

SUMMARY
N6-methyladenosine (m6A), the most abundant internal modification in mRNA, has been implicated in tumorigenesis. As an m6A demethylase, ALKBH5 has been shown to promote the development of breast cancer and brain tumors. However, in acute myeloid leukemia (AML), ALKBH5 was reported to be frequently deleted, implying a tumor-suppressor role. Here, we show that ALKBH5 deletion is rare in human AML; instead, ALKBH5 is aberrantly overexpressed in AML. Moreover, its increased expression correlates with poor prognosis in AML patients. We demonstrate that ALKBH5 is required for the development and maintenance of AML and self-renewal of leukemia stem/initiating cells (LSCs/LICs), but not essential for normal hematopoiesis. Mechanistically, ALKBH5 exerts tumor-promoting effects in AML by post-transcriptional regulation of its critical targets such as TACC3, a prognosis-associated oncogene in various cancers. Collectively, our findings reveal essential functions of ALKBH5 in leukemogenesis and LSC/LIC self-renewal/maintenance, and highlight the therapeutic potential of targeting the ALKBH5/m6A axis.
Graphical Abstract
ETOC BLURB:
Selectively eradicating cancer stem cells in acute myeloid leukemia (AML) remains a challenge. Shen et al. demonstrate that targeting ALKBH5, an m6A eraser, effectively inhibits AML development/maintenance and suppresses leukemia stem cell self-renewal while sparing normal hematopoiesis, highlighting the therapeutic potential of targeting the ALKBH5/m6A/TACC3 axis in treating AML.
INTRODUCTION
Acute myeloid leukemia (AML) is a fatal form of hematopoietic malignancy, characterized with the clonal expansion and differentiation block of myeloid progenitor cells (Döhner et al., 2017; Thomas and Majeti, 2017). Leukemia stem/initiating cells (LSCs/LICs), characterized by their unlimited self-renewal/repopulating potential, are considered to be the root cause for the initiation and progression of the disease, as well as for treatment failure and relapse of AML (Krause and Van Etten, 2007; Thomas and Majeti, 2017). With currently available therapeutics, over 70% of AML patients cannot survive more than 5 years (Döhner et al., 2017). Thus, there is still an unmet and urgent medical need to develop more effective novel therapeutic approaches to eliminate LSCs/LICs and cure AML.
N6-methyladenosine (m6A) is the most abundant internal mRNA modification, and the methylation of N6-adenosine is mainly deposited by the m6A methyltransferase (writer) complex composed of a METTL3 (methyltransferase-like 3) and METTL14 (methyltransferase-like 14) heterodimeric enzyme core and a co-factor, WTAP (Wilms’s tumor 1-associating protein) (Liu et al., 2014; Ping et al., 2014). The m6A demethylases (erasers), which include FTO (fat mass- and obesity-associated protein) and ALKBH5 (α-ketoglutarate-dependent dioxygenase AlkB homolog 5) can remove m6A methylation (Jia et al., 2011; Zheng et al., 2013). The dynamic m6A modification of mRNAs can be recognized by different reader proteins (e.g., YTHDF1/2/3, YTHDC1/2 and IGF2BP1/2/3), leading to regulation of mRNA stability (Huang et al., 2018; Wang et al., 2014a; Wang et al., 2014b), translation efficiency (Meyer et al., 2015; Wang et al., 2015), alternative polyadenylation and splicing (Xiao et al., 2016), and secondary structural switches (Liu et al., 2015). Emerging evidence also indicates that the mRNA m6A modification is involved in a plethora of physiological and pathological processes, including hematopoiesis and leukemogenesis (Deng et al., 2018; Huang et al., 2020a; Huang et al., 2020b; Weng et al., 2019).
We previously reported that FTO, the first identified m6A demethylase (Jia et al., 2011), is overexpressed and plays a critical oncogenic role in AML pathogenesis and drug response by post-transcriptionally regulating expression of a set of important targets (e.g., ASB2, RARA, MYC and CEBPA) (Li et al., 2017; Su et al., 2018), and is a druggable target in AML (Huang et al., 2019). In addition, two m6A writer genes (i.e., METTL3 and METTL14) and one m6A reader gene (i.e., YTHDF2) have also been reported to be overexpressed and play important tumor-promoting roles in AML development/maintenance, and are required for the self-renewal of LSCs/LICs (Barbieri et al., 2017; Paris et al., 2019; Vu et al., 2017; Weng et al., 2018). Interestingly, while knockout of Mettl14 and Mettl3 could notably affect self-renewal of mouse normal hematopoietic stem/progenitor cells (HSPCs) or early HSPC development (Weng et al., 2018; Yao et al., 2018; Zhang et al., 2017a), Ythdf2 depletion could significantly promote HSPC expansion (Li et al., 2018; Paris et al., 2019; Wang et al., 2018a). The role of FTO in self-renewal of LSCs/LICs and normal HSPCs has yet to be investigated. The observations that both m6A writer and eraser genes (i.e., METTL3/METTL14 and FTO) play oncogenic roles in AML might not be surprising, as it is also well known that DNMT3A (a DNA methyltransferase) and TET2 (a DNA demethylase) both function as tumor-suppressors and are frequently associated with loss-of-function mutations in AML (Delhommeau et al., 2009; Deng et al., 2018; Ley et al., 2010). Interestingly, another DNA demethylase, TET1, has been reported to play a critical oncogenic role (opposite than does TET2) in AML (Huang et al., 2013; Jiang et al., 2017). We thus became interested in exploring potential role of the other m6A eraser, AlkB homolog 5 (ALKBH5), in AML to see if it functions differently from FTO.
ALKBH5 was identified as the second RNA m6A demethylase (Zheng et al., 2013), which, similar to FTO, is also an Fe(II)/2OG-dependent dioxygenase (Thalhammer et al., 2011). Complete deletion of Alkbh5 in mice led to impaired spermatogenesis and male infertility (Zheng et al., 2013). Subsequently, ALKBH5 was reported to facilitate the development of several types of solid tumors and promote self-renewal of relevant cancer stem cells (CSCs). For instance, in breast cancer stem cells (BCSCs), hypoxia-induced ALKBH5 expression stabilizes pluripotency factor mRNAs by catalyzing m6A demethylation, which in turn promotes BCSC maintenance (Zhang et al., 2016). Similarly, in glioblastoma (GBM), overexpression of ALKBH5 promotes self-renewal and proliferation of GBM stem-like cells (GSCs) through upregulation of FOXM1 (Zhang et al., 2017b). However, the function of ALKBH5 in leukemogenesis, LSC/LIC self-renewal and normal hematopoiesis remains elusive.
A previous study (Kwok et al., 2017) reported that ALKBH5 is frequently deleted in AML patients, especially in TP53-mutant cases, based on the analysis of The Cancer Genome Atlas (TCGA) AML cohort dataset (Ley et al., 2013), which implies that ALKBH5 may play a tumor-suppressor role in AML (Deng et al., 2018). Surprisingly, however, here we show that ALKBH5 is actually overexpressed in human AML and that its increased expression is associated with poor prognosis in AML. We reanalyzed the TCGA AML dataset (Ley et al., 2013), along with several other independent AML cohort datasets, and found that ALKBH5 deletion is very rare in AML, and that its expression level is not correlated with TP53 mutations in cancer. We next conducted a series of functional and mechanistic studies, which revealed that ALKBH5 plays a critical role in promoting leukemogenesis and LSC/LIC self-renewal as an m6A demethylase by post-transcriptional regulation of its critical target transcripts (e.g., TACC3), but exhibits little effect on normal hematopoiesis. These data highlight ALKBH5 as a promising therapeutic target in AML.
RESULTS
ALKBH5 is Overexpressed in AML and Its Increased Expression Correlates with Poor Prognosis in Patients
It was reported (Kwok et al., 2017) that the copy number loss of ALKBH5 is high in AML patients and is significantly associated with TP53 mutations. However, in analysis of the TCGA AML cohort (n = 200) (Ley et al., 2013) and the TARGET AML cohort (n = 1,025) (Bolouri et al., 2018), we found that the copy number loss (deep deletion) rate of ALKBH5 was only 1% (2/200) and 0.1% (1/1,025), respectively (Figure S1A), much lower than that (6.3%) reported previously (Kwok et al., 2017). In addition, based on the gene expression profiling datasets of a large AML patient cohort (GSE13159, n = 542) and a normal donor cohort (GSE42519, n = 6), we observed that ALKBH5 is expressed at a significantly higher level in various subtypes of AML compared to normal hematopoietic stem cell (HSC) controls (Figure 1A). In analysis of another AML gene expression dataset (GSE68833) including TP53 wild-type (n = 136) and mutant patients (n = 14), we did not observe significantly differential expression of ALKBH5 between these two populations (Figure S1B, left panel). Similarly, ALKBH5 expression levels showed no significant difference between TP53 wild-type and mutant patients in either GBM (TCGA Glioblastoma; n = 143) or breast cancer (TCGA Breast Cancer; n = 980) cohorts (Figure S1B, middle and right panels).
Figure 1. Biological effects of forced expression or knockdown of ALKBH5 on human AML cells.
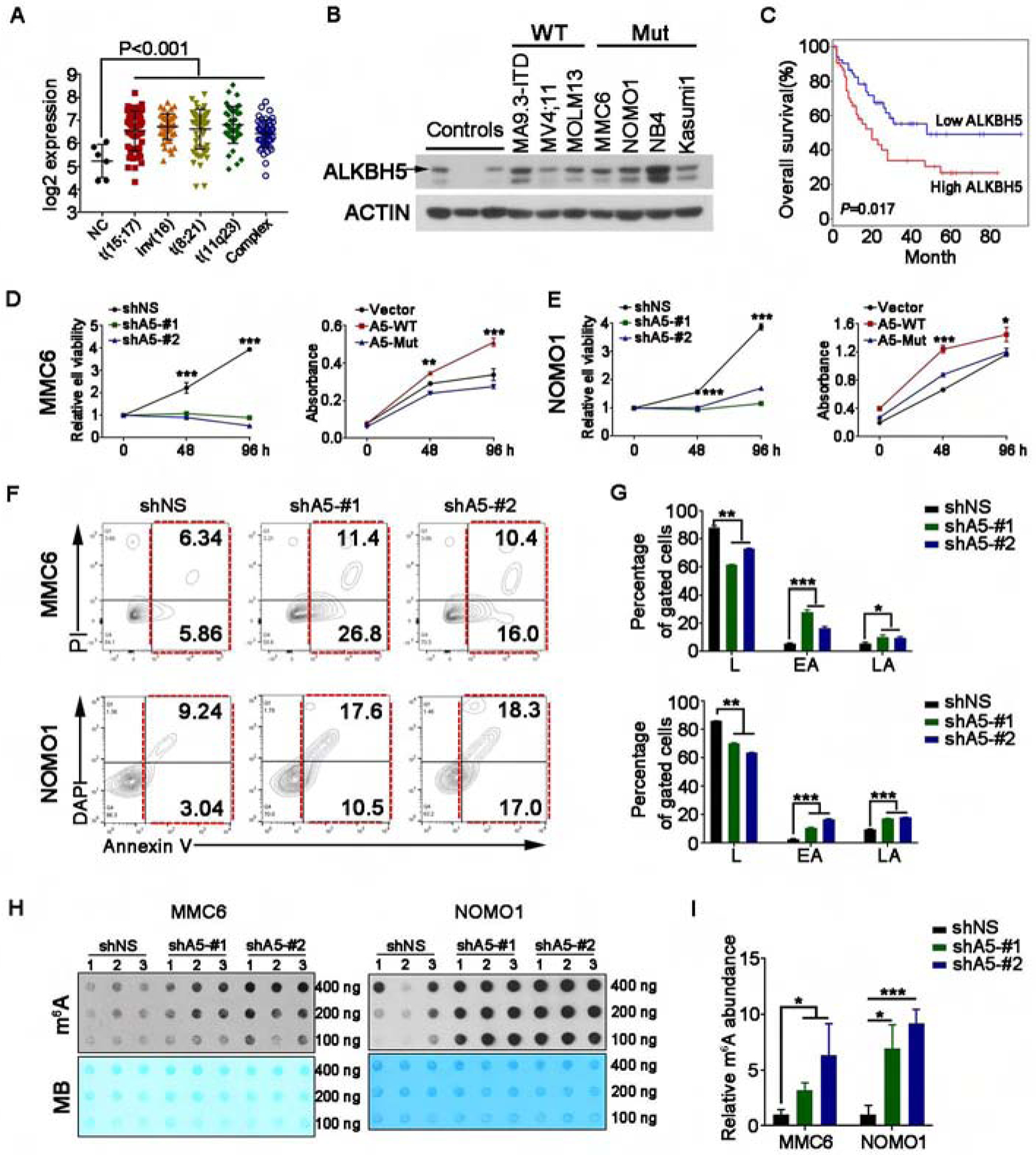
(A) Comparison of the expression levels of ALKBH5 in primary AML patients bearing various chromosomal translocations with those in BM hematopoietic stem cells (HSCs) collected from healthy donors (NC) based on the GSE13159 and GSE42519 datasets. The expression values (detected by Affymetrix exon arrays) were log2-transformed.
(B) Western blot of ALKBH5 protein in normal controls (human bone marrow mononuclear cells (MNCs)) as well as in TP53-wild-type and -mutant AML cell lines. ACTIN was used as a loading control.
(C) Kaplan-Meier survival analysis in TCGA-AML dataset (n=106). The patients were divided into two groups of equal size based on ALKBH5 levels. The p value was detected by the log-rank test.
(D and E) Cell growth/proliferation assays in MONOMAC6 (MMC6) (D) and NOMO1 (E) AML cell lines (TP53-mutant) transduced with lentiviruses expressing control shRNA (shNS) or two independent shRNAs targeting ALKBH5 (shA5-#1 and shA5-#2), as well as those expressing empty vector (Vector), wild-type ALKBH5 (A5-WT) and m6A demethylase-inactive ALKBH5 mutant (A5-Mut).
(F and G) Representative flow cytometry plots (F) and statistics (G) of the percentage of apoptotic cells in MMC6 and NOMO1 cells with shNS or ALKBH5 shRNAs.
(H and I) m6A dot blot assays (H) and quantitative comparison (I) of global m6A abundance in MMC6 and NOMO1 cells with shNS or ALKBH5 shRNAs (n=3 biological replicates). MB, methylene blue staining (as loading control).
Mean±SD values are shown for Figures 1A, D, E, G and I. *p < 0.05; **p <0.01; ***p < 0.001; t test. See also Figure S1.
Our western blot data also showed that ALKBH5 protein levels are comparable between TP53 wild-type and mutant AML cell lines, but both are higher than that in normal control cells (Figure 1B). Moreover, we also found that higher ALKBH5 expression is associated with shorter overall survival (OS) in AML patients according to the TCGA AML dataset (Ley et al., 2013) (Figure 1C). In contrast, although several other m6A regulatory genes such as METTL3, METTL14, WTAP, FTO and YTHDF2 have been reported to play oncogenic roles in AML (Bansal et al., 2014; Barbieri et al., 2017; Li et al., 2017; Paris et al., 2019; Su et al., 2018; Vu et al., 2017; Weng et al., 2018), their expression levels were not significantly associated with prognosis (see Figure S1C).
ALKBH5 Is Required for the Growth of Human AML Cells
To investigate the role of ALKBH5 in AML, both gain- and loss-of-function studies were conducted. ALKBH5 knockdown by shRNAs (Figure S1D, left panel) caused a substantial inhibition on human AML cell growth (Figures 1D–E, left panels), a significant induction of apoptosis (Figures 1F–G), and a noticeable increase in global m6A level (Figures 1H–I) in both MONOMAC-6 (MMC6) and NOMO1 AML cells. Conversely, forced expression (Figure S1D, right panel) of ALKBH5 wild-type (A5-WT), but not ALKBH5-H204A mutant (A5-Mut; a catalytically inactive mutant (Zheng et al., 2013)), significantly promoted growth of human AML cells (Figures 1D–E, right panels). Similar effects were observed in additional AML cell lines when ALKBH5 expression was manipulated (Figures S1E–G). Notably, manipulation of ALKBH5 expression exhibited similar effects between TP53-wild-type (e.g., MA9.3-ITD and MOLM13) and TP53-mutant (e.g., MMC6, NOMO1 and NB4) AML cell lines, suggesting the role of ALKBH5 in AML cells is not dependent on TP53 mutation status. We also established doxycycline-inducible ALKBH5 conditional knockdown MOLM13 cells (MOLM13-iKD) and knockout cells (MOLM13-iCas9). After doxycycline addition, ALKBH5 was sufficiently depleted (Figures S1H and S1K); similar to the stable knockdown of ALKBH5, conditional depletion of ALKBH5 also showed significant effects on AML cell growth and global m6A level (Figures S1I–J and S1L–M).
