

Introduction
The aorta is the largest artery and can be divided into several segments (Fig 1). The diaphragm divides the aorta into the thoracic and abdominal aorta. The thoracic aorta extends from the heart to the diaphragm and comprises the following 3 segments: the ascending thoracic aorta, the aortic arch, and the descending thoracic aorta. The ascending aorta originates at the aortic root, comprising the aortic valve and sinuses of Valsalva from which the coronary artery ostia arise, and ends just proximal to the origin of the innominate artery. The transverse aortic arch is the segment from which the innominate, left common carotid, and left subclavian arteries arise. The descending thoracic aorta extends from just beyond the origin of the left subclavian artery to the diaphragm. Below the diaphragm, the abdominal aorta is divided into suprarenal and infrarenal segments.
Fig. 1.
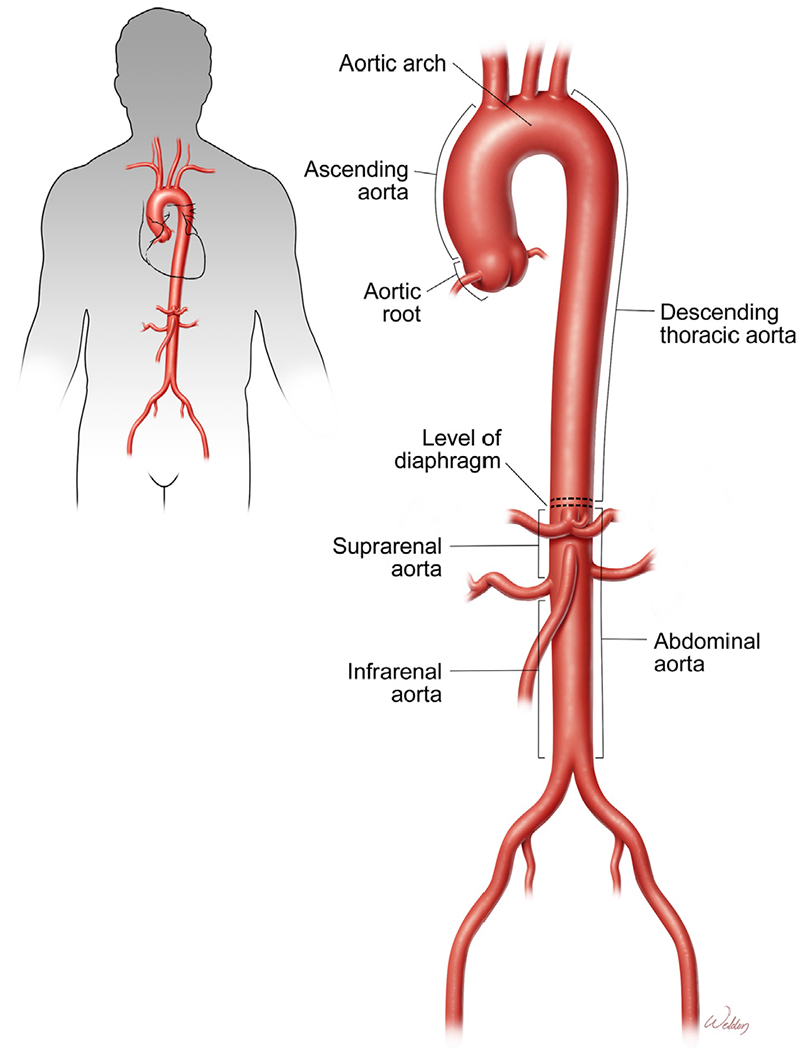
Aortic segments. The aorta comprises the ascending thoracic aorta, aortic arch, descending thoracic aorta, suprarenal aorta, and infrarenal aorta. The diaphragm divides the aorta into the thoracic and abdominal aorta. (Color version of figure is available online.)
Aortic aneurysms and dissections (AAD) are common diseases that can cause aortic rupture and other life-threatening complications. Aortic aneurysm occurs when the progressive weakening of the aortic wall causes the aorta to enlarge to a diameter of at least 1.5 times greater than normal (Fig 2A). Aortic dissection occurs when a tear forms within the aortic wall and causes blood to flow between the layers, thereby separating them and creating a false lumen with a severely weakened outer aortic wall (Fig 2B). Aneurysms and dissections may involve one or more aortic segments and are named accordingly. Thoracoabdominal aortic aneurysms are those that extend through the diaphragm, involving the descending thoracic aorta and the abdominal aorta in continuity.
Fig. 2.
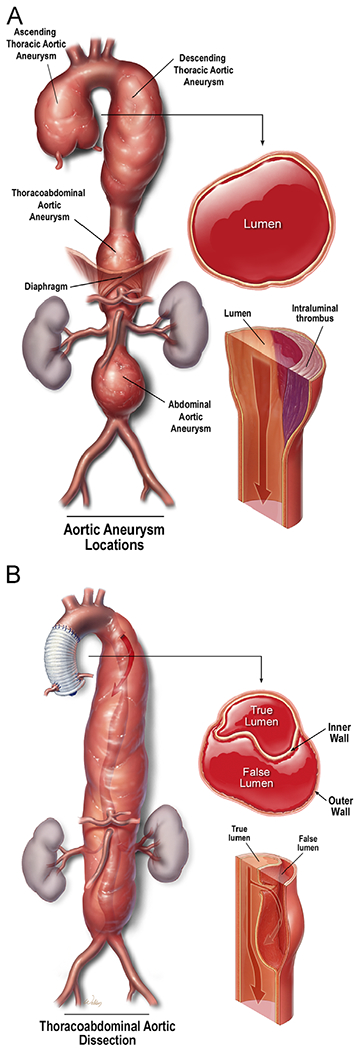
Aortic aneurysms and dissections. (A) Aortic aneurysm occurs when the progressive weakening of the aortic wall causes the aorta to enlarge to a diameter of at least 1.5 times greater than normal. (B) Aortic dissection occurs when a tear forms within the aortic wall and causes blood to flow between the layers, thereby creating a false lumen. (Color version of figure is available online.)
The incidence of thoracic AAD (TAAD) is estimated to be 9-16 cases per 100,000 individuals per year,1,2 with more cases occurring in men than in women (16.3 vs 9.1 cases per 100,000 individuals per year for men and women, respectively).2 Of all TAADs, 60% involve the aortic root, ascending aorta, or both; 10% involve the aortic arch; 40% involve the descending thoracic aorta; and 10% involve the thoracoabdominal aorta.3 Thoracic aortic dissection (TAD) is estimated to occur at a rate of 3 cases per 100,000 individuals per year.4–7 The prevalence of infrarenal abdominal aortic aneurysms (AAA) is estimated to be between 2.2% and 5% in men older than 55 years of age.3,8,9
AAD are highly lethal conditions that often necessitate surgical treatment. Operative treatment generally involves replacing the diseased segment with a prosthetic graft by using an open surgical, endovascular, or hybrid approach. Despite significant improvements in the surgical treatment of AAD, they cause more than 10,000 deaths in the United States each year. Although AAD are a leading cause of death in people 55 years of age or older,10,11 AAD are also a significant cause of morbidity and mortality in children and young adults.12 Recent reports indicate that the mortality rate of acute TAAD is 16%.2 AAD are particularly lethal when they involve the ascending aorta. The current incidence of in-hospital death is 24% for patients presenting with acute ascending aortic dissection (ie, DeBakey type I or II dissection and Stanford type A dissection).13
From an etiologic standpoint, TAAD can be classified as either genetically triggered or sporadic. Less than 30% of all TAAD cases are genetically triggered, whereas more than 70% are sporadic.14–16 Genetically triggered TAAD are caused by mutations in genes encoding proteins such as smooth muscle (SM) contractile proteins,17 extracellular matrix (ECM) proteins, and proteins involved in transforming growth factor beta (TGF-β) signaling.18,19 Sporadic TAAD are mainly associated with risk factors such as aging,16,20–24 male sex,21,25 smoking,20,22,23,26,27 and hypertension.21,24,27–29 Sporadic TAAD and AAA share similarities in risk factors and in pathogenesis. In both genetically triggered and sporadic AAD, the upregulation of common pathways such as reactive oxygen species (ROS) production and stress signaling activation can cause SM cell (SMC) dysfunction and death, ECM destruction, and aortic inflammation, which all contribute to the progression of these diseases.
Although surgical approaches for treating AAD have become more advanced and less invasive, an urgent need remains for new medical approaches that prevent disease progression. Although a few drugs, such as beta-blockers and angiotensin II receptor antagonists, can slow disease progression in some patients with genetically triggered aortopathy, no medications are widely effective in preventing or halting the disease. A better understanding of the molecular mechanisms underlying AAD initiation, progression, and rupture is important for developing effective medications to treat these diseases. In this review, we summarize the major progress that has been made in our understanding of the pathogenesis underlying genetically triggered and sporadic forms of AAD.
Normal structure and function of the aorta
Histology of the aortic wall
The aortic wall is composed of 3 layers (Fig 3A): a thin inner layer (the intima), a thick medial layer (the media), and a thin outer layer (the tunica adventitia). The intima is lined with a monolayer of endothelial cells that not only separates the aortic wall from circulating blood but also regulates vascular functions.30
Fig. 3.
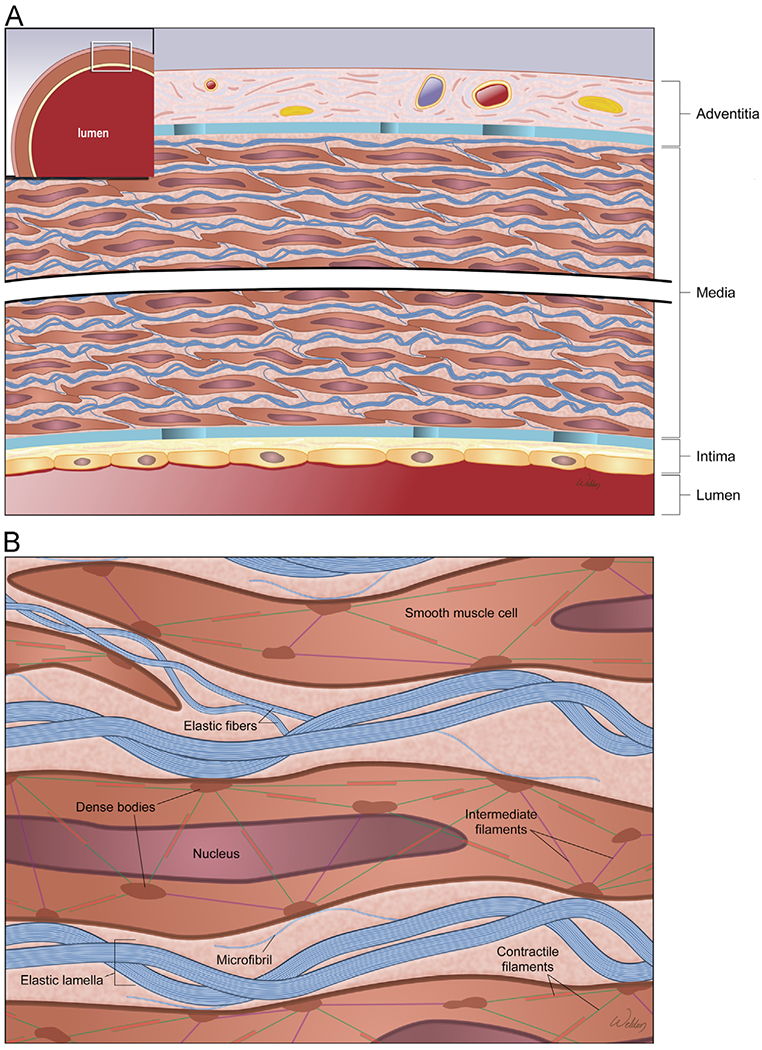
Structure of the aortic wall. (A) The aortic wall is composed of a thin inner layer (the intima), a thick medial layer (the media), and a thin outer layer (the tunica adventitia). (B) The lamellar unit is composed of smooth muscle cells (SMCs) sandwiched between 2 layers of elastic fibers and surrounded by collagen bundles. (Color version of figure is available online.)
The media is composed of SMCs within an ECM of elastic fibers, collagen, adhesive proteins (eg, laminin and fibronectin), and proteoglycans. The aortic media is critical in controlling aortic biomechanical functions, including elasticity and compliance (ie, the ability to distend and recoil), tensile stiffness (ie, changes in stress associated with changes in strain), and tensile strength (ie, maximum stress sustained without failure). Elastic fibers contribute to aortic compliance, whereas collagen fibers contribute to stiffness and strength.31
The aortic adventitia contains cells such as fibroblasts and adipocytes, ECM containing collagen and elastin, vasa vasorum and lymphatics, and perivascular nerves. The aortic adventitia not only provides nourishment to the aortic wall, but it is also an important gateway for inflammatory cell infiltration into the aortic wall. Recent studies have shown that the adventitia contains abundant stem cell or progenitor cell niches. Furthermore, a growing body of evidence supports a critical role of the adventitia in vascular injury, repair, inflammation, and remodeling.32
The lamellar unit, elastin-contractile unit, and mechanotransduction
The lamellar unit
The lamellar unit is the basic structural and functional unit of the aortic media. Each lamellar unit is composed of SMCs sandwiched between 2 layers of elastic fibers and surrounded by collagen bundles (Fig 3B). The connection between the SMCs and the surrounding ECM is important for maintaining the integrity of aortic structure and function.
The elastin-contractile unit
The elastin-contractile unit (Fig 4) is a unique configuration of elastic fibers, focal adhesions or dense plaques in the membrane, and contractile filaments inside the cell. The elastin-contractile unit33 transmits forces from the elastic fibers to the SMCs and serves as a mechanosensor unit of the aortic wall.31,34
Fig. 4.
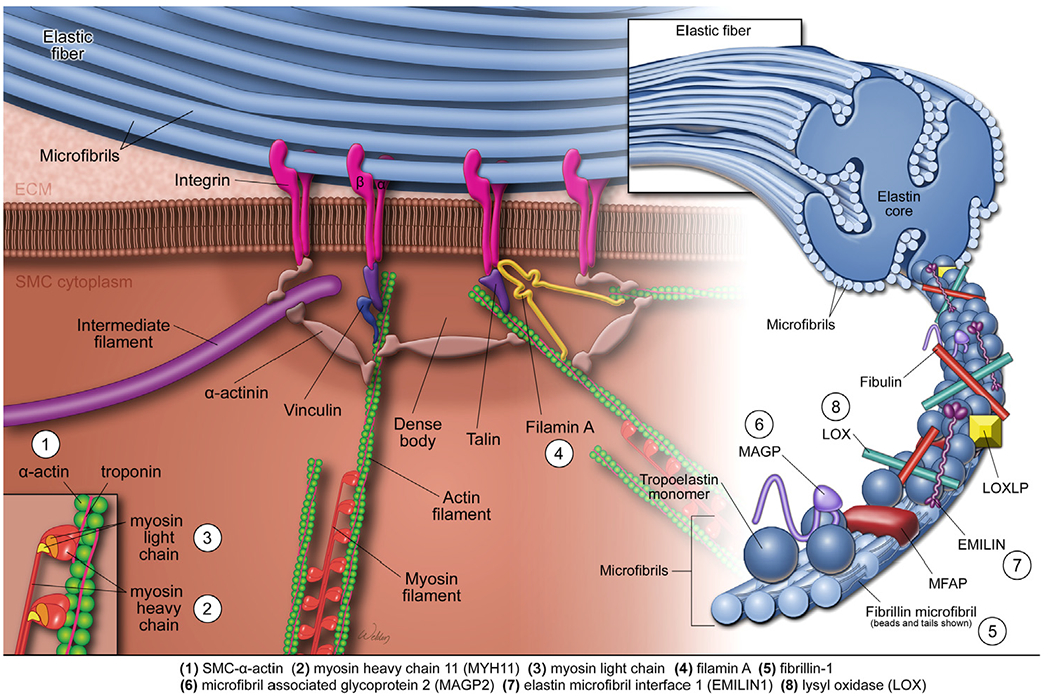
The elastin-contractile unit and mutations affecting aortic contractile function. The elastin-contractile unit is a unique configuration of elastic fibers, focal adhesions or dense plaques in the membrane, and contractile filaments inside the SMCs. The elastic fibers are organized as a core of elastin surrounded by microfibrils that are composed of fibrillin, microfibril-associated glycoproteins (MAGPs), elastin microfibril interfacer protein 1 (EMILIN1), fibulins, and other glycoproteins. An SMC contractile unit is composed of actin-containing thin filaments and myosin-containing thick filaments, along with regulatory proteins. Mechanical stimuli are transmitted from the elastic fiber, through the focal adhesions or dense plaques in the membrane, through the anchoring proteins or actin linkage proteins, and to the contractile unit, leading to the activation of SMC contraction. Genetic thoracic aortic aneurysms and dissections (TAAD) are associated with mutations in genes encoding proteins that control the structure and function of the elastin-contractile unit. These proteins include SM α-actin, myosin heavy chain 11 (MYH11), myosin light chain kinase (MYLK), filamin A, fibrillin-1, microfibril-associated glycoprotein 2 (MAGP2), EMILIN1, and lysyl oxidase (LOX). (Color version of figure is available online.)
Elastic fibers.
The elastic fibers (Fig 4) are organized as a core of elastin surrounded by microfibrils that are composed of fibrillin, microfibril-associated glycoproteins (MAGPs), elastin microfibril interfacer protein 1 (EMILIN1), fibulins, and other glycoproteins. Under normal conditions, elastin molecules are synthesized in SMCs and deposited into the ECM, where they are aligned and cross-linked to form elastic fibers. Elastin is believed to have a half-life of approximately 50 years in humans; most of the body’s elastic fibers are produced before adulthood.31,34,35
Focal adhesions or dense plaques.
Focal adhesions or dense plaques (Fig 4) on the SMC membrane contain integrin receptors composed of α and β subunits. Dense plaques connect to the fibrillin-containing microfibrils in elastin fibers.33,36 The connection of dense plaques in the SMC membrane to the fibrillins in elastic fibers contributes to the stability of elastic fibers.36,37 Dense plaques connect to the α-actin filaments in the contractile unit through actin linkage proteins or intracellular anchoring proteins, which are composed primarily of talin, vinculin, α-actinin, and filamin A.
SMC contractile unit.
A SMC contractile unit (Fig 4) is composed of actin-containing thin filaments and myosin-containing thick filaments, along with regulatory proteins. The thin filaments are composed of SM α-actin and regulatory proteins, including tropomyosin. Myosin thick filaments are composed of myosin heavy chains, essential light chains, and regulatory light chains. The signaling-induced phosphorylation of the regulatory light chains induces the formation of a cross-bridge between actin and myosin filaments, as well as the motion of the actin thin filaments, resulting in SMC contraction. The SMC contractile units are connected with cell membrane, cytoskeletal filaments, dense bodies, and nuclei. This arrangement of filaments intertwined with cellular components is necessary for the mechanotransduction properties of the SMC contractile unit.31 SMC contractile force requires SM α-actin assembly in the thin filaments38 and SM α-actin polymerization in the subcortical region.39
Mechanotransduction
The contractile activity of SMCs is fundamental to aortic functions.31 Mechanical stimuli, such as transmural pulse pressure, are transmitted from the elastic fiber, through the focal adhesions or dense plaques in the membrane, through the anchoring proteins or actin linkage proteins, and to the contractile unit, leading to the activation of SMC contraction (Fig 4).31 Mechanical signals also induce the influx of calcium; calcium then binds to calmodulin to form the calcium-calmodulin complex that leads to the activation of the myosin light chain kinase (MYLK), phosphorylation of the regulatory light chains in the myosin complex, and activation of actin-dependent ATPase activity in the myosin globular motor head, leading to SMC contraction.
Heterogeneity of the aorta in different segments
The heterogeneity of aortic structure
Aortic segments differ in structure and function.40–42 The ascending aorta is where the pressure is the highest and the thickness of the aortic media is the greatest. The aortic media becomes thinner as the aorta descends into the abdomen. The aortic root and ascending aorta are composed of an elastic wall that contains multiple elastic lamina and SMCs.40 The abundance of elastin in these segments contributes to the distensible properties of the aorta that are essential for responding to blood volume and pressure changes and withstanding the pulsatile force during the cardiac cycle.43 The blood velocity in the descending thoracic aorta is substantially decreased when compared to that in the ascending aorta and aortic arch. Likewise, the extracellular components in the descending thoracic aorta differ greatly from those in the ascending aorta.44,45
The heterogeneity of aortic SMC origin
Heterogeneity in the origin of aortic SMCs has also been observed among aortic segments. SMCs in the ascending aorta are derived from neural crest cells,46,47 whereas SMCs in the descending thoracic aorta are derived from the somitic mesoderm.48 There is abundant evidence showing that vascular SMCs with different embryonic origins have distinct gene expression profiles and characteristics.49–51 It has been suggested that the embryonic origin of vascular SMCs may play a role in their response to changes in the environment.49,50 Interestingly, a study recently showed that vascular SMCs from the ascending and descending thoracic aorta displayed distinct differences in migratory properties and contractile activity in embryonic mice; however, in adult mice, these functions were indistinguishable between the 2 domains.51 Thus, it remains to be determined whether the embryonic origin of SMCs dictates their phenotype and response to the environment in the adult vascular wall.40,47
Genetic TAAD
Proper aortic function requires the intact structure of the elastin-contractile unit, as well as the efficient signaling that controls the function of aortic cells. Defects in these structural and functional components result in compromised aortic homeostasis, rendering the aorta vulnerable to failure.31 A growing body of evidence supports a new theory that altered cell-matrix connections in the contractile-elastic unit play an important role in the development of TAAD.17,36,52–55 Genetic TAAD are associated with mutations in genes encoding proteins that control the structure and function of the elastin-contractile unit (Fig 4) and signaling pathway proteins (eg, TGF-β (Fig 5)) that control the maturation and functions of aortic cells.
Fig. 5.
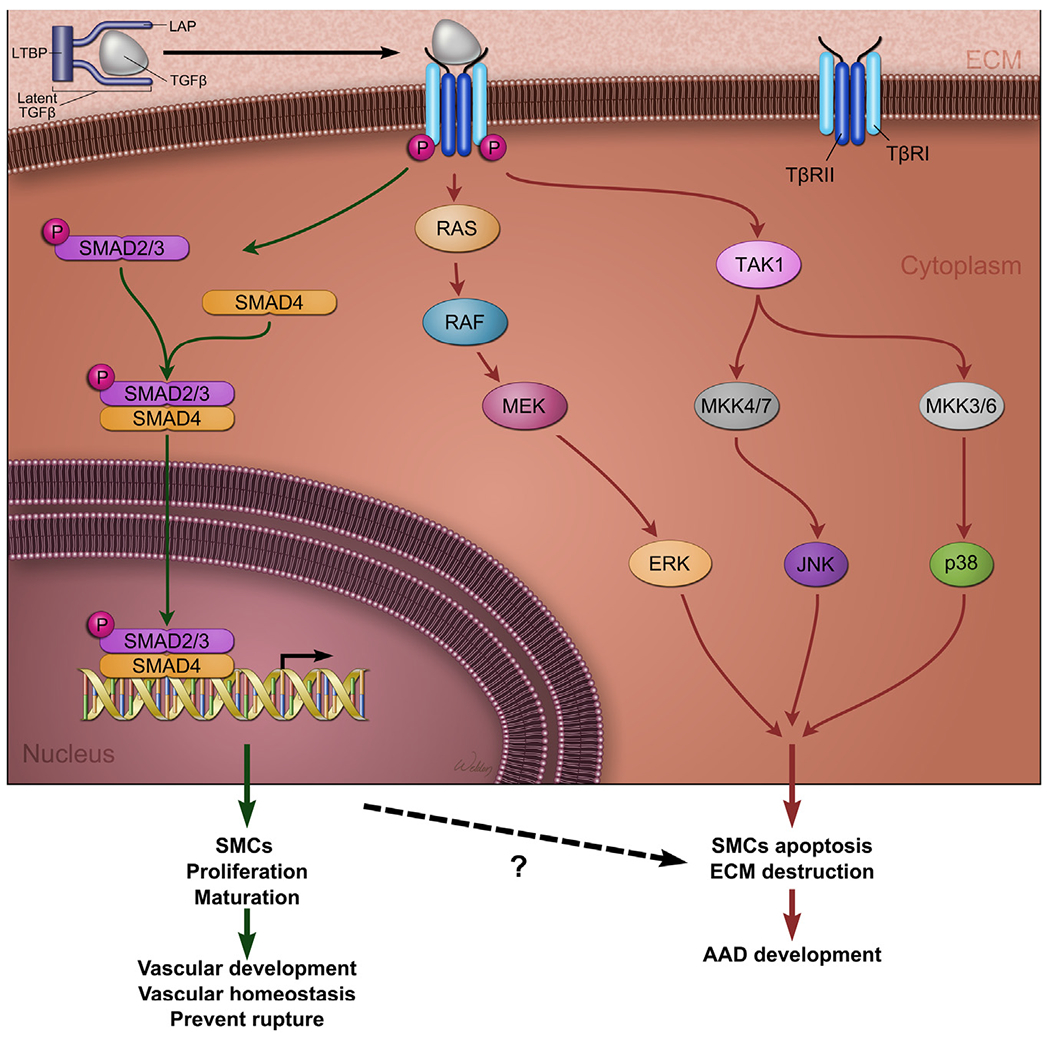
Transforming growth factor β (TGF-β) signaling in aortic aneurysms and dissections (AAD). TGF-β ligands are produced as a large latent TGF-β complex consisting of mature TGF-β, latency-associated peptide (LAP), and a latent TGF-β-binding protein (LTBP). TGF-β activates canonical and noncanonical signaling pathways. In the canonical pathway, TGF-β binds and induces heteromeric complex formation between TGF-β type (TβRI) and type II (TβRII) receptors, leading to TβRI phosphorylation. TβRI, in turn, phosphorylates and activates downstream transcription factors such as SMADs (ie, R-SMADs, SMAD2, and SMAD3). SMADs translocate to the nucleus, where they bind to the promoters of target genes and transactivate their gene expression. The canonical TGF-β signaling pathway promotes aortic development and maintains aortic wall homeostasis. Mutations in genes encoding TGF-β ligands, receptors, and downstream transcription factor SMAD cause TAAD. In the noncanonical pathways, the activated TGF-β receptor complex transmits signals through other pathways, such as TGF-β-activated kinase 1 (TAK1), p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase (ERK), JUN N-terminal kinase (JNK), and nuclear factor-κB (NF-κB). The noncanonical TGF-β pathways promote aortic destruction and AAD development. (Color version of figure is available online.)
Mutations affecting the function of the SMC contractile unit
SM α-actin gene (ACTA2)
ACTA2 encodes SMC-specific isoform of α-actin, a key component in the thin filaments of the SMC contractile unit. Mutations in ACTA238,56–58 are the primary cause of familial TAAD.58,59 Aortic tissues from affected individuals show medial degeneration with focal areas of SMC proliferation, hyperplasia, and disarray.38 ACTA2 mutations interfere with actin filament assembly38 and compromise actin filament movement and SMC contraction.39 ACTA2 mutations also affect actin polymerization,39,60 resulting in unstable actin that is susceptible to severing by cofilin.39 In addition, ACTA2 mutants have abnormal mitochondrial morphology, altered thermos stability, and a reduced ability to grow under stress conditions.61
Myosin heavy chain 11 gene (MYH11)
MYH11 encodes SMC-specific myosin heavy chain 11 (MYH11), a major component of the thick filaments of the SMC contractile unit. Heterozygous mutations in MYH11 have been identified in patients with nonsyndromic familial TAAD.62–64 The phenotypic features associated with MYH11 mutations include TAAD and patent ductus arteriosus.62,63 Mutations in MYH11 are predicted to prevent myosin from polymerizing into thick filaments, resulting in altered force generation.65 Mutations in MYH11 also cause changes in the phenotype of SMCs, such as reduced expression of SMC contractile proteins, increased proliferation and dedifferentiation of SMCs, and altered focal adhesions.65 In addition, MYH11 mutations compromise the stress or injury response and increase vulnerability to intramural damage.66
Myosin light chain kinase gene (MYLK)
MYLK encodes MYLK, which phosphorylates and activates myosin regulatory light chain, leading to an increase in actin-dependent myosin II ATPase activity and the initiation of SMC contraction.67 Loss-of-function mutations in the MYLK gene have been identified in a small percentage of patients with familial TAAD.68 MYLK mutations are predicted to decrease myosin regulatory light chain phosphorylation and activation, leading to compromised SMC contraction.68 Mice with SMC-specific Mylk deletion showed altered gene expression and medial degeneration in the aortic wall.68
Type I cGMP-dependent protein kinase gene (PRKG1)
PRKG1 encodes the type I cGMP-dependent protein kinase (PKG-1), which induces the activation of myosin light chain phosphatase. This leads to the dephosphorylation and inactivation of myosin regulatory light chain, thus promoting SMC relaxation. A gain-of-function mutation in the PRKG1 gene has been identified in patients with familial TAAD and has been associated with severe aortic dissection in the descending thoracic aorta.69 In patients with the gain-of-function mutation in PRKG1, PKG-1 is constitutively active, even in the absence of cGMP, leading to decreased phosphorylation of the myosin regulatory light chain, which is predicted to reduce SMC contraction.69
Filamin A gene (FLNA)
FLNA encodes filamin A, a large actin-binding protein that polymerizes cytoskeletal actin and anchors the actin cytoskeleton to transmembrane integrin molecules. These steps are important for maintaining cell shape and contraction.70 A loss-of-function mutation in the FLNA gene, located on the X chromosome, results in an X-linked dominant condition that is characterized by brain malformation (periventricular heterotopia) and an Ehlers-Danlos–like phenotype, as well as an increased risk of thoracic aortic disease.71
Together, these study findings have supported the notion that defective contractile function contributes to the pathogenesis of TAAD,38,39,56 highlighting the importance of SMC contractile function in preserving aortic structure and preventing TAAD development.17
Mutations affecting elastic fiber and collagen components
Fibrillin-1 gene (FBN1)
FBN1 encodes fibrillin-1, a 350-kDa cysteine-rich glycoprotein that is the major protein in microfibrils. Fibrillin-1 connects elastic fibers to dense plaques in the SMC membrane72,73; this connection is important for mechanotransduction and the stability of elastic fibers.36,37
FBN1 mutations cause Marfan syndrome.
