

Abstract
Background
Epidemiological studies have suggested an association between Helicobacter pylori (H pylori) infection and atherosclerosis through undefined mechanisms. Endothelial dysfunction is critical to the development of atherosclerosis and related cardiovascular diseases. The present study was designed to test the hypothesis that H pylori infection impaires endothelial function through exosome‐mediated mechanisms.
Methods and Results
Young male and female patients (18‐35 years old) with and without H pylori infection were recruited to minimize the chance of potential risk factors for endothelial dysfunction for the study. Endothelium‐dependent flow‐mediated vasodilatation of the brachial artery was evaluated in the patients and control subjects. Mouse infection models with CagA+ H pylori from a gastric ulcer patient were created to determine if H pylori infection‐induced endothelial dysfunction could be reproduced in animal models. H pylori infection significantly decreased endothelium‐dependent flow‐mediated vasodilatation in young patients and significantly attenuated acetylcholine‐induced endothelium‐dependent aortic relaxation without change in nitroglycerin‐induced endothelium‐independent vascular relaxation in mice. H pylori eradication significantly improved endothelium‐dependent vasodilation in both patients and mice with H pylori infection. Exosomes from conditioned media of human gastric epithelial cells cultured with CagA+ H pylori or serum exosomes from patients and mice with H pylori infection significantly decreased endothelial functions with decreased migration, tube formation, and proliferation in vitro. Inhibition of exosome secretion with GW4869 effectively preserved endothelial function in mice with H pylori infection.
Conclusions
H pylori infection impaired endothelial function in patients and mice through exosome‐medicated mechanisms. The findings indicated that H pylori infection might be a novel risk factor for cardiovascular diseases.
Keywords: cardiovascular risk factor, endothelial dysfunction, exosomes, Helicobacter pylori
Subject Categories: Vascular Disease, Atherosclerosis, Endothelium/Vascular Type/Nitric Oxide
Clinical Perspective
What Is New?
The present study demonstrated that Helicobacter pylori infection impaired endothelial function in patients and mice through exosome‐mediated mechanism(s).
The data revealed a potential novel mechanism for the development of vascular dysfunction in patients with Helicobacter pylori infection.
What Are the Clinical Implications?
The findings from the present study suggested that Helicobacter pylori infection could be a new risk factor for cardiovascular diseases, especially in young populations.
The data from the present study could help to explore new and effective strategies to preventing and treating cardiovascular diseases associated with Helicobacter pylori infection especially in young patients with premature atherosclerosis.
Nonstandard Abbreviations and Acronyms.
Ach acetylcholine
CagA Cytotoxin‐associated gene A
CVDs cardiovascular diseases
FMD flow‐mediated vasodilatation
H pylori Helicobacter pylori
NTG nitroglycerin
Introduction
The microaerophilic Gram‐negative bacterium Helicobacter pylori (H pylori) colonizes in the epithelium of human stomach in a significant portion of the general population ranging from 30% to 50% in developed countries up to 80% in developing countries, especially in Asia.1, 2 Most patients with H pylori infection have no symptoms clinically. However, H pylori could cause progressive damage to the gastric mucosa, and the bacterium is closely associated with various important gastric diseases including (but not limit to) chronic gastritis, peptic ulcers, and gastric cancer.3 Recent data indicate that H pylori infection could also contribute to important extragastrointestinal diseases such as cardiovascular diseases (CVDs), hematological diseases (especially idiopathic thrombocytopenia), neurological disorders, dermatological diseases, autoimmune diseases such as inflammatory bowel diseases, chronic liver disease, and diabetes mellitus.4, 5, 6, 7, 8, 9, 10, 11, 12
CVDs remain the leading cause of mortality and morbidity in developed countries.13 Atherosclerosis is an important contributing factor to CVDs, especially coronary artery diseases and stroke.14 Epidemiology studies have shown that, compared with those without H pylori infection, patients with H pylori infection, especially with the virulence factor Cytotoxin‐associated gene A (CagA)+, have increased incidence of atherosclerosis (29% versus 63%),15 and ischemic stroke (45% versus 77%).7 CagA may also contribute to destabilization of carotid atherosclerotic plaques.16 Recently, we analyzed the data from 17 613 adult patients with carotid ultrasound examination and a 13C‐urea breath test for H pylori and found that, after adjustment for age, sex, body mass index, lipid profile, hypertension, diabetes mellitus, and smoking, H pylori infection was an independent risk factor for carotid atherosclerosis in male patients ≤50 years of age.17 Previous studies with mice suggested that H pylori infection might enhance atherosclerosis,18, 19, 20 although 1 study showed that H pylori infection could not contribute to the development of atherosclerotic lesions in low‐density lipoprotein receptor–deficient mice.21 However, how H pylori infection could lead to atherosclerosis is unknown at this point. The underlying mechanisms for the pathogenesis of atherosclerosis are complex and have not been fully defined. Endothelial cells play a key role in maintaining the structural and functional integrity of vasculature,22 and endothelial dysfunction is considered the initial event leading to the development of atherosclerosis.
H pylori does not enter the blood circulation itself because of the gastric tissue barrier and a unique survival and growth environment.23 However, H pylori virulence factor CagA and H pylori DNA are present in human atherosclerotic lesions, aorta, and carotid and coronary arteries.16, 24, 25, 26 Many cells are known to release extracellular vesicles or exosomes with unique biophysical and biochemical properties.27, 28 Exosomes are found in various body fluids including blood, urine, saliva, and breast milk and play an important role in cell‐to‐cell communications through transport of a wide spectrum of bioactive constituents including proteins, lipids, and microRNAs.29, 30 Recent studies have demonstrated that exosomes are critically involved in protein transfers during infections, such as prion protein in neurodegenerative disease,31 human immunodeficiency virus–related proteins,32 and human T‐cell leukemia virus type‐1 proteins.33
It is unclear how H pylori infection could enhance atherosclerosis. In the present study both patients and mouse models with H pylori infection were used to test the hypothesis that H pylori infection impaired endothelial function through CagA‐containing exosome–mediated mechanisms.
Methods
Patient Selection and Ultrasound Assessment of Endothelial‐Dependent Flow‐Mediated Vasodilatation of the Brachial Artery in Human Subjects
The materials, methods, and data that supported the findings of this study are available from the corresponding author on reasonable request. The study with human subjects was conducted according to the principles of the Declaration of Helsinki, and the study protocol was reviewed and approved by the Clinical Research Ethics Committee of the Third Xiangya Hospital of Central South University, Changsha, China, and the Human Subjects Research Protections Program & Institutional Review Board at the University of Missouri School of Medicine, Columbia, MO (IRB Number: 2016112). Written informed consent was obtained from all subjects before their participation. Both male and female and age‐matched (within 5 years of age difference for each matched pair) subjects with the age range of 18‐35 years with (n=18) and without H pylori infection (n=13) were recruited for the study. No other confounding variables except the conditions listed in the exclusion criteria were considered for subject selection. Young patients were recruited to minimize the risk factors for endothelial dysfunction. Patients were excluded from the study if any of the following conditions were present: (1) history of H pylori eradication, (2) use of any medications including antibiotics, proton pump inhibitors, or H2‐receptor blockers 3 months before the study, (3) age <18 or >35 years, (4) connective tissue diseases or immunological diseases, (5) mental disorders, (6) asthma or COPD, (7) hematological disorders, (8) thyroid diseases, (9) malignancies, (10) recent (within 3 months) or chronic infection except H pylori infection, (11) congestive heart failure, (12) abnormal renal or liver function, (13) congenital heart diseases, (14) hypertension, (15) smoking, (16) diabetes mellitus, (17) lipid abnormalities, (18) stroke, (19) obesity, (20) sedentary life style, (21) alcohol use, (22) any use of energy drinks or coffee or tea within 48 hours, and (23) lack of response to anti–H pylori therapy. After 8 to 12 hours of fasting, brachial artery endothelium‐dependent flow‐mediated vasodilatation (FMD) was evaluated for patients and control subjects and presented as percentage change in postischemia diameter over baseline as described.34
H pylori Culture
CagA+ H pylori used in culture and mouse studies was isolated from the gastric specimens of gastric ulcer patients during gastroscopy as described.35 CagA+ H pylori was used for the present study because the vast majority of patients with H pylori infection were infected with CagA+ H pylori.36 The bacteria were grown on Columbia blood agar plates supplemented with antibiotics (10 mg/L vancomycin, 5 mg/L cefsulodin, 5 mg/L amphotericin B, 5 mg/L trimethoprim, and 10% sheep blood [Thermo Scientific R54008, Waltham, MA]) at 37°C under microaerophilic conditions (5% O2, 10% CO2, and 85% N2) for 3‐4 days. The concentration of H pylori was determined by measuring the optical density at OD600 nm, where 1 unit of OD600 nm corresponds to about 2×108 colony‐forming units/mL.35 The identity of CagA+ H pylori was confirmed using the complete sequence data of the H pylori 16s rRNA gene from GenBank data (sequence ID AP017362) and positive biochemical test reactions for oxidase, catalase, and urease (Figure S1). Killed or deactivated H pylori organisms were not used in the present study because dead bacteria were unable to interact actively with gastric epithelium and transport CagA into the cells. In addition, killed organisms have high immunogenicity and could release endotoxin and other substances that might trigger significant reactions in the gastric epithelial cells that could be very different from those evoked by live bacteria.