ALKBH5 is Required for Oncogene-Induced Cell Immortalization and Leukemogenesis
To examine the role of ALKBH5 in AML development, a targeted deletion of Alkbh5 in mice was created by deleting a part of exon 1 of the Alkbh5 gene using CRISPR-Cas9 technology (Hsu et al., 2014; Yuan and Hu, 2017) (Figure 2A). The Alkbh5 deficiency in mice was confirmed by genotyping, western blot, and Sanger sequencing analysis (Figures 2B–C and S2A). The Alkbh5 knockout (KO) (heterozygous or homozygous) mice exhibited normal development compared to the wild-type mice, and no obvious defects were observed in their growth and lifespan. Alkbh5 homozygous KO pups had a slightly higher female-to-male ratio compared to the heterozygous KO or wild-type pups. Consistent with a previous report (Zheng et al., 2013), Alkbh5 homozygous KO male mice are infertile.
Figure 2. The role of Alkbh5 in MLL-AF9 (MA9) mediated leukemogenesis.
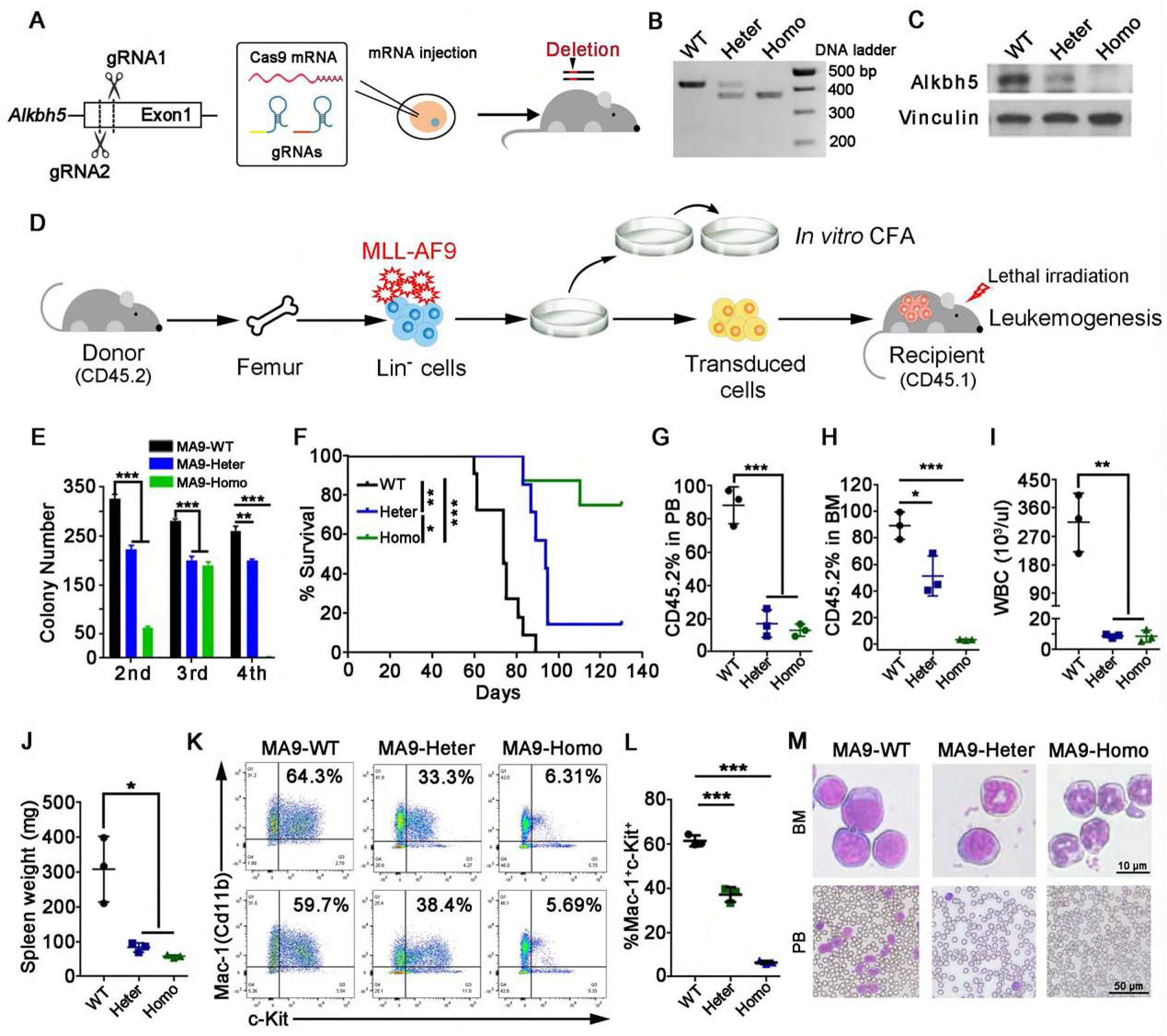
(A) Scheme of the design and procedures of generation of Alkbh5 knockout (KO) mice using the CRISPR-Cas9 technology.
(B and C) Representative DNA genotyping (B) and Western blot assay (C) data of the samples from Alkbh5 wild-type (WT), heterozygous (Heter) or homozygous (Homo) KO mice were shown.
(D) Bone marrow (BM) lineage negative (Lin−) cells collected from Alkbh5 WT, Heter or Homo KO mice were transduced with MA9 retrovirus and used for colony-forming/replating (CFA) assays. The MA9-transduced cells were also transplanted into lethally irradiated recipient mice after first round of colony formation for leukemogenesis.
(E) Colony forming cell counts at each round of plating were shown (n=3).
(F) Kaplan-Meier survival curves of recipient mice transplanted with MA9-transduced Alkbh5 WT (n=11), Heter (n=7) and Homo (n=8) HSPCs.
(G-M) Three mice from each transplant group were euthanized at the same time (day 61 post bone marrow transplantation (BMT)) for AML development analysis. (G-H) Percentage of CD45.2+ cells in peripheral blood (PB) (G) and BM (H) of recipient mice. (I) WBC count in PB of recipient mice. (J) Spleen weight of recipients. (K-L) Percentage of Mac-1+c-Kit+ cells in recipient mice. Representative flow cytometry plots (K) and statistics analysis (L) are shown. (M) Representative images of Wright-Giemsa staining of BM and PB from recipient mice.
*p < 0.05; **p <0.01; ***p < 0.001; t test (for Figures 2E, G–J and L; Mean±SD values are shown) or log-rank test (for Figure 2F). See also Figure S2.
We observed a significant increase in global m6A abundance in the BM of Alkbh5-deficient mice compared to that of their wild-type counterparts, as detected by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) (Figure S2B), consistent with the m6A demethylase activity of Alkbh5. To investigate the requirement of ALKBH5 in leukemic cell transformation and leukemogenesis, we employed MLL-AF9 (MA9)-mediated cell transformation (colony-formation/immortalization) and leukemogenesis models, coupled with the Alkbh5 KO mouse models (Figure 2D). MA9 is the most common form of MLL-rearranged fusion proteins in AML, which alone is sufficient to transform normal HSPCs and rapidly induce AML in mice, and thus MA9-induced AML has been widely used as a common AML model (Huang et al., 2020a; Krivtsov and Armstrong, 2007).
As shown in Figure 2E, genetic KO of Alkbh5 (Figure S2C), especially for the homozygous KO, significantly inhibited MA9-mediated cell immortalization as detected by the in vitro colony-forming/replating assays (CFAs). Conversely, forced expression of wild-type (A5-WT), but not mutated ALKBH5 (A5-Mut) significantly promoted MA9-mediated cell immortalization (Figures S2D–E).
To evaluate the role of Alkbh5 in leukemogenesis in vivo, we conducted mouse BM transplantation (BMT) assays. As shown in Figure 2F, depletion of Alkbh5 expression in mice significantly delayed leukemia onset and prolonged the survival in recipient mice in a dose-dependent manner. Notably, homozygous KO of Alkbh5 sufficiently inhibited leukemia onset in approximately 80% of the recipients (Figures 2F and S2G). We observed that deletion of Alkbh5 significantly inhibited the engraftment of MA9-transformed donor cells in peripheral blood (PB) (Figures 2G and S2H), BM and spleen (Figures 2H and S2I) of recipients (all euthanized simultaneously), leading to substantial reduction in white blood cell (WBC) count (Figures 2I and S2J), spleen weight (Figures 2J and S2K), liver weight (Figure S2L), immature blast cell population in PB and BM (Figures 2K–M and S2M), and leukemia infiltration in spleen and liver (Figure S2M).
ALKBH5 Expression Depletion Impairs AML Cell Repopulation and AML Maintenance
To determine if ALKBH5 is also required for AML maintenance, we transduced Alkbh5 shRNAs (shA5-#a and -#b) or a control shRNA (shNS) into leukemic BM cells collected from primary AML mice bearing MA9, MLL-AF10 (MA10), AML1-ETO9a (AE9a) or FLT3-ITD/NPM1-Mutant (FLT3-ITD/Mut)-induced AML (Jiang et al., 2017; Weng et al., 2018; Yan et al., 2006), and then performed CFAs. As shown in Figures 3A and S3A–B, Alkbh5 knockdown (associated with increased m6A abundance; Figure 3B) dramatically inhibited colony number, colony size, and cell number in all AML subtype models.
Figure 3. Knockdown of ALKBH5 affects the maintenance of human and murine AML.
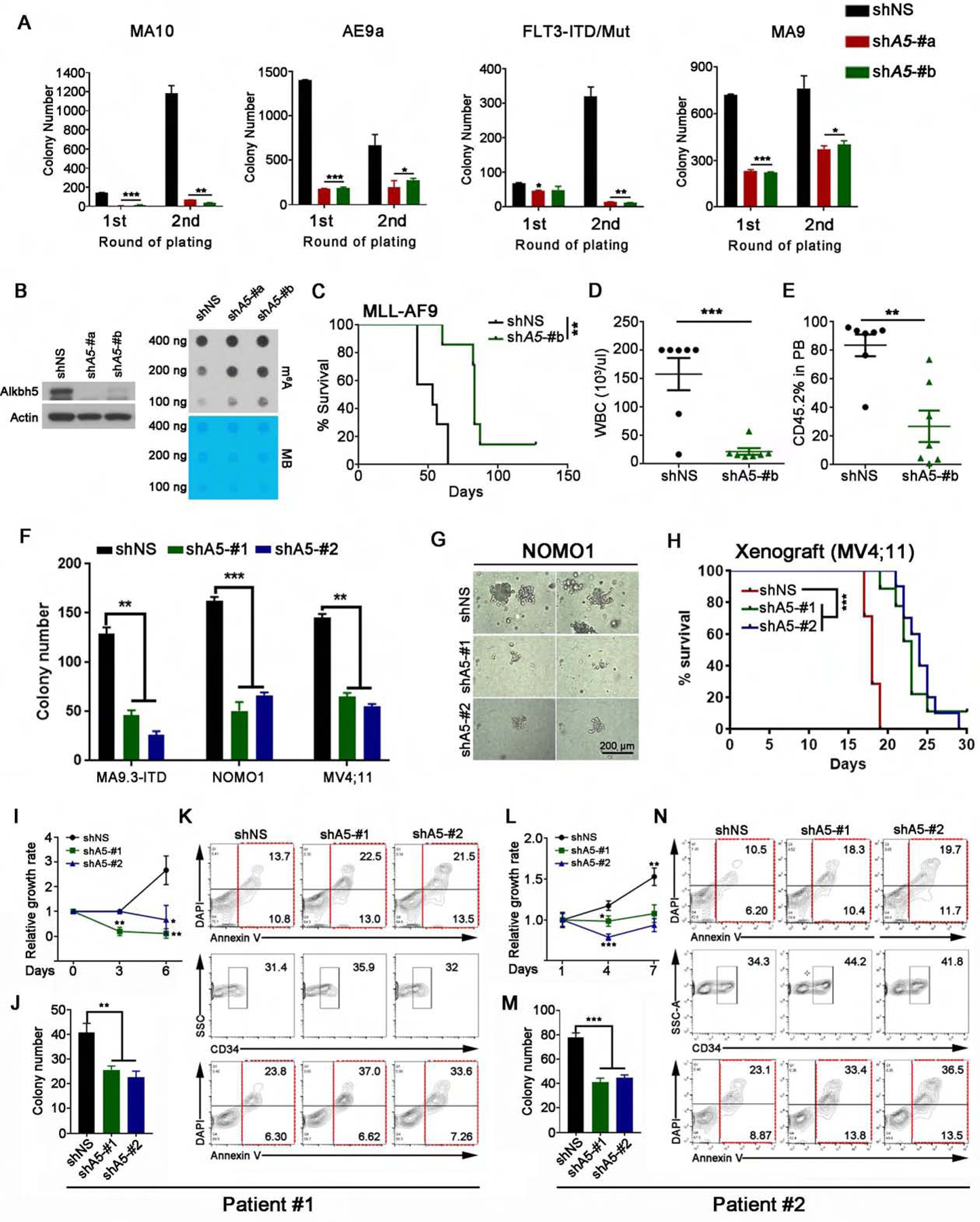
(A) Primary mouse leukemic BM cells were transduced with lentiviruses expressing control shRNA (shNS) and two independent shRNAs targeting Alkbh5 (shA5-#a and shA5-#b) and serially replated. Colony forming cell counts at each round of plating were shown (n=3).
(B) Western blot of Alkbh5 (left) and dot blot of global m6A abundance (right) in mouse MA9 AML cells transduced with shNS or Alkbh5 shRNAs. Actin was used as a loading control in Western blot. MB, methylene blue staining, was used as loading control in m6A dot blot.
(C) Kaplan-Meier survival curves of recipients transplanted with mouse MA9 AML cells with shNS or Alkbh5 shRNA (shA5-#b) (n=7 for each group).
(D-E) WBC count (D) and percentage of CD45.2+ cells (E) in the PB of recipient mice.
(F and G) Human AML cells were transduced with shNS or ALKBH5 shRNAs and then plated for colony forming assays. (F) Colony forming cell counts (n=3). (G) Representative pictures of colonies from NOMO1 cells. MA9.3-ITD (P53 wild-type), NOMO1 (P53 mutant) and MV4;11 (P53 wild-type) were used.
(H) Kaplan-Meier survival curves of NSGS mice transplanted with MV4;11 AML cells that were transduced with shNS (n=7) or ALKBH5 shRNAs (shA5-#1, n=9; shA5-#2, n=10) (0.1×106 donor cells/mouse).
(I-N) Primary leukemia cells from AML patients were transduced with shNS or ALKBH5 shRNAs and then seeded for experiments. (I, L) Cell growth/proliferation assays of transduced primary AML cells. (J, M) Colony forming cell counts of transduced AML cells. (K, N) Percentage of apoptotic cells in overall (top panel) or CD34+ (lower panel) transduced AML cells.
*p < 0.05; **p <0.01; ***p < 0.001; t test (for Figures 3A, D–F, I–J and L–M; Mean±SD values are shown) or log-rank test (for Figures 3C and H). See also Figure S3.
