

Abstract
The conversion of human fibroblasts into personalized induced neural stem cells (iNSCs) that actively seek out tumors and deliver cytotoxic agents is a highly promising approach for treating various types of cancer. However, the ability to generate iNSCs from the skin of cancer patients has not been explored. Here, we take an important step toward clinical application by generating iNSCs from skin biopsies of human patients undergoing treatment for the aggressive brain cancer, glioblastoma (GBM). We then utilized a panel of functional and genomic studies to investigate the efficacy and tumor-homing capacity of these patient-derived cells, as well as genomic analysis, to characterize the impact of interpatient variability on this personalized cell therapy. From the skin-tissue biopsies, we established fibroblasts and transdifferentiated the cells into iNSCs. Genomic and functional testing revealed marked variability in growth rates, therapeutic agent production, and gene expression during fibroblast-to-iNSC conversion among patient lines. In vivo testing showed patient-derived iNSCs home to tumors, yet rates and expression of homing-related pathways varied among patients. With the use of surgical-resection mouse models of invasive human cluster of differentiation 133+ (CD133+) GBM cells and serial kinetic imaging, we found that “high-performing” patient-derived iNSC lines reduced the volume of GBM cells 60-fold and extended survival from 28 to 45 days. Treatment with “low-performing” patient lines had minimal effect on tumor growth, but the anti-tumor effect could be rescued by increasing the intracavity dose. Together, these data show, for the first time, that tumor-homing iNSCs can be generated from the skin of cancer patients and efficaciously suppress tumor growth. We also begin to define genetic markers that could be used to identify cells that will contain the most effective attributes for tumor homing and kill in human patients, including high gene expression of the semaphorin-3B (SEMA3B), which is known to be involved in neuronal cell migration. These studies should serve as an important guide toward clinical GBM therapy, where the personalized nature of optimized iNSC therapy has the potential to avoid transplant rejection and maximize treatment durability.
Graphical Abstract
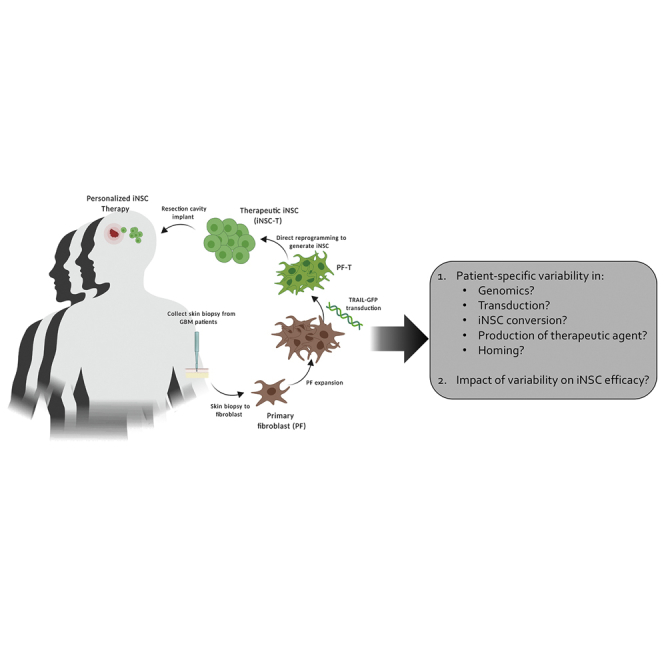
The conversion of fibroblasts into tumor-homing-personalized induced neural stem cells (iNSCs) is a promising approach for treating various types of cancer. We combine functional, genomic, and in vivo assays to investigate the efficacy and tumor-homing capacity of the first iNSCs derived from brain cancer patients, marking an important step toward clinical application.
Introduction
Almost 15,000 patients are diagnosed annually with glioblastoma (GBM), the most common primary brain tumor in adults.1 The clinical standard of care includes maximal surgical resection, chemoradiation therapy, and/or alternating tumor-treating electrical fields.2,3 Even with these options, the median survival has remained under 2 years, with a 5-year survival rate of less than five percent.2, 3, 4, 5, 6 Therapies that seek out disseminated GBM cells behind the blood-brain barrier are needed to prevent the inevitable recurrence in human patients.
