

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive malignancy characterized by extensive local invasion and systemic spread. In this study, we employed a three-dimensional organoid model of human pancreatic cancer to characterize the molecular alterations critical for invasion. Time lapse microscopy was used to observe invasion in organoids from 25 surgically resected human PDAC samples in collagen I. Subsequent lentiviral modification and small molecule inhibitors were used to investigate the molecular programs underlying invasion in PDAC organoids. When cultured in collagen I, PDAC organoids exhibited two distinct, morphologically defined invasive phenotypes, mesenchymal and collective. Each individual PDAC gave rise to organoids with a predominant phenotype, and PDAC that generated organoids with predominantly mesenchymal invasion showed a worse prognosis. Collective invasion predominated in organoids from cancers with somatic mutations in the driver gene SMAD4 (or its signaling partner TGFBR2). Re-expression of SMAD4 abrogated the collective invasion phenotype in SMAD4-mutant PDAC organoids, indicating that SMAD4 loss is required for collective invasion in PDAC organoids. Surprisingly, invasion in passaged SMAD4-mutant PDAC organoids required exogenous TGFβ, suggesting that invasion in SMAD4-mutant organoids is mediated through non-canonical TGFβ signaling. The Rho-like GTPases RAC1 and CDC42 acted as potential mediators of TGFβ-stimulated invasion in SMAD4-mutant PDAC organoids, as inhibition of these GTPases suppressed collective invasion in our model. These data suggest that PDAC utilizes different invasion programs depending on SMAD4 status, with collective invasion uniquely present in PDAC with SMAD4 loss.
Statement of Significance
Organoid models of PDAC highlight the importance of SMAD4 loss in invasion, demonstrating that invasion programs in SMAD4-mutant and SMAD4-wildtype tumors are different in both morphology and molecular mechanism.
Introduction
Pancreatic cancer, or pancreatic ductal adenocarcinoma (PDAC), is one of the most aggressive human malignancies with a five-year survival of only 8%, and it is predicted to soon be the second leading cause of cancer death in the United States (1, 2). Pancreatic cancer characteristically invades into adjacent organs, into blood vessels and around nerves, and ultimately disseminates throughout the body. Considering the critical importance of invasion in the morbidity and mortality of pancreatic cancer patients, relatively little is known about the specific molecular alterations that initiate and promote this behavior.
Recent comprehensive sequencing studies have characterized the genetic landscape of PDAC, the most common type of pancreatic cancer and the cause of the majority of patient suffering due to this disease (3–8). This landscape is dominated by four critical driver genes that are altered in the majority of pancreatic cancers, the oncogene KRAS and the tumor suppressor genes CDKN2A, TP53, and SMAD4. Analyses of non-invasive precancerous lesions have revealed the timing of these key genetic alterations in pancreatic tumorigenesis. Mutations in SMAD4, which occur in approximately half of PDACs, are the latest occurring of the major driver genes, and recent studies suggest SMAD4 mutations may be limited to invasive cancers and absent from precancerous lesions (9). These genetic data implicate inactivating SMAD4 mutations as a potential driver of invasion in PDAC. SMAD4 encodes a protein that is a critical component of the canonical transforming growth factor beta (TGFβ) signaling pathway (10). However, in addition to the SMAD4-dependent canonical pathway, multiple SMAD4-independent non-canonical pathways have also been described to respond to TGFβ activation, including ERK/MAP kinase signaling, PI3K/AKT signaling, and Rho-like GTPases (11). The determinants of the outcome of TGFβ stimulation via these complex and multifunctional pathways is currently a subject of active investigation, and it is clear that the outcome of TGFβ pathway activation is cell-type and likely context specific.
In vitro assays that interrogate invasion are critical for the empirical investigation of its underlying molecular basis. Although these two-dimensional models have provided a critical foundation (12), three-dimensional studies of invasion provide important advances, as they more closely recapitulate the environment in vivo (13). One such three-dimensional model is the use of organoids derived from fresh human tumors grown in extracellular matrix gels. Analyses of breast cancer organoids demonstrated that multiple different invasive phenotypes can occur and defined the molecular phenotype of a cellular subpopulation critical for invasion in breast cancer organoids (14, 15). Although an organoid model of murine and human pancreatic cancer has been reported and used for valuable studies of tumor pharmacotyping (16–18), this model has not yet been used to investigate the molecular and cellular determinants of invasion in PDAC.
In the present study, we employ an organoid model of fresh human PDAC in order to observe and empirically investigate invasion. We identify inactivation of SMAD4 as a driver of the collective invasion phenotype in our organoid model, and we highlight non-canonical TGFβ signaling as a likely pathway mediating this phenotype.
Materials and Methods
Organoids were isolated from 48 primary human PDAC samples acquired from surgical resection specimens at The Johns Hopkins Hospital – these included 25 PDAC samples (PCO1-PCO25) for immediate invasion analysis and 23 PDAC samples (PCO26-PCO48) for additional analyses, including immunofluorescence, lentiviral transduction, and pharmacological manipulation. This study was approved by the Institutional Review Board of The Johns Hopkins Hospital, and written informed consent was obtained from patients prior to sample acquisition. Organoids from 25 patients were isolated from fresh primary tumor samples as previously described (14). Briefly, PDAC tissue was digested and filtered; organoids were isolated by differential centrifugation. PDAC organoids were embedded into collagen I gels for time lapse imaging using a Nikon Eclipse Ti inverted microscope, with images collected at 30 minute intervals for 3–7 days. Immunofluorescence assays were used to assess expression of CK (1:100, #4545, Cell signaling Technologies), VIM (1:100, #5741, Cell signaling Technologies), SOX9 (1:100, #D8G8H, Cell signaling Technologies), Nkx6.1 (1:100, BDB563022, Fisher scientific), and GATA4 (1:100 #560327, BD Biosciences) in a subset of these organoids using established protocols with secondary antibodies coupled to Alexa Flour 488, 568 or 647 (1:1000, (A11001, A10042, A21235, Invitrogen). Primary PDAC tissue from these cases was analyzed by immunohistochemistry for SMAD4 (clone RBT-SMAD4, predilute; Bio SB), ECAD (clone EP700Y, predilute; Cell Marque), VIM (clone V9, predilute; Roche), P40 (clone BC28, 1:100; Biocare), and GATA6 (R&D Cat. Number AF1700,1:1000) using established protocols. We also isolated genomic DNA from the primary PDACs in our cohort for targeted sequencing of 11 PDAC driver genes using Ion AmpliSeq library preparation with a previously described primer panel (19), followed by sequencing on an Ion Torrent PGM.
For re-expression of SMAD4, the SMAD4 coding sequence was subcloned into pCW57.GFP-P2A-MCS for doxycycline-inducible expression, and lentivirus was produced from HEK-293T cells using the pLKO.1 lentiviral vector protocol. For lentiviral transduction, organoids from 3 patients were passaged in Matrigel and then digested into single cells, transduced, and re-established in Matrigel, followed by puromycin selection three days after transduction. To characterize the invasion of modified organoids, the organoids were transferred from Matrigel to collagen I and treated with doxycycline (5mg/L) for 3 days to induce the expression of SMAD4 followed by TGFβ1 treatment (5ng/mL) for an additional 3 days. Then, 30 organoids from each group were observed for 3–5 days by time-lapse microscopy, and the number of invasive organoids was quantified. Invasive organoids were divided into two groups based on the extent of their invasive protrusions into the surrounding collagen I gel: moderately invasive organoids (invasive protrusions 20–50 um) and extensively invasive organoids (invasive protrusions >50 um). Organoids with no protrusions or protrusions less than 20um were considered non-invasive. Protein expression in the treated organoids was assessed by immunofluorescence (CK, VIM – see details above) and Western blot (SMAD4 [1:50, #sc-7966, Santa Cruz], ECAD [1:1000, #3195, Cell Signaling Technologies], VIM [1:1000, #D21H3, Cell Signaling Technologies], total SMAD2 [1:500, #5339S, Cell Signaling Technologies], p-SMAD2 [1:500, #3108S, Cell signaling Technologies]) using established protocols. Secondary antibodies for Western blot included anti-Mouse IgG-HRP (1:10000, #NA931V, GE Healthcare) and anti-Rabbit IgG-HRP (1:10000, #NA934V, GE Healthcare). Proliferation and apoptosis of modified organoids were assessed by CellTiter-Glo® 3D Cell Viability Assay (Promega) and CellTiter-Glo® caspase 3/7 Assay (Promega) according to manufacturer’s instructions. We also treated passaged unmodified organoids of both SMAD4 genotypes with TGFβ1 (5ng/mL) and inhibitors of RAC1 (20uM, #553508, Calbiochem), CDC42 (50uM, #500503, Calbiochem), and ROCK1 (which inhibits signaling downstream of RHOA) (25uM, #688002, Calbiochem). After transfer from Matrigel to collagen I and treatment with TGFβ1 +/− inhibitors, 30 organoids of each group were observed by time-lapse microscopy for approximately 3 days to assess invasion. We analyzed activity of RAC1 and CDC42 by GTPase pull-down assay (Thermo Scientific) following TGFβ1 and inhibitor treatment.