Mutations in FBN1 have been identified in Marfan syndrome,74,75 a heritable autosomal-dominant disorder. More than 1000 FBN1 mutations have been identified in patients with Marfan syndrome, including missense mutations, nonsense mutations, splicing errors, and complete deletion of the FBN1 gene.76,77
FBN1 mutations result in reduced fibrillin-1 protein levels.
Mutational analysis of FBN1 has provided unique insight into the biologic and physiological role of fibrillin-1.78 Frameshift and nonsense mutations in FBN1 result in gene deletions that are predicted to decrease the normal amount of fibrillin-1 protein production by half.79 FBN1 mutations also cause fibrillin misfolding, impaired secretion, and disorganized elastin structure and assembly into microfibrils, rendering fibrillin-1 to proteolysis-mediated degradation.80,81 Fibroblasts explanted from patients with Marfan syndrome consistently show decreased deposition of fibrillin into the ECM, regardless of the underlying mutations.82,83
FBN1 mutations result in SMC dysfunction, phenotype alteration, and aortic destruction.
Mutations in FBN1 cause reduced SMC contraction53 that probably results from the disrupted connection between elastic fibers and SMCs and the compromised function of the elastin-contractile unit.31 Loss of fibrillin-1 alters SMC phenotype and induces ECM remodeling.36,84 Furthermore, mutations in FBN1 increase matrix metalloproteinase (MMP) activity and elastic fiber destruction.53 As a consequence, the aorta exhibits markedly abnormal SMC and elastic properties that lead to progressively increased dilatation.
FBN1 mutations result in altered TGF-β signaling.
Fibrillin-1 binds to latent TGF-β–binding protein (LTBP) and sequesters TGF-β in the ECM, thereby inhibiting TGF-β signaling.85–87 Mutations in FBN1 and fibrillin-1 deficiency alter the matrix sequestration of the latent TGF-β complex, leading to the uncontrolled release of active TGF-β from the ECM and enhanced TGF-β signaling.88–90 In a mouse model of Marfan syndrome, mice that lack LTBP have reduced aortic destruction and improved survival, suggesting the involvement of TGF-β signaling in aortic disease progression.91
Activation of noncanonical TGF-β signaling causes aortic destruction in Marfan syndrome.
Evidence has indicated that the role of TGF-β signaling in aortic disease progression is complex. Both SMAD-dependent canonical and SMAD-independent noncanonical TGF-β signaling pathways (including the extracellular signal–regulated kinase [ERK]1/2 pathway and the Jun N-terminal kinase [JNK] pathway) (Fig 5) have been shown to be increased in mice with Marfan syndrome.92,93 However, recent studies have suggested that only the noncanonical TGF-β signaling ERK1/2 and JNK pathways cause aortic destruction and drive the development of aortic disease in mice with Marfan syndrome.92,93
Angiotensin II type 1 receptor signaling is involved in aortic destruction in Marfan syndrome.
Angiotensin signaling has receptor-specific effects on disease progression in mice with Marfan syndrome. Deficiency of angiotensin II type 2 receptor (AT2R) was shown to accelerate aortic destruction,94 whereas the selective angiotensin II type 1 receptor (AT1R) blocker (ARB) losartan90,94,95 abrogated aneurysm progression in mice with Marfan syndrome, suggesting a protective role of AT2R signaling and a destructive role of AT1R signaling in the aorta of these mice. Notably, AT2R signaling was shown to attenuate aortic aneurysm formation in mice through ERK antagonism.94
Mutations in other genes encoding proteins in elastic fibers
Loss-of-function mutations in FBN2 (encoding fibrillin-2),96 MFAP5 (encoding microfibril-associated glycoprotein 2 [MAGP2]),97 and EMILIN1 (encoding elastin microfibril interfacer 1)98 also predispose individuals to TAAD. Mutations in the elastin-encoding gene ELN cause rare dominant cutis laxa, and aortic dilatation and rupture can develop in some patients.99
Fibulin-4 gene (FBLN4)
Fibulin-4 is an ECM protein that binds to elastin and promotes elastic fiber formation. Mutations in the gene encoding fibulin-4 also cause aortic aneurysms with arterial tortuosity and stenosis.100–102 Fibulin-4 deficiency in mice results in ascending thoracic aortic aneurysm (TAA) with perturbations of vascular homeostasis and aortic valve abnormalities.103 Mice with SMC-specific fibulin-4 deficiency develop ascending TAA. The aortic wall of these mice does not fully differentiate, featuring an immature SMC phenotype, reduced expression of SMC-specific contractile genes, medial degeneration, and focal SMC proliferation.104
Lysyl oxidase gene (LOX)
Lysyl oxidase (LOX), a copper-dependent amine oxidase that oxidizes lysine residues in elastin and collagen to form covalent cross-links, is an important enzyme in elastic fiber assembly and stabilization.105 Recently, mutations in LOX that cause LOX deficiency or reduce LOX activity have been identified in patients with familial TAAD.106,107 Individuals with LOX variants have enlargement of the aortic root and ascending aorta and associated aortic dissections.106 Transgenic mice heterozygous for the mutated human allele of LOX have fragmented elastic lamellae, whereas mice homozygous for the mutated human allele die shortly after parturition from ascending TAA.107 These findings suggest a critical role of LOX in maintaining aortic wall integrity and indicate the importance of LOX mutations in the development of genetic TAAD.
Collagen gene (COL)
Mutations in COL are associated with Ehlers-Danlos syndrome. Mutations in COL3A1108,109 encoding collagen III, COL5A1110 encoding collagen V, and COL1A2111 encoding collagen I have been identified in Ehlers-Danlos syndrome patients with TAAD.
Mutations affecting TGF-β signaling
TGF-β (Fig 5) plays a critical role in the regulation of cell growth, differentiation, and development in a wide range of biologic systems. Gene mutations affecting several signaling molecules in this pathway, including ligands, receptors, and downstream transcriptional factor SMAD, have been shown to cause TAAD.
TGF-β receptor genes (TGFBR1 and TGFBR2)
Mutations in TGFBR1 and TGFBR2 genes are associated with Loeys-Dietz syndrome and other TAAD. Mutations in the TGFBR genes TGFBR1 and TGFBR2 have been identified in patients who have Loeys-Dietz syndrome112–114 and phenotypic overlap with Marfan syndrome,113,115 and in patients with familial TAAD.113,116 The phenotype of transgenic mice with mutations in either Tgfbr1 or Tgfbr2 recapitulates the Loeys-Dietz syndrome phenotype.117
TGF-β ligand genes (TGFB2 and TGFB3)
Loss-of-function mutations in TGFB2 cause syndromic TAAD with the phenotype of Loeys-Dietz syndrome or with mild systemic features of Marfan syndrome.114,118 Mutations in the TGFB3 gene also cause syndromic TAAD.119 Mice deficient in Tgfb2 develop aortic aneurysm.114 Paradoxical upregulation of TGF-β signaling has been observed in aortic tissues from patients with mutations in TGFB2 or TGFB3.114,119
SMAD3 gene (SMAD3)
Mutations in SMAD3 cause syndromic familial TAAD.120–122 Mice with Smad3 deletion develop progressive aortic aneurysms123 and have elevated levels of granulocyte-macrophage colony-stimulating factor, which may contribute to the development of aortic aneurysms.123 Recently, the SMC-specific deletion of Smad4 in mice was shown to promote aortic aneurysm.124 Paradoxically, SMAD3 mutations lead to increased aortic expression of SMAD3 and other key proteins in the TGF-β pathway.120
Critical role of TGF-β signaling in aortic wall homeostasis
Several lines of evidence from mice suggest that TGF-β signaling protects against AAD development. Mice deficient in Tgfb2,114 Smad3,123 or Smad4124 develop aortic aneurysm. In addition, the SMC-specific deletion of Tgfbr2 in postnatal mice impairs aortic wall homeostasis125 and causes severe TAAD.125,126 Furthermore, aortas and explanted SMCs from patients with TGFBR2 mutations show decreased expression of contractile proteins, as well as compromised expression of contractile genes in response to TGF-β stimulation.127 Moreover, fibroblasts explanted from patients with TGFBR2 mutations fail to transform to mature myofibroblasts with TGF-β stimulation.127 Thus, it has been proposed that TGF-β signaling promotes aortic wall homeostasis125,127 and that mutations in this pathway cause AAD formation.
Activation of TGF-β signaling in aortas from patients with gene mutations affecting TGF-β signaling
Paradoxically, aortic tissues from patients with mutations in TGFBR1 or TGFBR2,112,128 TGFB2 or TGFB3,114,119 or SMAD3120 have consistently shown enhanced TGF-β signaling, as indicated by the upregulation of SMAD2 or SMAD3 and the increased production of TGF-β target genes such as collagen and connective tissue growth factor (CTGF). The mechanisms underlying this paradoxical elevation in TGF-β signaling are not completely understood. It is possible that decreased expression of TGF-β target genes leads to a secondary increase in TGF-β signaling.
Mutations in other genes
Forkhead transcription factor 3 gene (FOXE3)
Exome sequencing of families with TAAD has identified mutations in the FOXE3 gene that encodes the forkhead transcription factor FOXE3.129 Mice deficient for Foxe3 had a reduced number of aortic SMCs during development and increased rates of SMC apoptosis and aortic rupture in a model of transverse aortic constriction–induced TAAD. These phenotypes can be rescued by deleting the p53 gene or by inhibiting p53 with an inhibitor.129 Thus, FOXE3 may be important for aortic development and the stress response. Mutations in FOXE3 predispose individuals to TAAD.129
Methionine adenosyltranferase IIα gene (MAT2A)
Methionine adenosyltranferases (MATs) are the key enzymes involved in the production of S-adenosylmethionine, the principal methyl donor in cells. A loss-of-function mutation in MAT2A has been identified in patients with TAAD.130 The knockdown of mat2a in zebrafish was shown to disrupt cardiovascular development.130 These study findings suggest that loss-of-function mutations in MAT2A may cause TAAD.
MMP-17 gene (MMP17)
MMP-17/MT4-MMP is a membrane-bound MMP that is essential for vascular SMC maturation and function in the arterial vessel wall.131,132 A missense mutation in the MMP17 gene that prevents its expression has been identified in patients with genetic TAAD.132 Loss of function of MMP-17 in mice results in dysfunctional vascular SMCs and altered ECM in the vessel wall, leading to increased susceptibility to angiotensin II–induced TAA.132
Etiology of sporadic AAD
Sporadic AAD, including sporadic TAAD and AAA, are more common than genetically triggered TAAD and often occur in older patient populations.16,21,24 Sporadic AAD occur more frequently in men than in women.21,25 Risk factors for sporadic AAD include cigarette smoking,20,22,23,26,27 hypertension,21,24,27–29 and aging.16,20–24 More than 80% of all AAD cases and more than 70% of TAAD cases are sporadic.14–16 Understanding the risk factors and pathogenesis of sporadic AAD may provide opportunities for developing targets for the primary prevention of AAD.
Effect of age
Sporadic TAAD16,21,24 and AAA20,22,23 often occur in older patient populations. Aging is associated with changes in aortic structure and function that compromise the ability of the aorta to respond to biomechanical stress, predisposing these individuals to aortic destruction and dilatation. In people without known aortic disease, the descending thoracic aorta shows an age-related increase in fragility and susceptibility to permanent dilation after pressure distention.133 In older mice134 and monkeys,135 the aorta shows ECM disarray and stiffness, which are believed to contribute to TAA development.134,135 Aortic stiffness increases aortic wall stress, damage, and inflammation that precedes AAD growth.136
Effect of sex
Sex is a major determinant of sporadic TAAD21,25 and AAA23; the ratio of men to women who have sporadic AAD is 2:1.25 Although the incidence of AAA is higher in men,24 the prognosis of AAA is worse in women than in men.137 The mechanisms responsible for this difference between men and women are not well understood. It has been suggested that testosterone may play a role in promoting AAD development. Testosterone administration in neonatal female mice increases hydrogen peroxide generation and promotes angiotensin II–induced AAD.138 Testosterone upregulates AT1R, which may mediate the effects of testosterone in promoting AAD development.138,139
Genetic contributions
Recent studies have indicated that genetic variants contribute to sporadic AAD. Rare copy number variants in genes that regulate SMC adhesion and contractility have been identified in patients with sporadic TAAD.140 In addition, common genetic variants in FBN1 have been found in patients with sporadic TAAD.141,142 Genetic polymorphisms in several genes have been studied in patients with AAA. A recent meta-analysis showed that polymorphisms in genes such as LRP1 (low-density lipoprotein [LDL] receptor–related protein 1), APOA1 (apolipoprotein A), IL6R (interleukin 6 receptor), MMP3, AT1R (angiotensin II type I receptor), and ACE (angiotensin-converting enzyme) were associated with AAA.143 By performing the Sanger sequencing of all coding exons and exon-intron boundaries of the genes associated with genetic TAAD, van de Luijtgaarden and colleagues144 found potential pathogenic variants of COL3A and MYH11 of patients with familial AAA (with at least 1 first-degree relative with an aortic aneurysm) and a pathogenic variant in TGFBR2 in patients with sporadic AAA.
Hypertension
Hypertension is a risk factor for sporadic TAA,7,21,24,28,145 TAD,27 and AAA23 and is also associated with an increased rate of mortality from TAA.29 In mice, prolonged angiotensin II infusion produces aortic aneurysms in both the thoracic and the abdominal aorta.146–149 The effects of hypertension on the development of aortic disease are attributed, in part, to the mechanical effects of elevated blood pressure on the aortic wall. In an ex vivo study of aortas from patients with TAA, the mean circumferential wall stress was shown to increase linearly with increased intraluminal pressure and aortic diameter.150 It has been suggested that elevated systolic pressure significantly affects the structure and function of the aortic wall, thereby increasing aortic wall stress151 and promoting aortic dilatation.
Cigarette smoking
Smoking is a major risk factor for the development and progression of TAAD21,27 and AAA.20,22,23,26,27 Smoking is also a risk factor for aortic dissection.27 Several experimental studies in mice have shown that cigarette smoke exposure augments AAA induced by angiotensin II infusion or elastase perfusion.152,153 Smoking-induced effects have been attributed for alterations in aortic SMCs and in the inflammatory response.154,155 In patients with AAA, the cessation of cigarette smoke exposure did not produce an immediate decrease in aortic expansion,154 suggesting that the effects of cigarette smoking on the aortic wall may be long term.
Inflammatory diseases
AAD occur in several inflammatory diseases. Takayasu’s arteritis, a chronic inflammatory disease, is the most striking arteritis that affects the aorta.156 In patients with Takayasu’s arteritis, aortic dilatation and aneurysm formation157 can occur in the ascending aorta158,159 and aortic arch.156 Significant T-cell infiltration has been observed in the aorta of these patients.160 Because of the inflammatory process caused by Takayasu’s arteritis, severe narrowing, or even occlusion of the entire aorta and large arteries can develop in these patients. Another inflammatory disease that affects the aorta is giant-cell arteritis, which typically affects the temporal or cranial arteries but can also affect the aorta161–164 with serious outcomes.165 In addition, ankylosing spondylitis166 and psoriasis167 have been shown to be associated with ascending aortic aneurysms and AAA, respectively. Interestingly, a recent population-based case-control study showed that asthma is associated with AAA and rupture.168 In mouse models of AAA induced by angiotensin II and calcium chloride, the simultaneous induction of allergic inflammation in the lung increased aortic macrophage and mast cell infiltration, promoted SMC loss, and aggravated aortic enlargement.169 These findings suggest a potential association between allergic inflammation and AAD development.
Dyslipidemia and atherosclerosis
Ample evidence has shown that dyslipidemia and atherosclerosis are closely associated with the development of sporadic AAD.
Atherosclerosis in AAA
AAA are associated with the occurrence of cardiovascular events,170 atherosclerotic risk factors,23 carotid atherosclerosis and stenosis,20,22,26 and coronary artery disease.22 High LDL levels, low high-density lipoprotein (HDL) levels, and a high LDL/HDL ratio are common in patients with AAA.20,23,27,171 Furthermore, there is direct evidence showing the presence of atherosclerosis in patients with AAA. Atherosclerosis is associated with significantly increased aortic diameter and wall thickness.171
Atherosclerosis in sporadic TAAD
Atherosclerosis is also associated with sporadic TAAD and has been detected in the descending thoracic21,25 and thoracoabdominal aorta.172 One study showed that atherosclerosis risk factors and aortic atherosclerotic plaque are weakly associated with distal aortic dilatation, suggesting that atherosclerosis plays a minor role in aortic dilatation.21 However, other studies have shown that atherosclerosis in these distal segments is associated with aortic enlargement, suggesting that atherosclerosis is the predominant etiology of aneurysms in the descending thoracic aorta and thoracoabdominal aorta.25,172
Compared to descending TAA, ascending TAA have a lower incidence of grossly detectable atherosclerosis (9% in ascending TAA vs 88% in descending TAA).21,25 Although atherosclerosis is often not obvious in sporadic ascending TAAD on gross examination, histologic analysis has shown the presence of atherosclerosis in most cases.173 Advanced atherosclerosis (ie, denoted by the presence of atherosclerotic plaque with intraplaque hemorrhage, calcification, and intima hyperplasia) is present in more than 35% of sporadic ascending TAA cases and is associated with increased degeneration of the aortic media characterized by SMC depletion, elastic fiber destruction, and fibrotic remodeling.173 A recent study showed that increased cholesterol level is also associated with an increased rate of mortality from TAA.29
Role of atherosclerosis in sporadic AAD progression
Although studies in humans have shown an association of dyslipidemia and atherosclerosis with AAD incidence, the cause-effect relationship between atherosclerosis formation and aneurysm initiation and progression is less clear. It has been suggested that atherosclerosis may not be a causal event in AAA but, rather, develops in parallel with or secondary to aneurysmal dilatation.174 However, evidence from animal studies supports a role of dyslipidemia and atherosclerosis in the development of sporadic AAD. In mice, a high-fat diet produces hypercholesterolemia175 and progressively augments the incidence of angiotensin II–induced AAA175,176 in apolipoprotein E–deficient (ApoE−/−) mice in a time-dependent manner.176 Reducing plasma levels of lipoproteins containing apolipoprotein B attenuates angiotensin II–induced AAA formation in ApoE−/− mice.175 Furthermore, increasing plasma levels of HDL reduces angiotensin II–induced AAA formation in ApoE−/− mice or calcium chloride–induced AAA formation in C57BL/6 mice,177 presumably by reducing the activation of ERK1/2 and matrix remodeling.177 Thus, atherosclerosis may promote aortic destruction and disease progression in sporadic AAD.
Obesity and AAD
Body mass index has been associated with the incidence of TAA28 and AAA178 and the rate of mortality from TAA.29 Animal studies have shown that obesity induced by a high-fat diet increases macrophage accumulation in periaortic adipose tissue and enhances angiotensin II–induced AAA formation,176 supporting the potential link between obesity and aortic disease.
Diabetes
Although thoracic and abdominal aortic diseases have different clinical profiles,28 they both have a negative association with the incidence of diabetes.
Negative association between diabetes and sporadic TAAD
Consistently, diabetes has been shown to be associated with a reduced incidence of TAA, dissection, and rupture.179–182 Evidence from a community screening of 30,412 individuals has suggested that diabetes is not significantly associated with TAA.27 A recent study based on nationwide hospital discharge or inpatient data showed that TAAD among individuals diagnosed with diabetes occurred in 9.7 per 10,000, compared to 15.6 per 10,000 among all hospital discharges.179 The prevalence of diabetes has been reported to be lower in patients with TAAD (13%) than in general population controls (22%).179 Furthermore, study findings have indicated that aneurysms in patients with diabetes are less likely to develop TAD and rupture.182–184
Negative association between diabetes and AAA
Diabetes has also been negatively associated with AAA.22 In a recent systematic review of 17 population studies, the prevalence of AAA in individuals with diabetes was estimated to be lower than that in individuals without diabetes.185 Another meta-analysis of data from 19 studies of a total of 9777 patients confirmed that diabetes is associated with slower AAA growth.186 Thus, clinical observations have consistently shown a negative association between diabetes and the development, growth, and rupture of thoracic and abdominal aortic diseases.
The effects of diabetes on aortic structure
An important question remaining is, what are the mechanisms underlying the negative association between diabetes and thoracic and abdominal aortic diseases? Furthermore, what are the protective factors that could potentially be targeted for the prevention and treatment of sporadic aortic diseases? Interestingly, Golledge and colleagues187 found that aortic tissues from diabetic patients have decreased MMP-2 and MMP-9 activity. Furthermore, in vitro analysis showed that aortic tissue lysate from diabetic patients prevented monocyte activation187 and that glycated type I collagen lattices prevented monocyte activation and MMP and interleukin (IL)-6 secretion. The authors concluded that the cross-linking of type I collagen by glycation may inhibit inflammatory cell activation and MMP production, thus preventing excessive proteolysis.187
The effects of antidiabetic medication on AAD progression
Evidence has indicated that antidiabetic medications may have protective effects against thoracic and abdominal aortic diseases. Hsu and colleagues188 compared the use of antidiabetic medications between 4468 diabetic patients with aortic disease and 4468 matched diabetic patients without aortic disease. The use of metformin, sulfonylurea, or thiazolidinediones was found to be associated with a lower risk of developing aortic disease. The effects of metformin and sulfonylurea on the risk of developing aortic disease were dose-dependent. In another study of 58 patients, metformin use was negatively associated with AAA enlargement.189 In experimental models, metformin189,190 and rosiglitazone191 prevented elastin reduction, SMC destruction, and inflammatory cell infiltration in the aortic media and suppressed AAD formation and progression. Thus, AAD suppression in diabetes may be attributable to concurrent antidiabetic therapy.
Changes in aortic cells in AAD
The aortic wall is highly dynamic, performing sophisticated biomechanical functions to provide suitable compliance and adequate strength in response to hemodynamic changes. Dysregulation and destruction of the cellular and extracellular components of the aortic wall result in progressive SMC depletion, ECM destruction, and inflammation, which are pathologic changes that commonly lead to aortic aneurysm, dissection, and rupture (Fig 6).
Fig. 6.
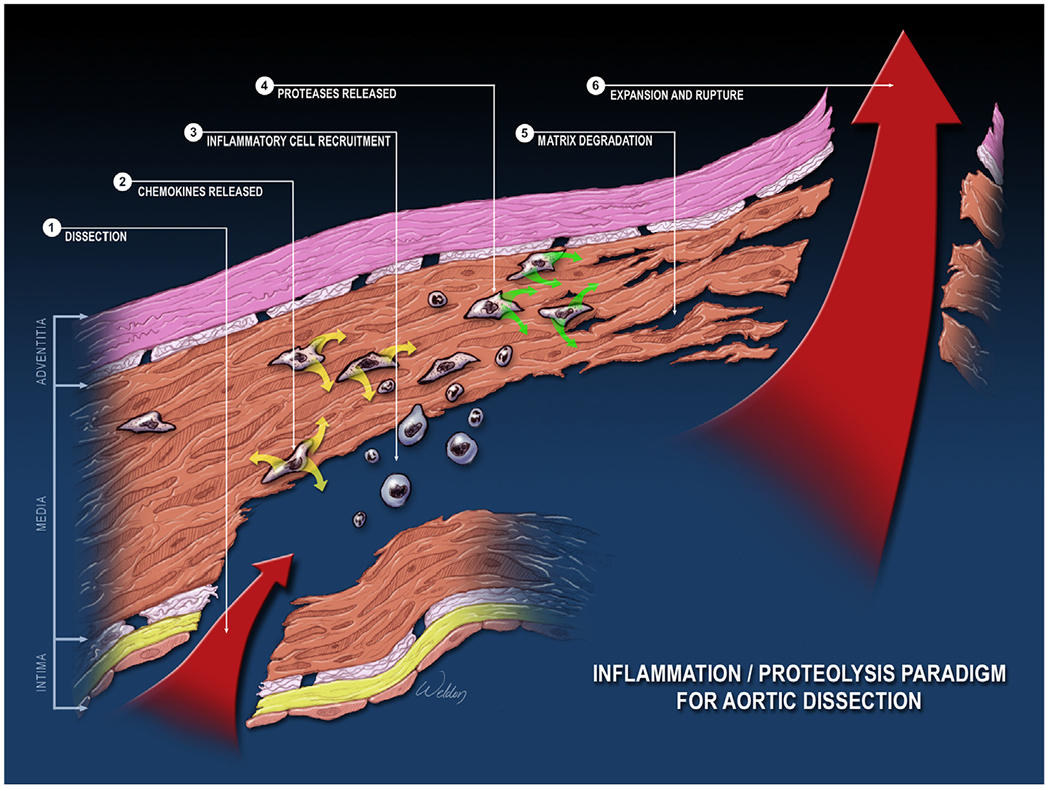
Common pathologic changes in aortic aneurysms and dissections (AAD). The common pathologic features of AAD are aortic degeneration, which is characterized by aortic smooth muscle cell (SMC) death and loss; elastic fiber destruction and depletion; and aortic inflammation. Aortic degeneration weakens the aortic wall, leading to the development of aortic aneurysm, dissection, and rupture. (Color version of figure is available online.)
SMC injury and death in AAD
SMC apoptosis in AAD
One of the key histopathologic features of AAD is progressive SMC depletion and aortic medial degeneration.192 A growing body of evidence has suggested that SMC injury and death contribute to medial degeneration during AAD development. SMC apoptosis occurs not only in the genetic form of AAD but also in sporadic AAD. SMC apoptosis, as evidenced by the presence of cells positive for terminal deoxynucleotidyl transferase dUTP nick end labeling and by the activation of caspase-mediated apoptotic pathways, has been detected in the aortas of patients with Marfan syndrome193,194 and in those of patients with sporadic TAA or TAD.195–198 The presence of apoptotic cells is also a prominent feature in the aortic wall of patients with AAA.199–201
Role of SMC apoptosis in aortic destruction and AAD development
Apoptosis has been suggested to play a major role in SMC depletion during AAD development. Animal studies have shown that caspase inhibitors diminished apoptosis and inflammation in the aortic media and prevented aneurysm progression in both ApoE−/− mice challenged with angiotensin II202 and mice with Marfan syndrome.203 Using a mouse model of inducible vascular SMC-specific apoptosis in ApoE−/− mice, Clarke and colleagues204 showed that vascular SMC apoptosis promoted elastic lamina breaks and abnormal matrix deposition, leading to medial degeneration and medial expansion. These findings support a critical role of SMC apoptosis in aortic destruction and AAD development.
Signaling pathways affecting SMC apoptosis in AAD development
Several pathways and factors have been shown to affect AAD development by regulating SMC apoptosis. Mechanical stretch induces endoplasmic reticulum (ER) stress and apoptosis that contribute to TAAD formation.198 Factors such as aldosterone,205 proinflammatory S100A12 protein,206 and mast cell–produced chymase and tryptase207 induce aortic SMC apoptosis and aneurysm formation. Signaling pathways such as TGF-β,194 protein kinase C,208 and p53209 are also involved in promoting SMC apoptosis during AAD development. In contrast, the elevation of HDL level inhibits angiotensin II–induced apoptosis and protects the aorta.177 AKT2 has also been shown to protect the aorta, in part, by inhibiting apoptosis.210 When challenged with angiotensin II, Akt2-deficient mice showed profound apoptotic cell death in the aortic media and developed AAD.210
Necroptosis in AAD development
Interestingly, a recent study showed that the activation of necroptosis, a programmed necrosis pathway, contributes to AAA. Receptor-interacting protein kinase 3 (RIP3) is a key regulator in promoting necroptosis. In mice, adenoviral overexpression of Rip3 was sufficient to trigger SMC necroptosis, whereas deficiency in Rip3 prevented SMC necrosis, inflammation, and AAA formation.211 Thus, SMC death contributes to aortic weakening and dilatation, dissection, and rupture.
Alteration of SMC phenotype in AAD
SMC phenotype changes
SMCs possess remarkable plasticity. Differentiated SMCs express SMC-specific contractile proteins and have characteristic myocyte morphology and contractile properties. Under conditions such as inflammation and injury, vascular SMCs can lose their contractile phenotype212 and exhibit phenotypes resembling those of inflammatory cells, fibroblasts, or osteogenic cells213–217–a phenomenon termed phenotypic switching (Fig 7). Although SMC phenotypic switching allows vessel growth, repair, and adaptation to the environment, it can also lead to vascular dysfunction and maladaptive remodeling.213–216
Fig. 7.