Recently, our team and others have shown the promise of neural stem cell (NSC) therapy for GBM.7, 8, 9, 10, 11, 12, 13, 14, 15 Genetically engineered neural stem cells have a unique tumor-homing capacity that allows them to deliver anticancer gene products directly into local and invasive GBM foci.9,16, 17, 18 Tumoricidal NSCs are proven to reduce human GBM xenografts significantly and suppress postsurgical recurrence in mice. Highly promising preclinical data allowed allogeneic NSC therapy recently to enter human patient testing, where encouraging results showed that the therapeutic cells are well tolerated in GBM patients.19
As this approach moves into human patient testing, the ideal NSC drug carrier should be easily isolated and autologous rather than allogeneic in order to avoid immune rejection. We recently discovered that transdifferentiation (TD), a process that transforms somatic cells into other adult cell types, can give rise to tumor-homing drug carriers capable of regressing GBM xenografts. With the use of a SOX2-based single-transcription factor strategy, we reprogrammed normal human fibroblasts into induced NSCs (iNSCs) that homed to GBM cells with the same velocity as brain-derived NSCs and migrated through the brain, tracking invasive human GBM cells.8 Therapeutic iNSCs (iNSC-Ts), genetically engineered to secrete the proapoptotic agent tumor necrosis factor-α (TNF-α)-related apoptosis-inducing ligand (TRAIL), did so at levels equal to cortical-derived NSCs and killed cocultured GBM cells. In orthotopic xenograft models, iNSC-Ts significantly reduced solid human GBM and extended median survival across multiple models of GBM.7,8 These findings demonstrate the potential of this novel approach and suggest that therapeutic iNSCs could be generated from the skin of cancer patients rapidly enough for implantation in a clinical setting, where the autologous nature of the implant could allow the cells sufficient time to traffic to distant neoplasms and persist for extended durations to maximize tumor kill. However, the optimal manufacturing process to generate iNSCs from the skin of cancer patients, as well as the impact of variability associated with patient-derived cell therapy products (CTPs), has not been explored.
Here, we utilized our novel strategy to take a critical step toward the clinic, generating tumor-homing iNSC therapies from the skin of cancer patients for the first time. With the collection of skin biopsies from the surgical border of malignant glioma patients undergoing tumor resection, we generated patient-derived primary fibroblasts (PFs) and subsequently transduced and transdifferentiated them into iNSC-Ts (Figure 1A). We then used a combination of molecular and functional profiling to define interpatient differences in phenotypes that are vital to optimal tumor kill between both fibroblasts and iNSCs derived from different patients. These proposed critical quality attributes (CQAs) include growth rate, transduction efficiency, expression of therapeutic transgenes, and tumor-homing capacity. With the use of a combination of molecular imaging in vitro and ex vivo and mouse models of GBM resection/recurrence, we also showed that iNSCs home to tumors and induce killing of patient-derived GBM cells ex vivo and postsurgical GBM foci in mice. Together, the combination of genomic and anti-tumor data demonstrates the ability to generate iNSCs from the skin of cancer patients, while elucidating phenotypic differences that can serve as a critical guide for establishing a quality target product profile (QTPP) and creating the most effective therapy for clinical use.
Figure 1.

Inter- and Intrapatient Variation of Primary Fibroblasts Isolated from Malignant Glioma (MG) Patient Biopsies
(A) Schematic depiction of the cell-engineering process. (B) White light photomicrographs of PF and NHF1 revealed morphology differences between samples. RNA was isolated from biological replicates (n = 3) to assess intra- and interpatient variability. (C) Principal component analysis (PCA) and a Pearson correlation heatmap, based on the entire gene-expression profile of all samples, suggested that interpatient variation exceeded intrapatient variation. Numbers represent biologic replicates. (D) Heatmap revealing the most highly differentially expressed genes between model fibroblast line NHF1 and each PF line. Samples are ordered according to unsupervised hierarchical clustering (p < 0.05, F < 0.0001).
