

SUMMARY
Autophagy is activated by prolonged fasting but cannot overcome the ensuing hepatic lipid overload, resulting in fatty liver. Here, we describe a peroxisome-lysosome metabolic link that restricts autophagic degradation of lipids. Acyl-CoA oxidase 1 (Acox1), the enzyme that catalyzes the first step in peroxisomal β-oxidation, is enriched in liver and further increases with fasting or high fat diet (HFD). Liver-specific Acox1 knockout (Acox1-LKO) protected mice against hepatic steatosis caused by starvation or HFD due to induction of autophagic degradation of lipid droplets. Hepatic Acox1 deficiency markedly lowered the total cytosolic acetyl-CoA levels, which led to decreased Raptor acetylation and reduced lysosomal localization of mTOR, resulting in impaired activation of mTORC1, a central regulator of autophagy. Dichloroacetic acid treatment elevated acetyl-CoA levels, restored mTORC1 activation, inhibited autophagy, and increased hepatic triglycerides in Acox1-LKO mice. These results identify peroxisome-derived acetyl-CoA as a key metabolic regulator of autophagy that controls hepatic lipid homeostasis.
eTOC Blurb
Autophagy is activated by starvation but is not sufficient to prevent the resulting fatty liver. He et al. describe a peroxisome-lysosome crosstalk that restricts autophagic degradation of lipid droplets in the fasted state. They show that acetyl-CoA derived from peroxisomal β-oxidation inhibits lipophagy by promoting Raptor acetylation and mTORC1 activation.
Graphical Abstract
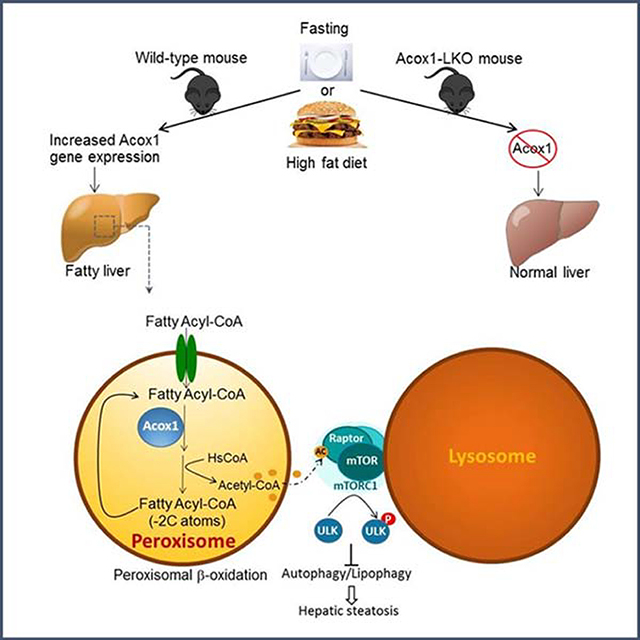
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease worldwide with the global prevalence of approximately 25% (Younossi et al., 2016). NAFLD is characterized by hepatic steatosis and can progress in a subset of patients to nonalcoholic steatohepatitis with inflammation and fibrosis or to hepatocellular carcinoma (Rinella, 2015). Commonly associated with obesity, NAFLD can be reversed with weight loss. However, drugs to specifically treat NAFLD are currently not available. Development of fatty liver involves dysregulation of lipid homeostasis, which is controlled by a coordinated regulation of various metabolic pathways involved in de novo synthesis, uptake, storage and catabolism of lipids. Thus, targeting lipid metabolism pathways could be an effective strategy to treat fatty liver.
Peroxisomes play an essential role in lipid metabolism, yet remain a neglected organelle. These versatile organelles are involved in various aspects of lipid metabolism, including ether lipid synthesis, bile acid synthesis, α-oxidation of branched chain fatty acids, and β-oxidation of very long chain fatty acids (VLCFA) (Lodhi and Semenkovich, 2014). The first step of peroxisomal β-oxidation is carried out by acyl-coenzyme A oxidase 1 (Acox1), which catalyzes the desaturation of acyl-CoA to 2-trans-enoyl-CoA. Acox1 preferentially catabolizes straight-chain fatty acids, while the related enzymes Acox2 and Acox3 are thought to utilize branched chain fatty acids and intermediates involved in bile acid synthesis as substrates (Van Veldhoven, 2010). Acox1 global knockout mice are viable but growth retarded (Fan et al., 1996). Notably, these mice exhibit steatohepatitis and develop hepatic carcinomas in the context of increased endoplasmic reticulum (ER) stress as they age (Huang et al., 2011). Consistent with these results, mice with a point mutation that affects a splice donor site in intron 11–12 of Acox1 (referred to as Acox1Lampe1 mice), resulting in partial deletion of the C-terminal domain of the protein, also exhibit hepatosteatosis that progresses to steatohepatitis and ultimately hepatocellular carcinoma (Sheridan et al., 2011). Bone marrow transplant from Acox1Lampe1 mice into wild-type recipients recapitulated the hepatocellular damage and systemic inflammation, suggesting that Acox1 inhibition in immune cells, rather than hepatocytes themselves, contributes to the phenotype (Moreno-Fernandez et al., 2018). Thus, the physiological significance of Acox1 in hepatocytes has remained unclear.
To study the role of hepatic peroxisomal β-oxidation, we generated mice with liver-specific knockout of Acox1. Here, we report that hepatic Acox1 deficiency surprisingly protects against fatty liver caused by starvation or high fat feeding due to induction of lipophagy, a subtype of macroautophagy that degrades intracellular lipid droplets (Cui et al., 2020; Singh et al., 2009). Autophagy is a homeostatic process of self-digestion that involves lysosomal degradation of cytoplasmic contents, including whole organelles, protein aggregates and lipid droplets through formation of a double membrane autophagosome around the cargo, which then fuses with lysosomes (Evans et al., 2017). Autophagy is regulated by mTORC1 (mechanistic target of rapamycin complex 1), a key growth and metabolism regulatory complex that includes the serine/threonine protein kinase mTOR and Raptor, an adaptor protein that regulates substrate recruitment to mTORC1 and its subcellular localization and activation (Saxton and Sabatini, 2017). The complex regulates autophagy by sensing nutrients. Under nutrient sufficiency, mTORC1 is activated and inhibits autophagy through phosphorylation of multiple autophagy-related proteins, including ULK1 (Unc-51 like autophagy activating kinase 1) (Kim et al., 2011), which promotes autophagy initiation and autophagosome biogenesis. The activation of mTORC1 also leads to inhibition of genes involved in lysosomal biogenesis and function, including lysosomal acid lipase (LAL), which mediates lipophagy (Zechner et al., 2017). Here, we report that acetyl-CoA derived from peroxisomal β-oxidation inhibits lipophagy by promoting activation of mTORC1. These findings suggest that inhibition of hepatic Acox1 may be an attractive strategy to treat NAFLD.
RESULTS
The generation of liver-specific Acox1 knockout mouse model
Gene expression analysis indicated that Acox1 is highly expressed in the liver (Figure 1A). However, its physiological role in the liver is unknown. Mice with a global knockout of Acox1 have been generated, but they exhibit growth retardation (Fan et al., 1996), which confounds metabolic analysis. Thus, we generated mice with liver-specific knockout of Acox1 (Acox1-LKO mice) by crossing Acox1 floxed mice (Park et al., 2019) to albumin-Cre mice (Figure 1B). Quantitative real-time PCR analysis in the liver showed that Acox1-LKO mice had significantly reduced expression of Acox1, without an effect on the expression of Acox2 or Acox3 (Figure 1C). Western blot analysis confirmed the knockout of Acox1 (Figure 1D). Disruption of VLCFA oxidation due to the Acox1 deficiency was confirmed by measuring catabolism of stable isotope-labeled docosanoic acid (D3-C22:0) to D3-C16:0 via mass spectrometric analysis in control and Acox1 knockout hepatocytes (Figure 1E). Consistent with the impaired fatty acid oxidation, VLCFAs accumulated in the livers and serum of Acox1-LKO mice (Figures 1F and S1A).
Figure 1. Generation of mice with liver-specific knockout of Acox1.

