

Abstract
Femtosecond lasers at fixed wavelengths above 1000 nm are powerful, stable, and inexpensive, making them promising sources for two-photon microscopy. Biosensors optimized for these wavelengths are needed for both next-generation microscopes and affordable turn-key systems. Here we report jYCaMP1, a yellow variant of the Calcium indicator jGCaMP7 that outperforms its parent in mice and flies at excitation wavelengths above 1000 nm and enables improved two-color calcium imaging with RFP-based indicators.
Two-photon (2P) microscopy has become the leading method for in vivo imaging owing to its optical sectioning capabilities and the increased depth-penetration of near-infrared light in scattering tissue1. However, the light sources commonly used for 2P imaging– tunable Titanium-Sapphire lasers and parametric oscillators – are costly, require frequent expert maintenance, and lack the output power needed for operating several microscopes simultaneously or for high-speed imaging methods that use extended focal patterns2–5.
Promising alternatives to these traditional light sources, such as high-power industrial Ytterbium-doped fiber lasers (YbFLs) and modelocked semiconductor lasers, have shown feasibility for in-vivo imaging2–7 and are becoming widely available at costs orders of magnitude lower and/or power outputs orders of magnitude higher than conventional tunable lasers (Supplementary Fig. 1). Since the bulk of a 2P microscope’s cost is the laser, these sources promise to make 2P imaging accessible to many more users. However, commercially-available lasers of this kind are largely limited to a fixed wavelength of approximately 1030–1080 nm, which poorly excites most green fluorescent protein (GFP)-based biosensors such as GCaMPs, the best-in-class genetically encoded fluorescent Ca2+indicator (GECI, Fig. 1b). To take advantage of inexpensive and powerful industrial lasers, high-performance biosensors that excite at wavelengths above 1000 nm are needed. 2P imaging of GECIs has become widespread for monitoring neuronal activity and is vital to modern neuroscience1. We therefore set out to identify spectral variants of the recently-developed jGCaMP7 family of GECIs8 with improved 2P excitation at fiber-laser wavelengths.
Fig. 1.
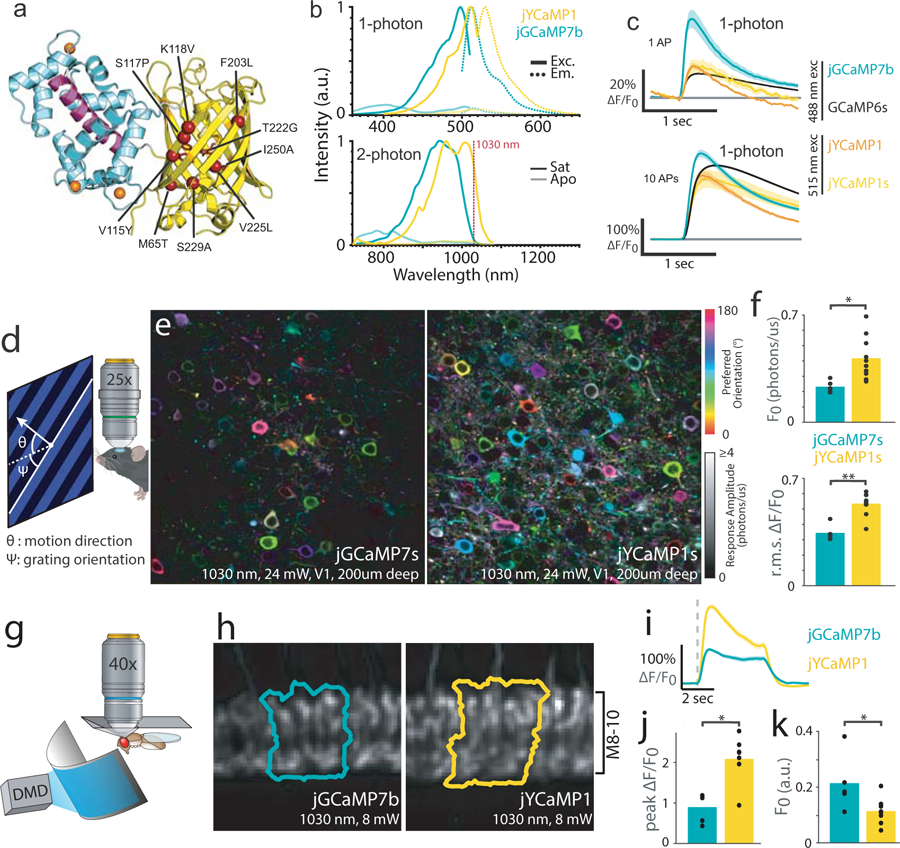
jYCaMP1 is a redshifted variant of jGCaMP7 capable of detecting single action potentials.