Additional details are provided in the Supplementary Methods.
Results
Organoids with ductal phenotype were cultured from human PDAC specimens.
In order to explore the molecular mechanisms of PDAC invasion, we derived and cultured organoids from 25 resected human PDAC specimens. All 25 patients in this study had a histologically confirmed diagnosis of PDAC – clinical characteristics are provided in Supplementary Table 1. Organoids were derived from PDAC tissue samples from each case within 2 hours of surgical resection and embedded into collagen I gels. We first sought to confirm the neoplastic origin of our isolated organoids. We immediately fixed and stained embedded organoids from three PDACs with fluorescent-labeled antibodies recognizing cytokeratin (CK), an epithelial marker, and SOX9, a marker of ductal differentiation in the pancreas. In this assay, 44% of PDAC organoids expressed CK, and 100% expressed SOX9 (Figure 1A, Supplementary Figure 1, Supplementary Table 2). We also fixed and stained organoids from normal pancreatic duct using the same CK and SOX9 immunofluorescence assay, which demonstrated consistent CK and SOX9 expression (Supplementary Figure 2). Confocal microscopy of cytokeratin and SOX9 immunofluorescence in PDAC and normal ductal organoids confirmed that the cells within both types of organoids had nuclear SOX9 expression, consistent with pancreatic ductal differentiation (Supplementary Figures 1 and 2). In order to compare this expression pattern with normal and malignant pancreatic tissue, we performed immunofluorescence analysis of CK and SOX9 on one section of normal pancreas (including normal pancreatic duct) and sections from five primary PDACs. In tissue from the normal pancreas, neither acinar cells nor stromal cells expressed SOX9, and SOX9 was strongly expressed in normal ductal epithelial cells (Figure 1B). In all five PDAC tissue sections, SOX9 was expressed in PDAC cells but not in the stromal cells (Figure 1C). We also stained organoids from three PDAC patients with GATA4 (an acinar cell marker) and NKX6.1 (an islet cell marker) - no analyzed organoids expressed GATA4 or NKX6.1. Taken together, these results demonstrate that our isolated organoids were derived from PDAC cells.
Figure 1. Expression of cytokeratin, vimentin, and SOX9 in human PDAC organoids and pancreatic tissue.
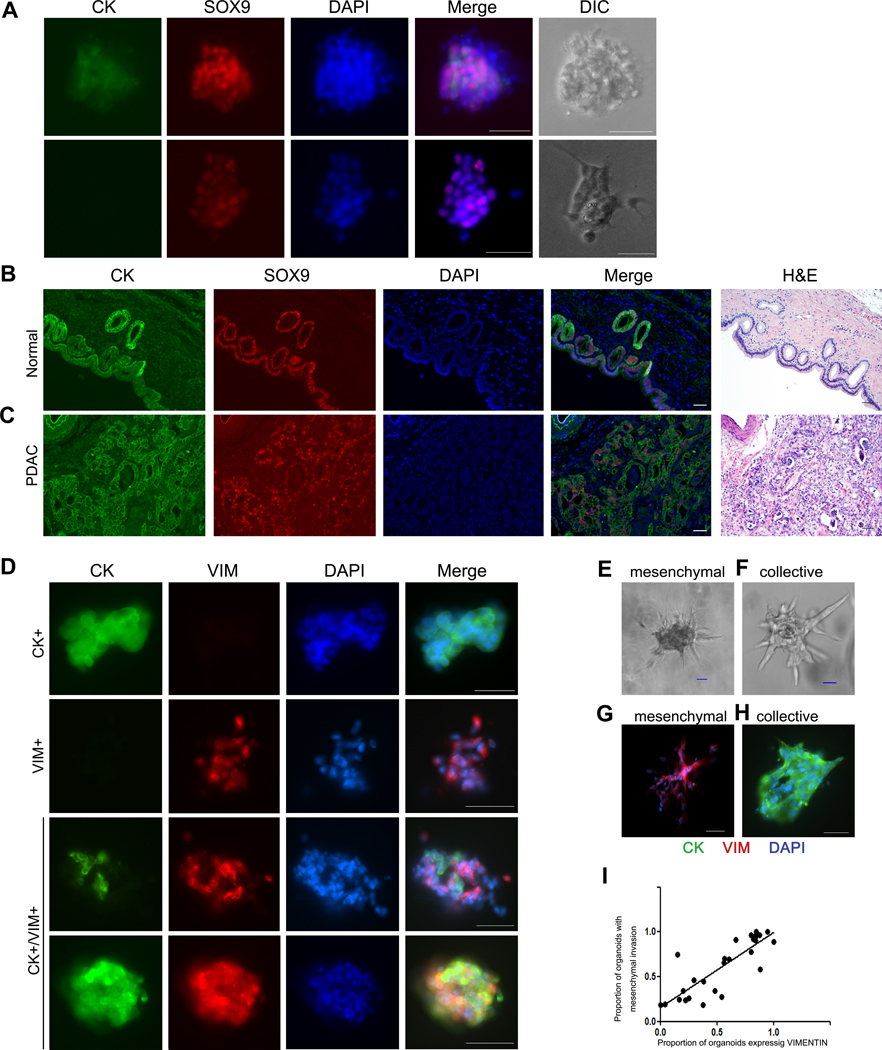
(A) Immunofluorescence co-staining of PDAC organoids for cytokeratin (green) and SOX9 (red) reveals that even cytokeratin-negative organoids express SOX9. Scale bars, 25 um. Images show two representative organoids (rows). (B-C) Immunofluorescence co-staining of normal pancreas and PDAC tissue with cytokeratin (green) and SOX9 (red). Scale bars, 50 um. (B) Representative normal pancreas sample shows SOX9 staining limited to ductal epithelium. (C) Representative PDAC sample shows SOX9 localization in malignant cells, but the stroma remains negative. (D) Immunofluorescence co-staining of PDAC organoids for cytokeratin (green) and vimentin (red) demonstrates variable expression patterns, including organoids expressing each marker alone, as well as organoids expressing both cytokeratin and vimentin. Scale bars, 25 um. (E-F) PDAC organoids invade with different phenotypes in collagen I gels: (E) mesenchymal invasion (F) collective invasion. (G-H) Immunofluorescence co-staining of PDAC organoids for cytokeratin (green) and vimentin (red) after 48 hours in culture in collagen I gels shows that organoids with mesenchymal invasion express vimentin, while those with collective invasion express cytokeratin. (I) Correlation of vimentin expression and mesenchymal invasion in PDAC organoids cultured in collagen I gel.
For 23 PDAC samples in our cohort, selected organoids were fixed and stained with antibodies targeting CK and VIM immediately after embedding into collagen I. As shown in Figure 1D, three expression patterns in the organoid samples were identified: (1) CK+/VIM- organoids, in which >90% of cells expressed CK; (2) CK-/VIM+, in which >90% of cells expressed vimentin; (3) CK+/VIM+. For each PDAC sample, the number of organoids with each of these three expression patterns was counted (Table 1). Intriguingly, the proportions of organoids with these patterns varied widely across the cohort (4.5%−100% for CK+/VIM-, 0%−82% for CK-/VIM+, 0–73% for CK+/VIM+).
Table 1.