A) Acox1 mRNA expression level was measured by quantitative real time PCR in various tissues from wild type C57 mice (n=2). B) Acox1 floxed mice were crossed with albumin-Cre mice to generate Acox1 liver specific knockout mouse. C) Quantitative real time PCR analysis of Acox1, Acox2 and Acox3 expression in the liver of control and Acox1-LKO mice; n = 5–7 per group, ***p < 0.001 (unpaired t-test). D) Western blot analysis of Acox1 knockout in the liver. E) Control and Acox1-KO hepatocytes were incubated with D3-C22:0, whose catabolism to D3-C16:0 was measured by mass spectrometry. FAO was expressed as ratio of D3-C16:0 to D3-C22:0; n = 4, ***p < 0.001 (unpaired t-test). F) Levels of VLCFA in the livers of Acox1-LKO and control mice; n = 6–8 per group), *p < 0.05; ***p < 0.00l (unpaired t-test). Data represent mean ± SEM. See also Figure S1.
Acox1-LKO mice were born at the expected Mendelian frequency and were overtly normal. There was no difference in body weight between control and knockout animals at 2 months or 12 months of age (Figure S1B). However, the liver-to-body weight ratio was modestly increased in Acox1-LKO mice at 2 months of age and did not become progressively worse at 12 months of age (Figure S1C). The liver glycogen content was significantly increased in Acox1-LKO mice (Figures S1D and S1E), which likely explains the increased liver weight in the mutant mice. Unlike Acox1Lampe1 mice, which develop hepatocellular carcinoma with dramatically increased expression of cancer markers at 8 weeks of age (Moreno-Fernandez et al., 2018), the livers in 12-month-old Acox1-LKO mice had a normal gross appearance (Figure S1F) and lacked the expression of cancer markers AFP, H19, and Ki67 (Figure S1G). The inflammatory markers IL-6 and TNFα were also barely detectable in the liver of Acox1-LKO mice, similar to control mice (Figure S1H). Consistent with the knockout mice possessing normal liver function, the serum levels of alanine transaminase (ALT) and aspartate transaminase (AST) were unchanged (Figure S1I). Taken together, these results suggest that liver-specific Acox1 inactivation does not cause liver damage or lead to hepatocellular carcinoma.
Acox1-LKO mice are protected from fatty liver induced by starvation
Peroxisomes likely have a role in β-oxidation that cannot be compensated for by mitochondria. Given that inhibition of mitochondrial fatty acid oxidation due to liver-specific knockout of carnitine palmitoyltransferase 2 (Cpt2) exacerbates starvation-induced fatty liver (Lee et al., 2016), we sought to determine the effect of starvation on the livers in Acox1-LKO and control mice (Figure 2). Twenty-four hour fasting resulted in fatty liver in control mice. However, Acox1-LKO mice were surprisingly protected from the starvation-induced fatty liver (Figure 2A). Bodipy staining and measurement of hepatic triglycerides confirmed the decreased lipid accumulation in Acox1-LKO mice (Figures 2B and 2C). These results, generated using female mice, were confirmed in male animals (Figure S2A). To determine if the protection against fatty liver was specifically mediated by the loss of Acox1, we treated mice with AAV expressing Acox1 or GFP. AAV-Acox1 increased the Acox1 protein levels in the knockout mice (Figure S2B) and reversed the protection against fatty liver (Figure S2C), resulting in increased accumulation of triglycerides (Figure S2D).
Figure 2. Acox1-LKO mice are protected from starvation-induced fatty liver.

A) Gross images of livers from 8–10 weeks old control and Acox1-LKO female mice that were fasted for 24 hours. B) Liver frozen sections from control and Acox1-LKO mice fasted for 24 hours were stained with Bodipy (Green) and DAPI (Blue). C) Liver homogenate from control or Acox1-LKO mice fed or fasted for 24 hours were used for triglyceride content measurement; n = 6–10, **p<0.0l, **p<0.00l (unpaired t-test). D) Non-esterified fatty acid levels measured in serum from control and Acox1-LKO female mice that were fed or fasted for 24 hours; n = 6, **p < 0.01 (unpaired t-test). E) Rate of triglyceride secretion calculated from the slope of the increase in plasma triglycerides after P-407 treatment in fasted female mice (n=7–10/group). F) Hepatic gene expression analysis in Acox1-LKO and control mice; n = 6–7, *p < 0.05 (unpaired t-test). G) Mitochondria fatty acid oxidation activity was measured by using l4C-labeled palmitate; n = 6, ***p < 0.001 (unpaired t-test). Data represent mean ± SEM. See also Figure S2.
The decreased hepatic lipid accumulation in Acox1-LKO mice could potentially reflect reduced adipose tissue lipolysis or increased triglyceride export as VLDL by the liver. Free fatty acids levels were comparable in the serum of fasting control and Acox1-LKO mice (Figure 2D), suggesting that adipose tissue lipolysis is not affected by the liver-specific inactivation of Acox1. Consistent with this possibility, there were no genotype-specific differences in body weight (Figure S2E) or fat mass in the fasted mice (Figures S2F). The rate of triglyceride secretion measured in fasted mice treated with the non-ionic detergent poloxamer-407, which inhibits lipoprotein lipase activity, was unchanged between Acox1-LKO and control mice (Figure 2E), suggesting that inhibition of peroxisomal β-oxidation does not increase hepatic triglyceride export. Serum cholesterol levels were also similar between control and Acox1-LKO mice (Figure S2G).
To explore the mechanism underlying the protection against starvation-induced fatty liver in Acox1-LKO mice, we next conducted gene expression analysis (Figure 2F). Hepatic expression of genes involved in de novo lipogenesis (ChREBP, SREBP, FASN, ACLY, and ACC1) and triglyceride synthetic pathway (AGPAT and DGAT) were unchanged (Figure 2F). Although the expression of genes involved in mitochondrial fatty acid oxidation (Cpt1a and Cpt2) and their upstream transcriptional regulator (PPARα) was unaltered, direct measurement of fatty acid oxidation using 14C-labeled palmitate, a mitochondrial substrate, indicated that disruption of peroxisomal β-oxidation does not impair, but rather increases mitochondrial β-oxidation (Figure 2G). Since peroxisomal β-oxidation is thought to contribute to the production of malonyl-CoA (Reszko et al., 2004), a potent inhibitor of Cpt1, we measured malonyl-CoA levels in the livers of Acox1-LKO and control mice. Although not statistically significant, the levels were slightly lower in the knockout mice (Figure S2H). Nevertheless, it is unlikely that the increased mitochondrial β-oxidation alone explains the protection against fatty liver in Acox1-LKO mice since previous studies demonstrate that increasing the liver mitochondrial fatty acid oxidation capacity through hepatic overexpression of malonyl-CoA-insensitive Cpt1 is not sufficient to prevent hepatic steatosis (Monsenego et al., 2012). This is presumably because fatty acids must first be released from triglycerides through lipolysis prior to their β-oxidation in mitochondria. Gene expression of lipolytic enzymes (ATGL and HSL) was unchanged between control and Acox1-LKO mice (Figure 2F). However, since the control of lipolysis occurs predominantly through the regulation of the recruitment of lipases to lipid droplets, rather than at the level of gene expression, we also assessed whether lipid droplet localization of ATGL was altered. Immunofluorescence analysis in the livers of fasted mice indicated that Acox1 inactivation does not influence the recruitment of ATGL to lipid droplets (Figures S2I and S2J). As noted above, lipophagy is an alternative process through which triglycerides stored in lipid droplets are hydrolyzed, generating free fatty acids and glycerol. Interestingly, the liver expression of LAL, which mediates the autophagic degradation of lipid droplets was modestly, but significantly, increased in Acox1-LKO mice (Figure 2F).
Hepatic Acox1 knockout promotes activation of autophagy
We next determined if the Acox1 knockout results in activation of autophagy in the liver (Figure 3). The protein levels of the autophagy markers p62 and LC3 in the Acox1 knockout liver were significantly lower, despite a lack of difference in the corresponding mRNA levels (Figures 3A and 3B), indicating induction of autophagy. Consistent with the notion that the decreased p62 and LC3 protein levels reflect increased autophagic flux, treatment of mice with leupeptin, an inhibitor of lysosomal proteases, resulted in accumulation of the autophagy makers in control and Acox1-LKO mice (Figure 3C). To further assess autophagy activation, we used hydrodynamic tail vein (HTV) injection to deliver a plasmid expressing mCherry-GFP dual tandem tagged LC3 (Ma et al., 2012) to control and Acox1 LKO mice, followed by treatment with or without chloroquine, which inhibits autophagic flux by increasing lysosomal pH and blocking fusion of autophagosomes with lysosomes (Mauthe et al., 2018). While autophagosomes exhibit dual mCherry and GFP fluorescence, autolysosomes primarily display red fluorescence since the GFP fluorescence is quenched in the acidic intralysosomal environment. Thus, a decrease in GFP fluorescence relative to mCherry fluorescence is indicative of increased autophagic flux (Ma et al., 2012). Interestingly, GFP fluorescence was decreased in the livers of Acox1-LKO mice (Figures 3D and 3E), consistent with activation of autophagy in these mice. As expected, choloroquine treatment blocked the autophagic flux in Acox1-LKO and control mice.