(A) Schematic of jYCaMP1 mutations (red spheres) overlaid on the GCaMP structure (PDB-ID: 3EVR11). (B) Normalized 1-photon excitation (solid lines) and emission (dotted lines) spectra (top) and 2-photon action cross-sections (bottom) in the presence and absence of calcium, for jYCaMP1 and jGCaMP7b. (C) Responses in rat primary hippocampal cultures to 1 and 10 action potentials for indicators at their respective excitation optima. GCaMP6s and jGCaMP7b data, acquired on the same apparatus as jYCaMP data, are reprinted from (8). Shading denotes SEM. N (# wells) =104 jGCaMP7b, 682 GCaMP6s,11 jYCaMP1, 10 jYCaMP1s. (D) Anaesthetized mice were shown visual stimuli while recording activity in layer 2/3 of visual cortex. (E) Orientation tuning maps in example jGCaMP7s and jYCaMP1s-expressing fields of view. Hue denotes preferred orientation; brightness denotes response amplitude. Some pixels are saturated. Similar results were obtained for N=5 (jGCaMP7s) and N=10 (jYCaMP1s) fields of view (F) Mean baseline intensity of responsive (∆F/F>1) pixels (top) and mean ∆F/F of bright (F0>0.5) pixels (bottom), for N=5 (jGCaMP7s) and N=10 (jYCaMP1s) fields of view. Dots denote individual fields of view, bars indicate mean. p=0.01 (F0), p=0.0001, (r.m.s. ∆F/F), two-sided 2-sample t-tests (G) Flies were head fixed to a pyramidal plate with the cuticle above the Mi1 neurons removed for imaging, and presented full-field visual stimuli. (H) Sample ROIs, typical of N=8 (jYCaMP1), N=5 (jGCaMP7b) flies, drawn around the columns in layers 8–10 of the medulla that showed the largest increase in intensity to the stimulation. (I) ∆F/F responses in Mi1 neurons expressing jYCaMP1 and jGCaMP7b. Grey dashed line presents stimulus onset (lights on). (J) Maximum ∆F/F reached after stimulation. p=1e-4, two-sided 2-sample t-test (K) Baseline fluorescence before stimulation. p=0.033, two-sided 2-sample t-test. (J-K) N=8 (jYCaMP1), N=5 (jGCaMP7b) flies. Dots denote individual fields of view, bars indicate mean.
Mutations can convert GFPs to yellow fluorescent proteins well-excited at YbFL wavelengths. Importantly, substitution of T115 (position 203 in GFP) with an aromatic amino acid allows this residue to form a π-stacking interaction with the phenolic ring of the GYG-chromophore (compared to the green TYG-chromophore) resulting in a shift towards longer wavelengths9. To redshift jGCaMP7, we first introduced mutations that convert GFP into mVenus10 ( “Venus-GCaMP”; jGCaMP7s + M65T, V115Y, K118V, F203L, T222G, V225L, S229A, I250A). Unfortunately, Venus-GCaMP did not exhibit the anticipated spectral shift, retaining excitation and emission spectra similar to its parent GCaMP (Supplementary Fig. 2). To find a truly yellow-fluorescent GCaMP variant, we randomly mutated Venus-GCaMP, and used fluorescence emission ratiometry to screen for spectral shift in bacterial colonies. We found a single amino acid mutation S117P (205 in GFP), close to T115, that produced a pronounced redshift. The resulting variant maintained sensor properties similar to those of the parent GCaMP while exhibiting 19 nm and 36 nm spectral shift in its one-photon (1P) and 2P excitation spectrum respectively (Fig. 1b, Supplementary Table 1). Similarly, jGCaMP7 variants containing YFP, YPET and Citrine mutations failed to produce yellow emission spectra but were rescued by additional introduction of the S117P mutation (data not shown). In GCaMP, the closed barrel structure of circularly-permuted GFP is opened within β-strand 7 to accommodate the calcium-sensing domains. Residues 115 and 117 lie on β-strand 10 of GFP, structurally adjacent to β-strand 7, and their position might be affected by the GCaMP permutation in a way that prevents the crucial π-stacking interaction (Fig. 1a). The substitution S117P might then reorient position 115 to rescue the π-stacking and yield the observed yellow fluorescence.
Among jGCaMP7 variants, jGCaMP7b and jGCaMP7s containing the mVenus and S117P mutations [M65T, V115Y, S117P, K118V, F203L, T222G, V225L, S229A, I250A] exhibited the largest fluorescence responses to field stimulation in neuron cultures (Fig. 1c, Supplementary Fig. 3). These variants, called “jYCaMP1” and “jYCaMP1s”, respectively, have slightly higher Ca2+affinity than their jGCaMP7b and 7s parents (Supplementary Fig. 4, Supplementary Table 1) and show responses similar to those of GCaMP6s at its 1-photon excitation optimum (Fig. 1c). Under red-shifted illumination, they showed significantly larger ∆F/F responses than GCaMP6s for 1 or 3 APs (4-fold and 2-fold larger respectively for both indicators, Supplementary Fig. 3). jYCaMP1 and jYCaMP1s also had 2.7-fold higher molecular brightness than jGCaMP7b when excited at 1030nm (Supplementary Fig. 5) and differed substantially from each other only in response kinetics. These properties suggested that jYCaMPs might outperform the best green GECIs in vivo when imaged using 2P excitation at or beyond 1030nm, so we further characterized them in mice and flies.