Secondary BMT assays demonstrated that knockdown of Alkbh5 significantly impaired progression of MA9-induced AML in recipient mice (Figures 3C–E). We next conducted in vitro (CFA) and in vivo (xenograft model) assays to evaluate the potential role of ALKBH5 in the maintenance of human AML. As expected, either stable or inducible ALKBH5 knockdown significantly decreased the colony number and size of human AML cells (Figures 3F–G, S3C–D). Moreover, ALKBH5 knockdown significantly delayed human AML progression and prolonged survival in xenograft recipient mice (Figures 3H and S3E–G). ALKBH5 knockdown also significantly inhibited human primary AML cell growth and colony-forming ability, and promoted apoptosis in human bulk AML cells as well as a CD34+ blast population (Figures 3I–N and S3H–I). Collectively, our data suggest that ALKBH5 is required for the maintenance of AML.
Depletion of ALKBH5 Expression Impairs LSC/LIC Self-Renewal
In AML patients, drug resistance and relapse have been linked with the existence of LSCs/LICs (Krause and Van Etten, 2007; Thomas and Majeti, 2017). As shown in Figure 1C, higher ALKBH5 expression correlates with shorter survival in AML patients. Moreover, ALKBH5 is expressed at a significantly higher level in LSCs than in AML whole blast cells (Figure S4A), and its protein level is also higher in CD34+ LSCs/LICs than in CD34− bulk AML cells (Figure S4B). In contrast, its expression level in normal CD34+ HSPCs is even lower than that in more mature normal CD34− cells (Figures S4C–D). These data imply that ALKBH5 may play an important role specifically in LSCs/LICs.
To assess the role of ALKBH5 in the self-renewal of LSCs/LICs, we virally transduced MLL-AF9-YFP into Lin− BM HSPCs and performed in vitro and in vivo assays. We showed that Alkbh5 KO significantly inhibited cell growth and promoted apoptosis in MA9-transformed HSPCs (Figures S4E–F), and caused a markedly decreased percentage of lin−c-kit+ (LK) cells and increased apoptosis in the LK cells (Figures S4G–H) in vitro. In the in vivo BMT assays, we observed that the ratio of the GMP-like (L-GMP) leukemic cell population (YFP+Lin−c-Kit+Scal1−CD34+FcrRII/III+) (Krivtsov et al., 2006) in BM was significantly lower in the Alkbh5 KO group than in the WT group (Figures 4A–B), accompanied with a higher apoptosis rate in L-GMP cells of the former group (Figure 4C). Similar patterns were observed in the Gr-1−c-Kit+ cell population (Figures S4I–J), which was also recognized as LSCs/LICs (Wang et al., 2010).
Figure 4. Effects of Alkbh5 depletion on self-renewal/repopulation of leukemia stem/initiating cells (LSCs/LICs) and normal hematopoiesis.
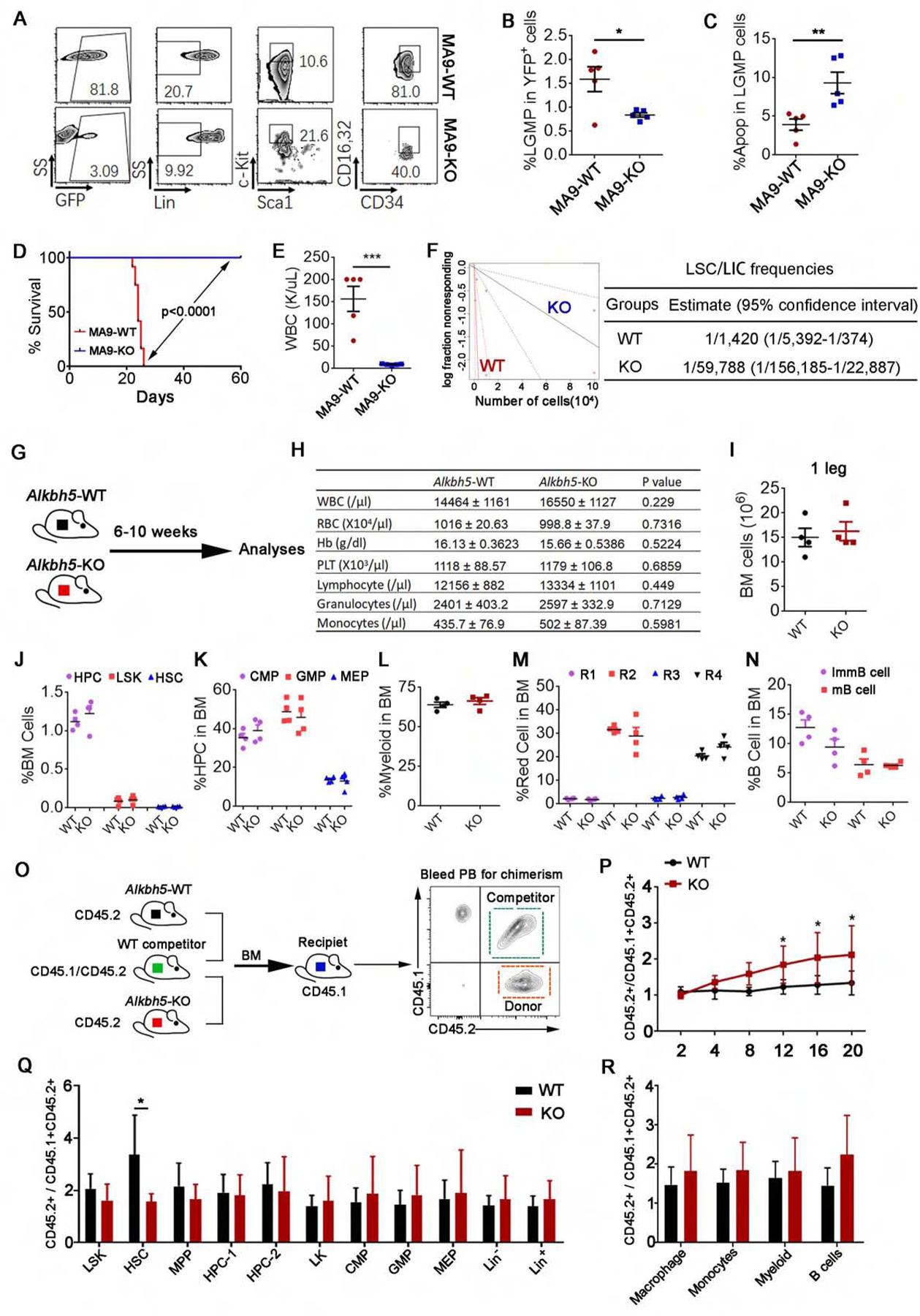
(A-C) BM lin- cells from Alkbh5 WT or homozygous KO mice were transduced with MA9-YFP retrovirus and then transplanted into primary recipient mice. Five mice from each transplant group were euthanized at the same time (day 42 post-BMT) for LSC/LIC analysis. (A) Representative flow cytometry analysis of LSCs/LICs in BM. (B) Percentage of leukemic GMPs (LGMPs) in YFP+ cells. (C) Percentage of apoptotic LGMPs.
(D and E) YFP+ BM cells were sorted out from primary BMT mice and transplanted into secondary recipient mice for 2nd BMT assay. (D) Survival curves of the 2nd BMT mice (MA9-WT, n=12; MA9-KO, n=10). (E) WBC count from the 2nd BMT mice euthanized on day 25 post-BMT.
(F) The in vivo limiting dilution assay (LDA). Secondary recipients (n=5 for each group) were transplanted with different doses of BM cells collected from primary recipients (see Figure 2F) euthanized at the same time (Day 61 post BMT).
(G-N) Effects of Alkbh5 KO on mouse static normal hematopoiesis. (G) Regularly bred 6- to 10-weeks Alkbh5 WT (n=7) and KO (n=10) mice were included for the analysis. PB CBC counts were shown in (H). BM analysis of 4 pairs of the same sex/age littermates were shown in (I-N).
(O-R) The in vivo competition assay. (O) Schematic outline of experiment strategy of the in vivo competition assays. (P) Ratio of CD45.2+/CD45.1+CD45.2+ in the PB of recipients. (Q and R) Ratio of CD45.2+/CD45.1+CD45.2+ in the Lin+, Lin−, LK, LSK, HSC and other progenitor cell populations (Q) and differentiated cell compartments (R) in the BM of recipients.
*p < 0.05; **p <0.01; ***p <0.001; t test (for Figures 4B–C, E, H–N, P–R; Mean±SD values are shown) or log-rank test (for Figures 4D and F). See also Figure S4.
We next showed that Alkbh5 KO dramatically inhibited the repopulation capacity of primary AML cells in vivo and blocked AML onset in secondary BMT recipients (Figures 4D–E and S4K), suggesting ALKBH5 is required for LSC/LIC repopulation. To quantitatively assess the effect of Alkbh5 depletion on LSC/LIC self-renewal, we conducted in vitro limiting dilution assays (LDAs) (Somervaille and Cleary, 2006). We found that knockdown of Alkbh5 significantly decreased LSC/LIC frequency in mouse MA9 cells (1/197.4 versus 1/11.9, p<0.001) (Figure S4L). We further performed in vivo LDAs and showed that the LSC/LIC frequency in primary leukemic BM cells was significantly lower in the Alkbh5 KO group than in the WT control group (1/59,788 vs 1/1,420; p<0.01) (Figures 4F and S4M). Together, our data suggest that ALKBH5 is required for LSC/LIC self-renewal.
Alkbh5 Deletion Does Not Significantly Affect Normal Hematopoiesis
We next investigated whether Alkbh5 depletion could cause any detrimental effects on HSC functions and multilineage development. First, we analyzed the complete blood count (CBC) in PB from regularly bred WT or KO mice and found no significant changes in any differentiated lineage cells between KO and WT mice (Figures 4G–H). We also euthanized four pairs of WT and KO mice (same sex, littermates) and showed that Alkbh5 deletion did not cause significant changes in total BM cell number or percentages of different subpopulations of HSPCs and differentiated lineages in BM (Figures 4I–N). Our data suggest that Alkbh5 deletion has no obvious impact on HSPC homeostasis and multilineage hematopoiesis in the steady state.
We next showed that Alkbh5 KO did not significantly affect HSPC growth or apoptosis during long-term in vitro culture (Figures S4N–O). To further reveal the effect of Alkbh5 deletion on the repopulation capacity of HSPCs, we performed in vivo competition assays. Briefly, we competitively transplanted BM cells from WT or KO donor mice (CD45.2+) and competitor mice (CD45.1+CD45.2+) into lethally irradiated recipients (CD45.1+), and monitored the CBC and engraftment in PB (Figure 4O). CBC analysis showed that different lineages of blood cells between the two groups of recipient mice exhibited no significant difference at most time points (Figures S4P–V). BM cells from KO mice showed a slightly higher reconstitution capacity compared to the WT counterpart (Figure 4P). Interestingly, we found a moderate decrease in the HSC population, as well as a slight increase in some progenitor cell populations and in all differentiated populations in the KO group (Figures 4Q–R and S4W), suggesting Alkbh5 deletion caused a slight expansion of progenitor and differentiated cells, likely due to the increased differentiation of HSCs. Our data suggest ALKBH5 may play a minor role in maintaining normal HSC self-renewal in hematopoiesis in a stressed state (e.g., under competitive stress).
Identification of Potential Targets of ALKBH5 in AML
To reveal the mechanism(s) underlying ALKBH5 function in AML, we conducted transcriptome-wide RNA-seq, RNA immunoprecipitation-seq (RIP-seq) and m6A-seq. The RNA-seq revealed that 623 and 1,237 genes were significantly up- and down-regulated, respectively, upon ALKBH5 knockdown in both MOLM13 and NOMO1 cells (Figures 5A and S5A–C). Using the Molecular Signature Database (MSigDB) of Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005), we identified the top 10 pathways in which those up-regulated and down-regulated genes were enriched (Figure 5B). Notably, ALKBH5 knockdown activated apoptosis and p53 pathways, while causing significant suppression of E2F targets, G2M checkpoints, MYC targets and mitotic spindle pathways (Figures 5B–C and S5D), which was consistent with our findings that ALKBH5 knockdown increased apoptosis and inhibited cell growth in AML cells. The RIP-seq (Figures 5D) revealed that the vast majority of ALKBH5 binding sites are located in protein coding transcripts (Figure 5E), and identified 1,392 genes as potential direct targets of ALKBH5 whose transcripts were strongly bound by ALKBH5 in AML cells. These targets are enriched in cell cycle- and proliferation-related pathways (Figure 5F). Our m6A-seq data showed that the vast majority of m6A peaks are distributed in the protein-coding region (CDS) and 3’ untranslated region (3’UTR) of mRNA transcripts in AML cells (Figures 5G–H). We identified 510 genes whose transcripts are associated with significantly increased m6A (m6A-hyper) peaks upon ALKBH5 knockdown. The major signaling pathways enriched with the 510 genes were shown in Figure 5I. Notably, many pathways are commonly detected by RNA-seq, RIP-seq and m6A-seq (Figures S5E–F), suggesting they are the main pathways enriched with potential responsive direct targets of ALKBH5 in AML.
Figure 5. Transcriptome-wide identification of ALKBH5 potential targets in AML cells.
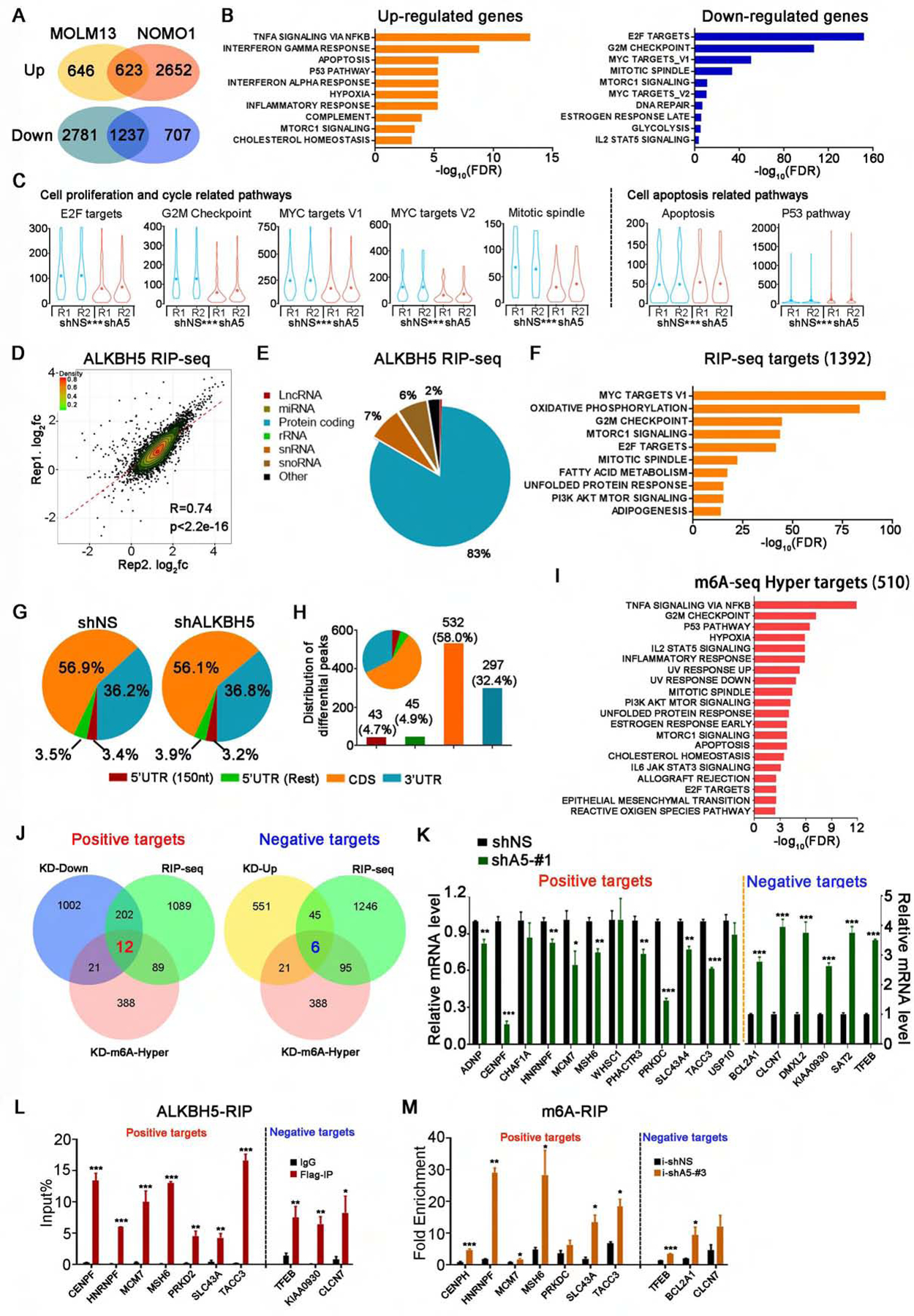
(A-C) RNA-seq analysis of gene expression profiles in ALKBH5 knockdown AML cells and control AML cells. (A) Venn diagram shows numbers of genes with significant changes in expression (RPKM>1, fold change>1.5) upon ALKBH5 knockdown. (B) GSEA of up- and down-regulated genes. (C) Violin plots showing the relative abundance of genes involved in the indicated pathways in ALKBH5 knockdown or control NOMO1 cells.