Figure 3. Acox1-LKO mice exhibit activation of autophagy/lipophagy in the liver.

A) Western blot analysis of autophagy markers p62 and LC3 for 8-l0 weeks old female mice fasted for 4 hours. B) Protein levels, but not the mRNA levels, of p62 and LC3 were decreased in the Acox1-LKO liver; n = 4–7, *p < 0.05 (unpaired t-test). C) Western blot analysis of autophagic flux in control and Acox1-LKO mice treated with leupeptin for four hours without diet. D-E) Assessment of autophagy flux using lentiviral mCherry-GFP dual tandem-tagged LC3 in the presence or absence of chloroquine (Chq) in the livers of Acox1-LKO and control mice. Images are representative of at least three independent experiments; n = 12, *p < 0.05, ***p < 0.001 (unpaired t-test). F-G) Colocalization between lipid droplet (PLIN2) and lysosome (LAMP2) was increased in Acox1-LKO liver as compared to the control liver of mice fasted for three hours. Images are representative of at least three independent experiments; n = 10, **p < 0.01 (unpaired t-test). Data represent mean ± SEM. See also Figure S3.
Given the protection against fatty liver in Acox1-LKO mice, we next determined whether lipid droplets in the mutant liver were associated with lysosomes, which would suggest activation of lipophagy. Immunofluorescence analysis indicated that lipid droplets stained using an antibody against perilipin 2 (PLIN2) colocalized with lysosomes stained using an antibody against LAMP2 (Figures 3F and 3G). Together, these results suggest that inactivation of peroxisomal β-oxidation protects against fatty liver by promoting hydrolysis of lipid droplets through lipophagy, followed by fatty acid oxidation in mitochondria.
To determine if the phenotype of Acox1-LKO mice was mediated by activation of autophagy, we generated mice with liver-specific knockout of Atg5 on Acox1-LKO or wild-type background (Figure S3). As expected, Atg5 knockout resulted in a dramatic accumulation of p62 and LC3 and abolished the decrease of these autophagy markers in Acox1-LKO mice (Figure S3A), confirming that the decrease was mediated by induction of autophagy. Surprisingly, Atg5 single knockout also markedly decreased Acox1 levels (Figure S3A), perhaps reflecting a feedback mechanism. The knockout of Atg5 alone or in the context of Acox1 inactivation resulted in a massive hepatomegaly (Figures S3B and S3C), consistent with previous reports suggesting that genetic inactivation of autophagy results in a severe liver injury, characterized by several pathologies, including fibrosis, liver inflammation, increased serum ALT and AST levels, dramatic protein accumulation, and tumorigenesis (Komatsu et al., 2007; Ni et al., 2014). The hepatic triglyceride content in Atg5-LKO mice was paradoxically decreased (Figure S3D), consistent with recent studies suggesting that in addition to regulating degradation of lipid droplets, autophagy might be required for biogenesis of lipid droplets (Li et al., 2018). The combined liver-specific Atg5 and Acox1 knockout did not further decrease the triglyceride content (Figure S3D). Overall, while inhibition of macroautophagy in the liver results in severe abnormalities and a complicated phenotype due to pleiotropic effects of the process, multiple lines of evidence support our notion that activation of lipophagy through pharmacological (DeBosch et al., 2016; Lin et al., 2013; Sinha et al., 2012) or genetic (Xiong et al., 2012; Yang et al., 2010) means protects against hepatic steatosis.
Peroxisomal β-oxidation is a major source of acetyl-CoA that regulates the mTORC1-autophagy axis
We next sought to understand how disruption of peroxisomal β-oxidation in the liver might promote autophagic degradation of lipids (Figure 4). Autophagy is induced under starvation conditions through activation of AMPK, which in turn phosphorylates and activates ULK1 (Kim et al., 2011). Therefore, we determined whether AMPK activation in the liver was affected by Acox1 inactivation. Western blot analysis indicated that Thr172 phosphorylation of AMPK, which is required for its activation, was unchanged in Acox1-LKO mice (Figure S4A).
Figure 4. Hepatic Acox1 inactivation decreases acetyl-CoA levels, resulting in impaired mTORC1 activation.

A) Peroxisomal β-oxidation pathway. B) Acetyl-CoA measurement in the livers of 8–10 weeks old control and Acox1-LKO female mice that were fasted overnight; n = 6–7, *p < 0.05 (unpaired t-test). C) Hepatic gene expression analysis of ACLY, ACSS2 and Acox1 in wild-type C57 mice that were subjected to a 24 hour fasting. Ct values are shown on histograms; n = 6, *p < 0.05 (unpaired t-test). D) Western blot analysis of Acox1, ACSS2 and ACLY expression in the livers of fed and fasted mice (n=3/group). E) FLAG-Raptor plasmid was delivered to mouse liver by hydrodynamic tail vein injection, followed by anti-FLAG immunoprecipitation and immunoblotting using anti-acetylated lysine (AcK) and FLAG antibodies (n=3/group). F-G) Lysine acetylation of endogenous Raptor and histone H3 was analyzed in livers of fasted Acox1-LKO and control mice by immunoprecipitation using an anti-AcK antibody, followed by Western blot analysis using anti-Raptor and H3 antibodies; n=3, *p < 0.05 (unpaired t-test). H-I) Immunofluorescence analysis using antibodies against PMP70 and LAMP2 suggesting that fasting promotes contact between peroxisomes and lysosomes in liver of mice fasted for 4 hours. n = 11, *p < 0.05 (unpaired t-test). J-K) The localization of mTOR to lysosome was inhibited in Acox1-LKO liver, as assessed by the decreased colocalization between mTOR and LAMP2. Images are representative of at least three independent experiments; n=10, ** p < 0.01 (unpaired t-test). L) mTOR activation was inhibited in the Acox1-LKO liver as shown by the decreased phosphorylation of mTOR (Ser2448), S6K (Thr389) and ULK1 (Ser757); n=4–5/group. M) Acox1 knockout promotes autophagic flux of PLIN2 in liver of mice treated with leupeptin for 4 hours without diet (n=2/condition). N) Effect of Acox1 knockout on phosphorylation of mTOR targets under fed or fasting conditions (n=2/group). Data represent mean ± SEM. See also Figure S4.
Thus, we explored other potential mechanisms. One of the products of peroxisomal β-oxidation is H2O2 (Figure 4A), a reactive oxygen species (ROS) reported to induce autophagy through activation of the tuberous sclerosis complex (TSC) signaling node at the peroxisome and the subsequent inhibition of mTORC1 (Zhang et al., 2013). To understand the impact of Acox1 knockout on ROS in the liver, we generated a peroxisome-targeted form of RoGFP (RoGFP-SKL), a redox-sensitive GFP that exhibits a second excitation peak at 400 nm (reflective of oxidative state), in addition to the regular excitation wavelength (~484 nm) of GFP (Waypa et al., 2010). Using HTV injection, we ectopically expressed RoGFP-SKL in the livers of Acox1-LKO and control mice and detected fluorescence by confocal microscopy (Figure S4B). Quantification of the fluorescence ratio after excitation at the two different wavelengths suggested that peroxisomal ROS levels were significantly decreased in the Acox1 knockout liver (Figure S4C). Mitochondria are also a major contributor of ROS. However, ROS levels were also lower using a mitochondria-targeted RoGFP (mito-RoGFP) in the Acox1 knockout liver (Figures S4D and S4E), which presumably may be a secondary effect of the decreased peroxisomal ROS production, since H2O2 can diffuse freely across organelle membranes (Reth, 2002). Since inactivation of Acox1 decreased H2O2 production, the induction of autophagy in Acox1-LKO mice is likely not caused by increased ROS levels.
