

Abstract
Mutations in LRRK2 cause autosomal dominant and sporadic Parkinson’s disease, but the mechanisms involved in LRRK2 toxicity in PD are yet to be fully understood. We found that LRRK2 translocates to the nucleus by binding to seven in absentia homolog (SIAH-1), and in the nucleus it directly interacts with lamin A/C, independent of its kinase activity. LRRK2 knockdown caused nuclear lamina abnormalities and nuclear disruption. LRRK2 disease mutations mostly abolish the interaction with lamin A/C and, similar to LRRK2 knockdown, cause disorganization of lamin A/C and leakage of nuclear proteins. Dopaminergic neurons of LRRK2 G2019S transgenic and LRRK2 −/− mice display decreased circularity of the nuclear lamina and leakage of the nuclear protein 53BP1 to the cytosol. Dopaminergic nigral and cortical neurons of both LRRK2 G2019S and idiopathic PD patients exhibit abnormalities of the nuclear lamina. Our data indicate that LRRK2 plays an essential role in maintaining nuclear envelope integrity. Disruption of this function by disease mutations suggests a novel phosphorylation-independent loss-of-function mechanism that may synergize with other neurotoxic effects caused by LRRK2 mutations.
Introduction
Parkinson’s disease (PD) leads to progressive degeneration of neurons, especially of dopaminergic neurons in the substantia nigra (1). Several genes are mutated in families with PD, including α-synuclein, LRRK2, parkin and PINK1 (2).
Mutations in the LRRK2 (leucine-rich repeat kinase 2) gene cause autosomal dominant (3,4) and sporadic PD (5,6). LRRK2 is a protein kinase that associates with membranes of different intracellular organelles, including mitochondria, endosomes and lysosomes, suggesting that it may regulate the activity of various intracellular processes, including autophagy and mitophagy (7–11). Notably, LRRK2 interacts with several members of Rab GTPases, suggesting that LRRK2 regulates the vesicular transport and other Rab-dependent processes (12–14).
LRRK2 kinase activity increases by several disease mutations, and this is associated with neuronal toxicity (15–17), mitochondrial depolarization (10), reduction in neurite length (18) and increased α-synuclein propagation (19). However, it is still not clear if increased LRRK2 kinase activity mediates all impairments seen with mutant LRRK2 (20,21). For instance, LRRK2 R1441C mutation interferes with the interaction of LRRK2 with Sec16A and affects ER-Golgi transport in a kinase-independent manner (22). Also, targeted deletion of LRRK2 and its homolog LRRK1 in mice cause dopaminergic degeneration, indicating that LRRK2 normal function is required for survival of dopaminergic neurons (23).
While most recent LRRK2 studies focus on phosphorylation-dependent regulation of Rab GTPases (12,24), two studies previously linked LRRK2 mutations to nuclear abnormalities (25,26). LRRK2 G2019S mutant neuronal stem cells display decreased nuclear circularity at late culture passages, a process ascribed to the higher kinase activity of the LRRK2 G2019S mutant (25). LRRK2 R1441C transgenic mice display progressive nuclear abnormalities in dopaminergic neurons, which were ascribed to neuronal aging (26). While these studies highlight the nucleus as an organelle affected in PD, they did not consider a normal role of wild-type LRRK2 at the nuclear envelope and did not consider loss-of-function mechanisms regarding LRRK2 mutants.
We now hypothesize that wild-type LRRK2 plays important roles in nuclear maintenance, and disruption of this normal role by disease mutations underlies the nuclear alterations previously observed in LRRK2 disease mutant models (25,26). We now demonstrate that wild-type LRRK2 binds lamin A/C, which is crucial to maintaining nuclear lamina organization and nuclear membrane integrity. LRRK2 knockdown causes nuclear envelope pathology. SIAH proteins associate with LRRK2 and promote its ubiquitination and nuclear translocation. Similar to that observed with LRRK2 knockdown, different LRRK2 disease mutations virtually abolish the interaction with lamin A/C, promoting nuclear envelope disruption by a kinase-independent mechanism. Similar nuclear abnormalities were present in LRRK2 −/− mice, LRRK2 G2019S transgenic mice and substantia nigra and cortex of LRRK2 G2019S and idiopathic PD. Our observations indicate that LRRK2 normal function is required to stabilize the nuclear lamina and maintain nuclear envelope homeostasis, a process that is disrupted in LRRK2 mutations.
Results
LRRK2 is present in the nucleus
We carried out subcellular fractionation of rat brains and found endogenous LRRK2 not only in the cytosol but also in the purified nuclear fraction (Fig. 1A). The presence of LRRK2 in the nuclear fraction is not based on non-specific adsorption since LRRK2 was not extracted by treatment with Triton X-100 or sodium carbonate, which remove loosely bound membrane proteins (Fig. 1A). The specificity of the anti-LRRK2 antibody was confirmed using brain lysates of LRRK2 −/− mice as controls (Supplementary Material, Fig. S1A). In addition, the endoplasmic reticulum protein BiP was not detected in the nuclear fraction (Supplementary Material, Fig. S1B), indicating that the presence of LRRK2 in the nucleus is not based on cross-contamination with other organelles, such as endoplasmic reticulum. Ectopically expressed LRRK2 G2019S, LRRK2 R1441C and LRRK2 kinase-dead (KD) mutants were present at the nuclear fraction at levels similar to the wild-type LRRK2 protein (Fig. 1B). No significant changes in the basal LRRK2 phosphorylation state (WT or G2019S mutant) were observed between cytosolic and nuclear-associated LRRK2 species (Supplementary Material, Fig. S2). In addition, the middle and C-terminal regions of LRRK2 translocate to the nucleus (Supplementary Material, Fig. S3), suggesting that different LRRK2 regions may be involved in its translocation to the nucleus.
Figure 1.
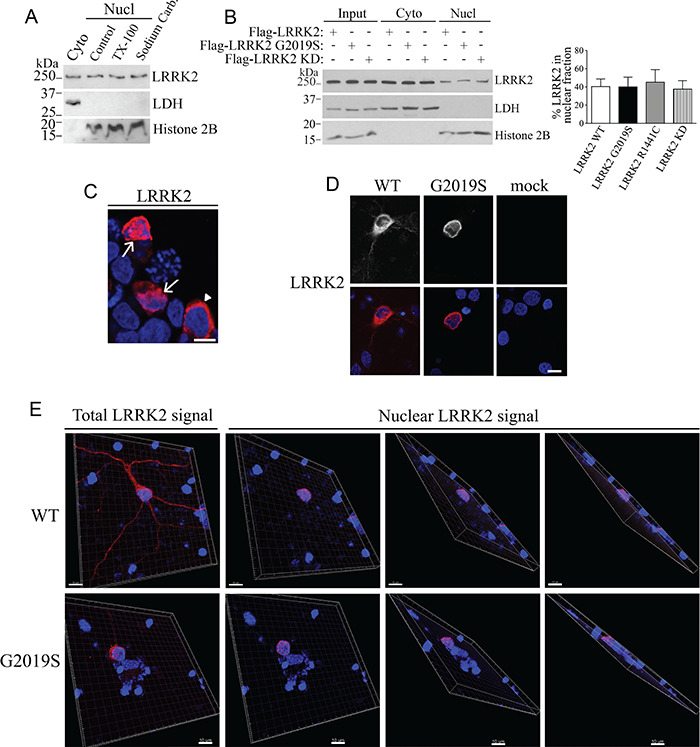
Presence of LRRK2 in the nucleus. (A) Rat brain lysates were fractionated into cytosolic and nuclear fractions. Nuclear fractions were further treated with 1% Triton X-100 or sodium carbonate (pH 11.5). Levels of endogenous LRRK2 were determined with anti-LRRK2 (first panel). The purity of cytosolic and nuclear fractions was monitored with anti-LDH and anti-histone H2B, respectively. (B) Transfected wild-type LRRK2, G2019S and R1441C disease mutants, and LRRK2 kinase-dead (KD) are equally present in the nuclear fraction of HEK293 cells. The distribution of LRRK2 was determined using anti-Flag (upper panel). The graph represents the percent of LRRK2 present in the nuclear fractions relative to the total LRRK2 levels. Values represent the average ± S.E.M. of three independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test (non-significant P < 0.999). (C) HEK293 cells with nuclear Flag-LRRK2 (arrows) according to co-localization with Hoescht 33342. Arrowhead indicates a cell with Flag-LRRK2 present only at the cytosol. HEK293 cells were processed for immunocytochemistry with anti-Flag antibody (red) and the nuclear marker Hoechst 33342 (blue) and analyzed by confocal microscopy. Scale bar, 10 μm. (D) Nuclear LRRK2 in primary cultured neurons were transfected with Flag-LRRK2 (wild-type or G2019S) or mock-transfected. At DIV 7 cells were analyzed by confocal microscopy regarding the co-localization of Flag-LRRK2 (red, lower panels) with Hoechst 33342 (blue, lower panels). Upper panels depict the correspondent LRRK2 signal without pseudocolor. Scale bar, 10 μm. (E) Cortical neurons were transfected and processed as in (D). Confocal z-series were acquired for transfected neurons, and images of serial sections were reconstituted by Imaris into 3D images. The first panels (upper and lower) represent the total LRRK2 3D signal while subsequent panels show the LRRK2 3D signal present only in the nucleus and rotated at different angles. The LRRK2 signal that did not co-localize with the nuclear marker Hoechst 33342 (cytosolic LRRK2) was digitally subtracted by Imaris. Scale bar, 10 μm.
Immunocytochemical experiments demonstrate that LRRK2 is present in the nuclei of transfected cells (Fig. 1C). Also, LRRK2 wild-type and G2019S disease mutants were consistently present in the nucleus of primary cortical neurons (Fig. 1D). Reconstitution of confocal z-series acquisitions into 3D projection at different angles, including more transversal projections, shows the spatial co-localization of LRRK2 with the nuclear marker Hoechst 33342 (Fig. 1E, lower panels) and confirms therefore the presence of LRRK2 immunoreactivity in the nucleus of cortical neurons (Fig. 1E).
Interaction of LRRK2 with lamin A/C
Nuclear subfractionation reveals that endogenous LRRK2 is present in the nucleoplasm (soluble nuclear fraction) and the insoluble nuclear fractions of untransfected HEK293 cells (Fig. 2A) and primary cultured neurons (Fig. 2B). Insoluble nuclear fraction contains nuclear lamina components, such as lamin A/C (Fig. 2A and B). Upon separation of insoluble nuclear material into chromatin and cytoskeletal fractions, we found that most endogenous LRRK2 is present in the nuclear cytoskeleton fraction, which contains lamin A/C (Fig. 2A and B).
Figure 2.
