

Abstract
BRCA1 gene mutations impair homologous recombination (HR) DNA repair, resulting in cellular senescence and embryonic lethality in mice. Therefore, BRCA1-deficient cancers require adaptations that prevent excessive genomic alterations from triggering cell death. RNF168-mediated ubiquitination of γH2AX at K13/15 (ub-H2AX) serves as a recruitment module for the localization of 53BP1 to DNA break sites. Here, we found multiple BRCA1 mutant cancer cell lines and primary tumors with low levels of RNF168 protein expression. Overexpression of ectopic RNF168 or a ub-H2AX fusion protein induced cell death and delayed BRCA1 mutant tumor formation. Cell death resulted from the recruitment of 53BP1 to DNA break sites and inhibition of DNA end resection. Strikingly, re-introduction of BRCA1 or 53BP1 depletion restored HR and rescued the ability of cells to maintain RNF168 and ub-H2AX overexpression. Thus, downregulation of RNF168 protein expression is a mechanism for providing BRCA1 null cancer cell lines with a residual level of HR that is essential for viability. Overall, our work identifies loss of RNF168 ubiquitin signaling as a proteomic alteration that supports BRCA1 mutant carcinogenesis. We propose that restoring RNF168-ub-H2AX signaling, potentially through inhibition of de-ubiquitinases, could represent a new therapeutic approach.
Keywords: BRCA1, RNF168, homologous recombination, breast cancer, ovarian cancer, DNA damage, DNA repair, 53BP1, RNF8
Introduction
BRCA1 mutation-driven carcinogenesis initiates when epithelial cells undergo loss of heterozygosity (LOH), where the wild-type copy of BRCA1 is lost and the remaining mutation-containing allele lacks tumor suppressor activity (1, 2). BRCA1 is a critical component of HR DNA repair, consequently, loss of BRCA1 activity and HR fosters genomic instability, propagating favorable conditions for epithelial cell transformation and the development of cancer (3, 4). HR repair is required for the repair of double stranded DNA breaks (DSBs) that arise during DNA synthesis and for the progression of replication forks. Thus, non-transformed cells are negatively impacted by loss of BRCA1 activity, and homozygous mutations often induce embryonic lethality in mice (5). Although cancers favor genome instability, highly mutagenic DNA repair can also trigger senescence and death (6, 7).
DSBs are initially detected by the MRE11-RAD50-NBS1 (MRN) complex (8), which subsequently recruits and activates ATM-mediated phosphorylation of H2AX (γH2AX) and MDC1 (9). Phosphorylated MDC1 and H2AX promote the recruitment of the RNF8 ubiquitin ligase that adds lysine (K) 63-linked chains to histone H1 and serves as a scaffold for the recruitment of ubiquitin binding proteins (10, 11). RNF168 interacts with K63-linked ubiquitin chains that are conjugated to histone H1 (12, 13). After binding to the ubiquitin chain, RNF168 monoubiquitinates histone H2A/H2AX on K13/15 (14). Ubiquitin binding proteins recruited to monoubiquitinated γH2AX (ub-γH2AX) include 53BP1, as well as RNF168 itself (15, 16). 53BP1 is an inhibitor of DNA end resection and HR (17, 18).
HR DNA repair depends on the initial resection of DSBs to form single stranded DNA (ssDNA) overhangs that are coated by RPA proteins. RAD51 subsequently displaces RPA and forms RAD51-ssDNA filaments. RAD51 coated ssDNA is required for the strand invasion step of HR, where D-loops are formed and homologous template DNA sequences are sought for DNA synthesis (19). BRCA1 promotes DNA end resection, potentially through displacing the end resection inhibitory protein 53BP1 from chromatin surrounding breaks, providing CtIP and MRE11 with access to DSBs (20, 21). Downstream of DNA end resection, the BRCA1-PALB2-BRCA2-RAD51 complex recruits and loads RAD51 onto ssDNA (22).
BRCA1 mutant cancers invariably harbor TP53 mutations, and loss of p53 activity is an established mechanism that abrogates DNA damage-induced senescence. In mouse genetic experiments, Brca1Δ11/Δ11; Tp53+/+ mice die in utero, but Brca1Δ11/Δ11; Tp53−/− mice are born at near Mendelian ratios (5). Interestingly, p53 KO was unable to rescue the embryonic viability of another Brca111- mutant allele (23). Notably, Brca1Δ11 generates a truncated but hypomorphic protein product, referred to as Brca1-Δ11, capable of promoting residual HR (24, 25). In contrast, Brca111- , which was unable to be rescued by p53 KO, fails to generate a Brca1 protein. These findings raise the possibility that p53 loss of function may be insufficient to support the viability of cancers that lack BRCA1 hypomorphic protein expression. In the current study, we found that multiple BRCA1 mutant cancer cell lines and primary tumors have reduced RNF168 protein expression. Moreover, that downregulation of RNF168 protein expression is a mechanism for providing residual HR repair in BRCA1 null cancers, and is necessary for cell and tumor viability.
Materials and methods
cDNA constructs and lentivirus production
The cDNA constructs was obtained as follows: ub-H2AX (Dr. Thanos Halazonetis), RNF168 and RNF8 (Dr. Daniel Durocher). The cDNA were PCR amplified and ligated into the Gateway entry vector pENTR1A (ThermoFisher Scientific). pENTR1A-RNF168 constructs were generated with and without mCherry tags. RNF168 mutagenesis was performed using Platinum SuperFi DNA Polymerase (ThermoFisher , catalog# 12358-010). BRCA1 cDNA was shuttled into vector pDest-IRES-GFP (ThermoFisher Scientific) using the LR Clonase II Enzyme Mix (ThermoFisher Scientific). Vector pCW57.1 was used for doxycycline inducible expression of GFP, Ub-H2AX, RNF168, and RNF8. RNF168 was shuttled into PLX304 for constitutive expression of mCherry-tagged constructs as well as GFP and mCherry controls. To generate lentivirus HEK293T cells were transfected with pxPAX2 packaging plasmid, VSV-G envelope plasmid, and cDNA containing expression plasmids using TransIT-LT1 transfection reagent. Cell culture media was changed 18h post-transfection to DMEM + 30% FBS and was collected 48h later then filtered with a 0.45 μm filter. Cell lines and dissociated tumor cells were infected with lentivirus in media containing polybrene and selected using FACS or antibiotic selection. Cell lines containing a dox-inducible expression construct were maintained in media containing TET-free FBS (Atlanta Biologicals). Expression of the cDNAs were assessed by Western blot.
Cell culture
Cell lines were obtained from ATCC or Asterand. All cell lines were maintained in media with 10% FBS and pen/strep. MDA-MB-231, MCF7, and MDA-MB-436 cell lines were cultured in RPMI. SUM149PT cell lines were grown in Ham’s F-12 with 5 μg/ml insulin, 1 μg/ml hydrocortisone, and 10 mM HEPES. SUM1315MO2 cell lines were cultured in Ham’s F-12 with 5 μg/ml insulin, 10 ng/ml EGF, and 10 mM HEPES. HCC1395 cells were cultured in RPMI with 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 10 mM HEPES, 1 mM sodium pyruvate. 293T cells were cultured in DMEM and MEFs were cultured in DMEM with 1 mM sodium pyruvate, 5 μg/ml insulin, and 2 mM L-glutamine. Cell line authentication was carried out by IDEXX using the short-tandem repeat (STAR) profiling method. Cells were tested for mycoplasma 2 weeks after thawing new vials, the last date of testing was 12/9/19 using MycoAlert (Lonza). Cells were passaged for a maximum of three months before returning to new frozen vials.
Western blotting
Nuclear extracts were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Kit following the manufacturer’s instructions (Thermo Scientific). Whole cell extracts were obtained using RIPA buffer. Protease and phosphatase inhibitors were added for nuclear and whole cell extracts. Proteins were separated by SDS-PAGE and transferred to a PVDF membrane. Membranes were blocked with 5% nonfat milk in PBST at room temperature for 1h. Primary antibodies were incubated overnight at 4 degrees and HRP-conjugated secondary antibodies were incubated for 1h at room temperature. The following primary antibodies were used: BRCA1 (EMD Millipore, catalog# OP92), 53BP1 (EMD Millipore, catalog# MAB3802), RNF8 (EMD Millipore, catalog# 09-813, Santa Cruz Biotechnology, catalog# sc-271462 ), RNF168 (EMD Millipore, catalog# ABE367 and #06-1130-I, R&D Systems, catalog# AF7217), Tubulin (Cell Signaling, catalog# 2148), GFP (Santa Cruz Biotechnology, catalog# sc-9996), RFP (ChromoTek, catalog# 6g6-20), FLAG (Cell Signaling, catalog# 14793), RPA32 (Santa Cruz Biotechnology, catalog# sc-28709), phospho-RPA32 S4/S8 (Bethyl Laboratories, catalog# A300-245A), PALB2 (Bethyl Laboratories, catalog# A301-246A), and γH2AX (EMD Millipore, catalog# 05-636).