In mice, we imaged jYCaMP1s activity in layer 2/3 of primary visual cortex (V1) while displaying moving grating visual stimuli to the mouse’s contralateral eye. Compared to jGCaMP7s, recordings from jYCaMP1s-labeled neurons were 1.8-fold as bright (p=0.01, 2-sample t-test) and showed 1.6-fold ∆F/F (p = 1e-4, 2-sample t-test) under 1030 nm illumination (jYCaMP1s: n= 10 fields of view (FOVs) in 4 independent animals, jGCaMP7s: n=5 FOVs, in 3 independent animals, Fig. 1d–f, Supplementary Fig. 6). jYCaMP’s brightness and sensitivity at 1030 nm enabled simultaneous high-speed imaging of multiple dendritic branches using YbFL-based two-photon tomography (Supplementary Fig. 7), and multi-area imaging using 1040 nm excitation (Supplementary Fig. 8).
In flies, we measured jYCaMP1 activity in Mi1 or Tm3 neurons in the medulla of the optic lobe while presenting whole-field flashing light stimuli (Fig 1g–k, Supplementary Fig. 9). Light increments produce stereotyped calcium transients in both neuron types12–14. In Mi1 (medulla layers 8–10), jYCaMP1 was 0.53-fold as bright as its parent jGCaMP7b at baseline (p=0.033, 2-sample t-test; Fig 1k) and showed 2.3-fold ∆F/F (p=1e-4, 2-sample t-test, Fig 1j) under 1030 nm excitation (jYCaMP1: n=8 flies; jGCaMP7b: n=5 flies). In Tm3 (lobula plate layer 1), jYCaMP1 was 0.62-fold as bright as jGCaMP7b at baseline (p=0.011, 2-sample t-test) and showed 2.3-fold ∆F/F responses (p=5e-3, 2 sample t-test; jYCaMP1: n=6 flies and jGCaMP7b=5 flies, Supplementary Fig. 9).
The ability to record in distinct spectral channels is an important strength of fluorescence imaging. For example, targeting spectrally-resolved sensors to different compartments enables simultaneous recording from compartments that are too close together to resolve spatially15, such as pre- and post-synapses. The 2P excitation spectrum of GCaMP overlaps poorly with state-of-the-art red GECIs such as jRGECO1a, complicating two-color Ca2+-imaging16. The shifted excitation spectrum of jYCaMP1 greatly improves this overlap while retaining well-separated emission with red sensors, making these GECIs an ideal combination for simultaneous dual-color 2P Ca2+-imaging with a single excitation laser. To test the capabilities of jYCaMP for two-color imaging with red GECIs, we performed simultaneous Ca2+-imaging of thalamocortical projections labeled with axon-targeted17 jYCaMP1s and cortical dendrites labeled with jRGECO1a (Fig. 2a,b see Methods).
Fig. 2.
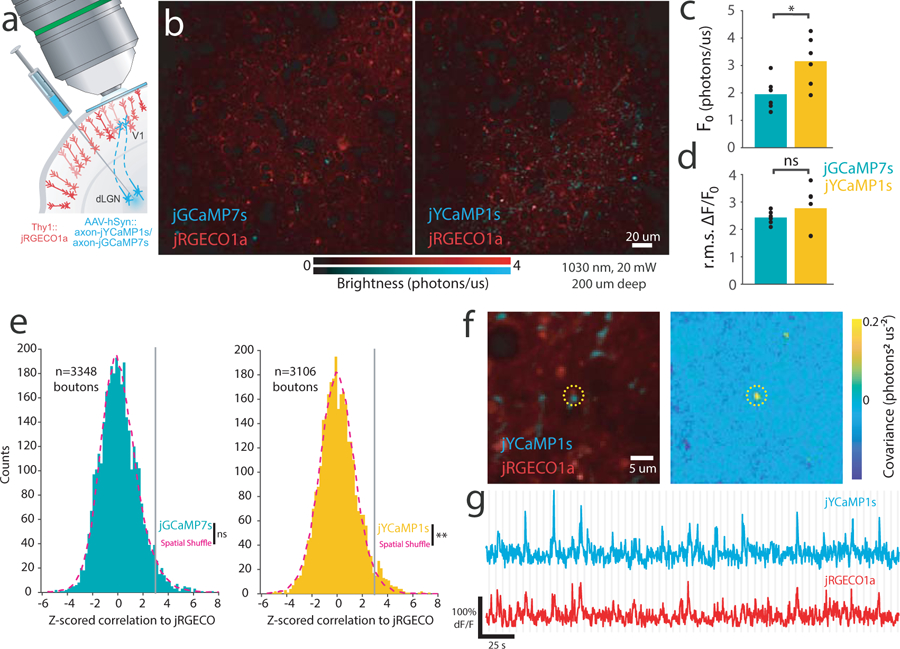
jYCaMP1 enables brighter two-color calcium imaging together with red GECIs, improving correlation analysis of overlapping compartments. (A)Schematic of two-color cortical labeling. Virus encoding axon-targeted jGCaMP7s or jYCaMP1s was injected into the dLGN of thalamus in transgenic Thy1:jRGECO1a mice, and visually-evoked activity recorded in cortex. (B) Example FOVs expressing jGCaMP7s and jYCaMP1s. Similar results were obtained for N=6 FOVs each sensor. (C, D) Mean brightness (C) and rms ∆F/F response (D) of putative boutons (N=6 FOVs, 3106 boutons jYCaMP1s, N=6 FOVs, 3348 boutons, jGCaMP7s). *:p=0.02, two-tail, two-sample t-test. Markers denote individual FOVs. (E) Distribution of z-scored correlations (Pearson’s r) between unmixed jRGECO and jGCaMP7s- or jYCaMP1s- responses at individual putative boutons, and corresponding null distributions obtained by shuffling bouton locations. p=0.484 (jGCaMP7s) p<0.004 (jYCaMP1s) Kolmogorov-Smirnov test. N=3348 boutons (jGCaMP7s), N=3106 boutons (jYCaMP1s). (F-G) An example jYCaMP1s-labeled putative bouton highly correlated to dendritic jRGECO response. Similar correlated sites were found in 5 of 6 FOVs. (F) (left) Average image of each channel and (right) pixelwise covariance of response amplitudes across channels. Dotted circle denotes the same area in both images. (G) jYCaMP and jRGECO recordings from the bouton site. Gray lines denote stimulus onsets (F-G)