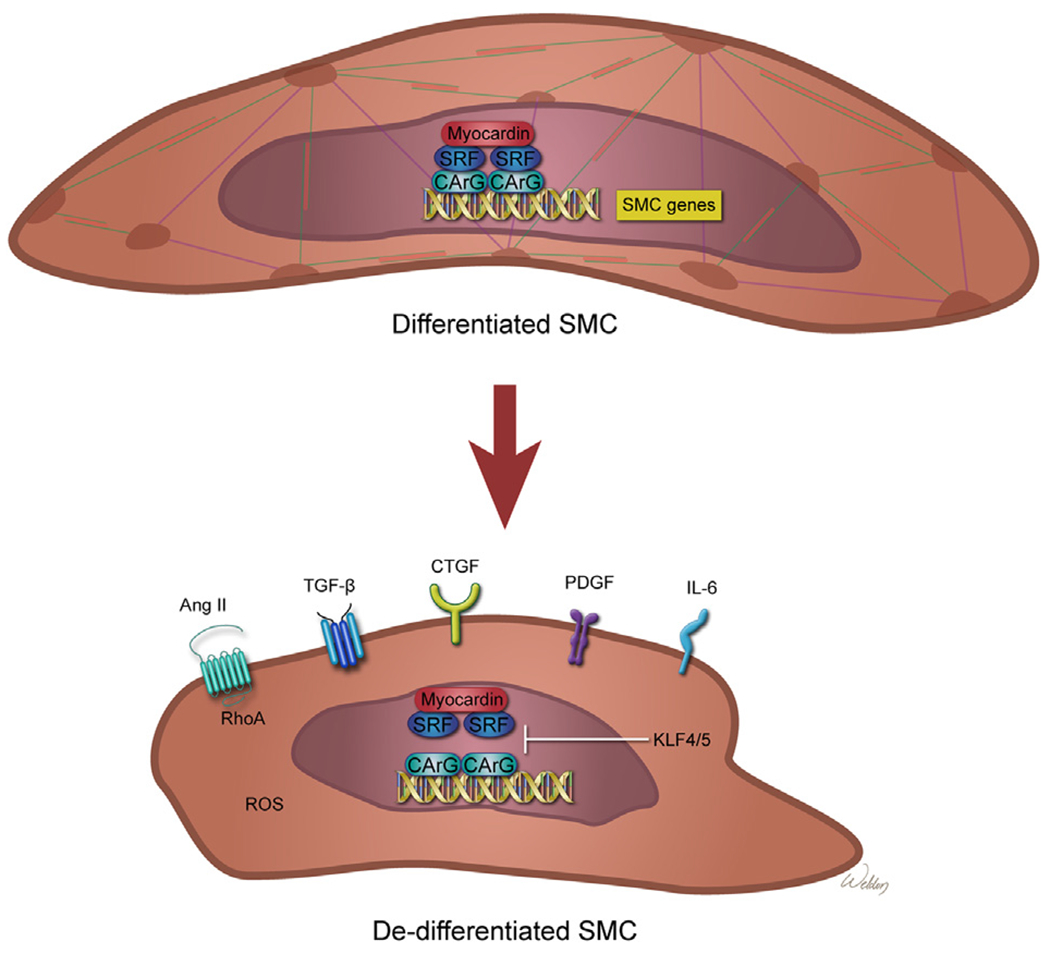
Smooth muscle cell (SMC) phenotype changes in aortic aneurysms and dissections (AAD). SMCs possess remarkable plasticity. Differentiated SMCs express SMC-specific contractile proteins and have characteristic myocyte morphology and contractile properties. In differentiated contractile SMCs, transcription factors myocardin and serum response factor (SRF) bind to the CArG motif in the promoters of SMC genes and induce the expression of SMC-specific contractile proteins. Under conditions such as inflammation and injury, SMCs can lose their contractile phenotype and exhibit phenotypes resembling those of inflammatory cells, fibroblasts, or osteogenic cells–a phenomenon termed phenotypic switching. In dedifferentiated SMCs, transcription factors such as Krupple-like factor 4 (KLF4) inhibit myocardin or SRF-mediated SMC gene expression, thereby inducing SMC phenotype alterations. Several pathways such as those involving angiotensin II (Ang II), transforming growth factor β (TGF-β), connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), interleukin 6 (IL-6), and reactive oxygen species (ROS) have a role in promoting the SMC phenotype switch. (Color version of figure is available online.)
Heterogeneity of SMC phenotype in AAD
The alteration of SMC phenotype is a critical event in AAD pathology. However, the phenotypes of SMCs vary significantly in different pathogeneses (eg, genetic vs sporadic), in different focal areas (eg, degeneration vs compensational hyperplasia or proliferation), and at different disease stages. SMC phenotype alterations can be characterized either by increased expression of SMC genes and ECM proteins or by decreased expression of SMC genes and ECM proteins. Regardless of whether the expression of SMC genes is increased or decreased, the feature of increased expression of inflammatory and destructive factors such as MMPs is often observed in these dedifferentiated SMCs.
SMC phenotype with increased expression of SMC and ECM proteins in AAD
A recent study showed that aortic SMCs in patients with Marfan syndrome have an altered phenotype characterized by the increased expression of contractile proteins, actin stress fibers, focal adhesion components, and collagen I, leading to increased cellular and ECM stiffness.84 A study in a mouse model of Marfan syndrome also showed that SMCs retained the expression of vascular SMC markers but had increased expression of matrix elements and MMP-9, and the SMC phenotypic alteration preceded elastolysis.36 It has been suggested that the TGF-β pathway may be involved in this type of phenotypic switch, given that the pharmacologic inhibition of the TGF-β pathway reduced alterations in gene expression.84
SMC phenotype alteration with decreased expression of SMC proteins and increased expression of inflammatory proteins in AAD
The SMC phenotype characterized by the reduced expression of SMC genes (eg, genes encoding SM22-α and SM α-actin) and the increased expression of inflammatory genes (eg, MMP-2 and MMP-9) has also been observed in aortas from patients with genetic and sporadic AAD.127,218,219 SMCs in the aortas of patients with TGFBR2 mutations showed decreased expression of contractile proteins.127 SMC phenotype modulation characterized by decreased SMC proteins and increased MMPs has been observed in aortas from patients with sporadic TAA.218,219 In a study of an elastase-induced model of AAA, SMC phenotypic modulation was observed as an early event during the development of AAA.218
Hyperplastic cellular remodeling
Hyperplastic cellular remodeling of the media has been observed in patients with ascending TAA.220 SMCs in the aortic media from patients with MYH11 mutations also showed phenotypic alterations, including SMC disarray and focal SMC hyperplasia.63
Factors involved in SMC phenotype alteration in AAD
An important question that remains is, what triggers SMC phenotypic changes in AAD? In a mouse model of angiotensin II–induced AAD, findings have indicated that increased peak wall stress induces a synthetic phenotype by increasing the production of ROS and the expression of CTGF.219 In addition, bone marrow–derived monocyte chemotactic protein 1 has been shown to be involved in the reduction of contractile protein levels and in the induction of MMP expression.221 Krupple-like factor 4 (KLF4) is a key transcription factor involved in inflammatory responses after vascular injury or disease. It has been shown that KLF4 binds to SMC gene promoters, suppresses SMC gene expression, and induces SMC phenotype alterations.222,223 Furthermore, KLF4 inhibition reduces SMC phenotypic switching and prevents angiotensin II–induced AAD development.223 Interestingly, a recent study showed that RNA editing controls messenger RNA (mRNA) splicing, which is an important mechanism underlying SMC phenotypic switching.224 Whether this mechanism is involved in SMC phenotype switching in AAD remains to be determined.
Thus, phenotypic changes in SMCs occur in both genetic and sporadic AAD. The dedifferentiation and phenotypic modulation of SMCs can cause SMC dysfunction, loss of a functional SMC population, and matrix remodeling–all of which contribute to aortic degeneration and biomechanical failure.84
Endothelial cell dysfunction in AAD
Endothelial cells play a fundamental role in controlling blood flow and blood pressure by regulating SMC functions. Endothelial cells also maintain aortic homeostasis by providing barrier functions and preventing inflammation and thrombosis. Endothelial dysfunction is known to be involved in the initiation of many cardiovascular diseases.
Protective role of endothelial cells against AAD
Evidence has indicated that intact endothelial functions are required for protecting the aorta against AAD. The endothelial-specific deletion of C-type natriuretic peptide, a fundamental factor that modulates vascular tone, leads to hypertension and aneurysm formation.225 In addition, the endothelial-specific deficiency of phosphoinositide 3-kinase-C2α (PI3K-C2α), an upstream kinase of the AKT pathway, impairs vascular barrier function and increases susceptibility to angiotensin II–induced AAD formation.226 Interestingly, a recent study showed that endothelial cells participate in aortic repair.227 In a rat xenograft model, the endovascular transplantation of endothelial cells in the aorta prevents AAA formation and stabilizes AAA.227 Rather than directly participating in aortic repair, the transplanted endothelial cells promote aortic repair through paracrine mechanisms and the recruitment of resident vascular cells, leading to the reconstitution of new aortic wall rich in SMCs and ECM.227
Endothelial dysfunction in AAD development
In pathologic conditions, abnormal activation of endothelial cells may have destructive effects in the aorta. The endothelial-specific deficiency of the AT1R receptor attenuates the formation of angiotensin II–induced aortic aneurysms in mice,228 suggesting that endothelial cells may transduce angiotensin II signals and mediate angiotensin II–induced AAD formation. Evidence has also indicated that endothelial dysfunction promotes AAD development. Endothelial cell–specific ROS production induced by endothelium-specific NOX2 overexpression increases susceptibility to angiotensin II–induced aortic dissection in mice.229 The aortas from these mice show increased adhesion molecule-1 expression, inflammatory cell infiltration, and MMP activity.229 By releasing cyclophilin A, endothelial cells induce the activation of the ERK1/2 pathway and ROS production in SMCs, thereby increasing susceptibility to angiotensin II–induced AAD formation.229 Finally, it has been shown that the immunosuppressive drug azathioprine reduces aneurysm progression through the inhibition of Ras-related C3 botulinum toxin substrate 1 (Rac1) and c-Jun N-terminal kinase (JNK) in endothelial cells.230
These findings illustrate a pivotal role of endothelial cells and endothelial cell–derived factors in protecting the aorta against AAD formation. Dysregulation of endothelial functions may contribute to increased susceptibility to stress-induced aortic destruction.
Activation of fibroblasts and myofibroblasts in AAD
Activation of myofibroblasts in AAD
Myofibroblasts are specialized cells that have a more contractile phenotype and produce more ECM proteins than fibroblasts. They can be derived from multiple cells such as fibroblasts, bone marrow cells, mesenchymal cells, epithelial cells, and endothelial cells.231,232 Myofibroblasts are activated in response to stress and tissue injury and contribute to tissue repair and structural integrity, as well as fibrotic remodeling.232–236
Recent studies have drawn attention to the possible role of myofibroblasts in aortic diseases.237 Activated myofibroblasts have been found in aortic tissues in human TAA238and AAA.239 In a calcium chloride–induced rodent model of TAA, Jones and colleagues240 observed a time-dependent increase in the number of fibroblasts and fibroblast-derived myofibroblasts in the aortic wall. In a transverse aortic constriction mouse model, Kuang and colleagues241 reported that biomechanical pressure induced significant adventitial hyperplasia, which was accompanied by an increase in myofibroblast accumulation and collagen production.241
Potential roles of myofibroblasts in aortic wall homeostasis and destruction
The precise roles of myofibroblasts in regulating aortic structure and function during the different stages of AAD development remain to be determined. Myofibroblasts can be activated in response to mechanical stress241,242 and injury.240 These cells can proliferate rapidly, migrate to the injured area, contract, and produce large amounts of ECM proteins, thereby maintaining the structural integrity of tissues. By producing reparative growth factors, myofibroblasts can participate in tissue repair.233,237 After acute aortic injury, the quick, substantial increase in myofibroblasts and ECM proteins may increase aortic strength and prevent aortic rupture, thus playing a protective role.
However, myofibroblasts can also produce proinflammatory cytokines, activate inflammatory cells, and induce MMP production and activation,233,234,236,237 thus promoting aortic inflammation and destruction. Additionally, by driving inflammatory responses and producing collagens, these cells are key players in maladaptive fibrotic remodeling,233,234,236,237 which can progress to aortic dysfunction and, ultimately, to aortic biomechanical failure. Thus, myofibroblasts may represent a compensatory response that maintains aortic structural integrity during acute injury, but their concurrent participation in ECM proteolysis and collagen deposition promotes vascular fibrotic remodeling, leading to aortic failure. Therefore, precise and timely control of myofibroblast activation and inactivation is critical for maintaining aortic homeostasis.
Regulation of myofibroblast activation in AAD
Myofibroblasts can be derived from the transdifferentiation of fibroblasts and mesenchymal cells. In addition, myofibroblasts can be derived from epithelial and endothelial cells through the epithelial-to-mesenchymal transition and endothelial-to-mesenchymal transition (EMT), respectively.231,232 Several signaling pathways and factors, including TGF-β, angiotensin II, CTGF, platelet-derived growth factor, endothelin-1, Wingless/int, and yes-associated protein 1, have been linked to the activation and maintenance of myofibroblasts and fibrotic remodeling.235 Altered TGF-β signaling is a key feature in AAD development in both genetically triggered and sporadic AAD. Fibroblasts explanted from patients with TGFBR2 mutations fail to transform into mature myofibroblasts when stimulated with TGF-β,127 suggesting that deficient TGF-β signaling can result in impaired myofibroblast activation in AAD. Angiotensin II signaling is also involved in myofibroblast activation in pressure-induced aortic remodeling. Blocking angiotensin II signaling with losartan, an AT1R antagonist, partially prevented pressure-induced myofibroblast activation, collagen accumulation, adventitial hyperplasia, and aortic dilatation.241
Despite the critical importance of myofibroblasts in aortic diseases, our understanding of aortic myofibroblasts, fibroblasts, and fibrotic remodeling in aortic homeostasis is limited. Improving our understanding of the regulation of myofibroblast activation and inactivation may help to develop therapies designed to manipulate these cells, thereby promoting their beneficial effects during acute injury but preventing their detrimental effects on aortic disease progression.
Stem cells and aortic repair
Aortic degeneration can result from a combination of excessive destruction and insufficient repair. When cells are injured, they release signals that trigger stem cell–mediated reparative responses. Stem cells proliferate and self-renew, differentiate into other cell types, and secrete factors to create a favorable microenvironment, thus promoting tissue repair or regeneration and maintaining tissue homeostasis. Insufficient tissue repair can cause sustained chronic inflammation that triggers further injury and tissue destruction. Stem cells play an important role in the repair and regeneration of various cardiovascular tissues.243–246
Activation of stem cells in AAD
Activation of stem cells has been seen in patients with AAD. Circulating endothelial progenitor cells are increased in patients with ascending TAA247 and in those with AAA.248 Moreover, significant amounts of mesenchymal cells have been detected in the aortic tissue of patients with TAA, TAD,249 and AAA.250
Mesenchymal stem cell–based therapy in AAD
Mesenchymal stem cells (MSCs) are promising candidates for cell therapy. Bone marrow–derived MSCs (BM-MSCs) and adipose-derived MSCs can be readily isolated and expanded in vitro and are therefore suitable for allogeneic cell-based therapy.251 The therapeutic effects of BM-MSCs in AAD have been tested extensively in animal models.252–258 These studies have shown that BM-MSC therapy–administered directly to the aortic adventitial surface253 by endovascular seeding254 or via intravenous injection255,256,258–reduced aortic destruction, decreased inflammation, and attenuated AAD development. Recent studies have shown that adipose tissue–derived MSCs have similar effects.259–261
Mechanisms of stem cell–mediated protection
MSCs exert their protective effects through several mechanisms. When co-cultured with SMCs, MSCs promote the secretion of elastin from SMCs.262 In addition, MSC treatment can inhibit the activation of proinflammatory cells and the secretion of proinflammatory factors, and reduce the production and activity of MMPs.253,257,263 These effects prevent aortic inflammation and destruction. In a recent study, MSCs activated anti-inflammatory cells such as M2 macrophages and T regulatory (Treg) cells and inhibited proinflammatory cells such as neutrophils.261
Regulation of stem cells in AAD
Stem cells receive signals from surrounding cells such as vascular and inflammatory cells.264 The communication and dynamic interplay among these cells are important in determining the fate and function of stem cells. Identifying the factors and signaling pathways involved in regulating stem cell recruitment, survival, proliferation, growth factor production, and differentiation in the aortic wall is important. Understanding the regulation of stem cell–mediated aortic repair and remodeling will be a critical step toward designing strategies to promote aortic repair and prevent adverse remodeling.
Inflammatory cells and inflammatory components
Inflammatory cells actively participate in tissue injury, repair, and remodeling. After tissue injury, inflammatory cells undergo significant phenotypic and functional changes. They play critical roles in the clearance of tissue debris, the initiation and maintenance of tissue repair and regeneration, and the resolution of inflammation. These steps require communication and interaction between inflammatory cells and aortic cells (eg, endothelial cells, SMCs, fibroblasts or myofibroblasts, and stem or progenitor cells). Dysregulation of inflammatory cell functions can lead to the insufficient clearance of damaged tissues, impaired tissue repair and regeneration, deficient anti-inflammatory responses and inflammation resolution, and the uncontrolled production of inflammatory mediators and growth factors. These things ultimately add to tissue injury and contribute to maladaptive fibrotic remodeling. A variety of inflammatory cells accumulate in TAAD and AAA tissue,265–269 including lymphocytes, macrophages, mast cells, and neutrophils. Each cell type has unique features and functions in the aorta (Fig 8).
Fig. 8.
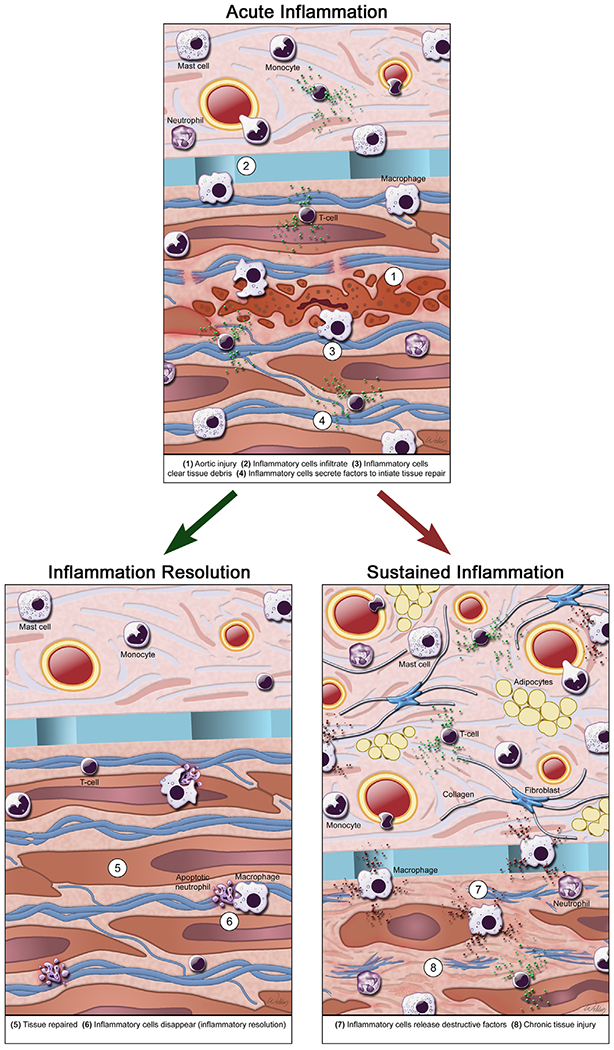
Inflammatory cells in aortic aneurysms and dissections (AAD). In AAD, a variety of inflammatory cells accumulates, including lymphocytes, macrophages, mast cells, and neutrophils. Inflammatory cells actively participate in tissue injury, repair, and remodeling in AAD. After tissue injury, inflammatory cells infiltrate the aortic wall, clear tissue debris, and initiate tissue repair and regeneration. Once the tissues are repaired, inflammatory cells are removed and the inflammation is resolved. Insufficient clearance of damaged tissues, deficient tissue repair and regeneration, and impaired anti-inflammatory responses and inflammation resolution can lead to sustained inflammation with uncontrolled production of destructive inflammatory mediators and growth factors, which aggravate tissue injury and contribute to maladaptive fibrotic remodeling. (Color version of figure is available online.)
T lymphocytes (T cells) in AAD
Activation of T cells in AAD
T cells are a heterogeneous group of lymphocytes with diverse classifications and functions. The major types of T cells are CD4+ cells and CD8+ cells. CD8+ cells perform cell-mediated cytotoxicity,270 whereas CD4+ cells regulate immune responses.271 A significant amount of CD4+ cells has been observed in aortas from patients with TAAD220,272,273 and AAA.265,268,274,275 Direct evidence supports a critical role of CD4+ cells in the development of AAD. Xiong and colleagues276 showed that aneurysm formation was attenuated in a mouse model of AAD when mice were deficient in CD4+ cells. Furthermore, CD4+ cells were shown to exacerbate disease progression by producing cytokines and MMPs and by promoting aortic inflammation and destruction.276
CD4+ T cells can be classified according to the unique profile of cytokines, transcription factors, cell markers, secreted products, and effector actions that they induce.271,277–280 Classifications of CD4+ T cells include T effector (Teff) cells and T helper (Th) cells, which can be further classified as T-helper 1 lymphocytes (Th1),277 T-helper 2 lymphocytes (Th2),277 T-helper 17 lymphocytes (Th17),278 and Treg cells.279
Th1 and Th2 in AAD
Th1 cells are proinflammatory, proatherogenic cells that are involved in several chronic inflammatory disorders.271,280 Th1 cells are activated by IL-12, which activates signal transducer and activator of transcription 4 (STAT4) and T-bet pathways, stimulates Th1 differentiation, and induces the expression of proinflammatory cytokines including interferon gamma (IFN-γ) and tumor necrosis factor-α (TNF-α). In turn, these cytokines activate inflammatory cells such as macrophages that can produce additional IL-12 and further promote Th1 activation.271,280 Th2 cells can be induced by IL-4, IL-13, or both, which activate STAT6 and GATA-binding protein 3 (GATA-3) transcription factors to stimulate Th2 differentiation and the production of IL-4, IL-5, and IL-13. Th2 cells have complex roles in the regulation of inflammation, repair, and regeneration. When Th2 cells are dysregulated, they participate in the pathogenesis of several allergic and fibrotic disorders.271,277,280,281
Activation of Th1 and Th2 in AAD.
Several complexities have been observed regarding the presence of Th1 and Th2 cells in AAD tissues and their roles in AAD development. Increased expression of the Th1 cytokine IFN-γ and the usually undetectable Th2 cytokines has been observed in ascending TAA tissues.282 In addition, Th1 immune responses have been positively correlated with aortic remodeling and the intimal expansion of TAA.220 Moreover, Th1 cells were found to be abundant in aneurysmal tissue from patients with AAA.268 However, a different study has shown that levels of Th1-related cytokines (particularly IFN-γ) were decreased and that Th2 cytokine levels were increased in human AAA tissues.274
Role of Th1 and Th2 in AAD development.
Th2 cells have been implicated in aneurysm development283,284; this has been supported by studies showing that the inhibition of the Th2 cytokine IL-4283 or IL-5284 reduces aortic destruction and AAD formation. However, the role of Th1 in AAD development may be more complex than that of Th2. In a mouse model of calcium chloride–induced AAA, the deletion of the gene encoding Th1 cytokine IFN-γ attenuated MMP expression and aneurysm formation, indicating a destructive role of IFN-γ in matrix remodeling and AAA development.276 Interestingly, in an aorta transplantation experiment, profound aortic aneurysm formation was observed in recipient mice deficient in IFN-γ receptor or IFN-γ signaling. The blockade of IL-4 prevented aortic aneurysm formation in these mice.283 These studies illustrate the complex role of Th1 in AAD development and highlight the importance of the Th1/Th2 balance in matrix remodeling and aortic aneurysm formation.283,285
Th17 cells in AAD
Th17 cells are an important proinflammatory cell type stimulated by IL-1, IL-6, and IL-23, which activate retinoic acid receptor–related orphan receptor γt and STAT3 to induce Th17 differentiation and the secretion of IL-17 and IL-23. These cytokines, in turn, promote the infiltration and activation of macrophages and are involved in several inflammatory diseases.278,286–288 The levels of Th17 cytokines IL-17 and IL-23 were also found to be significantly increased in aortic tissues of patients with AAA.257 A growing body of evidence has indicated an important role of Th17 cells in promoting aortic inflammation and aneurysmal disease development. Inhibition of the Th17 cytokine IL-17 or IL-23 by genetic deletion257 reduced cytokine production and inflammatory cell infiltration in the aorta and attenuated aneurysm development in an elastase perfusion-induced experimental model. Moreover, the inhibition of the Th17 inducer IL-6 attenuated angiotensin II–induced AAD development.289 A recent study showed that inhibiting the IL-17A inflammatory response with digoxin reduced aortic disease development and increased survival in a mouse model of AAD.290 Finally, giving mice MSCs has been shown to decrease IL-17 production and reduce aortic dilation.257 These studies support a destructive role of the Th17 response in AAD development.
Treg cells in AAD
Treg cells are a unique subclass of CD4+ T cells that function as anti-inflammatory cells.279,291,292 Treg cells are activated by IL-2 and TGF-β, which stimulate the forkhead box P3 (FOXP3) pathway to produce IL-10 and TGF-β. Treg cells prevent further inflammation and tissue damage by limiting the activation of proinflammatory cells and by inhibiting the secretion of proinflammatory cytokines such as TNF-α and IFN-γ.279,291,292 Treg cells also play a critical role in promoting tissue repair.279,291,292 Reduced Treg cell population size and impaired Treg cell function have been implicated in chronic inflammation279,291,292 and have also been observed in aortas from patients with AAA.293,294 Furthermore, evidence supports a protective role of Treg cells against AAD formation. Ait-Oufella and colleagues295 showed that depleting Treg cells increased the inflammatory response and significantly enhanced susceptibility to AAD formation and rupture in a mouse model of AAD. In contrast, the reconstitution of Treg cells in Treg-deficient mice limited inflammation and tissue damage and attenuated aortic aneurysm, dissection, and rupture in a mouse model of AAD.293,295
T cells play critical roles in inflammation, either as proinflammatory cells or antiinflammatory cells. In chronic inflammatory processes such as AAD, the imbalance between the proinflammatory and anti-inflammatory responses leads to chronic inflammation, progressive tissue destruction, and AAD development.
Monocytes and macrophages
Activation of macrophages in AAD
Macrophages are important components in the inflammatory response.296–299 The macrophage is a predominant cell type in the aorta of patients with TAAD273,300,301 and AAA302,303 and in mice with AAD.304,305 Because of their role in producing proinflammatory factors such as IL-6, TNF-α, and MMPs306–308 and in promoting inflammation, macrophages have been implicated in the development of AAD.
M1 and M2 populations
Macrophages display plasticity and have the ability to polarize to various phenotypes, such as the classically activated (M1) phenotype or the alternatively activated (M2) phenotype. M1 macrophages produce proinflammatory cytokines and initiate tissue destruction, whereas M2 macrophages promote inflammation resolution and tissue repair.296–299 M1 macrophages can be activated by proinflammatory cytokines including IFN-γ, lipopolysaccharides, and TNF-α, which activate the STAT1-, nuclear factor–κB (NFκB)-, and IRF5-mediated induction of the M1 phenotype. Through the production of proteolytic enzymes and proinflammatory mediators, M1 macrophages promote tissue inflammation and damage. The markers for M1 macrophages include inducible nitric oxide synthase; proinflammatory TNF-α, IL-1β, IL-6, and IL-23; and cell surface proteins such as CD80 and CD86, which are associated with antigen presentation.296–299 M2 macrophages are activated by IL-4 or IL-13, which antagonize IFN-γ to promote the STAT6-and peroxisome proliferator-activated receptor γ (PPARγ)-mediated induction of the M2 phenotype.298,299 M2 macrophages produce anti-inflammatory cytokines including IL-10 and promote tissue repair and healing.298,299 M2 macrophages also have unique markers such as mannose receptor, arginase I, CD206, and CD163. The M1/M2 balance is vital to the outcomes of sustained inflammation vs inflammation resolution and of tissue injury vs repair.
M1 and M2 macrophages in AAD
Both M1 and M2 cells are present in human AAA tissue303,309 and in mouse AAD tissue.305 A high M1/M2 ratio with predominantly M1 cells has been observed in AAA.310 Recent study findings have indicated that the M1/M2 imbalance is critical for aneurysm formation,310–313 and the manipulation of the M1 and M2 cell populations may affect AAD progression. Elastin-derived peptides, which act as proinflammatory mediators, induce M1 polarization. The neutralization of elastin-derived peptides was shown to attenuate aortic dilation. Importantly, the injection of M2-polarized macrophages reduced aortic dilation after aneurysm induction.310 In addition, the inhibition of Notch311 and STAT3312 signaling was shown to decrease the M1/M2 ratio and reduce aneurysm formation. Furthermore, administering D-series resolvins in mice increased M2 macrophage polarization, decreased the levels of proinflammatory cytokines, and inhibited AAA formation in a model of elastase infusion–induced AAA.313 Thus, these findings support that targeting the M1/M2 imbalance by reducing the M1 response and inducing the M2 response may be a useful therapeutic approach to reducing chronic inflammation and tissue destruction in AAA.310,313
Monocytes in AAD
Monocytes give rise to macrophages. In patients with AAA, circulating monocytes were shown to enhance adhesion to the endothelial cell wall and increase MMP-9 production.314 Circulating monocytes have been suggested to contribute to the development of AAA.315 Monocytes or macrophages are recruited to sites of injury by numerous chemokines, including monocyte chemotactic protein (MCP)-1, IL-8, and TNF-α.296 One of the main sources promoting the attraction or chemotaxis of monocytes to the aortic wall may be the presence of aortic ECM breakdown products, such as the peptide sequence VGVAPG found in elastin.316 Blocking the VGVAPG sequence with a monoclonal antibody reduces monocyte or macrophage recruitment and limits additional ECM breakdown.317,318
Neutrophils
Activation of neutrophils in AAD
Neutrophils are an important population of inflammatory cells that not only comprises key effectors of acute inflammation but also contributes to chronic inflammation.319,320 By interacting with innate and adaptive immune cells319 and releasing proteolytic enzymes and reactive intermediates,320 neutrophils contribute to immune responses, tissue injury, and repair.319,320 A significant increase in neutrophil infiltration has been observed in aortas from patients with TAA, TAD,301 and AAA.321–323 Neutrophils are also abundant in the aorta of experimental mouse models of AAD.324–327
Role of neutrophils in AAD development
Neutrophils play a significant role in AAD development. Neutrophil depletion by anti-neutrophil antibody328 or by anti–granulocyte-differentiation antigen-1 (anti–Gr-1) antibody326 significantly decreased the incidence of AAD in an experimental mouse model of AAD. Furthermore, preventing neutrophil recruitment to the aortic wall alleviated elastase-induced experimental AAA.325
Mechanisms of neutrophil-mediated aortic destruction
Neutrophils may promote aortic inflammation and destruction through several mechanisms. They produce destructive proteases such as MMPs326,328 and elastase- and neutrophil-derived proteases.327 In addition, neutrophils release cytokines such as IL-6329 and also express chemokines such as C-X-C motif ligand 1 (CXCL1) and their receptors, including chemokine receptor type 2 (CXCR2),303,329 which are important in inducing inflammation.