(D-F) RIP-seq analysis of ALKBH5 overexpressing NOMO1 cells. (D) Scatter plots of ALKBH5 RIP-seq replicates showing the correlation of enriched genes. (E) Pie charts showing the distribution of RIP-seq reads in RNA classes. (F) GSEA of significantly enriched genes in RIP samples (RPKM>1, immunoprecipitation/input>2).
(G-I) m6A-seq analysis of ALKBH5 knockdown NOMO1 cells. (G) The distribution of total m6A peaks in the indicated regions of mRNA transcripts in the control and ALKBH5-knockdown cells. (H) The distribution of differential m6A peaks (i.e., those with significant changes upon ALKBH5 manipulation). 5’UTR (150 nt) represents the first 150 nt of 5’ end of 5’UTR, while 5’UTR (Rest) represents the remaining regions of 5’UTR. (I) GSEA of the genes with significantly increased m6A abundance in ALKBH5 knockdown cells (p<0.05).
(J) Integrative analysis to identify transcriptome-wide potential targets of ALKBH5 in AML. Left: potential positive targets of ALKBH5. Right: potential negative targets of ALKBH5. KD-Down and KD-Up: genes with significantly decreased and increased expression, respectively, upon ALKBH5 knockdown in both NOMO1 and MOLM13 cells as detected by RNA-seq (RPKM>1, fold change >1.5). RIP-seq: genes with significant enrichment in RIP samples (RPKM>1, immunoprecipitation/input>2). KD-m6A-Hyper: genes with significantly higher m6A abundance in ALKBH5 knockdown cells (p<0.05).
(K) Expression change validation of potential positive and negative targets of ALKBH5 by qPCR.
(L) ALKBH5-RIP qPCR validation of ALKBH5 binding of representative positive and negative targets.
(M) Gene-specific m6A-RIP qPCR validation of m6A level changes of representative positive targets and negative targets.
Mean±SD values are shown for Figures 5C, K, L and M.*p < 0.05; **p <0.01; ***p < 0.001; t test. See also Figure S5.
Through integrative analysis of the RNA-seq, RIP-seq and m6A-seq data, we identified 18 highly confident potential targets of ALKBH5 in AML, of which 12 and 6 are significantly positively and negatively regulated by ALKBH5, respectively (Figure 5J and Table S1). Indeed, our RIP-qPCR, gene-specific m6A-qPCR, and qPCR results confirmed that most of these transcripts were strongly bound by ALKBH5, and were associated with significantly increased m6A abundance and expected expression level changes in AML cells upon ALKBH5 knockdown (Figures 5K–M).
As our previous studies showed that FTO, another m6A eraser, also plays an important oncogenic role in AML (Li et al., 2017; Su et al., 2018), it would be interesting to compare the targets and pathways affected by the two m6A erasers in AML. Based on the RNA-seq data, we found ALKBH5 knockdown (A5-KD) caused more genes to be down-regulated (Down vs. Up: 1,237 vs. 623) than did FTO knockdown (FTO-KD) (Down vs. Up: 888 vs. 2,279). Among these genes, only 119 down-regulated and 251 up-regulated genes were shared by ALKBH5 and FTO (Figure S5G, top panel). Nevertheless, although they shared a relatively small fraction of potential targets, the pathways affected by the two m6A erasers substantially overlapped (Figure S5G, bottom panel), suggesting that knockdown of either m6A eraser affects multiple similar pathways in AML. Similar findings were also observed in analysis of the m6A-seq data (Figure S5H). Notably, among the 18 highly confident potential targets of ALKBH5 (Table S1), only MCM7 and TFEB are also potential targets of FTO based on the above data analysis. Overall, it appears that ALKBH5 and FTO target more distinct transcripts than shared ones, although they target many shared pathways.
TACC3 Is a Direct and Functionally Important Target of ALKBH5 in AML
As a positive target of ALKBH5, TACC3 displayed an expected adverse prognostic impact in AML (Figure 6A), similar to that of ALKBH5 (Figure 1C), whereas the other highly potential targets of ALKBH5 showed either non-significant or unexpected prognostic impact in AML (Table S1). TACC3 exhibited a significantly (p<0.05) positive correlation in expression with ALKBH5 across primary AML samples (Figure S6A). In fact, among all the candidate targets tested, TACC3 transcripts are also associated with the greatest enrichment of ALKBH5 (Figure 5L). Consistent with the RNA-seq and m6A-seq data (Figure 6B), our qPCR validations confirmed that TACC3 transcripts are associated with significantly decreased expression level and increased m6A abundance upon ALKBH5 knockdown (Figures 5K and 5M). Moreover, TACC3 has been reported to be overexpressed and exhibit an adverse prognostic impact in various types of cancers (e.g., brain tumor, and breast, prostate, liver, lung, and pancreatic cancers), and play a critical oncogenic role in tumorigenesis and CSC self-renewal in a wide range of cancers (Jung et al., 2006; Song et al., 2018; Sun et al., 2017; Wang et al., 2018b; Yao et al., 2012; Yun et al., 2015; Zhou et al., 2015). Such characteristics of TACC3 are largely similar to those of ALKBH5 (see Refs. (Cho et al., 2018; Zhang et al., 2016; Zhang et al., 2019; Zhang et al., 2017b) and data shown herein). Thus, we decided to focus on TACC3 for further studies.
Figure 6. ALKBH5 regulates TACC3 expression via affecting its mRNA stability.
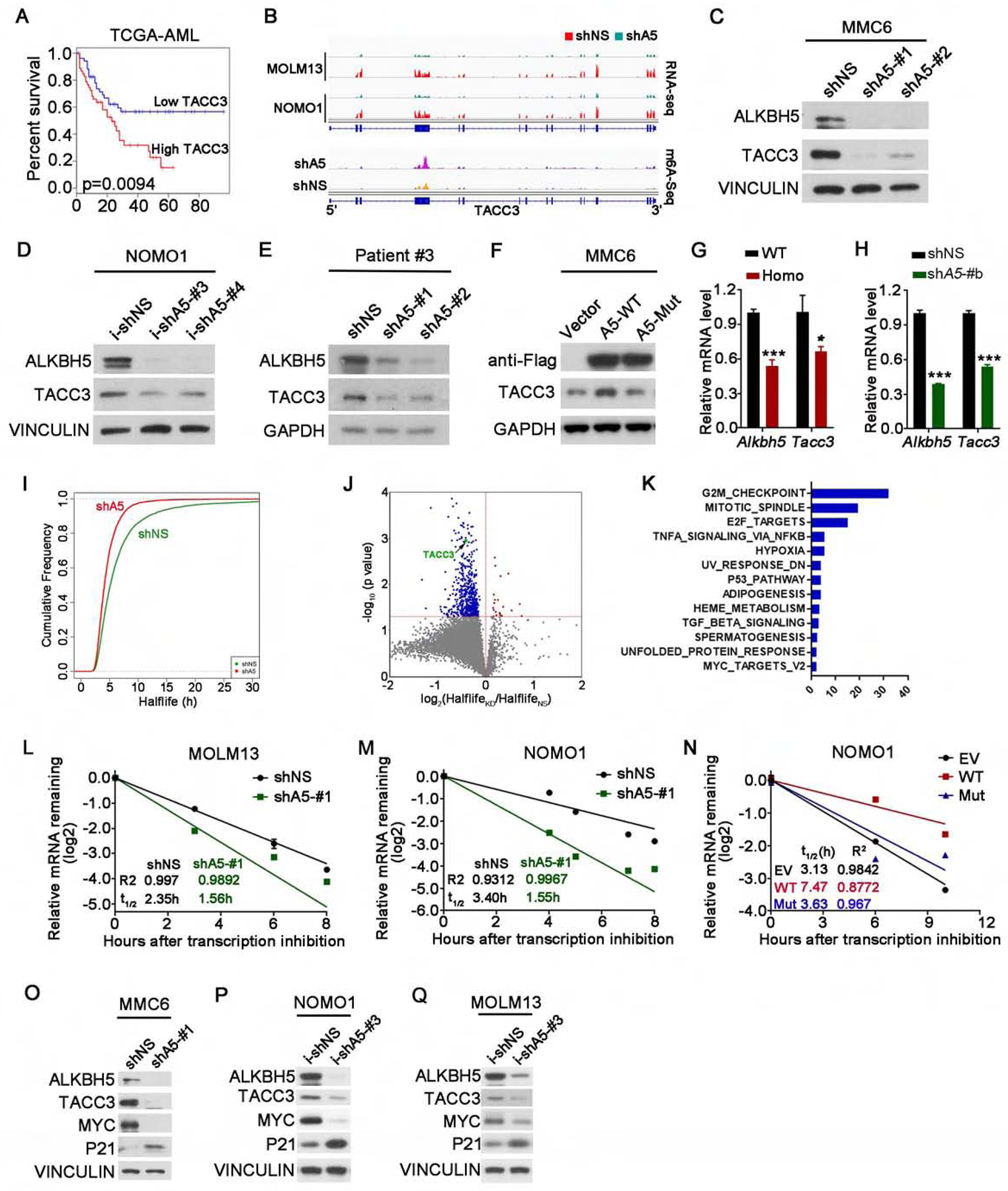
(A) Kaplan-Meier survival analysis of TACC3 in the TCGA AML dataset. The p value was detected by the log-rank test.
(B) The RNA (top) and m6A (bottom) abundance in TACC3 mRNA transcripts in ALKBH5 knockdown and control AML cells as detected by RNA-seq and m6A-seq.
(C-E) Western blots of ALKBH5 and TACC3 in ALKBH5 stable knockdown MMC6 cells (C), ALKBH5 inducible knockdown NOMO1 cells (Dox induction for 4 days) (D) and ALKBH5 stable knockdown primary AML cells (E). VINCULIN or GAPDH was used as a loading control.
(F) Western blots of ALKBH5 and TACC3 in MMC6 cells transduced with lentiviruses expressing empty vector (Vector) or wild-type ALKBH5 protein (A5-WT) or m6A demethylase-inactive mutant (A5-Mut). GAPDH was used as a loading control.
(G-H) qPCR detection of Tacc3 expression in Alkbh5 WT or Homo KO mouse BM cells (G) and in primary mouse MA9 AML cells with shNS or Alkbh5 shRNA (shA5-#b) (H).
(I-K) mRNA stability profiling. (I) Cumulative distribution of global transcript stability changes in shNS or shA5-#1 transduced NOMO1 cells. (J) Distribution of genes with significant half-life change in ALKBH5 knockdown cells compared to control cells. (K) Pathway analysis by GSEA showing the major pathways in which the genes with significantly shortened half-lives upon ALKBH5 knockdown are enriched.
(L-N) The mRNA half-life (t1/2) of TACC3 in MOLM13 cells (L) and NOMO1 cells (M) transduced with shNS or ALKBH5 shRNA (shA5-#1), and in NOMO1 cells transduced with empty vector (EV) or wild-type ALKBH5 (A5-WT) or ALKBH5 mutant (A5-Mut) (N).
(O-Q) Western blots of ALKBH5, TACC3, MYC and P21 in AML cells transduced with shNS or shALKBH5 (shA5-#1) (O) or with inducible shNS (i-shNS) or shALKBH5 (i-shA5-#3) (P and Q). VINCULIN was used as a loading control.
Mean±SD values are shown for Figures 6G–H and L–N. *p < 0.05; ***p<0.001, t test. See also Figure S6.
We first confirmed that ALKBH5 knockdown significantly decreased TACC3 level in human AML cell lines and primary AML cells (Figures 6C–E and S6B–D). Conversely, forced expression ofA5-WT but not A5-Mut increased TACC3 expression (Figures S6E and 6F). Consistently, Alkbh5 depletion also significantly decreased Tacc3 level in primary mouse BM cells (Figure 6G) and murine MA9 AML cells (Figure 6H). Furthermore, we found that TACC3 expression is not significantly suppressed by FTO knockdown in AML cells (Figure S6F). Thus, our data suggest that TACC3 is a specific target of ALKBH5 in AML.
The RNA m6A modification has been reported to affect mRNA stability and translation (Deng et al., 2018; Huang et al., 2018; Wang et al., 2014a; Wang et al., 2015). Strikingly, we found that ALKBH5 knockdown caused globally shorter half-lives of mRNA transcripts in AML cells (Figures 6I–J), with the trend being even more evident among transcripts of the potential targets of ALKBH5 (i.e., those detected by RIP-seq) (Figure S6G). Notably, around 600 hundred transcripts (including TACC3) showed significantly decreased half-lives, whereas only a few transcripts had increased half-lives (Figure 6J). GSEA showed that pathways related to cell cycle and cell growth/proliferation were also significantly enriched with these genes (Figure 6K). We confirmed that ALKBH5 knockdown significantly decreased TACC3 mRNA half-life in both MOLM13 (2.35 to 1.56 h) and NOMO1 (3.40 to 1.55 h) cells (Figures 6L–M), while overexpression of ALKBH5 wild-type, but not mutant, significantly increased TACC3 mRNA half-life (Figure 6N). We next performed polysome profiling and showed that ALKBH5 knockdown caused only a moderate drop in transcript levels in polysome fractions (Figure S6H). We did not observe a significant difference in TACC3 mRNA level in the translating pool between the control and ALKBH5 knockdown AML cells (Figure S6I). Thus, our data suggest that ALKBH5 regulates its targets’ expression level more likely by affecting mRNA stability rather than translation.
TACC3 has been reported previously to regulate MYC and P21 levels in normal or cancer cells (Piekorz et al., 2002; Schneider et al., 2008; Suhail et al., 2015) (Figure S6J). Consistently, we found that the ALKBH5 knockdown not only led to TACC3 suppression but also concordant changes in MYC (decrease) and P21 (increase) levels in AML cells (Figures 6O–Q). These data could indicate the ALKBH5/m6A/TACC3 axis also regulates P21 and MYC pathways in AML cells.
Thus, we next sought to investigate the functional importance of TACC3, especially as a direct target of ALKBH5, in AML. Consistent with the effects of ALKBH5 knockdown (Figures 1D–G and S1E–G), TACC3 knockdown also significantly inhibited cell growth and induced apoptosis in human AML cells (Figures 7A–B and S7A–B). Western blotting demonstrated that knockdown of TACC3 caused a significantly decreased MYC level and increased P21 level in human AML cells (Figures 7C–D and S7C), which confirmed MYC and P21 as downstream targets of TACC3 in AML. We next showed that knockdown of Tacc3 greatly inhibited mouse MA9 AML cell growth and colony-forming/replating capacity (Figures 7E–G and S7D–E). Our in vitro LDAs demonstrated that Tacc3 knockdown significantly reduced LSC/LIC frequency in mouse MA9 AML cells (Figures 7H and S7F), which also mimicked effects of Alkbh5 depletion (Figures 4F and S4L). Moreover, we showed that the inhibitory effects of ALKBH5 knockdown on cell growth can be largely rescued by forced expression of TACC3 (Figure 7I), which is accompanied by the restoration of MYC expression and reduction of P21 (Figure 7J). Taken together, our results demonstrate that TACC3 is a bona fide functionally important target of ALKBH5 in AML.
Figure 7. TACC3 is a functionally important target of ALKBH5 in AML.