Under 1030 nm excitation, jYCaMP1s enabled us to routinely record axonal responses with 20 mW of excitation power (Fig. 2b), showing 1.6-fold baseline brightness and similar response amplitudes to jGCaMP7s (jYCaMP1s: 6 FOVs, 5 mice, 3106 boutons; jGCaMP7s: 6 FOVs; 4 mice, 3348 boutons; Fig. 2c,d). Simultaneous 1030 nm excitation of jYCaMP1s and jRGECO1a enabled recording of distinct Ca2+ dynamics in spatially-overlapping axonal and dendritic compartments, which has been used to identify putative synapses16,18,19. We compared jGCaMP7s to jYCaMP1s for such detection of coactive axons and dendrites, based on trial-to-trial correlations to dendritic jRGECO responses at putative boutons. Using jGCaMP7s, at false positive rate of 1%, 1.01% of boutons showed significant correlations, and the observed distribution did not vary significantly from the null (p=0.484, two-sample Kolmogorov-Smirnov test). Using jYCaMP1s, at false positive rate of 1%, 2.22% of boutons showed significant correlations, and the distribution differed significantly from the null (p=0.004, two-sample Kolmogorov-Smirnov test, Fig. 2e). Significantly-correlated sites contained bright structures in both channels and highly-coordinated dynamics (Fig. 2f–g). These results demonstrate that jYCaMP’s improved coexcitation with RFPs facilitates challenging two-color experiments, which previously required high excitation powers16,18,19 that can damage neurites20.
In summary, we generated jYCaMP1, a variant of jGCaMP7 with a redshifted excitation spectrum. This yellow fluorescent GECI inherits the high Ca2+ affinity and excellent in vitro and in vivo performance characteristics of its optimized parent protein. Its redshifted excitation spectrum is compatible with inexpensive 1030 and 1040 nm lasers, enabling high-performance in vivo calcium imaging, high-speed methods requiring large pulse energies, and efficient single-laser two-color imaging alongside red GECIs. Other potential applications include compact optical systems incorporating miniaturized low-cost lasers, or multiplexing many microscopes using a single high-power source. In combination with other labels2,5 and functional indicators16 excitable at these wavelengths, jYCaMP1 will enable a rich variety of experiments using only inexpensive fixed-wavelength lasers without losses in signal quality.
Online Methods:
Animal Care and Use Statement:
All surgical and experimental procedures were in accordance with protocols approved by the HHMI Janelia Research Campus Institutional Animal Care and Use Committee and Institutional Biosafety Committee. This research has complied with all relevant ethical regulations.
Molecular biology
Bacterial expression plasmids for jYCaMP1 and jYCaMP1s were created by replacing the FP portion in pRSET-jGCaMP7 (gift from Douglas Kim) through restriction digest (SacI & AflII) and isothermal assembly of synthesized gene fragments (GBlocks). Randomized libraries were created using isothermal assembly of a Mutazyme (Agilent) PCR according to the manufacturer’s suggestions. Depletion of individual mutations was performed using a QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies Inc.) according to the manufacturer’s suggestions. Plasmid sequences were confirmed via Sanger sequencing.
Bacterial colony screen
Bacterial colonies on agar plates were screened for bathochromic fluorescence shift on a modified fluorescence stereo microscope (Leica M165 FC, Leica Microsystems) with a coupled mercury metal halide light source (Leica EL6000, Leica Microsystems). Illumination and emitted light was initially split by the microscope dichroic (GFP-LP, Leica) followed by a custom filter cube (ET510/20m, ET537/29m, T525LPXR; Chroma) to produce two narrow-band channels for emission ratiometry. These images were detected by CMOS cameras (Blackfly S Mono 5.0 MP, GigE Vision). A custom MATLAB script displayed the two channels overlaid in false color at a user defined ratio, allowing the user to rapidly visualize subtle spectral variations. Colonies that appeared red-shifted were picked for further analysis.