Immunofluorescence
Cells were exposed to 10 Gy γ-irradiation or mock irradiated and fixed at 8h after treatment unless otherwise noted. Pre-extraction was performed prior to fixation for RNF168, RPA, and RAD51 foci analysis. For pre-extraction, we treated cells with cold cytoskeleton buffer (10mM Pipes pH 6.8, 100mMNaCl, 300 mM sucrose, 3mM MgCl2,1mM EGTA, 0.5% Triton X-100) for 5 min on ice followed by 5 min incubation with cold cytoskeleton stripping buffer (10mM TrisHCl pH 7.4, 10mMNaCl, 3mM MgCl2, 1% Tween 40(v/v), 0.5% sodium deoxycholate). Cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton-X100 in PBS for 10 min. Primary antibody incubation was performed overnight at 4 degrees in 5% goat serum. FITC or Texas Red conjugated secondary antibodies (Jackson ImmunoResearch) were incubated for 1h at room temperature and coverslips were mounted with mounting media containing DAPI (Vector Laboratories Inc.). The following primary antibodies were used: RNF168 (EMD Millipore, catalog# ABE367), Ubiquitin conjugations clone FK2 (EMD Millipore, catalog# 04-263), 53BP1 (EMD Millipore, catalog# MAB3802), RPA32 (EMD Millipore, catalog# NA18), RAD51 (Genetex, catalog# GTX100469), Geminin (Abnova, catalog# 110-209 and Proteintech, catalog#10802-1-AP), γH2AX (EMD Millipore, catalog# 05-636), and BRCA1 (EMD Millipore, catalog# OP92). Z-stack images were captured using a Nikon NIU Upright Fluorescence microscope and generated images using Nikon NIS Elements software. Foci quantifications were reported as either the number of foci per nuclei or the percentage of nuclei that contain foci. Percent positivity was determined based on nuclei containing 5 or more foci for RAD51 and 10 or more foci for all other proteins and a minimum of 200 cells from 5 fields of view were quantified for each sample. For the number of foci per nuclei count analyses, ImageJ was used for a minimum of 100 random nuclei.
Colony assays
Cells were seeded in 6 well plates at differing cell densities or PARPi concentrations, grown for 2 weeks, then fixed in 4:1 methanol:acetic acid and assessed for colony formation using crystal violet staining. For experiments examining the effects of siRNA, cells were pretreated for 72h in 24 well plates prior to seeding for colony assays. PARPi sensitivity was assessed at seeding densities of 1000 and 500 cells per well and cells were maintained in rucaparib with media changes every 96h. Effects of RNF168, Ub-H2AX, and GFP expression on colony formation were assessed at seeding densities of 2000, 1000, 500, 250, 125, and 63 cells per well. Experiments using inducible constructs were maintained in 4 μg/ml doxycycline and normalized to dox-free wells. In experiments using mCherry or mCherry-tagged RNF168, cells were sorted for mCherry expression, counted, and immediately seeded for colony formation assays.
Comet assay
The alkaline comet assay was conducted according to the manufacturer’s instructions (Trevigen, catalog #4250-050-K). Briefly, doxycycline-treated cells were trypsinized, embedded in agarose, lysed, electrophoresis performed, and DNA stained with SYBR gold. Images were captured using a Nikon NIU Upright Fluorescence microscope and generated images using Nikon NIS Elements software. Analysis was performed in ImageJ using the OpenComet (v1.3) plugin. Data is presented as DNA% in the tail of the comet.
Cell cycle analysis
Exponentially growing cells were treated with doxycycline for 5 days then trypsinized and fixed in 50% ethanol at 4 degrees overnight. Fixed cells were then washed with PBS and incubated for 30 min in FxCycle PI/RNase staining solution (ThermoFisher Scientific, catalog #F10797). Cell cycle profiles were acquired using a BD FACScan flow cytometer and analyzed using FlowJo software.
Annexin V staining
Extracellular Annexin V staining was performed on cell lines treated for 5 days with doxycycline according to the manufacturer’s instructions (BD Biosciences, catalog #559763). Flow cytometry was performed with a BD LSR II flow cytometer and analyzed using FlowJo software.
Xenograft studies
Studies were approved by the FCCC Institutional Animal Care and Use Committee (IACUC). Cancer cell lines or PDX cell suspensions were subcutaneously injected into 6-week-old female NSG mice in a 1:1 suspension to Matrigel ratio. PDXs were first extracted and dissociated into cell suspensions, and transduced with RNF168 or control lentivirus prior to injection. Dissociation was performed by incubation in media containing 1.7 mg/ml collagenase (Sigma Aldrich) and 3 U/ml dispase (Sigma Aldrich) for 3h at 37 degrees with shaking at 175 rpm. Cells were washed with media and forced through a 0.70 μm cell strainer to obtain a cell suspension from the PDX. Two million PDX cells per T25 flask were plated then infected using 10 μg/ml polybrene and lentivirus containing GFP-tagged RNF168 or GFP control constructs. After 24 h of lentivirus exposure the media was replaced, retaining both suspension and adherent PDX cells in culture for an additional 24 h. Infected cells were then sorted by FACS, washed three times with PBS, then 1 million cells were injected into NSG mice at a ratio of 1:1 suspension to Matrigel. MDA-MB-436 xenograft experiments were conducted using cell lines with dox-inducible GFP or RNF168 expression. Mice were fed doxycycline-containing food for one week prior to cell injection and for the duration of the experiment. Tumors were measured twice weekly with calipers and volume calculated using (length x width2)/2. Mice were euthanized and tumors collected once established tumors reached 1500 mm3 in accordance with institutional guidelines. The established tumors were snap frozen in liquid nitrogen for Western blot analysis or fixed in 10% neutral buffered formalin prior to paraffin block embedding for IHC analysis.
Immunohistochemistry
Slides were deparaffinized with xylene and rehydrated. Antigen retrieval was performed at 100 degrees for 45 min in pH 9.0 Tris/EDTA buffer (Dako Target Retrieval Solution). Endogenous peroxide was quenched with 3% hydrogen peroxide for 20 min. Blocking was performed at room temperature for 30 min (Background Sniper, Biocare). Primary anti-RNF168 antibody (EMD Millipore, catalog# 06-1130-I) was diluted 1:300 (Da Vinci Green diluent, Biocare) and incubated overnight at 4 degrees in a humidified chamber. Slides were then washed and incubated for 1h at room temperature with EnVision+ Labelled Polymer HRP (Dako). Slides were again washed, developed with DAB (Dako DAB+ Chromogen), counterstained with Mayer’s hematoxylin, dehydrated and coverslips mounted. Tissue microarrays were compiled and provided by the Fox Chase Cancer Center Biosample Repository from triple-negative breast cancer and serous ovarian cancer patients and approved by the FCCC Institutional Review Board (IRB). Blinded immunohistochemistry scoring for nuclear RNF168 staining was performed by pathologist Dr. Kathy Cai from the Fox Chase Cancer Center Histopathology Core Facility. Some tissue cores were available in duplicate and triplicate and were independently scored then averaged to present a single case per data point.
siRNA and shRNA Experiments
siRNA experiments were conducted by reverse transfection with Lipofectamine RNAimax (ThermoFisher Scientific, catalog# 13778030) according to manufacturer instructions. The following siRNA were used: AllStars Negative Control siRNA (scrambled control) (Qiagen, catalog# SI03650318), 53BP1 SmartPool (Dharmacon) contains four unique sequences: #1 GAAGGACGGAGUACUAAUAUU, #2 GCUAUAUCCUUGAAGAUUUUU, #3 GAGCUGGGAAGUAUAAAUUUU, # 4GGACUCCAGUGUUGUCAUUUU, RNF168 #1 GACACUUUCUCCACAGAUAUU, RNF168 #2 CAGUCAGUUAAUAGAAGAAAUU, RNF8 #1 UGCGGAGUAUGAAUAUGAAUU, RNF8 #2 CAGAGAAGCUUACAGAUGUUU, PALB2 #1 GGUGUACAUAAAGCUUCAAUU, PALB2 #2 GGAUAUAUUGGGCCUCUUAUU. The plko.1 vector was used to produce lentivirus and generate shRNA cells with a non-targeting sequence or targeting of 53BP1 with the sequence AATCAATACTAATCACACTGG. We previously established the efficacy of two independent 53BP1 shRNAs and confirmed no off-target effects in the same BRCA1 null cancer cell lines as used here(26). Stable shRNA cell lines were selected with puromycin.
Statistical Analysis
Statistical tests were performed as indicated in each figure legend. Comparisons of foci positivity were performed with unpaired t-tests due to similar variances and sample sizes between conditions. IHC scoring between BRCA1 mutant and wild-type patient tissue sections was assessed for significance by Welch’s t-test due to unequal sample sizes to account for the larger wild-type BRCA1 sample size. Statistical tests were performed using Graphpad Prism software. Statistically significant p values are indicated in figure legends.
Results
BRCA1 mutant cells have reduced RNF168-mediated ubiquitin signaling
To characterize BRCA1 mutant cancers for alterations in DNA damage response proteins, we measured the expression levels of BRCA1, 53BP1, RNF8 and RNF168 proteins in a panel of BRCA1 wild-type (WT) and mutant cancer cell lines. MDA-MB-231 and MCF7 cell lines are BRCA1 WT and expressed the full-length and BRCA1-Δ11q isoforms. SUM149PT cells harbor a BRCA1 exon 11 mutation and express the BRCA1-Δ11q splice isoform that retains partial function (25). In contrast, SUM1315MO2, HCC1395, and MDA-MB-436 cells have RING or BRCT domain mutations and had undetectable BRCA1 protein expression. Across cell lines, we found that 53BP1 and RNF8 levels were similar. However, RNF168 expression was markedly lower in SUM1315MO2, HCC1395, and MDA-MB-436 cell lines (Fig. 1A and S1A). Because RNF168 directly ubiquitinates γH2AX(14), we subjected cells to mock or γ-irradiation (IR) treatments and measured γH2AX gel migration patterns. As expected, the levels of γH2AX increased in all IR treated cell lines. However, IR-induced mono- and di- ubiquitinated forms of γH2AX were abundant in MDA-MB-231, MCF7 and SUM149PT cells, but were markedly lower in SUM1315MO2, HCC1395, and MDA-MB-436 cells (Fig. 1B).
