

Abstract
Background
Spontaneously hypertensive rats of the stroke‐prone line (SHR‐A3) develop hypertensive renal disease as a result of naturally occurring genetic variation. Our prior work identified a single‐nucleotide polymorphism unique to SHR‐A3 that results in truncation of the carboxy terminus of STIM1. The SHR‐B2 line, which is also hypertensive but resists hypertensive renal injury, expresses the wild‐type STIM1. STIM1 plays a central role in lymphocyte calcium signaling that directs immune effector responses. Here we show that major defects in lymphocyte function affecting calcium signaling, nuclear factor of activated T cells activation, cytokine production, proliferation, apoptosis, and regulatory T‐cell development are present in SHR‐A3 and attributable to STIM1.
Methods and Results
To assess the role of Stim1 variation in susceptibility to hypertensive renal injury, we created a Stim1 congenic line, SHR‐A3(Stim1‐B2), and STIM1 function was rescued in SHR‐A3. We found that Stim1 gene rescue restores disturbed lymphocyte function in SHR‐A3. Hypertensive renal injury was compared in SHR‐A3 and the SHR‐A3(Stim1‐B2) congenic line. Histologically assessed renal injury was markedly reduced in SHR‐A3(Stim1‐B2), as were renal injury biomarker levels measured in urine. Stim1 deficiency has been linked to the emergence of antibody‐mediated autoimmunity. Renal glomerular immunoglobulin deposition was greater in SHR‐A3 than SHR‐B2 and was reduced by Stim1 congenic substitution. Serum anti–double‐stranded DNA antibody titers in SHR‐A3 were elevated compared with SHR‐B2 and were reduced in SHR‐A3(Stim1‐B2).
Conclusions
Stim1 deficiency in lymphocyte function originating from Stim1 truncation in SHR‐A3 combines with hypertension to create end organ disease and may do so as a result of antibody formation.
Keywords: autoimmunity, hypertension, immunoglobulin, renal disease, spontaneously hypertensive rat
Subject Categories: Genetics, Hypertension
Clinical Perspective
What Is New?
A single base change in Stim1 creates susceptibility to hypertensive renal injury in spontaneously hypertensive rats.
The Stim1 mutation disrupts calcium signaling in lymphocytes and impairs T‐cell effector and regulatory function.
Defects in T‐cell function may promote autoantibody formation and antibody‐mediated renal injury.
What Are the Clinical Implications?
Identification of genetic variants that enhance susceptibility to hypertensive renal disease in rodent models can uncover functional pathways that may be involved in disease pathogenesis in humans.
Introduction
Hypertension increases the risk of progressive renal disease. This risk is strongly influenced by heritable factors.1, 2, 3 Hypertension is common, but not all hypertensives will experience accelerated loss of renal function. Presence of a first‐degree relative with end‐stage renal disease significantly enhances the risk that hypertensive nephrosclerosis will culminate in end‐stage renal disease.3 Among blacks, the best predictor of individual dialysis risk is the existence of a close relative who has experienced end‐stage renal disease.4 These observations indicate the role of heritable genetic variation in disease pathogenesis. An independent genetic susceptibility to risk of renal disease in hypertension is also demonstrated in the spontaneously hypertensive rat (SHR) model in which similarly hypertensive SHR lines differ in their susceptibility to renal disease.5, 6 The pathogenic mechanism of hypertensive renal injury is poorly understood. Consequently, efforts are under way to dissect out the causative genetic variation that creates disease risk in the expectation that such insight may uncover disease mechanisms.7
In this study we investigated the genetic mechanism of hypertensive renal injury in the SHR by investigation of 2 SHR lineages, injury‐susceptible SHR‐A3 (also known as the stroke‐prone SHR) and SHR‐B2, a line that resists hypertensive end‐organ disease.5, 8, 9 Although these lines have a divergent heritable risk for progressive renal disease, they are 87% genetically identical and share in common alleles that create hypertension.7 Our previous studies of this model have suggested a multigene inheritance of risk of hypertensive renal injury involving genetic variation affecting immune function.5, 8 The immunoglobulin heavy chain gene is highly divergent between these SHR lines, and transfer of this immunoglobulin genetic variation from resistant to susceptible SHR lines reduces hypertensive renal injury, indicating a role for antibodies in this disease.9, 10
Whole‐genome sequence analysis of SHR‐A3 and SHR‐B2 lines led to the discovery of a deleterious mutation affecting the gene Stim1 in SHR‐A3.7 STIM1 is an endoplasmic reticulum (ER) Ca2+ sensor that plays a central role in lymphocyte effector and regulatory function.11 Stimulation of T‐ (TCR) or B‐cell receptors induces the rapid emptying of Ca2+ from lymphocyte ER stores. Ca2+ store depletion activates STIM1, which triggers opening of the plasma membrane (PM) calcium channel encoded by Orai1, thereby facilitating a large and sustained influx of extracellular Ca2+.12, 13 This process of capacitative, or store‐operated, Ca2+ entry is required for activation of the protein phosphatase calcineurin. Calcineurin activation results in dephosphorylation and nuclear localization of nuclear factor of activated T cells (NFAT), a key transcription factor that modulates cytokine production and coordinates T‐cell immune functions and proliferation.14 STIM1 activation is also necessary for the cellular metabolic reprogramming associated with lymphocyte activation.15 In the present article we characterize the immune phenotypes arising from Stim1 mutation. We rescued the Stim1 mutation in SHR‐A3 by replacement of the defective allele with wild‐type Stim1 and determined the effects of rescue on immune phenotypes and the emergence and extent of hypertensive renal injury. Because autoantibody‐mediated immunity has been reported in Stim1 deletion,16, 17, 18 we have sought evidence of autoantibody formation in SHR‐A3.
Materials and Methods
Data Availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animals and Treatments
The Institutional Animal Welfare Committee prospectively reviewed and approved all animal experiments and protocols. Studies were performed on male animals from the injury‐susceptible spontaneously hypertensive‐A3 (SHR‐A3) and the injury‐resistant SHR‐B2 rat lines, previously rederived and maintained in our Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)‐approved specific pathogen‐free facility. These lines and their origins before transfer to our laboratory have been recorded at the Rat Genome Database (https://rgd.mcw.edu/rgdweb/search/strains.html), which has applied the following identifiers: SHR‐A3, RGD ID=8142383, Symbol=SHRSP/BbbUtx; SHR‐B2, RGD ID=8142385, Symbol=SHR/Utx. Animals were provided a standard rodent chow diet and drinking water ad libitum. Animals aged 18 to 20 weeks were used for in vitro immune phenotyping experiments.
Stim1 Congenic Line Creation and Confirmation
The Stim1 mutation resides in an extended haplotype block of identity by descent in which the same genetic background arising from a single common ancestor has been fixed in both SHR‐A3 and SHR‐B2. The Stim1‐encompassing block extends for ≈45 Mb on chromosome 1 (chr1:163‐208 Mb) and, except for the Stim1 mutation, contains no other nonsynonymous coding sequence variants between SHR‐A3 and SHR‐B2 as determined using Annovar software19 (Open Bioinformatics Foundation, Basel, Switzerland) to analyze variant call format files derived from alignment to the rat reference genome of SHR‐A3 and SHR‐B2 genome sequences. Congenic transfer across this block allows rescue of the defective SHR‐A3 Stim1 allele while avoiding transmission of any other coding sequence variation. Congenic line construction began by crossing of parental line animals (SHR‐A3 males and SHR‐B2 females) to generate the F1 progeny. These progeny were backcrossed into SHR‐A3 animals for 5 generations. Backcrossed animals were genotyped at each generation using a previously described panel of ≈200 single‐nucleotide polymorphism (SNP) markers to allow speedy congenic selection of optimal animals (highest loss of SHR‐B2 background alleles while retaining the introgressed SHR‐B2 Stim1 haplotype block).9 The final congenic line was created by mating male and female animals from the previous backcross and selecting progeny that were homozygous for SHR‐B2 alleles at the Stim1 locus and for SHR‐A3 alleles at the other genotyped loci.