Protein Expression and in vitro Analysis
Recombinant sensor proteins were expressed in autoinduction medium according to known protocols1. Ca2+-saturated and Ca2+-free measurements were performed in 39 µM free Ca2+ (+Ca2+) buffer (30 mM MOPS, 10 mM CaEGTA in 100 mM KCl, pH 7.2) or 0 µM free Ca2+ (-Ca2+) buffer (30 mM MOPS, 10 mM EGTA in 100 mM KCl, pH 7.2) respectively.
1P photophysical measurements
Absorbance measurements were performed using a UV-Vis spectrometer (Lambda 35, Perkin Elmer). Quantum Yield measurements were performed using an integration sphere spectrometer (Quantaurus, Hamamatsu) for proteins in +Ca buffer. Extinction coefficients were determined using alkali denaturation method using extinction coefficient of denatured GFP as a reference (44,000M−1cm−1 at 447nm).
Ca2+-titrations as well as measurements of kinetic parameters were performed as previously described2–4.
2P measurements
2P excitation spectra were obtained as previously described5. Protein solutions of 2–4µM concentration in + Ca2+ or -Ca2+- buffer were prepared and measured using an inverted microscope (IX81, Olympus) equipped with a 60x, 1.2NA water immersion objective (Olympus). 2P excitation was obtained using an 80MHz Ti::Sapph laser (Chameleon Ultra II, Coherent) for spectra from 710nm to 1080 nm. Fluorescence collected by the objective was passed through a short pass filter (720SP, Semrock) and a band pass filter (550BP88, Semrock), and detected by a fiber-coupled Avalanche Photodiode (APD) (SPCM_AQRH-14, Perkin Elmer). The obtained 2P excitation spectra was normalized for 1 µM concentration.
Fluorescence correlation spectroscopy (FCS) was used to obtain the 2P molecular brightness of the proteins. The peak molecular brightness was defined by the rate of fluorescence obtained per total number of emitting molecules6. 50 – 100nM protein solutions were prepared in +Ca2+ buffer and excited with 1030nm wavelength at various power ranging from 2–30mW for 200sec. The obtained fluorescence at was collected by an APD and fed to an autocorrelator (Flex03LQ, Correlator.com). The obtained autocorrelation curve was fit on a diffusion model through an inbuilt Matlab function6 to determine the number of molecules <N> present in the focal volume. The 2P molecular brightness at each laser power was calculated as the average rate of fluorescence <F> per emitting molecule <N>, defined in kilocounts per second per molecule (kcpsm).
Characterization in neuronal culture
E18 rat cortical neurons nucleofected with pAAV-Synapsin1-jYCaMP1 (or jGCaMP respectively) variants were stimulated and imaged at 18 days in vitro in the presence of synaptic blockers as described previously7. 35Hz imaging was performed using a 10x 0.4NA objective, a X-cite exacte mercury lamp and an ex500/30, T515LP, em535/30 filter cube at 3.5mW at the sample plane. Cell bodies were segmented, the background subtracted and the relevant parameters extracted as described previously7.
Mouse surgical procedures
All vertebrate procedures were in accordance with protocols approved by the HHMI Janelia Research Campus Institutional Animal Care and Use Committee (IACUC 17–155). Emx1-Cre (B6.129S2-Emx1tm1(cre)Krj/J, Jackson Laboratories) and GP8.50 (Thy1::jRGECO1a, provided by the GENIE project, HHMI Janelia Research Campus) mice (either gender) were intracranially injected with AAV2/1-Synapsin1-FLEX-jYCaMP1 or AAV2/1-Synapsin1-FLEX-jGCaMP7s respectively and AAV2/1-Synapsin1-FLEX-axon-jYCaMP1s or AAV2/1-Synapsin1-FLEX-axon-jGCaMP7s respectively together with AAV2/1-Synapsin1-Cre through a craniotomy and a 4 mm cranial window was placed over the visual cortex: Mice were anaesthetized using isoflurane in oxygen (3–4% for induction, 1.5–2% for maintenance), placed on a 37°C heated pad, administered Buprenorphine HCl (0.1 mg/kg) and ketoprofen (5 mg/kg). The skin covering the skull was removed, the sutures of the frontal and parietal bones were sealed with a thin layer of cyanoacrylate glue and a titanium headbar was glued over the left visual cortex. A ~4.5mm craniotomy (centered 3.5mm lateral and 0.5mm rostral of lambda) was performed leaving the dura intact. Glass capillaries (Drummond Scientific, 3–000-203-G/X) pulled and beveled to 30° angle, 20μm outer diameter loaded into a precision injector (Drummond Scientific, Nanoject III) were used for injections. For experiments comparing axon-jGCaMP7s and axon-jYCaMP1s, mice of the same gender from the same litter were injected on the same day.
For V1 injections, the virus was diluted to 2x1012 GC/ml and slowly injected (1 nL/s) in 6–8 different injection sites around L 2.7 mm; 0.2 mm anterior to lambda; 300µm deep and 30nL per site.