Results
Generating Fibroblasts from GBM Patients and Defining Intra- and Interpatient Variability
To generate iNSCs from individual cancer patients, we first collected skin-punch biopsies from a panel of patients diagnosed with malignant glioma, which will be referred to by unique patient number (UPN). In the operating room, tissue biopsies were excised from the incision border as patients underwent surgical resection for treatment of their brain tumor. The biopsies were dissociated and PFs were isolated, expanded, and banked before further processing (Figure 1A). Microscopic analysis of six fibroblast lines from different GBM patients and a well-characterized normal human neonatal fibroblast line (NHF1) revealed that each line exhibited a distinct morphology, with differences in size, shape, and polarity (Figure 1B). We next performed RNA sequencing (RNA-seq) to analyze the transcriptome of fibroblasts derived from different patients (n = 3 biologic replicates) to determine the extent of inter- and intrapatient variation within our samples. Principal-component analysis (PCA) and a Pearson correlation heatmap (Figure 1C), along with hierarchical clustering (Figure 1D), revealed greater variation between patients than between replicates, indicating that intrapatient variation is minimal.
The process of generating personalized therapeutic iNSCs begins by expanding the patient fibroblasts to the sufficient clinical dose and transducing the fibroblasts with lentiviruses encoding specific transcription factors and therapeutic agents to induce conversion. We first sought to determine if there were differences in the growth and expansion rate of fibroblasts from different patients. Cell viability assays performed at different times postseeding revealed variability in the growth rate between patient fibroblast lines. PF-07 and NHF1 fibroblasts increased approximately 3-fold over 6 days, a subset referred to as “fast-growing” cells. PF-22, PF-23, and PF-24 increased approximately 1.5-fold in the same time frame, comprising a subset with an intermediate growth rate. A third subset of “slow-growing” cells, PF-08 and PF-02, showed only a slight trend toward proliferation compared to day 1 (Figure 2A).
Figure 2.

Interpatient Variation of Quality Attributes for PF Prior to TD
(A) Summary graph showing growth of all six PF samples compared to NHF1. Samples were plated in triplicate, and viability was measured at each time point. Patient-specific cells were stratified into groups based on significant differences in viability compared to day 1, 1 week later. ∗∗p < 0.01, ∗∗∗∗p < 0.0001 by Student’s t test. (B) Representative fluorescent photomicrographs of PF engineered to express GFP. Flow cytometric analysis revealed patient-specific differences in transduction efficiency based on GFP expression (C), mean fluorescent intensity of GFP (D), and GFP histograms (E) compared to nontransduced cells (n = 3).
Efficient transduction and expression of lentiviral gene products are key to generating therapeutic iNSCs. To examine the key step of lentiviral transduction in patient cells, PFs were transduced with lentivirus encoding an inducible SOX2 and a second virus encoding green fluorescent protein (GFP) at multiplicities of infection (MOIs) of 2 and 50, respectively. We then assessed transduction efficiency and reporter expression by quantitative analysis. Fluorescent microscopy images captured 48 h after transduction showed the presence of GFP in the majority of the patient lines tested. However, images revealed variability in the number of GFP+ cells and the intensity of GFP expression between patient lines (Figure 2B). Flow cytometry analysis revealed nearly 90% of PF-02 were GFP+, and this line showed the highest intensity of GFP expression with a mean fluorescent intensity (MFI) of nearly 300,000 units. In contrast, PF-08 showed that less than 20% of cells were GFP+, and the expression in those lines was significantly lower, with a MFI of less than 20,000 units (Figures 2C–2E). Linear regression analysis revealed that the percentage of positive cells and fluorescent intensity correlated at R2 = 0.801 and p = 0.065. Together, these data support our hypothesis that interpatient variability can impact proposed CQAs and should be considered as we develop an upstream processing workflow for iNSC-T generation.
Generation and Characterization of iNSCs Derived from GBM Patient Skin Fibroblasts
After the characterization of GBM patient fibroblasts, we next sought to convert the gene-edited PFs into therapeutic iNSCs using our established rapid conversion protocol.8 Briefly, to induce conversion, we activated a tetracycline-inducible promoter to initiate SOX2 overexpression in the TRAIL-transduced fibroblasts (PF-T) and cultured the cells in iNSC-inducing medium (NIM) for 5 days (Figure 3A). Differential expression analysis of the parental PF and the resulting patient iNSCs revealed substantial transcriptional changes, as PFs were directly reprogrammed during the rapid 5-day conversion (Figure 3B). We found that the level of global gene changes varied markedly across patient sources. More than 2,500 genes differentially expressed as PF-22 were converted to iNSCs. In contrast, only 152 genes were differentially expressed between the two cell types generated from UPN08. Interestingly, only 23 genes were differentially expressed across all lines (Figure 3C).