Another product of peroxisomal β-oxidation is acetyl-coenzyme A (acetyl-CoA) (Figure 4A). Each cycle of β-oxidation results in an acyl chain that is shorter by 2 carbons, with the thiolytic cleavage of terminal acetyl-CoA group. Cytosolic acetyl-CoA is considered a key metabolic regulator of autophagy and its depletion is sufficient to induce general autophagy (Mariño et al., 2014). Recent studies suggest that acetyl-CoA derived from leucine catabolism promotes acetylation of Raptor, resulting in lysosomal localization and activation of mTORC1 (Son et al., 2018). However, whether Raptor acetylation mediated by this or any other pool of acetyl-CoA regulates autophagy has not been determined. We thus asked whether peroxisome-derived acetyl-CoA regulates autophagy of lipids in the liver through activation of mTORC1. First, we determined the effect of inhibiting peroxisomal β-oxidation on acetyl-CoA levels in the liver. Remarkably, the total cytosolic acetyl-CoA pool in fasted Acox1-LKO mice was less than 50% of the levels in control mice (Figure 4B). This was surprising given that cytosolic acetyl-CoA is thought to be derived mainly from citrate through the action of ATP citrate lyase (ACLY) (Mariño et al., 2014). Acyl-CoA synthetase short-family member 2 (ACSS2), which converts acetate to acetyl-CoA also contributes to the cytosolic pool. However, the hepatic gene expression of both ACLY and ACSS2 markedly decreased with fasting, while the Acox1 gene expression increased. Moreover, based on the cycle threshold (Ct) value of quantitative real-time PCR, Acox1 might be more highly expressed than both ACLY and ACSS2 in the liver (Figure 4C). Western blot analysis confirmed that Acox1 expression increases, while ACLY and ACSS2 decrease, in the liver with fasting (Figure 4D). Thus, Acox1-mediated fatty acid oxidation is a major source of cytosolic acetyl-CoA in the liver, especially in the fasted state.
Next, we determined if Acox1 inactivation affects Raptor acetylation. To this end, we used HTV injection to express a FLAG-Raptor plasmid in the livers of Acox1-LKO and control mice. Immunoprecipitation using an anti-FLAG antibody, followed by Western blot analysis using an antibody against acetylated-lysine (AcK) indicated that Raptor acetylation was markedly reduced in the mutant mice (Figure 4E). To determine if Acox1 inactivation affects acetylation of endogenous Raptor and other proteins, we subjected liver lysates from fasted mice to immunoprecipitation using an AcK antibody. Western blot analysis indicated that Raptor acetylation was significantly decreased, while acetylation of histone H3 was unaffected by Acox1 knockout (Figures 4F and 4G), suggesting that peroxisome-derived acetyl CoA might have a selective role in acetylation. In this regard, previous studies suggest that lysosomes form membrane contacts with peroxisomes (Chu et al., 2015). Notably, immunofluorescence analysis using antibodies against peroxisomal and lysosomal markers indicated that fasting promotes colocalization of peroxisomes with lysosomes in the liver (Figures 4H and 4I), raising the possibility that lysosome-associated Raptor might undergo acetylation mediated by locally-generated peroxisomal acetyl-CoA.
Consistent with the impaired Raptor acetylation, lysosomal localization of mTOR was blocked in the Acox1 knockout liver (Figures 4J and 4K). Given that distribution of mTOR to lysosomes is required for its activation (Sancak et al., 2010), Acox1 inactivation resulted in inhibition of mTOR, as assessed by downregulation of Ser2448 phosphorylation of mTOR and the decreased phosphorylation of its downstream targets (Figure 4L). Importantly, the mTORC1 inhibition led to decreased Ser757 phosphorylation of ULK1, which regulates autophagy (Kim et al., 2011). Interestingly, PLIN2 levels were also decreased in Acox1-LKO mice (Figures 4L and S4F). Consistent with the possibility that this reflects an increased autophagic degradation of lipid droplets, intraperitoneal (IP) treatment of fasted control and Acox1-LKO mice with leupeptin resulted in accumulation of PLIN2 (Figures 4M).
Since mTORC1 is regulated by the energy status, we also determined whether its activation is affected by Acox1 knockout under fed and fasting conditions (Figure 4N). S6K phosphorylation, which controls the anabolic arm of the mTORC1 pathway, was dramatically inhibited by fasting and the residual phosphorylation was further decreased in the Acox1 knockout liver. In contrast, ULK1 phosphorylation, which regulates the catabolic arm, was only partially inhibited by fasting, with a more complete inhibition occurring with the Acox1 knockout. These data suggest that Acox1 maintains a basal level of mTORC1 activation and the inhibitory ULK1 phosphorylation, even under fasting conditions.
Increasing acetyl-CoA levels restores mTORC1 activation, inhibits autophagy and elevates triglyceride levels in Acox1-LKO mice
To determine if the impaired mTORC1 activation and decreased hepatic triglyceride accumulation in Acox1-LKO mice were due to reduced cytosolic acetyl-CoA levels (Figure 5), we treated Acox1-LKO mice with dichloroacetic acid (DCA) (Figure 5A), which increases cellular acetyl-CoA levels by inhibiting pyruvate dehydrogenase kinase (PDK) and thus promoting activation of pyruvate dehydrogenase (PDH) (Whitehouse and Randle, 1973). As expected, DCA treatment rescued the liver acetyl-CoA levels in Acox1-LKO mice (Figure 5B). Remarkably, this intervention restored Raptor acetylation in livers of Acox1-LKO mice to levels similar to those of control mice (Figures 5C and 5D). Consequently, lysosomal localization of mTOR was increased by DCA treatment in Acox-LKO mice to levels comparable to control mice (Figures 5E and 5F). This led to increased mTOR activation and ULK1 phosphorylation, resulting in inhibition of autophagy, as reflected by accumulation of p62 and LC3 in Acox1-LKO liver (Figure 5G). Consistent with suppression of lipophagy, DCA treatment decreased colocalization of PLIN2 with LAMP2 in the livers of Acox1-LKO mice (Figures 5H and 5I) and partially recued the hepatic triglyceride levels in the mutant mice (Figure 5J) without affecting the expression of genes involved in the triglyceride synthetic pathway (Figure S5). Together, these results suggest that acetyl-CoA derived from peroxisomal β-oxidation inhibits autophagic degradation of lipids by mediating mTORC1 activation.
Figure 5. Increasing acetyl-CoA levels restores mTORC1 activation, inhibits autophagy and elevates triglyceride levels in Acox1-LKO mice.

A) Schematic illustrating the role of dichloroacetic acid (DCA) in acetyl-CoA production through pyruvate dehydrogenase (PDH) activation. B) Liver acetyl-CoA levels in 8–10 weeks old female mice i.p. treated with 250 mg/kg DCA or vehicle for 3 consecutive days; n = 5–7, *p < 0.05 (unpaired t-test). C-D) Endogenous Raptor was immunoprecipitated from liver homogenates using an anti-Raptor antibody, followed by Western blot analysis using antibodies against acetylated-lysine (AcK), Raptor or actin; n = 3, *p < 0.05 (unpaired t-test). E-F) DCA restores lysosomal localization of mTOR in Acox1-LKO liver. n = 13–16, *p < 0.05 (unpaired t-test). G) Western blot analysis of mTORC1 activation and autophagy inhibition in the livers of mice treated with or without DCA as indicated (n=3/group). H-I) DCA inhibits lysosomal localization of PLIN2 in Acox1-LKO liver; n=12, *p < 0.05 (unpaired t-test). J) Liver TG content in female mice treated with or without DCA; n = 8, *p <0.05 (unpaired t-test). Data represent mean ± SEM. See also Figure S5.