For thalamic injections the sensor encoding virus was diluted to 2x1012 GC/ml, mixed 1:1 with 2x109 GC/ml AAV2/1-Synapsin1-Cre and the mix slowly injected (1 nL/s) in the dLGN (~2.1mm posterior to Bregma, ~2.3mm left of midline) at two depths (0.5mm apart, centered at ~2.55 mm deep) and 80 nL per site. Adjusted coordinates for each litter were established via test injections of fluorescent beads in sex-matched littermates, followed by sectioning and microscopic analysis.
The craniotomy was then covered with a 4 mm round #1.5 cover glass that was fixed to the skull with cyanoacrylate glue. The animals were imaged 3–6 weeks after surgery. For experiments comparing jGCaMP7b and jYCaMP1s, mice were imaged using the same excitation laser powers, broadband filter sets, and detectors settings.
In vivo imaging of visual responses in mouse visual cortex
2–4 weeks after viral injection, the mice were anesthetized using isoflurane, head fixed and restrained inside a custom-built heated holder to restrict movement and maintain a body temperature of 37°C. A vertically-oriented screen (ASUS PA248Q LCD monitor, 1920x1200 pixels), with a high-extinction 500 nm shortpass filter (Wratten 47B-type) was placed 17cm from the right eye of the mouse, centered at approximately 65 degrees of azimuth and −10 degrees of elevation. Moving bar stimuli were generated in MATLAB using the Psychtoolbox (20 repetitions of 0.153 cycles per cm, 1 cycle per second, 22 degrees of visual field per second, 2sec duration) in 8 equally-spaced directions spaced by equal time periods of mean luminance, and were shown to the animal synchronized with the acquisition.
Imaging was performed on a home-built 2P microscope equipped with an Insight DS Dual 120 femtosecond-pulse laser (Spectra-Physics, Santa Clara, CA) at 1030nm, a XLPLN25XWMP2 25x 1.05NA water immersion lens (Olympus) and two Silicon Photomultiplier (SiPM) detectors (MPPCs; Hamamatsu, custom part, see8 for details) with 540/80 and 650/90 bandpass filters. 512x512 Images were acquired at 3.41Hz and 21–24 mW post objective power using ScanImage software (Vidrio Technologies).
Raster recordings were aligned using custom MATLAB scripts as described previously8. For Figs. 1e–f and 2c,d, analyses were performed pixelwise. F0 was calculated as the mean pixel brightness during the 4 frames prior to each stimulus onset. 8-point tuning curves were calculated as the mean pixel brightness during the stimulus period, minus F0. The preferred orientation (hues in Figs. 1e and 2f) was calculated by vector summation of the tuning curve over the 4 stimulus orientations (i.e. 0, 90, 180, 270, 0, 90, 180, 270 degrees for the 8 stimulus directions). The response amplitude is the 2-norm of the 8-point tuning curve, and r.m.s. ΔF/F0 (Figs. 1f and 2c) is the response amplitude divided by F0. We plotted the mean F0 for pixels with response amplitude>1 photon/µs (jGCaMP7s: 6.9e5 responsive pixels, 5 FOVs, 3 mice. jYCaMP1: 1.5e6 responsive pixels, 10 FOVs, 4 mice); and the r.m.s. dFF for pixels with F0>1 photon/us (jGCaMP7s: 5.0e5 bright pixels; jYCaMP1: 1.0e6 bright pixels).
SLAP imaging8 was performed in the V1 of a Chrna2-OE25/C57Bl6J heterozygous mouse (a line that expresses Cre recombinase in a sparse subset of L5 pyramidal neurons) injected with AAV2/1.hSynapsin1.FLEX.jYCaMP1s. Imaging was performed at 500Hz using a 1030 nm YbFL (BlueCut, Menlo Systems GMBH) with the abovementioned visual stimuli. Segments of interest (Fig S7) were manually drawn, and remaining regions of the 3D reference image were segmented automatically using the SLAP software package (www.github.com/KasparP/SLAP). Reconstructions were performed using the SLAP software package using a time constant of 20ms, baseline of 0.8, and default parameters otherwise.
Dual color in vivo Ca2+-imaging in the mouse cortex
Dual-color cortical imaging experiments (Fig. 2) were performed using the same hardware, stimuli, microscope settings as one-color cortical imaging. We used GP8.50 Thy1:jRGECO1a transgenic mice, in which approximately 50% of layer 2/3 and layer 5 neurons are labeled. All recordings were performed 200 µm below the surface of the pia using 20 mW of post-objective laser power. Automated detection of boutons was used to quantify brightness and responsiveness of axon-targeted jGCaMP7s and jYCaMP1.
Putative boutons were detected as local maxima in the Green channel average intensity image that were brighter than ¼ of the 95th percentile of the image intensity, after smoothing with a sigma=0.5 µm Gaussian kernel. We plotted the mean F0 and r.m.s. ΔF/F0 treating field of view as the unit of variation (GCaMP: 6 FOVs; 4 mice, 3348 boutons; jYCaMP: 6 FOVs, 5 mice, 3106 boutons).
