 s reduces the disulfide crosslink, leading to release of DnaGC and faster dissociation.
(B, C) Destabilized leading-strand rolling-circle reactions in the absence of β2, SSB and χψ were supplemented with DnaBF102, DnaBF102 and 2 μM DnaGC, and DnaB~GC (as indicated) and the products separated on an agarose gel following 80 min reaction.
(D, E) Full leading-strand rolling-circle reactions, stopped at indicated time points, with either DnaBF102C or DnaB~DnaGC; products are resolved on a native gel.
We next used DnaB~GC in our leading-strand assay with destabilized replisomes (Figure 4A), comparing its activity to a DnaBF102C control (± DnaGC; Figure 5B, C). We reasoned that if the activity of the DnaB~GC-driven replisome is found to be similar to or stronger than those of controls, it would indicate that DnaB remains an active helicase even when primase is bound to it. The control lanes show that DnaBF102C is active (Figure 5C, lane 1) while the presence of DnaGC makes no difference to yield and product length (Figure 5C, lane 2). No change in activity is expected because of the much weaker affinity of the mutant helicase for DnaGC. However, the presence of DnaB~GC in the reaction results in higher replication efficiency compared to controls (Figure 5C, lanes 3 cf. 1 and 2). Considering that the template consumption was consistent across reactions, our results show that DnaB is an active helicase when it is bound to three DnaGC, confirming again that the dilated-like state on DNA is functional. Interestingly, similar leading-strand assays in the absence of SSB (to prevent SD) but with β2 and χψ revealed that replisomes with DnaB~GC are not more efficient than those with DnaBF102C (Figure 5C, D). This finding suggests that if Pol III on the leading strand is stabilized by interactions with β2, the strengthening of the DnaB–CLC interaction becomes less critical for the stability of β2–Pol IIIlead–CLC–DnaB. Taken together, we conclude that DnaB shows helicase activity in both modes of CLC–DnaB interaction, even when associated with its principal binding partners within the replisome.
DnaG Concentration Controls the Number of Pol III* Associated with the Replisome
The replication assays described above indicated toggling between different conformations of the DnaB N-tier on DNA depending on [DnaGC]. Next, we examined the influence of the conformational switch in the helicase on full replisome activity by real-time single-molecule (sm) observations. We previously reported use of a sm rolling-circle assay to image fluorescent Pol III* complexes associated with the replisome and visualize dynamic exchange of replisome-bound Pol III* with those in solution on the seconds time scale (Lewis et al., 2017). We hypothesized that DnaG-induced strengthening of the DnaB–CLC interaction could contribute to the accumulation of Pol III* at the replication fork by slowing down its exchange.
We employed sm-FRAP (fluorescence recovery after photobleaching) experiments (Lewis et al., 2017) to measure turnover of fluorescent Pol III* (3 nM) at the replication fork at different [DnaG] (30–300 nM) as both leading and lagging strands are replicated (Figure 6A). The lagging strand of the rolling circle template is surface-attached through its 5’-tail, so the dsDNA circle, stretched in the buffer flow moves away from the anchor point as dsDNA is synthesized. Replication is visualized by real-time near-TIRF imaging of the growing tip of the rolling circle using both dsDNA intercalating dye and fluorescent Pol III*. Labeled Pol III* in the field of view (TIRF volume) was photobleached every 20 s with pulses at high laser power (Figure 6B, top) and the recovery of fluorescence intensity as the labeled Pol III* from solution exchanges into active replisomes at the fork (confirmed by co-localization of the intercalated dye front at the tip of the rolling circle with fluorescent spot recovery) was measured over time (Figure 6B, bottom). Fluorescence intensities were converted to numbers of Pol III* by calibrating the intensity of a single labeled Pol III* (Lewis et al., 2017). To analyze the data, every recovery interval in which replication was confirmed was pooled for a particular [DnaG] and fit to their averaged fluorescence intensity values (Figure 6C) with a FRAP recovery equation (Equation 3, Method Details). This procedure was repeated for each [DnaG] (Figures 6D and S4A–C). We note that given that catalytically-dead αD403E exchanges into the replisome to stall forks preassembled with wild-type Pol III* (Yuan et al., 2016), and our previous (Lewis et al., 2017) and current reports, it cannot be concluded that FRAP assays necessarily report only on true Pol III* ‘exchange’ events whereby fluorescent Pol III* substitutes for photobleached Pol III* during active DNA synthesis; they also report on ‘recruitment’ of additional Pol III* onto the helicase for potential subsequent delivery of some onto DNA. Since neither term most appropriately describes the observable we measure, we use ‘apparent exchange’ to encapsulate phenomena that we cannot spatially or temporally resolve at the diffraction-limited spot.
Figure 6. DnaG Stimulates Polymerase Accumulation and Slows Its Exchange at the Replication Fork.
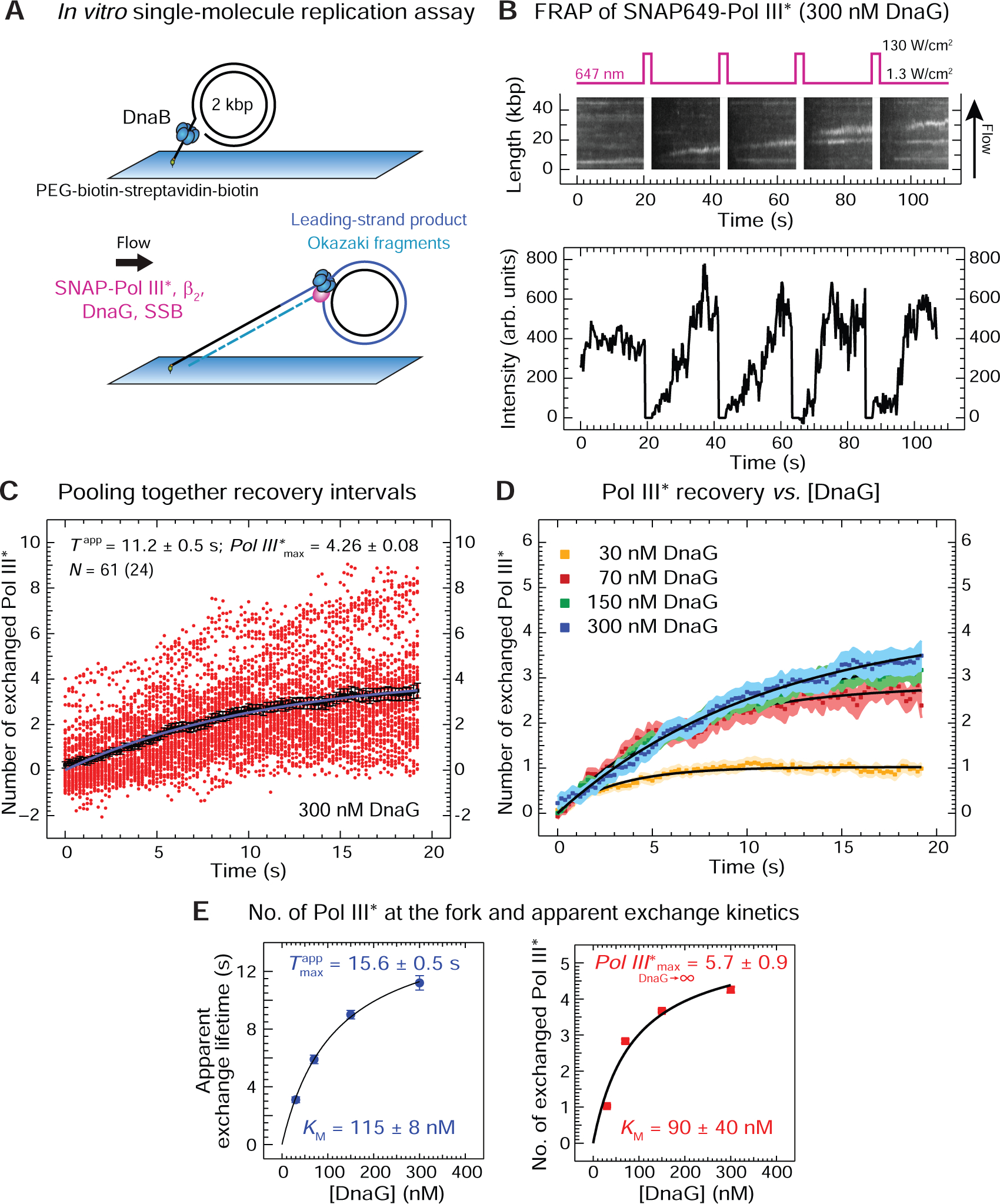
(A) Two stages of the single-molecule leading- and lagging-strand rolling-circle DNA synthesis assay. First, the rolling-circle substrate with DnaB is loaded with its 5’-tail bound to a coverslip. Then SNAP-Pol III* (3 nM), β2 (40 nM), SSB (250 nM) and DnaG (30–300 nM, as indicated) are introduced to initiate replication.
(B) Top: representative kymograph of SNAP-Pol III* at the fork in the presence of DnaG. Every 20 s, a 2-s high-power laser pulse photobleaches SNAP-Pol III* in the field of view. Bottom: recovery of SNAP-Pol III* intensities over time as unbleached polymerases from solution bind to an individual active replisome.