Lymphocyte Isolation
Peripheral blood was collected from the abdominal aorta of isoflurane‐anesthetized rats (16‐18 weeks old). Lymphocytes were isolated from whole blood using lymphocyte separation medium (Lonza, Allendale, NJ) according to the manufacturer's directions. Briefly, whole blood was diluted 1:1 with sterile buffered saline and layered on top of lymphocyte separation medium at a ratio of 3:2 followed by centrifugation at 400g for 20 minutes at 4°C. Lymphocytes were collected and washed twice followed by centrifugation at 70g for 10 minutes to remove platelets. For isolation of splenocytes, spleens were cut into small fragments and passed through a 70‐μm cell strainer into a 50‐mL conical tube. Collected cells were washed, the pellet was resuspended in 5 mL erythrocyte lysis buffer, and lysis was carried out for 10 minutes at 25°C with gentle shaking. After red cell lysis, splenocytes were collected by centrifugation. Lymphocytes were collected from abdominal aortic lymph nodes in a similar manner. For renal lymphocyte counts, we followed the isolation method detailed by Martina et al.20 Cells were either resuspended in complete Roswell Park Memorial Institute (RPMI)‐1640 medium (RPMI containing 10% FBS, 100 U/mL penicillin‐streptomycin, 4 mmol/L l‐glutamine, 1 mmol/L sodium pyruvate, 1% nonessential amino acids, 1% RPMI vitamins, 10 mmol/L HEPES, and 50 μmol/L β‐mercaptoethanol), or in flow cytometry staining buffer, based on downstream applications.
Flow Cytometry
Lymphocytes were stained according to standard protocols. Briefly, for staining surface markers, cells were resuspended in fluorescence‐activated cell‐sorting (FACS) buffer at a concentration of 2×106 cells/mL. Nonspecific antibody binding was blocked with anti–rat CD32 antibody (BD Biosciences, Franklin Lakes, NJ) followed by staining with specific antibodies (1:1000, all from BioLegend, San Diego, CA) for 60 minutes on ice in the dark. Antibodies used for flow cytometry included FITC‐CD3, APC‐CD3, PECy7‐CD4, PE‐CD25, FITC‐CD8, APC‐CD45RA, and Alexa Fluor 647‐Foxp3. Intracellular Foxp3 staining was performed after cell surface staining using the True Nuclear transcription factor staining kit (BioLegend) according to the manufacturer's protocol. Stained cells were washed twice and resuspended in FACS buffer and analyzed or sorted on a BD FACSAria II flow cytometer (BD Biosciences).
Western Blotting for STIM1 Protein Expression
Lymphocyte lysates were prepared by sonication in 300 μL of cell lysis buffer (Cell Signaling Technologies, Danvers, MA) with protease and phosphatase inhibitors (Sigma Aldrich, St. Louis, MO). Protein concentration was determined using the bicinchoninic acid assay kit (ThermoFisher Scientific, Waltham, MA). Samples were diluted with lysis buffer, denatured, and reduced in Laemmli buffer supplemented with β‐mercaptoethanol at 5% final concentration for 10 minutes at 100°C to obtain the final protein concentration of 2 μg/μL. Lysate proteins (30 μL/lane) were separated on 4% to 20% polyacrylamide gels at 70 V for 4 hours and transferred to nitrocellulose membrane for 1.5 hours at 100 V. Membranes were incubated with primary antibodies for 2 hours at room temperature. We used primary antibodies against the N‐terminus of STIM1 (1:3000 dilution, Sigma‐Aldrich, #S6072) and anti–β‐actin (1:2000, BioLegend). Membranes were washed 3 times in PBS‐Tween and incubated with peroxidase‐conjugated goat anti–rabbit secondary antibodies (1:5000, Santa Cruz Biotechnology, Dallas, TX) for 1 hour at room temperature. Blots were quantified using ImageJ 1.48 software (NIH, Bethesda, MD). The intensities of the studied protein bands were normalized to the intensities of the corresponding actin bands.
Store‐Operated Calcium Entry
Flow cytometry with the nonratiometric dye Fluo‐3AM (Promokine, Heidelberg, Germany) was used to assess store‐operated calcium entry (SOCE) in peripheral blood lymphocytes. Lymphocytes (2×106 cells/mL) were stained for surface CD3 and CD4 as described above and then loaded with Fluo‐3AM (final concentration 1 μmol/L) for 30 minutes at 37°C in complete RPMI. Cells were washed and resuspended in 2 mL Ca2+‐free Ringer solution. Fluorescence measurements were acquired on a FACSCalibur (BD Biosciences) flow cytometer. CD4+ T cells were gated using cell surface markers. Baseline Ca2+ measurements were made for 2 minutes followed by the addition of the ER Ca‐ATPase inhibitor thapsigargin (2 μmol/L) for 5 minutes. SOCE was induced by the addition of 2 mmol/L Ca2+, and data were recorded for 5 minutes. For TCR‐induced SOCE, labeled lymphocytes were incubated with biotin–anti‐CD3 (1:1000, BD Biosciences) for 5 minutes before baseline measurements were done. After recording of the baseline for 2 minutes, CD3 was cross‐linked by streptavidin (1:3000, ThermoFisher Scientific) followed by the addition of 2 mmol/L Ca2+ for 5 minutes to induce SOCE. Data were analyzed using FlowJo (Treestar, Ashland, OR) software. EGTA and ionomycin (2 μmol/L) were used to calculate maximal and minimal fluorescence values of Ca2+, and actual [Ca2+]i was calculated according to the equation: [Ca²⁺]i = Kd × {[F ‐ (Fmin)] × [(Fmax ‐ F)]} where F is the fluorescence of the indicator at the experimental concentration, Fmin is the fluorescence in the absence of calcium, and Fmax is the fluorescence of the indicator at saturated calcium concentration. The Kd for Fluo‐3 under our experimental conditions was 325 nM.
NFAT Nuclear Translocation
Peripheral blood lymphocytes (1×106 cells/mL) were stimulated with PMA (phorbol myristate acetate, 10 nmol/L) and ionomycin (2 μmol/L) for 1 hour at 37°C. Following treatment, cells were fixed with 4% paraformaldehyde in 0.1 phosphate buffer (Electron Microscopy Sciences, Hatfield, PA) for 10 minutes. Lymphocytes were then centrifuged in a cytospin for 5 minutes at 90g onto slides. Cells were then permeabilized by a 5‐minute incubation with 0.5% Triton X‐100 in PBS, followed by washing in PBS for 5 minutes. Nonspecific binding was blocked with Protein Block (BioGenex, San Ramon, CA) for 1 hour at room temperature. Cells were incubated with anti‐NFATc1 clone‐7A6 (BioLegend) overnight at 4°C followed by washing and a 1‐hour incubation at room temperature with Alexa 488‐conjugated goat anti–mouse IgG (BioLegend 405319). Nuclear counterstaining was performed with 5 mmol/L Draq5 (Cell Signaling Technology, Danvers, MA; 4084S) for 1 hour. Images were acquired with a ×63 oil‐immersion objective (NA1.4) of a Leica TCS SP5 confocal microscope. Single optical sections were obtained with high–numerical aperture lens (×63 with an additional ×2 software zoom) to determine the percentage of NFATc1‐nuclear cells. At least 200 cells were analyzed from each group with 3 independent replicates.
Cytokine Production in Stimulated T Cells
CD4+ T cells (2×105 cells) sorted by FACS from peripheral blood were seeded into 96‐well plates and stimulated with PMA (10 nmol/L) and ionomycin (2 μmol/L) (both from eBiosciences, San Diego, CA) for 4 hours at 37°C. For TCR‐induced cytokine production, cells were similarly seeded onto wells precoated with 5 μg/mL anti‐CD3 (clone G4.18, eBiosciences). Soluble anti‐CD28 (2.5 μg/mL, clone JJ319, BD Biosciences) was added for 24 hours at 37°C to activate T cells. Cells were also pretreated with the ORAI1 inhibitor Pyr6 (5 μmol/L, Tocris Biosciences, Bristol, UK) for 15 minutes before the addition of anti‐CD28. ELISA was used to quantify cytokine levels in the cell culture supernatants according to standard protocols. Briefly, plates were coated with primary antibodies to interferon‐γ (BioLegend) at a concentration of 2 μg/mL followed by incubation with cell culture supernatants for 2 hours at 25°C. The total bound antigen was quantified using biotinylated secondary antibodies (1:1000) and visualized using the horseradish peroxidase/streptavidin‐TMB system (Bethyl Laboratories, Montgomery, TX). Interleukin (IL)‐2 was quantified in a similar manner using a kit‐based assay (R&D Systems, Minneapolis, MN).