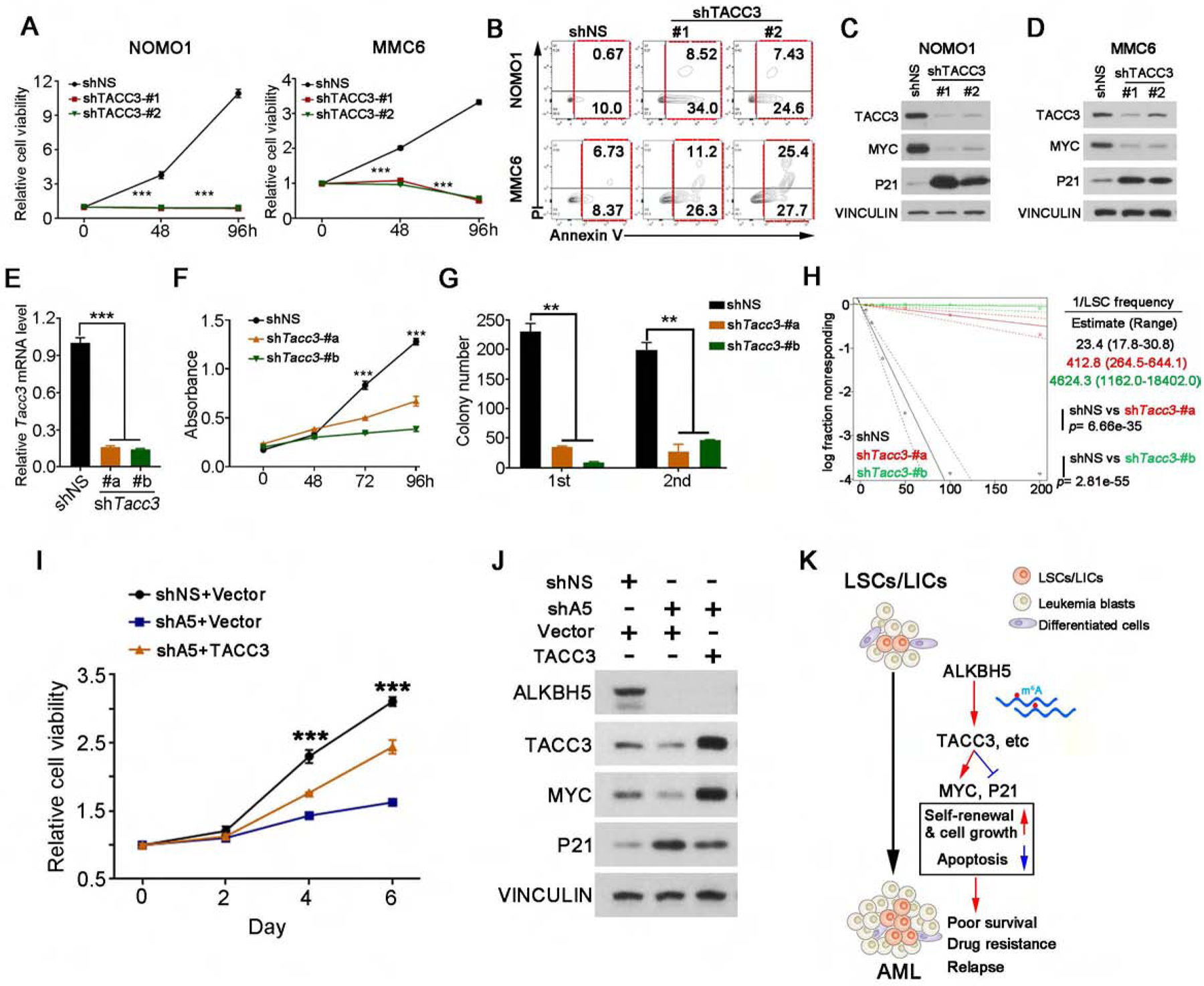
(A-B) Effects of ALHBK knockdown on cell growth/proliferation assays (A) and apoptosis (B) in AML cells.
(C-D) Western blots of TACC3, MYC and P21 in NOMO1 cells (C) and MMC6 cells (D) expressing shNS or TACC3 shRNAs. VINCULIN was used as a loading control.
(E) Relative levels of Tacc3 mRNA in mouse MA9 AML cells transduced with shNS or individual Tacc3 shRNAs (shTacc3-#a and shTacc3-#b).
(F) Effects of Tacc3 knockdown on the viability/proliferation of mouse MA9 AML cells.
(G) Effects of Tacc3 knockdown on the colony-forming/replating capacity of Mouse MA9 AML cells. Colony forming cell counts at each round of plating are shown (n=3).
(H) In vitro limiting dilution assays (LDAs). Logarithmic plot showing the percentage of non-responding wells at different doses. Non-responding wells, wells not containing colony forming cells. The estimated LSC/LIC frequency is calculated by ELDA and shown on the right. The p value was detected by the log-rank test.
(I-J) MMC6 cells were transduced with shNS or ALKBH5 shRNA (shA5), together with an empty (vector) or TACC3-encoding lentivirus as indicated. After drug selection, those co-transduced cells were seeded into 96-well plates for cell growth/proliferation assays (I). (J) Western blots of ALKBH5, TACC3, MYC and P21. VINCULIN was used as a loading control.
(K) Proposed model demonstrating the role and underlying mechanism(s) of ALKBH5 in AML pathogenesis and LSC/LIC self-renewal.
Mean±SD values are shown for Figures 7A, E–G and I. **p <0.01; ***p<0.001, t test. See also Figure S7.
DISCUSSION
While two m6A writer genes (METTL3 and METTL14) and an m6A eraser gene (FTO) have all been reported to be overexpressed and play improtant oncogenic roles in AML (Barbieri et al., 2017; Li et al., 2017; Su et al., 2018; Vu et al., 2017; Weng et al., 2018), frequent deletion or copy-number loss of ALKBH5 (the other m6A eraser gene besides FTO) was reported in AML (Kwok et al., 2017), implying that ALKBH5 may function as a tumor suppressor in AML. Here, however, we report that ALKBH5 actually functions as an oncoprotein, rather than a tumor suppressor, in AML. We found that the deletion frequency of ALKBH5 is low in AML and that it is instead overexpressed in AML relative to normal controls. Additionally, we found that the expression and function of ALKBH5 in AML was not TP53-dependent. We also showed that depletion of ALKBH5 inhibited cell growth and induced apoptosis in both TP53 wild-type and mutant AML cell lines, suggesting the role of ALKBH5 in AML is likely TP53-independent, distinct from the reported P53-dependent function of Alkbh5 in spermatogenesis (Zheng et al., 2013). Furthermore, our in vitro and in vivo functional studies showed that ALKBH5 is required for leukemic cell transformation and AML development and maintenance.
In contrast to METTL3, METTL14, WTAP, FTO and YTHDF2, whose expression levels were not significantly associated with prognosis in AML, increased expression of ALKBH5 correlates with poor prognosis in AML patients. Our observations, together with other reports (Cho et al., 2018; Zhang et al., 2017b), demonstrate a broad adverse prognostic impact of ALKBH5 expression levels in patients with AML and solid tumors. CSCs (including LSCs/LICs) have been implicated in the treatment failure and cancer relapse due to their roles in cancer initiation, progression, repopulation, and drug resistance (Batlle and Clevers, 2017; Krause and Van Etten, 2007). We showed here that ALKBH5 plays an essential role in the self-renewal of LSCs/LICs. Thus, the poor prognosis of AML patients with higher levels of ALKBH5 expression is likely due to enhanced LSC/LIC self-renewal capacity.
Moreover, we investigated the role of ALKBH5 in normal hematopoiesis and showed that depletion of Alkbh5 exhibited no significant effects on normal hematopoiesis in mice in the steady state, and had slight effect on HSC self-renewal and differeantiation in the stress state (e.g., under competitive repopulation). While the role of FTO in normal hematopoiesis remains unclear, depletionof Mettl3 or Mettl14 could significantly inhibit normal HSPC repopulation in mice (Weng et al., 2018; Yao et al., 2018; Zhang et al., 2017a), and the opposite is true when Ythdf2 is depleted (Li et al., 2018; Paris et al., 2019; Wang et al., 2018a). Thus, ALKBH5 appears to be uniquely hijacked to play an essential role in AML pathogenesis and LSC/LIC self-renewal, as it is dispensable for normal hamatopoiesis. Overall, our data suggest that ALKBH5 is a feasible target for AML therapy.
Through transcriptome-wide RNA-seq, RIP-seq and m6A-seq, we identified a set of potential targets of ALKBH5, which were directly bound by ALKBH5 and significantly responded to ALKBH5 knockdown in mRNA levels and m6A abundance in AML cells. We also identified pathways which could be positively or negatively regulated by ALKBH5. Interestingly, cell cycle- and cell growth/proliferation-related pathways such as E2F targets, G2/M checkpoints and apoptosis pathways are commonly detected by all three sequnecing methods. By comparing the sequencing data of ALKBH5 and FTO, we found that ALKBH5 and FTO have more distinct than shared targets. Next, we identified TACC3 as a direct target of ALKBH5 in AML cells. We showed that ALKBH5 positively regulates the mRNA stability but not translation efficiency of TACC3 transcripts, which leads to increased TACC3 expression through an m6A-dependent mechanism. Importantly, similar to ALKBH5, TACC3 has also been reported to be aberrantly overexpressed in various cancer types and play a critical oncogenic role in promoting tumorigenesis and CSC self-renewal/mainteance; moreover, its increased expression levels also indicate poor prognosis in patients with various types of cancers (Jung et al., 2006; Song et al., 2018; Sun et al., 2017; Wang et al., 2018b; Yao et al., 2012; Yun et al., 2015; Zhou et al., 2015). Our further functional studies demonstrate that TACC3 is an essential target of ALKBH5 in AML. As critical downstream targets of TACC3, MYC and P21 levels can also be indirectly regulated by ALKBH5 in AML. Through this axis, increased expression of ALKBH5 and TACC3 in cancer patients promoted LSC/LIC self-renewal and confers drug resistance or relapse, leading to poor prognosis (Figure 7K). Of course, besides TACC3, other potential targets of ALKBH5 identified herein might also be important downstream targets of ALKBH5 and may partially mediate the overall function/effects of ALBKH5 in AML (and other cancer types), which warrants further systematic investigation.
In conclusion, our studies demonstrate that ALKBH5 plays critical roles in leukemic cell transformation, AML development and maintenance, and LSC/LIC self-renewal through post-transcriptional regulation of critical targets (e.g., TACC3) via m6A-dependent mechanism(s), but minimally affects normal hematopoiesis. Mechanistically, we found ALKBH5 knockdown could globally reduce mRNA stability of its potential targets in AML cells. Our work also revealed a previously unrecognized signaling axis involving ALKBH5/m6A/TACC3 /MYC-p21 in AML pathogenesis and LSC/LIC biology, highlighting the functional importance of ALKBH5-mediated modulation of mRNA m6A methylation in leukemogenesis and LSC/LIC self-renewal. Notably, although several other m6A regulatory genes (e.g., METTL3, METTL14, WTAP, FTO, and YTHDF2) have also been reported to play oncogenic roles in AML, ALKBH5 is the only gene whose increased expression level is signficantly associated with a poor prognosis in AML patients. Given the essential roles of ALKBH5 in AML pathogenesis and LSC/LIC maintenance, with little effect on normal hematopoiesis, targeting ALKBH5 signaling represents a very promising therapeutic strategy for the treatment of AML patients (especially those who are resistant to currently available therapeutics) by eliminating LSCs/LICs and overcoming drug resistance, while sparing normal hematopoietic system. In particular, given the broad adverse prognostic imapcts of high ALKBH5 and TACC3 expression levels in patients with various types of cancers, targeting ALKBH5 and/or TACC3 by effective small-molecule compound inhibitors or agents that specifically degrade their proteins (e.g., proteolysis-targeting chimeras (PROTACs) (Chi, 2016; Sakamoto et al., 2001)), alone or in combination with other therapeutic agents, holds potent therapeutic potential in treating a wide variety of cancers in the clinic in the near future.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Jianjun Chen (jianchen@coh.org).
Materials Availability
All the materials generated in this manuscript are available from the Lead Contact under a complete Materials Transfer Agreement.
Data and Code Availability
The m6A-seq, RNA-seq, RIP-seq and mRNA stability profiling datasets obtained in this study have been deposited in the gene expression omnibus (GEO) repository and made accessible under accession numbers GSE144984.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and Animal Housing
The Alkbh5 (Embryonic knockout (KO))/B6 mouse model with the background of C57BL/6 was created by the Transgenic and Genome Editing Core (Cincinnati Children’s Hospital Medical Center). In brief, two gRNAs were designed to disrupt early exon 1. gRNAs were chosen according to the off-target scores from http://www.genome-engineering.org and in vitro transcribed using MEGAshortscript T7 kit (Thermo Fisher Scientific), followed by MEGAclear Kit (Thermo Fisher Scientific) for purification. Cas9 mRNA was in vitro transcribed using mMESSAGE mMACHINE T7 ULTRA kit (Thermo Fisher Scientific), according to manufacturer’s instruction. gRNA and Cas9 mRNA were mixed at concentration of 50 and 100 ng/ul, respectively, and injected to the cytoplasm of one-cell-stage embryos of C57BL/6 genetic background. Injected embryos were immediately transferred into the oviductal ampulla of pseudopregnant CD-1 females. Four mice tested positive for the excised allele. These four mice, heterozygous for the constitutive allele (Alkbh5+/−), were analyzed by Sanger sequencing and backcrossed with C57BL/6 mice for more than 6 generations and then used for further breeding to generate Alkbh5−/− mice.
C57BL/6 (CD45.2) and B6.SJL (CD45.1) mice were purchased from Envigo (Indianapolis, IN, USA) and the Charles River Laboratories (Wilmington, MA), respectively. NSGS mouse were purchased from the Jackson Laboratory (Bar Harbor, ME). Both male and female mice were used for the experiments. All laboratory mice were maintained in the animal facility at City of Hope or at the University of Florida. All experiments on mice in our research protocol were approved by Institutional Animal Care and Use Committee (IACUC) of City of Hope or the University of Florida.
Leukemic patient samples and normal hematopoietic cell samples
The leukemic samples were obtained at the time of diagnosis or relapse and with informed consent at City of Hope (COH) or Department of Medicine, University of Florida, and were approved by the institutional review board of the institutes/hospitals. The leukemic samples were stored in liquid nitrogen until used. Leukemia blasts and mononuclear cells (MNCs) were purified using NycoPrep 1.077A (Axis-Shield, Oslo, Norway) or Ficoll-Paque PLUS (GE Healthcare Life Sciences). AML CD34+ leukemia stem/initiating cells (LSCs/LICs) and CD34− cells were isolated using CD34+ beads (130-046-702, Miltenyi Biotec). Normal CD34+ hematopoietic stem/progenitor cells (HSPCs), and CD34− cells were purified from bone marrow cells of healthy donors from COH using CD34+ beads (Miltenyi Biotec).
Cell culture
For leukemia cells, U937, THP1, MV4–11 were obtained from America Type Culture Collection (ATCC) and cultured in endotoxin-free RPMI-1640 supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products), 1% HEPES and 1% penicillin-streptomycin; NOMO-1, MOLM13 and NB4 were obtained from DSMZ and kept in RPMI-1640 with 10% FBS, 1% HEPES and 1% penicillin-streptomycin; MA9.3ITD (MLL-AF9-transformed human CD34+ cord blood cell plus FLT3-ITD), was established by Dr. James Mulloy (Wunderlich et al., 2013) and cultured in IMDM supplemented with 20% FBS, 1% HEPES and 1% penicillin-streptomycin. For MonoMac-6 (MMC6) cells, 2 mM L-Glutamine, 1×Non-Essential Amino Acid, 1 mM sodium pyruvate, and 9 μg/ml insulin (12585014, Thermo Fisher Scientific) were added to the regular RPMI-1640. HEK-293T cells were grown in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin. IMDM supplemented with 20% FBS, 10 ng/ml human cytokines SCF, TPO, FLT3L, IL-3 and IL-6 (PeproTech) was used for culture of primary patient cells; All the cells are not among commonly misidentified cells lines and have been tested for mycoplasma contamination quarterly using a PCR Mycoplasma Detection Kit (Applied Biological Materials).