(C) Intensities (red circles) obtained from 61 recovery-interval trajectories of 24 replisomes are converted into the number of exchanged Pol III* and displayed, together with their average values (black squares). Fitting the evolution of average recovery intensities in time with the FRAP recovery equation (Equation 3, Method Details) provides the apparent exchange time Tapp, the maximum number of exchanged Pol III* (Pol III*max), and the remaining background intensity y0, converted to number of Pol III* (y0 = 0.72 ± 0.04), was then subtracted from every other curve.
(D) Averaged recovery intensities at each [DnaG] and their FRAP recovery fit curves (black). The corresponding values for Tapp, Pol III*max, and y0 are presented in Figure S4.
(E) Tapp (left) and Pol III*max (right), plotted as a function of [DnaG], fit with a steady-state equation (Equation 4, Method Details), providing indicated values of KM and the maximum number of exchanged Pol III* as [DnaG] approaches infinity.
See also Figure S4.
Our analysis revealed that the characteristic apparent exchange lifetime (Tapp) increases with [DnaG] in a physiologically relevant range (Figure 6E, left). Pol III* stabilization at the fork with increasing [DnaG] can readily be explained by DnaB spending more time in a dilated-like state as the amount of DnaG increases, leading to progressively slower exchange. In addition, the observed tendency of Tapp to trend towards zero at [DnaG] = 0 implies that the principal state of translocating DnaB as it unwinds dsDNA is not dilated-like in the absence of DnaG–DnaB contacts. Further, our data reveal that the number of replisome-associated Pol III* at 100 nM DnaG, the concentration found in the cell (Rowen and Kornberg, 1978), is two to three, increasing to four copies at 300 nM (Figure 6E, right). Fitting the number of apparently exchanged Pol III* against [DnaG] to a steady-state equation (Equation 4, Method Details) shows that the maximum number of associated Pol III* can be as high as six, and the KM extracted from these data is 90 ± 30 nM (Figure 6E, right; discussed below).
DISCUSSION
In this report, we present evidence and conclude that:
-
i
There are at least two functional modes of interaction of Pol III* and DnaB on DNA as a result of transient interaction of DnaG with DnaB. Binding of DnaG induces a conformational change in the N-tier of DnaB from a state with low affinity for the CLC to one with high affinity, both on and off DNA, slowing down the dynamics of CLC–DnaB interaction. One possibility is that upon DnaG binding, the DnaB N-tier domains transition on DNA from the broken lock-washer state (Figure 1B, bottom) to a planar dilated state as seen for the N-tiers in structures of G. stearothermophilus DnaB (Figure 1B, top left) and E. coli DnaBdilated (Strycharska et al., 2013) off DNA. This structural transition would suggest that the two tiers in DnaB on DNA have to physically decouple upon DnaG binding. However, if this were true, DnaBdilated would maintain the strong CLC–DnaB binding mode on DNA and the DNA replication that is dependent on it would not be stimulated by DnaGC; this was not the case (Figure 4D, lanes 1 and 2). Given the still insufficient data on structural dynamics of DnaB as it moves on DNA, it could be that the N-domains transit through disordered states with low affinity for the CLC that simply become more ordered upon DnaG binding, adopting with only minor adjustments a broken spiral dilated-like organization similar to that seen in the structure of DnaB on DNA (Figure 1B, bottom), with high affinity for the CLC. The principal difference between these scenarios is in the number of DnaG molecules that DnaB can harbor: while the planar dilated conformation can host up to three (Figure S2A), the spiral state can bind a maximum of two, since one of the three DnaG-binding interfaces on the three pairs of neighboring DnaB subunits that host DnaG is “broken”. In support of the broken-like DnaB•DnaGC structure of the N-terminal tier in DnaB, we note that two DnaG bound to DnaB were found to be sufficient to modulate primer initiation and length (Corn et al., 2005).
-
ii
DnaB does not pause when it is bound to DnaG. The mechanism by which the E. coli replisome coordinates repetitive lagging-strand priming with DNA synthesis on both leading and lagging strands is not known (Dixon, 2009). The helicase may either pause during primer synthesis on the lagging strand or continue unimpeded unwinding of dsDNA at the fork as it remains in contact with the primase. This latter mechanism would result in the formation of a “priming loop” that collapses as the new primer is handed off from primase to polymerase (Manosas et al., 2009; Pandey et al., 2009). However, priming loops have not yet been observed in E. coli DNA replication. A critical difference between these mechanisms is whether DnaB pauses when it is bound to primase. To answer this question, we developed a cross-linked DnaB~GC construct, ensuring that three primases and helicase always remain associated. After confirming that DnaB~GC is in the dilated conformation in solution with high affinity for CLC (Figure 5A), we found that the construct remains an active helicase in DNA replication, arguing that DnaG–DnaB contact does not stall the helicase (Figure 5B, C). Our findings thus suggest that the formation of priming loops in E. coli is possible, and if it does not happen, there must be a rather specific mechanism in place to prevent it.
Note that in DnaB~GC, each DnaGC contacts a pair of DnaB subunits, whereas crosslinking is achieved through only one of them. Hence it is still possible to crack open one of the interfaces in the N-terminal tier upon loading of DnaB~GC onto DNA to adopt a spiral dilated-like organisation with high affinity for CLC whereby only two of the crosslinked DnaGC are proficiently bound to DnaB.
-
iii
Multiple Pol III* can reside at the fork depending on the status of primase–helicase interaction. The working model describing replication of the E. coli chromosome has suggested for over 30 years that only one Pol III* operates at the replication fork, resulting in the incorporation of hundreds of thousands of nucleotides per single DNA-binding event. In agreement with this model, previous in vivo measurements identified one or two Pol III* at each fork, with two Pol III* arising from converging replisomes in cells that are about to complete replication (Reyes-Lamothe et al., 2010). However, the resolution of the technique was insufficient to directly confirm the latter hypothesis, so the additional Pol III* could as well be part of the same replisome. Other problems inherent in the technique are the slow maturation of fluorescent protein probes in vivo as well as the population of tagged proteins whose probes never mature. While the authors went to great lengths to account for these potential issues, the question remains if the applied corrections perfectly address all of them. Moreover, recent measurements in live E. coli cells showed that Pol III* frequently disengages from the replisome during DNA synthesis and exchanges with free copies from solution (Beattie et al, 2017), while in vitro measurements showed that τ1Pol III* can participate as efficiently as τ2 and τ3Pol III* in DNA synthesis, with similar lengths and abundance of Okazaki-fragment synthesis (Yuan et al., 2016), suggesting that at least two different Pol III* can simultaneously operate on leading and lagging strands.
Here we measured multiple Pol III* in the diffraction limited spot at the growing tip of a rolling-circle template using a single-molecule technique and estimated that at physiological concentrations of replication proteins there are more than three Pol III* residing at or near the replication fork (Figure 6E, right). While the spatial resolution (with the diffraction limit corresponding to ~0.6 kb) in these measurements limits our conclusion, we believe that at least some of the Pol III* detected at higher [DnaG] are recruited onto DnaB due to an increase in DnaB–CLC affinity during priming, which requires DnaG–DnaB contact (Johnson et al., 2000). Indeed, the KD measured for the DnaG‒DnaB interaction (2.8 μM; Oakley et al., 2005) is 30-fold higher than the KM (90 ± 40 nM) we observe for the DnaG-dependent increase in the measured number of Pol III* (Figure 6E, right). That the fit KM is much closer to the KM for primer utilization (17 ± 3 nM; Graham et al., 2017) underscores the critical role of interaction between DnaG and the DNA template for the primase-induced conformational switch in the helicase and Pol III* recruitment. Nevertheless, we cannot exclude that the association of some of Pol III* within the diffraction-limited spot is caused by weak interactions between Pol III* and other replisomal components; measured numbers of Pol III* may represent overestimation of their true values at the fork.
-
iv
A single copy of τ in the CLC is responsible for binding DnaB. There are two possibilities that may lead to this experimental outcome: either each τ in the CLC can bind a single DnaB hexamer or there is a location-specific interaction of a particular τ subunit within the CLC; in either case the probability of finding τ in the interaction-proficient location within the τnCLC cf. τ1CLC is n-fold higher. Since progressively more time is needed to reach equilibrium with increasing number of τ in τnCLC–DnaBwt•DnaGC interactions (Figures 3A and S2G, H), likely due to mass transfer limitations arising from steric obstacles for multiple DnaB hexamers to pack against the single τ2/τ3CLC close to the chip surface, we favor the first possibility that each τ in a single CLC can bind a single DnaB. Nevertheless, we conclude in both cases that in the context of the CLC, the C-terminal domains unique to τ subunits are much less flexible than thought, unable to simultaneously reach multiple binding sites on a single DnaB hexamer. This offers an explanation how the coordination between replication events on the leading and lagging strands by a single Pol III HE could be more relaxed than previously thought (Graham et al., 2017).
A reason for this feature could be to accelerate dynamics of Pol III* traffic on and off DnaB, orchestrated by the transience of the DnaB–DnaG interaction, for timely engagement of new Pol III* in DNA synthesis at the replication fork. In favor of this proposal, in the background of the measured capacity of DnaG to control the number of associated Pol III* molecules and measured apparent exchange time Tapp in the diffraction-limited spot using sm-FRAP experiments, we carried out the following analysis: at 70 nM DnaG, a concentration approximate to that in the cell (Rowen and Kornberg, 1978), and sub-physiological 3 nM Pol III*, Tapp was found to be ~6 s (Figure S4B). We used 3 nM Pol III* to enable experimental access to apparent exchange times across the entire range of [DnaG] tested (30–300 nM), while the physiological ~25 nM fluorescent Pol III* (Lewis et al., 2017) was inaccessible due to low signal/background ratio. Previous measurements showed that Tapp is reduced ~6-fold upon increase in [Pol III*] from 3 to 13 nM (at fixed 300 nM DnaG; Lewis et al., 2017), so a reduction of Tapp to around 1 s and an increase in number of exchangeable Pol III*s to more than three would be expected at physiological Pol III* and DnaG concentrations. These numbers would suggest that the apparent exchange lifetime per individual associated Pol III* (Tapp divided by Pol III*max) is well below 1 s and on par with the cycle time of Okazaki fragment synthesis.