Figure 3.

Interpatient Variation in iNSC Gene-Expression Profile Following Reprogramming Protocol
(A) Schematic depiction of therapeutic iNSC transdifferentiation from PF-T. Differential expression analysis (B) showed that our rapid transdifferentiation protocol induced significant alterations in the transcriptomic profile compared to PFs (p < 0.05). A list of genes that changed by 2-fold or greater was compiled for each patient, and an Edward’s Venn diagram was generated from all six lists (C). (D) PCA of all PF and iNSC generated, as well as wild-type NSCs and NHF1-derived fibroblasts and iNSCs, again demonstrated patient-specific variation in reprogramming. The beginning of arrows indicates PF coordinates, and arrowheads indicate iNSC coordinates following reprogramming. (E) Summary graph of mean Euclidean distances between PFs and iNSCs generated from the same unique patient. Vector direction was determined based on principal component 1 (PC1).
Principal component analysis (Figure 3D) across all patients revealed variation due to both cell type (PF versus iNSC) as well as patient source, with patient-specific differences impacting the extent of reprogramming (Figure 3E). Together, these data begin to reveal that gene changes occurring as we use our rapid reprogramming strategy on PFs are variable between patient sources, and interpatient variation should be considered for optimal clinical trial design.
Assessing Genomic Variability and Differences in Tumor-Homing Capacity of Patient-Derived iNSCs
Next, we further explored differences in the genomic landscape of iNSCs, as well as performed functional in vivo testing to examine the homing capacity of each patient-derived iNSC line. PCA and a Pearson correlation heatmap (Figure 4A), as well as hierarchical clustering of differentially expressed genes (DEGs) between control NHF1-derived iNSCs and iNSCs derived from each patient (Figure 4B), revealed that whereas interpatient variation is consistently present regardless of cell type, intrapatient variation is also increased postreprogramming. We also investigated the fate of iNSCs as they were converted from patient fibroblasts. RNA-seq data showed a trend toward upregulation of the NSC marker Sox2. Importantly, pluripotency markers, such as NANOG and POU5F1, were not elevated (Figure 4C). This supports our previous findings and further suggests that iNSCs do not express markers of a potentially carcinogenic pluripotent intermediate. Genes associated with differentiation were also not upregulated, suggesting that the iNSCs were not further differentiating down a specific lineage.
Figure 4.

Characterization of Patient-Derived Therapeutic iNSCs
(A) Following transdifferentiation, marked patient-specific variation was again revealed by PCA and a Pearson correlation heatmap. (B) A heatmap revealing the most highly differentially expressed genes between model iNSC line derived from NHF1 and each patient-specific iNSC line. (C) Boxplot comparison of fibroblasts and induced neural stem cell lines. The PF represent the log2 signal across patient-derived fibroblasts and the NHF1 cell line. The iNSC represents the log2 signal across patient-derived iNSCs and the iNSC cell line for SOX2, NEUROG2, NANOG, ASCL1, POU5F1, and PAX6.
In the clinical setting, the tumor-homing capacity of iNSCs is critical. The cells actively seek out GBM foci embedded throughout the brain and deliver genetically engineered cytotoxic agents. We investigated the tumor-tropic nature of iNSCs derived from 4 of the GBM patients using in vivo model systems. Human GBM cells expressing mCherry were implanted into the frontal lobe of mice. 3 days later, patient iNSCs, labeled with Qdot 655 (Thermo Fisher Scientific), were implanted 1.5 mm lateral to the established tumors (Figure 5A). After 1 week, mice were sacrificed, and microscopic analysis was performed to determine the colocalization of iNSCs with the GBM foci. We detected iNSC migration from the implantation site in all 4 samples, with the most significant migration in iNSC-7s and iNSC-22s, and numerous iNSCs colocalized with the solid GBM foci (Figure 5B). From this, we stratified patients into fast (iNSC-7s, iNSC-22s)- and slow (iNSC-8s, iNSC-24s)-migrating categories. These results provide the first evidence that iNSCs derived from cancer patients are tumor-homing cells that exhibit both genomic and phenotypic interpatient heterogeneity depending on parental cell source.