Cell Culture and Cell‐Bacteria Coculture
The human gastric epithelial cell line GES‐1, human umbilical vein endothelial cells (HUVECs), and mouse brain microvascular endothelial cells (bEND.3; ATCC, Manassas, VA) were cultured in RPMI‐1640, DMEM/F‐12, and DMEM (Gibco/Life Technologies, Grand Island, NY), respectively, supplemented with 10% fetal bovine serum (Biological Industries, Cromwell, CT), 100 U/mL penicillin, and 100 mg/mL streptomycin in a controlled humidified incubator with 5% CO2 as described.35 To evaluate the effect of exosomes on the function of endothelial cells, exosomes (50 μg/mL) from the serum of human subjects or mice with and without CagA+ H pylori infection and from conditioned media of GES‐1 cocultured with or without CagA+ H pylori for 2 hours were added into the culture system of HUVECs or bEND.3 (see details below for exosome preparation). After culture for 24 hours, HUVECs or bEND.3 were collected for studies on cell migration, tube formation, and proliferation, using a transwell culture system (MCEP24H48; Merck Millipore, Burlington, MA), a Matrigel basement membrane matrix (Cat. No. 356234; Corning Life Sciences, Tewksbury, MA), and a cell‐Light EdU imaging kit (Cat.No.C10310‐1; RiboBio, Guangzhou, China), respectively. GES‐1 was mixed and cultured with H pylori at a multiplicity of infection of 100 for 2, 6, or 12 hours as described.35
H Pylori Eradication
To further investigate the effect of H pylori infection on endothelial function, the patients and mice with H pylori infection were treated with anti–H pylori therapy to determine if eradication of H pylori could restore endothelial function. The H pylori–infected patients (n=10) were treated with BIS‐based quadruple oral anti–H pylori therapy (100 mg furazolidone, 100 mg doxycycline, 5 mg ilaprazole, and 220 mg colloidal bismuth tartrate twice a day for 2 weeks) as described.37 The CagA+ H pylori–infected mice at 12 weeks after infection were treated with triple therapy (123.30 mg/kg bismuth potassium citrate, 102.75 mg/kg tinidazole, and 51.38 mg/kg clarithromycin in a 0.2‐mL volume once daily by intragastric gavage for 2 weeks).38, 39 The effectiveness on eradication of H pylori was verified in both infected mice with a rapid urease test and Giemsa staining and in patients with a 13C‐urea breath test. The subjects were excluded from the study if resistant to standard anti–H pylori therapy.
Animal Model and GW4869 Administration
All the animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health. The experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Missouri School of Medicine, Columbia, MO, and the Third Xiangya Hospital of Central South University, Changsha, China. Specific‐pathogen‐free 4‐ to 6‐week‐old male C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were allowed to have free access to rodent diet and water. The mice were randomly divided into 2 groups: CagA+ H pylori and normal control group. After fasting for 24 hours, mice were infected with 0.2 mL of CagA+ H pylori inocula (approximate 4×109 colony‐forming units/mL) by intragastric gavage once per day for 3 days. If the infection rate with H pylori did not reach 100%, the H pylori inocula via intragastric gavage once per day continued for another 3 days after a 1‐day break as shown in Figure S2A. The mice were tested for H pylori infection using a rapid urease test and Giemsa staining with the goal of achieving 100% infection rate on both tests 7 days after the last intragastric gavage. The cycle of intragastric gavage with H pylori inoculum (3 days on and 1 day off) continued for the mice until 100% infection rate was achieved. Animals were euthanized at weeks 1 and 24 after the last gavage to collect thoracic aorta to determine endothelium‐dependent and endothelium‐independent vascular relaxation as described below and stomach to examine the presence of H pylori.
To evaluate the role of exosomes in mediating the effect of H pylori infection on endothelial function, a stock solution (2 mg/mL) for the inhibitor of exosome biogenesis/release in vivo, GW4869 (Selleck Chemicals, Houston, TX),40, 41 was prepared in DMSO. Mice were treated with GW4869 (0.05 mg/mL in PBS, final DMSO concentration 2.5%) via intraperitoneal injection once daily at a dose of 1.25 mg/[kg/d] for 11 days, while CagA+ H pylori inocula were gavaged simultaneously once daily for 3 days starting 24 hours after the first dose of GW4869. The same volumes of CagA+ H pylori inoculum and 2.5% DMSO in PBS were given to the control mice the same way as shown in Figure S2B. Animals were euthanized 24 hours after the final injection of GW4869 to collect blood for preparation of serum exosomes and thoracic aorta for evaluation of endothelium‐dependent and independent vascular relaxation.
Evaluation of Vascular Contraction and Relaxation in Mice
Thoracic aorta was isolated from mice to evaluate the contractility and endothelium‐dependent and endothelium‐independent vascular relaxation. After careful isolation, thoracic aorta was cut into 2‐ to 3‐mm segments in cold buffer on ice and was mounted onto a 4‐channel Wire Myograph System (610M; DMT, Aarhus, Denmark) for vascular function studies. The aortic segments were equilibrated with a resting tension of 4.9 mN for 45‐60 minutes in a temperature‐controlled tissue bath filled with 5 mL of Krebs solution and bubbled continuously with 95% O2 and 5% CO2 at 37°C. The aortic preparations were then tested for maximal contraction with 50 mmol/L KCl and dose‐dependent vasocontractile response to phenylephrine. After adequate washout with Krebs solution and equilibration, the aortic tissues were examined for endothelium‐dependent relaxation to cumulative doses of acetylcholine (Ach, 10−9 to 10−5 mol/L) and endothelium‐independent relaxation to cumulative doses of nitroglycerin (NTG, 10−9 to 10−5 mol/L) after submaximal contraction with phenylephrine as described.42, 43 Two aortic segments from each mouse were used for vascular function evaluation, and the average response from the 2 segments was used for data analysis for each mouse.
Exosome Isolation and Identification
To prepare exosomes from conditioned media, human gastric epithelium cells GES‐1 were cultured with or without CagA+ H pylori at a multiplicity of infection of 100 for 2, 6, and 12 hours as described above.35 The conditioned media were then collected from the coculture systems to isolate the exosomes as described.44 Briefly, cells and large cell debris in the conditioned media were eliminated by successive centrifugations (4°C) at increasing speeds (300g for 10 minutes, 2000g for 20 minutes, and 10 000g for 30 minutes). Then the supernatant was transferred to an ultracentrifuge tube and centrifuged at 100 000g at 4°C for 70 minutes twice (Beckman Coulter, Indianapolis, IN). Exosome pellets were resuspended in a small volume of PBS for further analysis. Serum from patients or mice with CagA+ H pylori infection was collected for exosome isolation and identification similar to conditioned media as described above.44The protein level in the exosomes was determined by the BCA protein assay (Cat.No.23235; Thermo Fisher, Waltham, MA). The morphologies, size distribution, and biomarkers (HSP70 and CD9) of the exosomes were examined using a transmission electron microscopy (TECNAI G2 Spirit; FEI, Hillsboro, OR), dynamic light scattering with a particle and molecular size analyzer (Zetasizer Nano ZS; Malvern Panalytical, Malvern, Worcestershire, UK), and Western blotting, respectively, as described.45
Entrance of Exosomes Into Endothelial Cells
To determine if the exosomes entered into the endothelial cells, the exosomes were labeled with fluorescent PKH67 (green) as described.46 Exosomes were prepared and labeled with a PKH67 Green Fluorescent Cell Linker Kit (Sigma‐Aldrich, St. Louis, MO) according to manufacturer's protocol. To evaluate the entry and distribution of exosomes in endothelial cells, PKH67‐labeled exosomes were incubated with HUVECs (30 μg protein/5×104 cells) for 12 hours. Then F‐actin in cytoskeleton and cell nuclei were stained with Alexa Fluor555 Phalloidin (Cat. No. A34055; Thermo Fisher, Waltham, MA) and DAPI fluorescent stain (Cat.No.D1306; Thermo Fisher, Waltham, MA), respectively. The cells were mounted with antifade reagent (Cat.No.13800; Thermo Fisher, Waltham, MA) and examined using a 3D confocal laser scanning microscope (Leica TCS SP8, Buffalo Grove, IL).