So why would there be a need for DnaG priming as a signal that triggers a helicase conformational change, which in turn recruits Pol III* to the vicinity of the replication fork? One obvious possibility is that a Pol III* newly recruited to DnaB could participate downstream in the primase-to-polymerase switch (Yuzhakov et al., 1999) and subsequent OF synthesis, operating in conjunction with a different Pol III* that is already replicating the leading strand. Such a model with multiple Pol III* complexes acting at the fork would allow for a large number of scenarios describing polymerase behavior in replication, including Pol III* being left behind on a nascent OF and the simultaneous synthesis of multiple OFs, as has already been observed for the T7 replisome (Geertsema et al., 2014; Duderstadt et al., 2016).
Taken together, we propose a model whereby DnaB serves as a mediator of Pol III* delivery onto primer-template DNA. In support, Yuan et al. (2016) showed that catalytically dead αD403E exchanged into the replisome, but only in the context of αD403E Pol III* and not αD403E Pol III that lacks the associated CLC that is needed for strong association with DnaB. Having one or more Pol III* associated with DnaB upon DnaG binding, ready to take over replication, is in accord with the recent paradigm shift in the field proposing a rather stochastic behavior of Pol III* at the replication fork, with new Pol III*s dynamically exchanging in the replisome while DnaB remains stably associated at the fork (Geertsema and van Oijen, 2013; van Oijen and Dixon, 2015; Yuan et al., 2016; Beattie et al., 2017; Lewis et al., 2017; Graham et al., 2017; Monachino et al., 2017). Consistent with our previous work (Geertsema et al., 2014), it is notable that three polymerases are detected in the cryo-EM structure of the T7 replisome, two engaged on DNA and one bound to the helicase-primase awaiting engagement on the lagging strand (Gao et al., 2019). Further work is necessary to deduce whether the proposed primase-to-polymerase switch via the primase-induced conformational change in the helicase represents a mechanism that is regularly utilized in OF synthesis, or whether it serves as a backup mechanism to handle roadblocks and obstructions, i.e., when priming on the leading strand becomes necessary or when there are delays in the recycling of the lagging-strand polymerase.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Antoine van Oijen (vanoijen@uow.edu.au).
Materials Availability
We are glad to share all unique/stable reagents with reasonable compensation by requestor for its shipping.
Data and Code Availability
Raw data are available upon request. All home-built ImageJ plugins used in this study are freely available on the Github repository for Single-molecule/Image analysis tools (https://github.com/SingleMolecule) or are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains
Recombinant Escherichia coli K12 proteins were from sources listed in the Key Resources Table or as described in Method Details.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| Escherichia coli BL21(λDE3)/pLysS | Studier et al., 1990 | N/A |
| Chemicals and Recombinant Proteins | ||
| EcoRI | New England Biolabs | R0101 |
| NdeI | New England Biolabs | R0111 |
| T4 DNA ligase | New England Biolabs | M0202 |
| dNTPs mix (dATP, dCTP, dGTP, dTTP) | Bioline | BIO-39044 |
| rNTPs mix (ATP, CTP, GTP,TTP) | Bioline | Discontinued |
| ATP | Sigma-Aldrich | A2383–10G |
| ADP | Sigma-Aldrich | A2754–1G |
| dithiothreitol (DTT) | Astral Scientific | C-1029–25G |
| 2-mercaptoethanol (β-mercaptoethanol) | Bio-Rad | 161–0710 |
| ethylenediaminetetra-acetic acid (EDTA) | Ajax-Finechem | AJA663–500G |
| EDTA disodium salt (Na2EDTA) | Ajax-Finechem | AJA180–500G |
| magnesium chloride (MgCl2) | Ajax-Finechem | AJA296–500G |
| magnesium acetate (Mg(OAc)2) | Sigma-Aldrich | M0631–100G |
| sodium chloride (NaCl) | Sigma-Aldrich | S9888–5KG |
| potassium glutamate (K-glutamate) | Sigma-Aldrich | G1501–1KG |
| ammonium sulfate ((NH4)2SO4) | Ajax-Finechem | AJA56–500G |
| hydrochloric acid 36% (HCl) | Ajax-Finechem | AJA1367–2.5L |
| glacial acetic acid (HOAc) | Ajax-Finechem | AJA1–2.5L |
| formic acid | Sigma-Aldrich | F0507–500ML |
| sodium hydroxide (NaOH) | Ajax-Finechem | AJA482–500G |
| urea | Sigma-Aldrich | U6504–1KG |
| glycerol | ChemSupply | GA010–2.5L |
| sodium dodecyl sulfate (SDS) | Sigma-Aldrich | L3771–1KG |
| Trizma base (Tris) | Sigma-Aldrich | T1503–1KG |
| bicine | Sigma-Aldrich | B3876–250G |
| agarose | Bioline | BIO-41026 |
| (±)-6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma-Aldrich | 238813–5G |
| catalase | Sigma-Aldrich | C9322–1G |
| D-(+)-glucose (glucose) | Sigma-Aldrich | G8270–1KG |
| glucose oxidase (GOx) | Sigma-Aldrich | G2133–50KU |
| mPEG-succinimidyl valerate, MW5000 (mPEG-SVA) | Laysan Bio, Inc. | MPEG-SVA-5000–5g |
| biotin-PEG-SVA, MW5000 | Laysan Bio, Inc. | Biotin-PEG-SVA-5000–100mg |
| polydimethylsiloxane (184 Silicone Elastomer; PDMS) | Sylgard | 500GM184KIT |
| ethidium bromide (EtBr) | Sigma | E1510–10ML |
| isopropyl-β-D-thiogalactoside (IPTG) | Astral Scientific | AST-0487–25G |
| spermidine trihydrochloride (spermidine-3HCl) | Sigma-Aldrich | S2501–25G |
| Tween20 | Sigma-Aldrich | P1379–100ML |
| surfactant P20 | GE Healthcare | BR100054 |
| SYBR Gold nucleic acid gel stain, 10,000x | Life Technologies | S11494 |
| GeneRuler DNA ladder mix, ready-to-use | ThermoFisher Scientific | SM0331 |
| agarose gel loading dye (6x, alkaline) | Boston BioProducts | BM-100AL |
| DNA gel loading dye (6x) | ThermoFisher Scientific | R0611 |
| λ DNA/HindIII marker | ThermoFisher Scientific | SM0103 |
| bovine serum albumin (BSA) | Sigma-Aldrich | 05470–5G |
| thymine | Sigma-Aldrich | T0895–25G |
| ampicillin | Sigma-Aldrich (Roche) | 10835269001 |
| chloramphenicol | Sigma-Aldrich | C0378–100G |
| D-biotin | Sigma-Aldrich | B4501–1G |
| biotin ligase | Jergic et al., 2007 | N/A |
| Pol III core assemblies: αεθ and SNAP649-αεθ (90%-labeled) | Lewis et al., 2017 | N/A |
| clamp loader subassemblies: χψτ3δδ’ and τ3δδ’ | Tanner et al., 2008 | N/A |
| minimal clamp loader subassembly: γ3δδ’ | Jergic et al., 2013 | N/A |
| Pol III holoenzyme sliding clamp: β2 | Oakley et al., 2003 | N/A |
| helicase and helicase loader: DnaB and DnaC | San Martin et al., 1995 | N/A |
| helicase-helicase loader complex: DnaBC | Jergic et al., 2013 | N/A |
| modified helicases: DnaBconstr and DnaBdilated | Strycharska et al., 2013 | N/A |
| single-stranded binding protein: SSB | Mason et al., 2013 | N/A |
| primase: DnaG | Stamford et al., 1992 | N/A |
| helicase-binding domain of primase: DnaGC | Loscha et al., 2004 | N/A |
| modified helicase / DnaGC primase domain / cross-linked complex: DnaB6F102C, DnaGC3R568C/C492L, DnaB6F102C~DnaGC3R568C/C492L (DnaB~GC) | This paper | N/A |
| biotinylated Pol III HE χ subunit: bioχ | This paper | N/A |
| biotinylated clamp loader subassembly: bioχψ | This paper | N/A |
| biotinylated clamp loader subassemblies: bioχψτ3δδ’, bioχψτ2γ1δδ’, bioχψτ1γ2δδ’ |
This paper and Tanner et al., 2008 | N/A |
| Oligonucleotides | ||
| oligo 108: forward primer for the design of bioholC gene coding for bioχ, see Method Details | GeneWorks | N/A (custom order) |
| oligo 109: reverse primer for the design of bioholC gene coding for bioχ, see Method Details | GeneWorks | N/A (custom order) |
| Recombinant DNA | ||
| biotinylated flap primed 2-kb circular DNA template | Monachino et al., 2018 | N/A |
| pET-χ: plasmid for preparation of χ | Xiao et al., 1993 | N/A |
| pKO1274: biotinylation vector | Jergic et al., 2007 | N/A |