Figure 5.

Characterization of iNSC Homing
Four patients were selected to assess the migration of iNSCs to distant, established GBM. U87-mCherry were implanted into the parenchyma of mice (n = 5), and iNSC-Cy5s were implanted ipsilaterally 1.5 mm away (A). Immunofluorescent analysis of postmortem tissue sections, 8 days postimplant, was used to stratify patients by tumor tropism (B). Blue arrows indicate day 1 iNSC injection site, and white arrows indicate iNSCs closest to tumor at day 7 postinjection.
Assessing the Anticancer Activity of Patient-Derived iNSCs
We next investigated the efficacy of GBM patient iNSC therapy by engineering the cells to express the proapoptotic agent TRAIL. Having observed variability in transduction and gene expression of the GFP reporter across PF lines, we first analyzed the transduction efficiency of the lentivirus encoding a secreted variant of TRAIL upstream of an internal ribosome entry site (IRES)-GFP element, levels of TRAIL release, and the potential impact of any variability on GBM suppression. Following transduction of PFs with lentiviruses encoding TRAIL and SOX2 (MOI 50 and 2, respectively), GFP imaging revealed variability in the percentage of TRAIL+ fibroblasts, which was confirmed by flow cytometry (Figures 6A and 6B) as well as the ELISA assay, to quantify the levels of TRAIL protein secretion (Figure 6C).
Figure 6.

Interpatient Variation in iNSC Potency
(A–C) Summary graph (A) and histograms (B) of flow cytometric analysis of iNSC-Ts (n = 3 biologic replicates) revealing differences in TRAIL expression quantified using GFP reporter signal, with interpatient variation in TRAIL secretion validated and quantified by ELISA, and one-way ANOVA indicated significant patient-specific differences at p < 0.0001 (C). iNSC-Ts or iNSC-GFPs from 4 different patients were mixed with GBM8-mCherry at 6 ratios, and a small foci of GBM/iNSCs were added to living organotypic rat brain tissue (n = 4). Tissue was imaged by both fluorescent microscopy and IVIS Kinetic immediately following seeding and again 24 h later. (D) Representative fluorescent images and fluorescent photomicrographs are shown at a ratio of 1:10 iNSC:GBM at 24 h. Changes in tumor volume from over 2 h revealed patient-specific potency profiles accounting for differing doses (E) when compared to each other and to control (iNSCs expressing GFP only). Data in (E) were analyzed via one-way ANOVA, with p = 0.0022 (mean + SEM).
We next investigated the potential impact in generating cytotoxic agents that this variability may have on iNSC therapy. Previous studies have shown that coculturing cells on living organotypic tissue-slice explants better recapitulates tumor growth, cell migration, and cell–cell interactions than traditional 2D assays performed on polystyrene tissue-culture plates.20 Thus, we turned to this model to test the efficacy of the patient-derived iNSCs against GBM. Four of the TRAIL+ PF lines were converted into iNSC-Ts as before, mixed with primary human GBM cells expressing mCherry and firefly luciferase (Fluc) at six different ratios, and seeded on living rat brain slices. A comparison of fluorescent images and quantification of fluorescence 0 and 24 h showed that all iNSC-T patient lines significantly reduced the volume of GBM tumor foci in a dose-dependent manner (Figure 6D). In agreement with the ELISA data, iNSC-7s and iNSC-22s expressed the highest levels of TRAIL and decreased tumor volumes below 50% at the lowest iNSC:GBM cell ratio (Figure 6E). Altogether, these data begin to reveal patient-specific differences in the efficacy of iNSC therapies.