T‐Cell Proliferation and Activation‐Induced Cell Death
To assess T‐cell proliferation, CD3+ lymphocytes (1×106 cells/mL) were incubated with 5 μmol/L carboxyfluorescein succinimidyl ester (CFSE, BioLegend) for 20 minutes at 37°C protected from light before costimulation with CD3/CD28. CFSE fluorescence was quenched by the addition of 5 mL complete RPMI, and cells were collected by centrifugation at 400g for 5 minutes. CFSE‐loaded cells were resuspended in prewarmed complete RPMI medium and were stimulated with CD3/CD28 for 48 hours as described above. At the end of the proliferation protocol, T cells were stained for surface CD4, and CFSE dilution was assessed by flow cytometry (BD FACSAria II). For activation‐induced cell death, T cells were costimulated with CD3/CD28 followed by a second stimulation with CD3/CD28 for 24 hours. At the end of the restimulation, apoptotic cells were identified using the FITC Annexin V Apoptosis detection kit (BioLegend) according to the manufacturer's protocol.
Assessment of Renal Injury
Periodic acid Schiff–stained kidney sections were scored for glomerular and tubular injury in 40‐week‐old animals as previously described.5, 8 Kidneys were collected by ventral laparotomy under isoflurane anesthesia at the end of the treatment period. Kidneys were cut into radial sections, fixed in 4% buffered formalin, and embedded in paraffin using standard techniques. Four‐micrometer sections were stained with periodic acid Schiff stain and were evaluated for renal injury by an experienced renal pathologist who was blinded to the treatment groups. Injury scores were obtained by randomly sampling 20 glomeruli and 10 cortical tubular fields per animal. Scores were graded according to degree of injury on a scale from 1 to 5. Glomerular injury was graded as follows: (0) no histologic abnormality; (1) mild mesangial thickening only; (2) moderate mesangial expansion without thickened capillary loops; (3) severe mesangial expansion, thickened capillary loops, or segmental sclerosis; (4) global or >50% segmental sclerosis. Tubulointerstitial disease was graded as (0) no injury; (1) mild patchy fibrosis, infiltrate <10 cells/high‐power field; (2) mild to moderate fibrosis, patchy tubular atrophy, infiltrate >10 cells/high‐power field; (3) diffuse tubular atrophy, moderate fibrosis, proteinaceous casts, diffuse interstitial infiltrate; (4) severe interstitial fibrosis with strong diffuse cellular infiltrates or tubular atrophy.
Urinary albumin excretion was assessed as previously described.5 Urine was collected directly from the bladder at the time of the telemetry probe implantation surgery and again at the end of the study period. Urinary albumin excretion was assessed as the ratio of urinary albumin:creatinine. Urinary creatinine concentrations were quantified by high‐performance liquid chromatography. A rat‐specific ELISA kit (Bethyl Laboratories) was used to determine urinary albumin concentrations according to the manufacturer's protocols.
For urinary biomarker analysis, we investigated the excretion of KIM‐1 (kidney injury molecule 1, Havcr1 [hepatitis A virus cell receptor 1]), Lcn2 (lipocalin 2, also known as NGAL [neutrophil gelatinase‐associated lipocalin]), and OPN (osteopontin) using the Kidney Injury Panel 1 (rat) Assay Kit manufactured by Meso Scale Diagnostics (Gaithersburg, MD). The multiplex assay plate was read on a Meso Scale Diagnostics SECTOR Imager 2400 electrochemiluminescence plate reader. Biomarkers were determined in 8 animals per group and normalized to urine creatinine levels measured by high‐performance liquid chromatography as described above.
Anti–Double‐Stranded DNA ELISA
Activated calf thymus DNA (Sigma, D4522) was bound to UV‐activated Immunlon 2B ELISA plates and washed 5 times. Diluted serum samples (1:30) were incubated for 1 hour at room temperature and subsequently washed, and horseradish peroxidase–conjugated anti–rat IgM or IgG was added to the wells for a further 1 hour before rinsing and developing with TMB substrate. Reactions were stopped with sulfuric acid, and the developed color measured at 450 nmol/L by plate reader.
Renal Immunofluorescence
Detection of glomerular immunoglobulin deposition was performed on frozen tissue sections. Tissue was incubated with AlexaFluor488‐tagged goat anti–rat IgM (Southern Biotech, Inc, Birmingham, AL) or biotin‐tagged goat anti–rat IgG (Bio‐Rad Laboratories, Inc, Hercules, CA) for 1 hour at a dilution of 1:500. FITC‐streptavidin (1:1000 for 1 hour) was used as the detection antibody. Twenty glomeruli from each rat were assessed by an investigator blinded to the rat genotypes. Images were captured on a Nikon Eclipse TE2000E wide‐field fluorescence microscope at a magnification ×400 and with an exposure time of 500 ms for all images.
HuProt Antigen Microarray Studies
These studies were performed in the Genomics and Microarray Core Facility, University of Texas Southwestern Medical School, Dallas, TX. HuProt v3.1 arrays (CDI Laboratories, Mayaguez, PR) spotted in duplicate with more than 20 000 full‐length recombinant human proteins representing >80% of the human proteome were used according to the manufacturer's published protocol. Each individual array was incubated with pooled serum collected from 6 individual animals of each SHR line (SHR‐A3, SHR‐B2, SHR‐A3(Stim1‐B2) congenic) at 40 weeks of age. Arrays were developed with detection antibodies targeting rat IgM and rat IgG (goat polyclonal anti–rat IgM‐heavy chain conjugated with Alexa Fluor 647 and goat polyclonal anti–rat IgG [H+L] conjugated with Cyanine3, both from Invitrogen, Carlsbad, CA) and read on a fluorescent slide reader (GenePix 4400A Microarray Scanner, Molecular Devices, Sunnyvale, CA). Z score normalization was used to permit comparison of autoantibody signals across arrays. Intra‐array reproducibility was assessed by analyzing correlation of signals between duplicate spots.
Blood Pressure Measurements
At 16 to 17 weeks of age, male SHR‐A3 and SHR‐A3(Stim1‐B2) rats were implanted with radiotelemetry devices (Data Sciences, St. Paul, MN) to record blood pressure (BP), as described.21 Animals were allowed to recover for at least 1 week before initiating BP recordings. BP was measured by continuous sampling for 30 seconds every 30 minutes for 24 hours at 18 weeks of age, before the emergence of histological renal injury,8 to determine whether the Stim1 mutation affects baseline (pre–renal injury) BP.
Statistical Analyses
Comparison of data from SHR‐A3 and SHR‐B2 rats was done by Student t test using Prism 7 software (GraphPad Software, La Jolla, CA). ANOVA with Tukey post‐hoc test was used to compare data from multiple groups. A value of P<0.05 was considered statistically significant, with n=5 to 7 independent replicates per group.
Results
Stim1‐Rescued SHR‐A3 Congenic Line Creation
Figure 1 indicates the location of identical genomic blocks that are shared by descent in SHR‐A3 and SHR‐B2 and the position of the Stim1 gene. These haplotype blocks were originally located by high‐density SNP analysis. Subsequently we have obtained whole‐genome sequence information for these rat lines that, in addition to identifying the Stim1 mutation, allowed us to confirm the distribution of identity by descent in the genomes of the 2 rat lines. Table S1 provides a list of coding sequence variations from the rat reference genome sequence that are present in SHR‐A3 and SHR‐B2. This listing was computed by Annovar software19 from Illumina short‐read resequencing of the SHR‐A3 and SHR‐B2 genomes with >45X read depth coverage.9 In this block from chr1:156 969 388‐198 720 287 (Rn6 assembly of the rat reference genome), the only coding base location in which SHR‐A3 and SHR‐B2 differ from each other is in Stim1 at chr1:167 531 867 (Rn6) in which SHR‐A3 has a SNP creating a premature stop codon. All other variations from the rat reference sequence in this block are shared by both SHR‐A3 and SHR‐B2.
Figure 1.