PDAC Organoid Invasion and Protein Expression Patterns
| Time Lapse Invasion Analysis | Immunofluorescence Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number (%) of Organoids with Invasion Pattern | Number (%) of Organoids with Expression Pattern | |||||||||
| Case | SMAD4 Mutation Status | SMAD4 IHC Status | Collective | Mesenchymal | Non-invasive | Organoid Invasion Pattern | CK+ | VIM+ | CK+ / VIM+ | Organoid Protein Expression Pattern |
| PCO1 | wild-type | intact | 0 (0) | 45 (83) | 9 (17) | mesenchymal | ND | ND | ND | ND |
| PCO2 | wild-type | lost | 5 (12) | 34 (83) | 2 (5) | mesenchymal | ND | ND | ND | ND |
| PCO3 | frameshift | lost | 21 (72) | 5 (17) | 3 (10) | collective | 24 (96) | 1 (4) | 0 (0) | epithelial |
| PCO4 | deletion | lost | 35 (76) | 8 (17) | 3 (7) | collective | 8 (100) | 0 (0) | 0 (0) | epithelial |
| PCO5 | wild-type | intact | 0 (0) | 15 (52) | 14 (48) | mesenchymal | 1 (5) | 18 (82) | 3 (14) | mesenchymal |
| PCO6 | wild-type | intact | 0 (0) | 48 (89) | 6 (11) | mesenchymal | 2 (15) | 9 (69) | 2 (15) | mesenchymal |
| PCO7 | deletion | lost | 17 (31) | 9 (16) | 29 (53) | collective | 4 (80) | 1 (20) | 0 (0) | epithelial |
| PCO8 | wild-type | intact | 3 (4) | 58 (73) | 18 (23) | mesenchymal | 6 (17) | 11 (31) | 18 (51) | mesenchymal |
| PCO9 | wild-type | lost | 12 (20) | 36 (61) | 11 (19) | mesenchymal | 17 (85) | 3 (15) | 0 (0) | epithelial |
| PCO10 | deletion | lost | 49 (62) | 17 (22) | 13 (16) | collective | 21 (75) | 1 (4) | 6 (21) | epithelial |
| PCO11 | wild-type | intact | 1 (3) | 29 (81) | 6 (17) | mesenchymal | 2 (13) | 12 (75) | 2 (13) | mesenchymal |
| PCO12 | deletion | lost | 3 (5) | 28 (50) | 25 (45) | mesenchymal | 4 (16) | 18 (72) | 3 (12) | mesenchymal |
| PCO13 | wild-type | lost | 1 (2) | 25 (51) | 23 (47) | mesenchymal | 2 (20) | 7 (70) | 1 (10) | mesenchymal |
| PCO14 | deletion | lost | 9 (20) | 31 (67) | 6 (13) | mesenchymal | 6 (20) | 18 (60) | 6 (20) | mesenchymal |
| PCO15 | wild-type | intact | 10 (12) | 24 (28) | 52 (60) | mesenchymal | 13 (43) | 0 (0) | 17 (57) | mesenchymal |
| PCO16 | wild-type* | intact | 19 (38) | 5 (10) | 26 (52) | collective | 14 (78) | 4 (22) | 0 (0) | epithelial |
| PCO17 | wild-type | intact | 1 (4) | 11 (44) | 13 (52) | mesenchymal | 2 (18) | 1 (9) | 8 (73) | mesenchymal |
| PCO18 | missense | intact | 25 (51) | 8 (16) | 16 (33) | collective | 25 (83) | 1 (3) | 4 (13) | epithelial |
| PCO19 | wild-type | intact | 19 (24) | 37 (47) | 23 (29) | mesenchymal | 11 (44) | 11 (44) | 3 (12) | mesenchymal |
| PCO20 | wild-type | intact | 1 (2) | 28 (61) | 17 (37) | mesenchymal | 2 (14) | 10 (71) | 2 (14) | mesenchymal |
| PCO21 | wild-type | lost | 15 (31) | 23 (48) | 10 (21) | mesenchymal | 8 (31) | 14 (54) | 4 (15) | mesenchymal |
| PCO22 | deletion | lost | 31 (53) | 25 (42) | 3 (5) | collective | 18 (62) | 10 (34) | 1 (3) | epithelial |
| PCO23 | deletion | lost | 26 (43) | 10 (17) | 24 (40) | collective | 13 (54) | 8 (33) | 3 (13) | epithelial |
| PCO24 | wild-type | intact | 0 (0) | 62 (78) | 18 (23) | mesenchymal | 3 (11) | 11 (39) | 14 (50) | mesenchymal |
| PCO25 | wild-type | intact | 3 (8) | 32 (80) | 5 (13) | mesenchymal | 2 (33) | 4 (67) | 0 (0) | mesenchymal |
Although wild-type for SMAD4, a mutation was identified in TGFBR2.
PDAC organoids exhibit two distinct patterns of invasion into collagen I.
In order to investigate the invasion patterns of the organoids derived from the 25 PDACs in our cohort, organoids from each sample were cultured in collagen I gels and observed by time-lapse microscopy. We noted multiple patterns of invasion, as has been previously described in breast cancer organoids (14). In some organoids, single cells with an elongated spindle morphology disseminated into the surrounding collagen matrix (Figure 1E, Supplementary Videos 1–2). In addition, single cells also disseminated from the organoids with an amoeboid morphology, in which rounded cells appeared to roll or squeeze through the collagen I matrix (Supplementary Video 3). Intriguingly, cells with these mesenchymal and amoeboid morphologies often disseminated from the same organoids and thus we grouped them together as “mesenchymal invasion” in subsequent analyses. In addition to these single-cell invasion morphologies, we also identified some organoids in which cells invaded into the collagen matrix as cohesive multicellular units, which has previously been described as “collective invasion” (Figure 1F, Supplementary Video 4) (14). The number of organoids with mesenchymal and collective invasion patterns were quantified for each PDAC in our cohort by review of the time-lapse videos (Table 1). Organoids from each tumor exhibited a predominant invasive phenotype, with 8 tumors generating organoids with predominantly collective invasion and 17 tumors generating organoids with predominantly mesenchymal invasion. Pearson correlation analysis revealed PDACs with a large proportion of organoids expressing VIM (including CK-/VIM+ and CK+/VIM+) also had a large proportion of organoids with mesenchymal invasion (p<0.001) (Figure 1I). We also performed immunofluorescence analysis of CK and VIM expression in the organoids after 48 hours cultured in Collagen I (n=4). As shown in Figure 1G and 1H, we identified invasion in both CK+/VIM- organoids and CK-/VIM+, indicating that both expression phenotypes undergo invasion in our model. Moreover, when we assessed CK and VIM expression in organoids from one PDAC after culture in collagen for 10 days, we saw expression of vimentin was more prevalent in organoids with mesenchymal invasion compared to organoids with collective invasion from the same tumor (Supplementary Figure 3). We also confirmed the neoplastic origin of both collective and mesenchymal organoids using KRAS pyrosequencing. In organoids derived from four primary PDACs with KRAS codon 12 hotspot mutations (including PCO46 with collective invasion and PCO44, PCO47, and PCO48 with mesenchymal invasion), high frequency mutations were identified in all unpassaged organoid samples, indicating a neoplastic cellularity of 80–100%. A fifth PDAC (PCO45) was assessed but excluded from this analysis due to wildtype KRAS in both organoids and primary tumor tissue. In keeping with previous reports, we also found that PDAC organoids cultured in Matrigel were less invasive than those from the same patient cultured in collagen I (Supplementary Figure 4) (14).
Invasion pattern of PDAC organoids is associated with clinical outcome and protein expression in the primary tumor.
We next sought to determine whether the invasion pattern observed in our organoid model was correlated with clinical or pathological features of the resected tumors. Importantly, we found that the patterns of invasion did not differ based on neoadjuvant therapy (Figure 2A, p=0.69, t test). There was also no significant difference in grade of differentiation between PDACs giving rise to organoids with predominantly mesenchymal or collective invasion. Specifically, poorly differentiated PDACs accounted for 50% of the PDACs with predominantly mesenchymal invasion and 58% of the PDACs with predominantly collective invasion (p=1.00, chi-square test). In addition, it is important to note that a sizable proportion of PDACs giving rise to mesenchymal organoids were moderately differentiated primary tumors. To analyze primary tumor morphology in depth, histological sections of each primary PDAC were reviewed by a pancreatic pathologist, who estimated the percentage of the tumor area with glandular differentiation (representative images in Figure 2B). We found that all of the tumors analyzed, even those diagnosed as poorly differentiated, had a significant component with glandular morphology (i.e. groups of malignant cells adherent to each other and organized around a lumen). In 24 of 25 primary PDACs in our cohort, the majority of tumor cells had a glandular architecture (Table 2). The percent of the primary tumor with glandular differentiation was comparable between the PDACs that gave rise to mesenchymal and collective organoids (mean of approximately 80% in both groups) (Table 2). There were no statistically significant differences in other pathological features of the primary tumor between the two groups – tumor size, lymph node metastasis, lymphovascular invasion, perineural invasion, and invasion of nearby structures (duodenum, common bile duct) were similar between the two groups. Still, as shown in Figure 2D, we observed a striking association between organoid invasion pattern and recurrence/death. The mesenchymal pattern conferred a poorer outcome (log-rank test, p<0.0001) and was associated with a significantly higher risk of recurrence/death after adjusting for potential clinical and pathological covariates in a multivariate analysis (HR=15.277; 95%CI: 2.000–116.689; p=0.009).
Figure 2. Pattern of invasion is associated with clinical outcome and SMAD4 mutation.