Figure 1. Assessment of RNF168 signaling in BRCA1-deficient cell lines.
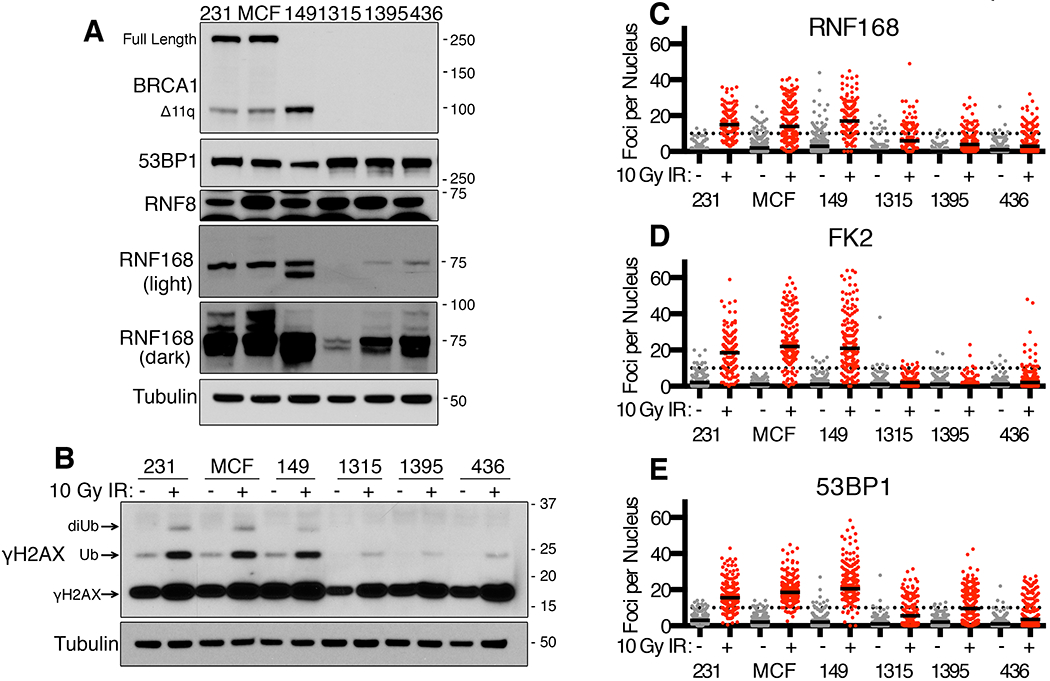
(A) MDA-MB-231 (231), MCF7 (MCF), SUM149PT (149), SUM1315MO2 (1315), HCC1395 (1395) and MDA-MB-436 (436) cell lines were assessed for BRCA1, 53BP1, RNF8 and RNF168 protein expression by Western blotting. BRCA1 full length and the BRCA1-Δ11q splice isoform are indicated as well as M.W. markers. Dark and light exposures of RNF168 are shown, also see Fig. S1A.
(B) Western blot analyses of γH2AX in cell lines from (A) at 8h after mock or 10 Gy IR. Mono- and di- ubiquitinated γH2AX forms are indicated.
(C) Cell lines from (A) were analyzed by immunofluorescence (IF) for RNF168 foci at 8h post mock (grey) or 10 Gy IR (red) treatments. The number of foci per nucleus are shown, a minimum of 100 nuclei were assessed. Black solid lines indicate median value; black dotted line indicate 10 foci per nucleus. See Fig. S1B for representative images.
(D) FK2/ubiquitin foci were assessed as described in (C)
(E) 53BP1 foci were assessed as described in (C)
In line with protein expression patterns, RNF168 formed robust foci in MDA-MB-231, MCF7, and SUM149PT cell lines, but γ-irradiation-induced foci (IRIF) were reduced in SUM1315MO2, HCC1395, and MDA-MB-436 cell lines (Fig. 1C and Fig. S1B). 53BP1 is a K13/15 γH2AX ubiquitin binding protein that form foci at sites of DNA damage (16). Ubiquitin (FK2) and 53BP1 foci were detected in all cell lines and there were minimal differences between untreated cells. However, FK2 and 53BP1 formed robust IRIF in MDA-MB-231, MCF7, and SUM149PT cell lines, but similar to RNF168, the number of IRIF were reduced in SUM1315MO2, HCC1395, and MDA-MB-436 cell lines (Fig. 1D–E and Fig. S1B). Cell cycle variations did not account for differences in foci as similar patterns were observed in G1 and S/G2-labelled populations (Fig. S1C). SUM1315MO2, HCC1395, and MDA-MB-436 cells were highly sensitive to PARPi treatment (Fig. S1D), suggesting that inefficient 53BP1 recruitment to DSBs, unlike 53BP1 gene knockout, is insufficient to support PARPi resistance. In summary, we found that several BRCA1 mutant cancer cell lines have reduced RNF168 protein expression and consequently H2A-ubiquitin associated DNA damage signaling.
RNF168 protein levels are reduced in subsets of BRCA1 mutant cancers
We aimed to determine whether RNF168 expression was reduced at the transcriptional or proteasomal level. RNF168 mRNA was measured in cell lines by quantitative (q) RT-PCR, and was reduced 4.1-, 2.7-, and 1.9-fold in SUM1315MO2, HCC1395 and MDA-MB-436 cells relative to MDA-MB-231 cells (Fig. 2A). Treatment with the proteasome inhibitor MG132 minimally impacted RNF168 in SUM1315MO2 cells, but increased protein expression in HCC1395 and MDA-MB-436 cells (Fig. 2B), indicating that both reduced mRNA expression and proteasomal degradation contribute to the reduction in RNF168 protein levels. Moreover, we assessed the levels of several proteasomal regulatory factors and DUBs reported to impact RNF168 expression (27, 28). However, we were unable to identify a single factor that could account for the reduction in RNF168 across the cell line panel (Fig. S2A).
Figure 2. Analyses of RNF168 expression in BRCA1 mutant cancers.
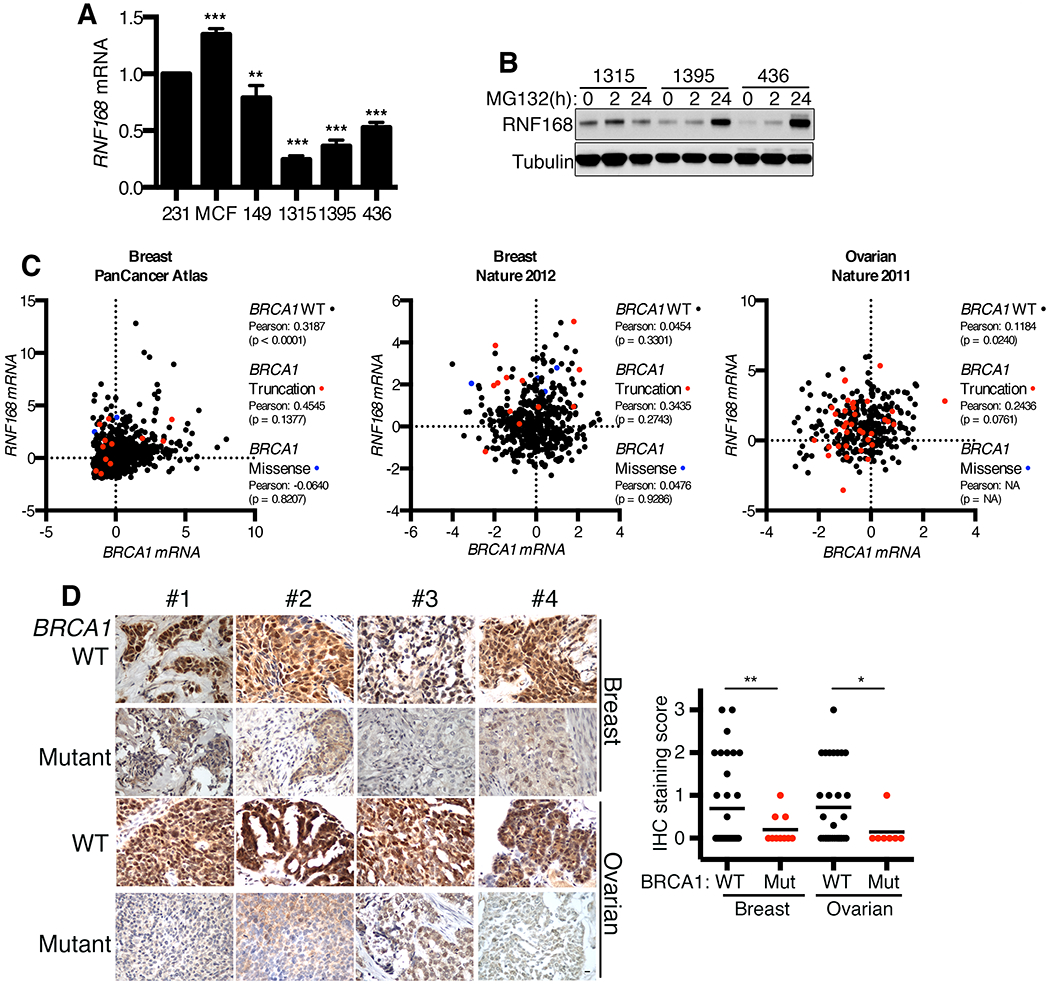
(A) Cell lines were analyzed for RNF168 mRNA levels by q-RT-PCR. Values normalized to HPRT gene expression and presented as a percentage of RNF168 mRNA expressed in MDA-MB-231 cells. Mean ± S.D. is shown, **p < 0.01, ***p < 0.001 (unpaired t-test).