Patient-Derived iNSC Therapy of Primary Human GBM Xenografts in Mice
In a future clinical treatment protocol, skin-derived fibroblasts from each GBM patient will be converted into his or her own tumor-homing iNSC treatment; these iNSCs will then be reimplanted back into the same patient at the time of tumor resection. To investigate the efficacy of GBM patient-derived iNSC therapy in vivo, we used an invasive patient-derived GBM line (GBM8) in our mouse model of surgical resection to mirror the clinical scenario as closely as possible. Surgical resection is part of the clinical standard of care for GBM and causes changes to the tumor microenvironment that affect therapeutic cell persistence at the site of injury.4,21 To mimic iNSC therapy for human patients with surgically resected GBM, GBM8 cells were implanted into the brain parenchyma of mice. We first chose to test UPN07, as the line expanded rapidly, migrated to GBM, and showed effective killing in brain-slice models. PF-07 was engineered with either a secreted variant of TRAIL or a GFP optical reporter and purified by fluorescence-activated cell sorting (FACS). PFs were then transdifferentiated to iNSCs (iNSC-7-Ts or iNSC-GFPs). 3 days after implant, established GBM8 tumors were partially resected, and iNSC-7-Ts or control iNSC-7-GFPs were delivered into the postsurgical cavity on clinically approved, resorbable hemostatic gelatin matrices (GEMs), which we have previously discovered maximizes stem cell transplant and efficacy (Figure 7A).22, 23, 24, 25, 26 Serial bioluminescence imaging (BLI) revealed that iNSC-7-T therapy attenuated GBM8 tumor recurrence, as residual GBMs were 60-fold smaller in the iNSC-7-T-treated animals compared to control-treated animals, 21 days postimplant (Figures 7B and 7C). iNSC treatment also extended median survival, with iNSC-7-T-treated animals surviving an average of 45 days postresection compared to only 28 days for control animals (Figures 7D and 7E). These data provide the first evidence that iNSCs can be generated from an optimal subset of glioma patients and engineered to slow GBM recurrence and extend survival following implantation.
Figure 7.

Patient-Derived iNSC Therapy for Diffuse Primary GBM
GBM8-mCherry-Fluc (mCFL) were implanted into the frontal lobe of mice and partially resected 3 days later. iNSC-7-Ts were implanted on GEM scaffolds into the resulting resection cavity at the time of surgery (A). Representative BLI (B) and summary data (C) demonstrated inhibition of GBM progression by iNSC-Ts compared to control-treated mice (n = 10). ∗∗p < 0.01 by one-way ANOVA. (D) Kaplan-Meier survival curves demonstrating the extension in survival in mice treated with iNSC-7-Ts compared to control. (E) Representative whole-brain and high-magnification images showing GBM8 by mCherry fluorescence, 21 days after delivering iNSC-GFPs or iNSC-7-Ts into the resection cavity. ∗p < 0.05 by log-rank test.
Selection of Candidate Markers for Quality Target Product Attributes
Based on the combination of functional and molecular data generated in these studies, we next developed a workflow for identification of candidate markers for high-quality iNSC-Ts (Figure 8A). Expansion rate, transduction efficiency, and tumor tropism are attributes that likely affect potency; therefore, we stratified patient-specific iNSC-Ts into groups based on these attributes. Once fast- and slow-expanding/migrating patient lines were identified, we compiled a list of DEGs between these groups. From there, we performed hierarchical clustering (Figures 8B and 8D) and Gene Ontology (GO) analysis (Figures 8C and 8E) of all DEGs with signal-expression profiles matching functional patient phenotype. This indicated that genes related to developmental process and regulation of cell proliferation were highly, differentially expressed in fast-expanding versus slow-expanding cells and that genes related to regulation of cell migration and biological adhesion were highly, differentially expressed in fast- and slow-migrating cells. From there, we selected DEGs with GO annotations related to cell proliferation (expansion) or cell migration (tumor tropism) and performed hierarchical clustering on the subset of genes with the highest fold change between patient iNSCs by quality-attribute category (Figures 8F and 8G). Example candidate quality assurance (QA) markers were selected from these lists based on known protein function and are listed in Figure 8H.
Figure 8.