Acox1-LKO mice are protected from high fat diet-induced fatty liver
We next investigated the clinical relevance of our findings (Figure 6). Obesity is associated with an increased risk of NAFLD (Fabbrini et al., 20l0). Interestingly, Acox1 gene expression in the liver trends higher in obese humans as compared to lean individuals (Figure 6A). In mice, high fat feeding resulted in a dramatic increase in the liver Acox1 gene expression (Figure 6B). Since Acox1-LKO mice are protected from fatty liver induced by starvation, we determined whether these mice are also protected from fatty liver induced by high fat diet (HFD). Interestingly, the Acox1-LKO liver was notably less pale as compared to the liver from control mice, suggesting protection against diet-induced steatosis (Figure 6C). Histologic analysis of liver sections revealed that lipid droplets were markedly smaller in Acox1-LKO mice (Figure 6D). Consistent with these results, Acox1-LKO mice had decreased hepatic triglyceride content compared to control mice (Figure 6E). We also assessed the effect of Acox1 inactivation on mTORC1 activation in the context of high fat feeding. Compared to control mice, Acox1-LKO exhibited impaired liver mTORC1 activation, as assessed by the decreased phosphorylation of mTOR, S6K and ULK1 (Figure 6F). Collectively, these data suggest that Acox1-LKO mice are protected from HFD-induced fatty liver through activation of autophagy/lipophagy resulting from inhibition of mTORC1.
Figure 6. Acox1-LKO mice are protected from HFD-induced fatty liver.

A) Liver gene expression of Acox1 in obese humans undergoing gastric bypass surgery or lean controls undergoing elective cholecystectomy. The data were extracted from GEO database (Reference ID: 71078077). n = 5–13 (unpaired t-test). B) Hepatic Acox1 gene expression in 8–10 weeks old wild-type C57 male mice fed normal chow diet or a HFD for 4 months; n = 5–7, **p < 0.01 (unpaired t-test). C) Gross liver images in HFD-fed mice. D) H&E staining of liver sections from HFD-fed male mice. E) Hepatic triglyceride content in HFD-fed mice; n = 6–7, *p < 0.05 (unpaired t-test). F) Western blot analysis of mTORC1 activation in HFD-fed Acox1-LKO and control male mice (n=4/group). Data represent mean ± SEM.
DISCUSSION
These studies suggest that peroxisomal β-oxidation is an essential regulator of hepatic lipid homeostasis. Using mice with liver-specific inactivation of Acox1, we demonstrate that disruption of peroxisomal β-oxidation protects mice against both starvation- and HFD-induced fatty liver. These mice do not exhibit the complications associated with prenatal global inactivation of the enzyme (Fan et al., 1996) and are healthy with normal liver function. Our results demonstrate that the conditional Acox1 knockout promotes hydrolysis of lipids through induction of autophagy. Mechanistically, our data suggest that acetyl-CoA derived from peroxisomal β-oxidation regulates autophagic degradation of lipid droplets in the liver. In agreement with previous studies suggesting that hydrolysis of triglycerides is closely linked to mitochondrial FFA uptake and oxidation (Cui et al., 2020; Zechner et al., 2017) and that the inhibition of hepatic autophagy impairs mitochondrial fatty acid oxidation (Singh et al., 2009), our results reveal that the activation of autophagy due to inhibition of hepatic peroxisomal β-oxidation is associated with increased mitochondrial β-oxidation.
The overall mechanisms of mitochondrial and peroxisomal β-oxidation processes are similar, with each cycle of fatty acid catabolism resulting in an acyl chain that is shorter by 2 carbons due to thiolytic cleavage of terminal acetyl-CoA group. The accumulation of VLCFAs in individuals with loss-of-function mutations in peroxisomal β-oxidation enzymes (Van Veldhoven, 2010) suggests that peroxisomes have a role in β-oxidation that cannot be replaced by mitochondria. It is thought that β-oxidation in peroxisomes is not carried to completion, generating chain-shorted fatty acids, which can be further catabolized in mitochondria. The fact that peroxisomal β-oxidation is not linked to ATP production raises the possibility that the peroxisomal pathway might have a signaling role. Surprisingly, the total cytosolic acetyl-CoA pool in the liver was reduced by a half in Acox1-LKO mice, demonstrating that peroxisomal catabolism of fatty acids is a major contributor of the cytosolic acetyl-CoA pool in the liver. Although peroxisomes are required for VLCFA oxidation, recent studies suggest that these organelles could also oxidize certain species of shorter fatty acids (Violante et al., 2018), but their relative contribution to catabolizing shorter fatty acids as compared to mitochondria is unclear. Nevertheless, it is noteworthy that for each mole of fatty acid oxidized, multiple moles of acetyl-CoA are generated. Moreover, acetyl-CoA generated by peroxisomal β-oxidation has been reported to be incorporated into lipids synthesized by peroxisomes (Hayashi and Oohashi, 1995), suggesting that catabolism and synthesis of lipids in peroxisomes might represent a futile cycle to generate acetyl CoA. Regardless of this, our results indicate that acetyl-CoA derived from peroxisomal fatty acid oxidation has an essential signaling role related to autophagic degradation of lipid droplets.
Acetyl-CoA is a key metabolic regulator of autophagy. Previous studies suggest that depletion of this metabolite is sufficient to induce autophagy (Mariño et al., 2014). Moreover, these authors demonstrated that nutrient deprivation decreases acetyl-CoA and restoring acetyl-CoA levels under such conditions inhibits the intracellular degradation process. Autophagy is suppressed by mTORC1, a master regulator of cell growth and metabolism. Recent studies suggest that acetyl-CoA derived from leucine catabolism promotes mTORC1 activation through acetylation of Raptor (Son et al., 2018). However, Raptor acetylation mediated by this or other sources of acetyl-CoA has not been studied in the context of autophagy. It is thought that cytosolic, rather than mitochondrial or nuclear, acetyl-CoA regulates autophagy (Mariño et al., 2014). Cytosolic acetyl-CoA is generated primarily from citrate by the lipogenic enzyme ACLY. Since fasting or high fat feeding dramatically inhibits the expression of hepatic ACLY (Carrer et al., 2017; Shimano et al., 1999), Acox1-mediated fatty acid oxidation might be the main source of cytosolic acetyl-CoA in the liver under such conditions. Our results suggest that this peroxisome-derived pool of acetyl-CoA maintains Raptor acetylation, resulting in a basal level of mTORC1 activation and the inhibitory ULK1 phosphorylation, even under fasting conditions, thus restricting autophagic degradation of lipid droplets. This might explain why autophagy/lipophagy cannot overcome the hepatic lipid overload that follows prolonged fasting, resulting in fatty liver. Acox1 inactivation relieves the suppression of lipophagy, resulting in protection against hepatic steatosis.
Lipophagy represents a process for intracellular lipid catabolism distinct from cytosolic lipolysis. Lipophagy is thought to contribute substantially to the hydrolysis of triglycerides in a variety of cell types, including hepatocytes, macrophages, neurons and brown adipocytes (Cingolani and Czaja, 2016). However, the relative contribution of lipophagy versus cytosolic lipolysis is unclear and likely varies under different cellular contexts. In a study that initially identified autophagic degradation of lipids, hepatocyte-specific inactivation of the autophagy factor Atg7 was reported to promote triglyceride accumulation (Singh et al., 2009). Subsequently, several studies (Settembre et al., 2013; Wang et al., 2010; Xiong et al., 2012; Yang et al., 2010), but not all (Liu et al., 2013), have shown that inhibition of hepatic autophagy promotes steatosis. Crosstalk between lipophagy and ATGL-mediated lipolysis exists (Zechner et al., 2017), and it is possible that disruption of autophagy results in compensation by lipolysis under certain metabolic contexts. Conversely, pharmacological activation of autophagy (DeBosch et al., 2016; Lin et al., 2013; Sinha et al., 2012) or the activation through liver-specific overexpression Atg7 (Yang et al., 2010), Atg14 (Xiong et al., 2012), or Transcription Factor EB (TFEB), a master transcriptional regulator of autophagy and lysosomal biogenesis (Settembre et al., 2013), results in protection against hepatic lipid accumulation. In line with these studies, our results suggest that production of cytosolic acetyl-CoA via activation of Acox1-mediated fatty acid oxidation inhibits autophagic degradation of lipids and promotes starvation- or HFD-induced fatty liver (Figure 7).
Figure 7.

A model depicting proposed molecular mechanism through which peroxisomal β-oxidation regulates mTORC1 activation to inhibit lipophagy.