| pSJ1376: plasmid for preparation of bioχ | This work | N/A |
| Software and Algorithms | ||
| Excel | Microsoft | N/A |
| ImageJ/Fiji (1.51w) | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Custom ImageJ/Fiji scripts | Karl Duderstadt (Max Planck Institute for Biochemistry) | N/A |
| ImageQuant TL (v. 8.1) | GE Healthcare | N/A |
| Quantity One (v. 4.6.9) | Bio-Rad | N/A |
| BIAevaluation (v. 4.0.1) | Biacore | N/A |
| PyMOL (v. 1.4.1) | DeLano Scientific | N/A |
| IgorPro (v. 6.11) | WaveMetrics | N/A |
| Adobe Illustrator CC | Adobe Inc. | N/A |
METHOD DETAILS
Buffers
ALEM buffer: 2x agarose gel loading dye (6x) alkaline, 200 mM EDTA; alkaline buffer: 50 mM NaOH, 1 mM EDTA; biomix buffer: 50 mM Tris.HCl, 250 mM bicine, pH 8.3, 50 mM ATP, 50 mM Mg(OAc)2, 250 mM D-biotin; buffer A: 30 mM Tris.HCl, pH 7.6, 1 mM dithiothreitol, 1 mM EDTA; buffer B: 20 mM Tris.HCl, pH 7.6, 100 mM NaCl, 2 mM dithiothreitol, 0.5 mM EDTA; buffer C: 25 mM Tris.HCl, pH 7.6, 90 mM NaCl, 2 mM dithiothreitol, 0.5 mM EDTA, 10% v/v glycerol; cross-linking buffer: 30 mM Tris.HCl, pH 7.6, 100 mM NaCl, 5 mM MgCl2, 100 μM ADP, 15% v/v glycerol; imaging buffer: 30 mM Tris.HCl, pH 7.6, 12 mM Mg(OAc)2, 50 mM K-glutamate, 0.5 mM EDTA, 0.0025% (v/v) Tween 20, 0.5 mg/ml BSA, 1 mM freshly made UV-aged Trolox, 0.45 mg/ml glucose oxidase, 0.024 mg/ml catalase, 0.8% (w/v) glucose monohydrate, 10 mM dithiothreitol, 1.25 mM ATP, 0.25 mM each UTP, CTP, and GTP, 50 μM each dATP, dTTP, dCTP and dGTP; LES buffer: 2x DNA gel loading dye, 200 mM EDTA, 2% SDS; lysis buffer: 50 mM Tris.HCl, pH 7.6, 2 mM dithiothreitol, 1 mM EDTA, 20 mM spermidine; neutralization buffer: 1 M Tris.HCl, pH 7.6, 1.5 M NaCl; replication buffer A: 30 mM Tris.HCl, pH 7.6, 12 mM Mg(OAc)2, 50 mM K-glutamate, 0.5 mM EDTA, 0.0025% (v/v) Tween 20; replication buffer B: 30 mM Tris.HCl, pH 7.6, 12 mM Mg(OAc)2, 50 mM K-glutamate, 0.5 mM EDTA, 0.0025% (v/v) Tween20, 0.5 mg/ml BSA; SPR buffer: 25 mM Tris.HCl, pH 7.6, 50 mM NaCl, 5 mM MgCl2, 0.25 mM dithiothreitol, 0.005% (v/v) P20; TAE buffer: 40 mM Tris, 20 mM acetic acid, 1 mM EDTA (final pH 8.3).
Plasmid Construction
pSJ1376 (bioholC): The plasmid pET-χ (Xiao et al., 1993) that directs overproduction of full-length χ was a kind gift of Dr. Mike O’Donnell. It was used as a template for PCR amplification of the holC gene using primer 108 (5’-AAA AAA AAC ATA TGA AAA ACG CGA CGT TCT ACC TTC TGG-3’), designed to incorporate a methionine start codon as part of the NdeI site, and primer 109 (5’-TTG AAT TCT TAT TTC CAG GTT GCC GTA TTC AGG-3’), that incorporates an EcoRI restriction site just following the TAA stop codon. The PCR product was isolated after digestion with NdeI and EcoRI and inserted between the same set of restriction sites in plasmid pKO1274 (Jergic et al., 2007). Vector pKO1274, a derivative of pETMSCI (Neylon et al., 2000), allows fusion of the gene in-frame behind a N-terminal biotinylation tag: MAGLNDIFEAQKIEWHEH (Beckett et al., 1999) using an NdeI restriction site. The resulting plasmid pSJ1376 places the bioholC gene under the transcriptional control of the bacteriophage T7 ϕ10 promoter, which directs bioχ protein overproduction on addition of IPTG.
Overproduction and Purification of bioχ
E. coli strain BL21(λDE3)/pLysS/pSJ1376 was grown at 37°C in LB medium supplemented with thymine (25 mg/l), ampicillin (100 mg/l), chloramphenicol (30 mg/l) and 25 μM D-biotin to A600 = 0.8. To induce overproduction of bioχ, 0.75 mM IPTG was added to the shaking culture. Cultures were grown for a further 3 h, and then chilled in ice. Cells were harvested by centrifugation (11,000 × g; 6 min), frozen in liquid N2 and stored at −80°C. After thawing, cells (4.27 g, from 2 l of culture) were resuspended in 65 ml lysis buffer and lysed by being passed twice through a French press (12,000 psi). Cell debris was removed from the lysate by centrifugation (30,000 × g; 30 min) to yield the soluble Fraction I. Proteins in Fraction I that were precipitated by addition of solid ammonium sulfate (0.45 g/ml) and stirring for 60 min, were collected by centrifugation (35,000 × g; 30 min) and dissolved in 30 ml buffer A+150 mM NaCl. The solution was dialyzed against two changes of 2 l of the same buffer to yield Fraction II.
Fraction II was applied at 1 ml/min onto a column (2.5 × 15 cm) of Toyopearl DEAE-650M resin that had been equilibrated against the buffer A+150 mM NaCl. Fractions containing χbio that did not bind to the resin were pooled and dialyzed against two changes of 2 l of buffer A+30 mM NaCl to yield Fraction III (50 ml), which was loaded at 1 ml/min onto the same DEAE column that had been equilibrated with buffer A+30 mM NaCl. The column was washed with the same buffer and bioχ eluted between 1–3 column volumes in a broad peak. Fractions containing bioχ were pooled and dialyzed against two changes of 2 l of buffer A+30 mM NaCl to give Fraction IV (120 ml), which was loaded at 1 ml/min onto a column (2.5 × 10 cm) of Toyopearl SuperQ that had been equilibrated with buffer A+30 mM NaCl. After washing with 60 ml of buffer A+30 mM NaCl, bioχ eluted in a linear gradient (300 ml) of 30–160 mM NaCl in buffer A, in a single peak at ~70 mM NaCl. Fractions containing bioχ were pooled and dialyzed against two changes of 2 l of buffer A+30 mM NaCl to give Fraction V (35 ml), which was loaded at 1 ml/min onto a column (2.5 × 15 cm) of heparin-Sepharose 4B (Wijffels et al., 2004) that had been equilibrated in buffer A+30 mM NaCl. The column was washed with the same buffer and bioχ eluted between 1–3 column volumes in a broad peak. This purification step did not contribute to improvement in the purity of the protein. Fractions containing bioχ were pooled to give Fraction VI.
Proteins in Fraction VI (80 ml) were then precipitated by addition of solid ammonium sulfate (0.45 g/ml) and stirring for 60 min. Precipitated proteins were collected by centrifugation (35,000 × g; 30 min), then dissolved in 5 ml buffer A+100 mM NaCl and finally dialyzed against 2 l of the same buffer to yield Fraction VII (6 ml, containing 8 mg of the pure protein).
The mol. wt. of purified bioχ determined by nanoESI-MS in 1% formic acid containing 1 mM β-mercaptoethanol (18,650.8 ± 0.2 Da and 18,779.1 ± 0.1 Da) indicated that the N-terminal methionine had been partially removed (in ~80% of proteins) and that biotinylation had not taken place, so the protein was biotinylated in vitro, as follows. One part of biomix buffer was mixed with three parts of substrate solution (Fraction VII, 70 μM bioχ) in buffer A+100 mM NaCl and one part of water, and biotin ligase added to 0.8 μM in final volume of 6.8 ml. This biotinylation mix was treated at 30°C for 3 h and then dialyzed in 2 l of buffer A+100 mM NaCl at 6°C for storage, yielding Fraction VIII (7.5 ml, containing ~6 mg of bioχ in the presence of some biotin ligase). Aliquots were stored at −80°C.
The mol. wt. of in vitro biotinylated bioχ determined by nanoESI-MS in 1% formic acid, 1 mM β-mercaptoethanol (18,877.1 ± 0.1 Da and 19,005.6 Da) compares well to the calculated value of 18,878 Da in the absence of initiating Met and 19,009 Da when Met is present.
Biotinylated Clamp Loader Complexes
Refolding of ψ in the presence of bioχ was carried out based on methods described by Tanner et al. (2008) with some modifications. About 6 ml of bioχ (~4.8 mg) in buffer A+100 mM NaCl was added to 9 ml buffer B while stirring, followed by dropwise addition of 1 ml (~10 mg) of ψ in 6 M urea. Consequently, the final concentration of urea in solution was ~0.4 M, a condition that allows ψ to fold and interact with χ. The solution was stirred for 4 h at 4°C and then dialyzed overnight in 2 l buffer C.