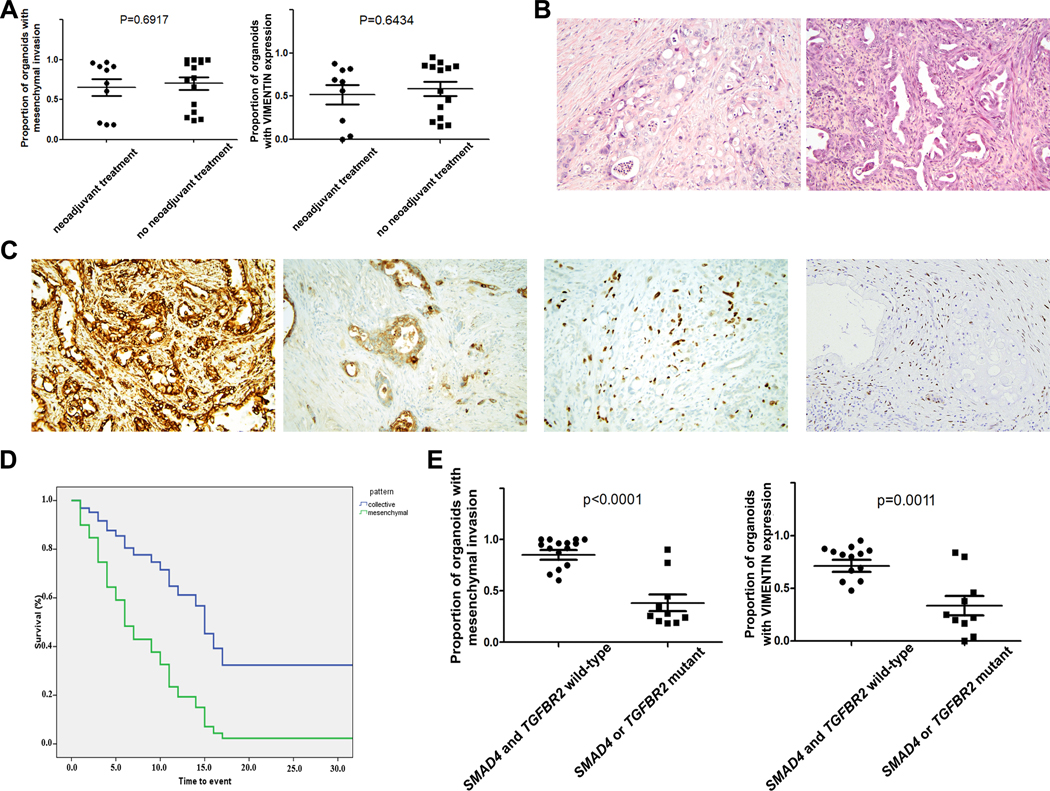
(A) There was no difference in pattern of invasion or VIM expression between patients who did or did not receive neoadjuvant chemotherapy prior to surgical resection. (B) Representative images of primary tumor morphology (H&E) showing predominantly glandular architecture: PCO18 (left) had predominantly collective invasion in our organoid model, while PCO5 (right) had predominantly mesenchymal invasion. (C) Multiple features of mesenchymal differentiation can be identified by immunohistochemistry in primary PDACs that give rise to mesenchymally invading organoids, including VIM expression (far left), ECAD loss (middle left), P40 expression (middle right), and GATA6 loss (far right). (D) Kaplan-Meier analysis of 25 resected PDAC patients shows increased risk of death associated with mesenchymal invasion (green) compared to collective invasion (blue) in our organoid culture model (HR=15.277; 95%CI: 2.000–116.689; p=0.009). (F) Collective invasion and lack of VIM expression in our organoid model were strongly associated with mutation in SMAD4 or TGFBR2 (p<0.001 and p=0.0011, t test).
Table 2.
Primary Tumor Features in PDAC Organoid Cohort
| Primary Tumor Morphological Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Growth Pattern | Expression by IHC | ||||||||
| SMAD4 Mutation Status | Organoid Invasion Pattern | Glandular (%) | Single Cell (%) | ECAD (%) | VIM (%) | P40 | GATA6 | Mesenchymal Score | |
| PCO1 | wild-type | mesenchymal | 80 | 20 | 100 | 0 | negative | high | 0 |
| PCO2 | wild-type | mesenchymal | 70 | 30 | 100 | 0 | focal | low | 2 |
| PCO3 | frameshift | collective | 90 | 10 | 100 | 0 | focal | moderate | 1 |
| PCO4 | deletion | collective | 70 | 30 | 100 | 0 | focal | high | 1 |
| PCO5 | wild-type | mesenchymal | 90 | 10 | 100 | 70 | focal | low | 3 |
| PCO6 | wild-type | mesenchymal | 90 | 10 | 100 | 10 | negative | low | 2 |
| PCO7 | deletion | collective | 80 | 20 | 100 | 0 | negative | high | 0 |
| PCO8 | wild-type | mesenchymal | 90 | 10 | 100 | 0 | negative | high | 0 |
| PCO9 | wild-type | mesenchymal | 100 | 0 | 100 | 0 | negative | high | 0 |
| PCO10 | deletion | collective | 80 | 20 | 100 | 0 | negative | high | 0 |
| PCO11 | wild-type | mesenchymal | 70 | 30 | 100 | 0 | focal | high | 1 |
| PCO12 | deletion | mesenchymal | 90 | 10 | 100 | 0 | focal | high | 1 |
| PCO13 | wild-type | mesenchymal | 90 | 10 | 100 | 0 | focal | high | 1 |
| PCO14 | deletion | mesenchymal | 70 | 30 | 90 | 0 | focal | high | 2 |
| PCO15 | wild-type | mesenchymal | 90 | 10 | 90 | 0 | negative | high | 1 |
| PCO16 | wild-type* | collective | 90 | 10 | 100 | 0 | negative | high | 0 |
| PCO17 | wild-type | mesenchymal | 100 | 0 | 100 | 0 | negative | high | 0 |
| PCO18 | missense | collective | 80 | 20 | 100 | 0 | negative | moderate | 0 |
| PCO19 | wild-type | mesenchymal | 60 | 40 | 90 | 10 | negative | high | 2 |
| PCO20 | wild-type | mesenchymal | 40 | 60 | 100 | 0 | negative | low | 1 |
| PCO21 | wild-type | mesenchymal | 90 | 10 | 80 | 0 | negative | low | 2 |
| PCO22 | deletion | collective | 50 | 50 | 90 | 0 | negative | high | 1 |
| PCO23 | deletion | collective | 90 | 10 | 100 | 0 | negative | moderate | 0 |
| PCO24 | wild-type | mesenchymal | 50 | 50 | 100 | 10 | focal | high | 2 |
| PCO25 | wild-type | mesenchymal | 90 | 10 | 90 | 0 | focal | high | 2 |
| p-value+ | - | 0.58 | 0.62 | 0.27 | 0.40 | 0.14 | 0.020 | ||
Although wild-type for SMAD4, a mutation was identified in TGFBR2. +p value comparing PDACs giving rise to predominantly collective vs mesenchymal organoids, calculated using Mann-Whitney U Test (growth pattern, mesenchymal score) or Fisher’s exact test (all others).
Considering the prominent expression of VIM in a subset of our PDAC organoids, we next analyzed VIM expression in primary PDAC tissue from each patient (Table 2). We identified VIM expression in only 4 of 25 primary PDACs, and in all but one tumor VIM expression was focal, involving only 10% of primary tumor cells (Figure 2C). The primary PDACs that expressed VIM all gave rise to predominantly mesenchymal organoids which expressed also VIM, demonstrating that at least in some cases the mesenchymal phenotype is present in the primary tumor at the level of protein expression. In addition, we analyzed expression of E-cadherin (ECAD) in each primary PDAC (Table 2). We identified focal loss of membranous ECAD expression in 6 of 25 primary PDACs, in each case involving <20% of tumor cells (Figure 2C). Most of the cases with focal ECAD loss did not express VIM, suggesting that these two potential markers of mesenchymal phenotype vary independently, and the PDACs with ECAD loss gave rise to organoids with predominantly mesenchymal invasion in all but one case. However, the differences in vimentin expression and ECAD loss between primary PDACs giving rise to predominantly mesenchymal versus collective organoids were not statistically significant (p=0.27 for VIM p=0.62 for ECAD, Fishers exact test). In addition, the majority of tumors generating predominantly mesenchymal organoids had no VIM expression or ECAD loss in the primary PDAC. The striking difference in clinical outcome in the mesenchymal invasion group suggests that our organoid model is capturing clinically relevant biological differences that are not evident through morphological analysis of the primary tumor alone. When ECAD and VIM expression in the primary tumor were added to the multivariate analysis, the impact of mesenchymal invasion on outcome remained statistically significant (HR=3.34; 95%CI: 1.21–9.28; p=0.02).