The Red and Green/Yellow emission bands of the fluorophores we used are well separated, and can be collected with only mild tradeoffs between collection efficiency and crosstalk in their tails. To remove residual crosstalk, Red and Green channels were linearly unmixed prior to analysis and display, using least squares unmixing (mixing proportions for our filters and detector sensitivities: jGCaMP/RGECO 0.022 Green->Red, 0.11 Red->Green; jYCaMP/RGECO 0.08 Green->Red, 0.11 Red->Green). To design effective analyses of correlations between axonal and dendritic compartments (Fig 2e–g), we performed simulations that assessed effects of bleed through and unmixing on measured correlations, and validated the generation of null distributions for statistical comparisons. These simulations showed that least squares spectral unmixing of Poisson-distributed measurements results in negative bias in computed sample correlations. We found that maximum likelihood unmixing of Poisson-distributed measurements results in positive bias, and did not use it in our studies. To compensate for bias and better normalize comparisons across fields of view and imaging conditions, sample correlations for each field of view were Z-scored according to the null distribution computed for that field of view. Z scores were computed by subtracting the median of the sample distribution and dividing by the standard deviation of the spatially-shuffled null distribution. The spatially-shuffled null distribution was also Z-scored. Detection rates reported were computed by pooling the Z-scored correlations across fields of view, and comparing to the pooled null distribution samples. Spatial shuffling consisted of randomly reassigning the bouton identities, but not the timeseries, for one channel (after unmixing) prior to computing the correlations between the channels. We validated that the computed and Z-scored null distributions accurately represent the Z-scored sample correlations for uncorrelated latents across a wide range of mean photon rates (1–1000 detected photons), mixtures of photon rates across boutons and channels, and a variety of activity distributions (Gaussian, Binary, Poisson, Spike and slab), and are robust to errors in unmixing coefficients of up to 50%. Matlab code that performs these simulations is available at www.github.com/KasparP/TwoColorUnmixing
Detection rates at 1% false positives reported were computed as the fraction of pooled correlations that exceeded the 99th percentile of the pooled null distribution. Two-sample Kolmogorov-Smirnov tests (Matlab kstest2, default alternative hypothesis) were performed to compare the pooled distributions.
Covariance maps (Fig 2f) were produced by computing mean unmixed ΔF/F response during each stimulus presentation for each pixel, and computing the covariance between the two channels across stimulus presentations.
Multi-area imaging in mouse somatosensory cortex.
In adult (6–8 week old) C57Bl6 mice, stereotaxic viral injections of AAV2/9.hSynapsin1.jYCaMP1s were performed in to express jYCaMP in primary (S1) and secondary (S2) somatosensory cortex (600 nL total volume, 6.8 x 1011 gc/ml). L2/3 and L5 of S1 was targeted at 1.1mm posterior to bregma, 3.3mm lateral, 300 and 500 µm below the pial surface. L2/3 and L5 of S2 was targeted at 0.7mm posterior to bregma, 4.2 lateral, 300 and 500 µm below the pial surface. Optical access over S1 and S2 was achieved by cranial window implantation. A metal headpost for head fixation was implanted on the skull surrounding the window. Imaging was performed with a custom-built resonant-scanning multi-area two-photon microscope controlled by custom-written Scope software based on a design previously described (Chen et al., 2016). The system consisted of a 31.25Mhz 1040 nm Ytterbium fiber laser (Spark Lasers) split into two temporally multiplexed beams positioned over S1 through a 16x/0.8NA water immersion objective (Nikon). Simultaneous imaging was carried out at 32.6 Hz frame rate. All image processing was performed in MATLAB (Mathworks). Two-photon images were motion-corrected using a piece-wise rigid motion correction algorithm. Regions of interest corresponding to individual active neurons were manually identified and calcium signals computed as (F-F0)/F0 where F0 represents the bottom 8th percentile of fluorescence across a 10 s sliding window.
Drosophila genetics
We generated w1118;; PBac{20XUAS-IVS-GECI-p10}VK00005 transgenic lines carrying jYCaMP1. Sensors were driven in the Mi1 neurons using the 19F01-GAL4 (attp2) and in Tm3 using the 59C10-GAL4 (attP2) drivers. Males from sensor lines were crossed with females containing the driver lines. Flies were raised at 25°C on standard cornmeal molasses media.
Imaging in Drosophila brain
Females 3–5 days after eclosure were anesthetized on ice. After transferring to a thermoelectric plate (4°C), legs were removed, and then facing down, the head was glued into a custom-made pyramid using UV-cured glue. The proboscis was pressed in and fixed using UV-cured glue. After adding saline (103 mM NaCl, 3 mM KCl, 1 mM NaH2PO4, 5 mM TES, 26 mM NaHCO3, 4 mM MgCl2, 2.5 mM CaCl2, 10 mM trehalose and 10 mM glucose, pH 7.4, 270–275 mOsm) to the posterior side of the head, cuticle was cut away above the right side creating a window above the target neurons. Tracheae and fat were removed. Muscles M1 and M6 were cut to minimize head movement.