For generation of iCas9 stable cell lines, cells were transduced with lentivirus by “spinoculation”, then refed with fresh medium with the selection drug. Infected cells were passaged every 2 days until uninfected control cells were completely killed. Killing took around 6 days for G418 and 4 days for puromycin. To generate single clones of MOLM13-iCas9 cells, single cells were seeded into 96 well plates. Cells grown up from a single cell were picked up and EGFP induction was tested upon 1ug/ml Doxycycline treatment for 24h.
METHODS DETAILS
Plasmid construction
The wild type ALKBH5-CDS and mutant ALKBH5-CDS were PCR-amplified from pF RT/TO/HIS/FLAG/HA-ALKBH5 plasmid (38073, addgene) and pFLAG CMV5.1-ABH 5-H204A (kindly provided by Dr. Chuan He), and then cloned into the pCDH lentivir al vector (CD513B-1, SBI, Mountain View, CA) using XbaI and BamHI enzyme sites. The TRC shRNAs targeting human ALKBH5 (shA5-#1: TRCN0000291838; shA5-#2: TRCN0000291769), mouse Alkbh5 (shA5-#a: TRCN0000201776; shA5-#b: TRCN00001 92524) were purchased from Sigma-Aldrich, the non-targeting control (pLKO.1) was fr om addgene. The inducible shRNA plasmids (TRIPZ-shA5-#3: V2THS_173653; TRIPZ-shA5-#4: V2THS_173654), as well as the non-targeting control shRNA, were all purchased from GE Dharmacon. The Lenti-iCas9-neo (doxycycline-inducible Cas9-EGFP ve ctor) and lenti-guide (gRNA expression vector) were purchased from Addgene. Lenti-s gALKBH5 was constructed as previously described (Ran et al., 2013). Stbl3™ E.coli (C7373–03, Thermo Fisher Scientific) and 5-alpha Competent E. coli (C29871, New E ngland Biolabs) were used in transformation.
Cell proliferation/growth and apoptosis assays
The cell proliferation/growth was assessed by MTT (G4100, Promega, Madison, WI) f ollowing the manufacturer’s instructions. Briefly, cells were seeded into a 96-well plat e in triplicates at the density of 5000–10000 cells/100 μL. Dye solution was added at indicated time points and incubated at 37°C for 3–4 hours before adding of solubiliz ation/stop to stop the reaction. The absorbance at 570nm (with reference at 630nm) w as read on the next day. For apoptosis assays, APC Annexin V apoptosis Detection Kit (88-8007-74, eBiosciences, San Diego, CA) was used following the manufacturer’s instructions.
Lentivirus preparation, precipitation, and infection
Lentivirus particles for overexpression and knockdown plasmids were all packaged with pMD2.G, psPAX2 (Addgene). Briefly, 5μg pMD2.G, 5μg psPAX2 and 5μg construct for overexpression or knockdown of specific genes were co-transfected into HEK-293T cells in 100 mm cell culture dish with Effectene Transfection Reagent (301427, Qiagen). The virus particles were harvested at 48 and 72 hours after transfection and concentrated with PEG-it virus precipitation solution (LV810A-1, SBI). For infection, the concentrated virus or the viral supernatant were directly added into cells in the presence of 4ug/ml polybrene (H9268, Sigma-Aldrich) and then spinoculation was conducted at 32°C, 600xg for 60 min. The positive infected cells were selected with 1ug/ml puromycin (P8833, Sigma-Aldrich) or 1mg/ml G418 (10131–027, Thermo Fisher Scientific) or 2.5ug/ml blasticidine (15205, Sigma-Aldrich). After selection, 1ug/ml Doxycycline (D9891, Sigma-Aldrich) was added to induce expression of TRIPZ-shRNAs or Cas9 protein.
Retrovirus preparation and in vitro colony-forming and replating (CFA) assay
These assays were conducted as described previously (Huang et al., 2013; Jiang et al., 2012; Li et al., 2012a; Li et al., 2012b; Li et al., 2013) with some modifications. Briefly, retroviruses were produced in 293T cells by co-transfection of individual retroviral construct with the pCL-Eco packaging vector (IMGENEX, San Diego, CA) with Effectene Transfection Reagent (301427, Qiagen). Bone marrow (BM) cells were collected from 6- to 8-week-old wild-type or Alkbh5 (Em-ko) mice five days after 5-fluorouracil (5-FU) treatment (150mg/kg), and BM progenitor (i.e., lineage negative, Lin−) cells were enriched with the Mouse Lineage Cell Depletion Kit (130-090-858, Miltenyi Biotec). BM progenitor cells were then co-transduced with different combinations of retroviruses or lentiviruses as indicated through two rounds of “spinoculation”. Thereafter, the transduced cells were then plated into ColonyGEL methylcellulose medium (ReachBio, Seattle, WA) supplied with 10 ng/ml of murine recombinant IL-3, IL-6, GM-CSF and 30 ng/ml of murine recombinant SCF, along with 1.0 mg/ml of G418 (Gibco BRL, Gaithersburg, MD) and/or 2.5 μg/ml of puromycin (Sigma-Aldrich). Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 for 6 to 7 days. Serial replating was then performed by collecting colony cells and replating them into new dishes/wells with methylcellulose medium every 7 days. Colony numbers were counted and compared for each passage.
For CFA assays using human AML cells, the cells were transduced with lentivirus and then seeded into MethoCult™ H4434 Classic medium (StemCell Technologies) with the addition of 2.5 μg/ml puromycin. Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 for 10 days before counting.
Mouse bone marrow transplantation (BMT)
These assays were conducted as described previously (Huang et al., 2013; Jiang et al., 2012; Li et al., 2012a; Li et al., 2012b; Li et al., 2013) with some modifications. Briefly, colony cells were collected from the colony-forming assays, washed with PBS once and transplanted via tail vein injection into lethally (900 cGy, 96 cGy/min, γ-rays) irradiated 8- to 10-week-old B6.SJL (CD45.1) or C57BL/6 (CD45.2) recipient mice. For each recipient mouse, 0.3–0.5×106 donor cells from CFA assays and a radioprotective dose of whole bone marrow cells (1×106) freshly harvested from a B6.SJL (CD45.1) or C57BL/6 (CD45.2) mouse were transplanted. For secondary BMT, BM cells from primary BMT mice (3 mice/group) were mixed and transplanted (a total of 1× 106 donor cells/recipient mouse) into sublethally (480 cGy, 96 cGy/min, γ-rays) irradiated 6- to 8-week-old C57BL/6 (CD45.2) recipient mice. Leukemic mice were euthanized by CO2 inhalation when they showed signs of systemic illness. Peripheral blood (PB) was collected for CBC test (the upper limit of the CBC machine used in Figures 3D and 4E is 200 k/ul). BM cells were isolated from both tibia and femur, and 100,000 cells were resuspended in 200 μl of cold MACS Buffer (1xPBS supplemented with 2 mmol/L EDTA and 0.5% BSA) and loaded for cytospin preparation. BM cytospin and blood smear slides were stained with Wright-Giemsa (Polysciences). Portions of the spleen and liver from leukemic mice were collected at the time of sacrifice, fixed in formalin and embedded in paraffin. The tissue samples were then sectioned and stained with haematoxylin and eosin (H&E) by the Molecular Pathology Core in University of Florida.
In vitro limiting dilution assays (LDAs)
BM leukemic cells collected from primary ALKBH5 wild-type leukemic mice that developed full-blown leukemia were stained with PE-CD45.2, sorted on a BD FACSAria III cell sorter (BD Biosciences), and transduced with shRNA virus targeting mouse Alkbh5. The infected cells were seeded into ColonyGEL methylcellulose medium (ReachBio, Seattle, WA) supplied with 10 ng/ml of murine recombinant IL-3, IL-6, GM-CSF and 30 ng/ml of murine recombinant SCF, along with 2.5 μg/ml of puromycin (Sigma-Aldrich). Seven days later, the colony cells were harvested and replated into 48-well plates with six different doses of cell number for each group. The number of wells developed MA9 clones was counted for each group with each dose of donor cells. ELDA software (Hu and Smyth, 2009) was used to estimate the frequency of leukemia stem/initiating cells (LSCs/LICs).
In vivo limiting dilution assays (LDAs)
BM cells collected from primary BMT mice (3 mice/group) which were euthanized at the same time were mixed and injected into lethally irradiated wild-type C57BL/6 mice through tail vein with three different doses of donor cells for each group. The number of recipient mice developed full-blown leukemia within six weeks post-transplantation was counted for each group with each dose of donor cells. ELDA software (Hu and Smyth, 2009) was used to estimate the frequency of leukemia stem/initiating cells (LSCs/LICs).
Competitive Repopulation Assay
BM cells (1×106, CD45.2+) from 7~8 weeks Alkbh5 wild-type (WT) or homozygous KO mice plus equal number of competitor BM cells (1×106, CD45.1+CD45.2+) from 7~8 weeks B6.SJL × C57BL/6 F1 mice were transplanted into lethally irradiated (900cGy) B6.SJL mice (CD45.1+) by tail vein injection. Two weeks after transplantation, Peripheral blood (PB) was collected by tail vein bleeding of the recipient mice and subjected to flow cytometric analysis with PE-CD45.1 and APC-CD45.2 antibodies. The CD45.2+/CD45.1+CD45.2+ chimeras in their PB was monitored by FACS analysis every 4 weeks for 20 weeks since week 4 post transplantation.
Flow cytometric analysis
Flow cytometry analysis of mouse BM cells were conducted as described previously (Huang et al., 2013; Jiang et al., 2012; Li et al., 2012a; Li et al., 2012b; Li et al., 2008) with some modifications. Cells from BM of transplanted mice were harvested for analysis of immunophenotypes. After blocking nonspecific binding with affinity-purified anti–mouse CD16/32 (eBioscience), cells were stained at 4°C with various antibodies diluted in Flow Cytometry Staining Buffer (eBioscience) for 30 minutes and resuspended in IC Fixation Buffer (eBioscience) before being loaded for flow cytometry analysis in BD FACS FortessaX-20. Antibodies used include anti-mouse CD11b-eflour 450 (Mac-1; 48-0112-82), anti-mouse Ly-6G (Gr-1) eFluor 450 (48-5931-82), anti-mouse CD117-APC (c-kit; 17-1171-83), anti-mouse CD45.2-PE (12-0454-82).
For the hematopoietic and leukemia stem cell and mature cell analysis, suspended single cells were prepared from bone marrow, spleen and peripheral blood. Red cells were lysed by ACK LYSING Buffer (VWR) before staining for all FACS analysis except for Red cell analysis. Cells were incubated with antibodies in FACS buffer (2% FBS in PBS) on ice for 20 minutes at dark. All antibodies were purchased from eBioscience except CD150. Anti-Gr-1-Biotin (13-5931-86), Ter119-Biotin (13-5921-85), B220-Biotin (13-0452-86), CD19-Biotin (13-0193-86), IgM-Biotin (13-4341-81), CD127-Biotin (13-1271-85), CD3e-Biotin (13-0033-86) and Streptavidin PE-Cy5 (15-4317-82) antibodies are for lineage markers, anti-Sca1-PE (12-5981-83), c-Kit-APC-eFluor 780 (47-1171-82), CD34-eFlour 660 (50-0341-82) and CD16,32-PE-Cy7 (25-0161-82) antibodies are for leukemic stem cell L-GMP population or hematopoietic progenitor cell (HPC) population analysis; anti-Sca1-PE (12-5981-83), c-Kit-APC-eFluor 780 (47-1171-82), CD150-APC (Biolegend, 115910) and CD48-PE-Cy7 (25-0481-80) antibodies were used for hematopoietic stem cell population (HSC) analysis; anti-c-Kit-PE-Cy7 (25-1171-82) and Gr1-APC-eFluor 780 (47-5931-82) are for c-Kit+ Gr1− population analysis. For competitive assay analysis, anti-CD45.1-PE (12-0453-82) and CD45.2-APC (17-0454-82) antibodies were introduced to separate Donor and Recipient mice cells. Gr1-APC-eFluor 780 (47-5931-82) and CD11b-PE (12-0112-83) are for myeloid markers; Ter119-APC (17-5921-82) is for Red Cells, B220-PE (12-0452-82) is for B Cells and CD3e-FITC (11-0031-82) is for T cells. For the detection of apoptosis, BM cells were stained with cell surface markers first, followed by Annexin V staining in its specific binding buffer, and DAPI was added at last. All cells were analyzed by Flow cytometry on LSR Fortessa SORP (BD) or FortessaX-20 (BD).
RNA extraction and quantitative RT-PCR analysis
Total RNA was purified using the miRNeasy mini kit (217004, Qiagen) following the manufacturer’s instructions and quantified by UV spectrophotometry. For detection of mRNA expression, 200–500 ng of total RNA was reverse-transcribed into cDNA in a total reaction volume of 20 μL with the QuantiTect Reverse Transcription Kit (205314, Qiagen). Quantitative real-time PCR analysis was then conducted with 4 μL diluted cDNA (with 7–10 fold dilution) using Maxima SYBR green qPCR master mix (Thermo Fisher Scientific) on the QuantStudio 7 Flex PCR system (Thermo Fisher Scientific). GAPDH or ACTB was used as endogenous control. Each sample was run in triplicate. The primers used for qPCR analysis were listed in Table S1.
m6A dot blot assay
Total RNA was extracted from different cells by miRNeasy Mini Kit (QIAGEN) according to the manufacturer’s instructions and quantified by UV spectrophotometry. The m6A dot blot assay was performed following a published protocol (Hodge, 1998) with some modifications(Jia et al., 2011). Briefly, the RNA samples were loaded onto the Amersham Hybond-N+ membrane (RPN119B, GE Healthcare) by vacuuming the Bio-Dot Apparatus (#170–6545, Bio-Rad), washed one time then crosslinked to the membrane by UV. The membrane was blocked with 5% nonfat dry milk (dissolved in 1X PBST) for 1–2 hours and incubated with a specific anti-m6A antibody (1:3000 dilution, Synaptic Systems, 202003) overnight at 4°C. Then the HRP-conjugated goat anti-rabbit IgG (sc-2030, Santa Cruz Biotechnology) was added to the blots for 1 hour at room temperature and the membrane was developed with Amersham ECL Prime Western Blotting Detection Reagent (RPN2232, GE Healthcare). The relative signal density of each dot was measured by Gel-Pro analyzer software (Media Cybernetics).
LC-MS/MS for determination of m6A/A ratio
Total RNA underwent two rounds of polyadenylated (poly-A) mRNA purification, using the Dynabeads mRNA DIRECT kit (61011, Thermo Fisher Scientific). The mRNA was digested by Nuclease P1 (1U, Sigma-Aldrich, St. Louis, MO) in 20 μl of buffer containing 20 mM NH4OAc (pH = 5.3) at 42°C for 4 h. After digestion, the nucleosides were dephosphorylated by adding FastAP Buffer (Thermo Fisher Scientific) and FastAP Thermosensitive Alkaline Phosphatase (1 U, Thermo Fisher Scientific) and incubating at 37°C for 4 h. The samples were then diluted to 50 μl and filtered (0.22 μm pore size, 4 mm diameter, Millipore), and 5 μl of the solution was injected into LC-MS/MS (three injections were performed per sample to serve as technical replicates). Nucleosides were separated by reverse phase ultra-performance liquid chromatography on a C18 column, followed by online mass spectrometry detection using an Agilent 6410 QQQ triple-quadrupole LC mass spectrometer in positive electrospray ionization mode. The nucleosides were quantified by using retention time and the nucleoside-to-base ion mass transitions of 282.1 to 150.1 (m6A), and 268 to 136 (A). The nucleosides of each sample were quantified by comparing the standard curve obtained from pure nucleoside standards that were run with the same batch of samples. The m6A level was calculated as the ratio of the calibrated concentrations of m6A to A (Jia et al., 2011).