(B) SUM1315MO2, HCC1395, and MDA-MB-436 cells were treated with 10 μM MG-132 proteasome inhibitor for 0, 2, or 24h and subject to Western blotting for RNF168 protein level.
(C) RNF168 and BRCA1 mRNA expression was examined in BRCA1 wild-type and mutant patient cohorts that were sub-divided into truncating and missense mutations in the following datasets using cBioPortal: Breast Invasive Carcinoma (TCGA, PanCancer Atlas), Breast Invasive Carcinoma (TCGA, Nature 2012), Ovarian Serous Cystadenocarcinoma. The results are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Additional details are in Fig. S2.
(D) RNF168 protein expression was assessed by IHC staining of human TNBC and HGSOC tissue microarrays generated by the Fox Chase Cancer Center Biosample Repository. Staining was scored 0–3 for nuclear RNF168 intensity. Representative images are shown, 10 μm scale bar. Tissue cores that were available in duplicate or triplicate were independently scored then averaged to present a single case per data point. The mean scores for each group are indicated with a black bar (WT-TNBC n = 34, Mut-TNBC n = 10, WT-HGSOC n = 31, Mut-HGSOC n = 7). *p < 0.05, **p < 0.01 (Welch’s t-test).
We next set out to measure RNF168 mRNA and protein expression in a panel of primary BRCA1 mutant cancers. We utilized TCGA datasets to examine potential relationships between BRCA1 mutations and RNF168 mRNA expression in breast and ovarian cancers (29–31). Overall, there were no clear patterns between RNF168 mRNA expression and BRCA1 wild-type or mutant cancers (Fig. 2C). However, at the genomic level, RNF168 copy number was unexpectedly increased in multiple BRCA1 mutant tumors (Fig. S2B). Although, the RNF168 gene is located (3q29) in close proximity to PIK3CA (3q26.32), and tumors frequently had co-occurring RNF168 and PIK3CA amplifications (Fig. S2C–D), giving rise to the possibility that RNF168 amplification is a passenger event.
Protein expression was examined in primary tumor samples using an RNF168 antibody optimized for IHC detection (Fig. S2E). Here, we observed low levels of RNF168 protein expression in the majority of primary BRCA1 mutant cancers examined, and expression was significantly lower than BRCA1 wild-type TNBC and HGSOCs (Fig. 2D). In conclusion, while RNF168 mRNA expression is variable, multiple BRCA1 mutant primary cancers demonstrated reduced RNF168 protein levels.
RNF168 is subject to negative selection
The above results give rise to the possibility that BRCA1 directly impacts RNF168 protein expression. To examine this possibility, we generated BRCA1 wild-type and mCherry control add-back SUM1315MO2, HCC1395, and MDA-MB-436 isogenic cell lines (Fig. S3A). Here, BRCA1 add-back restored BRCA1 and RAD51 foci formation (Fig. S3B), but did not impact RNF168 protein expression (Fig. S3A) or the efficiency of 53BP1 foci formation (Fig. S3C). Therefore, rather than the BRCA1 protein directly regulating RNF168 mRNA or protein stability, we hypothesized that RNF168 is subject to negative selection in BRCA1 mutant cancers. To test this idea, we examined the consequences of restoring RNF168 expression. Ectopic RNF168-mCherry or an mCherry control were expressed in isogenic BRCA1 deficient and wild-type complemented cell lines. mCherry positive cells were sorted and immediately subject to Western blotting analyses to confirm expression as well as seeded for colony formation experiments (Fig. 3A). Expression of mCherry alone had no impact on colony formation, but RNF168-mCherry resulted in a 9.1-fold and 4.0-fold reduction in the number of BRCA1 deficient SUM1315MO2 and MDA-MB-436 colonies forming. Strikingly, RNF168-mCherry had no effect on the colony formation of BRCA1 complemented cells (Fig. 3B).
Figure 3. Ectopic RNF168 expression decreases the viability of BRCA1 deficient cells.
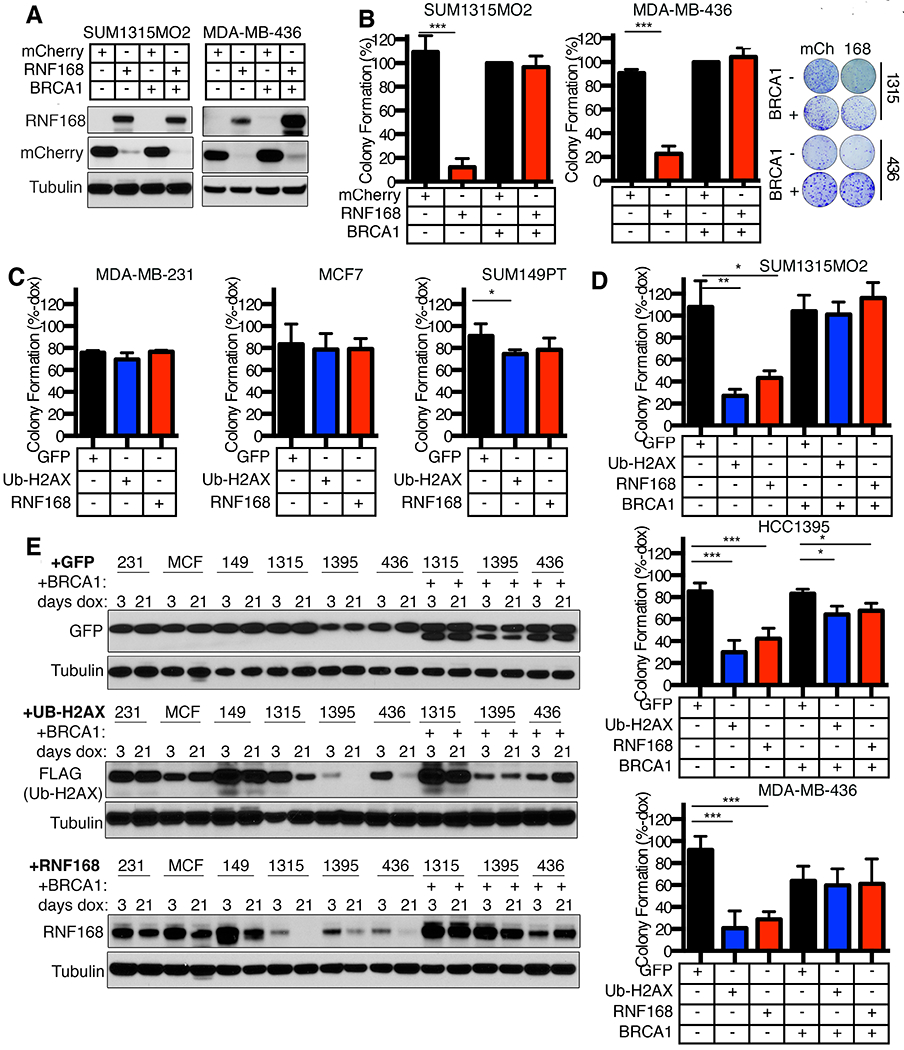
(A) SUM1315MO2 and MDA-MB-436 −/+ BRCA1 add-back cell lines were transduced with lentivirus for expression of mCherry-tagged RNF168 or an mCherry control. mCherry positive cells were sorted and subject to Western blotting for the indicated proteins.
(B) Cells that were mCherry positive from (A) were assessed for colony formation. Colony formation was calculated as a percentage of mCherry expressing BRCA1 add-back cells. Mean ± S.D. is shown, ***p < 0.001 (unpaired t-test). Representative plates and colonies are shown (right).
(C) Dox-inducible GFP, ub-H2AX, or RNF168 expressing MDA-MB-231, MCF7, and SUM149PT cell lines were assessed for colony formation in the absence or presence of dox. Colony formation of dox treated cells is shown as a percentage of colonies formed in the absence of dox. Mean ± S.D. is shown, *p < 0.05 (unpaired t-test). See Fig. S4A for more details.
(D) Dox-inducible GFP, ub-H2AX, or RNF168 expressing SUM1315MO2, HCC1395, and MDA-MB-436 cell lines, as well as isogenic BRCA1 add-back cell lines, were assessed for colony formation in the absence or presence of dox. Colony formation of dox treated cells is shown as a percentage of colonies formed in the absence of dox. Mean ± S.D. is shown, *p < 0.05, **p < 0.01, ***p < 0.001 (unpaired t-test)
(E) Cell lines from (C) and (D) were cultured in the presence of dox for 3 or 21 days and Western blotting performed assessing the indicated proteins.