Neutrophil extracellular traps in AAD
Neutrophils extrude molecular lattices of chromatin and granular materials that are termed neutrophil extracellular traps (NETs).330 NETs stimulate inflammatory cells and amplify inflammation.331 In addition, they promote vasculopathy and inflammasome activation.332,333 Interestingly, a recent study has shown that NETs are significantly increased in elastase-induced AAA.327 Furthermore, breaking NETs with DNase1 suppresses elastase-induced AAA,327 suggesting that NETs are involved in AAD development. The NETs contain cathelicidin-related antimicrobial peptide and self-DNA, which recruit dendritic cells and induce the production of inflammatory factors, leading to AAA formation.327
Regulation of neutrophil activation in AAD
Several factors are involved in the recruitment of neutrophils to the aortic wall. Dipeptidyl peptidase I (a protease critical for the activation of neutrophil elastase),325 CXCL1, and CXCR2329 are involved in local neutrophil recruitment in AAD. Deficiency in these factors by loss-of-function mutations or neutralization of these factors with antibodies diminishes neutrophil infiltration and reduces aneurysm formation and rupture in a mouse model of AAD.325,329 Diminished catalase expression and activity and increased oxidative stress in neutrophils are also responsible for neutrophil infiltration in AAA.334
Mast cells
Mast cells are a major inflammatory cell type involved in immediate hypersensitivity and chronic allergic reactions such as asthma and allergies.335 Through the rapid release of vasoactive mediators, mast cells regulate vascular permeability and recruit various inflammatory cells, initiating an acute inflammatory response.336 Evidence has indicated that mast cells also participate in chronic inflammation and tissue destruction in cardiovascular diseases.337 The granular content in mast cells includes heparin, histamine, and several enzymes (eg, tryptase, chymase, carboxypeptidase, and cathepsin G).338 Activated mast cells can cause cell apoptosis, matrix degradation, and inflammatory infiltration through the release of growth factors, histamine, chemokines, and proteases such as the mast cell–specific proteases chymase and tryptase.337
Role of mast cells in AAD development
Mast cells have been found in the aortic media and adventitia from patients with TAA, TAD,301 and AAA.269,339,340 Mast cells were also found in the aorta in mouse models of aneurysm formation.341–343 Mast cell–deficient mice showed reduced aortic elastic lamina degradation, inflammation, SMC apoptosis, and attenuated aneurysm formation.341 Reconstitution of these mice with bone marrow–derived mast cells from wild-type mice resulted in increased susceptibility to AAA formation.341 Furthermore, the inhibition of mast cells with the degranulation inhibitor tranilast attenuated AAA development in mice.269 It has been suggested that chymase342 and tryptase343 in mast cells have critical roles in MMP activation, ECM destruction, and SMC apoptosis, and thus promote AAA formation.344 Mast cell stabilizing agents and histamine receptor blockers may be effective therapeutic approaches in preventing the development and growth of AAA.338
Extracellular matrix
The ECM is a dynamic noncellular component of the aorta that provides structural support and controls the plasticity and strength of the aortic wall. The ECM also actively regulates cellular functions. ECM proteins bind to adhesion receptors (eg, integrins) on the cell membrane to mediate matrix and cell attachments and transduce biomechanical signals into cells. ECM proteins also bind to soluble growth factors (eg, TGF-β) and control their signaling in a spatially patterned and regulated fashion. The ECM gives rise to stem cell niches that host and reserve stem cells for tissue repair and regeneration. Although the ECM is constantly undergoing degradation and biosynthesis, abnormal degradation of ECM components can release bioactive fragments that serve as signaling molecules and actively affect cell functions.345–347 Thus, the integrity of the ECM is critical in maintaining aortic structure and function.
ECM degradation in AAD
Intact elastic fibers are vital for aortic structure and function. Elastic fiber fragmentation and depletion reduce aortic resilience that causes aortic dilatation.348,349 In addition, loss of elastic fiber integrity can decrease the undulation of collagen fibers, leading to decreased extensibility and increased stiffness of the aortic wall.350 Aortic dilatation and stiffness are characteristic of aortic disease.52,351 Additionally, functional elastic fibers contribute to the contractile phenotype of SMCs.352,353 Thus, the destruction of elastic fibers may result in aortic contractile dysfunction. Furthermore, microfibrils sequester latent TGF-β and inhibit TGF-β signaling. Elastic fiber damage can result in the release of TGF-β, leading to the overactivation of TGF-β signaling and SMC dysfunction.354,355
In contrast to elastic fibers, collagen fibers in the aorta have a short half-life.356 Appropriately regulated collagen turnover is critical for the aorta to adapt to altered hemodynamics or to respond to injury.357,358 Degradation or loss of collagen fiber integrity decreases aortic strength, rendering the aortic wall prone to rupture.
As discussed earlier, mutations in various genes encoding ECM proteins can cause genetically triggered AAD. However, in both genetic and sporadic aortic diseases, the excessive production of proteases that degrade elastic and collagen fibers can cause matrix destruction, leading to AAD formation.359,360 MMPs and cathepsins, which are involved in proteolysis, are the major classes of extracellular proteases affected in AAD.
Activation of MMPs in AAD
Matrix metalloproteinases
MMPs are a large family of Zn2+-dependent proteolytic proteases that are produced by endothelial cells, SMCs, fibroblasts, and inflammatory cells in the aortic wall.361,362 MMPs not only degrade ECM proteins and control ECM homeostasis,362 but they also regulate signal transduction.363,364 The activities of MMPs are regulated through transcription, zymogen activation, and inhibition by endogenous inhibitors.362–364 When the balance between MMP production or activation and inhibition is disrupted and favors MMP activation, ECM turnover is accelerated.363,364
Destructive role of MMP-9 in AAD
Elevated MMP-9 production and activity have been consistently seen in the aortas of patients with Marfan syndrome,365,366 TAA,367–369 and AAA.370,371 The gene-targeted disruption of MMP-9 in mice suppresses aortic destruction and TAA and AAA formation326,372–374 in models of aortic disease induced by elastase or angiotensin II infusion. Furthermore, the inhibition of MMP-9 activity with MMP inhibitors reduces AAD formation.326,375 Several inflammatory factors such as IL-6306 and TNF-α308 trigger AAD development by upregulating MMP-9. Doxycycline376 or cytokine antibodies such as TNF-α308 reduce AAD development through the downregulation of MMP-9. Thus, evidence from animal studies has indicated that MMP-9 is an important protease involved in aortic destruction and AAD formation. Other MMPs that may also be involved in TAA development are MMP-14 and MMP-19, which have increased expression in aortic tissues from patients with TAA.377
Dual role of MMP-2 in AAD
A recent meta-analysis of 8 studies (including a total of 106 patients and 30 controls) showed no change in MMP-2 levels among aortas from patients with TAA, although MMP-2 levels were higher in patients with TAA and bicuspid aortic valve (BAV) than in patients with TAA and tricuspid aortic valve.368 We observed reduced MMP-2 levels in the tissues of patients with chronic TAD.369 In contrast, MMP-2 has been reported to be upregulated in patients with AAA378 and has been suggested to have a dual role in AAD development. MMP-2 may contribute to AAD development in Marfan syndrome, as indicated by the finding that MMP-2 deficiency in mice with Marfan syndrome attenuated aortic destruction.53,93 Interestingly, a recent study showed that MMP-2 deficiency in mice suppressed angiotensin II–induced elastin and collagen production, thus increasing the severity of TAA.379 However, MMP-2–deficient mice were also shown to be protected against calcium chloride–induced ECM degradation and TAA formation. These findings suggest that MMP-2 has both protective (via ECM synthesis) and destructive (via ECM degradation) functions that affect aortic structure and AAD development.
Membrane-type MMPs
Membrane-type MMPs (MT-MMPs) form a subgroup of the MMP family. Because of their unique location in the cell membrane, MT-MMPs not only control matrix remodeling but also regulate cellular functions (eg, cell migration) and signaling activation (eg, Notch signaling). MT-MMPs are involved in various processes such as angiogenesis and inflammatory responses.380,381 The aortic expression of MT1-MMP, the most studied MT-MMP,380,381 was shown to be elevated in mouse models of AAA307 and TAA.382 In a mouse model of calcium chloride–induced AAA, chimeric mice with MT1-MMP−/− bone marrow cells had reduced aneurysm formation, and macrophage infiltration was unaffected. However, the infusion of MT1-MMP+/+ macrophages into chimeric mice with MT1-MMP−/− bone marrow cells induced AAA formation.307 Thus, MT1-MMP regulates macrophage proteolytic activity and plays an important role in aortic destruction and disease progression.307 In contrast to MT1-MMP, MMP-17/MT4-MMP has a protective role against AAD development. A missense mutation (R373H) in MMP17 causes inherited TAAD in humans.132 MMP-17 cleaves osteopontin, which is required for SMC maturation during aortic wall development. MMP-17 loss of function in mice was shown to result in dysfunctional vascular SMCs and altered ECM in the vessel wall, leading to increased susceptibility to angiotensin II–induced TAA.132
Tissue inhibitors of metalloproteinases
The tissue inhibitors of metalloproteinase (TIMP) family, which includes TIMP-1 to TIMP-4, are endogenous MMP inhibitors.361–363,383 Tissue MMP activity is determined by the balance of MMP and TIMP activation. A recent meta-analysis showed the significant induction of MMP-9 and reduction of TIMP-1 and TIMP-2 protein expression in human TAA tissues that in turn led to the elevation of MMP-9/TIMP-1 and MMP-9/TIMP-2 ratios.368 An imbalanced MMP and TIMP ratio was also observed in AAA tissues.384 Evidence supports a protective role of TIMPs against AAD development. Timp-1 deficiency in ApoE−/− mice increased atherogenic diet–induced atherosclerosis and promoted gelatinolytic activity, elastic lamellae degradation, and aortic destruction.385,386 Similar findings were observed in a model of calcium chloride–induced TAA.373 TIMP-3 deficiency also led to elevated proteolytic activities and inflammation and resulted in exacerbated aneurysm and aortic rupture in a mouse model of angiotensin II–induced AAA.387 Notably, the aortic diseases in these mouse models can be reversed by MMP inhibitors.373,387 These studies support critical roles of TIMP-1 and TIMP-3 in limiting MMP activity and highlight the importance of a low MMP to TIMP ratio in preventing aortic aneurysm formation, expansion, and rupture.
The role of cathepsins in AAD
Evidence has indicated that cathepsin L (CatL) and K (CatK) are involved in AAD development. Deficiency of Ctsl388 or Catk389 in mice significantly reduced aortic inflammation, elastin fragmentation, and the elastase perfusion-induced formation of AAAs. Cathepsin K has also been shown to cause medial SMC apoptosis in AAA.389
Proteoglycans and proteoglycan degradation enzymes
Syndecan-1
Syndecans are a family of TM heparan sulfate proteoglycans that are composed of a core protein and attached glycosaminoglycan chains. Through the glycosaminoglycan chains, syndecans can interact with a variety of extracellular molecules (eg, growth factors and cytokines) and regulate cell functions. Syndecans are involved in a variety of processes such as ECM assembly, tissue repair, and anti-inflammation.390 Syndecan-1, encoded by the SDC1 gene, has been shown to be important for maintaining aortic integrity. Protease activity, elastin degradation, and inflammatory cell infiltration were increased in mice deficient for Sdc1, and elastase- or angiotensin II–induced AAA formation was exacerbated.391 Syndecan-1 and syndecan-4 can be cleaved by MMPs392,393 and a disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4).394 Although this process has not been examined in the context of AAD, the possibility remains that activated MMPs and ADAMTS-4 may cleave and degrade syndecans in AAD, thereby inhibiting the protective actions of syndecans in the aortic structure.
Biglycan
Biglycan has tissue-specific affinities to structural components, and thus plays a crucial role in maintaining tissue integrity. Biglycan also acts as a signaling molecule that binds to its receptors and regulates cell functions.395 Decreased levels of biglycan have been observed in human AAA tissues.396 Furthermore, biglycan deficiency causes spontaneous aortic dissection and rupture in mice,397 indicating an essential role of biglycan in maintaining aortic wall integrity. Consistent with these findings, a recent study showed that loss-of-function mutations in the X-linked biglycan gene cause a severe early-onset syndromic form of TAAD.398 Thus, biglycan is important in maintaining aortic integrity, and genetic defects that alter its function or degradation may play a role in the pathogenesis of aortic dissection and rupture.
ADAMTS proteins in AAD
The ADAMTS family of extracellular proteinases has been shown to be involved in ECM turnover.399 ADAMTS-1 and ADAMTS-4 are particularly well-studied members of this family.400 ADAMTS-4 promotes ECM turnover by digesting proteoglycans including aggrecan, versican, decorin, and biglycan, and ECM proteins such as fibronectin.401,402 ADAMTS-1 digests versican403 and also inhibits cell proliferation by sequestering growth factors.404 The expression of ADAMTS-1,367,405,406 ADAMTS-4,405 ADAMTS-5, and ADAMTS-16406 has been shown to be increased in human TAA tissues. In human TAA tissues, ADAMTS-1 and ADAMTS-4 were identified in vascular SMCs and macrophages, concomitant with the degradation of versican, which is the main proteoglycan substrate of ADAMTS proteinases in the aorta.405 Given the activities of ADAMTS proteinases in ECM degradation, they may play a role in promoting aortic destruction during TAA development.
Plasmin, plasminogen, and plasminogen activators and thrombosis
The plasmin-plasminogen system plays a key role in modulating hemostasis, thrombosis, and inflammation. Plasmin is generated from plasminogen by tissue-type plasminogen activator (t-PA) and urokinase-type PA (u-PA). The major inhibitor of the plasmin-plasminogen system is PA inhibitor-1 (PAI-1). Apart from its key role in fibrinolysis, plasmin also functions as a powerful activator of MMPs,407–410 directly activating MMP-1 and MMP-3 and thereby inducing the activation of MMP-9.407 The plasmin-plasminogen system also plays a central role in the inflammatory response.411
Plasmin, plasminogen, and PAs in AAD
Significantly increased levels of t-PA and u-PA have been observed in aneurysmal tissues from patients with TAAD412,413 and AAA.414 Furthermore, u-PA expression is increased in mice challenged with angiotensin II infusion.415 Several studies have suggested that the plasmin-plasminogen system is essential for the progression of AAD development. The deletion of t-PA or u-PA in mice prevented medial destruction and aneurysm formation in experimental models of TAA and AAA.416–418 Plasminogen deficiency also reduced the formation of AAA in mice by reducing plasmin-dependent MMP activation and inflammation.419 However, another study showed that the deletion of u-PA in bone marrow–derived cells impaired thrombus resolution in the aortic wall and increased aortic rupture in an angiotensin II–induced mouse model of AAA.420 These studies have indicated that u-PA has potentially paradoxical roles in the development of AAD vs aneurysm rupture.421
PAI-1 in AAD
PAI-1 expression is increased in the aortic media of TAA tissues413 but is decreased in AAA tissues.422 Interestingly, the deficiency of Pai-1 in mice resulted in increased MMP-2 levels and promoted aneurysm formation.423 Conversely, the overexpression of PAI-1 in mice provided protection against aneurysm formation.424 Furthermore, PAI-1 overexpression in SMCs protected SMCs from plasmin-induced detachment and death.413 These findings suggest that PAI-1 has a protective role against aortic destruction and AAD development.
Platelet and prothrombin activation in AAD
Platelets are required for hemostasis but can also contribute to excessive thrombosis and inflammation. Platelet activation in plasma has been observed in patients with large ascending TAA425 and AAA.426 Significant platelet activation was observed in the luminal thrombus.426 In a rat model, the inhibition of platelets with inhibitors limited aortic aneurysm expansion, suggesting that platelet activation may enhance AAA development.426–428 The effect of platelet inhibitors on AAD progression has been examined in clinical studies, which is discussed in the medical treatment section.
Signaling pathways and cellular stress
The complex and precise regulation of aortic function and homeostasis involves highly coordinated actions of different aortic cells and the ECM. The aortic cells sense biomechanical and biologic signals and respond by adapting structurally and functionally through highly coordinated signaling pathways. Dysregulation of signaling pathways results in aortic dysfunction, aortic injury, uncontrolled aortic inflammation, and maladaptive remodeling, leading to aortic destruction and biomechanical failure. Elucidating the regulation of signaling pathways in aortic homeostasis and understanding the dysregulation of signaling pathways during AAD development will help us to identify novel targets for the treatment of AAD. Here, we focus on a few major pathways that are important in aortic homeostasis and AAD development.
TGF-β signaling
TGF-β signaling occurs through distinct pathways (Fig 5). The canonical TGF-β! signaling pathway involves receptor phosphorylation and the subsequent activation of SMAD2/3, which binds to SMAD4 and forms a complex that binds to the promoters of target genes. In the noncanonical pathways, the TGF-β receptor complex transmits signals through other pathways such as ERK1/2, JNK, and p38 mitogen-activated protein kinase (MAPK) pathways, which induce the production and release of MMP-2, MMP-9, MMP-13, and PA, thereby causing ECM disruption.
Protective role of TGF-β signaling in aortic homeostasis
The role of TGF-β in regulating aortic structure and function and the underlying molecular mechanisms of this regulation have proven to be complex. A more detailed discussion of this subject is available in another review.429 Numerous studies have supported a beneficial role of TGF-β signaling in aortic homeostasis. Loss-of-function mutations in the genes encoding TGF-β ligands TGFB2 and TGFB3,114,118,119 receptors TGFBR1 and TGFBR2,112,113,115,116 and target transcription factor SMAD3120–122 are associated with genetically triggered TAAD. In mice, the deletion of or loss-of-function mutations in genes that encode TGFB2,114 TGFBR1, TGFBR2,117,125,126 SMAD3,123 or SMAD4124 causes aortic destruction and AAD development. In a mouse model of angiotensin II–induced sporadic AAD, TGF-β signaling can be activated by angiotensin II. Systemically inhibiting TGF-β with neutralizing antibody enhanced inflammatory cell activation and MMP-12 activity and promoted AAD formation, dissection, and rupture.430,431 The results of these studies have clearly identified a protective role for TGF-β in preserving aortic integrity and homeostasis.
Mechanisms underlying the protective role of TGF-β signaling
TGF-β has a critical and fundamental role in the maturation and function of SMCs and in aortic development. Aortic tissue and explanted SMCs from patients with TGFBR2 mutations show reduced basal and TGF-β-induced expression of contractile proteins in SMCs and reduced transformation of fibroblasts into myofibroblasts,127 an important process in the postnatal aortic injury response and aortic homeostasis. Postnatal deletion of Tgfbr2 in the SMCs of mice altered SMC gene expression and caused severe AAD,125,126 suggesting that TGF-β is critical in regulating SMC functions and maintaining aortic wall homeostasis.
Destructive role of TGF-β signaling in AAD
TGF-β signaling has also been shown to have dimorphic effects on AAD formation. In a mouse model of Marfan syndrome, Ltbp-3 deficiency was associated with impaired TGF-β signaling, less aortic destruction, and greater survival,91 suggesting that TGF-β signaling is involved in aortic disease progression. It has also been shown that TGF-β is protective during the early stages of aortic development but is destructive at later stages.95
Mechanisms underlying the dimorphic roles of TGF-β signaling in AAD
One possible explanation for the dual roles of TGF-β in AAD is that TGF-β exerts protective and destructive effects through different pathways.92 Originally, only the canonical SMAD4-dependent TGF-β pathway was evaluated in aneurysmal disease. However, a recent study showed upregulation of both the canonical SMAD4-dependent and the noncanonical ERK1/2 and JNK pathways in mice with Marfan syndrome. Smad4 deficiency exacerbated aortic disease and caused premature death in mice with Marfan syndrome. In contrast, inhibiting ERK1/2 activation or JNK pathways ameliorated aortic dilation.92 Thus, it has been proposed that activation of the noncanonical TGF-β signaling ERK and JNK pathways drives aortic destruction and disease in mice with Marfan syndrome.92,93,95
Upregulation of TGF-β signaling in AAD
Paradoxically, increased TGF-β signaling has been consistently shown in aortic tissue from patients with familial TAAD,89 Marfan syndrome,88–90 or with mutations in TGFB2, TGFB3,114,119 TGFBR,112,128 or SMAD3.120 A recent study showed evidence of dysregulated TGF-β signaling in aortic tissue from patients with mutations in ACTA2 and MYH11.432 Canonical and noncanonical TGF-β signaling and target gene expression have also been shown to be upregulated in mice with AAD.92 The mechanisms underlying this paradoxical elevation are not completely understood. Evidence has indicated that deficiency or dysregulation in canonical TGF-β signaling causes the compensational elevation of TGF-β production,118 which, through the noncanonical pathway, causes aortic destruction and disease development.92
Angiotensin II signaling
The renin-angiotensin system is critically involved in regulating normal cardiovascular homeostasis. However, excessive stimulation of angiotensin II signaling promotes vascular destruction and dysfunction that contributes to a range of cardiovascular diseases. Angiotensin II is the most potent trigger of AAD formation, and it is widely used in experimental AAD models. Angiotensin II–mediated aortic dissection may occur independent of blood pressure changes.289 Angiotensin II has 2 distinct receptors, AT1R and AT2R, which counter-regulate each other and have opposite effects on vascular function (Fig 9).
Fig. 9.
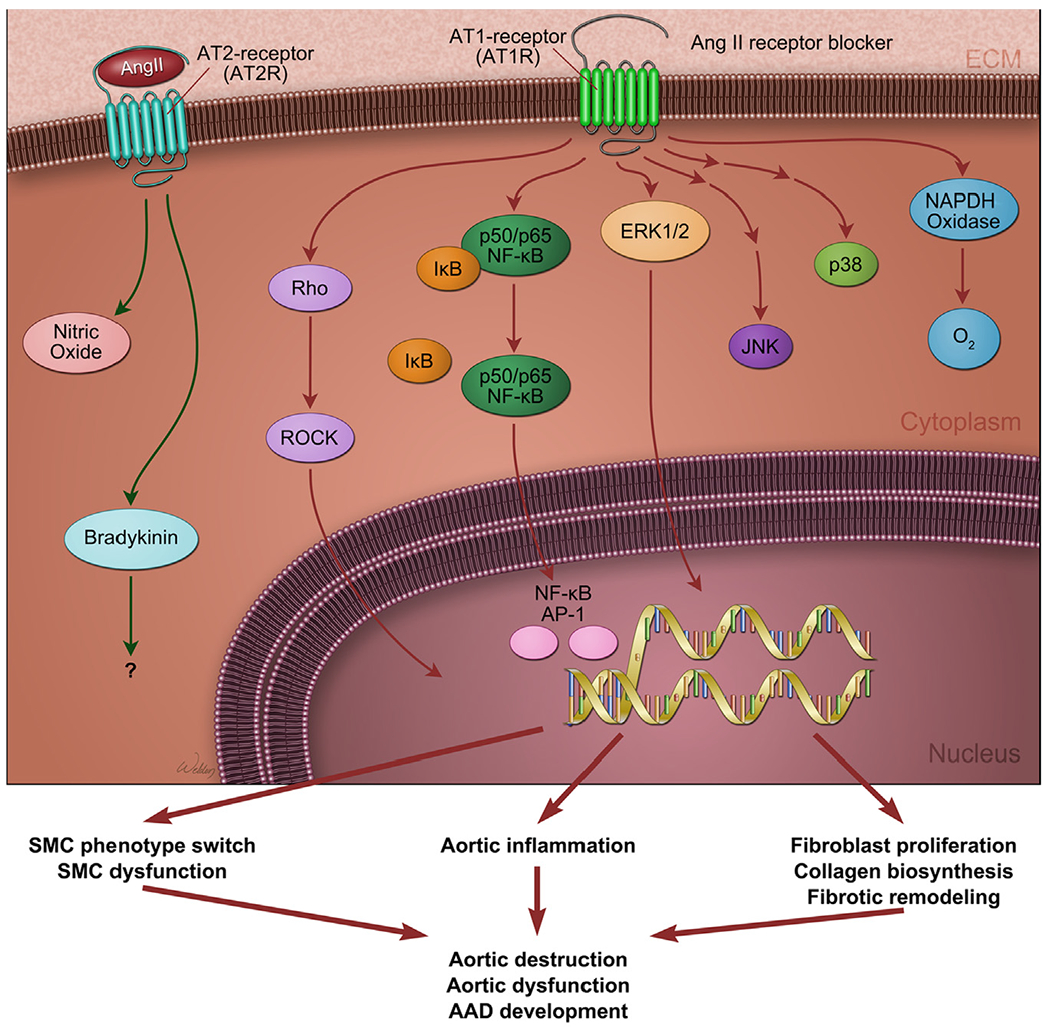
Angiotensin II signaling in aortic aneurysms and dissections (AAD). Angiotensin II (shown as Ang II) has 2 distinct receptors, AT1R and AT2R, which counter-regulate each other and have opposite effects on vascular function. The angiotensin system is critically involved in regulating the structure and function of aortic cells and in maintaining aortic homeostasis. However, excessive stimulation of angiotensin II signaling via AT1R activates several pathways involving rho or rho-associated protein kinase (ROCK), NFkB, ERK1/2, JNK, and p38 and induces NADPH oxidase activation and reactive oxygen species production. These signaling events promote aortic cell dysfunction, inflammation, and fibrotic remodeling, leading to aortic destruction and the formation and progression of AAD. (Color version of figure is available online.)
Destructive role of AT1R in AAD
AT1R has been suggested to have a destructive role in promoting AAD development. A polymorphism in AT1R was associated with AAA in clinical studies.433 In addition, mice deficient in At1r were protected from angiotensin II–induced aneurysm formation.228,434 Treating mice with the ARB losartan, which selectively blocks AT1R, abrogated aneurysm progression in mice with Marfan syndrome90 and partially prevented pressure-induced TAA in a transverse aortic constriction mouse model.241 AT1R may promote AAD formation through multiple mechanisms. It has been shown that blocking AT1R with losartan reduced angiotensin II–induced vascular inflammation and macrophage accumulation.241 Interestingly, AT1R deficiency in bone marrow–derived cells or in SMCs had no effect on ascending aneurysm formation, whereas AT1R deficiency in endothelial cells attenuated angiotensin II–induced TAA formation,228 indicating the importance of endothelial cells and endothelial-specific AT1R in aneurysm formation. Angiotensin II activates several signaling pathways such as the rho-kinase pathway that induces proteolysis and apoptosis, both of which are important activities associated with AAA. Fasudil, a rho-kinase inhibitor, attenuates angiotensin II–induced AAA in ApoE−/− mice by inhibiting apoptosis and proteolysis.435
Protective role of AT2R in AAD
In contrast to AT1R, AT2R has a protective role against AAD development. Deletion of Agt2r, the gene encoding AT2R, accelerated the aberrant growth and rupture of the aorta in a mouse model of Marfan syndrome.94 In addition, signaling through AT2R attenuates aortic aneurysm formation in mice through ERK antagonism.94
Notch signaling
Notch signaling regulates cell-fate determination during development and maintains adult tissue homeostasis. Notch ligands include the delta-like (eg, DLL1, DLL3, and DLL4) and jagged (eg, JAG1 and JAG2) protein families. Notch receptors are transmembrane proteins composed of functional extracellular, transmembrane, and intracellular domains. Upon ligand binding, the Notch intracellular domain is cleaved by γ-secretase, released from the membrane, and translocated into the nucleus, where it associates with the CSL (CBF1/Su[H]/Lag-1) transcription factor complex, thus activating Notch target genes such as the hairy and enhancer of split-1 (HES) and the hairy-related family of transcriptional repressors (HRT).436
Mutations of NOTCH1 in BAV disease
BAV is the most common congenital heart malformation, and it is associated with significant abnormalities in the left ventricular outflow tract and thoracic aorta. Recent studies suggest that Notch signaling regulates proper valve and outflow tract development.437 Notch signaling also appears to be important for inhibiting calcification.438 Loss-of-function mutations in NOTCH1 have been identified in patients with BAV and aortic valve calcification.438–441 NOTCH1 mutations cause early-stage developmental defects in the aortic valve and, at later stages, unsuppressed calcium deposition that leads to progressive aortic valve disease.438
Protective role of Notch signaling in the aortic valve and aorta
Mechanistically, the Notch signaling pathway suppresses the production of runt-related transcription factor 2 (RUNX2), a transcription factor critical for osteoblast cell differentiation, bone formation, and tissue calcification.438 The NOTCH1 signaling target HRT directly interacts with RUNX2 and represses RUNX2 transcriptional activity. It has been proposed that loss of Notch signaling results in the overactivation of RUNX2-mediated osteoblast cell differentiation and tissue calcification, leading to valve calcification. Notch signaling is also a key pathway in promoting EMT, a process that is important for tissue development and repair. Recently, Kostina and colleagues442 showed that Notch-dependent EMT was attenuated in patients with aortic aneurysm and BAV. Aortic endothelial cells from patients with aortic aneurysm and BAV had downregulated Notch signaling and failed to activate EMT in response to different Notch ligands.
Dysregulation of Notch signaling in sporadic TAAD
Notch signaling exhibits a complex pattern in sporadic TAA and TAD tissues.443 Notch signaling is impaired in SMCs of the aortic media but is activated in stem cells, fibroblasts, and macrophages of sporadic TAAD tissues.
Destructive role of Notch signaling in AAA
Although Notch signaling is protective against TAAD in BAV, it plays a destructive role in the abdominal aorta. Inhibiting Notch signaling by gene deletion of Notch1 in mice reduces macrophage-mediated inflammation and AAA formation.444 In addition, blocking the Notch signaling pathway inhibits vascular inflammation445 and AAD progression.311,444,446
Thus, Notch signaling has multiple functions in different cells in the aortic wall and may have both protective and destructive roles in AAD development. Further studies are needed to discern the various tissue-specific roles that Notch signaling plays in aortic homeostasis and disease development.
Other signaling pathways
β-Arrestin-2
β-Arrestin-2 is a multifunctional scaffolding protein that binds G-protein-coupled receptors such as AT1R and regulates numerous G-protein-coupled receptor-dependent and independent signaling pathways during physiological and pathophysiological processes. Recent studies have shown that βarr2 deficiency attenuates aortic aneurysm development in mouse models of Marfan syndrome447 and angiotensin II–induced AAA.448 The inhibition of disease development was attributed to the reduced aortic expression of MMP-2, MMP-9,447 and cyclooxygenase-2, and decreased inflammatory cell infiltration.448 These studies suggest that β-arrestin-2 is another player in promoting aortic destruction and AAD formation.448
Endothelin-1
Endothelin-1 has a role in aortic disease. In ApoE−/− mice, the selective overexpression of endothelin-1 in endothelial cells increased atherosclerosis and induced AAA development. This process was associated with increased ROS production, monocyte or macrophage infiltration, and MMP-2 activity in CD4+ T cells.449
Aldosterone and mineralocorticoid receptor signaling
Through the induction of inflammation, ROS production, and fibrosis, aldosterone and mineralocorticoid receptor signaling can have detrimental effects on the cardiovascular system.450,451 A recent study described a potential role of aldosterone in the pathogenesis of aortic aneurysm in mice.205 Administering aldosterone promoted salt-induced elastin degradation, SMC degeneration and apoptosis, inflammatory cell infiltration, and AAA and TAA formation and rupture in an age-dependent manner. These findings imply a role for the mineralocorticoid receptor in promoting aortic aneurysm development.