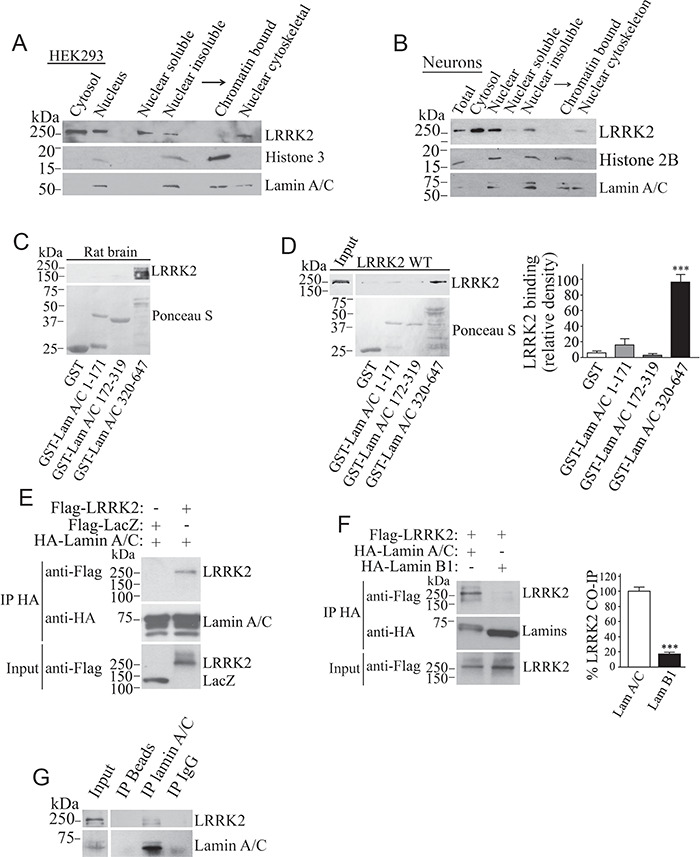
LRRK2 is present in the nuclear cytoskeleton and interacts with lamin A/C in vitro and in vivo. HEK293 cells (A) and primary neurons (B) were fractionated into cytosolic and nuclear fractions. Nuclear fractions were separated into soluble and insoluble material, and the insoluble fraction further separated into chromatin-bound and nuclear cytoskeletal fractions. Levels of endogenous LRRK2 were determined with anti-LRRK2 (upper panels). The blots were re-probed for lamin A/C, histone 3 (A) or 2B (B). (C) Interaction of endogenous LRRK2 from rat brain lysate with GST-lamin A/C (aa 320–647). Binding of endogenous LRRK2 was determined using anti-LRRK2 (upper panel). The lower panel depicts lamin A/C fragments stained by Ponceau S. (D) Binding of Flag-LRRK2 from cell lysate with GST-lamin A/C fragments. The binding was determined with anti-Flag (upper panel). The lower panel depicts lamin A/C fragments stained by Ponceau S. The graph depicts the relative binding of Flag-LRRK2 to the various GST-lamin A/C fragments. Values represent the average ± S.E.M. of three independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test (P < 0.001). (E) Co-immunoprecipitation of Flag-LRRK2 with HA-lamin A/C from transfected HEK293 cells. Co-immunoprecipitation of LRRK2 was detected with anti-Flag (upper panel) while levels of lamin A/C immunoprecipitation were determined with anti-HA (middle panel). (F) HEK293 cells were transfected with Flag-LRRK2, in the presence of HA-lamin A/C or HA-lamin B1. Lamins were immunoprecipitated from cell lysates with anti-HA antibody (middle panel), and co-immunoprecipitation with Flag-LRRK2 was detected with anti-Flag (upper panel). The graph represents the relative co-immunoprecipitation of wild-type LRRK2 with lamin A/C in contrast to lamin B1. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P < 0.001). (G) Lamin A/C was immunoprecipitated from rat brain using the anti-lamin A/C antibody (lower panel) and co-immunoprecipitation with endogenous LRRK2 determined with the anti-LRRK2 antibody (upper panel).
Next, we investigated if LRRK2 interacts with lamin A/C. Pull-down assays revealed that endogenous LRRK2 from rat brain (Fig. 2C), as well as Flag-LRRK2 from transfected HEK293 cell lysates (Fig. 2D), interact with the second half of GST-lamin A/C (amino acids 320–647), but not with fragments of the first half of the protein or GST alone. Flag-LRRK2 specifically co-immunoprecipitated with full-length lamin A/C from cells (Fig. 2E). In contrast, Flag-LRRK2 interacted to a much lesser extent with lamin B1 when in contrast to lamin A/C (Fig. 2F). Most importantly, LRRK2 from rat brain co-immunoprecipitates with lamin A/C (Fig. 2G), indicating that LRRK2 interacts with lamin A/C at endogenous levels.
Notably, we found that recombinant LRRK2 G2019S and R1441C mutants interact much less with purified lamin A/C (amino acids 320–647) in contrast to wild-type LRRK2 (Fig. 3A). The weaker interaction is not based on the higher kinase activity of the mutants since the kinase inhibitor LRRK2-IN-1 (27) did not increase the binding of LRRK2 G2019S to purified lamin A/C (Fig. 3B). Co-immunoprecipitation of Flag-LRRK2 with full-length myc-lamin A/C from cells was 70% lower with LRRK2 G2019S in contrast to the wild-type protein (Fig. 3C).
Figure 3.
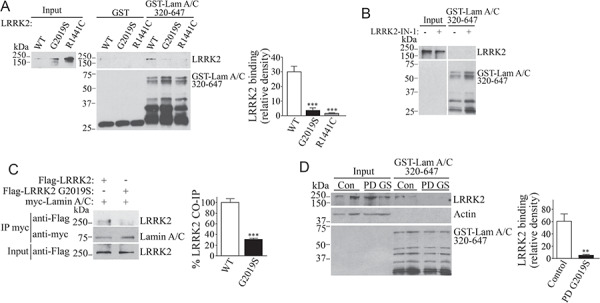
Disease mutations disrupt the interaction of LRRK2 with lamin A/C. (A) Purified recombinant LRRK2 (wild type, G2019S and R1441C) were incubated with GST or GST-lamin A/C fragment 320–647, and binding was determined using the anti-LRRK2 antibody. The lower panel depicts lamin A/C fragments detected by anti-GST. The graph depicts the relative binding of wild-type LRRK2, G2019S and R1441C to the GST-lamin A/C 320–647 fragment. Values represent the average ± S.E.M. of three independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test (P = 0.001 and <0.001 for G2019S and R1441C, respectively, in contrast to WT). (B) Purified recombinant LRRK2 G2019S was incubated with an ATP-containing medium, in the absence and presence of 2.5 μm LRRK2-IN-1, and subsequently incubated with GST-lamin A/C 320–647. The binding was determined with the anti-LRRK2 antibody. The lower panel depicts a lamin A/C fragment detected with anti-GST. (C) Full-length myc-lamin A/C was immunoprecipitated with anti-myc antibody (middle panel), and co-immunoprecipitation of transfected wild-type Flag-LRRK2 and G2019S mutant was detected with anti-Flag (upper panel). The graph depicts the relative co-immunoprecipitation of wild-type LRRK2 and G2019S mutant with myc-lamin A/C. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P < 0.001). (D) Postmortem human brain lysates from control and PD patient harboring the G2019S mutation were incubated with the GST-lamin A/C 320–647 fragment. Binding of LRRK2 was determined using the anti-LRRK2 antibody. The lower panel depicts lamin A/C fragments detected by anti-GST. The graph depicts the relative binding of the human LRRK2 from control and PD LRRK2 G2019S brains to lamin A/C. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.001).
We also monitored the binding of LRRK2 to lamin using extracts from PD patients harboring LRRK2 G2019S mutation and control individuals. The association of LRRK2 G2019S from autosomal dominant PD brain extracts with GST-lamin A/C was significantly lower in contrast to that observed for endogenous LRRK2 from control brains (Fig. 3D). Altogether, the data indicate that LRRK2 disease mutations disrupt the interaction withjb lamin A/C.
SIAH-1 facilitates LRRK2 nuclear translocation
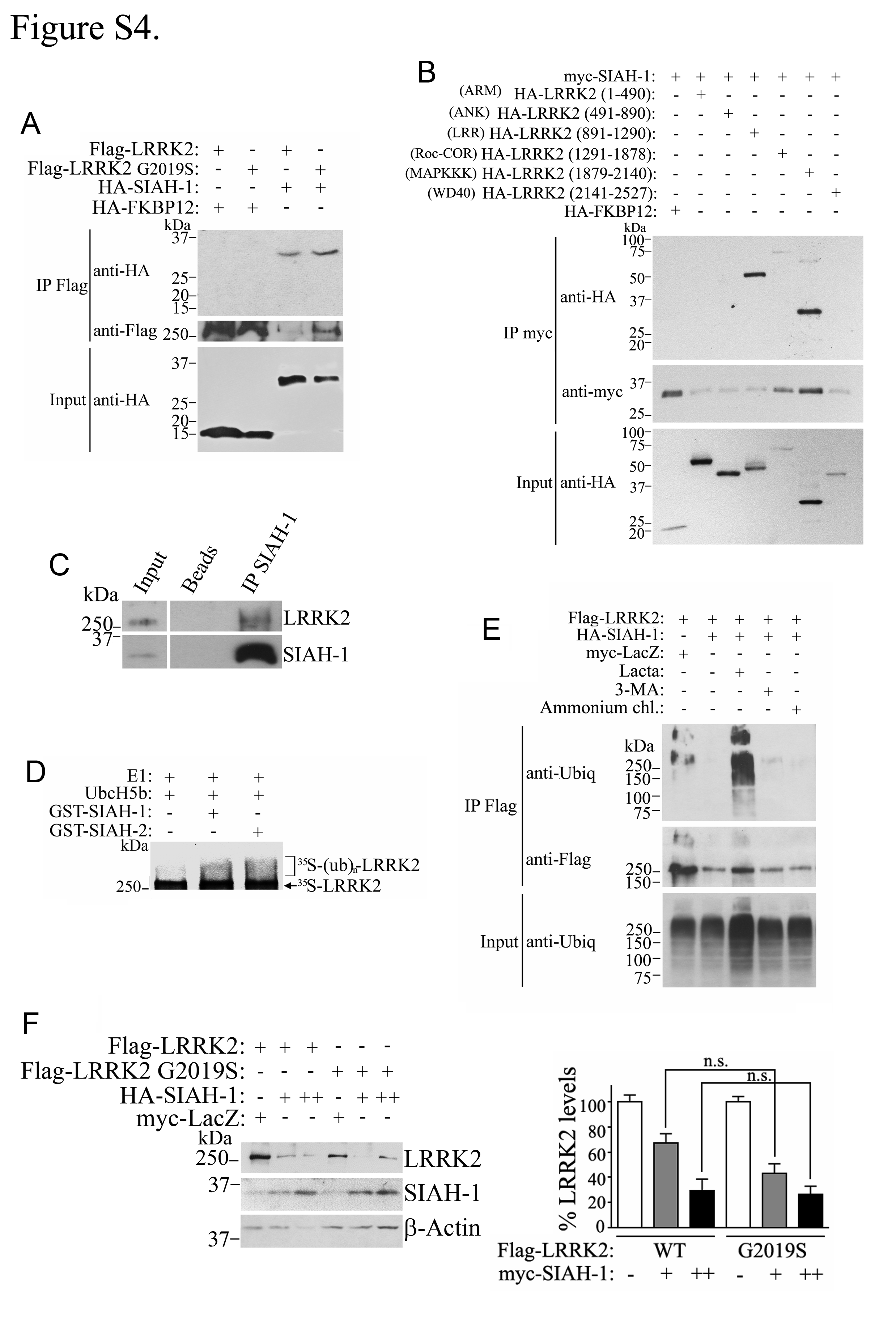
SIAH proteins are ubiquitin ligases that contain nuclear localization signals and can shuttle proteins to the nucleus (28). In addition to binding lamin A/C, LRRK2 also co-precipitated with SIAH-1 and SIAH-2, but not with FKBP12 control (Fig. 4A). The G2019S disease mutation did not alter the interaction with SIAH-1 (Supplementary Material, Fig. S4A). The LRR (leucine-rich repeat) and MAPKKK domains of LRRK2 were the regions of LRRK2 that more robustly interacted with SIAH-1 (Supplementary Material, Fig. S4B). Furthermore, LRRK2 from rat brain co-immunoprecipitated with SIAH-1, indicating interaction at endogenous levels (Supplementary Material, Fig. S4C).
Figure 4.