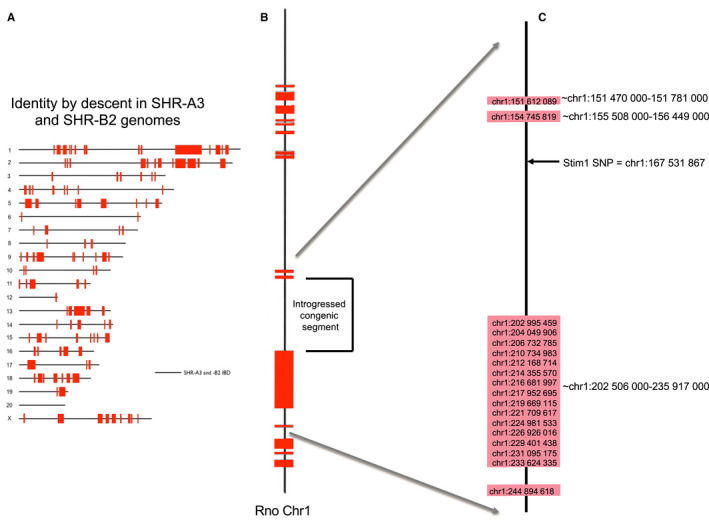
Chromosome scale view of congenic transfer of an identical‐by‐descent (IBD) haploblock containing wild‐type Stim1 from SHR‐B2 in the SHR‐A3 genetic background. A, Regions of genetic IBD sequences are shown for the SHR‐A3 and SHR‐B2 genomes (20 autosomes and X chromosome. This block was initially identified by genotyping ≈10 000 genome‐wide SNPs. It has been confirmed, and its boundaries precisely defined, by whole‐genome sequencing. All SHR lines are the progeny of a single progenitor male and therefore lack ancestral Y chromosome variation). Red blocks indicate regions of the genome at which the 2 rat lines are descended from different ancestors. Solid black lines represent the remaining 87% of the genome that is IBD. B, Enlarged view of the rat chromosome 1 (Rno chr1) indicating the target haplotype block transferred from SHR‐B2 into the SHR‐A3 background. C, Detailed view of the approximate beginning and end points of the haplotype blocks surrounding the transferred segment (indicated to the right of the blocks and determined by examination of whole‐genome sequence alignments of SHR‐A3 and SHR‐B2). The genomic position of single‐nucleotide polymorphisms in this region that were genotyped in speed congenic construction are indicated above the colored blocks. Inheritance of the Stim1 wild‐type allele was determined by PCR amplification of the sequence including the polymorphic site. Stim1 genotype was determined by restriction digestion of the PCR products with AluI, which digests the SHR‐A3 mutated sequence but not the wild‐type SHR‐B2 sequence. PCR indicates polymerase chain reaction; SHR, spontaneously hypertensive rat; SNP, single‐nucleotide polymorphism.
Genotypes of SNPs present on rat Chr1 in SHR‐A3 and SHR‐B2 and used to initially identify the Stim1‐containing block are provided in Table S2 and demonstrate the distribution of identical‐by‐descent sequences on this chromosome. The Stim1 polymorphism creates a premature stop codon and is located at chr1:167 531 867 (Rn6) in a 45‐Mb block that is otherwise identical by descent between SHR‐A3 and SHR‐B2. Our previously described strategy of marker‐assisted genotyping of SHR‐B2 animals backcrossed onto the SHR‐A3 genetic background9 was used to create a congenic line in which the Stim1 haploblock from SHR‐B2 was transferred to SHR‐A3.
Effect of Stim1 Gene Rescue on Renal Injury
We used histological assessments and urinary albumin and injury biomarker excretion to evaluate hypertensive renal injury in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats. Figure 2 indicates the presence of significant glomerular and tubulointerstitial injury in 40‐week‐old SHR‐A3 when compared with age‐matched SHR‐B2 rats. Histological measures of renal injury were reduced to SHR‐B2 levels in the Stim1 rescue congenic line (Figure 3A). Urinary biomarker excretion provides an opportunity to evaluate ongoing injury to the renal epithelium, particularly proximal tubular cells. We assessed urinary levels of 3 established markers of renal injury (OPN, NGAL, KIM‐1) at 30 weeks of age and found a high level of divergence between biomarker excretion in SHR‐A3 and SHR‐B2 rats (Figure 3B). Stim1 congenic rats had markedly reduced levels of these markers of renal injury. Albuminuria, however, remained high in SHR‐A3(Stim1‐B2) rats (Figure 3C), which suggests that this element of renal injury may arise from regions of the SHR‐A3 genome outside the Stim1 locus. BP at 18 weeks, before the emergence of renal injury, was comparable between the SHR‐A3 and SHR‐A3(Stim1‐B2) rats (Table), which implies that the Stim1 mutation contributes susceptibility to glomerular and tubulointerstitial injury in SHR‐A3 independent of BP effects.
Figure 2.
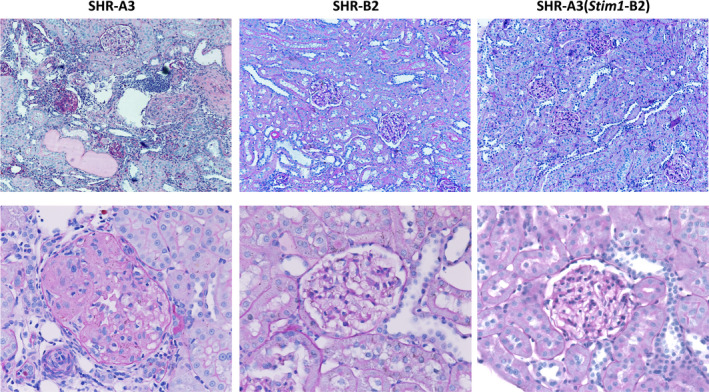
Effect of the Stim1 mutation on renal injury. Representative PAS‐stained kidney sections (upper panels, ×20; lower panels, ×40 magnification) from 40‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats. PAS indicates periodic acid Schiff stain.
Figure 3.
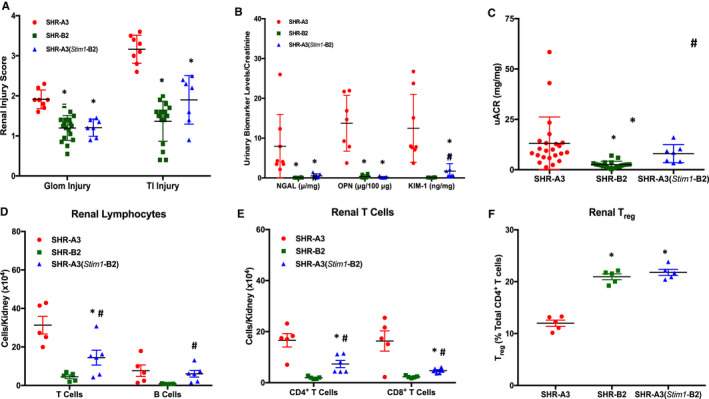
Renal injury and T‐cell infiltration in SHR‐A3, SHR‐B2 and SHR‐A3(Stim1‐B2). A, Glomerular (Glom) and tubulointerstitial (TI) injury scores, n=8 (SHR‐A3), 18 (SHR‐B2), 10 (SHR‐A3[Stim1‐B2]). B, Urinary renal injury biomarker levels for neutrophil gelatinase‐associated lipocalin (NGAL), osteopontin (OPN), and kidney injury molecule‐1 (KIM‐1) normalized to creatinine excretion in urine collected from 30‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats, n=8. C, Urinary albumin excretion normalized to urinary creatinine in 40‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats; n=23 (SHR‐A3), 19 (SHR‐B2), 14 (SHR‐A3[Stim1‐B2]). *P<0.05 vs SHR‐A3 and # P<0.05 vs SHR‐B2. Flow cytometric analysis of (D) T and B cells, (E) CD4+ and CD8+ T‐cell subsets, and (F) Tregs in kidneys from 40‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats; n=5 rats. *P<0.05 vs SHR‐A3 and # P<0.05 vs SHR‐B2. Treg indicates regulatory T cells; uACR, urinary albumin/creatinine ratio.
Table 1.
Blood Pressure in 18‐Week‐Old Rats
| Strain | SBP (mm Hg) | MBP (mm Hg) | DBP (mm Hg) | HR (bpm) |
|---|---|---|---|---|
| SHR‐A3 (n=10) | 194.6±6.6 | 170.1±5.8 | 145.3±6.4 | 318.7±7.9 |
| SHR‐A3(Stim1‐B2) (n=8) | 196.5±1.5NS | 166.8±1.8NS | 136.9±2.1NS | 310.5±4.9NS |
BP and HR were compared by Student t test where P<0.05 was considered statistically significant. HR indicates heart rate; NS, not statistically significant; SBP, MBP, DBP, systolic, mean, and diastolic arterial blood pressure.