Endothelial Cell Migration, Tube Formation, and Proliferation Assays
To evaluate the effect of exosomes on the function of endothelial cells, HUVECs and bEND.3 were used to determine the migration, tube formation, and proliferation. Cell migration was evaluated using a transwell culture system that could allow the cells to migrate from the upper chamber to the lower chamber via an 8‐μm pore. HUVECs or bEND.3 cultured with exosomes (50 μg/mL) for 24 hours were then plated (5×104 for HUVECs or 2×104 cells/well for bEND.3) in serum‐free medium in the upper chamber. The lower chamber was filled with 600 μL normal medium containing 10% fetal bovine serum. After 5 or 8 hours of incubation, the cells were fixed and stained with crystal violet for 10 minutes. The upper surface of transwell chambers was wiped with a cotton swab. The migrated cells were counted in 5 random microscopic fields as described.47
Tube formation was assessed in a 96‐well plate using a Matrigel basement membrane matrix as per manufacturer's instructions. Briefly, HUVECs or bEND.3 cultured with exosomes (50 μg/mL) for 24 hours and then were placed on the Matrigel (70 μL/well) with a cell density of 2×104 cells/well and cultured for 4 hours. The cell structures were examined using an inverted light microscope. The total capillary tube lengths were quantified using the software ImageJ. Five independent fields were assessed for each well, and the average number of tubes was calculated and analyzed.
Cell proliferation was evaluated using a cell‐Light Edu imaging kit as per manufacturer's instructions (RiboBio, Guangzhou, China). HUVECs or bEND.3 (1×104 cells/well) were cultured with exosomes (50 μg/mL) in serum‐depleted culture medium on 96‐well plates. After 24 hours of culture, 100 μL of 50 μmol/L EdU solution per well was added into the culture system for 2 more hours of incubation and then discarded. Cells were washed 2‐3 times with PBS and fixed with 4% paraformaldehyde, 2 mg/mL glycine, and 0.5% Triton X‐100 following the instructions. After incubation of the cells with 1× Apollo567 staining agent for 30 minutes at room temperature in dark, the staining agent was removed. The preparations were then exposed to 0.5% Triton X‐100 for cell membrane permeabilization. After washing with methanol and PBS, the cells were examined with a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Western Blotting
The levels of CagA protein and exosome markers (HSP70 and CD9) were determined using Western blotting. The protein preparations (30 μg proteins) were loaded on 5% and 10% SDS‐PAGE. After electrophoresis, the preparations were transferred to polyvinylidene fluoride membranes (Merck Millipore, Burlington, MA). After blocking with 5% skim milk in tris‐buffered saline with Tween for 1 hour, the membranes were incubated with the following primary antibodies: CagA (sc‐28368; diluted factor 1:200, Santa Cruz, Dallas, TX), Hsp70 (ab5439; dilution factor 1:1000, Abcam, Cambridge, MA), CD9 (ab92726; dilution factor 1:1000, Abcam, Cambridge, MA) at 4°C overnight. Then membranes were incubated with IRDye secondary antibodies (LI‐COR, dilution factor 1:10 000, Li‐Cor Biosciences, Lincoln, NE). The membranes were visualized using Odyssey CLx Imaging System (Li‐Cor Biosciences, Lincoln, NE).
Immunofluorescence Staining
After culturing with CagA+ H pylori at a multiplicity of infection of 100 for 12 hours, GES‐1 cells were washed, fixed, permeabilized, and incubated with 5% goat serum to block nonspecific staining, and then incubated with anti‐CagA primary antibody (sc‐28368 AF488; dilution factor 1:50, Santa Cruz, Dallas, TX) at 4°C overnight. After washing with PBS, F‐actins in cytoskeleton and cell nuclei were stained with Alexa Fluor555 Phalloidin (Molecular Probes, Eugene, OR) and DAPI fluorescent stain, respectively. The cells were mounted with antifade reagent and examined using a 3‐D confocal laser scanning microscope (Leica TCS SP8, Buffalo Grove, IL).
Statistical Analyses
Data were expressed as mean±SE and analyzed with GraphPad Prism 7.00 software (San Diego, CA). A 2‐tailed unpaired t test was used for the analysis of 2 groups of data with normal distribution and equal variance, and a 2‐tailed unpaired t test with Welch correction for analyzing 2 groups of data with normal distribution and unequal variance. A Mann‐Whitney U test was used for comparisons between 2 groups of data with abnormal distributions. ANOVA with post hoc Bonferroni test was used for 3 or more groups of data analysis with normal distributions and equal variance, and the Kruskal‐Wallis test with Dunn post hoc multiple‐comparison tests was used for 3 or more groups of data analysis with normal distributions and unequal variance or abnormal distributions. A difference was considered statistically significant when P<0.05.
Results
H Pylori Infection Impaired Endothelial Function in Patients
To determine if H pylori infection could impair endothelial function, we recruited 18 young patients (mean age 31.4±1.6 years, Table S1) with H pylori infection and without known risk factors for endothelial dysfunction to evaluate endothelium‐dependent FMD of the brachial artery. A group of 13 young healthy volunteers (mean age of 29.4±1.5 years) served as control. No significant difference in age was present between the patients and control subjects. Patients with H pylori infection exhibited a significant reduction in FMD compared with controls (Figure 1A).
Figure 1.
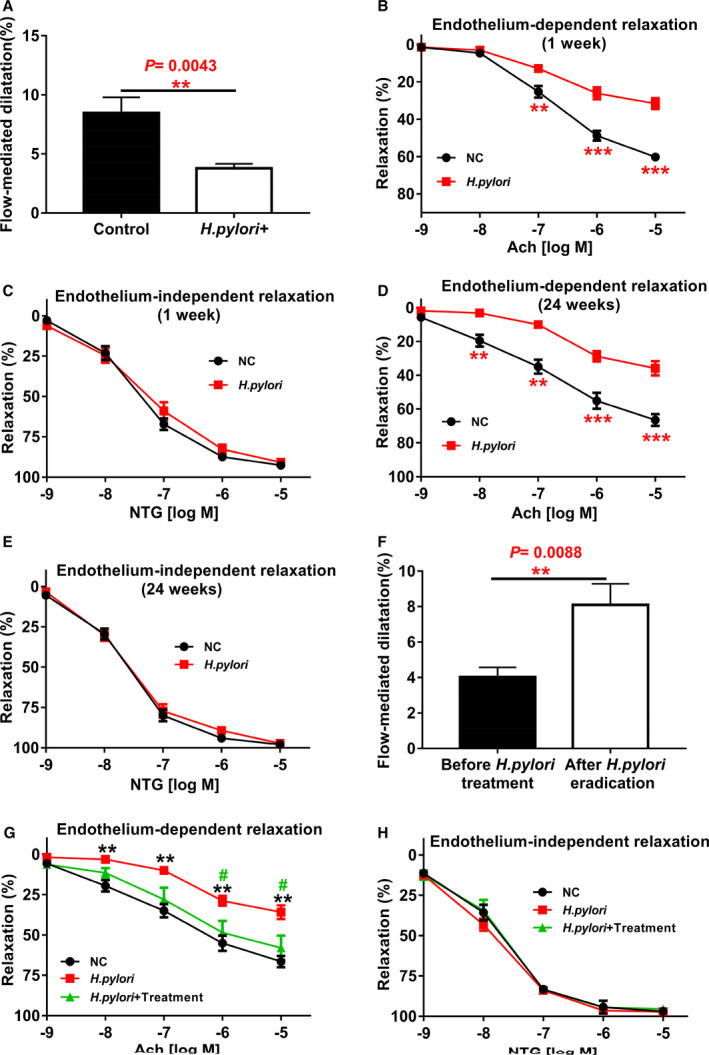
Helicobacter pylori infection significantly impaired endothelium‐dependent flow‐mediated dilatation (FMD) in human subjects and endothelium‐dependent vascular relaxation in mice.