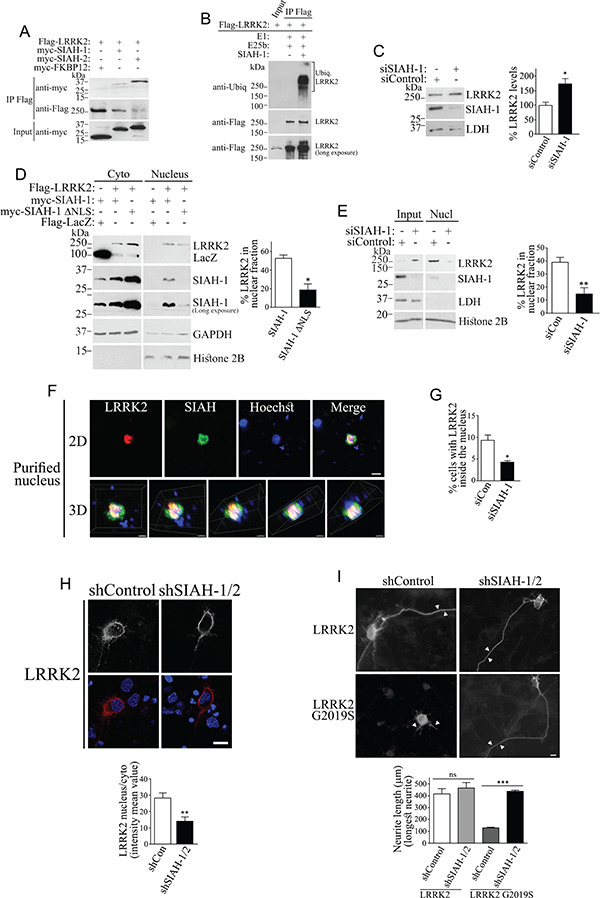
SIAH promotes the nuclear accumulation of LRRK2 and enhances LRRK2 G2019S toxicity. (A) Co-immunoprecipitation of SIAH-1 or SIAH-2 with Flag-LRRK2 from HEK293-transfected cells. Transfected LRRK2 was immunoprecipitated with anti-Flag (middle panel), and co-immunoprecipitation of SIAH-1 or SIAH-2 (upper panel) was detected with anti-myc (upper panel). (B) LRRK2 ubiquitination by SIAH-1. Flag-LRRK2 from transfected HEK293 cells was immunopurified with the anti-Flag antibody and further incubated with ubiquitination components and purified SIAH-1 (second and third lanes). Levels of ubiquitinated LRRK2 were determined using anti-ubiquitin (upper panel). Levels of immunoprecipitated LRRK2 were determined with anti-Flag (second and third lanes of middle and lower panels). The first lane represents the input of Flag-LRRK2 from transfected cells using anti-Flag (better visualized in lower panel). (C) SIAH-1 knockdown increases endogenous LRRK2 levels (upper panel) in HEK293 cells transfected with siRNA control or to SIAH-1. The graph depicts the percent of endogenous LRRK2 steady-state levels relative to LDH in the presence of siRNA control or siRNA to SIAH-1. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.022). (D) SIAH-1 increases the levels of Flag-LRRK2 in the nucleus of HEK293 cell, and this effect is disrupted by deletion of the SIAH-1 nuclear localization signal. The graph represents the percent of LRRK2 present in the nucleus relative to the total LRRK2 levels. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.01). (E) siRNA-mediated SIAH-1 knockdown decreased the levels of endogenous LRRK2 in the nuclear fraction of HEK293 cells. The purity of fractions was monitored with anti-LDH and anti-histone H2B. The graph represents the percent of LRRK2 present in the nucleus relative to the total LRRK2 levels with siRNA to SIAH-1 or control siRNA. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.008). (F) Purified nuclei from cells transfected with Flag-LRRK2 and myc-SIAH-1 were processed by immunocytochemistry with anti-Flag (red) and anti-myc (green) and analyzed by confocal microscopy (upper panels). Nuclei are shown by Hoechst 33342 staining. Scale bar, 10 μm. Lower panels show the confocal z-series reconstituted by Imaris into 3D images and rotated at different angles (Lower panels). Scale bar, 5 μm. (G) HEK293 transfected with Flag-LRRK2 and control siRNA or siRNA to SIAH-1 were processed for immunocytochemistry with anti-Flag antibody and analyzed by confocal microscopy regarding the co-localization of LRRK2 with Hoechst 33342. The graph represents the percentage of cells containing nuclear LRRK2 with different siRNAs. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.016). (H) Primary neurons were transfected with Flag-LRRK2, in the presence of control shRNA or shRNA to SIAH-1, and analyzed by confocal microscopy regarding the co-localization of Flag-LRRK2 (red, lower panels) with Hoechst 33342 (blue, lower panels). Upper panels depict a correspondent LRRK2 signal without pseudocolor: scale bar, 10 μm. The graph represents the mean intensity of nuclear LRRK2 relative to cytosolic LRRK2 in the presence of different shRNAs. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.004). (I) Primary neurons were transfected with Flag-LRRK2 (wild type and G2019S), and after 72 h, the cells were fixed and processed by immunocytochemistry with the anti-Flag antibody to assess the neurite length of transfected neurons. The end of the longest neurite in each field is indicated by arrowheads: scale bar, 10 μm. The graph represents the total length in microns of the longest neurite in transfected neurons, in the presence of different shRNAs. Values represent the average ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.468 and P < 0.001 for LRRK2 and LRRK2 G2019S, respectively).
Since SIAH-1 is an ubiquitin ligase, we investigated its ability to ubiquitinate LRRK2. We found that both SIAH-1 and SIAH-2 ubiquitinate in vitro translated LRRK2 (Supplementary Material, Fig. S4D) or LRRK2 immunoprecipitated from cells (Fig. 4B). Next, we carried out in vivo ubiquitination experiments and found that SIAH-1 ubiquitinates LRRK2 in cells treated with proteasome inhibitors but not with lysosomal and autophagy inhibitors (Supplementary Material, Fig. S4E). Even so, transfection of SIAH-1 only partially decreased the steady-state levels of wild-type LRRK2 or LRRK2 G2019S (Supplementary Material, Fig. S4F). Knockdown of endogenous SIAH-1 caused a moderate increase in the steady-state levels of endogenous LRRK2 (Fig. 4C), indicating that SIAH-1 has only a partial effect on LRRK2 degradation. The effect of SIAH-1, rather SIAH-2, was thereafter chosen to be investigated based on the paucity of reliable antibodies to SIAH-2 (29).
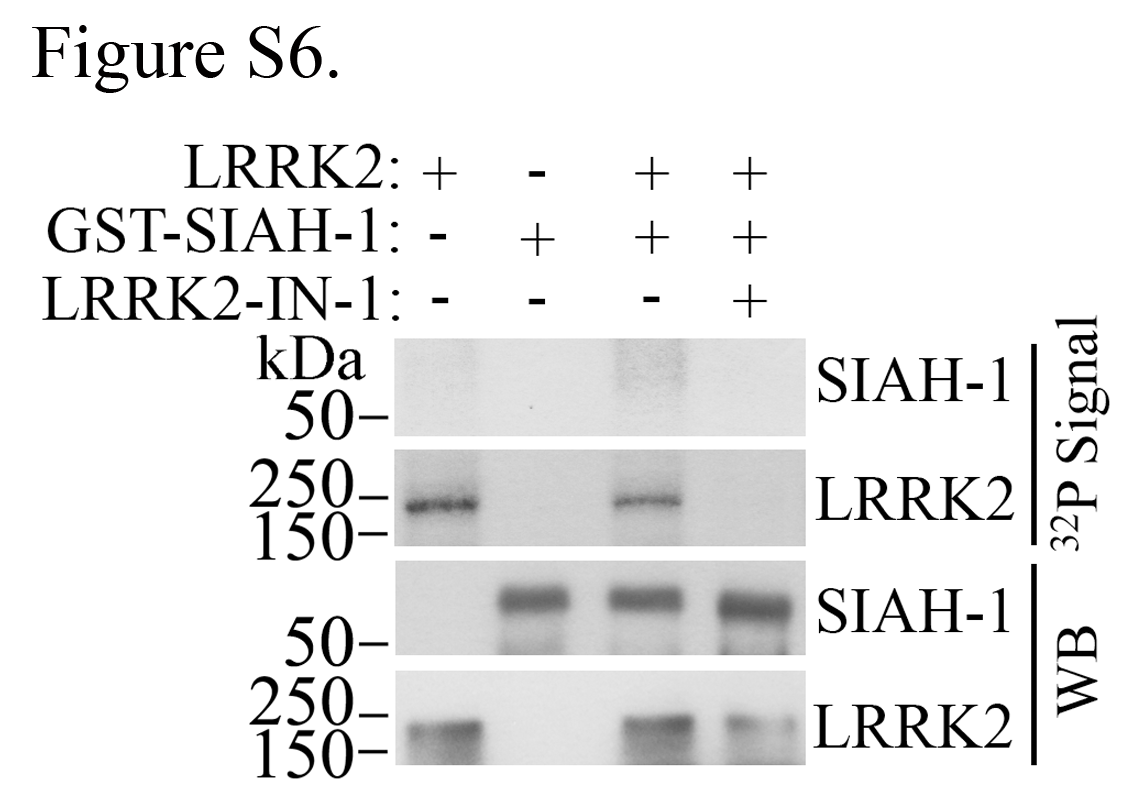
Like LRRK2, SIAH-1 partitions to the nuclear cytoskeleton fraction containing lamin A/C, with much lower levels associated with the chromatin (Supplementary Material, Fig. S5A and B). In contrast to LRRK2, however, SIAH-1 does not directly bind lamin A/C (Supplementary Material, Fig. S5C). Notably, SIAH-1 increased the nuclear levels of LRRK2 (Fig. 4D). Nuclear LRRK2 levels were consistently lower in the presence of a SIAH-1 construct devoid of nuclear translocation signals (SIAH-1 ∆NLS) (Fig. 4D, first panel). Surprisingly, LRRK2 also increased the nuclear levels of SIAH-1 (Fig. 4D, second and third panels), indicating that a LRRK2 and SIAH complex may facilitate the retention of both proteins in the nucleus. In vitro phosphorylation experiments showed negligible phosphorylation of SIAH-1 by LRRK2 (Supplementary Material, Fig. S6), suggesting that it does not represent a substrate for LRRK2.
Knockdown of endogenous SIAH-1 decreased the levels of endogenous LRRK2 in the nuclear fraction (Fig. 4E). Immunocytochemistry experiments confirmed the LRRK2 and SIAH-1 co-localization in purified nuclei (Fig. 4F), as better observed in z-series reconstituted into 3D projections at different angles (Fig. 4F, lower panels). SIAH-1 knockdown also decreased the percent of cells with nuclear LRRK2 by 50% in HEK293 cells (Fig. 4G) and in primary neurons (Fig. 4H). Strikingly, knockdown of SIAH prevented the toxicity of the LRRK2 G2019S mutant, as inferred from neurite length analysis (Fig. 4I). Therefore, nuclear accumulation of LRRK2 G2019S may be harmful to neurons.