Collectively, these studies identify a previously unrecognized peroxisome-lysosome metabolic link that restricts autophagic degradation of lipid droplets. Our results suggest that peroxisomal β-oxidation is a major source of cytosolic acetyl-CoA that maintains hepatic lipid homeostasis. Induction of lipid autophagy via inhibition of peroxisomal acetyl-CoA production might lead to a novel treatment option for NAFLD.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Irfan J. Lodhi (ilodhi@wustl.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal protocols were approved by the Washington University Institutional Animal Care and Use Committee. To generate a liver specific deletion of Acox1, Acox1lox/lox mice (Park et al., 2019) were crossed with albumin-Cre transgenic mice (Postic et al., 1999). ATG5lox/lox have been previously described (Hara et al., 2006) and were crossed with albumin-Cre transgenic mice to generate ATG5 liver specific knockout mice (ATG5-LKO). ATG5lox ox mice were mated with Acox1-LKO mice to generate ATG5 and Acox1 double knockout mice (double LKO). Mice were genotyped using previously described primer sets (Park et al., 2019) and fed either normal chow (Purina 5053) or a HFD (D12492, Research Diets). Animal of 6–10 weeks of age were used for experiments, unless noted otherwise. Both male and female were used, as indicated in figure legends.
METHOD DETAILS
Treatments and adeno-associated virus administration in mice
Unless otherwise noted, fasted condition in mice represents 24 hr food deprivation with ad libitum access to water. The fed condition represents mice that were fasted for 6 hrs and then re-fed, with the analysis performed 4 hrs after the refeeding. Dichloroacetic acid (DCA) was dissolved in saline and injected intra-peritoneally to mice daily at the dosage of 250 mg/kg for three consecutive days, and the mice were fasted for 24 hours after the last injection, unless noted otherwise. To assess autophagic flux, mice were deprived of food and IP injected with 40 mg/kg leupeptin dissolved in saline and then sacrificed for liver sampling after 4 hours. For re-expression of Acox1 in Acox1-LKO mice, AAV8-Alb-Acox1 or AAV8-Alb-eGFP was tail vein injected at the dose of 1e11gc/mouse. The mice were sacrificed for liver collection after three weeks.
Hydrodynamic tail vein injection
Thirty to fifty μg of plasmid were diluted into PBS and delivered to mouse liver through HTV injection, as previously described (Liu et al., 1999). For RoGFP-SKL and mito-RoGFP, the mice are fasted for 24 hours and sacrificed for frozen sectioning of liver. For autophagic flux analysis, mCherry-GFP-LC3 plasmid was delivered to liver by HTV injection. The mice were deprived of food overnight and then IP injected with 112mg/kg chloroquine dissolved in saline and sacrificed 4 hrs later. Livers were collected for sectioning following perfusion with PBS, followed by perfusion using 4% paraformaldehyde solution in 1X PBS.
Western blotting and immunoprecipitation
Protein samples were prepared as described previously (Park et al., 2019). Briefly, tissues were lysed using RIPA buffer and 40 μg protein samples were loaded for Western blotting. The analysis of acetylated Raptor in tissues was performed as previously described (Son et al., 2018). Briefly, liver tissues were homogenized using a lysis buffer (20 mM Tris-HCl (pH7.4), 5 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, 10 mM sodium butyrate, 1 mM TSA and protease/phosphatase inhibitors cocktail) and the lysates were used for immunoprecipitation.
Quantitative real time PCR
Total RNA was isolated by using Trizol (Life Technology, USA), 2 μg total RNA was reverse transcribed into cDNA using the iScript™cDNA Synthesis Kit (Bio-Rad, USA). PowerUp SYBR Green Master Mix was applied to conduct quantitative real time PCR. L32 was used as internal reference. Primer sequences are provided in Table S1.
Fatty acid oxidation assays
To measure peroxisomal β-oxidation, primary hepatocytes were isolated from Acox1lox/lox Cre negative mice and immortalized as previously described (Park et al., 2019). The hepatocytes were infected with either lacZ or Cre lentivirus followed by puromycin selection after 2 days infection. The cells were harvested for fatty acid isolation after the addition of D3-C22:0 for three days. Fatty acid isolation and mass spectrometry analysis were performed as previously described (Park et al., 2019).
To measure mitochondrial fatty acid oxidation, 200 mg freshly isolated liver was homogenized in 1ml STE buffer (0.25 M sucrose, 10 mM Tris-HCl, 1 mM EDTA, pH to 7.4) as previously described (Huynh et al., 2014). Briefly, liver homogenates were centrifuged at 400g for 10 min, and then 30 μl supernatant was added into 370 μl reaction mixture (100 mM Sucrose, 10 mM Tris-HCl, 5 mM KH2PO4, 0.2 mM EDTA, 80 mM KCl, 1 mM MgCl2, 2 mM L-Carnitine, 0.1 mM Malate, 0.05 mM Coenzyme A, 2 mM ATP, 1 mM DTT, 7% BSA, 5 mM palmitate and 0.4 μCi C14-palmitate). The reaction was stopped by the addition of 200 μl of 1M perchloric acid after incubating at 37 oC for 30 min. Carbon dioxide generated from mitochondria fatty acid oxidation was trapped by sodium hydroxide in a filter paper disc, which was placed in the lid of the reaction tube. Carbon dioxide trapped in the filter paper disc was transferred into scintillation vial with 4 ml scintillation fluid and the average counts per minute over 3 min was measured with a standard scintillation counter.
Very long chain fatty acid mass spectrometric analysis
Extraction of fatty acids from liver and serum samples was performed as previously described (Park et al., 2019). Briefly, 300 μg protein of liver homogenate or 10 μl serum with 60 pmol heptadecanoic acid was subjected to acid hydrolysis and fatty acids were extracted with hexane. The isolated fatty acids were analyzed by electrospray ionization mass spectrometry (ESI-MS) using a Thermo LTQ Orbitrap Velos mass spectrometer, operated using Xcalibur software. Five microliters of each sample were loop injected into the mass spectrometer with a built-in injector and syringe pump, which delivered a constant flow of 20 μl/min of methanol with 0.5% NH4OH. The instrument was operated with a resolution of 100,000 (at m/z 400) that allows accurate mass measurements of the VLCFA species and isolation of isobaric isomers to enable a very precise measurement of the intensities of all the FA species in the samples. The final mass spectra (scan range: 100–500 Da in the negative-ion mode) were a signal average of > 15 individual mass spectra from the eluted peaks, and a subtraction of the leading edge background mass spectrum. The peak list with measured m/z of all the ions including FA, the relative intensity, the theoretical m/z, the deviation (mDa), and the extracted elemental composition were exported to an Excel file for further data analysis.
Triglyceride secretion assay
Triglyceride secretion rate was determined as previously described (Singh et al., 2009). Briefly, mice were subjected to overnight fast, bled, and then IP injected with 1g per kg of poloxamer 407. The mice were bled again 5 hours after the injection. The serum TG content was measured and the rate of secretion was calculated.
Plasmids
pLVx-mCherry-GFP-LC3 plasmid was a generous gift from Abhinav Diwan (Ma et al., 2012). For the peroxisome targeted fluorescent ROS sensor construct, pLJM1-Matrix-RoGFP was used as template to amplify RoGFP by using primers RoGFP-Agel-Fwd: CGG CGA CCG GTG CCA CCA TGG TGA GCA AGG GCG AGG A and RoGFP-SKL-EcoRI-Rev: CGC CGG AAT TCT CAC AGC TTG CTG CGC TTG TAC AGC TCG TCC ATG CC. Purified RoGFP-SKL was inserted in place of GFP in pLJM1-EGFP (addgene #19319) after digestion with AgeI and EcoRI. To generate a FLAG- tagged Raptor construct, Raptor-GFP Flag fused ORF was amplified using pLJM1Raptor-GFP (addgene #112744) as template, and the primers used were NheI_Fwd: CGG CGG CTA GCG CCA CCA TGG AGT CCG AAA TGC TGC A, and EcoRI_Flag_Rev: CGC CGG AAT TCT TAC TTG TCG TCA TCG TCT TTG TAG TCC TTG TAC AGC TCG TCC ATG C. The EGFP of pLJM1-EGFP (addgene #19319) was replaced with Raptor-GFP-Flag to make a pLJM1-raptor-GFP-Flag plasmid. Acox1 ORF was amplified using primers Acox1_NheI_Fwd: GCC GCG CTA GCG CCG CCA CCA TGT ACC CAT and Acox1_EcoRI_Rev: GCG GCG AAT TCT CAA AGC TTC GAC TGC AGG and cloned into the backbone plasmid AAV-ALBp-3’iALB provided by Vector Biolabs. The virus packaging and production was performed by Vector Biolabs. All plasmids were prepared by using EndoFree Plasmid Maxi Kit (Qiagen, Germany).