Identifying Candidate QA Markers
(A) Flowchart describing the approach used to select candidate markers. DEGs with fold-change >2 and GO annotations related to proliferation and migration were compared among all patient cells and selected as candidate markers based on the similarity of their expression profile to functional phenotype. (B and C) Heatmap (B) and GO (C) analysis of most highly differentially expressed genes induced in fast-expanding versus slow-expanding PF revealed enrichment in genes associated with cell proliferation, cell adhesion, and extracellular matrix (ECM) organization. (D and E) Heatmap (D) and GO (E) analysis of most highly differentially expressed genes induced in fast-migrating and slow-migrating iNSC-Ts revealed differences in locomotion, biological adhesion, and regulation of cell migration. (F) Heatmap of the 14 most highly expressed candidate marker genes identified for cell growth, with 3 genes selected based on known function in fibroblasts proliferation or senescence. (G) Heatmap of 30 most highly expressed candidate marker genes identified for tumor tropism, with 3 genes selected based on known function in neural cell motility (n = 3, p < 0.05). Boxes indicate candidate genes selected. (H) Table indicating marker selection.
Candidate Markers Predict Patient-Specific Outcomes, and Anti-tumor Efficacy of Suboptimal Lines Can Be Improved by Increased Dose
Although several of our lines showed efficient transduction and tumor killing, some lines showed reductions in the key parameters described in Figure 8. Of these, UPN24 showed moderate cell growth, as well as reduced TRAIL production, tumor homing, and tumor kill when compared to UPN07 (Figures 9A and 9B). Thus, we hypothesized that iNSC-24 therapy would display suboptimal efficacy in vivo when compared to iNSC-7 therapy. To investigate tumor killing by a comparatively suboptimal line in vivo, we delivered FACS-purified control or TRAIL+ variants of the suboptimal line, iNSC-24, or the higher performing line, iNSC-7, into the postsurgical GBM cavity on GEM scaffolds at a dose of 4 × 105 cells/mouse. Serial kinetic imaging showed rapid tumor recurrence in control-treated animals. As hypothesized, treatment with suboptimal iNSC-24-Ts had minimal impact on tumor recurrence, with tumors rapidly redeveloping and showing no statistical difference in volume compared to control, 16 days post-treatment (Figures 9C and 9D). Additionally, only a slight statistical difference was observed in the survival of control- or iNSC-24-T-treated mice, with both groups showing a median survival of 22 and 25 days, respectively. In contrast, the higher functioning iNSC-7-T line stably suppressed tumor growth 16 days post-treatment and extended overall survival to 35 days post-treatment (Figure 9D).
Figure 9.

Patient-Derived iNSC Therapy for Diffuse GBM Informed by Candidate Quality Marker Expression
(A and B) Summary table (A) and summary plot (b) of candidate marker expression in PF-T and iNSC-Ts, generated from PF-07 and PF-24, predicts that iNSC-7-Ts would have improved potency compared to iNSC-24-Ts. (C) Representative BLI and (D) summary data of GBM-mCFL when iNSC-Ts from either patient was dosed at 4.0 × 105 cells/mouse or 1.0 × 106 cells/mouse (n = 5, ∗∗p < 0.01). (E) Kaplan-Meier survival curves at both doses demonstrating the extension in survival in mice receiving iNSC-Ts from both patients compared to iNSC-GFPs. ∗p < 0.05 and ∗∗p < 0.01 by log-rank test.
As treatment with iNSC-24-Ts had minimal impact on tumor recurrence at the above-mentioned dose, we tested whether an increase of the dose of therapeutic cells could enable these comparatively suboptimal cells to suppress postsurgical GBM regrowth effectively. We again implanted control or TRAIL+ iNSC-7s or iNSC-24s on GEM scaffolds into the postsurgical cavity, this time increasing the dose to 1 × 106 cells/mouse to account for the differences in TRAIL output. Kinetic imaging and survival analysis revealed that the previously suboptimal iNSC-24-T was able to suppress tumor regrowth significantly, reducing tumor volumes over 10-fold at 16 days post-treatment, although survival was not significantly improved compared to the lower dose (Figure 9E). This allowed the higher dose of iNSC-24-Ts to kill tumors almost as effectively as iNSC-7-Ts at the lower dose, but iNSC-24-T was still unable to achieve GBM suppression to the same level of iNSC-7-Ts when this high-functioning cell line was delivered at the higher dose of 1 × 106 cells/mouse. Thus, suboptimal tumor kill can be rescued through increasing the dose of therapeutic cells delivered into the surgical cavity, as predicted by candidate marker expression.