Kinin B2 receptor (B2R)
Activation of the kallikrein-kinin system, which signals through bradykinin B1 and B2 receptors, has both beneficial and deleterious effects on cardiovascular functions.452 Interestingly, a recent study has indicated that the activation of B2R signaling promotes aortic rupture.453 In a mouse model of angiotensin II–induced AAD, a B2R antagonist inhibited aortic rupture, whereas a B2R agonist promoted it. Administering a B2R antagonist reduced the expression of the angiotensin II–induced SMC inflammatory phenotype, inflammatory cell activation, and MMP-2 and MMP-9 activation. This study’s findings suggest that B2R signaling may promote a SMC inflammatory phenotype switch, aortic inflammation, and MMP production, leading to aortic destruction.
Apelin
Through its receptor, apelin induces vasodilation by releasing nitric oxide bioavailability from endothelial cells.454 Chun and colleagues455 showed that apelin induced nitric oxide production and inhibited angiotensin II–induced gene expression in SMCs, and it attenuated atherosclerosis and AAA in ApoE−/− mice.
LDL receptor–related protein 1
LDL receptor–related protein 1 (LRP1) is a member of a family of structurally related transmembrane proteins (which also includes LRP2, LDL receptor, and very low-density lipoprotein receptor) that participate in a wide range of physiological processes, including lipid metabolism and cardiovascular functions.456 Recent studies have indicated that LRP1 has a protective role against AAD.457,458 In mice, Lrp1 deficiency in vascular SMCs results in disruption of the elastic layer, SMC proliferation, and aneurysm formation.457,459 Inactivation of LRP1 in vascular SMCs increases elastic fiber fragmentation, which is associated with increased production of high-temperature requirement factor A1, a secreted serine protease involved in ECM degradation and elastogenesis inhibition.460
Kinase pathways and the inflammasome cascade
AKT signaling
The AKT kinase pathway regulates many cellular functions, including survival, proliferation, migration, cell growth, and metabolism; thus, this pathway plays diverse yet critical roles in the cardiovascular system. AKT2 has recently been shown to protect the aorta against AAD development.210 Akt2-deficient mice had abnormal elastic fibers and reduced medial thickness in the aortic wall. When challenged with angiotensin II, these mice developed aortic aneurysm, dissection, and rupture with features similar to those seen in humans, in both thoracic and abdominal segments. Aortas from these mice had profound aortic apoptosis and increased MMP production and inflammation. AKT2 was downregulated in aortic tissues from patients with sporadic TAAD. These results suggest that AKT2 maintains aortic integrity and protects the aorta against AAD formation by preventing aortic apoptosis and tissue destruction.210 Consistent with the protective role of AKT, the deletion of genes encoding upstream kinases of the AKT pathway, such as integrin-linked kinase,461 PI3K-C2α,226 and PI3K-δ,462 also results in aortic destruction and AAD formation.
ERK, JNK, and p38 mitogen-activated protein kinase pathways
The MAPK pathways include ERK, JNK, and p38 pathways. These pathways are activated by inflammatory cytokines and extracellular stresses, as well as by intracellular stress such as ROS and ER stress. They are involved in many stress responses, including apoptosis and inflammation. The JNK pathway is activated in the aortic wall of mice with Marfan syndrome. Inhibiting the JNK1 pathway abolishes aortic destruction in these mice, which suggests that JNK is involved in aortic destruction and disease progression.92 JNK has been shown to promote abnormal ECM degradation and AAA development.463 The ERK pathway is also activated in mice with Marfan syndrome92 and is likewise involved in aortic destruction and disease progression.92 Numerous studies have shown that ERK is critically involved in MMP activation, aortic destruction, and AAD development.93,464–467 Finally, p38 MAPK is also activated in mice with Marfan syndrome and promotes SMAD2/3 activation.468 p38 has been shown to potentiate MMP activation,466 indicating its potential role in aortic destruction.
Protein kinase C
Protein kinase C (PKC) regulates numerous cellular responses, including gene expression, protein secretion, cell proliferation, and the inflammatory response. It has been shown that PKC production is increased in ascending aortic aneurysms excised from patients with BAV.469 Elevated PKC-δ production has been shown to contribute to aneurysm pathogenesis by stimulating apoptosis and inflammatory signaling.208,470
NLRP3–caspase 1 inflammasome cascade
Inflammasomes are intracellular multiprotein complexes that are activated by various cell-stress signals471,472 and function as molecular platforms to mediate the activation of caspases and amplify inflammatory and stress responses.473,474 Recently, a study showed that the inflammasome is activated by mitochondrial oxidative stress in macrophages, leading to the development of angiotensin II–induced aortic aneurysm.475 Enhanced caspase activity was also shown to contribute to aortic wall remodeling and early aneurysm development in a mouse model of Marfan syndrome.203
Transcription factors
Peroxisome proliferator–activated receptor gamma
PPARγ is a ligand-activated nuclear receptor that regulates glucose and lipid metabolism, maintains endothelial function, and inhibits inflammation. Jones and colleagues191 showed that treating mice with the PPARγ activator rosiglitazone inhibited the angiotensin II–induced upregulation of AT1R and the inflammatory response and prevented aortic dilatation and fatal rupture. These findings suggest that PPARγ protects against AAD development.
Nuclear factor-κB
NFκB is a transcription factor that promotes the expression of inflammatory genes. Deficiency in the gene encoding MYD88, an upstream signaling molecule of NFκB, in bone marrow-derived cells inhibited angiotensin II–induced AAA formation, independent of signaling through Toll-like receptors 2 and 4,476 suggesting the involvement of NFκB in aneurysm formation.
Kruppel-like factor 4
KLF4 is a transcription factor that functions as both a repressor and an activator of transcription.477 KLF4 is essential for normal homeostasis and inflammation.477,478 Salmon and colleagues223 showed that KLF4 expression is increased in aortic SMCs in human and mouse aortic aneurysms. KLF4 binds to promoters of SMC genes and is involved in downregulating SMC gene expression. Deletion of Klf4 in SMCs reduced SMC gene suppression, elastin degradation, inflammation, and aneurysm formation in a mouse model of aortic disease induced by elastase infusion and angiotensin II infusion.223 These findings suggest that KLF4 plays a critical role in aneurysm formation.
MicroRNA
MicroRNAs (miRNAs) are small, noncoding RNAs that, by inducing mRNA degradation and translational repression, posttranscriptionally silence target mRNAs (Fig 10).479–481 Several miRNAs have been studied in aortic disease (for details, see the review by Raffort and colleagues482). The altered expression of miRNAs in TAA tissues has been reported and was shown to be associated with aortic enlargement and MMP activation.483
Fig. 10.
MicroRNAs (miRNAs) in aortic aneurysms and dissections (AAD). MiRNAs are small, noncoding RNAs that, by inducing mRNA degradation and translational repression, posttranscriptionally silence target mRNAs. MiRNA genes are transcribed by RNA polymerase II (Pol II). The precursor primary miRNA (pri-miRNA) is cleaved by the RNase III Drosha and DiGeorge syndrome critical region 8 (DGCR8) to form a hairpin-shaped precursor miRNA (pre-miRNA). The pre-miRNA is exported from the nucleus into the cytoplasm, where it is further cleaved by the RNase III enzyme Dicer, yielding an imperfect miRNA-miRNA* duplex. MiRNA is then incorporated into the RNA-induced silencing complex (RISC). Association of a miRNA with its mRNA targets results in degradation and translational inhibition of the target mRNAs. Stress conditions can influence miRNA biogenesis at multiple levels. Competing endogenous RNAs (ceRNAs) bind and sequester miRNAs, preventing them from binding to their mRNA targets and thereby regulating miRNA activity. Several miRNAs that target the extracellular matrix or tissue inhibitor of matrix metalloproteinase-3 (TIMP-3) have been implicated in aortic disease. lncRNA, long noncoding RNA; m7G, 7-methylguanosine (a modified form of guanosine attached to the 5’ ends of mRNAs); ORF, open reading frame. (Modified with permission from van Rooij E and Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860-72.) (Color version of figure is available online.)
MiR-29b
MiR-29b targets cell survival pathways, such as AKT signaling, and ECM proteins. MiR-29b has also been shown to induce apoptosis. MiR-29b levels were increased in the ascending aorta of mice with Marfan syndrome, and blocking miR-29b with antisense oligonucleotides prevented aortic wall apoptosis, ECM deficiencies, and aneurysm development in these mice.484 The overexpression of miR-29b augmented AAA expansion and increased the likelihood of aortic rupture.485 Thus, miR-29b promotes aortic destruction and rupture.484,485
MiR-195
MiR-195 also targets ECM proteins, including collagens, proteoglycans, elastin, and proteins associated with elastic microfibrils. MiR-195 was differentially expressed in the aortas of ApoE−/− mice with angiotensin II–induced AAA. Although silencing miR-195 had no effect on aortic dilation, it resulted in greater elastin expression, suggesting that miR-195 may reduce elastin production and contribute to the pathogenesis of aortic aneurysmal disease.486
MiR-205/MiR-712
MiR-205/miR-712 targets TIMP-3 and promotes MMP activity.487 MiR-205/miR-712 expression was shown to be increased in human AAA tissues and in the aneurysmal tissues of mice with angiotensin II–induced AAA. MiR-205/miR-712 reduces TIMP-3 expression and increases MMP activity in the aortic wall. Silencing miR-205/miR-712 in mice significantly decreased angiotensin II–induced MMP activity and inflammation and prevented AAA development.487
MiR-24
MiR-24 inhibits endothelial cell apoptosis and vascular inflammation.488 Maegdefessel and colleagues488 showed that, by targeting a proinflammatory chitinase-3-like and protein 1, miR-24 inhibited macrophage activation and the inflammatory response and attenuated AAA progression in mice.
Reactive oxygen species (ROS)
At low concentrations, ROS regulate signaling pathways and play important roles in various cell functions. However, the overproduction or accumulation of ROS causes cellular stress and is lethal to cells, thereby contributing to tissue dysfunction, inflammation, and destruction in patients with cardiovascular disease. In cardiovascular physiology and pathophysiology, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex constitutes a major source of ROS in cells.489
Increased ROS production in AAD
The expression of ROS-generating NADPH oxidase and p22phox is elevated in aortic tissues from patients with TAA, particularly in areas where monocytes or macrophages have accumulated.490 Metallothionein, a metal-binding protein that protects against heavy metal and oxidant damage,491 is dysregulated in ascending aortic SMCs of patients with BAV.492 ROS production is also increased in aortic tissues from patients with AAA.493,494
Role of ROS in AAD development
Numerous studies have established the role of ROS in the pathogenesis of AAD.493 Reducing ROS generation in mice by inhibiting NADPH oxidase with the inhibitor apocynin495 or by inhibiting cytochrome P450 by gene deletion496 prevented aneurysm formation in mouse models of AAD. Additionally, promoting ROS removal by administering the antioxidant enzyme catalase or by overexpressing catalase in SMCs decreased SMC apoptosis, aortic inflammation, and aneurysm formation in a model of calcium chloride-induced AAA.497 In contrast, inducing ROS generation in endothelial cells in mice by overexpressing NADPH oxidase NOX2 increased aortic inflammatory cell infiltration and enhanced angiotensin II–mediated aortic dissection.229 Furthermore, several triggers such as iron overload302 and cyclophilin A production498 in the aorta contribute to AAA development by causing vascular oxidative stress. ROS promote aortic destruction by inducing aortic inflammation, MMP production,302,498 and SMC apoptosis494 during AAD development.
Interestingly, a recent study showed that systemic Nox2 deficiency reduced the level of ROS in AAA lesions but promoted AAA development in mice.499 This study illustrates the complex role of NADPH oxidases and ROS in AAD development.
ER stress (unfolded protein response)
The cells of the aortic wall are constantly exposed to various stress-triggering factors such as hemodynamic disturbance, inflammatory factors, metabolic stress, and ROS. These factors can cause intracellular stress that interferes with normal cellular function. Protein folding in the ER is particularly vulnerable because stress can interfere with the complicated processes of concomitant glycosylation, disulfide bond formation, and membrane insertion. Protein misfolding, also called ER stress, not only causes protein dysfunction but also triggers signal-transduction events. These signaling events can amplify stress in a way that provokes an inflammatory response,500–503 causing cellular dysfunction and even cell death.500–503
ER stress in AAD
In a mouse model of aortic injury induced by the inhibition of LOX with inhibitor 3-aminopropionitrile fumarate, the expression of ER stress markers ATF4 and CHOP was induced. Chop deficiency reduced SMC apoptosis, inflammation, and AAD formation in mice.198 Additionally, the overexpression of SM Myh11 was shown to cause ER stress, which induced the autophagic degradation of thick filament-associated proteins.504
Medical treatment and drug development
A large number of therapeutic drugs has been tested in animal models of AAD. These drugs reduce aortic destruction and AAD development by targeting signaling pathways, inflammatory factors, and MMPs. Several of these drugs have also been examined in association studies or randomized trials in patients with AAD. The medical treatment of thoracic505–509 and abdominal510–513 AAD has been extensively reviewed in the literature.
β-Blockers
β-Blocker use in TAAD
β-Blockers have been the first-line medication for patients with Marfan syndrome and thoracic TAAD. Early studies showed that the β-blocker propranolol decreased aortic dilatation and mortality in patients with Marfan syndrome.514 However, its effect was not confirmed in later studies; large meta-analyses did not show the beneficial effects of β-blockers on the reduction of aortic dissection or mortality in patients with Marfan syndrome515 or chronic type B aortic dissection.516 Despite the limited evidence supporting their efficacy, β-blockers are still routinely provided to patients with Marfan syndrome and other causes of TAAD to prevent the progression of aortic disease.509
β-Blocker use in AAA
Early studies in rodents suggested that β-blockers may slow the progression of AAA. In later, large-scale randomized controlled trials (RCT), no significant reduction in AAA expansion was observed in patients receiving β-blockers.511,513,517 However, β-blockers may have a beneficial effect in patients with AAA during surgical repair. It has been shown that patients receiving β-blockers around the time of surgery have a reduced incidence of nonfatal myocardial infarction.518
ACE inhibitors
ACE inhibitor use in TAAD
ACE inhibitors limit the production of angiotensin II and thus reduce signaling through both AT1R and AT2R. Clinical studies have shown that the ACE inhibitors enalapril and perindopril reduce aortic stiffness and aortic root diameter in patients with Marfan syndrome.519,520 However, in a recent study in mice with Marfan syndrome, enalapril was less effective than losartan (an AT1R blocker) in reducing aortic enlargement.94
ACE inhibitor use in AAA
Animal studies have yielded promising results on the use of ACE inhibitors to treat AAA. ACE inhibitors (eg, captopril, lisinopril, and enalapril) attenuated elastin degradation and disease development in an elastase infusion-induced AAA model.521 However, the results from human studies have been conflicting. Associated studies have shown that patients taking ACE inhibitors had a reduced need for surgery522 and a lower risk of rupture.523 In contrast, a prospective cohort study showed an association between ACE inhibitor use and increased AAA expansion in patients with small AAAs.524 In a recent RCT, ACE inhibitor use effectively lowered systolic blood pressure but had no effect on AAA growth.525
Angiotensin receptor blockers
ARBs selectively inhibit AT1R but have no effect on AT2R.
ARB use in TAAD
Losartan, the best-studied ARB, has been shown to prevent aneurysm progression in a mouse model of Marfan syndrome.90,94 The effect was attributed to the reduction of TGF-β signaling and ERK activation.90,94 In small, prospective cohort studies, losartan slowed aortic root dilation in pediatric patients with Marfan syndrome.526,527 Furthermore, in a multicenter RCT of 233 adult patients, losartan treatment reduced aortic dilatation.528 Several trials are currently ongoing to further evaluate and compare the effects of losartan and β-blockers and their additive effects on disease progression in patients with Marfan syndrome.529–531 The use of ARBs in other genetic TAADs has been tested in mouse models. Recent studies have shown that losartan treatment ablates aortic aneurysms in mice with Loeys-Dietz syndrome.117,532 Additionally, in a transverse aortic constriction mouse model, losartan treatment significantly blocked pressure-induced vascular inflammation.241 These preliminary studies suggest that ARBs may have beneficial effects in treating genetically triggered and sporadic TAAD.
ARB use in AAA
Conflicting results have been reported regarding the use of ARBs in treating AAA. Although losartan completely inhibited angiotensin II–induced AAAs in ApoE−/− mice,533 another study showed that losartan had no significant effect on aneurysm formation in an elastase-induced model of AAA.521 These disparate results may be related to differences in the disease process in these different models. However, in a population-based case-control study, the protective effect of ARBs was not observed in patients with AAA.523 The efficacy of ARBs in limiting AAA growth is currently being studied in 2 randomized trials.534
Calcium channel blockers
Although calcium channel blockers (CCBs) are prescribed to patients with Marfan syndrome to prevent aneurysm progression,535 limited data are available on the efficacy and safety of their use for this condition. A small RCT showed that the use of perindopril, verapamil, and atenolol all reduced peripheral and central systolic pressure,536 but only atenolol delayed aortic expansion in patients with Marfan syndrome. However, in a recent study in mice, Doyle and colleagues535 observed unexpectedly that amlodipine and verapamil promoted aortic destruction and rupture in the aorta of mice with Marfan syndrome. In addition, patients with Marfan syndrome and other forms of inherited TAA who were taking CCBs had an increased risk of aortic dissection and need for aortic surgery compared with patients taking other antihypertensive agents.535 Further studies are needed to confirm this finding. For the time being, it is suggested that the use of CCBs warrants caution in patients with inherited TAAD.
Statins
Independent of their cholesterol-lowering effects, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) reduce inflammation and protect the vascular wall.537,538 Because they are relatively well tolerated for long-term use, statins may be useful for the treatment of aortic diseases.
Statin use in TAAD
In a mouse model of Marfan syndrome, statins were shown to preserve aortic structure and attenuate aortic root dilation.539 Moreover, several clinical studies have indicated that statin use was consistently associated with improved long-term outcomes in patients with TAA.540–543
Statin use in AAA
In a mouse model of elastase-induced AAA, statins have been shown to reduce MMP-9 production, decrease macrophage activation,544–547 and suppress AAA growth.547 In several clinical studies, statin use was associated with reduced AAA expansion, need for surgery, or incidence of rupture.541,548–550 However, other studies have not confirmed these findings.551,552 Conflicting results were reported in a recent review of 8 observational studies comprising a total of 4466 patients.553 One large trial is currently underway to examine the efficacy of statin use in limiting AAA growth (NCT01756833).554
Doxycycline
Doxycycline use in TAAD
The antibiotic doxycycline is a potent MMP inhibitor.555 Doxycycline has been shown to inhibit the production and activity of MMP-2 and MMP-9, reduce elastin degradation, and delay aneurysm rupture in mice with Marfan syndrome.93,376,556 Moreover, doxycycline and losartan have a synergistic effect on preventing the development of TAA in mice with Marfan syndrome.557 Additionally, long-term treatment with doxycycline prevented the development of spontaneous aortic lesions in a mouse model of vascular Ehlers-Danlos syndrome.558 However, no clinical data are available on its efficacy in patients with TAA.
Doxycycline use in AAA
In mouse models of AAA, doxycycline inhibited aneurysm development and progression.511 In preoperative patients with AAA, a short-term, 2-week regimen of doxycycline decreased inflammation in the aortic wall.559 In patients after endovascular AAA repair, doxycycline reduced circulating levels of MMPs.560 However, in a recent clinical trial, an 18-month regimen of doxycycline therapy did not reduce aneurysm growth or the need for AAA repair.561 Therefore, the beneficial effects of long-term doxycycline use for treating AAA remain to be determined.511–513,562 The efficacy of doxycycline in treating AAA is currently being evaluated in an ongoing clinical trial (NCT01756833).
Platelet inhibitors
Large thrombi containing significant amounts of inflammatory cells and increased levels of MMPs are often seen in descending TAAD and in AAA. Thrombus size has been shown to correlate with aneurysm growth in AAA.563 Thus, antiplatelet therapy has been studied for treating aortic diseases.
Platelet inhibitor use in AAA
In a mouse model of AAA, aspirin and clopidogrel reduced aortic inflammation, tissue destruction, and aneurysm rupture.428 A retrospective cohort study showed that aspirin use was associated with reduced AAA progression.564 Moreover, the use of aspirin or P2Y12 inhibitors has been associated with a reduced rate of death among patients with AAA.428 In a study of the potential mechanisms underlying the benefits of aspirin therapy, Bailey and colleagues565 showed that aspirin therapy in patients was associated with enhanced fibrinolysis and less compact fibrin networks. In a meta-analysis of a few trials, the use of aspirin was associated with lower expansion rates and less need for later surgical repair in patients with larger AAA ( > 35 mm).564 A clinical trial examining the efficacy of the antiplatelet agent ticagrelor in limiting AAA growth is currently underway (NCT02070653).
Experimental drugs for AAD treatment
A large number of therapeutic drugs has been tested in animal models for use in reducing AAD development.
Anti-inflammatory agents
Infliximab, a US Food and Drug Administration-approved TNF-α antagonist, has been shown to inhibit aneurysm growth in mice.308 Anakinra, an IL-1β antagonist, attenuated elastase-induced TAA formation in mice.566 Anakinra is currently being studied in a large clinical trial to determine its effects on atherosclerotic diseases (NTC02007252).567 The immunosuppressive drug azathioprine has been shown to reduce angiotensin II–induced inflammation and aneurysm progression in mice.230 Using an innovative experimental approach, investigators have injected nanoparticles containing the MMP inhibitor batimastat; they found that this approach prevented the progression of calcium chloride-induced AAAs in mice.568
Targeting other signaling pathways
In mice, the JNK inhibitor SP600125,463 the rho-kinase inhibitor fasudil,435 the rapamycin inhibitor everolimus,569 and the adenosine 2A receptor agonist570 have been shown to reduce aortic destruction, decrease inflammation, and attenuate AAD formation.
Conclusions
In the last decade, tremendous progress has been made toward improving our understanding of the molecular pathogenesis of AAD. The aortic wall is highly dynamic, performing sophisticated biomechanical functions to provide suitable compliance and adequate strength in response to hemodynamic changes. This requires the highly coordinated actions of different aortic cells and the ECM. Aortic function and homeostasis rely on intact cellular and extracellular proteins, as well as efficient signaling and communication among these components. Genetic defects in these structural components (eg, SMC contractile proteins and elastic microfibrils) or in their signaling pathways (eg, TGF-β signaling) cause aortic dysfunction and destruction. In the case of sporadic AAD, clinical risk factors for disease development (eg, smoking and hypertension) cause biologic insults, either directly or indirectly, that interfere with normal signaling and result in cell dysfunction. Both genetic defects and risk factors, through various shared stress pathways, disturb aortic homeostasis, cause SMC dysfunction and loss, promote ECM destruction, and induce uncontrolled inflammation and maladaptive fibrotic remodeling, leading to aortic aneurysm, dissection, and rupture.
Importantly, our improved understanding of the molecular pathogenesis of AAD has led to the development of new strategies for treating AAD. Several drugs have been tested that target signaling pathways (eg, with the AT1R blocker losartan), suppress inflammation (eg, with statins), or inhibit destructive proteases (eg, with doxycycline as an MMP inhibitor). These drugs have shown promising results from preclinical and early clinical studies and may be used to prevent aortic destruction and slow disease progression. However, larger scale and better controlled clinical studies are needed to confirm their efficacy, test combination therapy, and define the best suitable patient groups for each medical regimen. Future research focused on uncovering the precise regulation of aortic injury, repair, and remodeling will further improve our understanding of the disease process and facilitate the development of new drugs to treat these highly lethal diseases.
Acknowledgments
We gratefully acknowledge Scott A. Weldon, MA, CMI, for creating illustrations; and Nicole Stancel, PhD, ELS, and Rebecca Bartow, PhD, at the Texas Heart Institute for editorial support.