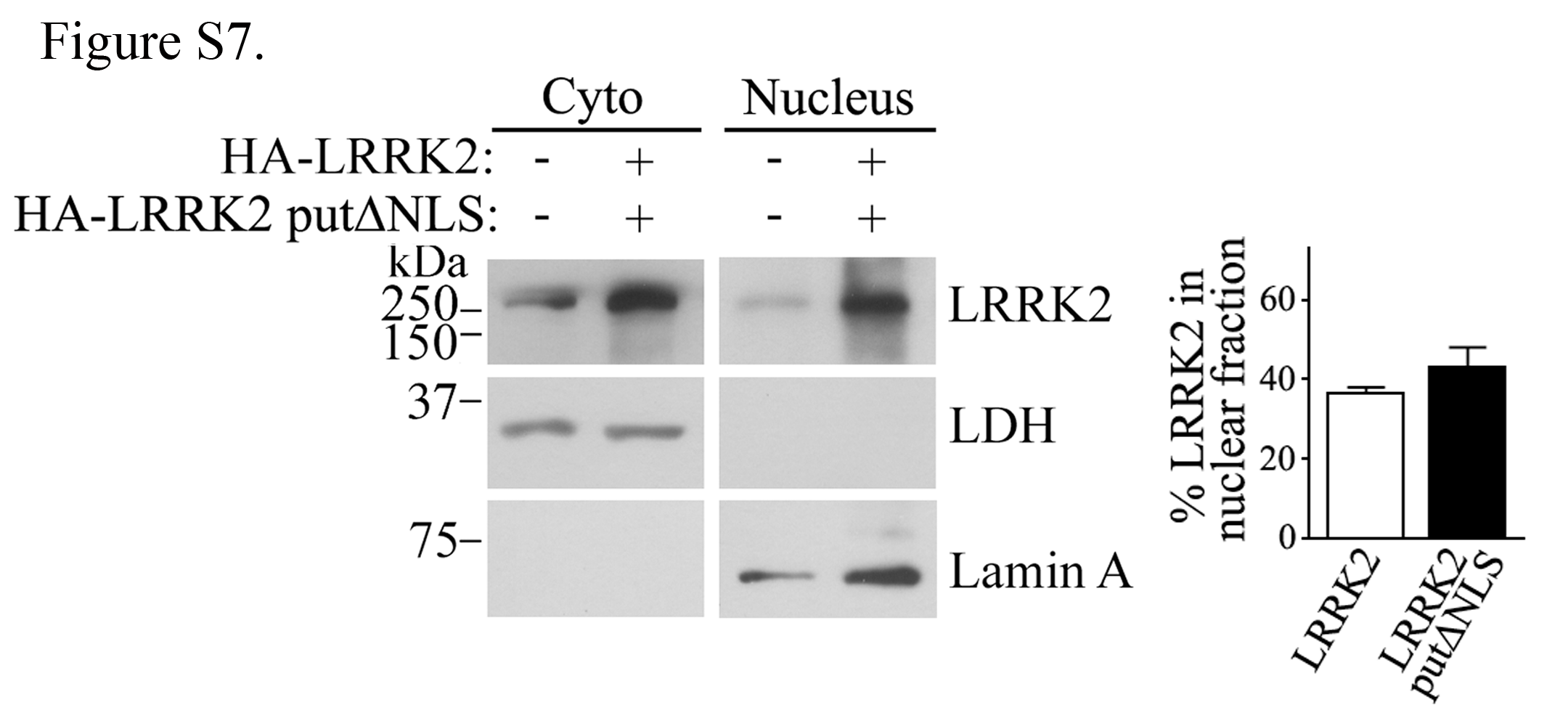
LRRK2 contains three putative monopartite NLS sites (amino acids 946–955; 1380–1389; 1634–1642) (30). We generated a LRRK2 construct where the lysines of the putative monopartite NLS sites were mutated to uncharged glutamine residues (K947,949,951Q; K1383,1385Q; K1637,1638,1640Q, which we called putΔNLS). We carried out nuclear fractionation experiments with LRRK2 putΔNLS but found no difference in its translocation to the nucleus in contrast to the wild-type protein (Supplementary Material, Fig. S7), suggesting that the predicted NLS sites are not functional.
Roles of nuclear LRRK2
Transfection of the LRRK2 G2019S disease mutant but not wild-type LRRK2 promoted disorganization and disruption of the lamin A/C network (Fig. 5A and D). The effect did not depend on LRRK2 kinase activity, as it was not rescued by KD LRRK2 (Fig. 5B) or the LRRK2 kinase inhibitors LRRK2-IN-1 and MLi-2 (27,31) (Fig. 5C). Additional LRRK2 disease mutants also led to similar disruption of the lamin A/C network (Fig. 5B).
Figure 5.
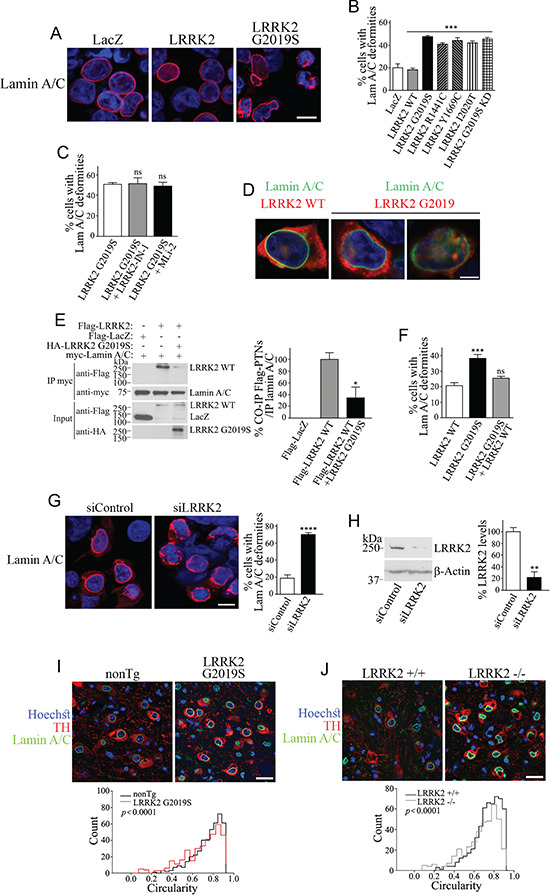
LRRK2 mutations or LRRK2 knockdown/deletion destabilize lamin A/C network in vitro and transgenic mice models. (A) HEK293 cells were transfected with Flag-LRRK2, Flag-LRRK2 G2019S or Flag-LacZ. Cells were processed for immunocytochemistry with anti-lamin A/C and Hoechst 33342 and analyzed by confocal microscopy. Scale bar, 10 μm. (B) HEK293 cells were transfected with Flag-LRRK2, Flag-LRRK2 disease mutants (G2019S, R1441C, Y1669C, I2020T), LRRK2 G2019S kinase-dead (KD) or Flag-LacZ. Cells were processed and analyzed as in (A). The graph represents the percentage of cells displaying lamin A/C deformities. Values represent the mean ± S.E.M. of six independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test comparing mutants to LRRK2 wild-type control (P < 0.001). (C) HEK293 cells were transfected with Flag-LRRK2 G2019S mutant and treated without or with LRRK2 inhibitors (1 μm LRRK2-IN-1 or 30 nM MLi-2) for 16 h. The graph represents the percentage of cells exhibiting lamin A/C deformities. Values represent the mean ± S.E.M. of three to six independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test comparing mutants to LRRK2 G2019S without LRRK2 inhibitors (P > 0.999). Unpaired two-tailed Student’s t test (non-significant P = 0.821). (D) HEK293 cells were transfected with Flag-LRRK2 or Flag-LRRK2 G2019S. Cells were processed for immunocytochemistry with anti-LRRK2, anti-lamin A/C and Hoechst 33342 and analyzed by confocal microscopy. Scale bar, 5 μm. (E) Co-expression of HA-LRRK2 G2019S inhibits the co-immunoprecipitation of Flag-LRRK2 wild type with myc-Lamin A/C. Co-immunoprecipitation of LRRK2 wild type with lamin A/C was detected with anti-Flag (first panel) while levels of lamin A/C immunoprecipitation were determined with anti-myc (second panel). The third panel shows similar expression of Flag-LRRK2 wild type in the absence and presence of HA-LRRK2 G2019S. The fourth panel shows the levels of LRRK2 G2019S with anti-HA. The graph depicts the relative co-immunoprecipitation of LRRK2 wild type with lamin A/C, in the absence and presence of LRRK2 G2019S. Values represent the mean ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test of LRRK2 WT co-immunoprecipitation in the presence of G2019S in contrast to that in the absence of G2019S (P = 0.038). (F) Percent of cells displaying lamin A/C deformities in HEK293 cells transfected with Flag-LRRK2 G2019S in the absence or presence of excess LRRK2 wild type. Values represent the mean ± S.E.M. of three independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test (P < 0.001 and = 0.344 for G2019S and G2019S plus WT, respectively, in contrast with WT alone). (G) HEK293 cells transfected with siRNA control or LRRK2 siRNA, processed and analyzed as in (A). Scale bar, 10 μm. The graph represents the percentage of cells with lamin A/C deformities. Values represent the mean ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P < 0.0001). (H) Control showing the extent of siRNA-mediated LRRK2 silencing on levels of endogenous LRRK2 in HEK293 cells. The graph depicts the quantification of endogenous LRRK2 in the presence of different siRNAs. Values represent the mean ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.003). (I) Substantia nigra from non-transgenic and G2019S overexpressing mice (12 months) was processed for immunohistochemistry with anti-lamin A/C, anti-tyrosine hydroxylase (TH) and Hoechst 33258 and analyzed by confocal microscopy. Scale bar, 25 μm. The histogram represents the distribution of lamin A/C circularity values in non-transgenic and LRRK2 G2019S Tg mice. Values represent the mean ± S.E.M. of three independent experiments with a total of 400 dopaminergic neurons analyzed per genotype. Mann–Whitney test (P < 0.0001). (J) Substantia nigra from non-transgenic and LRRK2 knockout mice (LRRK2 −/−) (12 months) were processed and analyzed as in (H). Scale bar, 25 μm. The histogram represents the distribution of lamin A/C circularity values in LRRK2 +/+ and LRRK2 −/− mice. Values represent the mean ± S.E.M. of three independent experiments with a total of 400 dopaminergic neurons analyzed per genotype. Mann–Whitney test (P < 0.0001).
Since the LRRK2 G2019S mutant had a much lower interaction with lamin A/C (Fig. 3A, B and D), we wondered if this mutation leads to disruption of lamin A/C by preventing the binding of the non-mutated LRRK2 to lamin A/C. In agreement, we found that LRRK2 G2019S decreases the amount of the wild-type LRRK2 that co-immunoprecipitates with lamin A/C despite their identical levels of LRRK2 (Fig. 5E). Overexpression of wild-type LRRK2 rescued the nuclear lamina deformities caused by LRRK2 G2019S (Fig. 5F), indicating that excess LRRK2 can overcome the deleterious effects of LRRK2 G2019S.
Similar to the LRRK2 G2019S mutant, siRNA-mediated LRRK2 knockdown caused lamin A/C network disruption (Fig. 5G and H), suggesting that the nuclear lamina integrity requires the interaction of LRRK2 with lamin A/C.
To verify in vivo effects of LRRK2, we analyzed the circularity of the nuclear membrane in dopaminergic neurons of the substantia nigra of LRRK2 G2019S transgenic mice (32). We found a decrease in circularity with a distribution toward lower values in the LRRK2 G2019S transgenic mice (Fig. 5I). A similar reduction in circularity was seen in dopaminergic neurons of the substantia nigra of LRRK2 −/− mice (33) (Fig. 5J). The similarities between LRRK2 G2019S and LRRK2 −/− mice suggest a normal role of LRRK2 in stabilizing the nuclear lamina in vivo and are compatible with a loss of function of the LRRK2 G2019S in this respect.
Next, we investigated the effect of LRRK2 in functional studies monitoring the nuclear membrane integrity by the distribution of the nuclear marker pdsRed-NLS. We found that pdsRed-NLS is retained in the nucleus when LacZ or LRRK2 wild type is expressed in cells (Fig. 6A–C). Expression of LRRK2 G2019S promoted a significant leakage of pdsRed-NLS from the nucleus to the cytosol, suggesting an increase in nuclear membrane permeability (Fig. 6A–C). This effect was not prevented by inhibition of LRRK2 kinase activity (Fig. 6B). An increase in the leakage of pdsRed-NLS to the cytosol was also observed with all tested LRRK2 disease mutants (Fig. 6B), suggesting they all disrupt the nuclear membrane. Similar to LRRK2 mutants, siRNA-mediated knockdown of LRRK2 increased the leakage of pdsRed-NLS to the cytosol (Fig. 6D), indicating that nuclear membrane leakage results from loss of normal LRRK2 activity.
Figure 6.