Following extensive dialysis, the solution was clarified by centrifugation (35,000 × g; 30 min) and the soluble fraction loaded at 1 ml/min onto a column (2.5 × 7 cm) of Toyopearl DEAE-650M that had been equilibrated in buffer C. Fractions containing bioχψ that did not bind to the column were pooled (12 ml, containing 4 mg of protein complex) and stored at −80°C.
Isolation of bioχψτ3δδ’, bioχψτ2γ1δδ’ and bioχψτ1γ2δδ’ (Figure S1A) followed methods for production of non-biotinylated CLCs (Tanner et al., 2008) except that biotinylated bioχψ was used in place of χψ during assembly of complexes.
Preparation of Crosslinked DnaB~GC
Cross-linking susceptible DnaBF102C and DnaGCR568C/C492L were isolated according to procedures used for the isolation of wild-type proteins (San Martin et al., 1995; Loscha et al., 2004). The facile disulfide cross-linking of three (every other) protomers in the DnaBF102C hexamer with three DnaGCR568C/C492L (each DnaGC crosslinks through only one of the two asymmetric contact points in the pair of neighboring DnaB protomers) was achieved by dialysis of DnaBF102C with excess of DnaGC3R568C/C492L (two DnaGCR568C/C492L per DnaBF102C monomer) in cross-linking buffer at 4°C, with four buffer changes over two days to remove dithiothreitol. Samples of DnaB~GC were frozen in liquid N2 and stored at −80°C.
Bulk DNA Replication Assays
Ensemble-averaged (bulk) DNA replication experiments aimed at comparing activities of DnaBdilated and DnaBconstr in the context of both leading-strand synthesis and simultaneous leading- and lagging-strand synthesis were commenced by mixing on ice: 3.8 nM biotinylated flap-primed 2-kb circular DNA template (Monachino et al., 2018), 1.25 mM ATP, 250 μM each UTP, CTP, and GTP, 200 μM each dATP, dTTP, dCTP, and dGTP, 30 nM χψτ3δδ’, 90 nM Pol III core, 200 nM β2, 50 nM SSB, 60 nM DnaBwt, DnaBconstr or DnaBdilated (as hexamers) and 360 nM DnaC in replication buffer A in 10 μl reaction volume. When specified, 300 nM DnaG was used for RNA priming on the lagging strand, allowing lagging-strand synthesis to proceed. Since DnaBconstr does not load efficiently in the presence of SSB (not shown), the reactions were initiated in a waterbath at 37°C for 1 min in absence of SSB to preload helicase. Following the addition of SSB, reactions were treated at 37°C for further 14 min. In this way, differences in efficiencies of DNA synthesis among the reactions containing different DnaB versions are mainly due to the elongation phase and not to the loading efficiency. Reactions were quenched by mixing equal volumes of replication solution with ALEM buffer, followed by heating in a waterbath at 70°C for 5 min and prompt cooling on ice for at least 3 min. Reaction products were then resolved by alkaline gel electrophoresis in a 0.5% (w/v) agarose gel. GeneRuler DNA Ladder mix (4 μl) in 1x agarose gel loading dye (6x, alkaline; final volume: 12 μl) was loaded as markers. The alkaline agarose gels were soaked for at least 1 h in alkaline buffer before the reaction products and the markers were loaded. Gels were electrophoresed at 15 V for ~15 h in a Mini-Sub Cell GT System (Bio-Rad). Then, gels were treated in neutralization buffer for ~2 h, and stained with 1x SYBR Gold in 2x TAE buffer for 7 h. The SYBR Gold-stained DNA molecules were imaged with a Bio-Rad Gel Doc XR instrument (302 nm trans-UV light; Figure 2F) using Quantity One software.
Bulk leading-strand replication reactions in the absence of β2 clamps, SSB and χψ were assembled by mixing on ice: 3.8 nM biotinylated flap-primed 2-kb circular DNA template, 1 mM ATP, 400 μM each dATP, dTTP, dCTP, and dGTP, 30 nM τ3δδ’ or γ3δδ’, 90 nM Pol III core, 30 nM DnaBwt, DnaBdilated, DnaBconstr, DnaBF102C or DnaB~GC, and 10 mM dithiothreitol in replication buffer A in 10 μl final volume (Figures 4B, C and S3B). DnaGC concentration is declared in each experiment. Dithiothreitol was omitted in the reactions that compare the activities of DnaBwt with DnaBdilated and DnaBconstr (Figure 4D) and of DnaB~GC with DnaBF102C (Figure 5B) to avoid reduction of the disulfide crosslinks in DnaBdilated or DnaB~GC. In case of (−χψ)Pol III* titration experiments, the complex was assembled by premixing identical volumes of Pol III core at 6.3 μM and τ3δδ’ at 2.1 μM for 5 min at room temperature (Figure S3A). Unless otherwise noted, reactions proceeded in a water bath at 30°C for 80 min, then were quenched by mixing equal volumes of replication solution and LES buffer. Reaction products were separated by gel electrophoresis in 0.66% (w/v) agarose gels in 2x TAE buffer for 100 min at 75 V in a Mini-Sub Cell GT System (Bio-Rad); λ/HindIII DNA digest was loaded as marker. Gels were stained with 1x SYBR Gold in 2x TAE buffer for 2 h and imaged as above.
Time courses of bulk leading-strand synthesis reactions with DnaBF102C and DnaB~GC in the presence of β2 used mixtures assembled as follows: a 60 μl-mix containing 3.8 nM biotinylated flap-primed 2-kb circular DNA template, 1 mM ATP, 400 μM each dATP, dTTP, dCTP, and dGTP, 30 nM χψτ3δδ’, 90 nM Pol III core, 200 nM β2, and 30 nM DnaBF102C or DnaB~GC was prepared by mixing components in replication buffer A on ice; 10 μl were removed for each condition and mixed with the same volume of LES buffer to provide the 0 min time-point. The remaining volumes were transferred in a water bath at 37°C. At indicated times, 10 μl were removed for each condition and mixed with the same volume of LES buffer to quench reactions. Products were separated by gel electrophoresis in 0.66% (w/v) agarose gels; GeneRuler DNA Ladder mixes were loaded as marker. Gels were run in 2x TAE buffer for 150 min at 60 V in a Wide Mini-Sub Cell GT System (Bio-Rad), followed by staining with 1x SYBR Gold in 2x TAE buffer for 2 h (Figure 5C).
Quantification of DNA bands in gels was done using GE Healthcare Life Sciences ImageQuant TL software. Lanes were manually identified. The “rubber band” background subtraction algorithm was used. The bands corresponding to the 2-kb DNA template were manually detected and their intensity was calculated by the software (Figures 4B, C, and 5B, C).
Surface Plasmon Resonance (SPR) Experiments
SPR experiments that enabled determination of the dissociation constants KD for CLC–DnaBwt and mutant DnaB versions, in the absence and presence of DnaGC, as well as CLC•DnaBwt–DnaGC used a BIAcore T200 (GE Healthcare) instrument at 20°C, unless stated otherwise. First, a streptavidin-coated (SA) sensor chip (GE Healthcare) was activated with three sequential injections of 1 M NaCl, 50 mM NaOH (40 s each at 5 μl/min). Then, bioχψτ1γ2δδ’ and bioχψτ3δδ’ were immobilized separately on two flow cells to 1400 and 1450 RU, respectively, in SPR buffer with 0.2 mM ATP for stabilization of CLCs on the sensor chip surface. Binding studies were initiated by sequential injections of solutions of serially-diluted DnaBwt (0.0625–8 μM, including zero) in SPR buffer+0.2 mM ADP at 30 μl/min for 30 s; all concentrations of DnaB are expressed as hexamers. While use of ADP (instead of ATP) did not influence measured values of binding at equilibrium (Req), it was found to reduce unspecific interactions at high [DnaB] and formation of species that were slow to dissociate, which becomes a critical contribution because CLCs on the chip surface cannot be regenerated. Sensorgrams were zero-subtracted and Req values, generated by averaging response values in the gray highlighted region (in Figures) from the appropriate DnaB concentration range, fit against [DnaB] using a 1:1 steady state affinity (SSA) model incorporated in the BIAevaluation software (Figures 2B and S1C):
| (Equation 1) |
where Rmax corresponds to the response when all the immobilized ligands on the surface are saturated with the analyte A, KD is the dissociation constant, and [A] is the concentration of analyte in solution. In general, raw sensorgrams and fit curves (where applicable) were exported using Excel, then plotted in IgorPro, and final Figures prepared in Adobe Illustrator CC.
The τ3CLC–DnaBconstr interaction was measured under similar conditions, except that the temperature was 25°C (Figure 2C). Binding studies for τ3CLC•DnaB–DnaGC interaction were also done under similar conditions by sequential injections of solutions containing fixed (125 nM) DnaB and serially-diluted DnaGC samples (0.5–8 μM, including zero; Figure S2D) at 30 μl/min for 30 s. Analyses of the data were performed using the SSA model as described above. In contrast, preliminary analysis showed that DnaBdilated binds much more strongly to τ3CLC, with much different kinetic parameters, so for illustrative purposes 250 nM DnaBdilated was injected for 400 s under the same conditions used for DnaBconstr, except that the slow dissociation was recorded for over 2,500 s (Figure 2D).