We next considered our primary tumors in light of the recently described transcriptional subtypes of PDAC into classical and basal/quasi-mesenchymal/squamous subtypes (20, 21). We analyzed the primary PDAC samples using immunohistochemistry for two markers previously correlated with transcriptional subtype: GATA6, which has been previously shown to have high expression in classical PDACs, and P40, a marker of squamous differentiation (Table 2) (21, 22). Ten PDACs focally expressed P40, including two with predominantly collective invasion and eight with predominantly mesenchymal invasion (Figure 2C). All PDACs showed some expression of GATA6, but all of the PDACs with low GATA6 expression gave rise to predominantly mesenchymal organoids (Figure 2C). Taken together, these data show that P40 expression and loss of GATA6 are both enriched in primary PDACs giving rise to organoids with predominantly mesenchymal invasion, though neither marker had a statistically significant difference between primary PDACs giving rise to predominantly mesenchymal versus collective organoids (p=0.40 for P40 and p=0.14 for GATA6, Fishers exact test). Intriguingly, these markers seem to vary independently of ECAD loss and VIM expression, highlighting multiple distinct primary tumor features that can suggest the mesenchymal phenotype which is more clearly evident in our organoid model. Finally, we developed a score with 1 point for each primary tumor mesenchymal feature (VIM expression, ECAD loss, P40 expression, low GATA6 expression) - the scores for primary PDACs giving rise to mesenchymal organoids were significantly higher than for those giving rise to collective organoids (mean 1.3 vs 0.4, p=0.020, Mann-Whitney U test).
Somatic mutations in SMAD4 or TGFBR2 are identified in PDACs with collective invasion.
We next assessed the possible correlation of invasive phenotype in our organoid model with somatic mutations in PDAC driver genes using deep targeted next generation sequencing (Supplementary Table 3). As expected, KRAS was the most commonly mutated gene in our PDAC cohort, with somatic mutations in 22 of 24 tumors. Somatic mutations in TP53 were identified in 14 of 24 PDACs, and CDKN2A was altered in 6 PDACs (Supplementary Table 4). Mutations in SMAD4, including both point mutations and deletions, were identified in 9 PDACs, and a single PDAC had a mutation in its signaling partner TGFBR2 (Supplementary Table 4). In addition, to analyze the SMAD4 mutation status of one PDAC sample which failed sequencing, we performed immunohistochemistry for SMAD4 protein expression on an FFPE section, demonstrating retained expression of SMAD4 indicative of wild-type status (23). Considering all PDACs in our cohort, all (8 of 8) PDACs which gave rise to organoids with predominantly collective invasion had somatic mutations in SMAD4 or the closely related gene TGFBR2, while only 2 of 17 PDACs which gave rise to organoids with predominantly mesenchymal invasion had SMAD4 mutations (Figure 2E, p<0.0001, t test; Table 1). No other mutated genes were associated with the invasive phenotype in our organoid model. As expected due to the correlation of SMAD4 mutation with collective invasion, lack of SMAD4 mutation was associated with a significantly poorer outcome, and the risk of poorer outcome persisted in multivariate analysis (HR=35.72; 95%CI: 3.29–387.33; p=0.003; Supplementary Figure 5).
Because characterization of deletions as heterozygous or homozygous in impure primary tumor samples can be difficult, we also assessed SMAD4 expression by immunohistochemistry, as loss of SMAD4 expression this assay has been previously shown to correlate closely with SMAD4 mutation (Table 1) (23). In the 2 cases with intact SMAD4 expression and predominantly collective invasion, we see clear explanations for these results – a missense SMAD4 mutation (which likely does not affect expression) and a mutation in TGFBR2 in the setting of wildtype SMAD4. In the 2 cases with SMAD4 deletions identified by sequencing but predominantly mesenchymal invasion, we see loss of SMAD4 expression by immunohistochemistry, consistent with homozygous deletion. Of note, we also see multiple primary tumors with loss of SMAD4 expression but predominantly mesenchymal invasion and no identification of SMAD4 mutation by sequencing, highlighting that this immunohistochemical assay is a less reliable correlate of organoid invasive phenotype than SMAD4 mutation at the DNA level.
The two PDACs with SMAD4 deletion but mesenchymal invasion are intriguingly outliers that might provide some insights into the link between these two features. With respect to clinical and pathological features, they did not stand out from the rest of the cohort. The primary tumors from both outlier cases showed focal P40 expression, and one had focal ECAD loss, suggesting some mesenchymal features in the primary tumors that are recapitulated in the organoid model.
Re-expression of SMAD4 can alter the invasion pattern of SMAD4-mutant PDAC organoids.
The previous results show a striking correlation between SMAD4 mutation and collective invasion in our PDAC organoid model. We next tested for a causal role of SMAD4 mutation in the promotion of collective invasion by re-expressing SMAD4 in SMAD4-mutant organoids and analyzing the invasive phenotype. In order to perform lentiviral transduction of our primary PDAC organoids, we cultured SMAD4-mutant PDAC organoids from three patients in Matrigel. After multiple passages in Matrigel with an enriched organoid media, the PDAC organoids grew as cohesive cystic structures, which is consistent with previous results (Figure 3A) (24). Consistent with previous studies in breast cancer, we did not observe any invasion into the Matrigel in these experiments (14). To investigate the role of SMAD4 in pancreatic cancer invasion, we used a tet-on inducible SMAD4 coding sequence; lentiviral transduction and puromycin selection did not change the morphology of the organoids in Matrigel (Figure 3B). To characterize the invasion of modified organoids, the organoids were transferred to collagen I, treated with doxycycline (to induce the expression of SMAD4) followed by TGFβ1, and observed by time-lapse microscopy. As shown in Figure 3C and 3D, most organoids were non-invasive in the group not treated with doxycycline or TGFβ1 (Supplementary Video 5). In the group treated with doxycycline to re-express SMAD4 without TGFβ1 (Dox+ TGFβ1-), the majority of organoids were still non-invasive (Supplementary Video 6). This result suggests that passaged organoids are minimally invasive without TGFβ1 stimulation, even after re-expression of SMAD4. When treated with TGFβ1 alone (Dox- TGFβ1+), more than 70% organoids invaded into the collagen I, almost exclusively with the collective invasion pattern (Supplementary Video 7). In the group treated with both doxycycline to re-express SMAD4 and TGFβ (Dox+ TGFβ1+), all of the organoids were extensively invasive with a mesenchymal pattern (Figure 3D, Supplementary Video 8).
Figure 3. Re-expression of SMAD4 leads to mesenchymal invasion in presence of TGFβ.

(A-B) Representative images showing morphology of unmodified (A) and lentivirally transduced (B) SMAD4-mutant PDAC organoids cultured in Matrigel. (C) Representative images of modified SMAD4-mutant PDAC organoids after treatment with doxycycline (Dox) and/or TGFβ and culture in collagen I. (D) Extent of invasion of modified SMAD4-mutant PDAC organoids after treatment with doxycycline (Dox) and/or TGFβ while cultured in collagen I. PCO26, PCO27, and PCO28 represent the organoids derived from 3 independent SMAD4-mutant PDACs. (E) Immunofluorescence co-staining of modified SMAD4-mutant PDAC organoids after treatment with doxycycline (Dox) and/or TGFβ while cultured in collagen I: cytokeratin (purple) and vimentin (red). (F) Expression of epithelial and mesenchymal markers in modified SMAD4-mutant PDAC organoids after treatment with doxycycline (Dox) and/or TGFβ as assayed by Western blot. (G-H) Effect of doxycycline (Dox) and/or TGFβ treatment on proliferation and apoptosis in modified SMAD4-mutant PDAC organoids: (G) proliferation as assessed by Celltiter assay. (H) apoptosis as assessed by caspase 3/7 activity. PCO26, PCO27, and PCO28 represent the organoids derived from 3 independent SMAD4-mutant PDACs.
In order to better characterize the epithelial vs mesenchymal phenotype of the modified organoids, we performed immunofluorescence analysis for CK and VIM. As shown in Figure 3E, Dox- TGFβ-, Dox+ TGFβ-, and Dox- TGFβ+ organoids expressed the epithelial marker CK and did not express the mesenchymal marker VIM. However, when treated with both Dox and TGFβ1 (Dox+ TGFβ+), the organoids lost CK expression and gained VIM expression, indicating that TGFβ1 stimulation and intact SMAD4 expression are required for the acquisition of mesenchymal features. The expression of key mesenchymal and canonical TGFβ pathway proteins was also measured by Western blot. As shown in Figure 3F, doxycycline treatment alone (Dox+ TGFβ-) significantly increased the expression of SMAD4 in modified organoids. However, the total expression of E-cadherin, VIM, and SMAD2 were not altered by the re-expression of SMAD4, nor was the phosphorylation of SMAD2. When the organoids were treated with TGFβ1 only (Dox- TGFβ+), phosphorylation of SMAD2 increased, but the total expression of E-cadherin, VIM, and SMAD2 were not altered. After treatment with both doxycycline (to restore SMAD4 expression) and TGFβ1 (Dox+ TGFβ+), phosphorylation of SMAD2 increased, while E-cadherin expression decreased and VIM expression increased dramatically. These data indicate that both SMAD4 expression and TGFβ1 stimulation are required for mesenchymal invasion in PDAC organoids, implicating canonical TGFβ signaling in this process. These data further suggest that the elimination of canonical TGFβ signaling by SMAD4 inactivation blocks mesenchymal invasion and enables collective invasion in response to TGFβ1. The requirement for TGFβ1 stimulation for invasion in SMAD4-mutant organoids raises the possibility that non-canonical TGFβ signaling may be a critical and specific mediator of collective invasion.