SHR‐A3 kidneys were characterized by extensive T‐ and B‐cell infiltration (Figure 3D) compared with SHR‐B2. Absolute CD4+ and CD8+ T‐cell counts were significantly higher in SHR‐A3 kidneys than in SHR‐B2 and were markedly reduced by Stim1 gene rescue in the congenic line (Figure 3E). On the other hand, the relative percentages of renal CD3+CD4+CD25+Foxp3+ Tregs were higher in SHR‐B2 and SHR‐A3(Stim1‐B2) rats (Figure 3F).
Effect of the Stim1 Mutation on Lymphocyte Calcium Signaling and T‐Cell Development
To assess the contribution of Stim1 mutation to susceptibility to renal injury in SHR‐A3, we created a congenic Stim1 rescue line, SHR‐A3(Stim1‐B2). in which the defective Stim1 present in SHR‐A3 was replaced with the wild‐type Stim1 allele from SHR‐B2. The SHR‐A3 Stim1 nonsense mutation leads to the truncation of 46 amino acids from the C‐terminal domain.7 The mutation does not activate non–sense‐mediated RNA decay because it is located in the terminal exon of the gene. Western blotting was used to investigate STIM1 protein expression using an antibody targeting the N‐terminus of STIM1. SHR‐A3 rats had the expected reduction in apparent molecular weight of STIM1 compared with SHR‐B2 and SHR‐A3(Stim1‐B2) rats, which express the “wild type” STIM1 (Figure 4A). Protein abundance of STIM1 was comparable among SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) lines (Figure 4B). STIM1 antibodies targeting the deleted C‐terminus of STIM1 detected protein in SHR‐B2 but not SHR‐A3.7
Figure 4.
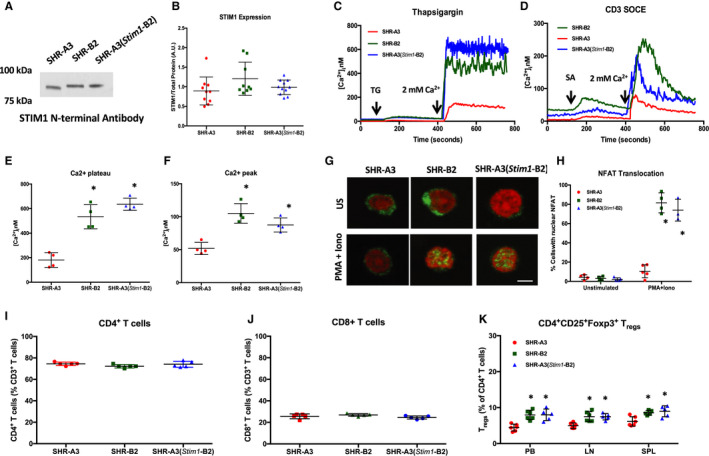
Effect of the Stim1 mutation on SOCE and NFAT activation in CD4+ T cells. A, Representative Western blot using N‐terminal–directed antibodies demonstrates the presence of STIM1 protein in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) T lymphocytes. B, Summary graph showing the results from densitometric analyses of STIM1 bands normalized to the intensities of total protein bands; n=5 per group. C, Average time course for [Ca2+]i influx in response to store depletion by thapsigargin (TG, 2 μmol/L) followed by Ca2+ readdition to induce SOCE. D, Average time course for [Ca2+]i influx in response to anti‐CD3 followed by CD3 cross‐linking with streptavidin (SA) and Ca2+ readdition to induce SOCE. E, Summary graph of sustained phases of TG‐induced Ca2+ influx. F, Summary graph showing peak [Ca2+]i levels in response to CD3 cross‐linking in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) CD4+ T cells. G, Confocal microscopy of NFATc1 nuclear translocation in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) lymphocytes stimulated for 60 minutes with PMA (10 nmol/L) and ionomycin (2 μmol/L). Green indicates NFATc1 staining, and red indicates nuclear stain using Draq5. H, Summary graph of 3 independent experiments showing the percentage of lymphocytes with NFAT nuclear translocation. Scale bar=1 μm. *P<0.05 vs SHR‐A3 with n=5 per group. Summary graphs of circulating (I) CD4+ and (J) CD8+ T‐cell frequencies as a percentage of total CD3+ T cells in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats. K, CD4+ CD25+Foxp3+ Treg cell frequencies as a percentage of total CD3+ T cells in peripheral blood (PB), aortic lymph nodes (LN) and spleens (SPL) from SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats. *P<0.05 vs SHR‐A3 with n=5 to 7 per group. Iono indicates ionomycin; NFAT, nuclear factor of activated T cells; PMA, phorbol myristate acetate; SOCE, store‐operated calcium entry; US, unstimulated.
The Stim1 mutation in SHR‐A3 deletes the downstream lysine‐rich polybasic carboxyl terminus of STIM1, but leaves intact the STIM1 region responsible for gating of ORAI1 capacitative calcium entry channel. The polybasic region has been shown to facilitate and stabilize coupling of STIM1‐ORAI1 and thus regulate the strength of the SOCE signal.22 Accordingly, we expected that SHR‐A3 T cells would demonstrate diminished SOCE in response to ER store depletion. Thapsigargin‐ and TCR‐induced SOCE were markedly reduced in CD4+ T cells from SHR‐A3 when compared with SOCE in SHR‐B2 and SHR‐A3(Stim1‐B2) T cells (Figure 4C through 4F). SOCE in SHR‐B2 and SHR‐A3(Stim1‐B2) CD4+ T cells was suppressed when opening of the ORAI1 channel was inhibited with Pyr6 (not shown), confirming that STIM1‐ORAI1 is the major pathway for SOCE in rat T cells. To test if deficient SOCE impairs calcineurin‐mediated dephosphorylation of NFAT, we examined NFAT nuclear translocation in response to activation of SOCE. Lymphocytes from SHR‐A3 rats have a significant impairment in NFAT activation compared with SHR‐B2 lymphocytes in response to ER Ca2+ depletion with PMA 10 nmol/L and ionomycin 2 μmol/L (Figure 4G and 4H). Defective NFAT activation was rectified by Stim1 gene rescue in the congenic line.
SOCE is dispensable for CD4+ and CD8+ T cell development.23 As expected, we found comparable levels of these T cell frequencies in our 3 rat strains (Figure 4I and 4J). In contrast, CD4+ T cell SOCE is required for T regulatory cell (Treg) differentiation.17, 23 CD4+CD25+Foxp3+ Treg numbers were significantly reduced in peripheral blood, spleens, and lymph nodes from SHR‐A3 (Figure 4K).
Effect of the Stim1 Mutation on CD4+ T Cell Effector and Regulatory Function
The production of cytokines, particularly IL‐2 and IFNγ, is an important SOCE‐dependent immune effector function of CD4+ T cells.23 SHR‐A3 CD4+ T cells produced markedly lower amounts of IL‐2 and IFNγ in response to TCR‐specific antibody stimulation using anti‐CD3/anti‐CD28 compared with SHR‐B2 rats (Figure 5A and 5B). This defect in cytokine production was rectified in T cells from the Stim1 rescued congenic line. Upregulation of IL‐2α (CD25) in response to TCR stimulation, a process largely under the transcriptional control of NFκB24 and independent of SOCE via STIM1‐ORAI1,23 was used as a marker of T‐cell activation.25 SHR‐A3 T cells were activated by CD3/CD28 costimulation for 24 hours. CD25 expression was comparable between SHR‐A3 and SHR‐B2 CD4+ T cells and was unaffected by inhibition of STIM1‐ORAI1 signaling with Pyr6 (Figure 5C).
Figure 5.