References
- 1.Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd Improved prognosis of thoracic aortic aneurysms: a population-based study. J Am Med Assoc. 1998;280(22):1926–1929. [DOI] [PubMed] [Google Scholar]
- 2.Olsson C, Thelin S, Stahle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation. 2006;114(24):2611–2618. [DOI] [PubMed] [Google Scholar]
- 3.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111(6):816–828. [DOI] [PubMed] [Google Scholar]
- 4.Meszaros I, Morocz J, Szlavi J, et al. Epidemiology and clinicopathology of aortic dissection. Chest. 2000;117(5):1271–1278. [DOI] [PubMed] [Google Scholar]
- 5.Clouse WD, Hallett JW Jr, Schaff HV, et al. Acute aortic dissection: population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc. 2004;79(2):176–180. [DOI] [PubMed] [Google Scholar]
- 6.Golledge J, Eagle KA. Acute aortic dissection. Lancet. 2008;372(9632):55–66. [DOI] [PubMed] [Google Scholar]
- 7.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8(2):103–113. [DOI] [PubMed] [Google Scholar]
- 8.Multicentre Aneurysm Screening Study G. Multicentre aneurysm screening study (MASS): cost effectiveness analysis of screening for abdominal aortic aneurysms based on four year results from randomised controlled trial. Br Med J. 2002;325(7373):1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svensjo S, Bjorck M, Gurtelschmid M, Djavani Gidlund K, Hellberg A, Wanhainen A. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation. 2011;124(10):1118–1123. [DOI] [PubMed] [Google Scholar]
- 10.Kung HC, Hoyert DL, Xu J, Murphy SL. Deaths: final data for 2005. Natl Vital Stat Rep. 2008;56(10):1–120. [PubMed] [Google Scholar]
- 11.Minino AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep. 2011;59(10):1–126. [PubMed] [Google Scholar]
- 12.Jain D, Dietz HC, Oswald GL, Maleszewski JJ, Halushka MK. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Pathol. 2011;20(1):15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Booher AM, Isselbacher EM, Nienaber CA, et al. The IRAD classification system for characterizing survival after aortic dissection. Am J Med. 2013;126(8):730.e719–724. [DOI] [PubMed] [Google Scholar]
- 14.Coady MA, Davies RR, Roberts M, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134(4):361–367. [DOI] [PubMed] [Google Scholar]
- 15.Coady MA, Rizzo JA, Goldstein LJ, Elefteriades JA. Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin. 1999;17(4):615–635;[vii]. [DOI] [PubMed] [Google Scholar]
- 16.Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections–incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82(4):1400–1405. [DOI] [PubMed] [Google Scholar]
- 17.Milewicz DM, Guo DC, Tran-Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302. [DOI] [PubMed] [Google Scholar]
- 18.Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ Res. 2013;113(3):327–340. [DOI] [PubMed] [Google Scholar]
- 19.Andelfinger G, Loeys B, Dietz H. A decade of discovery in the genetic understanding of thoracic aortic disease. Can J Cardiol. 2016;32(1):13–25. [DOI] [PubMed] [Google Scholar]
- 20.Alcorn HG, Wolfson SK Jr, Sutton-Tyrrell K, Kuller LH, O’Leary D. Risk factors for abdominal aortic aneurysms in older adults enrolled in The Cardiovascular Health Study. Arterioscler Thromb Vasc Biol. 1996;16(8):963–970. [DOI] [PubMed] [Google Scholar]
- 21.Agmon Y, Khandheria BK, Meissner I, et al. Is aortic dilatation an atherosclerosis-related process? Clinical, laboratory, and transesophageal echocardiographic correlates of thoracic aortic dimensions in the population with implications for thoracic aortic aneurysm formation. J Am Coll Cardiol. 2003;42(6):1076–1083. [DOI] [PubMed] [Google Scholar]
- 22.Madaric J, Vulev I, Bartunek J, et al. Frequency of abdominal aortic aneurysm in patients > 60 years of age with coronary artery disease. Am J Cardiol. 2005;96(9):1214–1216. [DOI] [PubMed] [Google Scholar]
- 23.Forsdahl SH, Singh K, Solberg S, Jacobsen BK. Risk factors for abdominal aortic aneurysms: a 7-year prospective study: the Tromso Study, 1994-2001. Circulation. 2009;119(16):2202–2208. [DOI] [PubMed] [Google Scholar]
- 24.Hultgren R, Larsson E, Wahlgren CM, Swedenborg J. Female and elderly abdominal aortic aneurysm patients more commonly have concurrent thoracic aortic aneurysm. Ann Vasc Surg. 2012;26(7):918–923. [DOI] [PubMed] [Google Scholar]
- 25.Vapnik JS, Kim JB, Isselbacher EM, et al. Characteristics and outcomes of ascending versus descending thoracic aortic aneurysms. Am J Cardiol. 2016;117(10):1683–1690. [DOI] [PubMed] [Google Scholar]
- 26.Lederle FA, Johnson GR, Wilson SE, et al. The aneurysm detection and management study screening program: validation cohort and final results. Aneurysm detection and management veterans affairs cooperative study investigators. Arch Intern Med. 2000;160(10):1425–1430. [DOI] [PubMed] [Google Scholar]
- 27.Landenhed M, Engstrom G, Gottsater A, et al. Risk profiles for aortic dissection and ruptured or surgically treated aneurysms: a prospective cohort study. J Am Heart Assoc. 2015;4(1):e001513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito S, Akutsu K, Tamori Y, et al. Differences in atherosclerotic profiles between patients with thoracic and abdominal aortic aneurysms. Am J Cardiol. 2008;101(5):696–699. [DOI] [PubMed] [Google Scholar]
- 29.Sidloff D, Choke E, Stather P, Bown M, Thompson J, Sayers R. Mortality from thoracic aortic diseases and associations with cardiovascular risk factors. Circulation. 2014;130(25):2287–2294. [DOI] [PubMed] [Google Scholar]
- 30.Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116(8):1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Majesky MW, Dong XR, Hoglund V, Mahoney WM Jr, Daum G. The adventitia: a dynamic interface containing resident progenitor cells. Arterioscler Thromb Vasc Biol. 2011;31(7):1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis EC. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest. 1993;68(1):89–99. [PubMed] [Google Scholar]
- 34.Arribas SM, Hinek A, Gonzalez MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther. 2006; 111(3):771–791. [DOI] [PubMed] [Google Scholar]
- 35.Davis EC. Elastic lamina growth in the developing mouse aorta. J Histochem Cytochem. 1995;43(11):1115–1123. [DOI] [PubMed] [Google Scholar]
- 36.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88(1):37–43. [DOI] [PubMed] [Google Scholar]
- 37.Pereira L, Lee SY, Gayraud B, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96(7):3819–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488–1493. [DOI] [PubMed] [Google Scholar]
- 39.Lu H, Fagnant PM, Bookwalter CS, Joel P, Trybus KM. Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc Natl Acad Sci U S A. 2015;112(31):E4168–E4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfaltzgraff ER, Bader DM. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev Dyn. 2015;244(3):410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Meurs-van Woezik H, Klein HW, Markus-Silvis L, Krediet P. Comparison of the growth of the tunica media of the ascending aorta, aortic isthmus and descending aorta in infants and children. J Anat. 1983;136(Pt 2):273–281. [PMC free article] [PubMed] [Google Scholar]
- 42.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59:89–144. [DOI] [PubMed] [Google Scholar]
- 43.Shirwany NA, Zou MH. Arterial stiffness: a brief review. Acta Pharmacol Sin. 2010;31(10):1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gadson PF Jr, Rossignol C, McCoy J, Rosenquist TH. Expression of elastin, smooth muscle alpha-actin, and c-jun as a function of the embryonic lineage of vascular smooth muscle cells. In Vitro Cell Dev Biol Anim. 1993;29A(10):773–781. [DOI] [PubMed] [Google Scholar]
- 45.Ruckman JL, Luvalle PA, Hill KE, Giro MG, Davidson JM. Phenotypic stability and variation in cells of the porcine aorta: collagen and elastin production. Matrix Biol. 1994;14(2):135–145. [DOI] [PubMed] [Google Scholar]
- 46.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–1616. [DOI] [PubMed] [Google Scholar]
- 47.Gittenberger-de Groot AC, DeRuiter MC, Bergwerff M, Poelmann RE. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler Thromb Vasc Biol. 1999;19(7):1589–1594. [DOI] [PubMed] [Google Scholar]
- 48.Wasteson P, Johansson BR, Jukkola T, et al. Developmental origin of smooth muscle cells in the descending aorta in mice. Development. 2008;135(10):1823–1832. [DOI] [PubMed] [Google Scholar]
- 49.Zhang H, Gu S, Al-Sabeq B, et al. Origin-specific epigenetic program correlates with vascular bed-specific differences in Rgs5 expression. FASEB J. 2012;26(1):181–191. [DOI] [PubMed] [Google Scholar]
- 50.Cheung C, Bernardo AS, Trotter MW, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol. 2012;30(2):165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pfaltzgraff ER, Shelton EL, Galindo CL, et al. Embryonic domains of the aorta derived from diverse origins exhibit distinct properties that converge into a common phenotype in the adult. J Mol Cell Cardiol. 2014;69:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Hamamsy I, Yacoub MH. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat Rev Cardiol. 2009;6(12):771–786. [DOI] [PubMed] [Google Scholar]
- 53.Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and −9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101(5):512–522. [DOI] [PubMed] [Google Scholar]
- 54.Lindsay ME, Dietz HC. The genetic basis of aortic aneurysm. Cold Spring Harb Perspect Med. 2014;4(9):a015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moltzer E, te Riet L, Swagemakers SM, et al. Impaired vascular contractility and aortic wall degeneration in fibulin-4 deficient mice: effect of angiotensin II type 1 (AT1) receptor blockade. PLoS One. 2011;6(8):e23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milewicz DM, Ostergaard JR, Ala-Kokko LM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A(10):2437–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Regalado ES, Guo DC, Prakash S, et al. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet. 2015;8(3):457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol. 2016;32(1):26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Malloy LE, Wen KK, Pierick AR, et al. Thoracic aortic aneurysm (TAAD)-causing mutation in actin affects formin regulation of polymerization. J Biol Chem. 2012;287(34):28398–28408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bergeron SE, Wedemeyer EW, Lee R, et al. Allele-specific effects of thoracic aortic aneurysm and dissection alpha-smooth muscle actin mutations on actin function. J Biol Chem. 2011;286(13):11356–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu L, Vranckx R, Khau Van Kien P, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38(3):343–349. [DOI] [PubMed] [Google Scholar]
- 63.Pannu H, Tran-Fadulu V, Papke CL, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16(20):2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuang SQ, Guo DC, Prakash SK, et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011;7(6):e1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuang SQ, Kwartler CS, Byanova KL, et al. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circ Res. 2012;110(11):1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bellini C, Wang S, Milewicz DM, Humphrey JD. Myh11(R247C/R247C) mutations increase thoracic aorta vulnerability to intramural damage despite a general biomechanical adaptivity. J Biomech. 2015;48(1):113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276(7): 4527–4530. [DOI] [PubMed] [Google Scholar]
- 68.Wang L, Guo DC, Cao J, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87(5):701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo DC, Regalado E, Casteel DE, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93(2):398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murrell M, Oakes PW, Lenz M, Gardel ML. Forcing cells into shape: the mechanics of actomyosin contractility. Nat Rev Mol Cell Biol. 2015;16(8):486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheen VL, Jansen A, Chen MH, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64(2):254–262. [DOI] [PubMed] [Google Scholar]
- 72.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103(6 Pt 1):2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Milewicz DM, Grossfield J, Cao SN, Kielty C, Covitz W, Jewett T. A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. J Clin Invest. 1995;95(5):2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. [DOI] [PubMed] [Google Scholar]
- 75.Dietz HC, Valle D, Francomano CA, Kendzior RJ Jr, Pyeritz RE, Cutting GR. The skipping of constitutive exons in vivo induced by nonsense mutations. Science. 1993;259(5095):680–683. [DOI] [PubMed] [Google Scholar]
- 76.Collod-Beroud G, Le Bourdelles S, Ades L, et al. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22(3):199–208. [DOI] [PubMed] [Google Scholar]
- 77.Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81(3):454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sood S, Eldadah ZA, Krause WL, McIntosh I, Dietz HC. Mutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat Genet. 1996;12(2):209–211. [DOI] [PubMed] [Google Scholar]
- 79.Dietz HC, Saraiva JM, Pyeritz RE, Cutting GR, Francomano CA. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF-like domains. Hum Mutat. 1992;1(5):366–374. [DOI] [PubMed] [Google Scholar]
- 80.Whiteman P, Hutchinson S, Handford PA. Fibrillin-1 misfolding and disease. Antioxid Redox Signal. 2006;8(3–4): 338–346. [DOI] [PubMed] [Google Scholar]
- 81.Suk JY, Jensen S, McGettrick A, et al. Structural consequences of cysteine substitutions C1977Y and C1977R in calcium-binding epidermal growth factor-like domain 30 of human fibrillin-1. J Biol Chem. 2004;279(49): 51258–51265. [DOI] [PubMed] [Google Scholar]
- 82.Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med. 1990;323(3):152–159. [DOI] [PubMed] [Google Scholar]
- 83.Milewicz DM, Pyeritz RE, Crawford ES, Byers PH. Marfan syndrome: defective synthesis, secretion, and extracellular matrix formation of fibrillin by cultured dermal fibroblasts. J Clin Invest. 1992;89(1):79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Crosas-Molist E, Meirelles T, Lopez-Luque J, et al. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):960–972. [DOI] [PubMed] [Google Scholar]
- 85.Dallas SL, Miyazono K, Skerry TM, Mundy GR, Bonewald LF. Dual role for the latent transforming growth factor-beta binding protein in storage of latent TGF-beta in the extracellular matrix and as a structural matrix protein. J Cell Biol. 1995;131(2):539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dallas SL, Keene DR, Bruder SP, et al. Role of the latent transforming growth factor beta binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J Bone Miner Res. 2000;15(1):68–81. [DOI] [PubMed] [Google Scholar]
- 87.Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278(4):2750–2757. [DOI] [PubMed] [Google Scholar]
- 88.Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33(3):407–411. [DOI] [PubMed] [Google Scholar]
- 89.Ng CM, Cheng A, Myers LA, et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114(11):1586–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci U S A. 2015;112(45):14012–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res. 2012;110(12):e92–e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332(6027):361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook JR, Clayton NP, Carta L, et al. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takeda N, Morita H, Fujita D, et al. Congenital contractural arachnodactyly complicated with aortic dilatation and dissection: case report and review of literature. Am J Med Genet A. 2015;167A(10):2382–2387. [DOI] [PubMed] [Google Scholar]
- 97.Barbier M, Gross MS, Aubart M, et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95(6): 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Capuano A, Bucciotti F, Farwell KD, et al. Diagnostic exome sequencing identifies a novel gene, EMILIN1, associated with autosomal-dominant hereditary connective tissue disease. Hum Mutat. 2016;37(1):84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Szabo Z, Crepeau MW, Mitchell AL, et al. Aortic aneurysmal disease and cutis laxa caused by defects in the elastin gene. J Med Genet. 2006;43(3):255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hucthagowder V, Sausgruber N, Kim KH, Angle B, Marmorstein LY, Urban Z. Fibulin-4: a novel gene for an autosomal recessive cutis laxa syndrome. Am J Hum Genet. 2006;78(6):1075–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dasouki M, Markova D, Garola R, et al. Compound heterozygous mutations in fibulin-4 causing neonatal lethal pulmonary artery occlusion, aortic aneurysm, arachnodactyly, and mild cutis laxa. Am J Med Genet A. 2007;143A(22): 2635–2641. [DOI] [PubMed] [Google Scholar]
- 102.Renard M, Holm T, Veith R, et al. Altered TGFbeta signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur J Hum Genet. 2010;18(8):895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hanada K, Vermeij M, Garinis GA, et al. Perturbations of vascular homeostasis and aortic valve abnormalities in fibulin-4 deficient mice. Circ Res. 2007;100(5):738–746. [DOI] [PubMed] [Google Scholar]
- 104.Huang J, Davis EC, Chapman SL, et al. Fibulin-4 deficiency results in ascending aortic aneurysms: a potential link between abnormal smooth muscle cell phenotype and aneurysm progression. Circ Res. 2010;106(3):583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88(4):660–672. [DOI] [PubMed] [Google Scholar]
- 106.Guo DC, Regalado ES, Gong L, et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118(6):928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee VS, Halabi CM, Hoffman EP, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A. 2016;113(31):8759–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263(13):6226–6232. [PubMed] [Google Scholar]
- 109.Kontusaari S, Tromp G, Kuivaniemi H, Ladda RL, Prockop DJ. Inheritance of an RNA splicing mutation (G + 1 IVS20) in the type III procollagen gene (COL3A1) in a family having aortic aneurysms and easy bruisability: phenotypic overlap between familial arterial aneurysms and Ehlers-Danlos syndrome type IV. Am J Hum Genet. 1990;47(1): 112–120. [PMC free article] [PubMed] [Google Scholar]
- 110.Monroe GR, Harakalova M, van der Crabben SN, et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am J Med Genet A. 2015;167(6):1196–1203. [DOI] [PubMed] [Google Scholar]
- 111.Schwarze U, Hata R, McKusick VA, et al. Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway. Am J Hum Genet. 2004;74(5):917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355(8):788–798. [DOI] [PubMed] [Google Scholar]
- 113.Attias D, Stheneur C, Roy C, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120(25):2541–2549. [DOI] [PubMed] [Google Scholar]
- 114.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36(8):855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tran-Fadulu V, Pannu H, Kim DH, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46(9):607–613. [DOI] [PubMed] [Google Scholar]
- 117.Gallo EM, Loch DC, Habashi JP, et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124(1):448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44(8):916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bertoli-Avella AM, Gillis E, Morisaki H, et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121–126. [DOI] [PubMed] [Google Scholar]
- 121.Regalado ES, Guo DC, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109(6): 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.van de Laar IM, van der Linde D, Oei EH, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49(1):47–57. [DOI] [PubMed] [Google Scholar]
- 123.Ye P, Chen W, Wu J, et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest. 2013;123(5):2317–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang P, Hou S, Chen J, et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ Res. 2016;118(3):388–399. [DOI] [PubMed] [Google Scholar]
- 125.Li W, Li Q, Jiao Y, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124(2):755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hu JH, Wei H, Jaffe M, et al. Postnatal deletion of the type II transforming growth factor-beta receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler Thromb Vasc Biol. 2015;35(12):2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Inamoto S, Kwartler CS, Lafont AL, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res. 2010;88(3):520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. [DOI] [PubMed] [Google Scholar]
- 129.Kuang SQ, Medina-Martinez O, Guo DC, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. 2016;126(3):948–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guo DC, Gong L, Regalado ES, et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet. 2015;96(1):170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rikimaru A, Komori K, Sakamoto T, et al. Establishment of an MT4-MMP-deficient mouse strain representing an efficient tracking system for MT4-MMP/MMP-17 expression in vivo using beta-galactosidase. Genes Cells. 2007; 12(9):1091–1100. [DOI] [PubMed] [Google Scholar]
- 132.Martin-Alonso M, Garcia-Redondo AB, Guo D, et al. Deficiency of MMP17/MT4-MMP proteolytic activity predisposes to aortic aneurysm in mice. Circ Res. 2015;117(2):e13–e26. [DOI] [PubMed] [Google Scholar]
- 133.Groenink M, Langerak SE, Vanbavel E, et al. The influence of aging and aortic stiffness on permanent dilation and breaking stress of the thoracic descending aorta. Cardiovasc Res. 1999;43(2):471–480. [DOI] [PubMed] [Google Scholar]
- 134.Zhang J, Zhao X, Vatner DE, et al. Extracellular matrix disarray as a mechanism for greater abdominal versus thoracic aortic stiffness with aging in primates. Arterioscler Thromb Vasc Biol. 2016;36(4):700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qiu H, Zhu Y, Sun Z, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107(5):615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Raaz U, Zollner AM, Schellinger IN, et al. Segmental aortic stiffening contributes to experimental abdominal aortic aneurysm development. Circulation. 2015;131(20):1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Norman PE, Powell JT. Abdominal aortic aneurysm: the prognosis in women is worse than in men. Circulation. 2007;115(22):2865–2869. [DOI] [PubMed] [Google Scholar]
- 138.Zhang X, Thatcher SE, Rateri DL, et al. Transient exposure of neonatal female mice to testosterone abrogates the sexual dimorphism of abdominal aortic aneurysms. Circ Res. 2012;110(11):e73–e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Henriques T, Zhang X, Yiannikouris FB, Daugherty A, Cassis LA. Androgen increases AT1a receptor expression in abdominal aortas to promote angiotensin II–induced AAAs in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28(7):1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prakash SK, LeMaire SA, Guo DC, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87(6): 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.LeMaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43(10):996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guo J, Cai L, Jia L, et al. Wide mutation spectrum and frequent variant Ala27Thr ofFBN1 identified in a large cohort of Chinese patients with sporadic TAAD. Sci Rep. 2015;5:13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bradley DT, Badger SA, McFarland M, Hughes AE. Abdominal aortic aneurysm genetic associations: mostly false? A systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2016;51(1):64–75. [DOI] [PubMed] [Google Scholar]
- 144.van de Luijtgaarden KM, Heijsman D, Maugeri A, et al. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum Genet. 2015;134(8):881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ince H, Nienaber CA. Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat Clin Pract Cardiovasc Med. 2007;4(8):418–427. [DOI] [PubMed] [Google Scholar]
- 146.Moltzer E, Essers J, van Esch JH, Roos-Hesselink JW, Danser AH. The role of the renin-angiotensin system in thoracic aortic aneurysms: clinical implications. Pharmacol Ther. 2011;131(1):50–60. [DOI] [PubMed] [Google Scholar]
- 147.Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci (Lond). 2012;123(9):531–543. [DOI] [PubMed] [Google Scholar]
- 148.Owens AP 3rd, Subramanian V, Moorleghen JJ, et al. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ Res. 2010;106(3):611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond). 2010;118(11):681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Okamoto RJ, Wagenseil JE, DeLong WR, Peterson SJ, Kouchoukos NT, Sundt 3rd TM. Mechanical properties ofdilated human ascending aorta. Ann Biomed Eng. 2002;30(5):624–635. [DOI] [PubMed] [Google Scholar]
- 151.Rabkin SW, Janusz MT. Aortic wall stress in hypertension and ascending thoracic aortic aneurysms: implications for antihypertensive therapy. High Blood Press Cardiovasc Prev. 2013;20(4):265–271. [DOI] [PubMed] [Google Scholar]
- 152.Stolle K, Berges A, Lietz M, Lebrun S, Wallerath T. Cigarette smoke enhances abdominal aortic aneurysm formation in angiotensin II-treated apolipoprotein E-deficient mice. Toxicol Lett. 2010;199(3):403–409. [DOI] [PubMed] [Google Scholar]
- 153.Bergoeing MP, Arif B, Hackmann AE, Ennis TL, Thompson RW, Curci JA. Cigarette smoking increases aortic dilatation without affecting matrix metalloproteinase-9 and −12 expression in a modified mouse model of aneurysm formation. J Vasc Surg. 2007;45(6):1217–1227. [DOI] [PubMed] [Google Scholar]
- 154.Jin J, Arif B, Garcia-Fernandez F, et al. Novel mechanism of aortic aneurysm development in mice associated with smoking and leukocytes. Arterioscler Thromb Vasc Biol. 2012;32(12):2901–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Norman PE, Curci JA. Understanding the effects of tobacco smoke on the pathogenesis of aortic aneurysm. Arterioscler Thromb Vasc Biol. 2013;33(7):1473–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Watanabe Y, Miyata T, Tanemoto K. Current clinical features of new patients with Takayasu arteritis observed from cross-country research in Japan: age and sex specificity. Circulation. 2015;132(18):1701–1709. [DOI] [PubMed] [Google Scholar]
- 157.Ishikawa K Natural history and classification of occlusive thromboaortopathy (Takayasu’s disease). Circulation. 1978;57(1):27–35. [DOI] [PubMed] [Google Scholar]
- 158.Nakano T, Okano H, Konishi T, Takezawa H. Aneurysm of the left aortic sinus caused by Takayasu’s arteritis: compression of the left coronary artery producing coronary insufficienc. y. J Am Coll Cardiol. 1986(3):7:696–700. [DOI] [PubMed] [Google Scholar]
- 159.Suzuki A, Amano J, Tanaka H, Sakamoto T, Sunamori M. Surgical consideration of aortitis involving the aortic root. Circulation. 1989;80(3 Pt 1):I222–I232. [PubMed] [Google Scholar]
- 160.Seko Y, Sato O, Takagi A, et al. Restricted usage of T-cell receptor Valpha-Vbeta genes in infiltrating cells in aortic tissue of patients with Takayasu’s arteritis. Circulation. 1996;93(10):1788–1790. [DOI] [PubMed] [Google Scholar]
- 161.Spence RK, Estella F, Gisser S, Schiffman R, Camishion RC. Thoracic aortic aneurysm secondary to giant cell arteritis: a reappraisal of etiology, treatment and possible prevention. J Cardiovasc Surg (Torino). 1985;26(5):492–495. [PubMed] [Google Scholar]
- 162.Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med. 1995;122(7):502–507. [DOI] [PubMed] [Google Scholar]
- 163.Nuenninghoff DM, Hunder GG, Christianson TJ, McClelland RL, Matteson EL. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48(12):3522–3531. [DOI] [PubMed] [Google Scholar]
- 164.Garcia-Martinez A, Hernandez-Rodriguez J, Arguis P, et al. Development of aortic aneurysm/dilatation during the followup of patients with giant cell arteritis: a cross-sectional screening of fifty-four prospectively followed patients. Arthritis Rheum. 2008;59(3):422–430. [DOI] [PubMed] [Google Scholar]
- 165.Evans JM, Bowles CA, Bjornsson J, Mullany CJ, Hunder GG. Thoracic aortic aneurysm and rupture in giant cell arteritis. A descriptive study of 41 cases. Arthritis Rheum. 1994;37(10):1539–1547. [DOI] [PubMed] [Google Scholar]
- 166.Takagi H, Mori Y, Umeda Y, et al. Abdominal aortic aneurysm with arteritis in ankylosing spondylitis. J Vasc Surg. 2003;38(3):613–616. [DOI] [PubMed] [Google Scholar]
- 167.Khalid U, Egeberg A, Ahlehoff O, Smedegaard L, Gislason GH, Hansen PR. Nationwide study on the risk of abdominal aortic aneurysms in patients with psoriasis. Arterioscler Thromb Vasc Biol. 2016;36(5):1043–1048. [DOI] [PubMed] [Google Scholar]
- 168.Liu CL, Wemmelund H, Wang Y, et al. Asthma associates with human abdominal aortic aneurysm and rupture. Arterioscler Thromb Vasc Biol. 2016;36(3):570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu CL, Wang Y, Liao M, et al. Allergic lung inflammation aggravates angiotensin II–induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2016;36(1):69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Freiberg MS, Arnold AM, Newman AB, Edwards MS, Kraemer KL, Kuller LH. Abdominal aortic aneurysms, increasing infrarenal aortic diameter, and risk of total mortality and incident cardiovascular disease events: 10-year follow-up data from the Cardiovascular Health Study. Circulation. 2008;117(8):1010–1017. [DOI] [PubMed] [Google Scholar]
- 171.Naydeck BL, Sutton-Tyrrell K, Schiller KD, Newman AB, Kuller LH. Prevalence and risk factors for abdominal aortic aneurysms in older adults with and without isolated systolic hypertension. Am J Cardiol. 1999;83(5):759–764. [DOI] [PubMed] [Google Scholar]
- 172.Shakeri AB, Tubbs RS, Shoja MM, Ghabili K, Rahimi-Ardabili B, Loukas M. Screening for thoracoabdominal aortic aneurysms in patients with aortoiliac atherosclerosis: a preliminary study. Folia Morphol (Warsz). 2008;67(1): 78–83. [PubMed] [Google Scholar]
- 173.Albini PT, Segura AM, Liu G, et al. Advanced atherosclerosis is associated with increased medial degeneration in sporadic ascending aortic aneurysms. Atherosclerosis. 2014;232(2):361–368. [DOI] [PubMed] [Google Scholar]
- 174.Johnsen SH, Forsdahl SH, Singh K, Jacobsen BK. Atherosclerosis in abdominal aortic aneurysms: a causal event or a process running in parallel? The Tromso study. Arterioscler Thromb Vasc Biol. 2010;30(6):1263–1268. [DOI] [PubMed] [Google Scholar]
- 175.Liu J, Lu H, Howatt DA, et al. Associations ofApoAI and ApoB-containing lipoproteins with AngII–induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2015;35(8):1826–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II–induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29(10): 1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Torsney E, Pirianov G, Charolidi N, et al. Elevation of plasma high-density lipoproteins inhibits development of experimental abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2012;32(11):2678–2686. [DOI] [PubMed] [Google Scholar]
- 178.Golledge J, Clancy P, Jamrozik K, Norman PE. Obesity, adipokines, and abdominal aortic aneurysm: Health in men study. Circulation. 2007;116(20):2275–2279. [DOI] [PubMed] [Google Scholar]
- 179.Prakash SK, Pedroza C, Khalil YA, Milewicz DM. Diabetes and reduced risk for thoracic aortic aneurysms and dissections: a nationwide case-control study. J Am Heart Assoc. 2012;1(2):e000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jimenez-Trujillo I, Gonzalez-Pascual M, Jimenez-Garcia R, et al. Type 2 diabetes mellitus and thoracic aortic aneurysm and dissection: an observational population-based study in Spain from 2001 to 2012. Medicine (Baltimore). 2016;95(18):e3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Takagi H, Umemoto T, Group A. Negative association of diabetes with thoracic aortic dissection and aneurysm. Angiology. 2016;68(3):216–224. [DOI] [PubMed] [Google Scholar]
- 182.Theivacumar NS, Stephenson MA, Mistry H, Valenti D. Diabetes mellitus and aortic aneurysm rupture: a favorable association? Vasc Endovascular Surg. 2014;48(1):45–50. [DOI] [PubMed] [Google Scholar]
- 183.Theivacumar NS, Stephenson MA, Mistry H, Valenti D. Diabetics are less likely to develop thoracic aortic dissection: a 10-year single-center analysis. Ann Vasc Surg. 2014;28(2):427–432. [DOI] [PubMed] [Google Scholar]
- 184.Tsai CL, Lin CL, Wu YY, Shieh DC, Sung FC, Kao CH. Advanced complicated diabetes mellitus is associated with a reduced risk of thoracic and abdominal aortic aneurysm rupture: a population-based cohort study. Diabetes Metab Res Rev. 2015;31(2):190–197. [DOI] [PubMed] [Google Scholar]
- 185.De Rango P, Farchioni L, Fiorucci B, Lenti M. Diabetes and abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2014;47(3):243–261. [DOI] [PubMed] [Google Scholar]
- 186.Takagi H, Umemoto T, Group A. Diabetes and abdominal aortic aneurysm growth. Angiology. 2016;67(6):513–525. [DOI] [PubMed] [Google Scholar]
- 187.Golledge J, Karan M, Moran CS, et al. Reduced expansion rate of abdominal aortic aneurysms in patients with diabetes may be related to aberrant monocyte-matrix interactions. Eur Heart J. 2008;29(5):665–672. [DOI] [PubMed] [Google Scholar]
- 188.Hsu CY, Su YW, Chen YT, et al. Association between use of oral-antidiabetic drugs and the risk of aortic aneurysm: a nested case-control analysis. Cardiovasc Diabetol. 2016;15(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fujimura N, Xiong J, Kettler EB, et al. Metformin treatment status and abdominal aortic aneurysm disease progression. J Vasc Surg. 2016;64(1):46–54. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64(6):2028–2041. [DOI] [PubMed] [Google Scholar]
- 191.Jones A, Deb R, Torsney E, et al. Rosiglitazone reduces the development and rupture of experimental aortic aneurysms. Circulation. 2009;119(24):3125–3132. [DOI] [PubMed] [Google Scholar]
- 192.Guo DC, Papke CL, He R, Milewicz DM. Pathogenesis of thoracic and abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:339–352. [DOI] [PubMed] [Google Scholar]
- 193.Nataatmadja M, West M, West J, et al. Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation. 2003;108:II329–II334. [DOI] [PubMed] [Google Scholar]
- 194.Nataatmadja M, West J, West M. Overexpression of transforming growth factor-beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aorticaneurysm. Circulation. 2006;114(Suppl 1): I371–I377. [DOI] [PubMed] [Google Scholar]