Patients with H pylori infection and healthy control subjects were evaluated for endothelium‐dependent FMD of the brachial artery with ultrasound. The diagnosis of H pylori infection was confirmed with gastric endoscopic biopsy and 13C‐urea breath test for each patient. Patients with H pylori infection (n=18 patients) displayed a significant reduction in their endothelium‐dependent FMD compared with the controls (n=13 subjects). A, **P<0.01 by t test. Mice infected with CagA+ H pylori for 1 week significantly decreased acetylcholine (Ach)‐induced endothelium‐dependent relaxation of thoracic aorta (B) without change in NTG‐induced endothelium‐independent vasorelaxation (C) after submaximal contraction with phenylephrine (PE) (10−6M). **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni test or Kruskal‐Wallis test with Dunn post hoc test, n=10 mice for each group. The impaired Ach‐induced endothelium‐dependent vasorelaxation persisted for as long as the infection was present for at least 24 weeks (D) without change in NTG‐induced endothelium‐independent vasorelaxation (E). **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni test or Kruskal‐Wallis test with Dunn post hoc test, n=10 mice for each group. Eradication of H pylori infection with anti–H pylori therapy effectively restored the endothelium‐dependent FMD in patients with H pylori infection (n=10 patients with anti–H pylori therapy) (F) (**P<0.01 by t test with Welch correction), and significantly improved Ach‐induced endothelium‐dependent vasorelaxation in mice with 12 weeks of chronic CagA+ H pylori infection (G), # P<0.05 (compared with CagA+ H pylori +Treatment mice); **P<0.01 (compared with NC mice) by one‐way ANOVA with Bonferroni's test or Kruskal‐Wallis test with Dunn's post hoc test, without change in NTG‐induced endothelium‐independent vasorelaxation (H) (n=8 mice for each group in G and H). Ach indicates acetylcholine; NC, normal control; NTG: nitroglycerin. Data were presented as mean±SE.
H Pylori Infection Impaired Endothelial Function in Mice
We then used male C57BL/6 mice to establish a mouse H pylori infection model to determine if endothelial dysfunction in patients with H pylori infection could be reproduced in mice using H pylori from patients. Mice were successfully infected with CagA+ H pylori (Table and Figure S3). Indeed, Ach‐induced endothelium‐dependent relaxation was significantly reduced in mice 1 week after H pylori infection (Figure 1B) without change in NTG‐induced endothelium‐independent relaxation (Figure 1C). Impaired Ach‐induced relaxation persisted for as long as the infection was present for at least 24 weeks (Figure 1D) without change in vascular contractions to either phenylephrine or potassium chloride (Figure S4), although NTG‐induced relaxation remained intact (Figure 1E).
Table 1.
Infection Rate of H pylori With Different Methods 1 Week After Last Intragastric Gavage
| Group | N | Rapid Urease Test Positive (n) | Geimsa Staining Positive (n) | Infection Rate (%) |
|---|---|---|---|---|
| Method 1a | ||||
| Control | 10 | 0 | 0 | 0 |
| CagA+ H pylori | 10 | 2 | 2 | 20 |
| Method 2b | ||||
| Control | 10 | 0 | 0 | 0 |
| CagA+ H pylori | 10 | 6 | 6 | 60 |
| Method 3c | ||||
| Control | 10 | 0 | 0 | 0 |
| CagA+ H pylori | 10 | 10 | 10 | 100 |
Method 1: Intragastric gavage once a day for 1 day.
Method 2: Intragastric gavage once a day for 2 days.
Method 3: Intragastric gavage once a day for 3 days.
H Pylori Eradication Significantly Improved Endothelium‐Dependent Vasodilation
We examined if H pylori eradication could improve endothelium‐dependent vasodilation. Indeed, when patients with H pylori infection were treated with oral anti–H pylori therapy, their endothelium‐dependent FMD of brachial artery was effectively restored (Figure 1F). H pylori elimination in mice with anti–H pylori therapy also significantly improved Ach‐induced vasorelaxation (Figure 1G) without change in NTG‐induced relaxation (Figure 1H).
CagA‐Containing Exosomes Entered Endothelial Cells and Impaired Endothelial Function
To determine how H pylori infection could impair endothelial function, we tested the hypothesis that H pylori infection impaired endothelial function through CagA‐containing exosome‐mediated mechanisms. Indeed, Western blotting analysis and immunofluorescence staining demonstrated that the unique H pylori virulence factor CagA entered into human GES‐1 after incubation with CagA+ H pylori (Figure 2A and 2B). We observed that characteristic exosomes were present in conditioned media of GES‐1 cultured with CagA+ H pylori as defined with specific morphology, size distribution, and specific biomarkers (HSP70 and CD9) using Western blotting (Figure 2C through 2E). Western blotting analysis demonstrated that exosomes from conditioned media of GES‐1 cultured with CagA+ H pylori contained the unique CagA protein, whereas exosomes from control conditioned media of GES‐1 (without culture with H pylori) had no CagA protein (Figure 2C). We also observed that a detectable amount of PKH67‐labeled (green) exosomes were present in HUVECs using 3D confocal microscopy at 12 hours after incubation with the labeled exosomes (Figure 2F), confirming the entry of exosomes into HUVECs. Treatment with GES‐1–derived CagA‐containing exosomes significantly inhibited the function of HUVECs in vitro with decreased proliferation, migration, and tube formation as compared with control exosomes (Figure 2G through 2I).
Figure 2.
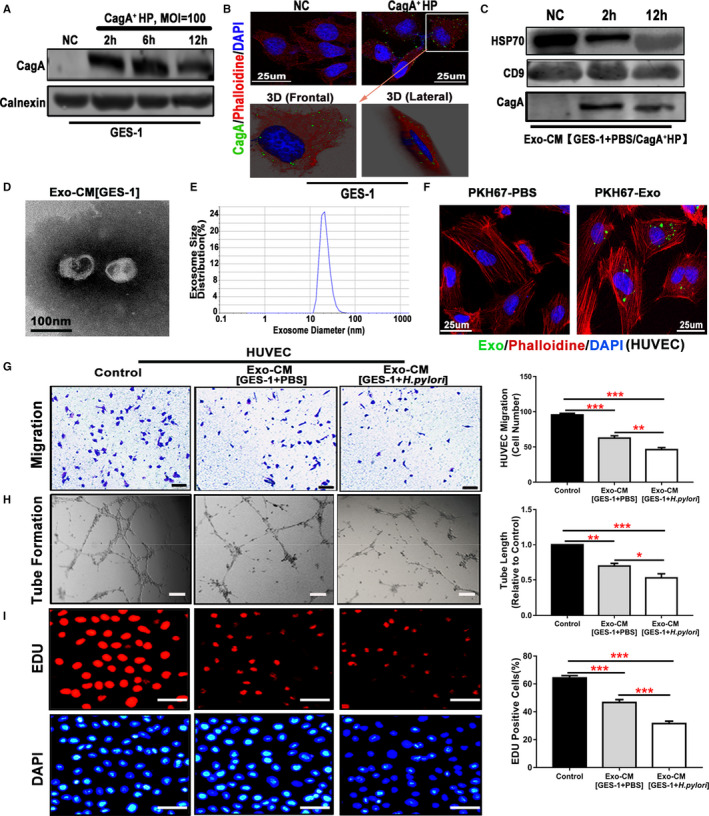
Exosomes from human gastric epithelial cells GES‐1 cultured with CagA+Helicobacter pylori significantly inhibited endothelial cell function in vitro.
Western blotting analysis (A) and immunofluorescence staining with a 3D confocal microscope (B) demonstrated that the unique H pylori virulence factor CagA entered into GES‐1 after culture with CagA+ H pylori. Exosomes isolated from the conditioned media of GES‐1 cultured with CagA+ H pylori displayed typical features for exosomes including characteristic biomarkers HSP70 and CD9 by Western blotting (C), morphologies on transmission electron microscopy (D), and size using a Zetasizer Nano ZS instrument (E). Western blotting analysis confirmed the presence of CagA protein in the exosomes from the conditioned media of GES‐1 cultured with CagA+ H pylori but not in the ones from GES‐1–conditioned media without CagA+ H pylori (C). PKH67‐labeled GES‐1–derived exosomes (green) were incubated with HUVECs (30 μg protein/5×104 cells), and a significant amount of PKH67‐labeled exosomes were detected inside the HUVECs as visualized using a 3D confocal microscope (F), confirming that the exosomes entered into the cells. Treatment of HUVECs with CagA protein‐containing exosomes (50 μg/mL) from GES‐1–conditioned media for 24 hours significantly inhibited the function of HUVECs with decreased migration (G, scale bars=200 μm), tube formation (H, scale bars=200 μm), and proliferation (I, scale bars=50 μm). CagA+ HP indicates CagA+ H pylori; Exo‐CM, exosomes derived from conditioned medium; GES‐1, human gastric epithelial cells; HUVEC, human umbilical vein endothelial cell; MOI, multiplicity of infection; NC, normal control. *P<0.05, **P<0.01, ***P<0.001 by 1‐way ANOVA with Bonferroni test or Kruskal‐Wallis test with Dunn post hoc test. Data were presented as mean±SE, n=3 independent experiments (experiment was repeated 3 times for every measurement).