RNA-seq
Total RNA was isolated from NOMO1 and MOLM13 cells with or without ALKBH5 knockdown using miRNeasy mini kit (Qiagen). Library construction of 250 ng total RNA for each sample was made using KAPA mRNA HyperPrep kit (Illumina Platforms) (Kapa Biosystems, Wilmington, USA). Libraries were purified using AxyPrep Mag PCR Clean-up kit (Thermo Fisher Scientific). Each library was quantified using a Qubit fluorometer (Thermo Fisher Scientific) and the size distribution assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). All samples were sequenced by Illumina HiSeq 2500 with a single-end 50-base pair (bp) read length.
m6A-seq
Total RNA was isolated from NOMO1 with or without ALKBH5 knockdown using QIAzol Lysis Reagent. Polyadenylated RNA was further enriched from total RNA using the Dynabeads mRNA DIRECT kit (Thermo Fisher Scientific). RNA fragmentation was performed by sonicating 1 μg mRNA in 100 μl RNase-free water using the Bioruptor Pico (Diagenode) with 30s on/30s off for 30 cycles at 4°C. m6A-IP and library preparation were performed per the reported protocol (Dominissini et al., 2012) with some modified instructions based on the EpiMark N6-Methyladenosine Enrichment Kit. Briefly, 25 μL Pierce Protein A/G Magnetic Beads (88803, Thermo Fisher Scientific) were washed twice with 1x IP buffer and mixed with 2 μL m6A antibody from the EpiMark N6-Methyladenosine Enrichment Kit (New England Biolabs, E1610S) and incubated with orbital rotation at 4°C for 30 min. The beads were washed twice with 1x IP buffer, and immunoprecipitation was performed by adding 1 μg sonicated RNA and mixing with orbital rotation for 3 h at 4°C. The beads were then separated and washed twice with 1x IP buffer, twice with low salt reaction buffer (50 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl, pH 7.4), and twice with high salt reaction buffer (500 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl, pH 7.4) before elution with Buffer RLT (Qiagen). The eluate was purified with the RNA Clean and Concentrator kit (Zymo, Orange, CA). The purified mRNA fragments were then used to construct libraries with the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA). Sequencing was carried out on Illumina HiSeq 4000 according to the manufacturer’s instructions with single-end 50-bp read length.
RNA immunoprecipitation (RIP) and RIP-seq
RNA immunoprecipitation was performed as previously described (Rinn et al., 2007) with some modifications. Briefly, after UV-crosslinking, 60 million cells per sample were harvested and washed with PBS. Cells were lysed with two volumes of lysis buffer consisting of 10 mM HEPES pH 7.6, 150 mM KCl, 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1X cOmplete Protease Inhibitor (Roche), and 400 U/mL SUPERase-In RNase Inhibitor (Thermo Fisher Scientific). Cell lysate was cleared through a 0.22 μm filter. Input sample for RNA sequencing was prepared by saving 5% of lysate and adding 1 mL TRIzol reagent. Samples were subjected to immunoprecipitation using anti-Flag M2 magnetic beads. Beads were washed 4 times and re-suspended with cold NT2 buffer (50 mM HEPES pH 7.6, 200 mM NaCl, 2 mM EDTA, 0.05% NP-40, 0.5 mM DTT, and 200 U/mL RNase inhibitor). Sample lysates were immunoprecipitated with orbital rotation at 4°C for 4 hours. Afterwards, beads were washed 8 times with cold NT2 buffer. Immunoprecipitated samples were subjected to Proteinase K digestion in NT2 buffer supplemented with 1% SDS and 1.2 mg/mL Proteinase K (Thermo Fisher Scientific) incubated with shaking at 1200 rpm at 55°C for 1 hour. Total RNA was extracted from both input and immunoprecipitated RNA by adding 5 volumes of TRIzol reagent, followed by Direct-zol RNA Miniprep (Zymo) and used for qPCR analysis or RNA-seq. For RIP-seq, RNA was then fragmented with an average length of 150 nucleotides using the Bioruptor Pico sonication device. Libraries for high-throughput sequencing were constructed using the TruSeq Stranded v2 mRNA Sample Prep Kit (Illumina), and were quantified by BioAnalyzer High Sensitivity DNA chip. RIP-seq libraries were sequenced on Illumina HiSeq 4000 according to the manufacturer’s instructions with single-end 50-bp read length.
Gene-specific m6A qPCR
To detect m6A modifications on specific genes, the Magna MeRIP m6A Kit (17–10499, Millipore, Billerica, MA) was used following the manufacturer’s instructions. Briefly, 200 μg of total RNA was sheared to approximately 100 nt in length by metal-ion induced fragmentation and purified, then incubated with m6A antibody- (#MABE1006, included in the kit) or mouse IgG-conjugated Protein A/G Magnetic Beads in 500 μl 1x IP buffer supplemented with RNase inhibitors at 4 °C for two hours. After washing four times with IP buffer, the m6A IP portion was eluted twice by 100 μl competitively binding free m6A, and recovered with the RNeasy kit (Qiagen). One tenth of fragmented RNA was saved as input control, and further analyzed by qPCR along with the MeRIP-ed RNAs using primers listed in Table S1. The related enrichment of m6A in each sample was calculated by normalizing Ct values of the sample immunoprecipitated with anti-m6A and the samples with negative control (IgG) to input (ΔCt): ΔCt = CtIP – (Ctinput-Log2 [Input Dilution Factor]) (Input dilution factor is 10 if using 10% input sample). To calculate the relative fold enrichment, the ΔCt values of sample with anti-m6A were normalized to the sample with negative control IgG (ΔΔCt): ΔΔCt =ΔCtm6A -ΔCtIgG). The fold enrichment of the sample with anti-m6A antibody over the negative control mouse IgG control was calculated: Fold enrichment=2− ΔΔCt.
RNA stability assays and mRNA stability profiling
Human AML cells with or without ALKBH5 knockdown were treated with actinomycin D (A9415, Sigma-Aldrich) at a final concentration of 5 μg/mL and collected at indicated time points. Total RNA was extracted by miRNeasy Kit (Qiagen) and analyzed by RT-PCR or RNA-seq. For RNA-seq, each RNA sample was spiked in with an appropriate amount of either Mix1 or Mix2 according to Life Technologies’ guidelines which would lead to about 1% of the total number of RNA-Seq reads mapping to the 92 ERCC control sequences, assuming the mRNA fraction in the total RNA is 2%. Library construction of 250 ng total RNA for each sample was made using KAPA mRNA HyperPrep kit (Illumina Platforms) (Kapa Biosystems, Wilmington, USA). Libraries were purified using AxyPrep Mag PCR Clean-up kit (Thermo Fisher Scientific). Each library was quantified using a Qubit fluorometer (Thermo Fisher Scientific) and the size distribution assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). All samples were sequenced by Illumina HiSeq 2500 with a single-end 50-base pair (bp) read length.
The turnover rate and half-life of mRNA was calculated according to a previously published paper (Chen et al., 2008). Since actinomycin D treatment results in transcription inhibition, the change of mRNA concentration at a given time (dC/dt) is proportional to the constant of mRNA decay (kdecay) and mRNA concentration (C) as shown in the following equation:
Thus the mRNA degradation rate kdecay was estimated by:
When 50% of mRNA is decayed (i.e., C/C0=1/2), the equation below can be used to calculate the mRNA half-life (t1/2):
from where:
Polysome profiling
We followed the reported protocols (Gandin et al., 2014; Wang et al., 2015) with the following modifications. NOMO1 cells were transduced with ishNS or ishA5-#3 lentivirus and selected with puromycin (1 μg/mL). Doxycycline was added into the culture to induce ALKBH5 knockdown and refreshed every 2 days for 6 days. Before collection, cycloheximide (CHX) (C4859, Sigma-Aldrich) was added to the culture media at 100 μg/mL for 7 min. Approximately 60–70 million AML cells from each group were harvested, rinsed in cold PBS with 100 μg/mL CHX and quickly frozen in liquid nitrogen before lysis. The lysis buffer was formulated as 20 mM HEPES (pH7.6), 100 mM KCl, 5 mM MgCl2, 100 μg/ml CHX, 1% Triton-X-100, with freshly added 1X cOmplete Protease Inhibitor (Roche) and 20 U/ml of SUPERase-In RNase inhibitor (Thermo Fisher Scientific). The cell lysate was then layered on top of a 5%-to-50% sucrose gradient containing 20 mM HEPES pH 7.6, 100 mM KCl, 5 mM MgCl2, 100 μg/mL cycloheximide, 1X protease inhibitor (Roche), and 20 U/mL RNase inhibitor (Thermo Fisher Scientific). The sucrose gradient was formed in an open-top polyclear tube (Seton) by the Gradient Maker on a Master unit from BioComp Instruments. The lysate and gradient were then centrifuged on an Optima L-100 XP Ultracentrifuge at 28,000 rpm for 3 hours at 4°C in order to separate components of the lysate. The sample was then fractionated into 30 fractions (0.5 mL per fraction), and analyzed by Gradient Station (BioComp Instruments) equipped with an ECONO UV monitor (BioRad, Hercules, CA) and Gilson FC203B fraction collector (Mandel Scientific, Guelph, Canada). RNA was purified from fractions 5–20 and subjected to RT-qPCR analysis. Expression of TACC3 in each fraction was normalized to GAPDH as well as Input.
Primary human AML patient derived samples
For cell growth/proliferation assays, CFA assays and cell apoptosis assays in Figures 3I–N and S3H–I, the following samples were used: AML patient #1 (karyotype 46, XX, t(9;11)(p22;q23)[20]), AML patient #2 (karyotype MLL rearrangement; mutation FLT3-ITD(−) and NPM-1(−)), AML patient #3 (karyotype 46, XY, Normal, mutation DNMT3A (+), FLT3-ITD (+) and NPM-1(−)).
For western blotting shown in Figures S4B and 6E, the following samples were used: AML1 (i.e., Patient #3 in Figures S3H–I and 6E) (karyotype 46, XY, Normal, mutation DNMT3A (+), FLT3-ITD (+) and NPM-1(+)); AML2 (karyotype 46, XY, t(6;9), mutation FLT3-ITD (+)).
Immunoblotting (Western blot)
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) and ruptured with RIPA buffer (Pierce, Rockford, IL) containing 5 mM EDTA, PMSF, cocktail proteinase inhibitors, and phosphatase inhibitor cocktail. Cell extracts were centrifuged at 12000 × g for 15 min and the supernatants were then collected. Cell lysates were resolved by SDS-PAGE and transferred onto PVDF membranes which were blocked with 5% non-fat milk (Bio-Rad) in Tris-buffered saline containing 0.1% Tween 20 for 1 hour and incubated sequentially with primary and secondary antibodies and detected by immunoblotting with the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) or Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare). Antibodies used for Western blotting were as follows: ALKBH5 (ab195377, abcam), ALKBH5 (HPA007196, Sigma-Aldrich), TACC3 (sc-376883, Santa Cruz Biotechnology), Flag (F3165, Sigma-Aldrich), c-Myc (9402S, Cell Signaling Technology), P21 (2947S, Cell Signaling Technology) GAPDH (sc-47724, Santa Cruz Biotechnology), β-Actin (3700, Cell Signaling Technology), Vinculin (sc-25336, Santa Cruz Biotechnology). GAPDH or β-Actin or Vinculin was used as a loading control.
Sequencing data analysis.
(1). RNA-seq data.
Samples were sequenced by Illumina HiSeq 2500 with a single-end 50-base pair (bp) read length. Reads were mapped to human genome version GRCh38 by STAR. Gene expression (RPKM) was calculated by RSEM. The average gene expressions of two biological replicates were used for the following analysis.
(2). RIP-seq data.
Samples were sequenced on Illumina HiSeq 4000 according to the manufacturer’s instructions with single-end 50-bp read length. Reads were mapped to human genome version GRCh38 by STAR. Gene expression (RPKM) was calculated by RSEM. The RIP targets were defined as genes with reads per kilobase, per million reads (RPKM) ≥1, immunoprecipitation/input ≥2.
(3). mRNA lifetime (stability) profiling data.
Samples were sequenced by Illumina HiSeq 2500 with a single-end 50-base pair (bp) read length. Reads were mapped to human genome version GRCh38 by STAR. Gene expression (RPKM) was calculated by RSEM. RPKM was converted to attomoles by linear fitting of the RNA spike-in. The degradation rate of RNA and the mRNA half-life were calculated according to the aforementioned formula. The final half-life was calculated by using the average value of 0 h, 8 h and 12 h.
(4). m6A-seq data.