For measurement of KD values of the bioχψτ3δδ’–DnaBwt•DnaGC, bioχψτ2γ1δδ’–DnaBwt•DnaGC and bioχψτ1γ2δδ’–DnaBwt•DnaGC interactions, a range of solutions of serially-diluted DnaBwt (0.5–64 nM, including zero) in the presence of 5 μM DnaGC were injected at 30 μl/min for 150 s over immobilized bioχψτ3δδ’ (1,830 RU), bioχψτ2γ1δδ’ (1955 RU) and bioχψτ1γ2δδ’ (1,630 RU), in SPR buffer containing 0.2 mM ATP and 0.2 mM EDTA, followed by flow of the same buffer (Figure S2F). Given that we varied [DnaB], whereas the DnaB•DnaGC complex and not DnaB titrates the immobilized ligand in the low-nM range of [DnaB], and that CLC does not affect the DnaB–DnaGC interaction, the concentration of DnaB•DnaGC in solution was obtained by solving the quadratic equation for x derived from Equation 2:
| (Equation 2) |
where x is solution [DnaB•DnaGC], [DnaB]0 and [DnaGC]0 are the initial concentrations of DnaB and DnaGC (a constant value, equal to 5 μM), and the KD(DnaB–DnaGC) is the dissociation constant for the interaction of DnaB with DnaGC (1.74 ± 0.09 μM; Figure S2D). This equation is an approximation derived based on 1:1 interaction between the DnaB and the first weakly-bound molecule of DnaGC, a property previously also observed by Oakley et al. (2005), in the background of positive cooperative interaction with the second and third DnaGC that each bind >10-fold more strongly compared to the first one (up to three DnaGCs allosterically bind to DnaB). Given the appropriate range of 0.5–32 nM of serially-diluted [DnaB]0 made to flow over τ3CLC and τ2CLC or 0.5–64 nM in the case of τ1CLC, Equation 2 was used to calculate the theoretical [DnaB•DnaGC] in samples injected over the immobilized ligand: 0, 0.4, 0.7, 1.5, 3.0, 5.9, 11.8 and 23.7 nM in case of τ3CLC and τ2CLC (Figures 3A and S2H), and an extra 47.1 nM injection in case of τ1CLC (Figure S2G).
To study how the presence of DnaGC affects dissociation of DnaB from the CLC, 32 nM DnaBwt with 5 μM DnaGC were injected over immobilized τ3CLC in SPR buffer with 0.2 mM ATP and 0.2 mM EDTA at 5 μl/min for 60 s, and then the solution of 5 μM DnaGC in the same buffer was “coinjected” for 1000 s immediately following the association phase to monitor dissociation of DnaB. Finally, the remaining proteins were washed with the SPR buffer (Figure 3B). In addition, similar conditions were used to test binding of DnaBF102C or disulfide-bond cross-linked DnaB~GC with immobilized τ3CLC, except that the proteins (0.5 μM each) were subsequently injected at 30 μl/min for 30 s, and the dissociation of DnaB~GC monitored for 1,720 s, followed by injection for 250 s of the buffer in use that had been additionally supplemented with 1 mM dithiothreitol to uncouple DnaBF102C from DnaGCR568C/C49L, stimulating the dissociation of DnaBF102C (Figure 5A).
Stoichiometry of CLC/DnaB•DnaGC Complexes
SPR responses (in response units, RU) are proportional to the mass of proteins bound at a chip surface, which allows measurement of the responses during immobilization of τ1CLC, τ2CLC and τ3CLC ligands (shown in Table 1) to first be normalized by the ratios of mol. wt. of the CLCs (Table 1) to determine relative ligand densities at the surface (LDrel). Next, fitting SPR responses at equilibrium for binding of DnaB•DnaGC to the immobilized CLCs using a 1:1 SSA model generates an Rmax value that corresponds to the theoretical response for binding at saturation, i.e., when all the immobilized ligand molecules (CLCs) have been bound to DnaB•DnaGC from the solution. In the case of the τ1CLC ligand, when only one helicase can bind per immobilized CLC, the fit Rmax was 163 ± 8 RU (Figure S2G), whereas in the case of τ2CLC and τ3CLC, Rmax values were 410 ± 10 RU (Figure S2H) and 464 ± 10 RU (Figure 3A), respectively. These values of Rmax were then adjusted to compensate for the differences in the corresponding LDrel to give normalized Rmax (Table 1). Finally, since only one helicase complex can bind per immobilized τ1CLC, the ratio of the normalized Rmax values for the τ2CLC and τ3CLC surfaces relative to that for τ1CLC estimates the maximum numbers of DnaB•DnaGC molecules capable of binding to each τ2CLC and τ3CLC (Table 1).
In Vitro Single-Molecule FRAP Experiments
In vitro single-molecule fluorescence recovery after photobleaching (sm-FRAP) experiments were carried out essentially as described previously (Lewis et al., 2017). Briefly, a microfluidic flow cell was built by positioning a PDMS flow chamber on top of a PEG-biotin-functionalized microscope coverslip. To reduce non-specific interactions of proteins and DNA molecules with the surface, the chamber was blocked with replication buffer B. The flow cell was placed on an inverted microscope (Nikon Eclipse Ti-E, Japan) with a CFI Ap TIRF 100x oil-immersion TIRF objective (1.49 NA, Nikon, Japan) and connected to a syringe pump (New Era Pump Systems, Adelab Scientific, Australia) for flow of buffer. Flow-cell temperature was maintained at 31°C by an electrically heated chamber (Okolab, Burlingame, CA).
Leading- and lagging-strand replication experiments were performed under continuous presence of all proteins except DnaBC and as a function of [DnaG] in the 30–300 nM range. Biotinylated flap-primed 2-kb circular DNA template (340 pM) was treated with 40 nM DnaBC in replication buffer B containing 10 mM dithiothreitol and 1 mM ATP for 3 min at 37°C. This mixture was then diluted 10 times to a final volume of 220 μl, and loaded into the flow cell first at 40 μl/min for 2.5 min, then at 10 μl/min for 7.5 min, and finally in absence of flow for few minutes (Figure 6A, top). During the loading process, the imaging buffer was made. SNAP-Pol III* was assembled in situ by treating 280 nM τ3CLC with 850 nM SNAP649-Pol III in imaging buffer for 90 s at 37°C. Finally, the replication solution, which contained 3 nM SNAP-Pol III*, 40 nM β2, 250 nM SSB, and 30, 70, 150, or 300 nM DnaG in imaging buffer, was loaded into the flow cell first at 20 μl/min for 3.5 min, then at 10 μl/min until the end of the experiment (Figure 6A, bottom).
The fluorescently-labeled Pol III* was visualized by excitation with a 647 nm laser (Coherent, Obis 647–100 CW) at 1.3 W/cm2 (photo-bleaching lifetime was 40 s) with an exposure time of 200 ms. Every 20 s, every Pol III* in the field of view was photo-bleached with a 2-s pulse at 130 W/cm2 (photo-bleaching lifetime was 0.7 s) (Figure 6B, top). Imaging was done with an Evolve 512 Delta EMCCD (Photometrics, Tucson, AZ). The camera allowed a resolution of 160 nm/px. Because of the high efficiency of E. coli DNA replication, only one field of view per experiment was recorded for 5 min. Each experimental condition was investigated at least twice.