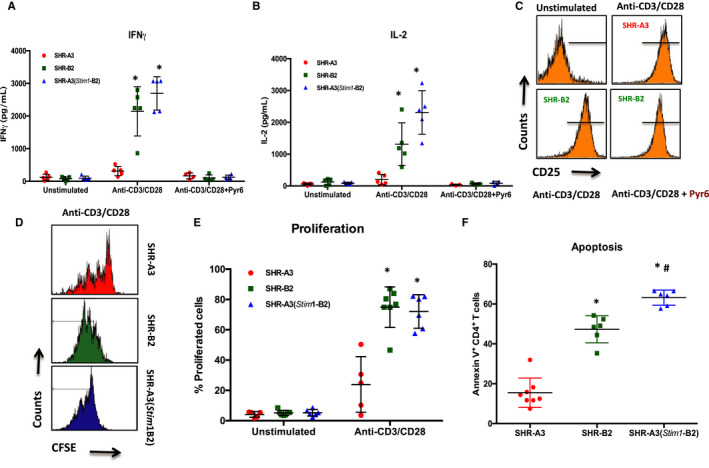
Effect of the Stim1 mutation on CD4+ T‐cell function. Quantification of (A) IFNγ and (B) IL‐2 production by CD4+ T cells from SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats in response to anti‐CD3/anti‐CD28 costimulation for 24 hours under nonpolarizing conditions. A subset of cells was pretreated with the ORAI1 channel blocker Pyr6 (5 μmol/L) for 15 minutes before the addition of CD28. C, Upregulation of the activation marker CD25 on CD4+ T cells after 18 hours of TCR stimulation with anti‐CD3/anti‐CD28. Pretreatment of T cells with Pyr6 did not prevent CD25 expression. Data are representative of 3 independent experiments. D, Histograms showing the intensity peaks of CFSE as an indicator of CD4+ T‐cell proliferation in response to TCR stimulation using anti‐CD3/anti‐CD28 for 72 hours in (upper panels) SHR‐A3, and (lower panels) SHR‐B2 and SHR‐A3(Stim1‐B2) cells. Each peak represents 1 cell division in stimulated cells. E, Summary graph of total percentage of CD4+ T cells undergoing at least 1 cell division. F, Summary graph of Annexin V+ CD4+ T cells from SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats in response to 24‐hour restimulation with anti‐CD3/anti‐CD28 after the initial 72‐hour TCR stimulation with anti‐CD3 and anti‐CD28. *P<0.05 vs SHR‐A3 and # P<0.05 vs SHR‐B2 with n=6 per group. CFSE indicates carboxyfluorescein succinimidyl ester; IFNγ, interferon‐γ; IL‐2, interleukin‐2; TCR, T‐cell receptor.
Proliferation induced in CD4+ T cells by CD3/CD28 costimulation for 72 hours was assessed by the CFSE dilution method23 (Figure 5D). At the end of the stimulation protocol, 70% to 80% of SHR‐B2 and SHR‐A3(Stim1‐B2) T cells had undergone at least 1 cell division. CD4+ T cells from SHR‐A3 exhibited a weak proliferative response, and only about 20% of cells underwent proliferation. SHR‐A3 CD4+ T cells failed to proliferate further in response to TCR activation, whereas SHR‐B2 and SHR‐A3(Stim1‐B2) cells continued to 2 or 3 cell divisions (Figure 5E). In addition to T‐cell effector functions, STIM1‐driven SOCE mediates activation‐induced T‐cell apoptosis.26, 27 To mimic sustained inflammatory conditions in vitro, CD4+ T cells were restimulated with anti‐CD3/anti‐CD28 for 24 hours after an initial 72‐hour TCR‐induced activation. Restimulation resulted in significantly lower counts of annexin V+ apoptotic cells among SHR‐A3 than SHR‐B2 and SHR‐A3(Stim1‐B2) CD4+ T cells (Figure 5F). Thus, the impaired proliferative responses to TCR stimulation in SHR‐A3 may be countered by suppression of apoptotic responses during sustained TCR stimulation.
Antibody Formation and STIM1 Function
STIM1 function is essential for antibody affinity maturation, and its deficiency is associated with the emergence of autoantibodies.17 Animals were studied at 40 weeks of age to determine whether renal injury could be linked to deposition of immunoglobulins in the glomerulus. Glomerular IgM and IgG were detectable in all 3 lines (Figure 6A); however, staining intensity for both immunoglobulins was higher in SHR‐A3 kidneys than in SHR‐B2. Glomerular staining for IgM and IgG in the SHR‐A3(Stim1‐B2) line was intermediate between the level observed in SHR‐A3 and that in SHR‐B2.
Figure 6.
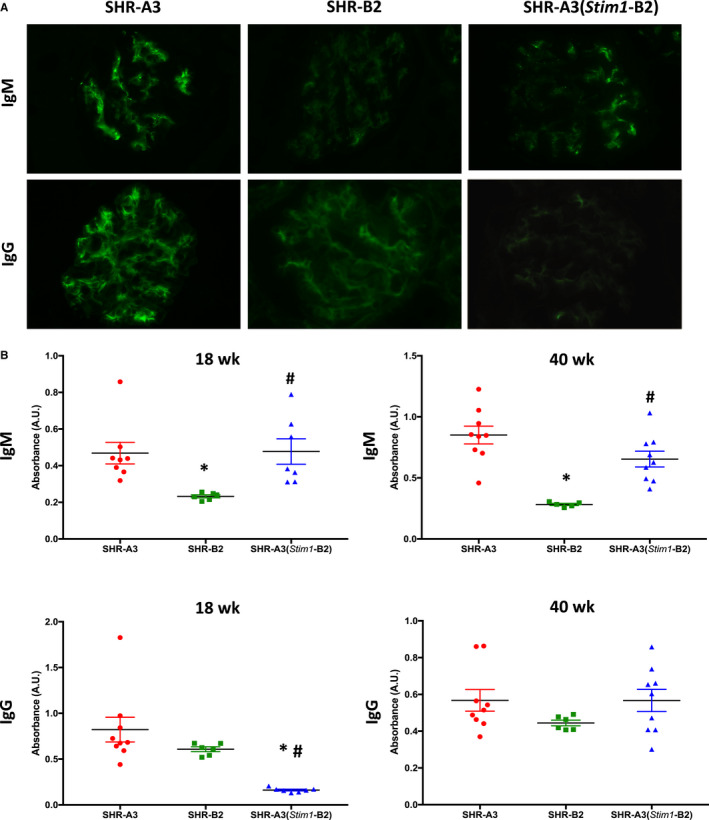
Renal IgM and IgG in SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2). A, Representative images of immunofluorescent detection of IgM (upper panels) and IgG (lower panels) in kidneys from 40‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats; n=5 to 6 rats. Original magnification ×400 and with an exposure time of 500 ms for all images. B, Serum ELISAs of anti–ds‐DNA for IgM (upper panels) and IgG (lower panels) in 18‐ and 40‐week‐old SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) rats; n=9 (SHR‐A3), 6 (SHR‐B2), 9 (SHR‐A3[Stim1‐B2]). *P<0.05 vs SHR‐A3 and # P<0.05 vs SHR‐B2.
Elevated levels of antinuclear antibodies (anti–double‐stranded [ds]‐DNA, anti–Ro60/SSA, anti–La/SSB) have been described in the serum from mice and humans with defective SOCE.17 We have examined the presence of anti–ds‐DNA antibodies in serum from the 3 rat lines. Serial dilutions of serum from 40‐week‐old SHR‐A3 and SHR‐B2 were strongly divergent with much greater binding observed in SHR‐A3 serum. Serum from SHR‐A3(Stim1‐B2) had significantly lower levels of anti–ds‐DNA IgG antibodies at 18 weeks of age, before the onset of renal injury (Figure 6B). At this age, IgM levels were comparable between SHR‐A3 and SHR‐A3(Stim1‐B2). However, at 40 weeks, when renal injury was established, IgG levels were comparable between SHR‐A3 and SHR‐A3(Stim1‐B2), whereas IgM levels in SHR‐A3(Stim1‐B2) were intermediate between those of SHR‐A3 and SHR‐B2 (Figure 6B).
We used HuProt 3.1 arrays to identify protein targets of autoantibody reactivity in the serum of SHR‐A3, SHR‐B2, and SHR‐A3(Stim1‐B2) congenic animals. After incubation with rat serum, arrays were interrogated with fluorescently conjugated anti–rat IgM and IgG detection antibodies. We sorted signals by highest intensity in SHR‐A3 using a Z score cutoff of >10. This identified 41 IgM and 34 IgG targets. Among these targets with high SHR‐A3 signals, 22 were common to both IgM and IgG. In 2 cases, unique array targets that represent different isoforms of the same protein generated signals with a Z score >10 for each of the isoforms (SSBP3, BCAR1). In addition to replication of the SSBP3 signal for 2 unique SSBP3 isoforms, SSBP2 also produced an SHR‐A3 signal with Z score >10 in both IgM and IgG. Full details of detected reactivities are provided as Table S3.