Two photon imaging took place under a 40xN.A. 0.8 water-immersion objective (Olympus) on a laser scanning microscope (BrukerNano, Middleton, WI) with GaAsP photomulitplier tubes (PMTs). Laser power was kept constant at 8 mW using Pockel cells. No bleaching was evident at this laser intensity. The emission dichroic was 580 nm and emission filters 515/30–25 nm. Images were 128x128 pixels with a frame rate at 9.64 Hz.
A Matlab script produced the visual stimulation via a digital micromirror device (DMD, LightCrafter) at 0.125 Hz onto a screen covering the visual field in front of the right eye. A blue led (474/23–25) emitting through a 474/23–25 bandpass filter provided illumination. At the fly’s position, when “ON”, irradiance was measured at 2.5 mW/m2.
Data analysis for Drosophila imaging
Using custom software written in python, regions of interest (ROIs) were segmented in the M8–10 medulla region for Mi1. When testing Tm3, columns were identified in the Lo1 region of the lobula plate. During testing, columns producing the maximum ΔF/F were identified by systematically testing layers until a maximum response was found. For each animal, the response over 2–3 columns was used to measure changes in fluorescence.
Statistics
Two-sided two-sample t-tests (matlab ttest2, default settings) were used to compare brightness and ΔF/F responses across indicators, with individual FOVs as replicates (Fig 1f,j,k, 2c,d, and S8b,d,g). Two-sample Kolmogorov-Smirnov tests (matlab kstest2, default settings) were performed to compare correlations between unmixed 2-channel indicator responses to corresponding null distributions generated by bootstrapping, described above.
Reagent distribution
DNA constructs with jYCaMP1 variants are available at Addgene (plasmids 135420–135424). Fly lines are available through the Bloomington Drosophila Stock Center (Stock # 84970). Sequences for jYCaMP and jYCaMP1s have been deposited in Genbank (MN808514; MN808515). Complete sequences for viral vectors are available from Addgene. Limited quantities of virus particles are available from the corresponding author.
Supplementary Material
Acknowledgments:
We thank H. Davies, C. Morkunas and M. Jeffries for logistical support, S. Di Lisio, K. Ritola, D. Walpita, J. Hasseman, and the GENIE Project Team for experimental support. We thank S. Vishwanathan for the gift of Emx1-Cre mice. This work was funded by the Howard Hughes Medical Institute. M.A.M was supported by the Janelia Graduate Research Fellowship Program. EJM, C-YL, and AA were supported by the Janelia Undergraduate Scholars Program. Work by JC and AA was supported by the National Science Foundation Neuronex Neurotechnology Hub (NEMONIC #1707287) and National Institutes of Health BRAIN Initiative Award (UF1NS107705).
Footnotes
Competing Interests Statement:
The authors declare no competing interests.
Data availability statement:
Example raw datasets and summary statistics are available through Figshare; DOI: 10.25378/janelia.12098361. All other data associated with this study are available from the corresponding author.
Code availability statement:
Custom code is available from the corresponding author, and at github.com/KasparP/twoColorUnmixing and github.com/KasparP/SLAP
References:
- 1.Grienberger C & Konnerth A Neuron 73, 862–885 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Voigt FF et al. Biomed. Opt. Express 8, 3213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kazemipour A et al. Nat. Methods 778–786 (2019). [DOI] [PMC free article] [PubMed]
- 4.Wu J et al. bioRxiv 1–21 (2019).
- 5.Perillo EP et al. Biomed. Opt. Express 7, 324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim DU et al. Opt. Express 20, 12341 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Tilma BW et al. Light Sci. Appl 4, e310–e310 (2015). [Google Scholar]
- 8.Dana H et al. Nat. Methods 649–657 (2019). [DOI] [PubMed]
- 9.Ormö M et al. Science 273, 1392–1395 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Nagai T et al. Nat. Biotechnol 20, 87–90 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Shui B, Kotlikoff MI & Sondermann H Structure 16, 1817–1827 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behnia R, Clark DA, Carter AG, Clandinin TR & Desplan C Nature 427–430 (2014). [DOI] [PMC free article] [PubMed]
- 13.Strother JA et al. Neuron 94, 168–182.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Strother JA, Nern A & Reiser MB Curr. Biol 24, 976–983 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Valm AM et al. Nature 162–167 (2017). [DOI] [PMC free article] [PubMed]
- 16.Dana H et al. Elife 5, e12727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broussard GJ et al. Nat. Neurosci 21, 1272–1280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue M et al. Cell 177, 1346–1360.e24 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Sun Y et al. Soc. Neurosci (2016).
- 20.Podgorski K & Ranganathan GJ Neurophysiol (2016). [DOI] [PMC free article] [PubMed]
Methods only References:
- 1.Studier FW Methods Mol. Biol 1091, 17–32 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Chen T-W et al. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dana H et al. Elife 5, e12727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akerboom J et al. Front. Mol. Neurosci 6, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akerboom J et al. J. Neurosci 32, 13819–13840 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mütze J et al. Biophys. J 102, 934–944 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardill TJ et al. PLoS One 8, e77728 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kazemipour A et al. Nat. Methods 778–786 (2019). [DOI] [PMC free article] [PubMed]
- 9.Gong S et al. Nature 425, 917–925 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.