In these experiments, we used previously described media for expansion and passage of our PDAC organoids in Matrigel prior to lentiviral transduction (16). This media contains the TGFβ inhibitors A83–01 and mNoggin, which could influence the TGFβ dependence we observed for invasion of SMAD4-mutant organoids. Thus, we next sought to test whether the dependence on exogenous TGFβ stimulation for invasion was the result of culture with these TGFβ inhibitors or a feature of passaged organoids independent of TGFβ inhibition. For six primary PDAC samples, we isolated organoids for culture in two distinct media, culturing half of the organoids in the previously described media and half in this media without A83–01 or mNoggin. SMAD4 status was assessed by immunohistochemistry of the primary tumor tissue. Two cases (PCO29 and PCO33) showed loss of SMAD4 expression indicative of mutation, while three cases (PCO32, PCO34, and PCO38) showed retained SMAD4 expression indicative of wildtype SMAD4. The final PDAC (PCO35) showed a mixed pattern, with focal SMAD4 loss. For the organoids derived from SMAD4-mutant PDACs, TGFβ stimulation resulted in a comparable increase in invasion in organoids cultured with both types of media (Supplementary Figure 6). Intriguingly, we saw little if any enhanced invasion after TGFβ treatment in the organoids derived from SMAD4-wildtype or mixed PDACs established in either media, suggesting that SMAD4 genotype influences the TGFβ-dependence of organoid invasion. Thus, the requirement for exogenous TGFβ stimulation after SMAD4-mutant organoid passaging is not a result of pathway inhibitors but rather a phenotypic shift with passaging in culture. In addition, the enhanced invasion in response to TGFβ1 treatment is a specific feature of SMAD4-mutant PDACs that is not shared by SMAD4-wildtype organoids.
Previous studies have demonstrated that in addition to its effects on cell phenotype, TGFβ can also induce apoptosis and cell cycle arrest (25). We next assessed whether these actions of TGFβ signaling are dependent on SMAD4 in our system. The proliferation and apoptosis of the modified organoids in collagen I (+/− doxycycline and TGFβ1) were assessed each day after TGFβ1 treatment. As shown in Figure 3G and 3H, the proliferation of Dox+ TGFβ1+ organoids of was significantly lower than that of Dox- TGF-β1+ organoids. In addition, the Dox+ TGFβ1+ group had a significantly higher rate of apoptosis than the Dox- TGFβ1+ organoids. These results indicate that SMAD4 expression in the presence of TGFβ1 inhibits proliferation and induces apoptosis.
RAC1 and CDC42 activation is required for TGFβ1-induced collective invasion.
The dependence of collective invasion on TGFβ1 stimulation in our SMAD4-mutant organoids raises the possibility that this process is mediated by non-canonical TGFβ signaling. Multiple downstream pathways have been proposed in non-canonical TGFβ signaling, including MAPK, PI3K, and Rho-like GTPases (11). Interestingly, members of the Rho family of small Rho GTPases (including RAC1, RHOA, and CDC42) have been recognized as regulators of cell migration through their actions on actin polymerization, actomyosin contractility, and microtubules (26, 27). As such, we hypothesized that these GTPases might be activated by non-canonical TGFβ signaling, leading to collective invasion in SMAD4-mutant organoids. In unmodified SMAD4-mutant organoids that were passaged in Matrigel and then transferred to collagen I, we found that collective invasion was significantly induced when the organoids were treated with TGFβ1 (Figure 4A). To investigate the role of RAC1, RHOA, and CDC42 in TGFβ1-induced collective invasion, SMAD4-mutant organoids from one patient were treated with TGFβ1 and inhibitors of RAC1, CDC42, and ROCK1 (which inhibits signaling downstream of RHOA). As shown in Figure 4A, treatment with either RAC1 inhibitor or CDC42 inhibitor abrogated TGFβ1-induced collective invasion. In contrast, treatment with a ROCK1 inhibitor (which inhibits signaling downstream of RHOA) led to drastic alterations in organoid morphology, with many bubble-like structures and cell protrusions. Invasion decreased with increasing concentrations of RAC1 and CDC42 inhibitors (Figures 4B and 4C). RAC1 and CDC42 GST pull down assays demonstrated that TGFβ1 treatment led to increased active RAC1 and CDC42 in these SMAD4-mutant organoids, and the specific inhibitors utilized could repress the activity of RAC1 and CDC42 (Figure 4D). These results suggest that RAC1 and CDC42 are activated by TGFβ1 and are required for the induction of collective invasion in SMAD4-mutant organoids.
Figure 4. RAC1 and CDC42 activation is required for TGFβ-induced collective invasion in SMAD4-mutant PDAC organoids.

(A) Representative images of organoids from a SMAD4-mutant PDAC cultured in collagen I and treated with TGFβ and/or RAC1, ROCK1, or CDC42 inhibitors. (B-C) Relative invasion of organoids from a SMAD4-mutant PDAC treated with different doses of RAC1 inhibitor (B) or CDC42 inhibitor (C). (D) Inhibition of RAC1 and CDC42 in PDAC organoids was confirmed by active GTPase pulldown followed by Western blot. (E) Representative images of organoids from additional SMAD4-wildtype (top) and SMAD4-mutant (bottom) PDACs cultured in collagen I and treated with TGFβ and/or RAC1 or CDC42 inhibitors. We identified a single SMAD4-wildtype PDAC (PCO36) with collective invasion in our organoid model (middle panels). Only SMAD4-mutant organoids decreased invasion after treatment with RAC1 or CDC42 inhibitors. (F) Impact of TGFβ and/or RAC1 or CDC42 inhibition on invasion in organoids with mesenchymal invasion. PDAC organoids with mesenchymal invasion (all SMAD4-wildtype) had little to no decrease in invasion after RAC1/CDC42 inhibition. (G) Impact of TGFβ and/or RAC1 or CDC42 inhibition on invasion in organoids with collective invasion. All organoids with collective invasion showed increased invasion after TGFβ treatment, but only those with SMAD4 mutations had decreased invasion after RAC1/CDC42 inhibition. *PCO36 is SMAD4-wildtype, while PCO29 and PCO37 are SMAD4-mutant. (H) Phalloidin labeling of F-actin in PDAC organoids in collagen I gels after treatment with TGFβ and/or RAC1 or CDC42 inhibitors.
We then sought to determine the effect of RAC1 and CDC42 inhibition on TGFβ-stimulated invasion in additional PDAC organoids of both SMAD4 genotypes, as assessed by immunohistochemistry. We cultured unmodified low-passage SMAD4-mutant and SMAD4-wildtype organoids in collagen I and treated with TGFβ1 +/− RAC1 or CDC42 inhibitors, followed by quantification of invasion by time lapse microscopy for 2–3 days. In two SMAD4-mutant PDACs (PCO29, PCO37), we saw collective invasion in response to TGFβ1 treatment which was abrogated by RAC1 and CDC42 inhibition (Figure 4E and 4G). These results are in keeping with the results of the initial SMAD4-mutant PDAC tested above. In three SMAD4-wildtype PDACs (PCO32, PCO38, PCO40), the organoids invaded with a mesenchymal pattern with little or no enhancement of invasion with TGFβ1 treatment and minimal change in invasion after treatment with RAC1 and CDC42 inhibitors (Figure 4E and 4F). The final assessed case (PCO36) showed an unexpected pattern, with striking collective invasion that was enhanced by TGFβ1 treatment but not decreased by RAC1 and CDC42 inhibition (Figure 4E and 4G). The primary tumor retained SMAD4 expression by immunohistochemistry, suggestive of wildtype status. Due to the unexpected collective invasion pattern in these organoids, we performed targeted sequencing of unpassaged organoids from this tumor, which revealed mutations in KRAS (p.G12V), CDKN2A (p.R80X), and TP53 (p.L308fs), but no mutations in SMAD4 or TGFBR2. Importantly, the KRAS mutant allele frequency in this sample was high (~45%), confirming the purity of the malignant cells in our organoid cultures. These results confirm the importance of non-canonical TGFβ signaling via RAC1 and CDC42 for collective invasion in SMAD4-mutant organoids. In addition, the lack of invasion in response to TGFβ1 in most SMAD4-wildtype organoids raises the possibility of a TGFβ-independent invasion mechanism in the setting of wildtype SMAD4. Intriguingly, these results are in contrast to the marked increase in mesenchymal invasion in SMAD4-mutant organoids after re-introduction of SMAD4, suggesting that TGFβ signaling is altered by other molecular mechanisms in PDACs that are natively SMAD4 wildtype. We also identify a SMAD4-wildtype PDAC with collective invasion that is enhanced by TGFβ1 treatment but not dependent on RAC1 and CDC42, suggesting an alternative as-yet-unidentified molecular mechanism allowing collective invasion that is not dependent on these GTPases in a small subset of SMAD4-wildtype PDACs.