Discussion
Susceptibility to hypertensive renal injury is not uniformly distributed in the hypertensive population and is strongly influenced by genetic factors. Large‐scale human population genetics studies intended to uncover the genetic variants that permit hypertension to initiate and perpetuate renal injury have resulted in limited progress. SHR lines share interesting features in common with human hypertensives: elevated BP has a genetic component that arises from natural genetic variants. These variants were rapidly fixed in SHR by selection on the trait of BP.28 The subsequent separation of distinct SHR lines has resulted in lines that also mirror the human hypertensive population: natural genetic variation contributes to susceptibility to hypertensive renal injury but is not uniformly distributed among the hypertensive population.29 The objective of genetic studies of hypertensive renal injury in SHR is to identify genetic variation that can interact with high BP to result in renal disease. Knowledge of the genes involved in SHR disease and their function may illuminate a pathogenic pathway that leads to progressive loss of renal function in hypertension that is generalizable beyond hypertensive rats. Our prior work in SHR implicates germ‐line genetic variation in the immunoglobulin heavy chain gene, a gene that encodes the preimmune repertoire, in determining susceptibility to renal injury.9, 30 Genetic defects of Stim1 are also associated with antibody‐mediated autoimmunity.16, 17, 18 These observations indicate a potential role for antibodies in hypertensive renal disease in SHR.
We have constructed the Stim1 rescue congenic line using high‐density SNP genotyping and whole genome sequencing data. We identified a large block on chromosome 1 in which SHR‐A3 and SHR‐B2 have inherited genomic sequence from a single shared ancestor. Within this block we identified a single coding sequence change that affects Stim1 in SHR‐A3 only. This is the only coding sequence mutation by which SHR‐A3 and SHR‐B2 differ in this block (Table S1) and appears to be a relatively recent mutation. Examination of SNP data from 40 inbred rat lines31 indicates it is found only in SHR‐A3. Hitchhiker effects normally limit the ability to assign traits to a single variant in a congenic block. However, transfer of wild‐type Stim1 from within a large block in which SHR‐A3 and SHR‐B2 are otherwise genetically identical eliminates the transfer of linked genetic variation.
The immune phenotype created by Stim1 mutation in SHR‐A3 provides further support to our earlier studies indicating that the genetic pathway to end‐organ injury in SHR‐A3 arises from altered immune function.5, 9, 30 We show here that Stim1 mutation disrupts lymphocyte calcium signaling, prevents NFAT nuclear localization, and blocks cytokine and proliferative responses to T‐lymphocyte activation. T‐lymphocyte functions include the provision of help to antibody‐producing B lymphocytes. Stim deficiency limits the ability of T cells to provide effective help to B‐cell antibody affinity maturation,17, 18, 32 and spontaneous autoantibody‐mediated disease occurs in humans lacking Stim1 16, 33 and in targeted mice in which SOCE is eliminated by T‐cell targeting of both Stim1 and Stim2.17, 32 These mice exhibit increased renal immunoglobulin deposits.17 Elevated levels of antinuclear antibodies (anti–ds‐DNA, anti–Ro60/SSA, anti–La/SSB) have been described in the serum from mice and humans with defective SOCE.17 The presence of higher levels of glomerular immunoglobulin depositions in SHR‐A3 than seen in SHR‐B2 or SHR‐A3(Stim1‐B2) and of increased levels of anti–ds‐DNA antibodies in the same pattern further suggests the emergence of antibody‐mediated autoimmune disease in SHR‐A3 and its amelioration by congenic substitution with functional STIM1. We have reported that differential susceptibility to renal injury in SHR‐A3 is also influenced by extensive variation in the immunoglobulin heavy chain (IgH) gene,9, 34 which encodes the preimmune antibody repertoire and may confer predisposition to autoreactive antibody formation. The contribution of impaired T‐cell effector function reported here indicates a defect in T‐cell help to B cells, an essential component of antibody affinity maturation that likely contributes, along with preexisting germline variation in IgH, to the pathogenesis of renal disease in this model. This interaction between 2 loci, both of which determine the expressed IgG and IgM repertoires but at different stages of autoantibody production, might explain why restoration of STIM1 function alone was not sufficient to reduce anti–ds‐DNA to SHR‐B2 levels. Interestingly autoantibody reactivity to single‐stranded DNA‐binding proteins (SSBP2, SSBP3) was detected in SHR‐A3 using the HuProt array, with lower levels of such signals in SHR‐B2 and SHR‐A3(Stim1‐B2). Autoantibodies against nucleic acid binding proteins are a common feature of systemic lupus erythematosus and suggest possible overlap in pathogenic mechanism in renal injury in lupus nephritis and in hypertensive renal injury in SHR‐A3.35 Our efforts to limit B‐cell function in SHR‐A3 using an anti‐CD20 antibody reactive to murine CD20 (Genentech, South San Francisco, CA; anti‐CD20 clone 5D2) failed to elicit sustained reductions in B‐cell counts. After an initial sharp fall in blood B‐cell count, SHR‐A3 B‐cell counts returned to normal, and no sustained B‐cell suppression during the development of renal injury was achieved (data not shown). Other approaches to blocking autoantibody production in SHR‐A3 are currently being investigated but have been limited by the occurrence of sepsis.
STIM1 deficiency disrupts Treg‐cell development, resulting in reduced Treg numbers.26, 32, 36 A similar finding in SHR‐A3 (Figure 3C) indicates reduced tolerance arising from Stim1 mutation. Reduced Treg counts appear to be a consequence of the dependence of expression of the T regulatory master phenotype transcription factor Foxp3 on NFAT function.37 Together our observations suggest involvement in SHR‐A3 end‐organ injury pathogenesis of a Stim1‐driven mechanism involving antibody‐mediated immunity and diminished tolerance. This is also supported by our previously reported studies showing that the immunosuppressant drug mycophenolate mofetil reduces end‐organ injury in SHR‐A3.38 Mycophenolate mofetil acts to reduce cell proliferation by inhibiting a nucleic acid synthesis pathway that is exclusive to proliferating T and B lymphocytes.39
Effects of Stim1 mutation on end‐organ injury are dependent on the presence of hypertension because pharmacological lowering of BP prevents end‐organ injury in SHR‐A3.40 We observed that Stim1 mutation has no effect on BP levels in 18‐week‐old animals before the emergence of renal injury.
Our model organism studies can be considered in light of heritability of risk of hypertensive end‐organ injury in humans. Stim1 mutation that causes a complete loss of SOCE produces a severe phenotype and occurs only rarely in humans. However, Stim deficiency disrupts T‐cell interactions with B cells.17, 18 Numerous examples of genetic variation in other genes have been identified in human populations that impair T cell–B cell interaction and have been shown to be involved in autoimmune disease.41 The pathogenic alleles are not rare in the population and contribute importantly to autoimmune disease risk. The effects of Stim1 mutation may reflect several functionally similar known immunopathogenic human gene variations that may contribute to end‐organ injury in the presence of hypertension, including common variants that affect lymphocyte receptor‐induced calcium signaling.42, 43
In conclusion, we have identified genetic variation in SHR‐A3 affecting Stim1. We have shown that this variation contributes to the risk of hypertensive renal disease. As with all germ‐line mutations, phenotypic effects may occur in any cell or tissue with STIM1 function. Congenic replacement of truncated Stim1 with the wild‐type allele restores a major defect in immune function in SHR‐A3. Concurrently, the ability of hypertension to elicit damage to the kidney is reduced. This may indicate that the major immune phenotype produced by the Stim1 mutation participates in renal injury in SHR‐A3, and this may include injury contributed by autoantibody formation that interferes with T‐cell help to B cells. However, other functional defects arising from Stim1 mutation may also participate in injury.
Sources of Funding
This work was supported by National Institutes of Health grants (NIH R01DK069632 and R01DK081866) to Doris and American Heart Association postdoctoral fellowship award (AHA 17OST33660779) to Dhande.
Disclosures
None.
Supporting information
Table S1. Whole Genome Sequencing of SHR‐A3 and SHR‐B2 Aligned to the Rat Reference Genome
Table S2. Single‐Nucleotide Polymorphisms in SHR‐A3 and SHR‐B2 Obtained by the STAR Consortium
Table S3. HuProt Array
(J Am Heart Assoc. 2020;9:e014142 DOI: 10.1161/JAHA.119.014142.)