Immunofluorescence and confocal microscopic imaging
Frozen sections were fixed with ethanol or 4% paraformaldehyde, followed by primary antibody and the corresponding secondary antibody incubation. The slides were imaged using a Nikon AIRsi Confocal Microscope. The fluorescent intensity and colocalization were calculated using ImageJ.
Metabolite measurements
Triglyceride (TR22421, Thermo Fisher Scientific), non-esterified fatty acid (NEFA) (FUJIFILM), Cholesterol (Cat# 999–02601, FUJIFILM), glycogen (K646, Biovision), and acetyl-CoA (K317, Biovision) were measured according to the instructions provided by the manufacturers. Where values of metabolites under fasting and fed conditions are reported, the mice were fasted for 24–48 hrs as indicated. The fed condition represents mice that were fasted for 6 hrs and then re-fed, with the analysis performed 4 hrs after the refeeding. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) measurements were performed by the Core Laboratory for Clinical Studies at Washington University Medical School using reagents from Roche Diagnostics. The assays were performed according to the manufacturer’s instructions and were run on a Roche Cobas C501 system. Both assays are coupled enzyme assays that measure a decrease in absorbance as NADH is oxidized, which is proportional to enzyme activity.
Glycogen staining
Livers were harvested from fed Acox1-LKO and control mice. Liver sections were subjected to Periodic Acid Schiff (PAS) staining, which was performed by the Musculoskeletal Histology and Morphometry Core at Washington University School of Medicine. Briefly, frozen sections were oxidized in 0.5% periodic acid solution for 5 minutes and then placed in Schiff reagent for 15 minutes after rinsing with distilled water. The sections were counter-stained in Mayer’s hematoxylin for 1 minute. The slides were dehydrated and coverslips were mounted using a mounting medium.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results with error bars express mean ± SEM. Statistical analysis was performed by using Student’s two-tailed t-test. A P value less than 0.05 was considered significant. Statistical significance is represented as follows: *p < 0.05, **p <0.01, ***p < 0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-phospho-P70S6K (Thr389) | Cell Signaling Technology | Cat# 9205; RRID:AB_2734746 |
| Rabbit monoclonal anti-p70 S6 Kinase | Cell Signaling Technology | Cat# 2708; RRID:AB_390722 |
| Rabbit monoclonal anti-phospho-mTOR (Ser2448) | Cell Signaling Technology | Cat# 2976; RRID:AB_490932 |
| Rabbit polyclonal anti-p62 | Cell Signaling Technology | Cat# 5114; RRID:AB_10624872 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Rabbit polyclonal anti-Acox1 | Proteintech | Cat# 10957-1-AP; RRID:AB_2221670 |
| Rabbit polyclonal anti-Perilipin2 | Proteintech | Cat# 15294-1-AP; |
| Rabbit monoclonal anti-Raptor | Cell Signaling Technology | Cat# 2280; RRID:AB_561245 |
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | Cat# 2983; RRID:AB_2105622 |
| Mouse monoclonal anti-LC3b | Cell Signaling Technology | Cat# 83506; RRID:AB_2800018 |
| Rabbit monoclonal anti-LC3b | Cell Signaling Technology | Cat# 2775; RRID:AB_915950 |
| Rabbit polyclonal anti-Perilipin2 | Thermo Fisher | Cat# PA1-1052; RRID:AB_2268558) |
| Rabbit monoclonal anti-phospho-ULK1(Ser757) | Cell Signaling Technology | Cat# 14202; RRID:AB_2665508 |
| Rabbit polyclonal anti-ULK1 | Sigma-Aldrich | Cat# A7481; RRID:AB_1840703 |
| Rabbit polyclonal anti-Raptor | Proteintech | Cat# 20984-1-AP |
| Rabbit polyclonal Anti-acetyl Lysine | Abcam | Cat# ab21623; RRID:AB_11182390 |
| Rat monoclonal anti-LAMP2 | Abcam | Cat# ab13524; RRID:AB_2134736 |
| Rabbit polyclonal anti-phospho-AMPK (Thr172) | Cell Signaling Technology | Cat# 2531; RRID:AB_330330 |
| Rabbit polyclonal anti-AMPK | Cell Signaling Technology | Cat# 2532; RRID:AB_330331 |
| Rabbit polyclonal anti-FASN | Abcam | Cat# ab22759; RRID:AB_732316 |
| Rabbit polyclonal anti-Actin | Santa Cruz Biotechnology | Cat# SC-1616-R |
| Rabbit polyclonal anti-Acss2 | Proteintech | Cat# 16087-1-AP |
| Rabbit polyclonal anti-Acly | Proteintech | Cat# 15421-1-AP; RRID:AB_2223741 |
| Rabbit polyclonal anti-Histone H3 | Cell Signaling Technology | Cat# 9715; RRID:AB_331563 |
| Rabbit monoclonal anti-ATG5 | Cell Signaling Technology | Cat# 12994; RRID:AB_2630393 |
| Mouse monoclonal anti-ATGL | Santa Cruz Biotechnology | Cat# sc-365278; RRID:AB_10859044 |
| Rabbit polyclonal anti-PMP70 | Sigma-Aldrich | Cat# P0090, RRID:AB_1841112 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher | Cat# A-11008; RRID: AB_143165 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher | Cat# A-11007, RRID:AB_141374 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher | Cat# A-11012, RRID:AB_141359 |
| Bacterial and Virus Strains | ||
| E.coli: Stbl3 | NEB | Cat# C3040I |
| AAV8-ALB-eGFP | Vector Biolabs | Cat# VB1586 |
| AAV8-ALB-Acox1 | Vector Biolabs | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chloroquine | Sigma-Aldrich | Cat# C6628, CAS: 50-63-5 |
| Leupeptin | Cayman | Cat# 14026, CAS: 103476-89-7 |
| Bodipy | Thermo Fisher | Cat# D3922, CAS: 121207-31-6 |
| DAPI | Sigma-Aldrich | Cat# 10236276001, CAS: 28718-90-3 |
| Dichloroacetic acid | Sigma-Aldrich | Cat# D54702, CAS: 79-43-6 |
| Sodium butyrate | Sigma-Aldrich | Cat# B5887, CAS: 156-54-7 |
| Trichostatin A | Sigma-Aldrich | Cat# T8552, CAS: 58880-19-6 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Protease and phosphatase inhibitor cocktail | Sigma-Aldrich | Cat# PPC1010 |
| Sucrose | Sigma-Aldrich | Cat# S8501, CAS: 57-50-1 |
| L-Carnitine | Cayman | Cat# 21489, CAS: 541-15-1 |
| Malate | Sigma-Aldrich | Cat# 02300, CAS: 636-61-3 |
| Coenzyme A | Cayman | Cat# 16147, CAS: 85-61-0 |
| ATP | Cayman | Cat# 14498, CAS: 987-65-5 |
| DTT | Cayman | Cat# 700416, CAS: 3483-12-3 |
| Palmitate | Sigma-Aldrich | Cat# P0500, CAS: 57-10-3 |
| Palmitic acid [1-14C] | American Radiolabeled Chemicals | Cat# ARC-0172A |
| Heptadecanoic acid | Cayman | Cat# 19722, CAS: 506-12-7 |
| Perchloric acid | Sigma-Aldrich | Cat# 244252, CAS: 7601-90-3 |
| Sodium hydroxide | Sigma-Aldrich | Cat# S0899, CAS: 1310-73-2 |
| Critical Commercial Assays | ||
| Triglyceride kit | Thermo Fisher | Cat# TR22421 |
| Non-esterified fatty acid kit | FUJIFILM | Cat# 999-34691 |
| Cholesterol kit | FUJIFILM | Cat# 999-02601 |
| Glycogen assay kit | Biovision | Cat# K646 |
| Acetyl-CoA kit | Biovision | Cat# K317 |
| Malonyl-CoA kit | Mybiosource | Cat# MBS705127 |
| Signal-Seeker™ Acetyl-Lysine Detection kit | Cytoskeleton | Cat# BK163 |
| Deposited Data | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Acox1Lox/Lox | (Park et al., 2019) | N/A |
| Mouse: Albumin-Cre | (Postic et al., 1999) Jackson Laboratories | Cat# 003574 |
| Mouse: ATG5Lox/Lox | (Hara et al., 2006) | N/A |
| Oligonucleotides | ||
| qRT-PCR primers, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pLVx-mCherry-GFP-LC3 | N/A | (Ma et al., 2012) |
| pLJM1 Matrix-RoGFP | N/A | (Park et al., 2019) |
| pLJM1 RoGFP-SKL | This paper | Expression construct generated in the lab |
| pLJM1 Raptor-GFP-FLAG | This paper | Expression construct generated in the lab |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Image Studio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/index |
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885-889.