We then isolated serum exosomes from patients with CagA+ H pylori infection and from healthy age‐ and sex‐matched volunteers. Serum exosomes from both patients and healthy subjects were characterized by morphology, size distribution, and unique biomarkers (HSP70 and CD9) (Figure 3A through 3C). CagA protein was detected in serum exosomes from patients and mice with CagA+ H pylori infection but not from control subjects using Western blotting analysis (Figure 3C). We then labeled human serum exosomes with PKH67 and cultured them with HUVECs and noticed that detectable amounts of PKH67‐labeled (green) exosomes were present in HUVECs using 3D confocal microscopy at 12 hours after incubation (Figure 3D), confirming the entry of serum exosomes into HUVECs. Treatment with serum‐derived CagA‐containing exosomes from patients with CagA+ H pylori infection significantly inhibited the function of HUVECs with decreased migration, proliferation, and tube formation (Figure 3E through 3G). Of note, culture with serum exosomes from healthy control subjects also moderately and yet significantly inhibited endothelial function, suggesting that some endogenous substances in normal serum exosomes could cause endothelial dysfunction. However, serum exosomes from patients with CagA+ H pylori infection exhibited significantly greater inhibitory effects on endothelial functions than exosomes from healthy subjects.
Figure 3.
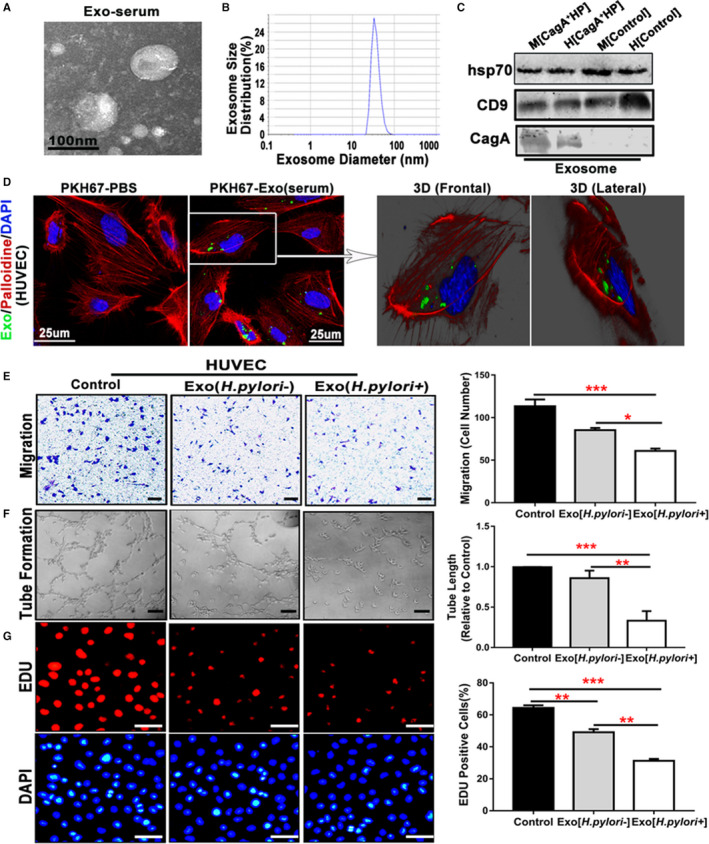
Serum exosomes from patients with CagA+Helicobacter pylori infection inhibited endothelial function in vitro.
Exosomes isolated from patients with CagA+ H pylori infection exhibited typical exosome characteristics with unique morphologies as shown on transmission electron microscopy (A), size distribution as demonstrated using a Zetasizer Nano ZS (Malvern Instruments, Malvern, Worcestershire, UK) instrument (B), and specific biomarkers (HSP70 and CD9) using Western blot analysis (C). Western blotting also showed that the serum exosomes from mice or patients with CagA+ H pylori infection contained specific CagA protein (C). PKH67‐labeled human serum–derived exosomes (green) were incubated with human umbilical vein endothelial cells (HUVECs) (30 μg protein/5×104 cells). A significant amount of PKH67‐labeled exosomes were detected inside the HUVECs as visualized using a 3‐D confocal microscope (D), confirming that the exosomes entered into the cells (HUVECs). Transwell culture showed that exposure to the serum exosomes from patients with CagA+ H pylori infection significantly inhibited the migration of HUVECs (E, scale bar=200 μm) as compared with the controls. Culture with the serum exosomes from patients with CagA+ H pylori infection also significantly attenuated tube formation (F, scale bar=200 μm), and proliferation (using EdU assay) (G, scale bar=50 μm) of HUVECs. Of note, culture with the serum exosomes from healthy control subjects also moderately and yet significantly inhibited endothelial function with decreased migration, tube formation, and proliferation (E through G); however, the serum exosomes from patients with CagA+ H pylori infection exhibited significantly greater inhibitory effects on endothelial functions than the ones from healthy subjects. CagA+ HP indicates CagA+ H pylori; Exo, Exosomes; H, Human; M, Mouse. *P<0.05, **P<0.01, ***P<0.001 by Kruskal‐Wallis test with Dunn post hoc test or 1‐way ANOVA with the Bonferroni test. Data were presented as mean±SE, n=3 independent experiments (experiment was repeated 3 times for every measurement).
Serum exosomes from mice with and without CagA+ H pylori infection were also isolated and characterized for morphology (Figure 4A), size distribution (Figure 4B), and unique biomarkers (HSP70 and CD9) (Figure 3C). Treatment with mouse serum CagA‐containing exosomes also significantly inhibited the function of mouse brain microvascular endothelial cells (bEND.3) in vitro with decreased migration, tube formation, and proliferation compared with control (Figure 4C through 4E).
Figure 4.
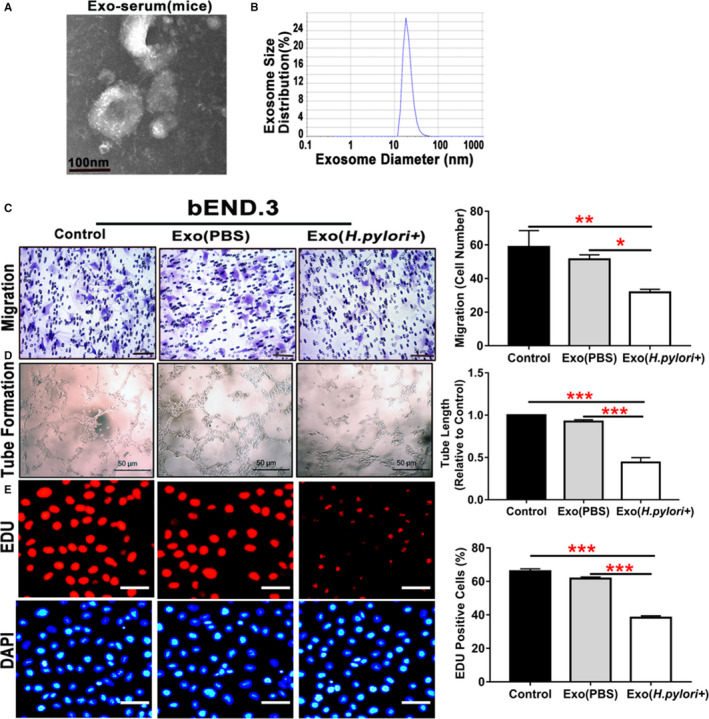
Serum exosomes from mice with Helicobacter pylori infection inhibited endothelial function in vitro.