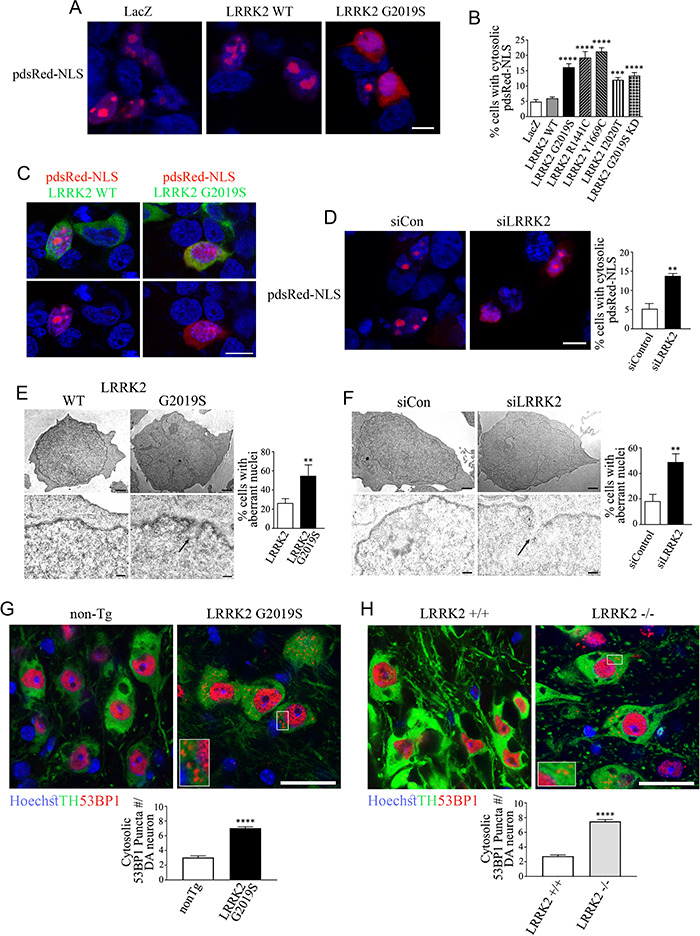
LRRK2 disease mutants or LRRK2 knockdown/deletion disrupt nuclear membrane and alter its permeability in vitro and in vivo. (A) HEK293 cells transfected with Flag-LacZ, Flag-LRRK2 or Flag-LRRK2 G2019S. Cells were processed for immunocytochemistry, and pdsRed-NLS intracellular distribution was analyzed by confocal microscopy. Scale bar, 10 μm. (B) HEK293 cells were transfected with Flag-LRRK2, Flag-LRRK2 disease mutants (G2019S, R1441C, Y1669C, I2020T), LRRK2 G2019S KD or Flag-LacZ. Cells were processed and analyzed as in (A). The graph represents the percentage of cells with cytosolic pdsRed-NLS. Values represent the mean ± S.E.M. of six independent experiments. Repeated-measures one-way ANOVA with Bonferroni post hoc test comparing mutants to LRRK2 wild-type control (P < 0.0001 for LRRK2 G2019S, R1441C, Y1669C and G2019S KD and P < 0.001 for I2020T). (C) HEK293 cells transfected with Flag-LRRK2 or Flag-LRRK2 G2019S. Cells were processed for immunocytochemistry with anti-LRRK2, and pdsRed-NLS intracellular distribution was analyzed by confocal microscopy. Upper panels show a merged image while lower panels show the same image but depicted with pdsRed-NLS and Hoechst 33342 only. Scale bar, 10 μm. (D) siRNA to LRRK2 increase the nuclear membrane permeability to pdsRed-NLS. Scale bar, 10 μm. The graph represents the percentage of cells with pdsRed-NLS present in the cytosol. Values represent the mean ± S.E.M. of three independent experiments. Unpaired two-tailed Student’s t test (P = 0.002). (E) Transmission electron microscopy of HEK293 cells transfected with Flag-LRRK2 or Flag-LRRK2 G2019S. Scale bar, 1 μm (upper panels) and 200 nm (lower panels). The arrow in the lower right panel depicts a site with rupture of the nuclear membrane. The graph represents the percentage of cells exhibiting aberrant nuclei. Values represent the mean ± S.E.M. of three independent experiments with at least 30 cells analyzed per condition. Unpaired two-tailed Student’s t test (P = 0.004). (F) Transmission electron microscopy of HEK293 cells transfected with siRNA control or LRRK2 siRNA and processed and analyzed as in (D). Scale bar, 1 μm (upper panels) and 200 nm (lower panels). The arrow in the lower right panel depicts a rupture of the nuclear membrane. The graph represents the percentage of cells exhibiting aberrant nuclei. Values represent the mean ± S.E.M. of three independent experiments with at least 30 cells analyzed per condition. Unpaired two-tailed Student’s t test (P = 0.008). (G) Increase in cytosolic 53BP1 occurrence in dopaminergic neurons of SN of LRRK2 G2019S transgenic mice (12 months). Tissues were processed for immunohistochemistry with anti-53BP1, anti-TH and Hoechst 33258 and analyzed by confocal microscopy. Inset depicts enlarged boxed area: scale bar, 25 μm. The graph represents the 53BP1 puncta number present in the cytosol per SN dopaminergic (DA) neurons. Values represent the mean ± S.E.M. of three independent experiments with a total of 400 dopaminergic neurons analyzed per genotype. Mann–Whitney test (P < 0.0001). (H) Increase in cytosolic 53BP1 in the SN of LRRK2 −/− mice. Mice (12 months) were processed and analyzed as in (F). Inset depicts enlarged boxed area: scale bar, 25 μm. The graph represents the 53BP1 puncta number present in the cytosol per SN DA neurons. Values represent the mean ± S.E.M. of three independent experiments with a total of 400 dopaminergic neurons analyzed per genotype. Mann–Whitney test (P < 0.0001).
We next investigated the alterations on nuclear membrane integrity by transmission electron microscopy. We found that the expression of LRRK2 G2019S or LRRK2 knockdown in HEK293 cells caused similar lobulation of nuclei and elicited nuclear membrane discontinuity compatible with small local disruptions on the nuclear membrane integrity (Fig. 6E and F).
To evaluate if nuclear membrane permeability is also altered in vivo, we analyzed the distribution of the nuclear protein 53BP1 in dopaminergic neurons of the substantia nigra. We found higher levels of 53BP1 in the cytosol of dopaminergic neurons of both LRRK2 G2019S transgenic and LRRK2 −/− mice in contrast to control non-transgenic mice (Fig. 6G and H), confirming the data in cultured cells.
We next investigated possible changes in the organization of nuclear lamina in the substantia nigra and frontal cortex of PD patients. We found evidence of abnormal nuclear lamina characterized by excess folding and localized thickening of the nuclear lamina in more than 70% of dopaminergic neurons in the substantia nigra of idiopathic PD patients (Fig. 7A and B). In addition, approximately 88% of nigral neurons show lamin A/C alteration in one PD patient with LRRK2 G2019S mutation (Fig. 7A). Moreover, more than 60% of cortical neurons of both idiopathic and LRRK2 G2019S PD have abnormalities in their nuclear lamina (Fig. 7C and D).
Figure 7.
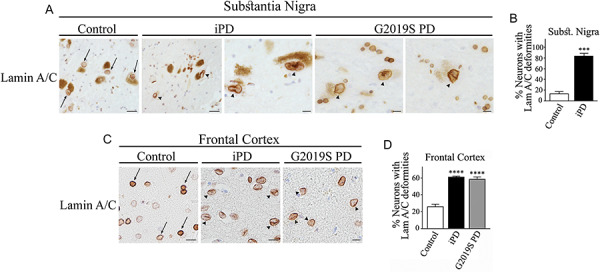
Abnormalities in the nuclear lamina structure in dopaminergic neurons of PD patients. (A) Substantia nigra from control, idiopathic PD and LRRK2 G2019S PD patients were processed for immunohistochemistry with anti-lamin A/C. Scale bar, 20 μm (first and second panels) and 10 μm (third to fifth panels). Arrows indicate the nuclear lamina of neuromelanin-containing neurons. Misshapen nuclear lamina of neuromelanin-containing neurons is indicated with arrowheads. (B) The graph represents the percent of dopaminergic neurons with abnormal lamina structure characterized by accentuated folds and localized thickness. Values represent the mean ± S.E.M. Substantia nigra of three control and three PD patients were analyzed, and 18–40 dopaminergic neurons were scored for every patient. Unpaired two-tailed Student’s t test (P < 0.001). (C) Frontal cortex from control, idiopathic PD and LRRK2 G2019S PD patients were processed for immunohistochemistry with anti-lamin A/C. Scale bar, 20 μm. Arrows indicate the nuclear lamina of neuromelanin-containing neurons. Misshapen nuclear lamina of neuromelanin-containing neurons is indicated with arrowheads. (D) The graph represents the percent of cortical neurons with abnormal lamina. Values represent the mean ± S.E.M. Cortici of four control, four idiopathic PD and three LRRK2 G2019S PD patients were analyzed, and an average of 90–105 cortical neurons were scored from each patient group. Repeated-measures one-way ANOVA with Bonferroni post hoc test (P < 0.0001).
Altogether, our data suggest that LRRK2 helps maintain the nuclear lamina and the nuclear membrane integrity and that the disease mutations interfere with this ability. Moreover, dopaminergic neurons of PD patients have widespread alterations of the lamina structure, supporting the notion that nuclear envelope disruption is a hallmark of the disease.
Discussion
Our study indicates that LRRK2 translocates to the nucleus and interacts with lamin A/C, independent of its kinase activity. LRRK2 knockdown promotes the disorganization of the lamin A/C network and the disruption of nuclear membrane integrity with leakage of nuclear marker to the cytosol. We also found decreased nuclear lamina circularity and increased release of 53BP1 to the cytosol in dopaminergic neurons of the substantia nigra in LRRK2 −/− mice. These data indicate that LRRK2 is essential to stabilizing the nuclear lamina and nuclear membrane in vivo.
LRRK2 disease mutants interact significantly less with lamin A/C, and this was observed in binding experiments using recombinant proteins, PD G2019S brain extracts and co-immunoprecipitation analyses. However, similar to LRRK2 −/− mice, LRRK2 G2019S expression leads to disruption of the lamin A/C network and nuclear membrane integrity with leakage of nuclear proteins to the cytosol. Evidence of altered nuclear permeability was observed in transfected cells, primary neurons and substantia nigra of LRRK2 G2019S transgenic mice. Our data is compatible with a loss of function with the LRRK2 G2019S mutation regarding LRRK2-mediated maintenance of nuclear stability. Our data may also be relevant for idiopathic PD as we observed substantial nuclear lamina abnormalities, characterized by increased folds and localized thickness in dopaminergic nigral and cortical neurons of both LRRK2 G2019S PD and idiopathic PD. Still, we did not observe significant exacerbation of the nuclear lamina abnormalities in LRRK2 G2019S carriers in contrast to idiopathic PD. This could be because these postmortem tissues were analyzed at late stages of the disease. Future studies using brains from patients at earlier stages are necessary to investigate such differences.
Studies analyzing a LRRK2 structural model, the crystal structure of its WD40 domain and low-resolution cryo-EM indicate that LRRK2 occurs as a dimer (34–36). LRRK2 WT/G2019S heterodimers behave differently than WT/WT or G2019/G2019S homodimers (37). In transfected cells, expression of LRRK2 wild type in excess of LRRK2 G2019S restores the normal function of the nuclear envelope, which is compatible to the notion that LRRK2 WT/WT homodimers are more efficient than WT/G2019S in maintaining the nuclear structure. Therefore, it is conceivable that LRRK2 G2019S disrupts normal LRRK2 function at the nuclear envelope by making heterodimers in a dominant negative-like effect.
The interaction of SIAH-1 with LRRK2 LRR and kinase domain fits with the model of the LRRK2 quaternary structure where the two domains are positioned in close proximity (34), further supporting their ability to interact. In addition to ubiquitinating and promoting partial LRRK2 degradation, SIAH-1 facilitates the translocation of LRRK2 to the nucleus. SIAH-1 and LRRK2 translocate to the nucleus as a complex, but phosphorylation of SIAH-1 by LRRK2 is negligible, suggesting that the mechanism involved in their translocation to the nucleus is not dependent on phosphorylation by LRRK2. On the other hand, translocation of LRRK2 was dependent on the presence of SIAH-1 NLS regions. In line with these findings, SIAH-1 promotes the translocation of GAPDH to the nucleus upon nitrosative stress (28). Activation of the SIAH/GAPDH pathway was also observed under non-apoptotic stimuli, such as the addition of the growth factors BDNF and NGF (38), and in proliferation and migration of liver cells (39).
Nuclear fractionation experiments with LRRK2 mutated at putative NLS sites failed to validate functional monopartite NLS sites in LRRK2. Although future mutagenesis studies may identify functional bipartite or non-convetional NLS sites (30), the lack of functional monopartite NLS sites within LRRK2 are in line with the role of SIAH-1 in promoting LRRK2 translocation to the nucleus. We also identified that both the middle and C-terminal regions of LRRK2 translocate to the nucleus, suggesting that multiple regions may be involved in LRRK2 translocation, including regions different from the regulatory N-terminus of the protein (40,41).