Ma, X., Liu, H., Foyil, S.R., Godar, R.J., Weinheimer, C.J., and Diwan, A. (2012). Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy 8, 1394-1396.
Park, H., He, A., Tan, M., Johnson, J.M., Dean, J.M., Pietka, T.A., Chen, Y., Zhang, X., Hsu, F.F., Razani, B., et al. (2019). Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J Clin Invest 129, 694-711.
Postic, C., Shiota, M., Niswender, K.D., Jetton, T.L., Chen, Y., Moates, J.M., Shelton, K.D., Lindner, J., Cherrington, A.D., and Magnuson, M.A. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274, 305-315.
Highlights.
Fasting or high fat diet increases the liver gene expression of Acox1
Acox1-LKO mice are protected against fatty liver through induction of lipophagy
Acetyl-CoA from Acox1-mediated β-oxidation activates mTORC1 and inhibits lipophagy
Pharmacologically increasing acetyl-CoA rescues the phenotype in Acox1-LKO mice
Acknowledgments
This work was supported by NIH grants DK115867 and DK118333 and by funds from the Washington University-Centene Corporation Personalized Medicine Initiative. The core services of the Washington University Diabetes Research Center (DK020579) and the Nutrition Obesity Research Center (DK056341) also provided support for this work. Mass spectrometric analysis was conducted by the Biomedical Mass Spectrometry Research Resource at Washington University, which is supported by GM103422 and DK020579. Confocal microscopy was performed through the use of Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CD-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770 and 4642).
Footnotes
Declaration of interests
The authors declare that there are no conflicts of interest.
Supplementary Material
Supplemental Information includes five figures and one table.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carrer A, Parris JL, Trefely S, Henry RA, Montgomery DC, Torres A, Viola JM, Kuo YM, Blair IA, Meier JL, et al. (2017). Impact of a High-fat Diet on Tissue Acyl-CoA and Histone Acetylation Levels. J Biol Chem 292, 3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu BB, Liao YC, Qi W, Xie C, Du X, Wang J, Yang H, Miao HH, Li BL, and Song BL (2015). Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306. [DOI] [PubMed] [Google Scholar]
- Cingolani F, and Czaja MJ (2016). Regulation and Functions of Autophagic Lipolysis. Trends Endocrinol Metab 27, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Sathyanarayan A, Lopresti M, Aghajan M, Chen C, and Mashek DG (2020). Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBosch BJ, Heitmeier MR, Mayer AL, Higgins CB, Crowley JR, Kraft TE, Chi M, Newberry EP, Chen Z, Finck BN, et al. (2016). Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal 9, ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TD, Sergin I, Zhang X, and Razani B (2017). Target acquired: Selective autophagy in cardiometabolic disease. Sci Signal 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini E, Sullivan S, and Klein S (2010). Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CY, Pan J, Chu R, Lee D, Kluckman KD, Usuda N, Singh I, Yeldandi AV, Rao MS, Maeda N, et al. (1996). Hepatocellular and Hepatic Peroxisomal Alterations in Mice with a Disrupted Peroxisomal Fatty Acyl-coenzyme A Oxidase Gene. Journal of Biological Chemistry 271, 24698–24710. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. [DOI] [PubMed] [Google Scholar]
- Hayashi H, and Oohashi M (1995). Incorporation of acetyl-CoA generated from peroxisomal beta-oxidation into ethanolamine plasmalogen of rat liver. Biochim Biophys Acta 1254, 319–325. [DOI] [PubMed] [Google Scholar]
- Huang J, Viswakarma N, Yu S, Jia Y, Bai L, Vluggens A, Cherkaoui-Malki M, Khan M, Singh I, Yang G, et al. (2011). Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. The American journal of pathology 179, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh FK, Green MF, Koves TR, and Hirschey MD (2014). Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol 542, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan K-L (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology 13, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Lee J, Choi J, Scafidi S, and Wolfgang MJ (2016). Hepatic fatty acid oxidation restrains systemic catabolism during starvation. Cell reports 16, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chao X, Yang L, Lu Q, Li T, Ding WX, and Ni HM (2018). Impaired Fasting-Induced Adaptive Lipid Droplet Biogenesis in Liver-Specific Atg5-Deficient Mouse Liver Is Mediated by Persistent Nuclear Factor-Like 2 Activation. The American journal of pathology 188, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, and Yin XM (2013). Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol 58, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Song Y, and Liu D (1999). Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther 6, 1258–1266. [DOI] [PubMed] [Google Scholar]
- Liu S, Brown JD, Stanya KJ, Homan E, Leidl M, Inouye K, Bhargava P, Gangl MR, Dai L, Hatano B, et al. (2013). A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 502, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, and Semenkovich CF (2014). Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab 19, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, and Diwan A (2012). Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy 8, 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, and Niso-Santano M (2014). Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular cell 53, 710–725. [DOI] [PubMed] [Google Scholar]
- Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, and Reggiori F (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsenego J, Mansouri A, Akkaoui M, Lenoir V, Esnous C, Fauveau V, Tavernier V, Girard J, and Prip-Buus C (2012). Enhancing liver mitochondrial fatty acid oxidation capacity in obese mice improves insulin sensitivity independently of hepatic steatosis. J Hepatol 56, 632–639. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernandez ME, Giles DA, Stankiewicz TE, Sheridan R, Karns R, Cappelletti M, Lampe K, Mukherjee R, Sina C, and Sallese A (2018). Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni HM, Woolbright BL, Williams J, Copple B, Cui W, Luyendyk JP, Jaeschke H, and Ding WX (2014). Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol 61, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, He A, Tan M, Johnson JM, Dean JM, Pietka TA, Chen Y, Zhang X, Hsu FF, Razani B, et al. (2019). Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J Clin Invest 129, 694–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL, Brunengraber H, and Des Rosiers C (2004). Peroxisomal fatty acid oxidation is a substantial source of the acetyl moiety of malonyl-CoA in rat heart. J Biol Chem 279, 19574–19579. [DOI] [PubMed] [Google Scholar]
- Reth M (2002). Hydrogen peroxide as second messenger in lymphocyte activation. Nature immunology 3, 1129. [DOI] [PubMed] [Google Scholar]
- Rinella ME (2015). Nonalcoholic fatty liver disease: a systematic review. Jama 313, 2263–2273. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. (2013). TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan R, Lampe K, Shanmukhappa SK, Putnam P, Keddache M, Divanovic S, Bezerra J, and Hoebe K (2011). Lampe1: an ENU-germline mutation causing spontaneous hepatosteatosis identified through targeted exon-enrichment and next-generation sequencing. PLoS One 6, e21979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, lizuka Y, Ohashi K, Harada K, et al. (1999). Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem 274, 35832–35839. [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. (2012). Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest 122, 2428–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son SM, Park SJ, Lee H, Siddiqi F, Lee JE, Menzies FM, and Rubinsztein DC (2018). Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Veldhoven PP (2010). Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 51, 2863–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violante S, Achetib N, van Roermund CW, Hagen J, Dodatko T, Vaz FM, Waterham HR, Chen H, Baes M, and Yu C (2018). Peroxisomes can oxidize medium-and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. The FASEB Journal, fj. 201801498R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Singh R, Xiang Y, and Czaja MJ (2010). Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 52, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, and Schumacker PT (2010). Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse S, and Randle PJ (1973). Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate (Short Communication). Biochem J 134, 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Tao R, DePinho RA, and Dong XC (2012). The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem 287, 39107–39114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li P, Fu S, Calay ES, and Hotamisligil GS (2010). Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 11, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, and Wymer M (2016). Global epidemiology of nonalcoholic fatty liver disease—meta - analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Zechner R, Madeo F, and Kratky D (2017). Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol 18, 671–684. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, and Han JM (2013). A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nature cell biology 15, 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.