- 195.Ihling C, Szombathy T, Nampoothiri K, et al. Cystic medial degeneration of the aorta is associated with p53 accumulation, Bax upregulation, apoptotic cell death, and cell proliferation. Heart. 1999;82(3):286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Della Corte A, Quarto C, Bancone C, et al. Spatiotemporal patterns of smooth muscle cell changes in ascending aortic dilatationwith bicuspid and tricuspid aortic valve stenosis: focus on cell-matrix signaling. J Thorac Cardiovasc Surg. 2008;135(1):8–18. 18.e1–2. [DOI] [PubMed] [Google Scholar]
- 197.Adiguzel Z, Arda N, Kacar O, et al. Evaluation of apoptotic molecular pathways for smooth muscle cells isolated from thoracic aortic aneurysms in response to oxidized sterols. Mol Biol Rep. 2014;41(12):7875–7884. [DOI] [PubMed] [Google Scholar]
- 198.Jia LX, Zhang WM, Zhang HJ, et al. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J Pathol. 2015;236(3):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Holmes DR, Lopez-Candales A, Liao S, Thompson RW. Smooth muscle cell apoptosis and p53 expression in human abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:286–287. [DOI] [PubMed] [Google Scholar]
- 200.Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8(10):623–631. [DOI] [PubMed] [Google Scholar]
- 201.Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99(1):96–104. [DOI] [PubMed] [Google Scholar]
- 202.Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30(4):702–707. [DOI] [PubMed] [Google Scholar]
- 203.Emrich FC, Okamura H, Dalal AR, et al. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(1):146–154. [DOI] [PubMed] [Google Scholar]
- 204.Clarke MC, Littlewood TD, Figg N, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102(12):1529–1538. [DOI] [PubMed] [Google Scholar]
- 205.Liu S, Xie Z, Daugherty A, et al. Mineralocorticoid receptor agonists induce mouse aortic aneurysm formation and rupture in the presence of high salt. Arterioscler Thromb Vasc Biol. 2013;33(7):1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Das D, Gawdzik J, Dellefave-Castillo L, et al. S100A12 expression in thoracic aortic aneurysm is associated with increased risk of dissection and perioperative complications. J Am Coll Cardiol. 2012;60(8):775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang Y, Shi GP. Mast cell chymase and tryptase in abdominal aortic aneurysm formation. Trends Cardiovasc Med. 2012;22(6):150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Morgan S, Yamanouchi D, Harberg C, et al. Elevated protein kinase C-delta contributes to aneurysm pathogenesis through stimulation of apoptosis and inflammatory signaling. Arterioscler Thromb Vasc Biol. 2012;32(10): 2493–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Leeper NJ, Raiesdana A, Kojima Y, et al. Loss of CDKN2B promotes p53-dependent smooth muscle cell apoptosis and aneurysm formation. Arterioscler Thromb Vasc Biol. 2013;33(1):e1–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yh Shen, Zhang L, Ren P, et al. AKT2 confers protection against aortic aneurysms and dissections. Circ Res. 2013; 112(4):618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang Q, Liu Z, Ren J, Morgan S, Assa C, Liu B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ Res. 2015;116(4):600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Shi N, Chen SY. Smooth muscle cell differentiation: model systems, regulatory mechanisms, and vascular diseases. J Cell Physiol. 2016;231(4):777–787. [DOI] [PubMed] [Google Scholar]
- 213.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100(23):13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26(12):2696–2702. [DOI] [PubMed] [Google Scholar]
- 215.Choi HY, Rahmani M, Wong BW, et al. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation. 2009;119(25):3223–3231. [DOI] [PubMed] [Google Scholar]
- 216.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129(15): 1551–1559. [DOI] [PubMed] [Google Scholar]
- 217.Lomashvili KA, Wang X, O’Neill WC. Role of local versus systemic vitamin D receptors in vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34(1):146–151. [DOI] [PubMed] [Google Scholar]
- 218.Ailawadi G, Moehle CW, Pei H, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138(6):1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Branchetti E, Poggio P, Sainger R, et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res. 2013;100(2):316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tang PC, Coady MA, Lovoulos C, et al. Hyperplastic cellular remodeling of the media in ascending thoracic aortic aneurysms. Circulation. 2005;112(8):1098–1105. [DOI] [PubMed] [Google Scholar]
- 221.Moehle CW, Bhamidipati CM, Alexander MR, et al. Bone marrow-derived MCP1 required for experimental aortic aneurysm formation and smooth muscle phenotypic modulation. J Thorac Cardiovasc Surg. 2011;142(6):1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Salmon M, Gomez D, Greene E, Shankman L, Owens GK. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111(6):685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Salmon M, Johnston WF, Woo A, et al. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. 2013;128(26 Suppl 1):S163–S174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fei J, Cui XB, Wang JN, Dong K, Chen SY. ADAR1-mediated RNA editing, a novel mechanism controlling phenotypic modulation of vascular smooth muscle cells. Circ Res. 2016;119(3):463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Moyes AJ, Khambata RS, Villar I, et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J Clin Invest. 2014;124(9):4039–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yoshioka K, Yoshida K, Cui H, et al. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med. 2012;18(10):1560–1569. [DOI] [PubMed] [Google Scholar]
- 227.Franck G, Dai J, Fifre A, et al. Reestablishment of the endothelial lining by endothelial cell therapy stabilizes experimental abdominal aortic aneurysms. Circulation. 2013;127(18):1877–1887. [DOI] [PubMed] [Google Scholar]
- 228.Rateri DL, Moorleghen JJ, Balakrishnan A, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor−/− mice. Circ Res. 2011;108(5):574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Fan LM, Douglas G, Bendall JK, et al. Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation. 2014;129(25):2661–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Marinkovic G, Hibender S, Hoogenboezem M, et al. Immunosuppressive drug azathioprine reduces aneurysm progression through inhibition of Rac1 and c-Jun-terminal-N-kinase in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(10):2380–2388. [DOI] [PubMed] [Google Scholar]
- 231.LeBleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosi. s. Nat Med. 2013;19 (8):1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–107. [DOI] [PubMed] [Google Scholar]
- 233.Ramkisoensing AA, de Vries AA, Atsma DE, Schalij MJ, Pijnappels DA. Interaction between myofibroblasts and stem cells in the fibrotic heart: balancing between deterioration and regeneration. Cardiovasc Res. 2014;102(2):224–231. [DOI] [PubMed] [Google Scholar]
- 234.Leask A Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116(7):1269–1276. [DOI] [PubMed] [Google Scholar]
- 235.Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF-beta, WNT, and YAP/TAZ converge. Front Med (Lausanne). 2015;2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119(1):91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Forte A, Della Corte A, De Feo M, Cerasuolo F, Cipollaro M. Role of myofibroblasts in vascular remodelling: focus on restenosis and aneurysm. Cardiovasc Res. 2010;88(3):395–405. [DOI] [PubMed] [Google Scholar]
- 238.Forte A, Della Corte A, Grossi M, et al. Differential expression of proteins related to smooth muscle cells and myofibroblasts in human thoracic aortic aneurysm. Histol Histopathol. 2013;28(6):795–803. [DOI] [PubMed] [Google Scholar]
- 239.Sakata N, Nabeshima K, Iwasaki H, et al. Possible involvement of myofibroblast in the development of inflammatory aortic aneurysm. Pathol Res Pract. 2007;203(1):21–29. [DOI] [PubMed] [Google Scholar]
- 240.Jones JA, Beck C, Barbour JR, et al. Alterations in aortic cellular constituents during thoracic aortic aneurysm development: myofibroblast-mediated vascular remodeling. Am J Pathol. 2009;175(4):1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kuang SQ, Geng L, Prakash SK, et al. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol. 2013;33(9):2172–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. 2016;93:133–142. [DOI] [PubMed] [Google Scholar]
- 243.Goodell MA, Rando TA. Stem cells and healthy aging. Science. 2015;350(6265):1199–1204. [DOI] [PubMed] [Google Scholar]
- 244.Xin T, Greco V, Myung P. Hardwiring stem cell communication through tissue structure. Cell. 2016;164(6): 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med. 2015;21(8):854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Karantalis V, Hare JM. Use of mesenchymal stem cells for therapy of cardiac disease. Circ Res. 2015;116(8): 1413–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Parietti E, Pallandre JR, Deschaseaux F, et al. Presence of circulating endothelial progenitor cells and levels of stromal-derived factor-1alpha are associated with ascending aorta aneurysm size. Eur J Cardiothorac Surg. 2011; 40(1):e6–12. [DOI] [PubMed] [Google Scholar]
- 248.Dawson J, Tooze J, Cockerill G, Choke E, Loftus I, Thompson MM. Endothelial progenitor cells and abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:327–330. [DOI] [PubMed] [Google Scholar]
- 249.Shen YH, Hu X, Zou S, Wu D, Coselli JS, LeMaire SA. Stem cells in thoracic aortic aneurysms and dissections: potential contributors to aortic repair. Ann Thorac Surg. 2012;93(5):1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ciavarella C, Alviano F, Gallitto E, et al. Human Vascular wall mesenchymal stromal cells contribute to abdominal aortic aneurysm pathogenesis through an impaired immunomodulatory activity and increased levels of matrix metalloproteinase-9. Circ J. 2015;79(7):1460–1469. [DOI] [PubMed] [Google Scholar]
- 251.Hare JM, Fishman JE, Gerstenblith G, et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. J Am Med Assoc. 2012;308(22):2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Turnbull IC, Hadri L, Rapti K, et al. Aortic implantation of mesenchymal stem cells after aneurysm injury in a porcine model. J Surg Res. 2011;170(1):e179–e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hashizume R, Yamawaki-Ogata A, Ueda Y, Wagner WR, Narita Y. Mesenchymal stem cells attenuate angiotensin II–induced aortic aneurysm growth in apolipoprotein E-deficient mice. J Vasc Surg. 2011;54(6):1743–1752. [DOI] [PubMed] [Google Scholar]
- 254.Schneider F, Saucy F, de Blic R, et al. Bone marrow mesenchymal stem cells stabilize already-formed aortic aneurysms more efficiently than vascular smooth muscle cells in a rat model. Eur J Vasc Endovasc Surg. 2013;45(6): 666–672. [DOI] [PubMed] [Google Scholar]
- 255.Fu XM, Yamawaki-Ogata A, Oshima H, Ueda Y, Usui A, Narita Y. Intravenous administration of mesenchymal stem cells prevents angiotensin II–induced aortic aneurysm formation in apolipoprotein E-deficient mouse. J Transl Med. 2013;11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yamawaki-Ogata A, Fu X, Hashizume R, et al. Therapeutic potential of bone marrow-derived mesenchymal stem cells in formed aortic aneurysms of a mouse model. Eur J Cardiothorac Surg. 2014;45(5):e156–e165. [DOI] [PubMed] [Google Scholar]
- 257.Sharma AK, Lu G, Jester A, et al. Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment. Circulation. 2012;126(11 Suppl 1):S38–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Davis JP, Salmon M, Pope NH, et al. Attenuation of aortic aneurysms with stem cells from different genders. J Surg Res. 2015;199(1):249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Blose KJ, Ennis TL, Arif B, Weinbaum JS, Curci JA, Vorp DA. Periadventitial adipose-derived stem cell treatment halts elastase-induced abdominal aortic aneurysm progression. Regen Med. 2014;9(6):733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Parvizi M, Harmsen MC. Therapeutic prospect of adipose-derived stromal cells for the treatment of abdominal aortic aneurysm. Stem Cells Dev. 2015;24(13):1493–1505. [DOI] [PubMed] [Google Scholar]
- 261.Xie J,Jones TJ, Feng D, et al. Human adipose-derived stem cells suppress elastase-induced murine abdominal aortic inflammation and aneurysm expansion through paracrine factors. Cell Transplant. 2016;26(2):173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Tian X, Fan J, Yu M, et al. Adipose stem cells promote smooth muscle cells to secrete elastin in rat abdominal aortic aneurysm. PLoS One. 2014;9(9):e108105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Sharma AK, Salmon MD, Lu G, et al. Mesenchymal stem cells attenuate NADPH oxidase-dependent high mobility group box 1 production and inhibit abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2016;36(5):908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013; 13(4):392–402. [DOI] [PubMed] [Google Scholar]
- 265.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35(8):1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Curci JA, Liao S, Huffman MD, Shapiro SD, Thompson RW. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J Clin Invest. 1998;102(11):1900–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Koch AE, Haines GK, Rizzo RJ, et al. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am J Pathol. 1990;137(5):1199–1213. [PMC free article] [PubMed] [Google Scholar]
- 268.Galle C, Schandene L, Stordeur P, et al. Predominance of type 1 CD4+ T cells in human abdominal aortic aneurysm. Clin Exp Immunol. 2005;142(3):519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Tsuruda T, Kato J, Hatakeyama K, et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res. 2008;102(11):1368–1377. [DOI] [PubMed] [Google Scholar]
- 270.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.DuPage M, Bluestone JA. Harnessing the plasticity of CD4( + ) T cells to treat immune-mediated disease. Nat Rev Immunol. 2016;16(3):149–163. [DOI] [PubMed] [Google Scholar]
- 272.Folkersen L, Wagsater D, Paloschi V, et al. Unraveling divergent gene expression profiles in bicuspid and tricuspid aortic valve patients with thoracic aortic dilatation: the ASAP study. Mol Med. 2011;17(11-12):1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.He R, Guo DC, Estrera AL, et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J Thorac Cardiovasc Surg. 2006;131(3):671–678. [DOI] [PubMed] [Google Scholar]
- 274.Schonbeck U, Sukhova GK, Gerdes N, Libby P. T(H)2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. 2002;161(2):499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ocana E, Bohorquez JC, Perez-Requena J, Brieva JA, Rodriguez C. Characterisation of T and B lymphocytes infiltrating abdominal aortic aneurysms. Atherosclerosis. 2003;170(1):39–48. [DOI] [PubMed] [Google Scholar]
- 276.Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of CD4+ T cells and IFN-gamma in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172(4):2607–2612. [DOI] [PubMed] [Google Scholar]
- 277.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15(5):271–282. [DOI] [PubMed] [Google Scholar]
- 278.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11(10): 763–776. [DOI] [PubMed] [Google Scholar]
- 279.Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. 2016;16(4):220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ait-Oufella H, Sage AP, Mallat Z, Tedgui A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res. 2014;114(10):1640–1660. [DOI] [PubMed] [Google Scholar]
- 281.Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. 2016;15(1):35–50. [DOI] [PubMed] [Google Scholar]
- 282.Tang PC, Yakimov AO, Teesdale MA, et al. Transmural inflammation by interferon-gamma-producing T cells correlates with outward vascular remodeling and intimal expansion of ascending thoracic aortic aneurysms. FASEB J. 2005;19(11):1528–1530. [DOI] [PubMed] [Google Scholar]
- 283.Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114(2):300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Xu J, Ehrman B, Graham LM, Eagleton MJ. Interleukin-5 is a potential mediator of angiotensin II–induced aneurysm formation in apolipoprotein E knockout mice. J Surg Res. 2012;178(1):512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26(5):987–994. [DOI] [PubMed] [Google Scholar]
- 286.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. [DOI] [PubMed] [Google Scholar]
- 287.Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. 2013;121(13):2402–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest. 2015;125(6):2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Ju X, Ijaz T, Sun H, et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2013;33(7):1612–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wei Z, Wang Y, Zhang K, et al. Inhibiting the Th17/IL-17A-related inflammatory responses with digoxin confers protection against experimental abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2014;34(11):2429–2438. [DOI] [PubMed] [Google Scholar]
- 291.Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38(3): 414–423. [DOI] [PubMed] [Google Scholar]
- 292.Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14(3):154–165. [DOI] [PubMed] [Google Scholar]
- 293.Yin M, Zhang J, Wang Y, et al. Deficient CD4+CD25+ T regulatory cell function in patients with abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2010;30(9):1825–1831. [DOI] [PubMed] [Google Scholar]
- 294.Zhou Y, Wu W, Lindholt JS, et al. Regulatory T cells in human and angiotensin II–induced mouse abdominal aortic aneurysms. Cardiovasc Res. 2015;107(1):98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ait-Oufella H, Wang Y, Herbin O, et al. Natural regulatory T cells limit angiotensin II–induced aneurysm formation and rupture in mice. Arterioscler Thromb Vasc Biol. 2013;33(10):2374–2379. [DOI] [PubMed] [Google Scholar]
- 296.Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17(1):34–40. [DOI] [PubMed] [Google Scholar]
- 297.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750–761. [DOI] [PubMed] [Google Scholar]
- 299.Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1): 153–166. [DOI] [PubMed] [Google Scholar]
- 300.He R, Guo DC, Sun W, et al. Characterization of the inflammatory cells in ascending thoracic aortic aneurysms in patients with Marfan syndrome, familial thoracic aortic aneurysms, and sporadic aneurysms. J Thorac Cardiovasc Surg. 2008;136(4):922–929. 929.e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wu D, Choi JC, Sameri A, et al. Inflammatory cell infiltrates in acute and chronic thoracic aortic dissection. Aorta (Stamford). 2013;1(6):259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sawada H, Hao H, Naito Y, et al. Aortic iron overload with oxidative stress and inflammation in human and murine abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(6):1507–1514. [DOI] [PubMed] [Google Scholar]
- 303.Boytard L, Spear R, Chinetti-Gbaguidi G, et al. Role of proinflammatory CD68(+) mannose receptor(-) macrophages in peroxiredoxin-1 expression and in abdominal aortic aneurysms in humans. Arterioscler Thromb Vasc Biol. 2013;33(2):431–438. [DOI] [PubMed] [Google Scholar]
- 304.Nahrendorf M, Keliher E, Marinelli B, et al. Detection of macrophages in aortic aneurysms by nanoparticle positron emission tomography-computed tomography. Arterioscler Thromb Vasc Biol. 2011;31(4):750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Rateri DL, Howatt DA, Moorleghen JJ, Charnigo R, Cassis LA, Daugherty A. Prolonged infusion of angiotensin II in apoE(−/−) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol. 2011;179(3):1542–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Tieu BC, Lee C, Sun H, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119(12):3637–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Xiong W, Knispel R, MacTaggart J, Greiner TC, Weiss SJ, Baxter BT. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J Biol Chem. 2009;284(3): 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Xiong W, MacTaggart J, Knispel R, Worth J, Persidsky Y, Baxter BT. Blocking TNF-alpha attenuates aneurysm formation in a murine model. J Immunol. 2009;183(4):2741–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Dutertre CA, Clement M, Morvan M, et al. Deciphering the stromal and hematopoietic cell network of the adventitia from non-aneurysmal and aneurysmal human aorta. PLoS One. 2014;9(2):e89983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Dale MA, Xiong W, Carson JS, et al. Elastin-derived peptides promote abdominal aortic aneurysm formation by modulating M1/M2 macrophage polarization. J Immunol. 2016;196(11):4536–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Cheng J, Koenig SN, Kuivaniemi HS, Garg V, Hans CP. Pharmacological inhibitor of Notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J Am Heart Assoc. 2014;3(6):e001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Qin Z, Bagley J, Sukhova G, et al. Angiotensin II–induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3. J Mol Cell Cardiol. 2015;87:160–170. [DOI] [PubMed] [Google Scholar]
- 313.Pope NH, Salmon M, Davis JP, et al. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016;30(12):4192–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Samadzadeh KM, Chun KC, Nguyen AT, Baker PM, Bains S, Lee ES. Monocyte activity is linked with abdominal aortic aneurysm diameter. J Surg Res. 2014;190(1):328–334. [DOI] [PubMed] [Google Scholar]
- 315.Mellak S, Ait-Oufella H, Esposito B, et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe−/− mice. Arterioscler Thromb Vasc Biol. 2015;35(2):378–388. [DOI] [PubMed] [Google Scholar]
- 316.Hance KA, Tataria M, Ziporin SJ, Lee JK, Thompson RW. Monocyte chemotactic activity in human abdominal aortic aneurysms: role of elastin degradation peptides and the 67-kD cell surface elastin receptor. J Vasc Surg. 2002;35(2): 254–261. [DOI] [PubMed] [Google Scholar]
- 317.Houghton AM, Quintero PA, Perkins DL, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Guo G, Booms P, Halushka M, et al. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG-containing fibrillin-1 fragment. Circulation. 2006;114(17):1855–1862. [DOI] [PubMed] [Google Scholar]
- 319.Neutrophils Nathan C. and immunity: challenges and opportunities. Nat Rev Immunol. 2006;6(3):173–182. [DOI] [PubMed] [Google Scholar]
- 320.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. [DOI] [PubMed] [Google Scholar]
- 321.Fontaine V, Touat Z, Mtairag el M, et al. Role of leukocyte elastase in preventing cellular re-colonization of the mural thrombus. Am J Pathol. 2004;164(6):2077–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Houard X, Touat Z, Ollivier V, et al. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc Res. 2009;82(3):532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Folkesson M, Kazi M, Zhu C, et al. Presence of NGAL/MMP-9 complexes in human abdominal aortic aneurysms. Thromb Haemost. 2007;98(2):427–433. [PubMed] [Google Scholar]
- 324.Pagano MB, Zhou HF, Ennis TL, et al. Complement-dependent neutrophil recruitment is critical for the development of elastase-induced abdominal aortic aneurysm. Circulation. 2009;119(13):1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Pagano MB, Bartoli MA, Ennis TL, et al. Critical role of dipeptidyl peptidase I in neutrophil recruitment during the development of experimental abdominal aortic aneurysms. Proc Natl Acad Sci U S A. 2007;104(8):2855–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Kurihara T, Shimizu-Hirota R, Shimoda M, et al. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126(25):3070–3080. [DOI] [PubMed] [Google Scholar]
- 327.Yan H, Zhou HF, Akk A, et al. Neutrophil proteases promote experimental abdominal aortic aneurysm via extracellular trap release and plasmacytoid dendritic cell activation. Arterioscler Thromb Vasc Biol. 2016;36(8): 1660–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Eliason JL, Hannawa KK, Ailawadi G, et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112(2):232–240. [DOI] [PubMed] [Google Scholar]
- 329.Anzai A, Shimoda M, Endo J, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116(4):612–623. [DOI] [PubMed] [Google Scholar]
- 330.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663): 1532–1535. [DOI] [PubMed] [Google Scholar]
- 331.Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. 2011;7(12):691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Sorensen OE, Borregaard N. Neutrophil extracellular traps–the dark side of neutrophils. J Clin Invest. 2016;126(5): 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Ramos-Mozo P, Madrigal-Matute J, Martinez-Pinna R, et al. Proteomic analysis of polymorphonuclear neutrophils identifies catalase as a novel biomarker of abdominal aortic aneurysm: potential implication of oxidative stress in abdominal aortic aneurysm progression. Arterioscler Thromb Vasc Biol. 2011;31(12):3011–3019. [DOI] [PubMed] [Google Scholar]
- 335.Gurish MF, Boyce JA. Mast cells: ontogeny, homing, and recruitment of a unique innate effector cell. J Allergy Clin Immunol. 2006;117(6):1285–1291. [DOI] [PubMed] [Google Scholar]
- 336.Kunder CA, St John AL, Abraham SN. Mast cell modulation of the vascular and lymphatic endothelium. Blood. 2011;118(20):5383–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Bot I, Shi GP, Kovanen PT. Mast cells as effectors in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(2): 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Swedenborg J, Mayranpaa MI, Kovanen PT. Mast cells: important players in the orchestrated pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2011;31(4):734–740. [DOI] [PubMed] [Google Scholar]
- 339.Ihara M, Urata H, Kinoshita A, et al. Increased chymase-dependent angiotensin II formation in human atherosclerotic aorta. Hypertension. 1999;33(6):1399–1405. [DOI] [PubMed] [Google Scholar]
- 340.Mayranpaa MI, Trosien JA, Fontaine V, et al. Mast cells associate with neovessels in the media and adventitia of abdominal aortic aneurysms. J Vasc Surg. 2009;50(2):388–395. [discussion 395–396]. [DOI] [PubMed] [Google Scholar]
- 341.Sun J, Sukhova GK, Yang M, et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest. 2007;117(11):3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Sun J, Zhang J, Lindholt JS, et al. Critical role of mast cell chymase in mouse abdominal aortic aneurysm formation. Circulation. 2009;120(11):973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhang J, Sun J, Lindholt JS, et al. Mast cell tryptase deficiency attenuates mouse abdominal aortic aneurysm formation. Circ Res. 2011;108(11):1316–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Furubayashi K, Takai S, Jin D, et al. Chymase activates promatrix metalloproteinase-9 in human abdominal aortic aneurysm. Clin Chim Acta. 2008;388(1–2):214–216. [DOI] [PubMed] [Google Scholar]
- 345.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Fan D, Creemers EE, Kassiri Z. Matrix as an interstitial transport system. Circ Res. 2014;114(5):889–902. [DOI] [PubMed] [Google Scholar]
- 347.Duca L, Blaise S, Romier B, et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res. 2016;110(3):298–308. [DOI] [PubMed] [Google Scholar]
- 348.Valentin A, Humphrey JD, Holzapfel GA. A multi-layered computational model of coupled elastin degradation, vasoactive dysfunction, and collagenous stiffening in aortic aging. Ann Biomed Eng. 2011;39(7):2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Wilson JS, Baek S, Humphrey JD. Importance of initial aortic properties on the evolving regional anisotropy, stiffness and wall thickness of human abdominal aortic aneurysms. J R Soc Interface. 2012;9(74):2047–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Ferruzzi J, Collins MJ, Yeh AT, Humphrey JD. Mechanical assessment of elastin integrity in fibrillin-1-deficient carotid arteries: implications for Marfan syndrome. Cardiovasc Res. 2011;92(2):287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Lakatta EG, Wang M, Najjar SS. Arterial aging and subclinical arterial disease are fundamentally intertwined at macroscopic and molecular levels. Med Clin North Am. 2009;93(3):583–604, [table of contents]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Li DY, Brooke B, Davis EC, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393 (6682):276–280. [DOI] [PubMed] [Google Scholar]
- 353.Karnik SK, Brooke BS, Bayes-Genis A, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003;130(2):411–423. [DOI] [PubMed] [Google Scholar]
- 354.ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8(11):857–869. [DOI] [PubMed] [Google Scholar]
- 355.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS Lett. 2012; 586(14):2003–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Nissen R, Cardinale GJ, Udenfriend S. Increased turnover of arterial collagen in hypertensive rats. Proc Natl Acad Sci U S A. 1978;75(1):451–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Rodriguez-Feo JA, Sluijter JP, de Kleijn DP, Pasterkamp G. Modulation of collagen turnover in cardiovascular disease. Curr Pharm Des. 2005;11(19):2501–2514. [DOI] [PubMed] [Google Scholar]
- 358.Dajnowiec D, Langille BL. Arterial adaptations to chronic changes in haemodynamic function: coupling vasomotor tone to structural remodelling. Clin Sci (Lond). 2007;113(1):15–23. [DOI] [PubMed] [Google Scholar]
- 359.Hornebeck W, Emonard H. The cell-elastin-elastase(s) interacting triade directs elastolysis. Front Biosci (Landmark Ed). 2011;16:707–722. [DOI] [PubMed] [Google Scholar]
- 360.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12):a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–839. [DOI] [PubMed] [Google Scholar]
- 362.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. [DOI] [PubMed] [Google Scholar]
- 363.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803(1):55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48(3):222–272. [DOI] [PubMed] [Google Scholar]
- 365.Segura AM, Luna RE, Horiba K, et al. Immunohistochemistry of matrix metalloproteinases and their inhibitors in thoracic aortic aneurysms and aortic valves of patients with Marfan’s syndrome. Circulation. 1998;98(Suppl 19): II331–II337;[discussion II337–338]. [PubMed] [Google Scholar]
- 366.Ikonomidis JS, Jones JA, Barbour JR, et al. Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation. 2006;114(Suppl 1):I365–I370. [DOI] [PubMed] [Google Scholar]
- 367.Taketani T, Imai Y, Morota T, et al. Altered patterns of gene expression specific to thoracic aortic aneurysms: microarray analysis of surgically resected specimens. Int Heart J. 2005;46(2):265–277. [DOI] [PubMed] [Google Scholar]
- 368.Rabkin SW. Differential expression of MMP-2, MMP-9 and TIMP proteins in thoracic aortic aneurysm—comparison with and without bicuspid aortic valve: a meta-analysis. Vasa. 2014;43(6):433–442. [DOI] [PubMed] [Google Scholar]
- 369.Zhang X, Wu D, Choi JC, et al. Matrix metalloproteinase levels in chronic thoracic aortic dissection. J Surg Res. 2014;189(2):348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Thompson RW, Holmes DR, Mertens RA, et al. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96(1):318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Thompson RW, Parks WC. Role of matrix metalloproteinases in abdominal aortic aneurysms. Ann N Y Acad Sci. 1996;800:157–174. [DOI] [PubMed] [Google Scholar]
- 372.Pyo R, Lee JK, Shipley JM, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105(11):1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Ikonomidis JS, Gibson WC, Butler JE, et al. Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. 2004;110(11 Suppl 1):II268–II273. [DOI] [PubMed] [Google Scholar]
- 374.Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109(11):1408–1414. [DOI] [PubMed] [Google Scholar]
- 375.Moore G, Liao S, Curci JA, et al. Suppression of experimental abdominal aortic aneurysms by systemic treatment with a hydroxamate-based matrix metalloproteinase inhibitor (RS 132908). J Vasc Surg. 1999;29(3):522–532. [DOI] [PubMed] [Google Scholar]
- 376.Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;47(1):166–172;[discussion 172]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Jackson V, Olsson T, Kurtovic S, et al. Matrix metalloproteinase 14 and 19 expression is associated with thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2012;144(2):459–466. [DOI] [PubMed] [Google Scholar]
- 378.Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104(3):304–309. [DOI] [PubMed] [Google Scholar]
- 379.Shen M, Lee J, Basu R, et al. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(4):888–898. [DOI] [PubMed] [Google Scholar]
- 380.Spinale FG, Janicki JS, Zile MR. Membrane-associated matrix proteolysis and heart failure. Circ Res. 2013;112(1): 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Itoh Y Membrane-type matrix metalloproteinases: their functions and regulations. Matrix Biol. 2015;44-46: 207–223. [DOI] [PubMed] [Google Scholar]
- 382.Jones JA, Ruddy JM, Bouges S, et al. Alterations in membrane type-1 matrix metalloproteinase abundance after the induction of thoracic aortic aneurysm in a murine model. Am J Physiol Heart Circ Physiol. 2010;299(1):H114–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13(9):649–665. [DOI] [PubMed] [Google Scholar]
- 384.Knox JB, Sukhova GK, Whittemore AD, Libby P. Evidence for altered balance between matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation. 1997;95(1):205–212. [DOI] [PubMed] [Google Scholar]
- 385.Silence J, Collen D, Lijnen HR. Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res. 2002;90(8):897–903. [DOI] [PubMed] [Google Scholar]
- 386.Lemaitre V, Soloway PD, D’Armiento J. Increased medial degradation with pseudo-aneurysm formation in apolipoprotein E-knockout mice deficient in tissue inhibitor of metalloproteinases-1. Circulation. 2003;107(2): 333–338. [DOI] [PubMed] [Google Scholar]
- 387.Basu R, Fan D, Kandalam V, et al. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II. J Biol Chem. 2012;287(53):44083–44096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Sun J, Sukhova GK, Zhang J, et al. Cathepsin L activity is essential to elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2500–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Sun J, Sukhova GK, Zhang J, et al. Cathepsin K deficiency reduces elastase perfusion-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2012;32(1):15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Xian X, Gopal S, Couchman JR. Syndecans as receptors and organizers of the extracellular matrix. Cell Tissue Res. 2010;339(1):31–46. [DOI] [PubMed] [Google Scholar]