Finally, we examined the effect of these GTPase inhibitors on actin polymerization in SMAD4-mutant PDACs. SMAD4-mutant organoids were stained with Alexa 488 labeled Phalloidin after they were treated with TGFβ1 and RAC1 and CDC42 inhibitors. As shown in Figure 4H, there were few large F-actin fibers evident in untreated organoids. After treatment with TGFβ1, we observed enrichment of F-actin specifically in the invading cells (Figure 4H). The invasion and actin polymerization were abrogated in the organoids treated with TGFβ1 and RAC1 or CDC42 inhibitor. Taken together, these data show that RAC1 and CDC42 activation is required for TGFβ1-induced collective invasion in SMAD4-mutant PDAC organoids, highlighting this aspect of non-canonical TGFβ signaling as specifically important in PDACs with this genotype.
Discussion
Taken together, our results suggest that SMAD4 inactivation enables a collective invasion program in PDAC organoids, while organoids with wild-type SMAD4 invade with a mesenchymal phenotype. SMAD4 encodes a critical component of the TGFβ signaling pathway (10). Previous work has highlighted the importance of TGFβ signaling in the acquisition of mesenchymal features in epithelial cancers (epithelial-to-mesenchymal transition or EMT), and previous analyses of murine pancreatic cancers have demonstrated abrogation of the mesenchymal phenotype with SMAD4 inactivation (28). Previous studies also suggest that in addition to stimulating EMT, TGFβ signaling can decrease proliferation and induce apoptosis in pancreatic cancer cells (28, 29). Our results are consistent with these previous studies, in that restoration of SMAD4 expression in our pancreatic cancer organoids resulted in decreased proliferation, increased apoptosis, and a switch to mesenchymal invasion upon TGFβ stimulation. However, an important caveat of our SMAD4 re-expression experiments is that expression was not under the control of the native promoter, and thus SMAD4 is likely expressed at non-physiological levels in this system. Still, our data suggest that in addition to abrogating the TGFβ-stimulated phenotypes proliferation arrest and apoptosis, loss of SMAD4 also activates a novel collective invasion strategy that is dependent on TGFβ stimulation. These observations suggest that SMAD4 inactivation provides multiple advantages to enable cancer cell survival in addition to promoting a unique invasion program. They also suggest that rather than being a universal feature of cancer cell invasion, the presence of EMT may be genotype-specific.
After passaging in culture, we found that some PDAC organoids required stimulation with exogenous TGFβ1 to invade into collagen I gels. Surprisingly, this requirement for TGFβ1 occurred uniquely in SMAD4-mutant organoids, while most SMAD4-wildtype organoids did not increase their invasion in response to TGFβ1 in the conditions used in this study. In addition to the SMAD4-dependent canonical pathway, multiple SMAD4-independent non-canonical pathways have also been described to respond to TGFβ activation, including ERK/MAP kinase signaling, PI3K/AKT signaling, and Rho-like GTPases (11). Because SMAD4 inactivation abrogates canonical TGFβ signaling, we hypothesized that non-canonical TGFβ signaling plays a role in invasion in SMAD4-mutant organoids. We show that TGFβ1 treatment of SMAD4-mutant organoids results in activation of the Rho-like GTPases RAC1 and CDC42, and inhibition of these GTPases leads to decreased invasion in SMAD4-mutant organoids. These results are consistent with the previously characterized role of these GTPases in migration in multiple cell types, though the link between SMAD4 loss, TGFβ signaling and RAC1/CDC42 in cancer cell invasion has not been previously reported (30). In addition, we found that inhibition of RAC1 and CDC42 did not decrease invasion in SMAD4-wildtype organoids. Thus, non-canonical TGFβ signaling via RAC1 and CDC42 is critical for invasion in SMAD4-mutant PDACs, but SMAD4-wildtype PDACs do not rely on this pathway and instead likely employ a TGFβ-independent invasion mechanism. This highlights unique molecular programs underlying invasion based on SMAD4 genotype, suggesting that distinct strategies may be required to inhibit invasion (and thus metastasis) in these two types of PDAC. SMAD4 mutation status may become a key biomarker as therapies targeting these specific invasion programs are developed. However, our identification of an outlier SMAD4-wildtype case, in which TGFβ stimulates collective invasion that is not dependent on RAC1/CDC42, highlights that other molecular mechanisms likely influence invasion in a small subset of PDACs.
Our analyses revealed striking differences in the organoid invasive phenotype between PDACs from different patients, with SMAD4 genotype as a critical determinant. However, while the organoid morphology was markedly different, the morphology of the tumors on standard hematoxylin-and-eosin (H&E) stained histological sections was similar. The PDACs with mesenchymal invasion were not significantly enriched for poorly differentiated tumors, and the vast majority of the tumors also had sizable components with glandular differentiation. In keeping with previous studies, we found that VIM was not highly expressed in the primary tumors, and loss of membranous ECAD expression was not common in primary PDAC cells and was focal when it did occur (31, 32). However, of the 8 primary PDACs that had either focal ECAD loss or focal VIM expression, 7 resulted in organoids with predominantly mesenchymal invasion. In addition, focal expression of P40 and loss of GATA6 were also more frequent in primary tumors giving rise to predominantly mesenchymal organoids. In keeping with this, a score describing mesenchymal features was significantly higher in primary PDACs giving rise to mesenchymal organoids compared to those giving rise to collective organoids. This suggests that mesenchymal features may be identified by immunohistochemistry in some primary tumors that give rise to mesenchymally invading organoids, highlighting that the phenotypes in organoid culture are likely capturing relevant in vivo tumor features. However, it is also important to note that four PDACs (~25%) that generated mesenchymally invading organoids did not exhibit these expression features in the primary tumor. One possible explanation is that the tissue samples used to derive organoids contained more poorly differentiated or mesenchymal tumor cells than the tissue samples submitted for histological examination. However, such random sampling differences are unlikely to explain the consistent pattern of enhanced mesenchymal phenotype in the organoid model. Another possible explanation is that the process of organoid derivation and culture selects for a population with a mesenchymal phenotype when present in the harvested tumor sample. Alternatively, it is also possible that there are factors in the in vivo microenvironment that constrain mesenchymal features, and our organoid culture conditions remove these constraints, allowing the emergence of the cell-intrinsic invasive phenotype. Relatedly, it is possible that the mesenchymal program is a dynamic process that is difficult to reliably identify on a single static biopsy and thus organoid culture is a more sensitive assay for this phenotype. Importantly, the strong correlation of invasive phenotype with molecular features of the primary tumor suggests that we are observing a true difference in tumor cell behavior.
Our study also demonstrated a correlation of invasive phenotype with clinical features, with PDACs giving rise to organoids with predominantly mesenchymal invasion having a significantly increased risk of death. This observation is in line with recent efforts to develop a molecular classification for PDAC. Multiple studies have categorized PDACs using gene expression signatures, and although some classifications differ, a shared theme is the distinction between a group with a classical/progenitor signature and a group with mesenchymal/basal features (8, 20, 21). In multiple studies, PDACs with a mesenchymal gene expression phenotype also displayed a worse clinical outcome (8, 20, 21). A recent comprehensive study by The Cancer Genome Atlas (TCGA) confirmed the equivalence of these expression groups across studies (33). Analysis of the TCGA data demonstrates that SMAD4 mutations were enriched in the classical/progenitor group, while the majority of PDACs in the mesenchymal/basal group were SMAD4 wild-type. The enrichment of SMAD4 mutations in PDACs of the classical gene expression subtype was also recently confirmed in an large independent cohort analyzed by parallel genomic and transcriptomic profiling (34). These studies support the conclusion suggested by our organoid model: SMAD4 mutations disrupt the acquisition of mesenchymal features, resulting in more classical epithelial behavior. Still, comprehensive molecular profiling of primary PDACs and correlation with invasive phenotype in organoids from the same tumors represents an important future direction. In addition, these results highlight a discrepancy in the field: SMAD4 mutations are enriched in PDACs with a classical gene expression phenotype and collective invasion, both of which are expected to have improved outcomes. In contrast, previous studies have shown an association of SMAD4 mutation with worse prognosis in resected PDAC samples (35–39). There are several possible explanations for this discrepancy. In one study, sequencing analysis was performed on cell lines and xenografts, which may not be representative of all resected PDACs (35). In addition, multiple studies define SMAD4 status by immunohistochemistry, which in our cohort was less tightly correlated with organoid invasive phenotype than mutation identified by DNA sequencing (36–40). Finally, changes in clinical practice in the years since initial studies were performed, including changes in timing and composition of chemotherapy, may impact the correlation of SMAD4 mutation and survival, as could other clinical differences between analyzed cohorts, such as stage of disease – one of the key studies focused on end stage disease at autopsy, while ours focused on surgically resectable tumors (37). It is likely that invasive phenotype is one of several factors driving outcome, and analyses in larger cohorts incorporating SMAD4 mutation status, organoid invasive phenotype, and other clinical, pathological, and molecular features will be necessary to more conclusively determine the clinical impact of our results.