Lamin C is a shorter isoform of lamin A that is widely expressed in neurons (42). Expression of lamin A in the brain is suppressed by miRNA-9 (43), indicating that lamin C is the main interacting partner for LRRK2 in neurons. We found that LRRK2 co-immunoprecipitates with lamin B1 to a much lower extent, supporting the preferential interaction of LRRK2 with lamin A/C. While lamin B1 plays essential roles in neurodevelopment and survival of neurons (42), no mutations of lamin B1 were associated with neurological diseases to date. On the other hand, a point mutation in lamin A/C (R298C) causes peripheral axonal degeneration in Charcot–Marie–Tooth disease (44) and the point mutation E31N causes muscular dystrophy associated with cognitive impairment (45). Lamin A/C is disrupted by expanded CGG-repeat FMR1 and is present in FMR1 neuronal inclusions in fragile X-associated tremor/ataxia syndrome, supporting a role in neurodegeneration mechanisms (46,47). In this framework, our findings that LRRK2 interacts more prominently with lamin A/C than with lamin B1 suggest that disruption of the lamin A/C network by LRRK2 disease mutations may be instrumental for disruption of the nuclear lamina of neurons in PD.
Aging is a risk factor for PD (48) and is associated with lamin A/C-dependent nuclear defects, including ruffling of the nuclear lamina (49). Expression of progerin, a short lamin A transcript associated with the Hutchinson–Gilford progeria syndrome, accelerates disease-related phenotypes in inducible pluripotent cells derived from PD patients, including dendrite degeneration, mitochondrial swelling and inclusion bodies (50). Human neural stem cells harboring LRRK2 G2019S mutation display passage-dependent disorganization of nuclear lamina ascribed to phosphorylation of lamin B1 by the mutant LRRK2 (25). Hippocampal tissue from PD brains with G2019S mutation and LRRK2 R1441C transgenic mice exhibit abnormalities of nuclear envelope as well (25,26). However, none of the studies previously mentioned considered a normal role of LRRK2 along with lamin A/C in the nucleus. Our data indicate that nuclear abnormalities with LRRK2 mutants are likely based on LRRK2 loss of function.
Some studies demonstrated partial loss-of-function mechanisms associated with some aspects of LRRK2 toxicity. Targeted deletion of both LRRK2 and LRRK1 in mice impairs the autophagy-lysosomal pathway and causes degeneration of dopaminergic neurons (23). Also, knockout of the Drosophila LRRK2 orthologue causes severe reduction of locomotor activity and neurodegeneration (51), while the knockout of the C. elegans LRRK2 orthologue increases the vulnerability to mitochondrial dysfunction (52). Further supporting loss-of-function mechanisms, the LRRK2 R1441C mutation decreases its interaction with Sec16A (22), while the LRRK2 G2385R mutant interacts less strongly with the 14–3-3 protein and synaptic vesicles (53–55).
Although increased kinase activity of LRRK2 disease mutants has been strongly linked to their toxicity (15,56), additional mechanisms were suggested to contribute to the toxicity of LRRK2 disease mutants. For instance, we and others have shown that unconventional ubiquitination may contribute to LRRK2 toxicity (20,21). Since inhibiting LRRK2 kinase activity did not prevent lamina disorganization or nuclear membrane leakage by the LRRK2 G2019S mutant, we now raise the possibility that part of LRRK2 disease mutation toxicity could be mediated by a kinase-independent manner. Therefore, we hypothesize that LRRK2 mutations represent a gain of function regarding kinase activity but that in the context of the nucleus, LRRK2 mutations may act as a dominant negative by preventing wild-type LRRK2 to interact with lamin A/C.
Compatible with a nuclear role in PD, α-synuclein was shown to translocate to the nucleus and to promote severe transcriptional deregulation (57). Unlike α-synuclein, we found no evidence for LRRK2 interaction with chromatin in subnuclear fractionation experiments. Nevertheless, we cannot discard the possibility that LRRK2 G2019S or some stressors may prompt LRRK2 to interact with chromatin. As some LRRK2 G2019S transgenic mouse models were shown to accelerate α-synuclein pathology (58–60), it is possible that nuclear lamina disruption may contribute to further transcriptional deregulation by α-synuclein. Electron microscopy studies of idiopathic PD and LRRK2 G2019S PD brains at different stages of disease will be of great value to correlate the damage of nuclear lamina and α-synuclein pathology in patients.
In summary, our study has uncovered a new role of LRRK2 in interacting with SIAH proteins and lamin A/C to maintain nuclear envelope integrity. We raise the possibility that nuclear envelope disorganization by LRRK2 disease mutations is at least in part a consequence of loss of function that may synergize with the neurotoxic mechanisms caused by the activation of LRRK2 kinase activity.
Materials and Methods
Cell culture and transfections
HEK293 cells were grown in DMEM containing 10% fetal bovine serum in a 5% CO2 atmosphere. HEK293 cells were transiently transfected with N-terminal-tagged pRK5 plasmids utilizing Lipofectamine 2000 (Invitrogen). LRRK2 with mutations K1906A and D1994N was used as KD (16).
For experiments using small-interference RNAs, cells were transfected with Lipofectamine 2000 as previously described (61) using siSIAH-1 (5′-CGCCCAUUCUUCAAUGUCA-3′), LRRK2 (5′- GCUGUGCCUUAUAACCGAA-3′) or scrambled siRNA control (Ambion).
Primary neuronal cultures
E18 primary cortical cultures were prepared from Sprague-Dawley rats according to the protocol approved by the committee for animal experimentation at the Technion-Israel Institute of Technology. Briefly, after decapitation, the cortices were retrieved and maintained in Hank’s balanced salt solution. Then, the samples were digested by incubation for 10 min with trypsin (Beit-Haemek Biological Industries) at 37°C. This was followed by physical trituration with Pasteur pipettes, and the samples were briefly centrifuged for 10 s at 1000g to remove debris. The supernatant was filtered through a 0.7 μm filter (BD Biosciences) followed by centrifugation at 900g for 4 min. The cell pellet was resuspended in a Neurobasal medium supplemented with B27 (Invitrogen) and 0.5 mm L-glutamine (Beit-Haemek Biological Industries). Neurons were plated in 12- or 6-well plates covered with poly-D-Lysine (Sigma) at a density of 0.45 and 2 × 106 cells per well, respectively. Transfections were carried out at DIV 4 using the calcium phosphate method, as previously described (62).
Antibodies and western blot analysis
Samples were homogenized as previously described (29). Blots were probed with the antibodies mouse anti-HA (901501, BioLegend); mouse anti-actin (SKU 08691001, MP Biomedicals); rabbit anti-myc (sc-789), rabbit anti-HA (sc-805), mouse anti-LDH (sc-133123), mouse anti-GAPDH (sc-47724), mouse anti-GST (sc-138), mouse anti-ubiquitin (sc-8017), rabbit anti-lamin A/C (sc-20681), mouse anti-lamin A/C (sc-376248), goat anti-lamin B1 (sc-6216) and rabbit anti-histone 2b (sc-10808) (Santa Cruz); mouse anti-myc (M4439), mouse anti-Flag (F1804) and rabbit anti-Flag (F7425) (Sigma); rabbit anti-ubiquitin (Z0458, Dako); rabbit anti-histone 3 (ab1791), goat anti-SIAH-1 (ab2237), rabbit anti-53BP1 (ab175933), rabbit anti-LRRK2 (ab133474) and rabbit anti-Phospho-LRRK2 S1292 (ab203181) (Abcam); and mouse anti-LRRK2 (N241A/34, NeuroMab). More specifically, for endogenous brain and HEK293 lysates, anti-LRRK2 antibody (1:100) was incubated overnight at 4°C in 5% non-fat dried milk and 0.1% Tween 20. Quantifications of enhanced chemiluminescence reactions were carried out according to ImageMaster analysis.
Co-immunoprecipitation assays
For the co-immunoprecipitations, transfected cells were lysed in buffer containing 50 mm Tris (pH 7.4), 140 mm NaCl, 1% Triton X-100, 0.1% SDS, 30 μm MG132, 20 mm NaF, 2 mm Na3VO4, 10 mm PPi, 20 mm β-glycerol phosphate and protease inhibitor cocktail (MiniComplete, Roche). Cell extracts were clarified by centrifugation and incubated for 4 h with anti-HA (E6779), anti-myc (A7470) or anti-Flag (F2426) coupled to protein G beads (Sigma) (29). Immunoprecipitates were washed with lysis buffer containing 500 mm NaCl and detected by western blot.
For endogenous co-immunoprecipitation assays, rat brains were homogenized in lysis buffer as previously mentioned. Brain homogenates were clarified by centrifugation at 13000g for 5 min. The antibody to SIAH-1 or lamin A/C was coupled to protein G beads (29) and incubated for 4 h with brain homogenate (2 mg/ml). Immunoprecipitates were washed with lysis buffer and detected by western blot using the anti-LRRK2 antibody.
In vitro ubiquitination assays
LRRK2 was translated using TnT wheat germ in vitro translation kit (Promega) in the presence of [35S] methionine (Amersham). In vitro translated proteins were incubated in a reaction medium containing 40 mm Tris (pH 7.6), 5 mm MgCl2, 2 mm DTT, 1 mm ATP, 10 mm phosphocreatine, 0.1 mg/ml creatine kinase, 7.5 μg ubiquitin, 1 μg ubiquitin aldehyde, 100 ng E1, 200 ng UbcH5b, in the absence and presence of 500 ng of GST-SIAH-1 or 500 ng of GST-SIAH-2. Reactions were incubated at 37°C for 1 h, loaded on a SDS-PAGE and then transferred to nitrocellulose membranes. 35S-labeled-LRRK2 was determined by PhosphoImager analysis.
In some experiments, LRRK2 was immunoprecipitated from transfected cells and incubated with components of the in vitro ubiquitination previously described, in the absence or in the presence of 500 ng GST-SIAH-1. Reactions were incubated at 37°C for 1 h, loaded on an SDS-PAGE and then transferred to nitrocellulose membranes.
In vivo ubiquitination assays
Transfected cells were processed as previously described (63). Briefly, cells were directly lysed in buffer containing 50 mm Tris (pH 7.4), 140 mm NaCl and 1% SDS and boiled at 100°C for 5 min. Next, the lysates were sonicated and 10 times diluted with buffer containing 50 mm Tris (pH 7.4), 140 mm NaCl, 1% Triton X-100, 30 μm MG132 and protease inhibitor cocktail (Mini Complete, Roche). The samples were centrifuged at 13000g for 5 min, and the supernatants were incubated with anti-Flag antibody coupled to beads (Sigma) for 4 h at 4°C. After incubation, beads were extensively washed with buffer containing 50 mm Tris (pH 7.4), 500 mm NaCl, 1% Triton X-100 and 0.1% SDS. Immunoprecipitates were loaded on an SDS-PAGE gel for western blot analysis.
In vitro phosphorylation assays
Purified recombinant LRRK2 (Thermo Scientific) (100 ng) and GST-SIAH-1 (200 ng) were incubated in the presence of 40 mm Tris (pH 7.6), 2 mm DTT, 5 mm MgCl2, 0.1 mm ATP and 2.5 μCi [32P] ATP per reaction. After 1 h of incubation at 37°C, reactions were stopped with SDS sample buffer and analyzed by SDS-PAGE. The amounts of 32P-labeled SIAH-1 and 32P-labeled LRRK2 were determined by PhosphorImager analysis. The total amount of SIAH-1 and LRRK2 was determined by western blot using anti-SIAH-1 and anti-LRRK2 antibodies.