Sequencing was carried out on Illumina HiSeq 4000 according to the manufacturer’s instructions with single-end 50-bp read length. Reads were mapped to human genome version GRCh38 by STAR. The longest isoform was retained if a gene has more than one isoforms. Differential m6A modified peaks between IP and input samples were identified using exomePeak (p < 0.05).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed and presented as mean±SD. Two-tailed Student’s t-test was used to compare means between groups as indicated; p < 0.05 was considered significant. For Figures 2F, S2G, 3C, 3H, S3E, 4D and S4K, Kaplan-Meier survival curves were generated using GraphPad Prism 6 and the p values were calculated using the log rank test. For western blots, representative figures from two biological replicates were shown.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| m6A (N6-methyladenosine) antibody | Synaptic Systems | Cat#202003; RRID: AB_2279214 |
| beta-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat#3700; RRID: AB_2242334 |
| GAPDH antibody (0411) | Santa Cruz Biotechnology | Cat#sc-47724; RRID: AB_627678 |
| Anti-ALKBH5 antibody, Rabbit | Sigma-Aldrich | Cat#HPA007196; RRID: AB_1850461 |
| Recombinant anti-ALKBH5 antibody | Abcam | Cat#ab195377; RRID: AB_2827986 |
| Vinculin (H-10) | Santa Cruz Biotechnology | Cat#sc-25336; RRID: AB_628438 |
| TACC3 | Santa Cruz Biotechnology | Cat#sc-376883; RRID: AB_2827987 |
| c-Myc | Cell Signaling Technology | Cat#9402S; RRID: AB_2151827 |
| P21 Waf1/Cip1 (12D1) Rabbit mAb | Cell Signaling Technology | Cat#2947S; RRID: AB_823586 |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat#F3165; RRID: AB_259529 |
| Anti-Mouse CD117 (c-Kit) APC | eBioscience | Cat#17-1171-83; RRID: AB_469431 |
| Mouse CD117 (c-Kit) APC-eFluor 780 | eBioscience | Cat#47-1171-82; RRID: AB_1272177 |
| Mouse CD117 (c-Kit) PE-Cy7 | eBioscience | Cat#25-1171-82; RRID: AB_469644 |
| Anti-mouse CD45.2-PE | eBioscience | Cat#12-0454-82; RRID: AB_465678 |
| Anti-Mouse CD45.1 PE | eBioscience | Cat#12-0453-83; RRID: AB_465675 |
| Anti-Mouse CD45.2 APC | eBioscience | Cat#17-0454-82; RRID: AB_469400 |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | Cat#12-5981-83; RRID: AB_466087 |
| Mouse TER-119 Biotin | eBioscience | Cat#13-5921-85; RRID: AB_466798 |
| Mouse TER-119 APC | eBioscience | Cat#17-5921-82; RRID: AB_469473 |
| Mouse CD150 APC | Biolegend | Cat#115910; RRID: AB_493460 |
| Mouse CD48 PE-CY7 | eBioscience | Cat#25-0481-80; RRID: AB_1724087 |
| Mouse CD34 Monoclonal Antibody (RAM34), eFluor 660 | eBioscience | Cat#50-0341-82; RRID: AB_10596826 |
| Streptavidin PE-Cy5 | eBioscience | Cat#15-4317-82; RRID: AB_10116415 |
| Mouse CD16/CD32 PE-Cy7 | eBioscience | Cat#25-0161-82; RRID: AB_469598 |
| Mouse CD19 Biotin | eBioscience | Cat#13-0193-86; RRID: AB_657655 |
| Mouse B220 PE | eBioscience | Cat#12-0452-82; RRID: AB_465671 |
| Mouse/Human CD45R (B220) Biotin | eBioscience | Cat#13-0452-86; RRID: AB_466451 |
| Anti-Mouse CD11b PE | eBioscience | Cat#12-0112-83; RRID: AB_465548 |
| Anti-Mouse CD11b eFluor 450 | eBioscience | Cat#48-0112-82; RRID: AB_1582236 |
| Mouse Ly-6G (Gr-1) eFluor 450 | eBioscience | Cat#48-5931-82; RRID: AB_1548788 |
| Mouse Ly-6G (Gr-1) APC-eFluor 780 | eBioscience | Cat#47-5931-82; RRID: AB_1518804 |
| Rat IgM Isotype Control Biotin | eBioscience | Cat#13-4341-81; RRID: AB_470086 |
| Mouse CD127 Biotin | eBioscience | Cat#13-1271-85; RRID: AB_466589 |
| Mouse CD3e Biotin | eBioscience | Cat#13-0033-86; RRID: AB_842773 |
| Mouse CD3e eFluor 450 | eBioscience | Cat#48-0031-82; RRID: AB_10735092 |
| Mouse Ly-6G (Gr-1) Biotin | eBioscience | Cat#13-5931-86; RRID: AB_466802 |
| Bacterial and Virus Strains | ||
| Stbl3™ E. coli | Thermo Fisher Scientific | Cat#C7373–03 |
| 5-alpha Competent E. coli | New England Biolabs | Cat#C29871 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin dihydrochloride | Sigma-Aldrich | Cat#P8833 |
| Insulin, human recombinant, zinc solution | Thermo Fisher Scientific | Cat#12585014 |
| Actinomycin D | Sigma-Aldrich | Cat#A9415 |
| Cycloheximide solution | Sigma-Aldrich | Cat#C4859 |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891–10G |
| Pierce™ Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat#88803 |
| Recombinant Human IL-6 | PeproTech | Cat#200–06 |
| Recombinant Human SCF | PeproTech | Cat#300–07 |
| Recombinant Human TPO | PeproTech | Cat#300–18 |
| Recombinant Human Flt-3 ligand | PeproTech | Cat#300–19 |
| Recombinant Human IL-3 | PeproTech | Cat#200–03 |
| Recombinant mouse IL-3 | PeproTech | Cat#213–13 |
| Recombinant murine Flt-3 ligand | PeproTech | Cat#250–31L |
| Recombinant mouse SCF | PeproTech | Cat#250–03 |
| Recombinant murine GM-CSF | PeproTech | Cat#315–03 |
| Critical Commercial Assays | ||
| QuantiTect Reverse Transcription Kit | QIAGEN | Cat#205314 |
| miRNeasy Mini Kit (50) | QIAGEN | Cat#217004 |
| Magna MeRIP m6A Kit | Millipore | Cat#17–10499 |
| CellTiter 96 Non-Radioactive Cell Proliferation Assay | Promega | Cat#G4100 |
| Dynabeads™ mRNA DIRECT™ Purification Kit | Thermo Fisher Scientific | Cat#61011 |
| APC Annexin V Apoptosis Detection Kit | eBiosciences | Cat#88-8007-74 |
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat#130-046-702 |
| Lineage Cell Depletion Kit | Miltenyi Biotec | Cat#130-090-858 |
| Deposited Data | ||
| m6A-seq (Raw and analyzed data) | This manuscript | GEO: GSE144984 |
| RNA-seq (Raw and analyzed data) | This manuscript | GEO: GSE144984 |
| RIP-seq (Raw and analyzed data) | This manuscript | GEO: GSE144984 |
| mRNA stability profiling (Raw and analyzed data) | This manuscript | GEO: GSE144984 |
| Experimental Models: Cell Lines | ||
| HEK-293T | ATCC | CRL-3216; RRID: CVCL_0063 |
| MonoMac 6 | DSMZ | ACC-124; RRID: CVCL_1426 |
| NOMO-1 | DSMZ | ACC-542; RRID: CVCL_1609 |
| MOLM-13 | DSMZ | ACC-554; RRID: CVCL_2119 |
| MV4;11 | ATCC | CRL-9591; RRID: CVCL_0064 |
| U-937 | ATCC | CRL-1593.2; RRID: CVCL_0007 |
| THP-1 | ATCC | TIB-202; RRID: CVCL_0006 |
| MA9.3ITD | A gift from Dr. James Mulloy | N/A |
| Kasumi-1 | ATCC | CRL-2724; RRID: CVCL_0589 |
| NB4 | DSMZ | ACC-207; RRID: CVCL_0005 |
| Experimental Models: Organisms/Strains | ||
| NSGS mouse | The Jackson Laboratory | Stock No: 013062; RRID: IMSR_JAX:013062 |
| C57BL/6 mice | Envigo | Stock No: 044 |
| B6.SJL-PtprcaPepcb/BoyCrCrl mice | Charles River Laboratories | Strain code: 564; RRID: IMSR_CRL:564 |
| Alkbh5(Em-ko)/B6 | This manuscript | N/A |
| Oligonucleotides | ||
| Primers for Real-time PCR, see Table S2 | Integrated DNA Technologies (IDT) | N/A |
| Recombinant DNA | ||
| pLKO.1 | (Stewart et al., 2003) | Cat#8453; RRID: Addgene_8453 |
| TRIPZ-shRNA | Dharmacon | Cat#RHS4696 |
| pCDH-CMV-EF1 | SBI | Cat#CD513B-1 |
| A5-WT-pCDH | This manuscript | N/A |
| A5-H204A-pCDH | This manuscript | N/A |
| PLX304-TACC3 | Harvard Medical school | Cat#HsCD00419397 |
| MSCV-MLL-AF9-Neo | A gift from Dr. Michael Thirman | N/A |
| MSCV-MLL-AF9-YFP | A gift from Dr. Scott Armstrong | N/A |
| Software and Algorithms | ||
| Bowtie2–2.2.9 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| cutadapt-1.9.1 | (Martin, 2011) | http://cutadapt.readthedocs.io/en/stable/index.html |
| MACS2–2.1.1. | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| samtools-1.3.1 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| tophat-2.1.1 | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/ |
| Venny −2.1 | (Oliveros, 2007) | http://bioinfogp.cnb.csic.es/tools/venny/ |
| Bedtools-2.25.0 | (Quinlan, 2014) | http://bedtools.readthedocs.io/en/latest/ |
| exomePeak-2.8.0 | (Meng et al., 2014) | http://bioconductor.org/packages/release/bioc/html/exomePeak.html |
| igv-2.3.72g | (Thorvaldsdottir et al., 2013) | http://software.broadinstitute.org/software/igv/ |
| GSEA-2.2.3 | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| ELDA | (Hu and Smyth, 2009) | http://bioinf.wehi.edu.au/software/elda/ |
| Bloodspot | (Bagger et al., 2016) | http://servers.binf.ku.dk/bloodspot/ |
| GEPIA | (Tang et al., 2017) | http://gepia.cancer-pku.cn/ |
| cBioPortal | (Cerami et al., 2012) (Gao et al., 2013) |
http://www.cbioportal.org/ |
HIGHLIGHTS:
ALKBH5 is overexpressed in AML and associated with poor prognosis in patients.
ALKBH5 is required for the development and progression of AML.
ALKBH5 is essential for LSCs/LICs self-renewal but not for normal hematopoiesis.
The ALKBH5/m6A/TACC3 axis contributes to the functions of ALKBH5 in AML.
ACKNOWLEDGEMENTS
We thank Dr. Hailing Shi for her help in polysome assays and thank the Genomics Core Facility of the University of Chicago for the next-generation sequencing. This work was supported in part by the National Institutes of Health (NIH) Grants R01 CA214965 (J.C.), R01 CA236399 (J.C.), R01 CA243386 (J.C.), R01 CA211614 (J.C.), R01 HL131444 (Z.Q.), R01 DK107615 (Z.Q.), RM1 HG008935 (C.H.), and Cancer Center Support Grant (P30CA33572) from City of Hope National Medical Center. J.C. and Z.Q. are Leukemia & Lymphoma Society (LLS) Scholars. C.H. is an Investigator of the Howard Hughes Medical Institute (HHMI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
C.H. is a scientific founder and a scientific advisor board member of Accent Therapeutics, Inc.; J.C. is a scientific founder and the President of Genovel Biotech Corp. Both hold equities with their corresponding company.
REFERENCES
- Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, et al. (2014). WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28, 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, and Clevers H (2017). Cancer stem cells revisited. Nature Medicine 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- Bolouri H, Farrar JE, Triche T Jr., Ries RE, Lim EL, Alonzo TA, Ma Y, Moore R, Mungall AJ, Marra MA, et al. (2018). The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 24, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CYA, Ezzeddine N, and Shyu AB (2008). Messenger Rna Half-Life Measurements in Mammalian Cells. Method Enzymol 448, 335–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi KR (2016). Drug developers delve into the cell’s trash-disposal machinery. Nat Rev Drug Discov 15, 295–297. [DOI] [PubMed] [Google Scholar]
- Cho SH, Ha M, Cho YH, Ryu JH, Yang K, Lee KH, Han ME, Oh SO, and Kim YH (2018). ALKBH5 gene is a novel biomarker that predicts the prognosis of pancreatic cancer: A retrospective multicohort study. Annals of hepato-biliary-pancreatic surgery 22, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. (2009). Mutation in TET2 in myeloid cancers. N Engl J Med 360, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Deng X, Su R, Weng H, Huang H, Li Z, and Chen J (2018). RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res 28, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. (2017). Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Gandin V, Sikstrom K, Alain T, Morita M, McLaughlan S, Larsson O, and Topisirovic I (2014). Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge R (1998). Preparation of RNA dot-blots. Methods Mol Biol 86, 73–75. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Huang H, Jiang X, Li Z, Li Y, Song CX, He C, Sun M, Chen P, Gurbuxani S, Wang J, et al. (2013). TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc Natl Acad Sci USA 110, 11994–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, and Chen J (2020a). m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 37, 270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Deng X, and Chen J (2020b). RNA modifications in cancer: functions, mechanisms, and therapeutic implications. Annual Review of Cancer Biology 4, 221–240. [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, et al. (2019). Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Hu C, Ferchen K, Nie J, Cui X, Chen CH, Cheng L, Zuo Z, Seibel W, He C, et al. (2017). Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat Commun 8, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S, Chen P, He C, You D, Zhang S, et al. (2012). Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 22, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CK, Jung JH, Park GS, Lee A, Kang CS, and Lee KY (2006). Expression of transforming acidic coiled-coil containing protein 3 is a novel independent prognostic marker in non-small cell lung cancer. Pathology international 56, 503–509. [DOI] [PubMed] [Google Scholar]
- Krause DS, and Van Etten RA (2007). Right on target: eradicating leukemic stem cells. Trends Mol Med 13, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, and Armstrong SA (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Kwok CT, Marshall AD, Rasko JE, and Wong JJ (2017). Genetic alterations of m(6)A regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. (2010). DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ, Laird PW, Baty JD, et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang H, Chen P, He M, Li Y, Arnovitz S, Jiang X, He C, Hyjek E, Zhang J, et al. (2012a). miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun 2, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang H, Li Y, Jiang X, Chen P, Arnovitz S, Radmacher MD, Maharry K, Elkahloun A, Yang X, et al. (2012b). Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood 119, 2314–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, et al. (2008). Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A 105, 15535–15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. (2018). Suppression of m(6)A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. (2017). FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 31, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhang Z, Li Y, Arnovitz S, Chen P, Huang H, Jiang X, Hong GM, Kunjamma RB, Ren H, et al. (2013). PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood 121, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA, Sepulveda C, et al. (2019). Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 25, 137–148 e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekorz RP, Hoffmeyer A, Duntsch CD, McKay C, Nakajima H, Sexl V, Snyder L, Rehg J, and Ihle JN (2002). The centrosomal protein TACC3 is essential for hematopoietic stem cell function and genetically interfaces with p53-regulated apoptosis. Embo Journal 21, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by Noncoding RNAs. Cell 129, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Essmann F, Kletke A, Rio P, Hanenberg H, Schulze-Osthoff K, Nurnberg B, and Piekorz RP (2008). TACC3 depletion sensitizes to paclitaxel-induced cell death and overrides p21WAF-mediated cell cycle arrest. Oncogene 27, 116–125. [DOI] [PubMed] [Google Scholar]
- Somervaille TCP, and Cleary ML (2006). Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 10, 257–268. [DOI] [PubMed] [Google Scholar]
- Song H, Liu C, Shen N, Yi P, Dong F, Li X, Zhang N, and Huang T (2018). Overexpression of TACC3 in Breast Cancer Associates With Poor Prognosis. Appl Immunohistochem Mol Morphol 26, 113–119. [DOI] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhail TV, Singh P, and Manna TK (2015). Suppression of centrosome protein TACC3 induces G1 arrest and cell death through activation of p38-p53-p21 stress signaling pathway. Eur J Cell Biol 94, 90–100. [DOI] [PubMed] [Google Scholar]
- Sun Y, Tian Y, Wang GZ, Zhao SH, Han B, Li YL, and Jiang CL (2017). Overexpression of Transforming Acidic Coiled CoilContaining Protein 3 Reflects Malignant Characteristics and Poor Prognosis of Glioma. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhammer A, Bencokova Z, Poole R, Loenarz C, Adam J, O’Flaherty L, Schodel J, Mole D, Giaslakiotis K, Schofield CJ, et al. (2011). Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha). PLoS One 6, e16210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, and Majeti R (2017). Biology and relevance of human acute myeloid leukemia stem cells. Blood 129, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, Liu B, Xiao F, Wang W, Huang G, et al. (2018a). Loss of YTHDF2-mediated m(6)A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res 28, 1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu ZK, Gomez A, Hon GC, Yue YN, Han DL, Fu Y, Parisien M, Dai Q, Jia GF, et al. (2014a). N-6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ding X, Hu H, He Y, Lu Z, Wu P, Tian L, Xia T, Yin J, Yuan H, et al. (2018b). Long non-coding RNA lnc-PCTST predicts prognosis through inhibiting progression of pancreatic cancer by downregulation of TACC-3. Int J Cancer 143, 3143–3154. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, and Armstrong SA (2010). The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014b). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, and Chen J (2019). RNA N (6)-Methyladenosine Modification in Normal and Malignant Hematopoiesis. Adv Exp Med Biol 1143, 75–93. [DOI] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 22, 191–205 e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, and Mulloy JC (2013). AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood 121, e90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 61, 507–519. [DOI] [PubMed] [Google Scholar]
- Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, Chen IM, Chen Z, Rowley JD, Willman CL, et al. (2006). A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med 12, 945–949. [DOI] [PubMed] [Google Scholar]
- Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, and Zhou BO (2018). Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res 28, 952–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R, Natsume Y, Saiki Y, Shioya H, Takeuchi K, Yamori T, Toki H, Aoki I, Saga T, and Noda T (2012). Disruption of Tacc3 function leads to in vivo tumor regression. Oncogene 31, 135–148. [DOI] [PubMed] [Google Scholar]
- Yuan CL, and Hu YC (2017). A Transgenic Core Facility’s Experience in Genome Editing Revolution. Adv Exp Med Biol 1016, 75–90. [DOI] [PubMed] [Google Scholar]
- Yun M, Rong J, Lin ZR, He YL, Zhang JX, Peng ZW, Tang LQ, Zeng MS, Zhong Q, and Ye S (2015). High expression of transforming acidic coiled coil-containing protein 3 strongly correlates with aggressive characteristics and poor prognosis of gastric cancer. Oncol Rep 34, 1397–1405. [DOI] [PubMed] [Google Scholar]
- Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y, et al. (2017a). m6A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273–276. [DOI] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, and Semenza GL (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A 113, E2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guo S, Piao HY, Wang Y, Wu Y, Meng XY, Yang D, Zheng ZC, and Zhao Y (2019). ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. Journal of physiology and biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O, et al. (2017b). m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 31, 591–606.e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DS, Wang HB, Zhou ZG, Zhang YJ, Zhong Q, Xu L, Huang YH, Yeung SC, Chen MS, and Zeng MS (2015). TACC3 promotes stemness and is a potential therapeutic target in hepatocellular carcinoma. Oncotarget 6, 24163–24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The m6A-seq, RNA-seq, RIP-seq and mRNA stability profiling datasets obtained in this study have been deposited in the gene expression omnibus (GEO) repository and made accessible under accession numbers GSE144984.