Exosomes isolated from mice with CagA+ H pylori infection exhibited typical exosome characteristics with unique morphologies as shown on transmission electron microscopy (A), and size distribution as demonstrated using a Zetasizer Nano ZS (Malvern Instruments, Malvern, Worcestershire, UK) instrument (B). Transwell culture showed that exposure to the serum exosomes from mice with CagA+ H pylori infection significantly inhibited the migration of mouse brain microvascular endothelial cells bEND.3 (C, scale bar=50 μm) as compared with the controls. Culture with the serum exosomes from mice with CagA+ H pylori infection also significantly attenuated tube formation (D, scale bar=50 μm) and proliferation (using EdU assay) (E, scale bar=50 μm) of bEND.3. bEND.3 indicates mouse brain microvascular endothelial cells; Exo (H pylori +), xosomes derived from mice treated with CagA+ H pylori infection; Exo (PBS), exosomes derived from control mice treated with PBS; Exo‐serum, exosomes isolated from mouse serum. *P<0.05, **P<0.01, ***P<0.001 by Kruskal‐Wallis test with Dunn post hoc test or 1‐way ANOVA with Bonferroni test. Data were presented as mean±SE, n=3 independent experiments (experiment was repeated 3 times for every measurement).
Blocking Exosome Release With GW4869 In Vivo Effectively Prevented Endothelial Dysfunction in Mice With H Pylori Infection
We then determined if blocking exosome release with GW4869 in vivo could prevent endothelial dysfunction in mice with CagA+ H pylori infection. Indeed, treatment with GW4869 significantly decreased serum exosome level in mice with H pylori infection (Figure 5A) and effectively preserved Ach‐induced aortic relaxation without changing NTG‐induced vasorelaxation (Figure 5B and 5C).
Figure 5.
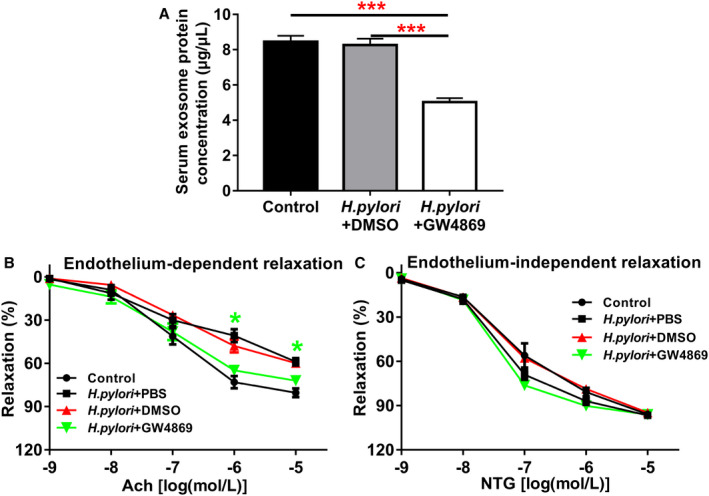
Inhibition of exosome secretion by GW4869 significantly improved endothelium‐dependent vascular relaxation in mice with CagA+Helicobacter pylori infection.
Treatment with GW4869 significantly decreased the serum exosome level in the mice with CagA+ H pylori infection (A) as reflected by the significantly decreased total exosome protein levels (***P<0.001 by 1‐way ANOVA with Bonferroni test) and significantly improved acetylcholine (Ach)‐induced endothelium‐dependent relaxation (B) of the aorta in the mice with CagA+ H pylori infection without change in nitroglycerin (NTG)‐induced endothelium‐independent relaxation (C). *P<0.05 by 1‐way ANOVA with Bonferroni test (when CagA+ H pylori+GW4869 group compared with CagA+ H pylori+DMSO group). Ach indicates acetylcholine; DMSO, dimethylsulfoxide (solubilizer of GW4869); NTG, nitroglycerin. Data shown were mean±SE. n=8 mice for control group, and n=10 mice for other groups.
Discussion
Endothelial dysfunction plays a critical role in CVDs especially atherosclerosis and related coronary artery diseases.48, 49 Previous studies (mostly clinical observations) have shown an inconsistent relationship between H pylori infection and endothelial dysfunction.50, 51, 52 In the present study we demonstrated that H pylori infection significantly decreased endothelium‐dependent FMD of brachial artery in young patients without known cardiovascular risk factors compared with age‐ and sex‐matched healthy volunteers. H pylori infection also significantly attenuated Ach‐induced aortic relaxation without change in NTG‐induced relaxation in mice. H pylori eradication significantly improved endothelium‐dependent vasodilation in both patients and mice with H pylori infection. Exosomes from conditioned media of human gastric epithelial cells cultured with CagA+ H pylori or serum exosomes from patients and mice with H pylori infection significantly decreased endothelial functions with decreased migration, tube formation, and proliferation in vitro. Inhibition of exosome secretion with GW4869 effectively preserved endothelial function in mice with H pylori infection. These data strongly suggested that H pylori infection impaired endothelial function in patients and mice through exosome‐medicated mechanisms. The data also indicated that impaired endothelium‐dependent relaxation was reversible in both patients and mice with H pylori infection.
CagA is considered the most important virulent factor for H pylori.53 CagA+ H pylori is believed to be associated with the development and instability of atherosclerotic plaques54 and with increased risk of recurrent atherosclerotic stroke.55 Aqueous extract of CagA+ H pylori is potent to induce apoptosis and necrosis of HUVECs, and CagA+ H pylori could cause significant endothelial dysfunction in vitro.56 Future studies are needed to determine if significant differences between CagA+ and CagA− H pylori infections on endothelial function could exist in vivo. In the present study we observed that infection with CagA+ H pylori significantly decreased Ach‐induced aortic relaxation without a change in NTG‐induced vasorelaxation in mice. The impaired endothelium‐dependent vasorelaxation persisted for as long as the infection was present for at least 24 weeks. These data were consistent with previous observations that relaxation responses were significantly decreased in ApoE‐knockout mice coinfected with H pylori and Chlamydia pneumoniae.57 VacA is another important H pylori virulence factor, and could inhibit endothelial function in vitro.57 Unlike CagA, VacA is present in all H pylori strains.53 Further studies are needed to evaluate the role of VacA in endothelial dysfunction in vivo.
During infection, H pylori invades and interacts with gastric mucosal epithelial layer. A small number of H pylori could be found in the gastric mucosa capillaries and postcapillary venules in the lamina propria of submucosa.58, 59 These H pylori could result in systemic dissemination; however, H pylori has not been detected in blood in patients with H pylori infection.58 How could H pylori impair endothelial function in vivo? It is known that exosomes, as lipid vesicles, are released from various cells, and carry a wide spectrum of biologically active substances including miRNA, DNA, and proteins or other molecules that could travel through blood, and interact with target cells with subsequent changes in the cell function.29, 60, 61 In the present study we demonstrated that CagA‐containing exosomes were present in serum of CagA+ H pylori‐infected patients and mice. This was consistent with recent findings that CagA existed in serum‐derived exosomes in patients with CagA+ H pylori infection.45
In the present study we tested the hypothesis that H pylori infection could impair endothelial function through an exosome‐mediated mechanism. CagA protein arises only from CagA+ H pylori and could serve as an ideal tracking molecule for the study. CagA+ H pylori is able to efficiently translocate CagA into gastric epithelial cells but not into HUVECs.62 We demonstrated that CagA‐containing exosomes were released into GES‐1–conditioned media cultured with CagA+ H pylori. CagA‐containing exosomes were present in the serum of patients and mice with CagA+ H pylori infection. Exosomes from both conditioned media and serum entered into endothelial cells and significantly impaired their function in vitro. Treatment of CagA+ H pylori–infected mice with GW4869 to inhibit exosome biogenesis/release in vivo40, 41 significantly decreased the level of serum exosomes and effectively preserved Ach‐induced aortic relaxation without change in endothelium‐independent relaxation. These data supported our hypothesis. Of note, culture of HUVECs with serum exosomes from healthy subjects without H pylori infection also moderately inhibited endothelial function with decreased migration, tube formation, and proliferation, indicating that endogenous exosomes in healthy individuals may impair endothelial function. Clearly, further studies are needed to identify the molecules in the exosomes responsible for endothelial dysfunction and related mechanisms associated with H pylori infection. It is also important to determine if H pylori infection could enhance the development and/or progression of atherosclerosis using animal models in future studies. H pylori infection could trigger a complex response in the host including local and systematic inflammation.9, 53 It is certainly possible that inflammatory factors could contribute to oxidative stress and endothelial dysfunction. In fact, eradication of H pylori could affect both oxidative stress and myeloperoxidase activity (an important biomarker for atherosclerosis).63 Nitric oxide is generated from l‐arginine by endothelial nitric oxide synthase and plays a crucial role in endothelial function.49 Asymmetric dimethylarginine is the most potent endogenous endothelial nitric oxide synthase inhibitor and is considered a risk factor for CVDs.64, 65 H pylori eradication could decrease the serum asymmetric dimethylarginine level.66 Further studies are needed to elucidate other potential mechanisms for H pylori infection‐induced endothelial dysfunction.