To determine if the reduction in colony formation resulted from γH2AX mono-ubiquitination, we expressed a ubiquitin N-terminal H2AX (ub-H2AX) fusion cDNA, previously shown to mimic K13/15 ub-γH2AX(32). Because RNF168 expression was detrimental to the viability of BRCA1 deficient cells, a doxycycline (dox) system was utilized to overexpress ub-H2AX, RNF168 or a GFP control in an inducible manner (Fig. S4A). Here, RNF168 and ub-H2AX had little impact on MCF7, MDA-MB-231 or SUM149PT cells that express BRCA1 full-length or BRCA1-Δ11q isoforms (Fig. 3C). However, RNF168 and ub-H2AX significantly reduced the colony formation of BRCA1 deficient MDA-MB-436, HCC1395 and SUM1315MO2 cell lines compared to GFP expressing control cells. Significantly, BRCA1 add-back rescued the ability of ub-H2AX and RNF168-expressing cells to form colonies (Fig. 3D).
We also measured the expression of ectopic proteins in cells that were incubated with dox for short and long-term cultures (Fig. 3E). As expected, GFP expression was similar at 3 and 21 days of dox in all cell lines. RNF168 and ub-H2AX expression was either similar or moderately reduced by 21 days in MCF7, MDA-MB-231 or SUM149PT cell lines. In contrast, RNF168 and ub-H2AX expression were lower at 3 days compared with BRCA1 proficient cells, and were significantly decreased by 21 days of dox culture in SUM1315MO2, HCC1395, and MDA-MB-436 cells. Interestingly, RNF168 and ub-H2AX expression was maintained in long-term cultures in isogenic BRCA1 add-back cell lines (Fig. 3E). The short- and long-term ectopic RNF168 expression status did not affect PARPi sensitivity in BRCA1 deficient cells (Fig. S4B).
Importantly, BRCA1-deficient cells were able to maintain constitutive expression of the RNF168 C16S and R57D mutant proteins that specifically disrupt the ability of RNF168 to ubiquitinate H2A (13, 14) (Fig. S4C). CRISPR/Cas9-mediated deletion of the BRCA1-Δ11q isoform, which retains partial functionality(25), in conjunction with ectopic RNF168 expression, resulted in loss of viability in SUM149PT cells, indicating that BRCA1 hypomorphic isoforms enable cells to tolerate RNF168 overexpression (Fig. S4D). Therefore, RNF168-mediated histone ubiquitination is detrimental to the viability of BRCA1-null human cancer cells, and is subject to negative selection.
RNF168 overexpression delays BRCA1 mutant tumor formation
We next examined the effects of ectopic RNF168 overexpression on BRCA1 mutant tumor formation and growth. Isogenic MDA-MB-436 control and BRCA1 add-back cells expressing either GFP or RNF168 cDNA were injected into NSG mice and the time required for tumors to form measured. MDA-MB-436 BRCA1- and BRCA1+ cells with GFP expression developed palpable (≥200 mm3) tumors at median of 52- and 46-days post-injection, respectively. In contrast, BRCA1- and BRCA1+ cells with ectopic RNF168 expression developed palpable tumors at median of 90- and 44-days post-injection, respectively (Fig. 4A). Similar to long-term cell growth cultures in vitro, GFP was equally abundant, but RNF168 expression was markedly lower in BRCA1-deficient compared with BRCA1 add-back established tumors (Fig. 4B).
Figure 4. RNF168 delays BRCA1 mutant tumor formation.
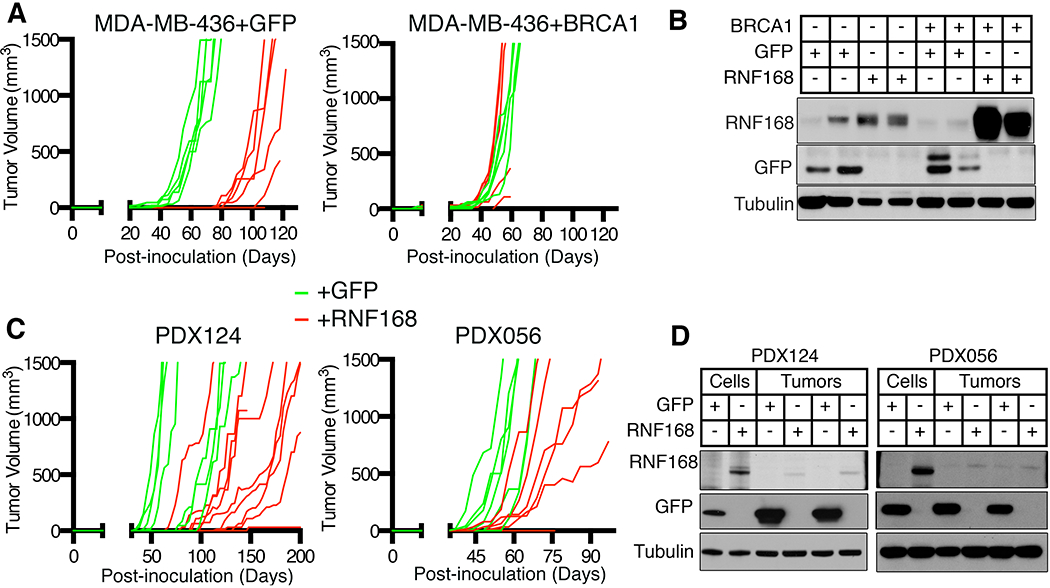
(A) Dox-inducible GFP (green lines) and RNF168 (red lines) MDA-MB-436 +/− BRCA1 cell lines were injected into mice and tumor sizes measured. Individual tumor volumes are displayed (n=5, minimum), including instances when injected cells did not form tumors
(B) Established tumors samples described in (A) were subjected to Western blotting for the indicated proteins. Two tumor samples were analyzed per group.
(C) BRCA1 mutant PDX124 and PDX056 were transduced with lentivirus to express GFP or GFP-tagged RNF168, sorted and injected into mice. Individual tumor volumes showing tumor growth are displayed (n=6 minimum), including include instances when injected cells did not form tumors. See Fig. S4E for more details.
(D) Established tumors from (C) were collected and compared with transduced cells that were generated prior to injection by Western blotting.
RNF168 expression was also assessed in patient-derived xenograft (PDX) tumors. Here, BRCA1 WT PDX036 (HGSOC) demonstrated abundant BRCA1 full-length and BRCA1-Δ11q isoform expression, as well as readily detectable RNF168 expression (Fig. S4E). As expected, full-length BRCA1 was undetectable in BRCA1 mutant PDX196 (HGSOC), PDX124 (TNBC), and PDX056 (HGSOC) tumors. Despite the latter tumors harboring BRCA1 exon 11 mutations, BRCA1-Δ11q isoform expression was relatively low compared with PDX036. Moreover, RNF168 protein expression was also markedly lower in BRCA1 mutant PDX tumors compared with BRCA1 WT PDX036 (Fig. S4E).
We assessed the effects of ectopic RNF168 overexpression in BRCA1 mutant PDX tumors. PDX tumors were disaggregated, infected with lentivirus containing GFP or RNF168-GFP cDNA, sorted, and an equal number of cells re-implanted into NSG mice. Of note, PDX196 tumors did not re-grow after disaggregation under any conditions. However, GFP and RNF168 expressing PDX124 cells formed tumors (≥200 mm3) at a median of 55- and 121-days, respectively. Similarly, GFP and RNF168 expressing PDX056 cells formed tumors at a median of 48- and 64-days, respectively (Fig. 4C). While tumors maintained GFP expression, cells initially expressed an abundance of ectopic RNF168 immediately prior to injection, but tumors that formed had selected against RNF168 expression (Fig. 4D). Thus, RNF168 is subject to negative selection and delays BRCA1 mutant tumor formation.
RNF168 augments 53BP1-mediated inhibition of HR
To gain mechanistic insight into the effects of RNF168 overexpression on the DNA repair activity of BRCA1-deficient cells, we measured RPA32 and RAD51 IRIF, which are markers for the DNA end resection and RAD51 loading steps of HR, respectively. We confirmed that RNF168 expression significantly increased the number of 53BP1 foci in dox-inducible MDA-MB-436 and SUM1315MO2 cells (Fig. 5A). Because BRCA1 mutant cell lines are defective for HR, few RAD51 foci positive cells were observed at early time points post-IR. However, quantifiable numbers of RAD51 foci positive cells could be detected at later time points (Fig. S5A). Subsequently, we found that RNF168 expression resulted in significant reductions in the number of RPA32 and RAD51 foci positive cells. In contrast, RNF168 expression did not significantly impact foci in BRCA1 add-back cells (Fig. 5B and Fig. S5A–B). RNF168-mediated inhibition of residual HR in BRCA1 deficient cells resulted in increased numbers of spontaneously arising γH2AX foci (Fig. 5C), comet tails (Fig. 5D), G2/M phase population (Fig. 5E), and Annexin-positive cells (Fig. 5F).
Figure 5. RNF168 regulates RPA32 and RAD51 IRIF.