Another important observation from these studies is the striking change in organoid invasive phenotype after passaging. Our initial time lapse experiments revealed that the majority of freshly derived organoids will invade when cultured in collagen I. However, lentiviral transduction required passage of the organoids in Matrigel, and following passaging and transduction, the organoids from three separate SMAD4-mutant PDACs no longer invaded when transferred back into collagen I. Treatment with TGFβ1 restored the ability of the passaged organoids to invade into collagen I. Overall, these results highlight the critical role of this pathway in invasion in SMAD4-mutant PDACs. In addition, they emphasize the importance of observing at least some organoid phenotypes as soon as possible after derivation, as the process of expansion in culture likely alters the organoids at the molecular level.
Although our experiments were performed entirely in the in vitro organoid system, they provide complementary data to several recently published studies in murine models. Assessing our data in the context of these studies also has implications for the role of epithelial-to-mesenchymal transition (EMT) in pancreatic cancer invasion. In the genetically engineered KPC murine model, Aiello et al describe two different EMT phenotypes distinguished by distinct gene expression, as well as several other phenotypic differences, including different invasive morphologies in a spheroid model, that are comparable to our collective and mesenchymal invasion (41). Both this study and ours suggest that EMT is not an all-or-none phenomenon, and different tumors may adopt different features of this process, potentially in a reproducible pattern that is based on molecular alterations. In a complementary study, Zheng et al suggest that EMT may not be required for metastasis in the KPC model, as mice with deletion of key EMT mediators Snail and Twist develop primary tumors and metastases as KPC mice (42). This finding is consistent with our data, in that some PDACs in our study lacked any evidence of mesenchymal features in the primary tumor and organoid model, though the completeness of the suppression of EMT programs in the murine models has been called into question (43). Also using a genetically engineered mouse model, Whittle et al explore the role of SMAD4 loss more directly in invasion. The authors find mice with homozygous disruption of SMAD4 in the KPC background had enhanced metastatic capability and are resistant to EMT in response to TGFβ. Consistent with our organoid results, this study highlights SMAD4 as a critical regulator of PDAC invasion in the murine model and suggests that PDACs with SMAD4 loss lack features of EMT (29). In addition, David et al and Huang et al explore EMT and apoptosis in response to TGFβ in murine and human samples, with results that are consistent with our findings, particularly that treatment with TGFβ results in both mesenchymal features and enhanced apoptosis in PDACs with restored SMAD4 expression (28, 44). Overall, these convergent findings in complementary model systems provide strong support for the existence of multiple invasive phenotypes in PDAC and for the importance of SMAD4 alteration in mediating these phenotypes. Future studies could more directly assess the behavior of modified human organoids in murine models.
Through observation and interrogation of an organoid model of human pancreatic cancer, we show that SMAD4 inactivation enables collective invasion that is stimulated by TGFβ via non-canonical signaling through RAC1 and CDC42. These results raise the possibility of unique molecular programs underlying invasion in PDACs based on SMAD4 genotype, and as such treatments that target these invasive programs will likely need to be genotype-specific. Future studies will investigate the potential of GTPase inhibition as a strategy to inhibit PDAC invasion and determine the molecular program that underlies mesenchymal invasion in SMAD4-wildtype PDAC organoids. Such studies will expand the possibility of personalized therapy for pancreatic cancer that targets the specific molecular programs of invasion in each patient’s tumor.
Supplementary Material
Acknowledgements
The authors thank Dr. Ralph Hruban for helpful discussions. The authors wish to dedicate this work to Dr. Michael Reiss, as our group’s work on the TGFβ pathway is one of his many scientific legacies.
The authors acknowledge the following sources of financial support: American Cancer Society RSG-18-143-01-CSM (LD Wood); NIH/NCI P50 CA62924 (LD Wood); NIH/NIDDK K08 DK107781 (LD Wood); NIH/NCI U01CA217846 (AJ Ewald); NIH/NCI U54 CA2101732 (AJ Ewald); Sol Goldman Pancreatic Cancer Research Center (LD Wood); Buffone Family Gastrointestinal Cancer Research Fund (LD Wood); Carol S. and Robert M. Long Pancreatic Cancer Research Fund (LD Wood); Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention (LD Wood); AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer (LD Wood); Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award (LD Wood); AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research, Grant Number 16-20-46-WOOD (LD Wood); Joseph C Monastra Foundation (LD Wood); The Gerald O Mann Charitable Foundation (LD Wood); Susan Wojcicki and Denis Troper; Ontario Institute for Cancer Research (PanCuRx Translational Research Initiative) through funding provided by the Government of Ontario (S Fischer); Wallace McCain Centre for Pancreatic Cancer supported by the Princess Margaret Cancer Foundation (S Fischer); Terry Fox Research Institute (S Fischer); Canadian Cancer Society Research Institute (S Fischer); Pancreatic Cancer Canada Foundation (S Fischer).
Footnotes
Conflict of Interest: LDW served as a consultant for Personal Genome Diagnostics and receives research funding from Applied Materials. AJE has pending patent applications on molecular markers for cancer invasion and antibody-based strategies to treat cancer, and his wife is an employee of ImmunoCore. The other authors report no conflict of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 3.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 2015;6:7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 9.Hosoda W, Chianchiano P, Griffin JF, Pittman ME, Brosens LA, Noe M, Yu J, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massague J TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012;13:616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 2009;19:128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menyhart O, Harami-Papp H, Sukumar S, Schafer R, Magnani L, de Barrios O, Gyorffy B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim Biophys Acta 2016;1866:300–319. [DOI] [PubMed] [Google Scholar]
- 13.Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol 2014;15:647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, Yaswen P, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A 2012;109:E2595–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013;155:1639–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, Froeling FEM, et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov 2018;8:1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Driehuis E, van Hoeck A, Moore K, Kolders S, Francies HE, Gulersonmez MC, Stigter ECA, et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci U S A 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pea A, Yu J, Rezaee N, Luchini C, He J, Dal Molin M, Griffin JF, et al. Targeted DNA Sequencing Reveals Patterns of Local Progression in the Pancreatic Remnant Following Resection of Intraductal Papillary Mucinous Neoplasm (IPMN) of the Pancreas. Ann Surg 2017;266:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature genetics 2015;47:1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tacha D, Bremer R, Haas T, Qi W. An immunohistochemical analysis of a newly developed, mouse monoclonal p40 (BC28) antibody in lung, bladder, skin, breast, prostate, and head and neck cancers. Arch Pathol Lab Med 2014;138:1358–1364. [DOI] [PubMed] [Google Scholar]
- 23.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, Yeo CJ, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas : a new marker of DPC4 inactivation. Am J Pathol 2000;156:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, Cantrell MA, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med 2014;20:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seoane J, Gomis RR. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeichi M Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol 2014;15:397–410. [DOI] [PubMed] [Google Scholar]
- 27.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 2010;11:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA, et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell 2016;164:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, Wood LD, et al. RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell 2015;161:1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol 2018;217:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong SM, Li A, Olino K, Wolfgang CL, Herman JM, Schulick RD, Iacobuzio-Donahue C, et al. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod Pathol 2011;24:1237–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Handra-Luca A, Hong SM, Walter K, Wolfgang C, Hruban R, Goggins M. Tumour epithelial vimentin expression and outcome of pancreatic ductal adenocarcinomas. Br J Cancer 2011;104:1296–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, Connor AA, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020. [DOI] [PubMed] [Google Scholar]
- 35.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res 2009;15:4674–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshima M, Okano K, Muraki S, Haba R, Maeba T, Suzuki Y, Yachida S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg 2013;258:336–346. [DOI] [PubMed] [Google Scholar]
- 37.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 2009;27:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamada S, Fujii T, Shimoyama Y, Kanda M, Nakayama G, Sugimoto H, Koike M, et al. SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas 2015;44:660–664. [DOI] [PubMed] [Google Scholar]
- 39.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, Adsay V, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res 2001;7:4115–4121. [PubMed] [Google Scholar]
- 40.Herman JM, Jabbour SK, Lin SH, Deek MP, Hsu CC, Fishman EK, Kim S, et al. Smad4 Loss Correlates With Higher Rates of Local and Distant Failure in Pancreatic Adenocarcinoma Patients Receiving Adjuvant Chemoradiation. Pancreas 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell 2018;45:681–695.e684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aiello NM, Brabletz T, Kang Y, Nieto MA, Weinberg RA, Stanger BZ. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017;547:E7–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang YH, Hu J, Chen F, Lecomte N, Basnet H, David CJ, Witkin MD, et al. ID1 mediates escape from TGF-beta tumor suppression in pancreatic cancer. Cancer Discov 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.