In summary, we demonstrated that H pylori infection significantly impaired endothelial function in patients and mice via exosome‐mediated mechanisms. The data indicated that H pylori infection might be a novel risk factor for CVDs, especially in young patients.
Sources of Funding
This work was supported by grants from the National Natural Scientific Foundation of China (No. 81570509), Hunan Natural Science Foundation of China (No. 2018JJ6136), New Xiangya Talent Project of the Third Xiangya Hospital of Central South University (No. 20150310), and US NIH grants HL148196 and ES026200 (Liu).
Disclosures
None.
Supporting information
Table S1 Figures S1A, S1–S4
Acknowledgments
We thank Ms Min Fang in the Division of Stem Cell Regulation and Application, Hunan University of Chinese Medicine, Changsha, China, for her technical help in the evaluation of vascular contraction and relaxation in mice, and Thomas E. Spencer, PhD, in the Division of Animal Sciences, University of Missouri College of Agriculture, Columbia, MO, for assistance on the characterization of exosomes from serum and conditioned medium. The human gastric epithelial cell line GES‐1 was obtained as a gift from Professor Xin Wang in the Department of Gastroenterology, Xijing Hospital of the Fourth Military Medical University, Xi'an, China.
(J Am Heart Assoc. 2020;9:e014120 DOI: 10.1161/JAHA.119.014120.)
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Zhenguo Liu, Email: liuzheng@health.missouri.edu.
Canxia Xu, Email: xucanxia2000@163.com.
References
- 1. Mentis A, Lehours P, Megraud F. Epidemiology and diagnosis of Helicobacter pylori infection. Helicobacter. 2015;20(suppl 1):1–7. [DOI] [PubMed] [Google Scholar]
- 2. Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014;19(suppl 1):1–5. [DOI] [PubMed] [Google Scholar]
- 3. Yamaoka Y, Graham DY. Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 2014;10:1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu Z, Li J, Wang H, Xu G. Helicobacter pylori infection and atherosclerosis: is there a causal relationship? Eur J Clin Microbiol Infect Dis. 2017;36:2293–2301. [DOI] [PubMed] [Google Scholar]
- 5. Lee M, Baek H, Park JS, Kim S, Kyung C, Baik SJ, Lee BK, Kim JH, Ahn CW, Kim KR, et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: a cross‐sectional study. PLoS One. 2018;13:e193646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kodama M, Kitadai Y, Ito M, Kai H, Masuda H, Tanaka S, Yoshihara M, Fujimura K, Chayama K. Immune response to CagA protein is associated with improved platelet count after Helicobacter pylori eradication in patients with idiopathic thrombocytopenic purpura. Helicobacter. 2007;12:36–42. [DOI] [PubMed] [Google Scholar]
- 7. Sawayama Y, Ariyama I, Hamada M, Otaguro S, Machi T, Taira Y, Hayashi J. Association between chronic Helicobacter pylori infection and acute ischemic stroke: Fukuoka Harasanshin Atherosclerosis Trial (FHAT). Atherosclerosis. 2005;178:303–309. [DOI] [PubMed] [Google Scholar]
- 8. Kronsteiner B, Bassaganya‐Riera J, Philipson C, Viladomiu M, Carbo A, Abedi V, Hontecillas R. Systems‐wide analyses of mucosal immune responses to Helicobacter pylori at the interface between pathogenicity and symbiosis. Gut Microbes. 2016;7:3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Izzotti A, Durando P, Ansaldi F, Gianiorio F, Pulliero A. Interaction between Helicobacter pylori, diet, and genetic polymorphisms as related to non‐cancer diseases. Mutat Res. 2009;667:142–157. [DOI] [PubMed] [Google Scholar]
- 10. Rocha M, Avenaud P, Menard A, Le Bail B, Balabaud C, Bioulac‐Sage P, de Magalhaes QD, Megraud F. Association of Helicobacter species with hepatitis C cirrhosis with or without hepatocellular carcinoma. Gut. 2005;54:396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franceschi F, Gasbarrini A, Polyzos SA, Kountouras J. Extragastric diseases and Helicobacter pylori . Helicobacter. 2015;20(suppl 1):40–46. [DOI] [PubMed] [Google Scholar]
- 12. Sonnenberg A, Genta RM. Helicobacter pylori is a risk factor for colonic neoplasms. Am J Gastroenterol. 2013;108:208–215. [DOI] [PubMed] [Google Scholar]
- 13. Jokinen E. Obesity and cardiovascular disease. Minerva Pediatr. 2015;67:25–32. [PubMed] [Google Scholar]
- 14. Torres N, Guevara‐Cruz M, Velazquez‐Villegas LA, Tovar AR. Nutrition and atherosclerosis. Arch Med Res. 2015;46:408–426. [DOI] [PubMed] [Google Scholar]
- 15. Shmuely H, Passaro DJ, Vaturi M, Sagie A, Pitlik S, Samra Z, Niv Y, Koren R, Harell D, Yahav J. Association of CagA+ Helicobacter pylori infection with aortic atheroma. Atherosclerosis. 2005;179:127–132. [DOI] [PubMed] [Google Scholar]
- 16. Rozankovic PB, Huzjan AL, Cupic H, Bencic IJ, Basic S, Demarin V. Influence of CagA‐positive Helicobacter pylori strains on atherosclerotic carotid disease. J Neurol. 2011;258:753–761. [DOI] [PubMed] [Google Scholar]
- 17. Zhang L, Chen Z, Xia X, Chi J, Li H, Liu X, Li R, Li Y, Liu D, Tian D, et al. Helicobacter pylori infection selectively increases the risk for carotid atherosclerosis in young males. Atherosclerosis. 2019;291:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen XH, Wang JB, Wang YS, Liu ZM, Li Y. Helicobacter pylori infection enhances atherosclerosis in high‐cholesterol diet fed C57BL/6 mice. Zhonghua Xin Xue Guan Bing Za Zhi. 2010;38:259–263. [PubMed] [Google Scholar]
- 19. Yang S, Xia YP, Luo XY, Chen SL, Li BW, Ye ZM, Chen SC, Mao L, Jin HJ, Li YN, et al. Exosomal CagA derived from Helicobacter pylori‐infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J Mol Cell Cardiol. 2019;135:40–51. [DOI] [PubMed] [Google Scholar]
- 20. Ayada K, Yokota K, Hirai K, Fujimoto K, Kobayashi K, Ogawa H, Hatanaka K, Hirohata S, Yoshino T, Shoenfeld Y, et al. Regulation of cellular immunity prevents Helicobacter pylori‐induced atherosclerosis. Lupus. 2009;18:1154–1168. [DOI] [PubMed] [Google Scholar]
- 21. Mach F, Sukhova GK, Michetti M, Libby P, Michetti P. Influence of Helicobacter pylori infection during atherogenesis in vivo in mice. Circ Res. 2002;90:E1–E4. [DOI] [PubMed] [Google Scholar]
- 22. Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buzas GM. Metabolic consequences of Helicobacter pylori infection and eradication. World J Gastroenterol. 2014;20:5226–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iriz E, Cirak MY, Engin ED, Zor MH, Erer D, Ozdogan ME, Turet S, Yener A. Detection of Helicobacter pylori DNA in aortic and left internal mammary artery biopsies. Tex Heart Inst J. 2008;35:130–135. [PMC free article] [PubMed] [Google Scholar]
- 25. Kilic A, Onguru O, Tugcu H, Kilic S, Guney C, Bilge Y. Detection of cytomegalovirus and Helicobacter pylori DNA in arterial walls with grade III atherosclerosis by PCR. Pol J Microbiol. 2006;55:333–337. [PubMed] [Google Scholar]
- 26. Kaplan M, Yavuz SS, Cinar B, Koksal V, Kut MS, Yapici F, Gercekoglu H, Demirtas MM. Detection of Chlamydia pneumoniae and Helicobacter pylori in atherosclerotic plaques of carotid artery by polymerase chain reaction. Int J Infect Dis. 2006;10:116–123. [DOI] [PubMed] [Google Scholar]
- 27. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. [DOI] [PubMed] [Google Scholar]
- 28. Borges FT, Reis LA, Schor N. Extracellular vesicles: structure, function, and potential clinical uses in renal diseases. Braz J Med Biol Res. 2013;46:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- 30. Mittelbrunn M, Gutierrez‐Vazquez C, Villarroya‐Beltri C, Gonzalez S, Sanchez‐Cabo F, Gonzalez MA, Bernad A, Sanchez‐Madrid F. Unidirectional transfer of microRNA‐loaded exosomes from T cells to antigen‐presenting cells. Nat Commun. 2011;2:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bellingham SA, Guo BB, Coleman BM, Hill AF. Exosomes: vehicles for the transfer of toxic proteins associated with neurodegenerative diseases? Front Physiol. 2012;3:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campbell TD, Khan M, Huang MB, Bond VC, Powell MD. HIV‐1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethn Dis. 2008;18:S2–S14. [PMC free article] [PubMed] [Google Scholar]