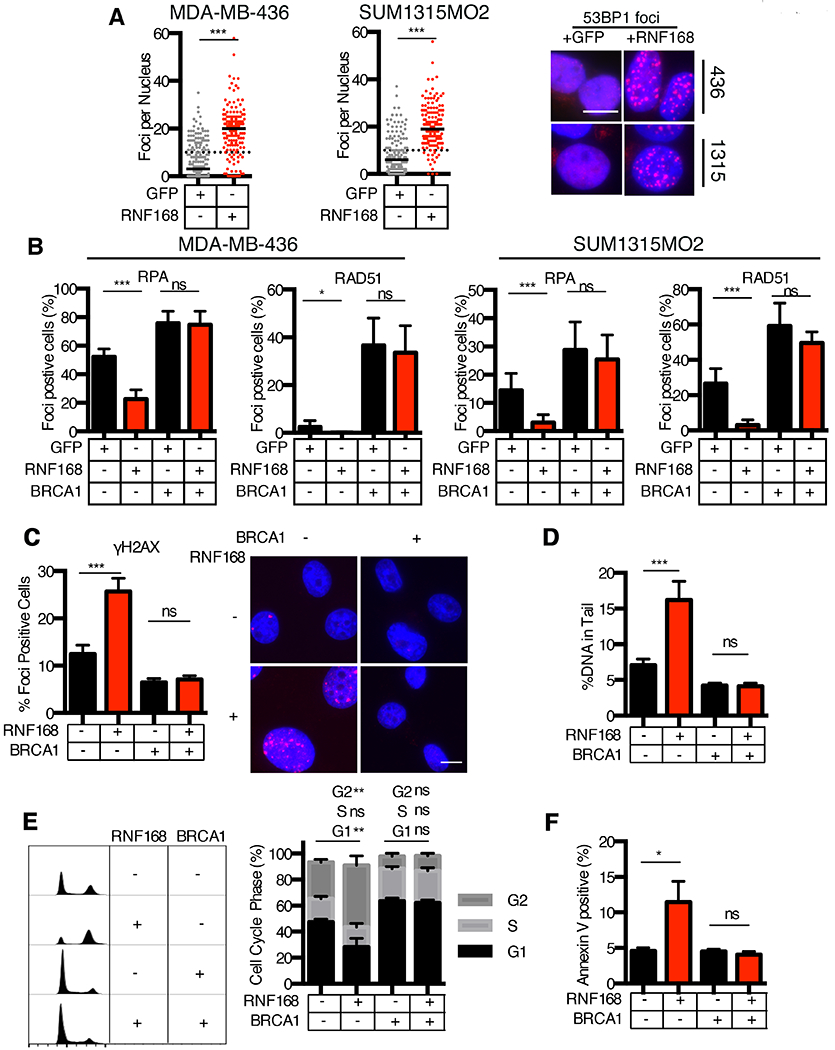
(A) Dox-inducible GFP and RNF168 MDA-MB-436 and SUM1315MO2 cells were analyzed for 53BP1 IRIF. The number of foci per nucleus is shown, a minimum of 150 nuclei were assessed. Black solid lines indicate median value; black dotted line indicate 10 foci per nucleus. Representative images are shown, 10 μm scale bar. ***p<0.001 (Mann-Whitney test).
(B) Dox-inducible GFP and RNF168 MDA-MB-436 and SUM1315MO2 +/− BRCA1 cells were assessed for RPA32 and RAD51 foci 18h post 10 Gy IR. Nuclei with 5 or more foci were scored positive. Mean ± S.D. foci positive cells are shown, *p < 0.05, **p < 0.01, ***p < 0.001, (ns), not significant (unpaired t-test). Representative images are shown in Fig. S5B.
(C) Dox-inducible GFP and RNF168 SUM1315MO2 cells were incubated with doxycycline for 5 days then fixed and assessed for γH2AX foci formation, inset, representative images. Mean ± S.E.M foci positive cells are shown, ***p < 0.001, (ns), not significant (unpaired t-test).
(D) Cells were treated as in (C) and assessed for DNA damage by comet assay. Mean ± S.E.M DNA% in tail cells are shown, ***p < 0.001, (ns), not significant (unpaired t-test) for a minimum of 70 comet tails per sample.
(E) Cells were treated as in (C), fixed, and cell cycle profile obtained. FlowJo software was utilized to quantify G1, S, and G2 phase populations based on DNA content. Left, representative histograms. Right, mean ± S.E.M for percent population within each cell cycle phase are shown, **p < 0.01, (ns), not significant (unpaired t-test).
(F) Cells were treated as in (C), and stained for extracellular Annexin V, assessed by flow cytometry. Mean ± S.E.M for percent of cells staining positive for Annexin V are shown, *p < 0.05, (ns), not significant (unpaired t-test).
53BP1 is an inhibitor of DNA end resection and HR. To verify that 53BP1 was responsible for RNF168 overexpression-induced inhibition of residual HR, MDA-MB-436 and SUM1315MO2 dox-inducible GFP, RNF168, or ub-H2AX overexpressing cells were incubated with scrambled or 53BP1-targeting siRNA. 53BP1 knockdown counteracted the RNF168 and ub-H2AX-induced reduction in RPA32 and RAD51 IRIF in MDA-MB-436 and SUM1315MO2 cells (Fig. 6A–B and Fig. S5C). Similarly, an RNF168-induced reduction in phospho(S4/S8)-RPA levels, another marker of active DNA end resection, was also abrogated with 53BP1 siRNA (Fig. S5D). Thus, in the absence of BRCA1, RNF168 inhibits residual HR activity via 53BP1 recruitment to DSBs.
Figure 6. Loss of viability is 53BP1-dependent.
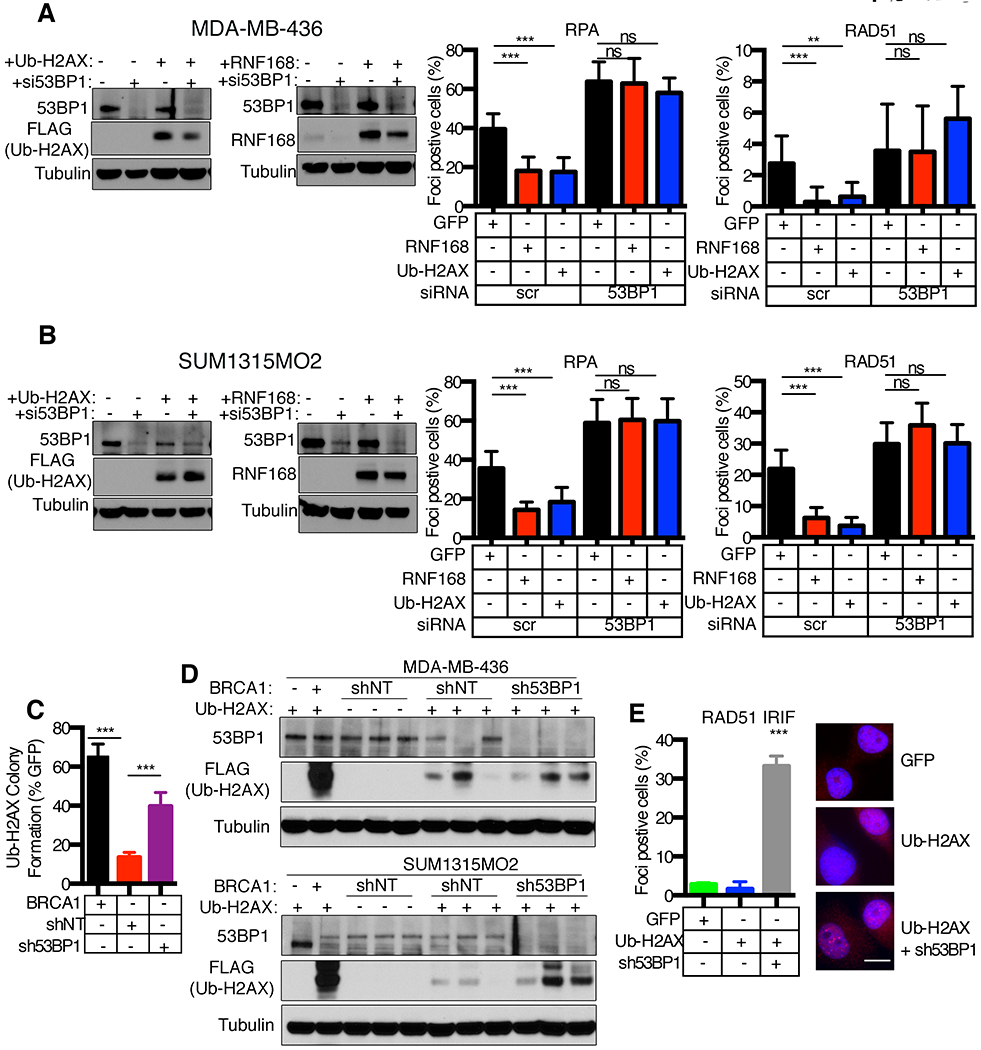
(A) Dox-inducible GFP, RNF168, ub-H2AX expressing MDA-MB-436 cells were subject to scrambled (sc) or 53BP1 siRNA and assessed for RPA32 and RAD51 foci at 18h post 10 Gy IR. Mean ± S.D. foci positive cells are shown, *p < 0.05, **p < 0.01, ***p < 0.001, (ns), not significant (unpaired t-test). Inset, Western blotting showing 53BP1 depletion. Representative images are shown in Fig. S5C.
(B) Dox-inducible GFP, RNF168, ub-H2AX SUM1315MO2 cells, as for (A).
(C) MDA-MB-436 cells with the indicated genetic manipulations were transduced with GFP or ub-H2AX lentivirus and assessed for colony formation under blasticidin selection. Ub-H2AX colony formation was normalized to GFP control colonies for each condition. Mean ± S.D. is shown, ***p < 0.001 (unpaired t-test).