References
- 1. Freedman BI, Soucie JM, McClellan WM. Family history of end‐stage renal disease among incident dialysis patients. J Am Soc Nephrol. 1997;8:1942–1945. [DOI] [PubMed] [Google Scholar]
- 2. Satko SG, Sedor JR, Iyengar SK, Freedman BI. Familial clustering of chronic kidney disease. Semin Dial. 2007;20:229–236. [DOI] [PubMed] [Google Scholar]
- 3. Skrunes R, Svarstad E, Reisaeter AV, Vikse BE. Familial clustering of ESRD in the Norwegian population. Clin J Am Soc Nephrol. 2014;9:1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Freedman BI, Spray BJ, Tuttle AB, Buckalew VM Jr. The familial risk of end‐stage renal disease in African Americans. Am J Kidney Dis. 1993;21:387–393. [DOI] [PubMed] [Google Scholar]
- 5. Braun MC, Herring SM, Gokul N, Monita M, Bell R, Zhu Y, Gonzalez‐Garay ML, Wenderfer SE, Doris PA. Hypertensive renal injury is associated with gene variation affecting immune signaling. Circ Cardiovasc Genet. 2014;7:903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Churchill PC, Churchill MC, Griffin KA, Picken M, Webb RC, Kurtz TW, Bidani AK. Increased genetic susceptibility to renal damage in the stroke‐prone spontaneously hypertensive rat. Kidney Int. 2002;61:1794–1800. [DOI] [PubMed] [Google Scholar]
- 7. Mamenko M, Dhande I, Tomilin V, Zaika O, Boukelmoune N, Zhu Y, Gonzalez‐Garay ML, Pochynyuk O, Doris PA. Defective store‐operated calcium entry causes partial nephrogenic diabetes insipidus. J Am Soc Nephrol. 2016;27:2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braun MC, Herring SM, Gokul N, Monita M, Bell R, Hicks MJ, Wenderfer SE, Doris PA. Hypertensive renal disease: susceptibility and resistance in inbred hypertensive rat lines. J Hypertens. 2013;31:2050–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dhande IS, Cranford SM, Zhu Y, Kneedler SC, Hicks MJ, Wenderfer SE, Braun MC, Doris PA. Susceptibility to hypertensive renal disease in the spontaneously hypertensive rat is influenced by two loci affecting blood pressure and immunoglobulin repertoire. Hypertension. 2018;71:700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez‐Garay ML, Cranford SM, Braun MC, Doris PA. Diversity in the preimmune immunoglobulin repertoire of SHR lines susceptible and resistant to end‐organ injury. Genes Immun. 2014;15:528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store‐operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gudlur A, Quintana A, Zhou Y, Hirve N, Mahapatra S, Hogan PG. STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat Commun. 2014;5:5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou Y, Srinivasan P, Razavi S, Seymour S, Meraner P, Gudlur A, Stathopulos PB, Ikura M, Rao A, Hogan PG. Initial activation of STIM1, the regulator of store‐operated calcium entry. Nat Struct Mol Biol. 2013;20:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A. Signalling to transcription: store‐operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium. 2007;42:145–156. [DOI] [PubMed] [Google Scholar]
- 15. Vaeth M, Maus M, Klein‐Hessling S, Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E, Berberich‐Siebelt F, Possemato R, Feske S. Store‐operated Ca2+ entry controls clonal expansion of T cells through metabolic reprogramming. Immunity. 2017;47:664–679.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux‐Laucat F, Rechavi G, Rao A, Fischer A, Feske S. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich‐Siebelt F, Clancy R, Unutmaz D, Feske S. Store‐operated Ca2+ entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity. 2016;44:1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaeth M, Muller G, Stauss D, Dietz L, Klein‐Hessling S, Serfling E, Lipp M, Berberich I, Berberich‐Siebelt F. Follicular regulatory T cells control humoral autoimmunity via NFAT2‐regulated CXCR5 expression. J Exp Med. 2014;211:545–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martina MN, Bandapalle S, Rabb H, Hamad AR. Isolation of double negative αβ T cells from the kidney. J Vis Exp. 2014;87:51192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bell R, Herring SM, Gokul N, Monita M, Grove ML, Boerwinkle E, Doris PA. High‐resolution identity by descent mapping uncovers the genetic basis for blood pressure differences between spontaneously hypertensive rat lines. Circ Cardiovasc Genet. 2011;4:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol. 2009;11:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oh‐Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Algarte M, Lecine P, Costello R, Plet A, Olive D, Imbert J. In vivo regulation of interleukin‐2 receptor alpha gene transcription by the coordinated binding of constitutive and inducible factors in human primary T cells. EMBO J. 1995;14:5060–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poulton TA, Gallagher A, Potts RC, Beck JS. Changes in activation markers and cell membrane receptors on human peripheral blood T lymphocytes during cell cycle progression after PHA stimulation. Immunology. 1988;64:419–425. [PMC free article] [PubMed] [Google Scholar]
- 26. Desvignes L, Weidinger C, Shaw P, Vaeth M, Ribierre T, Liu M, Fergus T, Kozhaya L, McVoy L, Unutmaz D, Ernst JD, Feske S. STIM1 controls T cell‐mediated immune regulation and inflammation in chronic infection. J Clin Invest. 2015;125:2347–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. [DOI] [PubMed] [Google Scholar]
- 28. Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. [DOI] [PubMed] [Google Scholar]
- 29. Okamoto K, Yamori Y, Nagaoka A. Establishment of the stroke‐prone spontaneously hypertensive rat (SHR). Circ Res. 1974;14:I143–I153. [Google Scholar]
- 30. Herring SM, Gokul N, Monita M, Bell R, Boerwinkle E, Wenderfer SE, Braun MC, Doris PA. Immunoglobulin locus associates with serum IgG levels and albuminuria. J Am Soc Nephrol. 2011;22:881–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hermsen R, de Ligt J, Spee W, Blokzijl F, Schäfer S, Adami E, Boymans S, Flink S, van Boxtel R, van der Weide RH, Aitman T, Hübner N, Simonis M, Tabakoff B, Guryev V, Cuppen E. Genomic landscape of rat strain and substrain variation. BMC Genomics. 2015;16:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vaeth M, Wang Y‐H, Eckstein M, Yang J, Silverman GJ, Lacruz RS, Kannan K, Feske S. Tissue resident and follicular Treg cell differentiation is regulated by CRAC channels. Nat Commun. 2019;10:1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, Boisson B, Picard C, Dewell S, Zhao C, Jouanguy E, Feske S, Abel L, Casanova J‐L. Whole‐exome sequencing‐based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dhande IS, Kneedler SC, Joshi AS, Zhu Y, Hicks MJ, Wenderfer SE, Braun MC, Doris PA. Germ‐line genetic variation in the immunoglobulin heavy chain creates stroke susceptibility in the spontaneously hypertensive rat. Physiol Genomics. 2019;51:578–585. DOI: 10.1152/physiolgenomics.00054.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mustelin T, Lood C, Giltiay NV. Sources of pathogenic nucleic acids in systemic lupus erythematosus. Front Immunol. 2019;10:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shaw PJ, Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Front Biosci (Elite Ed). 2012;4:2253–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. [DOI] [PubMed] [Google Scholar]
- 38. Dhande IS, Zhu Y, Braun MC, Hicks MJ, Wenderfer SE, Doris PA. Mycophenolate mofetil prevents cerebrovascular injury in stroke‐prone spontaneously hypertensive rats. Physiol Genomics. 2017;49:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. [DOI] [PubMed] [Google Scholar]
- 40. Stier CT, Benter IF, Ahmad S, Zuo HL, Selig N, Roethel S, Levine S, Itskovitz HD. Enalapril prevents stroke and kidney dysfunction in salt‐loaded stroke‐prone spontaneously hypertensive rats. Hypertension. 1989;13:115–121. [DOI] [PubMed] [Google Scholar]
- 41. Petersone L, Edner NM, Ovcinnikovs V, Heuts F, Ross EM, Ntavli E, Wang CJ, Walker LSK. T cell/B cell collaboration and autoimmunity: an intimate relationship. Front Immunol. 2018;9:1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Menard L, Saadoun D, Isnardi I, Ng Y‐S, Meyers G, Massad C, Price C, Abraham C, Motaghedi R, Buckner JH, Gregersen PK, Meffre E. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rieck M, Arechiga A, Onengut‐Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179:4704–4710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Whole Genome Sequencing of SHR‐A3 and SHR‐B2 Aligned to the Rat Reference Genome
Table S2. Single‐Nucleotide Polymorphisms in SHR‐A3 and SHR‐B2 Obtained by the STAR Consortium
Table S3. HuProt Array
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.