Nuclear preparations
Nuclear fractions were carried out as described previously (64,65). Briefly, cells were homogenized with a glass homogenizer in buffer containing 0.25 M sucrose, 20 mm HEPES (pH 7.4), 140 mm NaCl, 3 mm EDTA, 20 mm NEM, 30 μm MG132, 20 mm NaF, 2 mm Na3VO4, 10 mm PPi, 20 mm β-glycerol phosphate and protease inhibitor cocktail (Mini Complete, Roche). Homogenates were centrifuged for 10 min at 3000g, and supernatant (S1) and nuclear pellet (P1) were collected. The P1 fraction was washed in fresh buffer and centrifuged for 10 min at 3000g to remove contaminating cytosolic proteins and denoted WP1. S1 was centrifuged for 30 min at 200000g. The supernatant consisting of soluble cytosolic fraction was collected. WP1 pellet was then resuspended in buffer containing 1.32 M sucrose, 1 mm KP buffer (0.1 M KH2PO4 and 0.1 M K2HPO4), 0.25% Triton, 1 mm MgCl2, 30 μm MG132, 20 mm NaF, 2 mm Na3VO4, 10 mm PPi, 20 mm β-glycerol phosphate and protease inhibitor cocktail (Mini Complete, Roche) and was poured over with 1.7 M sucrose solution. Purified nuclear pellet was obtained after a 75-min centrifugation at 53000g.
Purification of sub-nuclear fractions was done as described (64) by disrupting purified nuclei with three rounds of freeze and thaw, followed by a 13 000g centrifugation for 10 min, generating a supernatant (nucleoplasm) and pellet (insoluble nuclear) fractions. The insoluble fraction was further separated into chromatin-bound and cytoskeletal fractions by incubating with 20 000 units micrococcal nuclease (New England Biolabs) for 30 min at 37°C and spinning at 13000g for 5 min. The supernatant consisted of chromatin fraction and the pellet of cytoskeletal fraction.
Lamin A/C-binding experiments
GST-lamin A/C constructs were expressed in E. coli BL21 bacteria and purified as described before (29). GST-lamin A/C proteins attached to glutathione beads were incubated with brain or cell lysates (2 mg/ml) in buffer containing 50 mm Tris–HCl (pH 7.4), 140 mm NaCl, 1% Triton X-100, 30 μm MG132, 20 mm NaF, 2 mm Na3VO4, 10 mm PPi, 20 mm β-glycerol phosphate and protease inhibitor cocktail (MiniComplete, Roche). Binding was carried out at 4°C for 30 min and then washed in lysis buffer containing 500 mm NaCl and analyzed by western blots. Binding of purified recombinant LRRK2 (Thermo Scientific) with GST-lamin A/C was determined by incubating 2 ng of LRRK2 with GST-lamin A/C or control GST in the same conditions as previously mentioned.
For the binding experiments where LRRK2 was previously phosphorylated, purified LRRK2 was preincubated in a medium containing 50 mm Tris–HCl (pH 7.4), 2 mm DTT, 5 mm MgCl2, 0.1 mm ATP and 2 mg/ml BSA, in the absence or in the presence of 2.5 μm LRRK2-IN-1, for 1 h at 37°C. Reactions were then incubated with GST-lamin A/C and subjected to binding assays as previously mentioned.
Immunocytochemistry
Transfected cells and neurons were fixed with 4% paraformaldehyde for 15 min and blocked in PBS containing 0.2% Triton X-100 and 5% normal goat serum. Cells were labeled with primary antibodies as previously described (29). Immunolabeling was detected using FITC- and Cy3-labeled secondary antibodies (Jackson Laboratories).
For the nuclear localization and lamin A/C integrity experiments, samples were examined under a Zeiss LSM 700 confocal microscope and optical sections were obtained under a ×63 immersion objective at a definition of 1024 × 1024 pixels with the pinhole diameter adjusted to 1 μm. Sections were acquired under the same laser parameters and image magnification.
To determine the percent of HEK293 cells containing LRRK2 in the nucleus, cells were counted for the co-localization of LRRK2 and the nuclear marker Hoechst 33342 in their best confocal plane. ZEN software (Zeiss) was used to determine the Manders’s co-localization coefficient (66). Cells were counted as positive for LRRK2 and Hoechst 33342 when the Manders’ coefficient of selected areas within the nucleus was at least 0.4. Approximately 100 cells were examined for each condition in every experiment.
Distribution of LRRK2 in the nucleus of neurons was analyzed 72 h after neuronal transfection and visualized by 3D reconstitution from confocal Z-series using Imaris Image Analysis Software 9.0.1. The Hoechst 33342 surface was generated in Imaris to highlight the specific LRRK2 signal throughout the nucleus.
To determine the integrity of lamin A/C in cells, three adjacent confocal planes were analyzed. Approximately 100 cells were examined for each condition in every independent experiment, and data are representative of at least three independent experiments.
LRRK2 neuronal toxicity
Primary neurons were transfected at DIV 4 with LRRK2, and 72 h after transfection, the cells were fixed with 4% paraformaldehyde and processed for immunocytochemistry, as previously described. Low magnification pictures were taken in an upright fluorescent microscope (Zeiss Axio Imager.Z2 upright) and length of processes determined according to ImageJ/FIJI. Approximately 100 cells were examined for each condition in every independent experiment, and data are representative of at least three independent experiments.
Animals
The use of the animals followed the National Institutes of Health guidelines and was approved by the Institutional Animal Care and Use Committee at Thomas Jefferson University. All efforts were made to minimize the number of animals used. LRRK2-G2019S transgenic (Tg) mice (stock #012467, The Jackson Laboratory) were originally obtained from Dr Zhenyu Yue at Icahn School of Medicine at Mount Sinai and maintained in Dr Zhang’s laboratory. LRRK2 −/− mice (33) were a kind gift from Dr Jie Shen (Harvard Medical School) and maintained in Dr Zhang’s laboratory. Three pairs of mice at the age of 9–14 months of each strain were used in the study.
Immunohistochemistry
Twelve-month-old LRRK2 G2019S transgenic mice, LRRK2 −/− mice and wild-type littermate controls were perfused with 4% paraformaldehyde, and 30 μm coronal sections of midbrain were obtained with a vibratome. The sections were blocked in 10% normal goat serum in PBS for 1 h at RT. Anti-53BP1 (ab175933, Abcam), anti-lamin A/C (sc-376 248, Santa Cruz) and anti-tyrosine hydroxylase (TH) (MAB5280, Millipore; P40101, Pel-Freez) antibodies were incubated at 4°C for 30 h, and sections were subsequently incubated with Alexa Fluor 488- and 594-conjugated secondary antibodies (Life Technologies). Sections were counterstained with the nuclear marker Hoechst 33342. Samples were examined under a Leica SP8 confocal microscope, and images were collected at the same laser power, gain and offset settings. Images were analyzed by Imaris software, and approximately 400 substantia nigra dopaminergic neurons were counted per genotype.
Human tissues
For immunohistochemistry, human midbrains were obtained from Queen Square Brain Bank (UCL, London). Paraffin-embedded sections (8 μm) of the midbrain from three idiopathic PD and three control cases were deparaffinized in xylene followed by graded rehydration. Endogenous peroxidase activity was blocked with methanol/0.3% H2O2. Sections were pretreated by pressure cooking in citrate buffer at pH 6.0 for 10 min, and non-specific protein binding was blocked in 10% normal goat serum in PBS for 30 min at room temperature. The primary antibody was incubated at 4°C for 16 h, immunostained by the avidin–biotin peroxidase complex method antibody (Santa Cruz). Sections were counterstained with Mayer’s hematoxylin.
For lamin A/C-binding experiments using PD brain homogenates, human corteci (control and PD G2019S) were obtained from Queen Square Brain Bank (UCL, London). Samples were homogenized in 10 volumes of buffer containing 50 mm Tris–HCl (pH 7.4), 140 mm NaCl, 1% Triton X-100, 30 μm MG132, 20 mm NaF, 2 mm Na3VO4, 10 mm PPi, 20 mm β-glycerol phosphate and protease inhibitor cocktail (MiniComplete, Roche). The binding was carried out at 4°C for 30 min and then washed in lysis buffer containing 500 mm NaCl and analyzed by western blots.
Electron microscopy
HEK293 cells were seeded in 100-mm plates and transfected with LRRK2 constructs or siRNAs (control and to LRRK2). After 36 h, cells were washed with preheated serum-free medium and fixed for 1 h at RT with 2% glutaraldehyde and 3% paraformaldehyde in 0.1 M sodium cacodylate buffer containing 5 mm CaCl2 and 3% sucrose. Cells were washed with sodium cacodylate buffer, scraped and embedded in agarose. Following post-fixation and staining with 1% osmium tetraoxide, 0.5% potassium hexacyanoferrate and 0.5% potassium dichromate in 0.1 M cacodylate buffer, the samples were en-block-stained with 1% uranyl acetate for 1 h at RT. Cells were then dehydrated in graded ethanol series, transferred to propylene oxide and embedded in Epon 812. Ultrathin sections (75 nm) were cut with an ultramicrotome UC7 (Leica), transferred to copper grids and viewed using Zeiss Ultra-Plus FEG-SEM equipped with a STEM detector at accelerating voltage of 30 kV.
Statistics
Statistical analysis was performed by repeated-measures one-way ANOVA with Bonferroni’s multiple comparison test, two-tailed Student’s t test or Mann–Whitney test using GraphPad Prism software version 6.03 (GraphPad Inc.). Differences were considered significant when P ≤ 0.05. All western blots shown in this study are representative of at least three independent experiments.
Funding
Reta Lila Weston Institute (to R.B.); National Institute of Neurological Disorders and Stroke (NINDS), United States (NS097530 to H.Z.); Israel Academy of Sciences; The Allen and Jewel Prince Center for Neurodegenerative Disorders of the Brain; Dears Foundation Research Fund; Hopkins-Technion Collaboration Fund; Technion Research funds (to S.E.)
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Edith Suss-Toby, Melia Gurewitz, Maya Holdengreber and Lior Kellerman for their help with microscopy and imaging analysis.
Conflict of Interest statement. None declared.