- 33. Jaworski E, Narayanan A, Van Duyne R, Shabbeer‐Meyering S, Iordanskiy S, Saifuddin M, Das R, Afonso PV, Sampey GC, Chung M, et al. Human T‐lymphotropic virus type 1‐infected cells secrete exosomes that contain Tax protein. J Biol Chem. 2014;289:22284–22305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard‐Herman M, Herrington D, et al. Guidelines for the ultrasound assessment of endothelial‐dependent flow‐mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39:257–265. [DOI] [PubMed] [Google Scholar]
- 35. Li R, Jiang XX, Zhang LF, Liu XM, Hu TZ, Xia XJ, Li M, Xu CX. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by Helicobacter pylori infection. Mediators Inflamm. 2017;2017:4927964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sahara S, Sugimoto M, Vilaichone RK, Mahachai V, Miyajima H, Furuta T, Yamaoka Y. Role of Helicobacter pylori CagA EPIYA motif and VacA genotypes for the development of gastrointestinal diseases in Southeast Asian countries: a meta‐analysis. BMC Infect Dis. 2012;12:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malfertheiner P, Megraud F, O'Morain CA, Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY, Rokkas T, et al. Management of Helicobacter pylori infection–the Maastricht IV/Florence Consensus Report. Gut. 2012;61:646–664. [DOI] [PubMed] [Google Scholar]
- 38. Dick‐Hegedus E, Lee A. Use of a mouse model to examine anti‐Helicobacter pylori agents. Scand J Gastroenterol. 1991;26:909–915. [DOI] [PubMed] [Google Scholar]
- 39. Cammarota G, Cannizzaro O, Tursi A, Papa A, Gasbarrini A, Cuoco L, Cianei R, Armuzzi A, Fedeli P, Fedeli G, et al. One‐week therapy for Helicobacter pylori eradication: ranitidine bismuth citrate plus medium‐dose clarithromycin and either tinidazole or amoxycillin. Aliment Pharmacol Ther. 1998;12:539–543. [DOI] [PubMed] [Google Scholar]
- 40. Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, Wang Y, Zingarelli B, Peng T, Fan GC. Blockade of exosome generation with GW4869 dampens the sepsis‐induced inflammation and cardiac dysfunction. Biochim Biophys Acta. 2015;1852:2362–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang X, Gu H, Huang W, Peng J, Li Y, Yang L, Qin D, Essandoh K, Wang Y, Peng T, et al. Hsp20‐mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes. 2016;65:3111–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spiers A, Padmanabhan N. A guide to wire myography. Methods Mol Med. 2005;108:91–104. [DOI] [PubMed] [Google Scholar]
- 43. Pojoga LH, Yao TM, Opsasnick LA, Garza AE, Reslan OM, Adler GK, Williams GH, Khalil RA. Dissociation of hyperglycemia from altered vascular contraction and relaxation mechanisms in caveolin‐1 null mice. J Pharmacol Exp Ther. 2014;348:260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;3:3–22. [DOI] [PubMed] [Google Scholar]
- 45. Shimoda A, Ueda K, Nishiumi S, Murata‐Kamiya N, Mukai SA, Sawada S, Azuma T, Hatakeyama M, Akiyoshi K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci Rep. 2016;6:18346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gomari H, Forouzandeh MM, Soleimani M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco Targets Ther. 2018;11:5753–5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Justus CR, Leffler N, Ruiz‐Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brunner H, Cockcroft JR, Deanfield J, Donald A, Ferrannini E, Halcox J, Kiowski W, Luscher TF, Mancia G, Natali A, et al. Endothelial function and dysfunction. Part II: association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J Hypertens. 2005;23:233–246. [DOI] [PubMed] [Google Scholar]
- 49. Gimbrone MJ, Garcia‐Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rasmi Y, Rouhrazi H, Khayati‐Shal E, Shirpoor A, Saboory E. Association of endothelial dysfunction and cytotoxin‐associated gene A‐positive Helicobacter pylori in patients with cardiac syndrome X. Biomed J. 2016;39:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blum A, Tamir S, Mualem K, Ben‐Shushan RS, Keinan‐Boker L, Paritsky M. Endothelial dysfunction is reversible in Helicobacter pylori‐positive subjects. Am J Med. 2011;124:1171–1174. [DOI] [PubMed] [Google Scholar]
- 52. Coskun S, Kasirga E, Yilmaz O, Bayindir P, Akil I, Yuksel H, Polat M, Sanlidag T. Is Helicobacter pylori related to endothelial dysfunction during childhood? Pediatr Int. 2008;50:150–153. [DOI] [PubMed] [Google Scholar]
- 53. Kalali B, Mejias‐Luque R, Javaheri A, Gerhard M. H. pylori virulence factors: influence on immune system and pathology. Mediators Inflamm. 2014;2014:426309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu Y, Tao Z, Song C, Jia Q, Bai J, Zhi K, Qu L. Overexpression of YKL‐40 predicts plaque instability in carotid atherosclerosis with CagA‐positive Helicobacter pylori infection. PLoS One. 2013;8:e59996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Diomedi M, Stanzione P, Sallustio F, Leone G, Renna A, Misaggi G, Fontana C, Pasqualetti P, Pietroiusti A. Cytotoxin‐associated Gene‐A‐positive Helicobacter pylori strains infection increases the risk of recurrent atherosclerotic stroke. Helicobacter. 2008;13:525–531. [DOI] [PubMed] [Google Scholar]
- 56. Kalani M, Hodjati H, GhamarTalepoor A, Samsami DA, Doroudchi M. CagA‐positive and CagA‐negative Helicobacter pylori strains differentially affect the expression of micro RNAs 21, 92a, 155 and 663 in human umbilical vein endothelial cells. Cell Mol Biol (Noisy‐le‐grand). 2017;63:34–40. [DOI] [PubMed] [Google Scholar]
- 57. Tobin NP, Henehan GT, Murphy RP, Atherton JC, Guinan AF, Kerrigan SW, Cox D, Cahill PA, Cummins PM. Helicobacter pylori‐induced inhibition of vascular endothelial cell functions: a role for VacA‐dependent nitric oxide reduction. Am J Physiol Heart Circ Physiol. 2008;295:H1403–H1413. [DOI] [PubMed] [Google Scholar]
- 58. Aspholm M, Olfat FO, Norden J, Sonden B, Lundberg C, Sjostrom R, Altraja S, Odenbreit S, Haas R, Wadstrom T, et al. SabA is the H. pylori hemagglutinin and is polymorphic in binding to sialylated glycans. PLoS Pathog. 2006;2:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, Ricci V, Solcia E. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori . Gastroenterology. 2007;132:1009–1023. [DOI] [PubMed] [Google Scholar]
- 60. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, Baty CJ, Gibson GA, Erdos G, Wang Z, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena‐Esteves M, Curry WJ, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tafreshi M, Guan J, Gorrell RJ, Chew N, Xin Y, Deswaerte V, Rohde M, Daly RJ, Peek RJ, Jenkins BJ, Davies EM, et al. Helicobacter pylori type IV secretion system and its adhesin subunit, CagL, mediate potent inflammatory responses in primary human endothelial cells. Front Cell Infect Microbiol. 2018;8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nazligul Y, Aslan M, Horoz M, Celik Y, Dulger AC, Celik H, Erel O. The effect on serum myeloperoxidase activity and oxidative status of eradication treatment in patients Helicobacter pylori infected. Clin Biochem. 2011;44:647–649. [DOI] [PubMed] [Google Scholar]
- 64. Boger RH. Asymmetric dimethylarginine (ADMA): a novel risk marker in cardiovascular medicine and beyond. Ann Med. 2006;38:126–136. [DOI] [PubMed] [Google Scholar]
- 65. Boger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. 2003;59:824–833. [DOI] [PubMed] [Google Scholar]
- 66. Aydemir S, Eren H, Tekin IO, Harmandar FA, Demircan N, Cabuk M. Helicobacter pylori eradication lowers serum asymmetric dimethylarginine levels. Mediators Inflamm. 2010;2010:685903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Figures S1A, S1–S4