(D) MDA-MB-436 and SUM1315MO2 cells with the indicated genetic manipulations were transduced for ub-H2AX as described in (C). Lysates were collected from colonies that survived blasticidin selection and subjected to Western blot for the indicated proteins.
(E) SUM1315MO2 cells with inducible GFP control or ub-H2AX expression, as well as cells that survived blasticidin selection and expressed 53BP1 shRNA and ub-H2AX cDNA from (D), were assessed for RAD51 IRIF at 8h post 10 Gy IR. Representative images are shown, 10 μm scale bar. Mean ± S.D. foci positive cells are shown, ***p < 0.001 (unpaired t-test) compared to +GFP cells.
We predicted that depletion of 53BP1 would increase tolerance to ub-H2AX expression in BRCA1 mutant cancer cells. To test this possibility, cells were infected with non-target or 53BP1 shRNA followed by ub-H2AX and colony formation assessed. In line with previous results, ub-H2AX expressing MDA-MB-436 cells with BRCA1 add-back formed robust colonies. Moreover, cells expressing 53BP1 shRNA formed significantly more colonies after infection with ub-H2AX expressing lentivirus compared non-target shRNA expressing cells (Fig. 6C). Western blotting analyses of surviving colonies revealed robust ub-H2AX expression in BRCA1 add-back cells, while 53BP1 depletion enabled cells to express moderate levels of ub-H2AX (Fig. 6D). BRCA1 deficient and non-target (NT) shRNA expressing-cells that survived infection had selected against ub-H2AX expression. Although, there were several colonies that also demonstrated detectable ub-H2AX expression in NT shRNA treated cells, and these cells had downregulated 53BP1 expression in the absence of exogenous manipulation (Fig. 6D). Additionally, 53BP1 shRNA treated cells that expressed ub-H2AX demonstrated RAD51 IRIF (Fig. 6E), suggesting restored HR. All in, in the absence of full-length or hypomorphic BRCA1 isoforms, RNF168-mediated ubiquitin signaling can be tolerated through loss of 53BP1 and restoration of HR DNA repair.
Residual RNF168 expression is required for BRCA1-independent RAD51 loading
In addition to countering HR via 53BP1 recruitment to DSB sites, RNF168 can also promote HR through directly recruiting PALB2-BRCA2-RAD51 to DSBs in a BRCA1-independent manner (33, 34). We speculated that residual levels of RNF168 expression remained present in BRCA1 null cells for the purpose of promoting PALB2-BRCA2-RAD51 loading. Indeed, RNAi-mediated depletion of RNF168 and PALB2, but less so RNF8, significantly reduced the residual RAD51 IRIF levels that could be detected in BRCA1 deficient MDA-MB-436 and SUM1315MO2 cells (Fig. 7A–B). Furthermore, while RNF8 depletion moderately reduced colony formation, RNF168 and PALB2 RNAi dramatically reduced the number of MDA-MB-436 and SUM1315MO2 cells that formed colonies relative to scrambled siRNA-treated control cells (Fig. 7C).
Figure 7. RNF168 is required for RAD51 loading in BRCA1 null cells.
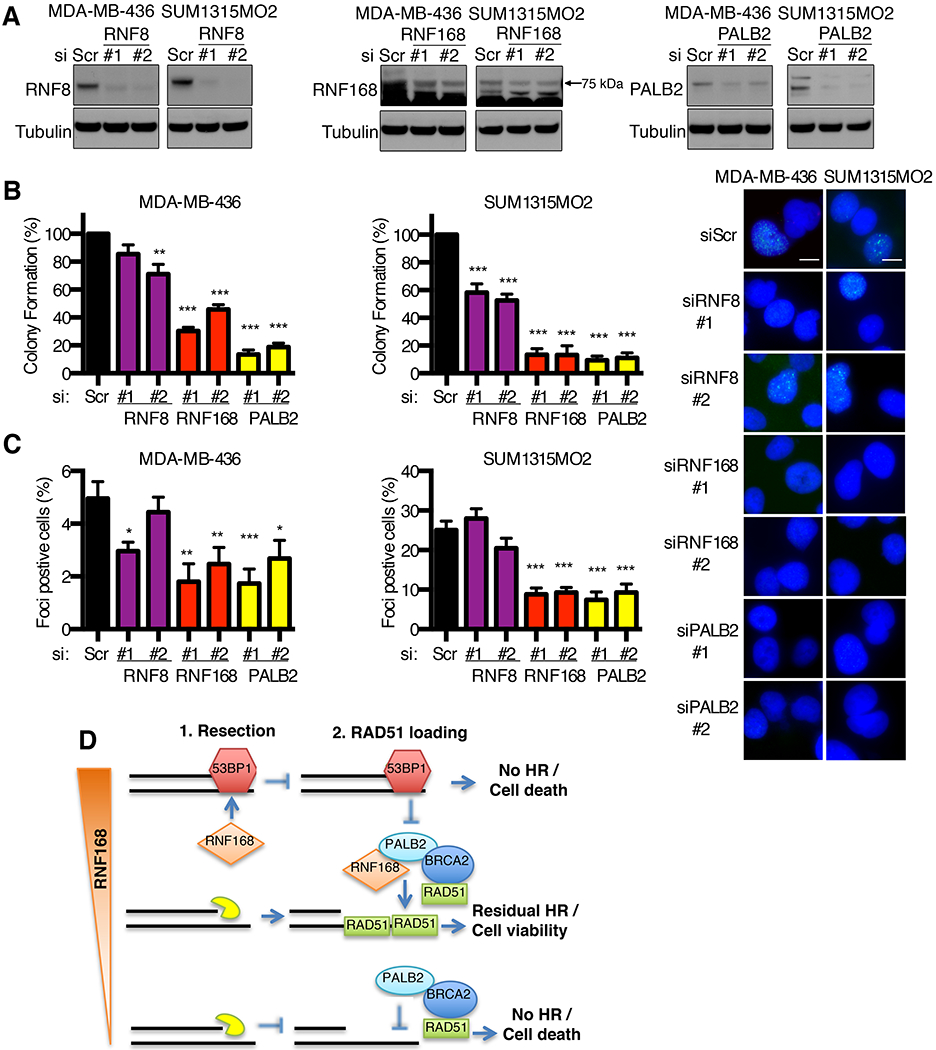
(A) Western blot showing the effects of scrambled (Sc) and two independent siRNAs targeting RNF8, RNF168 and PALB2 in MDA-MB-436 and SUM1315MO2 cell lines.
(B) Cells from (A) were assessed for RAD51 IRIF and quantified as described in Fig. 6A. Inset, representative images.
(C) Cells from (A) were assessed for colony formation. Mean ± S.E.M. are shown normalized to Sc treated cells, **p < 0.01, ***p < 0.001 (unpaired t-test) compared to Sc cells.
(D) Model for the role of RNF168 protein expression levels in regulating residual HR in BRCA1 null cancers.
Because RNF8 is required for RNF168 recruitment to DNA breaks, we examined the effects of RNF8 manipulation. Here, ectopic RNF8 was expressed and expression maintained and did not impact the colony formation of BRCA1 deficient cells (Fig. S6A). We found that RNF8 inducible expression resulted in a milder increase in 53BP1 IRIF compared to RNF168 (Fig. S6B), likely accounting for the ability of cells to maintain expression of the former. In line with RNF8 being required for RNF168 recruitment, depletion of RNF8 abrogated RNF168 foci, and consequently RNF168 overexpression-induced 53BP1 foci (Fig. S6C). These data confirm the role of RNF8 in recruiting RNF168 and indicate that RNF168-induced cell death is likely dependent on RNF8 activity.
In summary, we propose that when there is an abundance of RNF168, robust 53BP1 recruitment blocks DNA end resection, and in the absence of ssDNA, RNF168 is unable to promote PALB2-BRCA2-RAD51 loading, resulting in a complete shutdown of HR and cell death. Similarly, when both BRCA1 and RNF168 are entirely absent, DNA end resection occurs efficiently, but PALB2-BRCA2-RAD51 are unable to be loaded, resulting in loss of residual HR and cell death. In contrast, a reduction in basal RNF168 expression levels alleviates the 53BP1-mediated end resection block, and remaining RNF168 molecules promote PALB2-BRCA2-RAD51 loading, these events provide a residual level of HR that is sufficient to support cell viability (Fig. 7D).
Discussion
Genome instability drives the development and progression of cancer, with low fidelity DNA replication and repair responsible for generating mutations, translocations and rearrangements that provide a genomic niche favorable to carcinogenesis. Nonetheless, high mutational rates and improper DNA repair can also result in an inability to replicate the genome, and may trigger cellular senescence. Therefore, a balance between genome instability-driven carcinogenesis and maintaining a tolerable level of DNA damage is necessary for viability. Here, we describe a cellular adaptation involving the downregulation of RNF168 protein expression and ub-γH2AX signaling, which supports BRCA1 mutation-driven tumorigenesis.
BRCA1 and BRCA2 are essential mediators of HR. Because HR is critical for the viability of replicating cells, severely deleterious mutations or gene knockout results in cell death and embryonic lethality. Paradoxically, BRCA1/2 loss of function is beneficial to carcinogenesis, and mutation-containing cancer cells are highly malignant. Despite this accepted hallmark, in the context of established cancers, the absence of BRCA1/2 may not necessarily translate to loss of HR in its entirety; rather, we show here that cancer cells can acquire epi/genetic modifications that enable a residual level of HR to be maintained. In support of this notion, low numbers of RAD51 foci positive cells can often be detected in BRCA1 and BRCA2 mutant cancer cell lines (25, 35, 36), and it may be possible for limited RAD51 loading to occur below the threshold of experimental detection.