Data analysis was done with Fiji, using in-house built plugins and macros. Briefly, using Fiji, every field of view was corrected to account for background and beam profile. Individual replicating DNA molecules were manually located. Then, the position of the fluorescently-labeled Pol III* at the tip was semi-manually tracked (Figure 6B, top) and its integrated intensity calculated in a 5 px-by-5 px square, applying a local background subtraction (Figure 6B, bottom). By calibrating the intensity of a single SNAP649-Pol III, we could convert intensities into number of Pol III*s. At a fixed DnaG concentration, only those recovery intervals where replication occurred were averaged together and fit with the following FRAP recovery function using IgorPro (Equation 3):
| (Equation 3) |
where Pol III*max is the maximum number of exchanged Pol III*, Tapp is the characteristic apparent exchange time, and y0 accounts for incomplete background removal and was further subtracted from recovery intervals (Figure 6C, D and Figure S4A–C). We averaged recovery intervals from at least 21 individual DNA molecules for each DnaG concentration. Shown error bars are standard errors of the mean (normalized to the number of recovery intervals). The resulting Pol III*max and Tapp values, plotted against DnaG concentration, were fit with a steady-state affinity function (Equation 4):
| (Equation 4) |
where ymax represents the maximum value reached by y (either Pol III*max or Tapp) when the concentration of DnaG approaches infinity, and KM is a pseudo-Michaelis constant (Figure 6E).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of molecules or events analyzed is indicated in the text or figure legends. Errors reported in this study represent the standard error of the mean (SEM) or the error of the fit, as indicated in the text or figure legends. All SPR and single-moleculae data are representative of those from at least two technical replicates per experiment.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to Karl Duderstadt and Christiaan Punter for ImageJ plugins, Yao Wang for providing protein reagents, Lisanne Spenkelink for development of and assistance with the sm-FRAP assays, and Harshad Ghodke for fruitful discussions. This work was supported by the Australian Research Council (DP150100956 and DP180100858 to A.M.v.O. and N.E.D. and an Australian Laureate Fellowship, FL140100027 to A.M.v.O.), King Abdullah University of Science and Technology, Saudi Arabia (OSR-2015-CRG4–2644 to N.E.D. and A.M.v.O), Nederlandse Organisatie voor Wetenschappelijk Onderzoek (12CMCE03 to E.M.), and the National Institutes of Health (NIGMS R37–071747 to J.M.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alberts BM, Johnson A, Lewis J, Raff M, Roberts K, and Walter P (2007). Molecular Biology of the Cell, 5th Ed. (Ney York: Garland Science; ). [Google Scholar]
- Anderson SG, Williams CR, O’Donnell M, and Bloom LB (2007). A function for the ψ subunit in loading the Escherichia coli DNA polymerase sliding clamp. J. Biol. Chem 282, 7035–7045. 10.1074/jbc.M610136200 [DOI] [PubMed] [Google Scholar]
- Arias-Palomo E, Puri N, O’Shea Murray VL, Yan Q, and Berger JM (2019). Physical basis for the loading of a bacterial replicative helicase onto DNA. Mol. Cell 74, 173–184.e4. 10.1016/j.molcel.2019.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey S, Eliason WK, and Steitz TA (2007). Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318, 459–463. 10.1126/science.1147353 [DOI] [PubMed] [Google Scholar]
- Beattie TR, Kapadia N, Nicolas E, Uphoff S, Wollman AJ, Leake MC, and Reyes-Lamothe R (2017). Frequent exchange of the DNA polymerase during bacterial chromosome replication. eLife 6, e21763 10.7554/eLife.21763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E, and Schatz PJ (1999). A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8, 921–929. 10.1110/ps.8.4.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird LE, Pan H, Soultanas P, and Wigley DB (2000). Mapping protein–protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry 39, 171–182. 10.1021/bi9918801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujalowski W, Klonowska MM, and Jezewska MJ (1994). Oligomeric structure of Escherichia coli primary replicative helicase DnaB protein. J. Biol. Chem 269, 31350–31358. [PubMed] [Google Scholar]
- Corn JE, Pease PJ, Hura GL, and Berger JM (2005). Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell 20, 391–401. 10.1016/j.molcel.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Dallmann HG, Kim S, Pritchard AE, Marians KJ, and McHenry CS (2000). Characterization of the unique C terminus of the Escherichia coli τ DnaX protein. Monomeric C-τ binds α and DnaB and can partially replace τ in reconstituted replication forks. J. Biol. Chem 275, 15512–15519. 10.1074/jbc.M909257199 [DOI] [PubMed] [Google Scholar]
- Dixon NE (2009). DNA replication: prime-time looping. Nature 462, 854–855. 10.1038/462854a [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, and McHenry CS (2005). A bipartite polymerase–processivity factor interaction: only the internal β binding site of the α subunit is required for processive replication by the DNA polymerase III holoenzyme. J. Mol. Biol 350, 228–239. 10.1016/j.jmb.2005.04.065 [DOI] [PubMed] [Google Scholar]
- Dohrmann PR, Correa R, Frisch RL, Rosenberg SM, and McHenry CS (2016). The DNA polymerase III holoenzyme contains γ and is not a trimeric polymerase. Nucleic Acids Res. 44, 1285–1297. 10.1093/nar/gkv1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duderstadt KE, Geertsema HJ, Stratmann SA, Punter CM, Kulczyk AW, Richardson CC, and van Oijen AM (2016). Simultaneous real-time imaging of leading and lagging strand synthesis reveals the coordination dynamics of single replisomes. Mol. Cell 64, 1035–1047. 10.1016/j.molcel.2016.10.028 [DOI] [PubMed] [Google Scholar]
- Fernandez-Leiro R, Conrad J, Scheres SH, and Lamers MH (2015). Cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife 4, e11134 10.7554/eLife.11134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, and McHenry CS (2001a). τ binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of τ, binds the replication fork helicase, DnaB. J. Biol. Chem 276, 4441–4446. 10.1074/jbc.M009830200 [DOI] [PubMed] [Google Scholar]
- Gao D, and McHenry CS (2001b). τ binds and organizes Escherichia coli replication proteins through distinct domains. Partial proteolysis of terminally tagged τ to determine candidate domains and to assign domain V as the α binding domain. J. Biol. Chem 276, 4433–4440. 10.1074/jbc.M009828200 [DOI] [PubMed] [Google Scholar]
- Gao Y, Cui Y, Fox T, Lin S, Wang H, de Val N, Zhou ZH, and Yang W (2019) Structures and operating principles of the replisome. Science. 363, pii: eaav7003 10.1126/science.aav7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertsema HJ, Kulczyk AW, Richardson CC, and van Oijen AM (2014). Single-molecule studies of polymerase dynamics and stoichiometry at the bacteriophage T7 replication machinery. Proc. Natl. Acad. Sci. USA 111, 4073–4078. 10.1073/pnas.1402010111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geertsema HJ, and van Oijen AM (2013). A single-molecule view of DNA replication: the dynamic nature of multi-protein complexes revealed. Curr. Opin. Struct. Biol 23, 788–793. 10.1016/j.sbi.2013.06.018 [DOI] [PubMed] [Google Scholar]
- Glover BP, and McHenry CS (1998). The χψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem 273, 23476–23484. 10.1074/jbc.273.36.23476 [DOI] [PubMed] [Google Scholar]
- Graham JE, Marians KJ, and Kowalczykowski SC (2017). Independent and stochastic action of DNA polymerases in the replisome. Cell 169, 1201–1213.e17. 10.1016/j.cell.2017.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis JM, Kazmirski SL, Finkelstein J, Kelman Z, O’Donnell M, and Kuriyan J (2004). Crystal structure of the chi:psi sub-assembly of the Escherichia coli DNA polymerase clamp-loader complex. Eur. J. Biochem 271, 439–449. 10.1046/j.1432-1033.2003.03944.x [DOI] [PubMed] [Google Scholar]
- Itsathitphaisarn O, Wing RA, Eliason WK, Wang J, Steitz TA (2012). The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151, 267–277. 10.1016/j.cell.2012.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jergic S, Horan NP, Elshenawy MM, Mason CE, Urathamakul T, Ozawa K, Robinson A, Goudsmits JMH, Wang Y, Pan X, et al. (2013). A direct proofreader-clamp interaction stabilizes the Pol III replicase in the polymerization mode. EMBO J. 32, 1322–1333. 10.1038/emboj.2012.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jergic S, Ozawa K, Williams NK, Su X-C, Scott DD, Hamdan SM, Crowther JA, Otting G, and Dixon NE (2007). The unstructured C-terminus of the τ subunit of Escherichia coli DNA polymerase III holoenzyme is the site of interaction with the α subunit. Nucleic Acids Res. 35, 2813–2824. 10.1093/nar/gkm079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeruzalmi D, O’Donnell M, and Kuriyan J (2001). Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell 106, 429–441. 10.1016/S0092-8674(01)00463-9 [DOI] [PubMed] [Google Scholar]
- Johnson SK, Bhattacharyya S, and Griep MA (2000). DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry 39, 736–744. 10.1021/bi991554l [DOI] [PubMed] [Google Scholar]
- Kelman Z, and O’Donnell M (1995). DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu. Rev. Biochem 64, 171–200. 10.1146/annurev.bi.64.070195.001131 [DOI] [PubMed] [Google Scholar]
- Kim S, Dallmann HG, McHenry CS, and Marians KJ (1996). Coupling of a replicative polymerase and helicase: a τ-DnaB interaction mediates rapid replication fork movement. Cell 84, 643–650. 10.1016/S0092-8674(00)81039-9 [DOI] [PubMed] [Google Scholar]
- Kodaira M, Biswas SB, and Kornberg A (1983). The dnaX gene encodes the DNA polymerase III holoenzyme τ subunit, precursor of the γ subunit, the dnaZ gene product. Mol. Gen. Genet 192, 80–86. [DOI] [PubMed] [Google Scholar]
- Kong X-P, Onrust R, O’Donnell M, and Kuriyan J (1992). Three-dimensional structure of the β subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell 69, 425–437. [DOI] [PubMed] [Google Scholar]
- Kornberg A, and Baker TA, 1991. DNA Replication, 2nd Ed. (New York: W.H. Freeman and Co.). [Google Scholar]
- Lee J-B, Hite RK, Hamdan SM, Xie XS, Richardson CC, and van Oijen AM (2006). DNA primase acts as a molecular brake in DNA replication. Nature 439, 621–624. 10.1038/nature04317 [DOI] [PubMed] [Google Scholar]