References
- 1. Engelender S. and Isacson O. (2017) The threshold theory for Parkinson’s disease. Trends Neurosci., 40, 4–14. [DOI] [PubMed] [Google Scholar]
- 2. Trinh J. and Farrer M. (2013) Advances in the genetics of Parkinson disease. Nat. Rev. Neurol., 9, 445–454. [DOI] [PubMed] [Google Scholar]
- 3. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R.J., Calne D.B. et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607. [DOI] [PubMed] [Google Scholar]
- 4. Paisan-Ruiz C., Jain S., Evans E.W., Gilks W.P., Simon J., Brug M., Lopez de Munain A., Aparicio S., Gil A.M., Khan N. et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595–600. [DOI] [PubMed] [Google Scholar]
- 5. Gilks W.P., Abou-Sleiman P.M., Gandhi S., Jain S., Singleton A., Lees A.J., Shaw K., Bhatia K.P., Bonifati V., Quinn N.P. et al. (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet, 365, 415–416. [DOI] [PubMed] [Google Scholar]
- 6. Di Fonzo A., Rohe C.F., Ferreira J., Chien H.F., Vacca L., Stocchi F., Guedes L., Fabrizio E., Manfredi M., Vanacore N. et al. (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet, 365, 412–415. [DOI] [PubMed] [Google Scholar]
- 7. Biskup S., Moore D.J., Celsi F., Higashi S., West A.B., Andrabi S.A., Kurkinen K., Yu S.W., Savitt J.M., Waldvogel H.J. et al. (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol., 60, 557–569. [DOI] [PubMed] [Google Scholar]
- 8. Schapansky J., Nardozzi J.D., Felizia F. and LaVoie M.J. (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum. Mol. Genet., 23, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higashi S., Moore D.J., Yamamoto R., Minegishi M., Sato K., Togo T., Katsuse O., Uchikado H., Furukawa Y., Hino H. et al. (2009) Abnormal localization of leucine-rich repeat kinase 2 to the endosomal-lysosomal compartment in lewy body disease. J. Neuropathol. Exp. Neurol., 68, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Papkovskaia T.D., Chau K.Y., Inesta-Vaquera F., Papkovsky D.B., Healy D.G., Nishio K., Staddon J., Duchen M.R., Hardy J., Schapira A.H. et al. (2012) G2019S leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet., 21, 4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh A., Zhi L. and Zhang H. (2019) LRRK2 and mitochondria: recent advances and current views. Brain Res., 1702, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steger M., Tonelli F., Ito G., Davies P., Trost M., Vetter M., Wachter S., Lorentzen E., Duddy G., Wilson S. et al. (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beilina A., Rudenko I.N., Kaganovich A., Civiero L., Chau H., Kalia S.K., Kalia L.V., Lobbestael E., Chia R., Ndukwe K. et al. (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. U. S. A., 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eguchi T., Kuwahara T., Sakurai M., Komori T., Fujimoto T., Ito G., Yoshimura S.I., Harada A., Fukuda M., Koike M. et al. (2018) LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. U. S. A., 115, E9115–E9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. West A.B., Moore D.J., Biskup S., Bugayenko A., Smith W.W., Ross C.A., Dawson V.L. and Dawson T.M. (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U. S. A., 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith W.W., Pei Z., Jiang H., Dawson V.L., Dawson T.M. and Ross C.A. (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci., 9, 1231–1233. [DOI] [PubMed] [Google Scholar]
- 17. Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., Brug M.P., Beilina A., Blackinton J., Thomas K.J. et al. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis, 23, 329–341. [DOI] [PubMed] [Google Scholar]
- 18. MacLeod D., Dowman J., Hammond R., Leete T., Inoue K. and Abeliovich A. (2006) The familial parkinsonism gene LRRK2 regulates neurite process morphology. Neuron, 52, 587–593. [DOI] [PubMed] [Google Scholar]
- 19. Bae E.J., Kim D.K., Kim C., Mante M., Adame A., Rockenstein E., Ulusoy A., Klinkenberg M., Jeong G.R., Bae J.R. et al. (2018) LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nature Commun., 9, 3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nucifora F.C. Jr., Nucifora L.G., Ng C.H., Arbez N., Guo Y., Roby E., Shani V., Engelender S., Wei D., Wang X.F. et al. (2016) Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nature Commun., 7, 11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skibinski G., Nakamura K., Cookson M.R. and Finkbeiner S. (2014) Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J. Neurosci., 34, 418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho H.J., Yu J., Xie C., Rudrabhatla P., Chen X., Wu J., Parisiadou L., Liu G., Sun L., Ma B. et al. (2014) Leucine-rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER-Golgi export. EMBO J., 33, 2314–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giaime E., Tong Y., Wagner L.K., Yuan Y., Huang G. and Shen J. (2017) Age-dependent dopaminergic neurodegeneration and impairment of the autophagy-lysosomal pathway in LRRK-deficient mice. Neuron, 96(796–807), e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeong G.R., Jang E.H., Bae J.R., Jun S., Kang H.C., Park C.H., Shin J.H., Yamamoto Y., Tanaka-Yamamoto K., Dawson V.L. et al. (2018) Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener., 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu G.H., Qu J., Suzuki K., Nivet E., Li M., Montserrat N., Yi F., Xu X., Ruiz S., Zhang W. et al. (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature, 491, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tsika E., Kannan M., Foo C.S., Dikeman D., Glauser L., Gellhaar S., Galter D., Knott G.W., Dawson T.M., Dawson V.L. et al. (2014) Conditional expression of Parkinson’s disease-related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol. Dis, 71, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J.D., Patricelli M.P., Nomanbhoy T.K., Alessi D.R. et al. (2011) Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol, 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hara M.R., Agrawal N., Kim S.F., Cascio M.B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J.H., Tankou S.K., Hester L.D. et al. (2005) S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol, 7, 665–674. [DOI] [PubMed] [Google Scholar]
- 29. Liani E., Eyal A., Avraham E., Shemer R., Szargel R., Berg D., Bornemann A., Riess O., Ross C.A., Rott R. et al. (2004) Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A., 101, 5500–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kosugi S., Hasebe M., Tomita M. and Yanagawa H. (2009) Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U. S. A., 106, 10171–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fell M.J., Mirescu C., Basu K., Cheewatrakoolpong B., DeMong D.E., Ellis J.M., Hyde L.A., Lin Y., Markgraf C.G., Mei H. et al. (2015) MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J. Pharmacol. Exp. Ther., 355, 397–409. [DOI] [PubMed] [Google Scholar]
- 32. Li X., Patel J.C., Wang J., Avshalumov M.V., Nicholson C., Buxbaum J.D., Elder G.A., Rice M.E. and Yue Z. (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci., 30, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J. 3rd and Shen J. (2010) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U. S. A., 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guaitoli G., Raimondi F., Gilsbach B.K., Gomez-Llorente Y., Deyaert E., Renzi F., Li X., Schaffner A., Jagtap P.K., Boldt K. et al. (2016) Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. U. S. A., 113, E4357–E4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sejwal K., Chami M., Remigy H., Vancraenenbroeck R., Sibran W., Sutterlin R., Baumgartner P., McLeod R., Chartier-Harlin M.C., Baekelandt V. et al. (2017) Cryo-EM analysis of homodimeric full-length LRRK2 and LRRK1 protein complexes. Sci. Rep., 7, 8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang P., Fan Y., Ru H., Wang L., Magupalli V.G., Taylor S.S., Alessi D.R. and Wu H. (2019) Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. U. S. A., 116, 1579–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leandrou E., Markidi E., Memou A., Melachroinou K., Greggio E. and Rideout H.J. (2019) Kinase activity of mutant LRRK2 manifests differently in hetero-dimeric vs. homo-dimeric complexes. Biochem. J., 476, 559–579. [DOI] [PubMed] [Google Scholar]
- 38. Sen N. and Snyder S.H. (2011) Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc. Natl. Acad. Sci. U. S. A., 108, 20178–20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brauckhoff A., Malz M., Tschaharganeh D., Malek N., Weber A., Riener M.O., Soll C., Samarin J., Bissinger M., Schmidt J. et al. (2011) Nuclear expression of the ubiquitin ligase seven in absentia homolog (SIAH)-1 induces proliferation and migration of liver cancer cells. J.Hepatol., 55, 1049–1057. [DOI] [PubMed] [Google Scholar]
- 40. Antoniou N., Vlachakis D., Memou A., Leandrou E., Valkimadi P.E., Melachroinou K., Re D.B., Przedborski S., Dauer W.T., Stefanis L. et al. (2018) A motif within the armadillo repeat of Parkinson’s-linked LRRK2 interacts with FADD to hijack the extrinsic death pathway. Sci. Rep., 8, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez Carrion M., Pischedda F., Biosa A., Russo I., Straniero L., Civiero L., Guida M., Gloeckner C.J., Ticozzi N., Tiloca C. et al. (2018) The LRRK2 variant E193K prevents mitochondrial fission upon MPP+ treatment by altering LRRK2 binding to DRP1. Front.Mol Neurosci, 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Young S.G., Jung H.J., Lee J.M. and Fong L.G. (2014) Nuclear lamins and neurobiology. Mol. Cell. Biol., 34, 2776–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jung H.J., Coffinier C., Choe Y., Beigneux A.P., Davies B.S., Yang S.H., Barnes R.H., 2nd, Hong J., Sun T., Pleasure S.J. et al. (2012) Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. U. S. A., 109, E423–E431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chaouch M., Allal Y., De Sandre-Giovannoli A., Vallat J.M., Amer-el-Khedoud A., Kassouri N., Chaouch A., Sindou P., Hammadouche T., Tazir M. et al. (2003) The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul Disord, 13, 60–67. [DOI] [PubMed] [Google Scholar]
- 45. Bonati U., Bechtel N., Heinimann K., Rutz E., Schneider J., Frank S., Weber P. and Fischer D. (2014) Congenital muscular dystrophy with dropped head phenotype and cognitive impairment due to a novel mutation in the LMNA gene. Neuromuscul. Disord., 24, 529–532. [DOI] [PubMed] [Google Scholar]
- 46. Iwahashi C.K., Yasui D.H., An H.J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J. and Hagerman P.J. (2006) Protein composition of the intranuclear inclusions of FXTAS. Brain, 129, 256–271. [DOI] [PubMed] [Google Scholar]
- 47. Arocena D.G., Iwahashi C.K., Won N., Beilina A., Ludwig A.L., Tassone F., Schwartz P.H. and Hagerman P.J. (2005) Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet., 14, 3661–3671. [DOI] [PubMed] [Google Scholar]
- 48. Collier T.J., Kanaan N.M. and Kordower J.H. (2011) Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat. Rev. Neurosci., 12, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scaffidi P. and Misteli T. (2006) Lamin A-dependent nuclear defects in human aging. Science, 312, 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miller J.D., Ganat Y.M., Kishinevsky S., Bowman R.L., Liu B., Tu E.Y., Mandal P.K., Vera E., Shim J.W., Kriks S. et al. (2013) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell, 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee S.B., Kim W., Lee S. and Chung J. (2007) Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun., 358, 534–539. [DOI] [PubMed] [Google Scholar]
- 52. Saha S., Guillily M.D., Ferree A., Lanceta J., Chan D., Ghosh J., Hsu C.H., Segal L., Raghavan K., Matsumoto K. et al. (2009) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci., 29, 9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Carrion M.D.P., Marsicano S., Daniele F., Marte A., Pischedda F., Di Cairano E., Piovesana E., von Zweydorf F., Kremmer E., Gloeckner C.J. et al. (2017) The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci Rep, 7, 5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rudenko I.N., Kaganovich A., Hauser D.N., Beylina A., Chia R., Ding J., Maric D., Jaffe H. and Cookson M.R. (2012) The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J., 446, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nichols R.J., Dzamko N., Morrice N.A., Campbell D.G., Deak M., Ordureau A., Macartney T., Tong Y., Shen J., Prescott A.R. et al. (2010) 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J., 430, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smith W.W., Pei Z., Jiang H., Moore D.J., Liang Y., West A.B., Dawson V.L., Dawson T.M. and Ross C.A. (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. U. S. A., 102, 18676–18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pinho R., Paiva I., Jercic K.G., Fonseca-Ornelas L., Gerhardt E., Fahlbusch C., Garcia-Esparcia P., Kerimoglu C., Pavlou M.A.S., Villar-Pique A. et al. (2019) Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol Genet, 28, 31–50. [DOI] [PubMed] [Google Scholar]
- 58. Novello S., Arcuri L., Dovero S., Dutheil N., Shimshek D.R., Bezard E. and Morari M. (2018) G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol. Dis., 120, 21–33. [DOI] [PubMed] [Google Scholar]
- 59. Xiong Y., Neifert S., Karuppagounder S.S., Liu Q., Stankowski J.N., Lee B.D., Ko H.S., Lee Y., Grima J.C., Mao X. et al. (2018) Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. U. S. A., 115, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin X., Parisiadou L., Gu X.L., Wang L., Shim H., Sun L., Xie C., Long C.X., Yang W.J., Ding J. et al. (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron, 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Avraham E., Rott R., Liani E., Szargel R. and Engelender S. (2007) Phosphorylation of parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J. Biol. Chem., 282, 12842–12850. [DOI] [PubMed] [Google Scholar]
- 62. Eyal A., Szargel R., Avraham E., Liani E., Haskin J., Rott R. and Engelender S. (2006) Synphilin-1A: an aggregation-prone isoform of synphilin-1 that causes neuronal death and is present in aggregates from alpha-synucleinopathy patients. Proc. Natl. Acad. Sci. U. S. A., 103, 5917–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rott R., Szargel R., Haskin J., Shani V., Shainskaya A., Manov I., Liani E., Avraham E. and Engelender S. (2008) Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem., 283, 3316–3328. [DOI] [PubMed] [Google Scholar]
- 64. Kolodney G., Dumin E., Safory H., Rosenberg D., Mori H., Radzishevsky I. and Wolosker H. (2015) Nuclear compartmentalization of serine racemase regulates D-serine production: implications for N-methyl-D-aspartate (NMDA) receptor activation. J. Biol. Chem., 290, 31037–31050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mullen R.J., Buck C.R. and Smith A.M. (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development, 116, 201–211. [DOI] [PubMed] [Google Scholar]
- 66. Zinchuk V., Zinchuk O. and Okada T. (2005) Experimental LPS-induced cholestasis alters subcellular distribution and affects colocalization of Mrp2 and Bsep proteins: a quantitative colocalization study. Microsc. Res. Tech, 67, 65–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.