In mouse genetic experiments, 53BP1 knockout rescues HR and the development of BRCA1 mutant embryos (17). 53BP1-mediated inhibition of DNA end resection is dependent on RNF168 activity, with the latter ubiquitinating γH2AX and recruiting 53BP1 to sites of DNA damage. RNF168 depletion was previously shown to restore HR via loss of 53BP1 recruitment in BRCA1 mutant cancer cells (37). We found that decreased RNF168 expression provided enough HR activity to support basal cell viability, but was not sufficient to induce PARPi resistance. We previously reported a similar finding where 53BP1 KO rescued the embryonic viability of a BRCA1 null mouse allele, but derived cells had minimal RAD51 foci and remained sensitive to PARPi (26). Downstream of DNA end resection, BRCA1 is thought to be primarily responsible for recruiting PALB2-BRCA2-RAD51 to DSB sites. However, in the absence of BRCA1, RNF168 has a role in loading RAD51 via the PALB2-BRCA2 complex (33, 34). We show that while reduced RNF168 expression promotes end resection and HR, depleting RNF168 levels further resulted in loss of residual RAD51 loading activity in BRCA1 null cells.
BRCA1 mutant PDX tumors that had low levels of BRCA1-Δ11q expression were also intolerant to RNF168 overexpression. However, it is important to note that RNF168 expression was not universally downregulated in BRCA1 mutant cancers. SUM149PT BRCA1 mutant cells, which express an abundance of BRCA1-Δ11q, were able to readily tolerate ectopic overexpression. Indeed, the presence of wild-type or hypomorphic BRCA1 protein expression provided tolerance for RNF168 overexpression. Similarly, depletion of 53BP1 enabled cells to tolerate ectopic ub-H2AX expression. Thus, hypomorphic forms of BRCA1, loss of 53BP1 expression, and downregulation of RNF168 protein expression, are all mechanisms that BRCA1 mutant cancers employ to maintain residual HR pathway proficiency.
Overall, our work points to the retention of HR as a critical factor that is required for the viability and tumorigenicity of BRCA1 deficient cancers. Finally, de-ubiquitinases (DUBs) such as USP11 have been reported to specifically counteract RNF168-mediated ubiquitination of K13/15 γH2AX (38). We advocate that small molecule inhibitors of DUB enzymes could restore ub-H2AX signaling and consequently reduce HR activity to unsustainable levels, opening the possibility of a new synthetic lethal approach.
Supplementary Material
Statement of significance.
This study explores the concept that homologous recombination DNA repair is not an all-or-nothing concept but a spectrum, and that where a tumor stands on this spectrum may have therapeutic relevance.
Acknowledgments
This work was supported by US National Institutes of Health (NIH) Grant R01CA214799, Susan Komen CCRCR17499048, and OC130212 Department of Defense to N Johnson. J Krais was supported by an American Cancer Society - Tri State CEOs Against Cancer Postdoctoral Fellowship, PF-19-097-01-DMC, Ovarian Cancer Research Alliance and Phil and Judy Messing grant 597484, and T32 CA009035. Clovis Oncology supplied rucaparib. We are grateful to FCCC Genomics, Cell Culture and Cell Sorting facilities. We thank Dr. Daniel Durocher for providing the RNF168 and RNF8 plasmids, Dr. Thanos Halazonetis for providing the ub-H2AX plasmid, Dr. Stephen Sykes for providing the pLKO.1-RFP plasmid, and Drs. Clare Scott, Judith Balmaña and Violeta Serra for providing PDX samples.
Footnotes
The authors declare no conflicts of interest.
References
- 1.Stratton JF, Gayther SA, Russell P, Dearden J, Gore M, Blake P, Easton D, Ponder BA Contribution of BRCA1 mutations to ovarian cancer. N Engl J Med. 336:1125–1130, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Merajver SD, Frank TS, Xu J, Pham TM, Calzone KA, Bennett-Baker P, Chamberlain J, Boyd J, Garber JE, Collins FS, et al. Germline BRCA1 mutations and loss of the wild-type allele in tumors from families with early onset breast and ovarian cancer. Clin Cancer Res. 1:539–544, 1995. [PubMed] [Google Scholar]
- 3.Moynahan ME, Chiu JW, Koller BH, Jasin M Brca1 controls homology-directed DNA repair. Mol Cell. 4:511–518, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Roy R, Chun J, Powell SN BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 12:68–78, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evers B, Jonkers J Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 25:5885–5897, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Chen CC, Feng W, Lim PX, Kass EM, Jasin M Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol. 2:313–336, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng W, Jasin M BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature communications. 8:525, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stracker TH, Petrini JH The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 12:90–103, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiloh Y, Ziv Y The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 14:197–210, 2013. [PubMed] [Google Scholar]
- 10.Bekker-Jensen S, Mailand N Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst). 9:1219–1228, 2010. [DOI] [PubMed] [Google Scholar]
- 11.Nakada S Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in ubiquitination-dependent DNA double-strand break response signaling and DNA-repair pathway choice. J Radiat Res. 57 Suppl 1:i33–i40, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, Mailand N Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 527:389–393, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Pinato S, Scandiuzzi C, Arnaudo N, Citterio E, Gaudino G, Penengo L RNF168, a new RING finger, MIU-containing protein that modifies chromatin by ubiquitination of histones H2A and H2AX. BMC Mol Biol. 10:55, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell. 150:1182–1195, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, Lukas J, Lukas C RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 136:435–446, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Hu Q, Botuyan MV, Cui G, Zhao D, Mer G Mechanisms of Ubiquitin-Nucleosome Recognition and Regulation of 53BP1 Chromatin Recruitment by RNF168/169 and RAD18. Mol Cell. 66:473–487 e479, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 141:243–254, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, Haffty BG, Tommiska J, Blomqvist C, Drapkin R, Adams DJ, Nevanlinna H, Bartek J, Tarsounas M, Ganesan S, Jonkers J 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 17:688–695, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prakash R, Zhang Y, Feng W, Jasin M Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor perspectives in biology. 7:a016600, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, Xu D, Durocher D A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 49:872–883, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Chapman JR, Sossick AJ, Boulton SJ, Jackson SP BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of cell science. 125:3529–3534, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sy SM, Huen MS, Chen J PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 106:7155–7160, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen SX, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan XY, Ried T, Deng CX A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 17:3115–3124, 1998. [DOI] [PubMed] [Google Scholar]
- 24.Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng CX, Chodosh LA Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol Cell Biol. 21:4005–4015, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, Zhang Y, Harrell MI, Johnson SF, Candido Dos Reis FJ, Pharoah PD, Karlan B, Gourley C, Lambrechts D, Chenevix-Trench G, Olsson H, Benitez JJ, Greene MH, Gore M, Nussbaum R, Sadetzki S, Gayther SA, Kjaer SK, kConFab I, D’Andrea AD, Shapiro GI, Wiest DL, Connolly DC, Daly MB, Swisher EM, Bouwman P, Jonkers J, Balmana J, Serra V, Johnson N The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 76:2778–2790, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, DiMarcantonio D, Martinez E, Zong D, Handorf EA, Bellacosa A, Testa JR, Nussenzweig A, Gupta GP, Sykes SM, Johnson N BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell reports. 24:3513–3527 e3517, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bekker-Jensen S, Rendtlew Danielsen J, Fugger K, Gromova I, Nerstedt A, Lukas C, Bartek J, Lukas J, Mailand N HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat Cell Biol. 12:80–86; sup pp 81–12, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grofte M, Bartkova J, Poulsen M, Oka Y, Bekker-Jensen S, Mailand N, Neumann B, Heriche JK, Shearer R, Saunders D, Bartek J, Lukas J, Lukas C TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 150:697–709, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–404, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 6:pl1, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma Nature. 474:609–615, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kocylowski MK, Rey AJ, Stewart GS, Halazonetis TD Ubiquitin-H2AX fusions render 53BP1 recruitment to DNA damage sites independent of RNF8 or RNF168. Cell Cycle. 14:1748–1758, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, Couturier AM, Mullenders LH, Masson JY, van Attikum H A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. Elife. 6, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zong D, Adam S, Wang Y, Sasanuma H, Callen E, Murga M, Day A, Kruhlak MJ, Wong N, Munro M, Ray Chaudhuri A, Karim B, Xia B, Takeda S, Johnson N, Durocher D, Nussenzweig A BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol Cell. 73:1267–1281 e1267, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Kim HH, George E, Swisher EM, Simpkins F, Johnson N RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest. 126:3145–3157, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 59:3547–3551, 1999. [PubMed] [Google Scholar]
- 37.Munoz MC, Laulier C, Gunn A, Cheng A, Robbiani DF, Nussenzweig A, Stark JM Ring Finger Nuclear Factor RNF168 Is Important for Defects in Homologous Recombination Caused by Loss of the Breast Cancer Susceptibility Factor BRCA1. J Biol Chem. 287:40618–40628, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu M, Liu K, Mao Z, Luo J, Gu W, Zhao W USP11 Is a Negative Regulator to gammaH2AX Ubiquitylation by RNF8/RNF168. J Biol Chem. 291:959–967, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.