- 391.Xiao J, Angsana J, Wen J, et al. Syndecan-1 displays a protective role in aortic aneurysm formation by modulating T cell-mediated responses. Arterioscler Thromb Vasc Biol. 2012;32(2):386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Manon-Jensen T, Multhaupt HA, Couchman JR. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013;280(10):2320–2331. [DOI] [PubMed] [Google Scholar]
- 393.Endo K, Takino T, Miyamori H, et al. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J Biol Chem. 2003;278(42):40764–40770. [DOI] [PubMed] [Google Scholar]
- 394.Gao G, Plaas A, Thompson VP, Jin S, Zuo F, Sandy JD. ADAMTS4 (aggrecanase-1) activation on the cell surface involves C-terminal cleavage by glycosylphosphatidyl inositol-anchored membrane type 4-matrix metalloproteinase and binding of the activated proteinase to chondroitin sulfate and heparan sulfate on syndecan-1. J Biol Chem. 2004;279(11):10042–10051. [DOI] [PubMed] [Google Scholar]
- 395.Nastase MV, Young MF, Schaefer L. Biglycan: a multivalent proteoglycan providing structure and signals. J Histochem Cytochem. 2012;60(12):963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Theocharis AD, Karamanos NK. Decreased biglycan expression and differential decorin localization in human abdominal aortic aneurysms. Atherosclerosis. 2002;165(2):221–230. [DOI] [PubMed] [Google Scholar]
- 397.Heegaard AM, Corsi A, Danielsen CC, et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation. 2007;115(21):2731–2738. [DOI] [PubMed] [Google Scholar]
- 398.Meester JA, Vandeweyer G, Pintelon I, et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med. 2016. 10.1038/gim.2016.126, [ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997;272(1):556–562. [DOI] [PubMed] [Google Scholar]
- 400.Salter RC, Ashlin TG, Kwan Ap, Ramji DP. ADAMTS proteases: key roles in atherosclerosis? J Mol Med. 2010;88(12): 1203–1211. [DOI] [PubMed] [Google Scholar]
- 401.Sandy JD, Westling J, Kenagy RD, et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J Biol Chem. 2001;276(16):13372–13378. [DOI] [PubMed] [Google Scholar]
- 402.Gendron C, Kashiwagi M, Lim NH, et al. Proteolytic activities of human ADAMTS-5: comparative studies with ADAMTS-4. J Biol Chem. 2007;282(25):18294–18306. [DOI] [PubMed] [Google Scholar]
- 403.Torres-Collado AX, Kisiel W, Iruela-Arispe ML, Rodriguez-Manzaneque JC. ADAMTS1 interacts with, cleaves, and modifies the extracellular location of the matrix inhibitor tissue factor pathway inhibitor-2. J Biol Chem. 2006;281 (26):17827–17837. [DOI] [PubMed] [Google Scholar]
- 404.Krampert M, Kuenzle S, Thai SN, Lee N, Iruela-Arispe ML, Werner S. ADAMTS1 proteinase is up-regulated in wounded skin and regulates migration of fibroblasts and endothelial cells. J Biol Chem. 2005;280(25): 23844–23852. [DOI] [PubMed] [Google Scholar]
- 405.Ren P, Zhang L, Xu G, et al. ADAMTS-1 and ADAMTS-4 levels are elevated in thoracic aortic aneurysms and dissections. Ann Thorac Surg. 2013;95(2):570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Gunes MF, Akpinar MB, Comertoglu I, et al. The Investigation of a Disintegrin and Metalloproteinase with ThromboSpondin Motifs (ADAMTS) 1, 5 and 16 in thoracic aortic aneurysms and dissections. Clin Lab. 2016;62(3): 425–433. [PubMed] [Google Scholar]
- 407.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274(19):13066–13076. [DOI] [PubMed] [Google Scholar]
- 408.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2(9):657–672. [DOI] [PubMed] [Google Scholar]
- 409.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3(3):207–214. [DOI] [PubMed] [Google Scholar]
- 410.Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol. 2007;27(6):1231–1237. [DOI] [PubMed] [Google Scholar]
- 411.Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost. 2004;2(10):1798–1805. [DOI] [PubMed] [Google Scholar]
- 412.Borges LF, Gomez D, Quintana M, et al. Fibrinolytic activity is associated with presence of cystic medial degeneration in aneurysms of the ascending aorta. Histopathology. 2010;57(6):917–932. [DOI] [PubMed] [Google Scholar]
- 413.Gomez D, Kessler K, Borges LF, et al. Smad2-dependent protease nexin-1 overexpression differentiates chronic aneurysms from acute dissections of human ascending aorta. Arterioscler Thromb Vasc Biol. 2013;33(9):2222–2232. [DOI] [PubMed] [Google Scholar]
- 414.Schneiderman J, Bordin GM, Engelberg I, et al. Expression of fibrinolytic genes in atherosclerotic abdominal aortic aneurysm wall. A possible mechanism for aneurysm expansion. J Clin Invest. 1995;96(1):639–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Wang YX, Martin-McNulty B, Freay AD, et al. Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol. 2001; 159(4):1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Carmeliet P, Schoonjans L, Kieckens L, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368(6470):419–424. [DOI] [PubMed] [Google Scholar]
- 417.Carmeliet P, Moons L, Lijnen R, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17(4):439–444. [DOI] [PubMed] [Google Scholar]
- 418.Deng GG, Martin-McNulty B, Sukovich DA, et al. Urokinase-type plasminogen activator plays a critical role in angiotensin II–induced abdominal aortic aneurysm. Circ Res. 2003;92(5):510–517. [DOI] [PubMed] [Google Scholar]
- 419.Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118(9):3012–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Uchida HA, Poduri A, Subramanian V, Cassis LA, Daugherty A. Urokinase-type plasminogen activator deficiency in bone marrow-derived cells augments rupture of angiotensin II–induced abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2011;31(12):2845–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Rein CM, Cardenas JC, Church FC. The controversial role of the urokinase system in abdominal aortic aneurysm formation and rupture. Arterioscler Thromb Vasc Biol. 2011;31(12):2769–2771. [DOI] [PubMed] [Google Scholar]
- 422.Defawe OD, Colige A, Lambert CA, et al. TIMP-2 and PAI-1 mRNA levels are lower in aneurysmal as compared to athero-occlusive abdominal aortas. Cardiovasc Res. 2003;60(1):205–213. [DOI] [PubMed] [Google Scholar]
- 423.DiMusto PD, Lu G, Ghosh A, et al. Increased PAI-1 in females compared with males is protective for abdominal aortic aneurysm formation in a rodent model. Am J Physiol Heart Circ Physiol. 2012;302(7):H1378–H1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Qian HS, Gu JM, Liu P, et al. Overexpression of PAI-1 prevents the development of abdominal aortic aneurysm in mice. Gene Ther. 2008;15(3):224–232. [DOI] [PubMed] [Google Scholar]
- 425.Touat Z, Lepage L, Ollivier V, et al. Dilation-dependent activation of platelets and prothrombin in human thoracic ascending aortic aneurysm. Arterioscler Thromb Vasc Biol. 2008;28(5):940–946. [DOI] [PubMed] [Google Scholar]
- 426.Touat Z, Ollivier V, Dai J, et al. Renewal of mural thrombus releases plasma markers and is involved in aortic abdominal aneurysm evolution. Am J Pathol. 2006;168(3):1022–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Dai J, Louedec L, Philippe M, Michel JB, Houard X. Effect of blocking platelet activation with AZD6140 on development of abdominal aortic aneurysm in a rat aneurysmal model. J Vasc Surg. 2009;49(3):719–727. [DOI] [PubMed] [Google Scholar]
- 428.Owens AP 3rd, Edwards TL, Antoniak S, et al. Platelet inhibitors reduce rupture in a mouse model of established abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(9):2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Wang Y, Ait-Oufella H, Herbin O, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120(2):422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Chen X, Rateri DL, Howatt DA, et al. TGF-beta neutralization enhances AngII–induced aortic rupture and aneurysm in both thoracic and abdominal regions. PLoS One. 2016;11(4):e0153811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Renard M, Callewaert B, Baetens M, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFbeta signaling in FTAAD. Int J Cardiol. 2013;165(2):314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Jones GT, Thompson AR, van Bockxmeer FM, et al. Angiotensin II type 1 receptor 1166 Cpolymorphism is associated with abdominal aortic aneurysm in three independent cohorts. Arterioscler Thromb Vasc Biol. 2008;28(4):764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II–induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007; 27(2):380–386. [DOI] [PubMed] [Google Scholar]
- 435.Wang YX, Martin-McNulty B, da Cunha V, et al. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II–induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation. 2005;111(17):2219–2226. [DOI] [PubMed] [Google Scholar]
- 436.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2): 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Koenig SN, Bosse K, Majumdar U, Bonachea EM, Radtke F, Garg V. Endothelial Notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J Am Heart Assoc. 2016;5(4):e003075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056): 270–274. [DOI] [PubMed] [Google Scholar]
- 439.Mohamed SA, Aherrahrou Z, Liptau H, et al. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. 2006;345(4):1460–1465. [DOI] [PubMed] [Google Scholar]
- 440.McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM 3rd. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134(2):290–296. [DOI] [PubMed] [Google Scholar]
- 441.Kent KC, Crenshaw ML, Goh DL, Dietz HC. Genotype-phenotype correlation in patients with bicuspid aortic valve and aneurysm. J Thorac Cardiovasc Surg. 2013;146(1):158–165 e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Kostina AS, Uspensky Vcapital Ie C, Irtyuga OB, et al. Notch-dependent EMT is attenuated in patients with aortic aneurysm and bicuspid aortic valve. Biochim Biophys Acta. 2016;1862(4):733–740. [DOI] [PubMed] [Google Scholar]
- 443.Zou S, Ren P, Nguyen M, Coselli JS, Shen YH, LeMaire SA. Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS One. 2012;7(12):e52833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Hans CP, Koenig SN, Huang N, et al. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arterioscler Thromb Vasc Biol. 2012;32(12):3012–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Piggott K, Deng J, Warrington K, et al. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123(3):309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Zheng YH, Li FD, Tian C, Ren HL, Du J, Li HH. Notch gamma-secretase inhibitor dibenzazepine attenuates angiotensin II–induced abdominal aortic aneurysm in ApoE knockout mice by multiple mechanisms. PLoS One. 2013;8(12):e83310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Wisler JW, Harris EM, Raisch M, et al. The role of beta-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am J Physiol Heart Circ Physiol. 2015;309(9):H1516–H1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Trivedi DB, Loftin CD, Clark J, et al. Beta-arrestin-2 deficiency attenuates abdominal aortic aneurysm formation in mice. Circ Res. 2013;112(9):1219–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Li MW, Mian MO, Barhoumi T, et al. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2013;33(10):2306–2315. [DOI] [PubMed] [Google Scholar]
- 450.Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension. 2015;65(2):257–263. [DOI] [PubMed] [Google Scholar]
- 451.Lother A, Hein L. Vascular mineralocorticoid receptors: linking risk factors, hypertension, and heart disease. Hypertension. 2016;68(1):6–10. [DOI] [PubMed] [Google Scholar]
- 452.Couture R, Blaes N, Girolami JP. Kinin receptors in vascular biology and pathology. Curr Vasc Pharmacol. 2014;12(2): 223–248. [DOI] [PubMed] [Google Scholar]
- 453.Moran CS, Rush CM, Dougan T, et al. Modulation of kinin B2 receptor signaling controls aortic dilatation and rupture in the angiotensin II-infused apolipoprotein E-deficient mouse. Arterioscler Thromb Vasc Biol. 2016;36(5): 898–907. [DOI] [PubMed] [Google Scholar]
- 454.Japp AG, Cruden NL, Amer DA, et al. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. 2008;52(11): 908–913. [DOI] [PubMed] [Google Scholar]
- 455.Chun HJ, Ali ZA, Kojima Y, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118(10):3343–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Gonias SL, Campana WM. LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am J Pathol. 2014;184(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300(5617):329–332. [DOI] [PubMed] [Google Scholar]
- 458.Strickland DK, Au DT, Cunfer P, Muratoglu SC. Low-density lipoprotein receptor-related protein-1: role in the regulation of vascular integrity. Arterioscler Thromb Vasc Biol. 2014;34(3):487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Davis FM, Rateri DL, Balakrishnan A, et al. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II–induced superior mesenteric arterial and ascending aortic aneurysms. Arterioscler Thromb Vasc Biol. 2015;35(1):155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Muratoglu SC, Belgrave S, Hampton B, et al. LRP1 protects the vasculature by regulating levels of connective tissue growth factor and HtrA1. Arterioscler Thromb Vasc Biol. 2013;33(9):2137–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Shen D, Li J, Lepore JJ, et al. Aortic aneurysm generation in mice with targeted deletion of integrin-linked kinase in vascular smooth muscle cells. Circ Res. 2011;109(6):616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Zheng L, Xing L, Zeng C, et al. Inactivation of PI3Kdelta induces vascular injury and promotes aneurysm development by upregulating the AP-1/MMP-12 pathway in macrophages. Arterioscler Thromb Vasc Biol. 2015;35(2): 368–377. [DOI] [PubMed] [Google Scholar]
- 463.Yoshimura K, Aoki H, Ikeda Y, et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11(12):1330–1338. [DOI] [PubMed] [Google Scholar]
- 464.Zhang Y, Naggar JC, Welzig CM, et al. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler Thromb Vasc Biol. 2009;29(11): 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Ehrlichman LK, Ford JW, Roelofs KJ, et al. Gender-dependent differential phosphorylation in the ERK signaling pathway is associated with increased MMP2 activity in rat aortic smooth muscle cells. J Surg Res. 2010;160(1): 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Zhang F, Banker G, Liu X, et al. The novel function of advanced glycation end products in regulation of MMP-9 production. J Surg Res. 2011;171(2):871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Ghosh A, DiMusto PD, Ehrlichman LK, et al. The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J Am Coll Surg. 2012;215(5):668–680 e661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Carta L, Smaldone S, Zilberberg L, et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284(9):5630–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Jones JA, Stroud RE, Kaplan BS, et al. Differential protein kinase C isoform abundance in ascending aortic aneurysms from patients with bicuspid versus tricuspid aortic valves. Circulation. 2007;116(Suppl 11):I144–I149. [DOI] [PubMed] [Google Scholar]
- 470.Ren J, Wang Q, Morgan S, et al. Protein kinase C-delta (PKCdelta) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-kappaB subunit p65 in vascular smooth muscle cells. J Biol Chem. 2014;289(13):9013–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440(7081):228–232. [DOI] [PubMed] [Google Scholar]
- 472.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–140. [DOI] [PubMed] [Google Scholar]
- 473.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. [DOI] [PubMed] [Google Scholar]
- 475.Usui F, Shirasuna K, Kimura H, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II–induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(1): 127–136. [DOI] [PubMed] [Google Scholar]
- 476.Owens AP 3rd, Rateri DL, Howatt DA, et al. MyD88 deficiency attenuates angiotensin II–induced abdominal aortic aneurysm formation independent of signaling through Toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol. 2011;31(12):2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6(1): 11–23. [DOI] [PubMed] [Google Scholar]
- 478.Nemer M, Horb ME. The KLF family of transcriptional regulators in cardiomyocyte proliferation and differentiation. Cell Cycle. 2007;6(2):117–121. [DOI] [PubMed] [Google Scholar]
- 479.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121(8): 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14(8):475–488. [DOI] [PubMed] [Google Scholar]
- 481.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16(7):421–433. [DOI] [PubMed] [Google Scholar]
- 482.Raffort J, Lareyre F, Clement M, Mallat Z. Micro-RNAs in abdominal aortic aneurysms: insights from animal models and relevance to human disease. Cardiovasc Res. 2016;110(2):165–177. [DOI] [PubMed] [Google Scholar]
- 483.Jones JA, Stroud RE, O’Quinn EC, et al. Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circ Cardiovasc Genet. 2011;4(6):605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Merk DR, Chin JT, Dake BA, et al. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110(2):312–324. [DOI] [PubMed] [Google Scholar]
- 485.Maegdefessel L, Azuma J, Toh R, et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. 2012;122(2):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Zampetaki A, Attia R, Mayr U, et al. Role of miR-195 in aortic aneurysmal disease. Circ Res. 2014;115(10):857–866. [DOI] [PubMed] [Google Scholar]
- 487.Kim CW, Kumar S, Son DJ, Jang IH, Griendling KK, Jo H. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arterioscler Thromb Vasc Biol. 2014;34(7): 1412–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Maegdefessel L, Spin JM, Raaz U, et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat Commun. 2014;5:5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110(10):1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Ejiri J, Inoue N, Tsukube T, et al. Oxidative stress in the pathogenesis of thoracic aortic aneurysm: protective role of statin and angiotensin II type 1 receptor blocker. Cardiovasc Res. 2003;59(4):988–996. [DOI] [PubMed] [Google Scholar]
- 491.Coyle P, Philcox JC, Carey LC, Rofe AM. Metallothionein: the multipurpose protein. Cell Mol Life Sci. 2002;59(4): 627–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Phillippi JA, Klyachko EA, Kenny JPt, Eskay MA, Gorman RC, Gleason TG. Basal and oxidative stress-induced expression of metallothionein is decreased in ascending aortic aneurysms of bicuspid aortic valve patients. Circulation. 2009;119(18):2498–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.McCormick ML, Gavrila D, Weintraub NL. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2007;27(3):461–469. [DOI] [PubMed] [Google Scholar]
- 494.Delbosc S, Diallo D, Dejouvencel T, et al. Impaired high-density lipoprotein anti-oxidant capacity in human abdominal aortic aneurysm. Cardiovasc Res. 2013;100(2):307–315. [DOI] [PubMed] [Google Scholar]
- 495.Xiong W, Mactaggart J, Knispel R, et al. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis. 2009;202(1):128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Thirunavukkarasu S, Khan NS, Song CY, et al. Cytochrome P450 1B1 contributes to the development of angiotensin II–induced aortic aneurysm in male Apoe( — / —) mice. Am J Pathol. 2016;186(8):2204–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2013;33(10):2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Satoh K, Nigro P, Matoba T, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II–induced aortic aneurysms. Nat Med. 2009;15(6):649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Kigawa Y, Miyazaki T, Lei XF, et al. NaDPH oxidase deficiency exacerbates angiotensin II–induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2014;34(11):2413–2420. [DOI] [PubMed] [Google Scholar]
- 500.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013–1030. [DOI] [PubMed] [Google Scholar]
- 501.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6): 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Petrasek J, Iracheta-Vellve A, Csak T, et al. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110(41):16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Kwartler CS, Chen J, Thakur D, et al. Overexpression of smooth muscle myosin heavy chain leads to activation of the unfolded protein response and autophagic turnover of thick filament-associated proteins in vascular smooth muscle cells. J Biol Chem. 2014;289(20):14075–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117(21):2802–2813. [DOI] [PubMed] [Google Scholar]
- 506.Danyi P, Elefteriades JA, Jovin IS. Medical therapy of thoracic aortic aneurysms. Trends Cardiovasc Med. 2012;22(7): 180–184. [DOI] [PubMed] [Google Scholar]
- 507.Braverman AC. Medical management of thoracic aortic aneurysm disease. J Thorac Cardiovasc Surg. 2013;145(Suppl 3): S2–S6. [DOI] [PubMed] [Google Scholar]
- 508.Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2014;64(16):1725–1739. [DOI] [PubMed] [Google Scholar]
- 509.Attenhofer Jost CH, Greutmann M, Connolly HM, et al. Medical treatment of aortic aneurysms in Marfan syndrome and other heritable conditions. Curr Cardiol Rev. 2014;10(2):161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008; 117(14):1883–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Golledge J, Norman PE. Current status of medical management for abdominal aortic aneurysm. Atherosclerosis. 2011;217(1):57–63. [DOI] [PubMed] [Google Scholar]
- 512.Rughani G, Robertson L, Clarke M. Medical treatment for small abdominal aortic aneurysms. Cochrane Database Syst Rev. 2012;9:CD009536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Golledge J, Norman PE, Murphy MP, Dalman RL. Challenges and opportunities in limiting abdominal aortic aneurysm growth. J Vasc Surg. 2016. [DOI] [PubMed] [Google Scholar]
- 514.Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med. 1994;330(19):1335–1341. [DOI] [PubMed] [Google Scholar]
- 515.Gersony DR, McClaughlin MA, Jin Z, Gersony WM. The effect of beta-blocker therapy on clinical outcome in patients with Marfan’s syndrome: a meta-analysis. Int J Cardiol. 2007;114(3):303–308. [DOI] [PubMed] [Google Scholar]
- 516.Chan KK, Lai P, Wright JM. First-line beta-blockers versus other antihypertensive medications for chronic type B aortic dissection. Cochrane Database Syst Rev. 2014;(2):CD010426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Propanolol Aneurysm Trial I Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg. 2002;35(1):72–79. [DOI] [PubMed] [Google Scholar]
- 518.Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet. 2008;372(9654):1962–1976. [DOI] [PubMed] [Google Scholar]
- 519.Yetman AT, Bornemeier RA, McCrindle BW. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am J Cardiol. 2005;95(9):1125–1127. [DOI] [PubMed] [Google Scholar]
- 520.Ahimastos AA, Aggarwal A, D’Orsa KM, et al. Effect of perindopril on large artery stiffness and aortic root diameter in patients with Marfan syndrome: a randomized controlled trial. J Am Med Assoc. 2007;298(13):1539–1547. [DOI] [PubMed] [Google Scholar]
- 521.Liao S, Miralles M, Kelley BJ, Curci JA, Borhani M, Thompson RW. Suppression of experimental abdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg. 2001;33(5): 1057–1064. [DOI] [PubMed] [Google Scholar]
- 522.Kristensen KE, Torp-Pedersen C, Gislason GH, Egfjord M, Rasmussen HB, Hansen PR. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in patients with abdominal aortic aneurysms: nation-wide cohort study. Arterioscler Thromb Vasc Biol. 2015;35(3):733–740. [DOI] [PubMed] [Google Scholar]
- 523.Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet. 2006;368(9536):659–665. [DOI] [PubMed] [Google Scholar]
- 524.Sweeting MJ, Thompson SG, Brown LC, Greenhalgh RM, Powell JT. Use of angiotensin converting enzyme inhibitors is associated with increased growth rate of abdominal aortic aneurysms. J Vasc Surg. 2010;52(1):1–4. [DOI] [PubMed] [Google Scholar]
- 525.Bicknell CD, Kiru G, Falaschetti E, Powell JT, Poulter NR, AARDVARK Collaborators. An evaluation of the effect of an angiotensin-converting enzyme inhibitor on the growth rate of small abdominal aortic aneurysms: a randomized placebo-controlled trial (AARDVARK). Eur Heart J. 2016;37(42):3213–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358(26):2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Pees C, Laccone F, Hagl M, Debrauwer V, Moser E, Michel-Behnke I. Usefulness of losartan on the size of the ascending aorta in an unselected cohort of children, adolescents, and young adults with Marfan syndrome. Am J Cardiol. 2013;112(9):1477–1483. [DOI] [PubMed] [Google Scholar]
- 528.Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491–3500. [DOI] [PubMed] [Google Scholar]
- 529.Lacro RV, Dietz HC, Wruck LM, et al. Rationale and design of a randomized clinical trial of beta-blocker therapy (atenolol) versus angiotensin II receptor blocker therapy (losartan) in individuals with Marfan syndrome. Am Heart J. 2007;154(4):624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Lacro RV, Guey LT, Dietz HC, et al. Characteristics of children and young adults with Marfan syndrome and aortic root dilation in a randomized trial comparing atenolol and losartan therapy. Am Heart J. 2013;165(5): 828–835 e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Moberg K, De Nobele S, Devos D, et al. The Ghent Marfan Trial—a randomized, double-blind placebo controlled trial with losartan in Marfan patients treated with beta-blockers. Int J Cardiol. 2013;157(3):354–358. [DOI] [PubMed] [Google Scholar]
- 532.Davis F, Rateri DL, Daugherty A. Aortic aneurysms in Loeys-Dietz syndrome—a tale of two pathways? J Clin Invest. 2014;124(1):79–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Daugherty A, Manning MW, Cassis LA. Antagonism of AT2 receptors augments angiotensin II–induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134(4):865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Morris DR, Cunningham MA, Ahimastos AA, et al. TElmisartan in the management of abDominal aortic aneurYsm (TEDY): the study protocol for a randomized controlled trial. Trials. 2015;16:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Doyle JJ, Doyle AJ, Wilson NK, et al. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife. 2015;4:e08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Williams A, Kenny D, Wilson D, et al. Effects of atenolol, perindopril and verapamil on haemodynamic and vascular function in Marfan syndrome—a randomised, double-blind, crossover trial. Eur J Clin Invest. 2012;42(8):891–899. [DOI] [PubMed] [Google Scholar]
- 537.Schonbeck U, Libby P. Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 2004;109(21 Suppl 1):II18–II26. [DOI] [PubMed] [Google Scholar]
- 538.Davignon J Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109(23 Suppl 1):III39–III43. [DOI] [PubMed] [Google Scholar]
- 539.McLoughlin D, McGuinness J, Byrne J, et al. Pravastatin reduces Marfan aortic dilation. Circulation. 2011;124(Suppl 11):S168–S173. [DOI] [PubMed] [Google Scholar]
- 540.Goel SS, Tuzcu EM, Agarwal S, et al. Comparison of ascending aortic size in patients with severe bicuspid aortic valve stenosis treated with versus without a statin drug. Am J Cardiol. 2011;108(10):1458–1462. [DOI] [PubMed] [Google Scholar]
- 541.Jovin IS, Duggal M, Ebisu K, et al. Comparison of the effect on long-term outcomes in patients with thoracic aortic aneurysms of taking versus not taking a statin drug. Am J Cardiol. 2012;109(7):1050–1054. [DOI] [PubMed] [Google Scholar]
- 542.Stein LH, Berger J, Tranquilli M, Elefteraides JA. Effect of statin drugs on thoracic aortic aneurysms. Am J Cardiol. 2013;112(8):1240–1245. [DOI] [PubMed] [Google Scholar]
- 543.Angeloni E, Vitaterna A, Pirelli M, Refice S. Effects of statin therapy on ascending aorta aneurysms growth: a propensity-matched analysis. Int J Cardiol. 2015;191:52–55. [DOI] [PubMed] [Google Scholar]
- 544.Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;18(11):1671–1678. [DOI] [PubMed] [Google Scholar]
- 545.Nagashima H, Aoka Y, Sakomura Y, et al. A 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, cerivastatin, suppresses production of matrix metalloproteinase-9 in human abdominal aortic aneurysm wall. J Vasc Surg. 2002;36(1):158–163. [DOI] [PubMed] [Google Scholar]
- 546.Steinmetz EF, Buckley C, Shames ML, et al. Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysms in normal and hypercholesterolemic mice. Ann Surg. 2005;241(1): 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Kalyanasundaram A, Elmore JR, Manazer JR, et al. Simvastatin suppresses experimental aortic aneurysm expansion. J Vasc Surg. 2006;43(1):117–124. [DOI] [PubMed] [Google Scholar]
- 548.Mosorin M, Niemela E, Heikkinen J, et al. The use of statins and fate of small abdominal aortic aneurysms. Interact Cardiovasc Thorac Surg. 2008;7(4):578–581. [DOI] [PubMed] [Google Scholar]
- 549.Takagi H, Yamamoto H, Iwata K, Goto S, Umemoto T, ALICE Group. Effects of statin therapy on abdominal aortic aneurysm growth: a meta-analysis and meta-regression of observational comparative studies. Eur J Vasc Endovasc Surg. 2012;44(3):287–292. [DOI] [PubMed] [Google Scholar]
- 550.Takagi H, Umemoto T. Reply to ‘Comment on effects of statin therapy on abdominal aortic aneurysm growth: a meta-analysis and meta-regression of observational comparative studies’. Eur J Vasc Endovasc Surg. 2013;45(1): 98–99. [DOI] [PubMed] [Google Scholar]
- 551.Ferguson CD, Clancy P, Bourke B, et al. Association of statin prescription with small abdominal aortic aneurysm progression. Am Heart J. 2010;159(2):307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.van der Meij E, Koning GG, Vriens PW, et al. A clinical evaluation of statin pleiotropy: statins selectively and dose-dependently reduce vascular inflammation. PLoS One. 2013;8(1):e53882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Dunne JA, Bailey MA, Griffin KJ, Sohrabi S, Coughlin PA, Scott DJ. Statins: the holy grail of abdominal aortic aneurysm (AAA) growth attenuation? A systematic review of the literature. Curr Vasc Pharmacol. 2014;12(1): 168–172. [DOI] [PubMed] [Google Scholar]
- 554.Baxter BT, Matsumura J, Curci J, et al. Non-invasive treatment of abdominal aortic aneurysm clinical trial (N-TA(3) CT): design of a phase IIb, placebo-controlled, double-blind, randomized clinical trial of doxycycline for the reduction of growth of small abdominal aortic aneurysm. Contemp Clin Trials. 2016;48:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Lee HM, Ciancio SG, Tuter G, Ryan ME, Komaroff E, Golub LM. Subantimicrobial dose doxycycline efficacy as a matrix metalloproteinase inhibitor in chronic periodontitis patients is enhanced when combined with a non-steroidal anti-inflammatory drug. J Periodontol. 2004;75(3):453–463. [DOI] [PubMed] [Google Scholar]
- 556.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and −9. CircRes. 2008;102(8):e73–e85. [DOI] [PubMed] [Google Scholar]
- 557.Yang HH, Kim JM, Chum E, van Breemen C, Chung AW. Effectiveness of combination of losartan potassium and doxycycline versus single-drug treatments in the secondary prevention of thoracic aortic aneurysm in Marfan syndrome. J Thorac Cardiovasc Surg. 2010;140(2):305–312 e302. [DOI] [PubMed] [Google Scholar]
- 558.Tae HJ, Marshall S, Zhang J, Wang M, Briest W, Talan MI. Chronic treatment with a broad-spectrum metalloproteinase inhibitor, doxycycline, prevents the development of spontaneous aortic lesions in a mouse model of vascular Ehlers-Danlos syndrome. J Pharmacol Exp Ther. 2012;343(1):246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Lindeman JH, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm: doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119(16):2209–2216. [DOI] [PubMed] [Google Scholar]
- 560.Hackmann AE, Rubin BG, Sanchez LA, Geraghty PA, Thompson RW, Curci JA. A randomized, placebo-controlled trial of doxycycline after endoluminal aneurysm repair. J Vasc Surg. 2008;48(3):519–526. [discussion 526]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Meijer CA, Stijnen T, Wasser MN, et al. Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann Intern Med. 2013;159(12):815–823. [DOI] [PubMed] [Google Scholar]
- 562.Dodd BR, Spence RA. Doxycycline inhibition of abdominal aortic aneurysm growth: a systematic review of the literature. Curr Vasc Pharmacol. 2011;9(4):471–478. [DOI] [PubMed] [Google Scholar]
- 563.Parr A, McCann M, Bradshaw B, Shahzad A, Buttner P, Golledge J. Thrombus volume is associated with cardiovascular events and aneurysm growth in patients who have abdominal aortic aneurysms. J Vasc Surg. 2011;53 (1):28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Lindholt JS, Sorensen HT, Michel JB, Thomsen HF, Henneberg EW. Low-dose aspirin may prevent growth and later surgical repair of medium-sized abdominal aortic aneurysms. Vasc Endovascular Surg. 2008;42(4):329–334. [DOI] [PubMed] [Google Scholar]
- 565.Bailey MA, Aggarwal R, Bridge KI, et al. Aspirin therapy is associated with less compact fibrin networks and enhanced fibrinolysis in patients with abdominal aortic aneurysm. J Thromb Haemost. 2015;13(5):795–801. [DOI] [PubMed] [Google Scholar]
- 566.Johnston WF, Salmon M, Pope NH, et al. Inhibition of interleukin-1beta decreases aneurysm formation and progression in a novel model of thoracic aortic aneurysms. Circulation. 2014;130(11 Suppl 1):S51–S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162(4):597–605. [DOI] [PubMed] [Google Scholar]
- 568.Nosoudi N, Nahar-Gohad P, Sinha A, et al. Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles. Circ Res. 2015;117(11): e80–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Moran CS, Jose RJ, Moxon JV, et al. Everolimus limits aortic aneurysm in the apolipoprotein E-deficient mouse by downregulating C-C chemokine receptor 2 positive monocytes. Arterioscler Thromb Vasc Biol. 2013;33(4):814–821. [DOI] [PubMed] [Google Scholar]
- 570.Bhamidipati CM, Mehta GS, Moehle CW, et al. Adenosine 2a receptor modulates inflammation and phenotype in experimental abdominal aortic aneurysms. FASEB J. 2013;27(6):2122–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]