- Lewis JS, Jergic S, and Dixon NE (2016). The E. coli DNA replication fork. The Enzymes 39, 31–88. 10.1016/bs.enz.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Lewis JS, Spenkelink LM, Jergic S, Wood EA, Monachino E, Horan NP, Duderstadt KE, Cox MM, Robinson A, Dixon NE, and van Oijen AM (2017). Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife 6, e23932 10.7554/eLife.23932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loparo JJ, Kulczyk AW, Richardson CC, and van Oijen AM (2011). Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc. Natl. Acad. Sci. USA 108, 3584–3589. 10.1073/pnas.1018824108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscha K, Oakley AJ, Bancia B, Schaeffer PM, Prosselkov P, Otting G, Wilce MCJ, and Dixon NE (2004). Expression, purification, crystallization, and NMR studies of the helicase interaction domain of Escherichia coli DnaG primase. Protein Expr. Purif 33, 304–310. 10.1016/j.pep.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Maki H, and Kornberg A (1985). The polymerase subunit of DNA polymerase III of Escherichia coli. II. Purification of the α subunit, devoid of nuclease activities. J. Biol. Chem 260, 12987–12992. [PubMed] [Google Scholar]
- Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, and Croquette V (2009). Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat. Chem. Biol 5, 904–912. 10.1038/nchembio.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason CE, Jergic S, Lo ATY, Wang Y, Dixon NE, and Beck JL (2013). Escherichia coli single-stranded DNA-binding protein: nanoESI-MS studies of salt-modulated subunit exchange and DNA binding transactions. J. Am. Soc. Mass Spectrom 24, 274–285. 10.1007/s13361-012-0552-2 [DOI] [PubMed] [Google Scholar]
- Mok M, and Marians KJ (1987). The Escherichia coli preprimosome and DNA B helicase can form replication forks that move at the same rate. J. Biol. Chem 262, 16644–16654. [PubMed] [Google Scholar]
- Monachino E, Spenkelink LM, and van Oijen AM (2017). Watching cellular machinery in action, one molecule at a time. J. Cell Biol 216, 41–51. 10.1083/jcb.201610025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monachino E, Ghodke H, Spinks RR, Hoatson BS, Jergic S, Xu Z-Q, Dixon NE, and van Oijen AM (2018). Design of DNA rolling-circle templates with controlled fork topology to study mechanisms of DNA replication. Anal. Biochem 557, 42–45. 10.1016/j.ab.2018.07.008 [DOI] [PubMed] [Google Scholar]
- Mullin DA, Woldringh CL, Henson JM, and Walker JR (1983). Cloning of the Escherichia coli dnaZX region and identification of its products. Mol. Gen. Genet 192, 73–79. [DOI] [PubMed] [Google Scholar]
- Naktinis V, Turner J, and O’Donnell M (1996). A molecular switch in a replication machine defined by an internal competition for protein rings. Cell 84, 137–145. 10.1016/S0092-8674(00)81000-4 [DOI] [PubMed] [Google Scholar]
- Neylon C, Brown SE, Kralicek AV, Miles CS, Love CA, and Dixon NE (2000). Interaction of the Escherichia coli replication terminator protein (Tus) with DNA: a model derived from DNA-binding studies of mutant proteins by surface plasmon resonance. Biochemistry 39, 11989–11999. 10.1021/bi001174w [DOI] [PubMed] [Google Scholar]
- Oakley AJ, Loscha KV, Schaeffer PM, Liepinsh E, Pintacuda G, Wilce MCJ, Otting G, and Dixon NE (2005). Crystal and solution structures of the helicase-binding domain of Escherichia coli primase. J. Biol. Chem 280, 11495–11504. 10.1074/jbc.M412645200 [DOI] [PubMed] [Google Scholar]
- Oakley AJ, Prosselkov P, Wijffels G, Beck JL, Wilce MCJ, and Dixon NE (2003). Flexibility revealed by the 1.85 Å crystal structure of the β sliding-clamp subunit of Escherichia coli DNA polymerase III. Acta Crystallogr. D. Biol. Crystallogr 59, 1192–1199. 10.1107/S0907444903009958 [DOI] [PubMed] [Google Scholar]
- Olson MW, Dallmann HG, and McHenry CS (1995). DnaX complex of Escherichia coli DNA polymerase III holoenzyme. The χ.ψ complex functions by increasing the affinity of τ and γ for δ.δ’ to a physiologically relevant range. J. Biol. Chem 270, 29570–29577. 10.1074/jbc.270.49.29570 [DOI] [PubMed] [Google Scholar]
- Onrust R, Finkelstein J, Naktinis V, Turner J, Fang L, and O’Donnell M (1995). Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. I. Organization of the clamp loader. J. Biol. Chem 270, 13348–13357. 10.1074/jbc.270.22.13348 [DOI] [PubMed] [Google Scholar]
- Pandey M, Syed S, Donmez I, Patel G, Ha T, and Patel SS (2009). Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 462, 940–943. 10.1038/nature08611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park AY, Jergic S, Politis A, Ruotolo BT, Hirshberg D, Jessop LL, Beck JL, Barsky D, O’Donnell M, Dixon NE, and Robinson CV (2010). A single subunit directs the assembly of the Escherichia coli DNA sliding clamp loader. Structure 18, 285–292. 10.1016/j.str.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz RT, and O’Donnell M (2010). Direct restart of a replication fork stalled by a head-on RNA polymerase. Science 327, 590–592. 10.1126/science.1179595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard AE, Dallmann HG, Glover BP, and McHenry CS (2000). A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of δδ’ with DnaX4 forms DnaX3δδ’. EMBO J. I, 6536–6545. 10.1093/emboj/19.23.6536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Lamothe R, Sherratt DJ, and Leake MC (2010). Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328, 498–501. 10.1126/science.1185757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowen L, and Kornberg A (1978). Primase, the dnaG protein of Escherichia coli. An enzyme which starts DNA chains. J. Biol. Chem 253, 758–764. [PubMed] [Google Scholar]
- San Martin MC, Stamford NPJ, Dammerova N, Dixon NE, and Carazo JM (1995). A structural model for the Escherichia coli DnaB helicase based on electron microscopy data. J. Struct. Biol 114, 167–176. 10.1006/jsbi.1995.1016 [DOI] [PubMed] [Google Scholar]
- Scheuermann RH, and Echols H (1984). A separate editing exonuclease for DNA replication: the ε subunit of Escherichia coli DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. USA 81, 7747–7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetta KR, Kazmirski SL, Goedken ER, Cantor AJ, Kelch BA, McNally R, Seyedin SN, Makino DL, O’Donnell M, and Kuriyan J (2009). The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 137, 659–671. 10.1016/j.cell.2009.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha NK, Morris CF, and Alberts BM (1980). Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J. Biol. Chem 255, 4290–4293. [PubMed] [Google Scholar]
- Stamford NP, Lilley PE, and Dixon NE (1992). Enriched sources of Escherichia coli replication proteins. The dnaG primase is a zinc metalloprotein. Biochim. Biophys. Acta 1132, 17–25. 10.1016/0167-4781(92)90047-4 [DOI] [PubMed] [Google Scholar]
- Strycharska MS, Arias-Palomo E, Lyubimov AY, Erzberger JP, O’Shea VL, Bustamante CJ, and Berger JM (2013). Nucleotide and partner-protein control of bacterial replicative helicase structure and function. Mol. Cell 52, 844–854. 10.1016/j.molcel.2013.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ and Dubendorff JW (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89. 10.1016/0076-6879(90)85008-c [DOI] [PubMed] [Google Scholar]
- Studwell-Vaughan PS, and O’Donnell M (1993). DNA polymerase III accessory proteins. V. θ encoded by holE. J. Biol. Chem 268, 11785–11791. [PubMed] [Google Scholar]
- Taft-Benz SA, and Schaaper RM (2004). The θ subunit of Escherichia coli DNA polymerase III: a role in stabilizing the ε proofreading subunit. J. Bacteriol 186, 2774–2780. 10.1128/JB.186.9.2774-2780.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NA, Hamdan SM, Jergic S, Loscha KV, Schaeffer PM, Dixon NE, and van Oijen AM (2008). Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol 15, 170–176. 10.1038/nsmb.1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tougu K, and Marians KJ (1996). The extreme C terminus of primase is required for interaction with DnaB at the replication fork. J. Biol. Chem 271, 21391–21397. 10.1074/jbc.271.35.21391 [DOI] [PubMed] [Google Scholar]
- Tsuchihashi Z, and Kornberg A (1990). Translational frameshifting generates the γ subunit of DNA polymerase III holoenzyme. Proc. Natl. Acad. Sci. USA 87, 2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen AM, and Dixon NE (2015). Probing molecular choreography through single-molecule biochemistry. Nat. Struct. Mol. Biol 22, 948–952. 10.1038/nsmb.3119 [DOI] [PubMed] [Google Scholar]
- Wijffels G, Dalrymple BP, Prosselkov P, Kongsuwan K, Epa VC, Lilley PE, Jergic S, Buchardt J, Brown SE, Alewood PF, et al. (2004). Inhibition of protein interactions with the β2 sliding clamp of Escherichia coli DNA polymerase III by peptides from β2-binding proteins. Biochemistry 43, 5661–5671. 10.1021/bi036229j [DOI] [PubMed] [Google Scholar]
- Xiao H, Crombie R, Dong Z, Onrust R, and O’Donnell M (1993). DNA polymerase III accessory proteins. III. holC and holD encoding χ and ψ. J. Biol. Chem 268, 11773–11778. [PubMed] [Google Scholar]
- Yu X, Jezewska MJ, Bujalowski W, and Egelman EH (1996). The hexameric E. coli DnaB helicase can exist in different quaternary states. J. Mol. Biol 259, 7–14. 10.1006/jmbi.1996.0297 [DOI] [PubMed] [Google Scholar]
- Yuan Q, Dohrmann PR, Sutton MD, and McHenry CS (2016). DNA polymerase III, but not polymerase IV, must be bound to a τ-containing DnaX complex to enable exchange into replication forks. J. Biol. Chem 291, 11727–11735. 10.1074/jbc.M116.725358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, and McHenry CS (2009). Strand displacement by DNA polymerase III occurs through a τ–ψ–χ link to single-stranded DNA-binding protein coating the lagging strand template. J. Biol. Chem 284, 31672–31679. 10.1074/jbc.M109.050740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzhakov A, Kelman Z, and O’Donnell M (1999). Trading places on DNA – a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 96, 153–163. 10.1016/S0092-8674(00)80968-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available upon request. All home-built ImageJ plugins used in this study are freely available on the Github repository for Single-molecule/Image analysis tools (https://github.com/SingleMolecule) or are available upon request.