Discussion
Although preclinical studies of iNSC-based cell therapy have been promising, iNSC characterization has been specific to cells generated from immortalized mouse or human cell lines.2 By taking an important step toward the clinic, we have provided the first evidence that therapeutic iNSCs can be generated from the skin of cancer patients. With the use of genetic analysis and clinically relevant mouse models of GBM, we show that GBM patient-derived iNSCs home to GBM in mice and suppress regrowth of postsurgical tumors. We also begin to uncover the heterogeneity in growth, release of therapeutic agents, and migration that must be accounted for in clinical-grade, patient-derived iNSC cell products. Together, these data should serve as an important early guide for generating high-quality iNSCs and for cell-quality optimization in the future.
We isolated and characterized six PF samples with respect to variability in expansion and transduction in order to stratify them qualitatively into groups based on these attributes. With the use of global gene-expression profiling by RNA-seq, we saw transcriptome-wide variation between patients, with significantly less variation between biological replicates from the same patient. This trend held true with respect to PF growth and transduction efficiency. We hypothesized that these characteristics could directly impact cell product quality, as the short time frame for cell manufacturing necessitated by this therapeutic approach requires fast expansion of phenotypically desirable cells. We believe that both the degree of cell expansion and the degree of transduction will affect whether a clinically meaningful cell dose is achievable in this short time frame. Transduction efficiency could affect potency 2-fold. First, reprogramming and therefore, tumor tropism may depend on the extent of exogenous SOX2 overexpression. Second, the successful integration of genes encoding for exogenous cytotoxic agents would correlate directly with anticancer activity once iNSCs home to cancerous foci.
iNSCs were stratified into groups based on functional expansion rate, migratory capacity, and anticancer activity. Based on functional data, we predicted that iNSC-7-Ts would have the most favorable balance of potency and rapid expansion. We found that treatment with iNSC-7-Ts, as opposed to sham treatment using iNSC-GFPs, markedly reduced tumor volumes in vivo and extended median survival 1.6-fold, which is consistent with our previous work, treating with the optimized NHF1-derived iNSC-Ts.8 This is advantageous compared with preclinical studies of the clinical standard of care against an established immortalized cell line, which extends median survival similarly (1.7-fold) but with notable off-target toxicity.27 Bolstered by this proof of concept, we investigated differences in gene expression of patient-specific iNSCs based on the functional stratification described above. DEGs between fast- and slow-expanding or -migrating cells were compiled, and GO analysis was conducted in order to identify specific genes related to expansion and tumor tropism. From there, DEGs were selected as candidate quality markers based on patient phenotype (i.e., if DEGs were highly expressed in fast-expanding cells and minimally expressed in slow-expanding cells, then they would only be selected if the signal for medium-expanding cells was between the two). As transduction efficiency varied significantly, not only between patients but also between viral vector, genetic markers could not be identified, and functional markers were suggested instead.
Example candidate markers for rapid PF expansion were nicotinamide phosphoribosyl transferase (NAMPT) and interleukin-1 (IL1). NAMPT is a protein that regulates cell metabolism, and its expression diminishes as cells senesce.28,29 IL-1 alpha and beta (IL-1α and IL-1β) have both been shown to modulate dermal fibroblast proliferation, and mRNA levels of both are lower in postmitotic or growth-inhibited fibroblasts.30 Importantly, expansion was tested in this study by measuring viability of cells seeded in polystyrene-well plates over time. In order to identify candidate quality markers for expansion in a clinical setting, stratification of patients in terms of proliferative activity should be validated in good manufacturing practice (GMP)-compliant bioreactors to maximize scalability. Example candidate markers identified for tumor tropism were semaphorin-3B (SEMA3B), phosphatidylinositol 3-kinase (PI3K) catalytic subunit delta (PIK3CD), and WWC1. SEMA3B encodes semaphorin-3B, a member of the semaphorin class of proteins involved in neuronal migration-guidance signaling. SEMA3s are secreted, chemotropic proteins that interact with neuropilin and plexin receptors and can have both autocrine and paracrine function, initiating an intracellular signaling cascade that leads to rearrangements in the actin cytoskeleton.31, 32, 33 PIK3CD, on the other hand, was selected because it encodes a catalytic subunit of PI3K, and its overexpression significantly promotes cell migration in multiple cell types. It is a downstream effector of C-X-C chemokine receptor type 4 (CXCR4), the most well-established mediator of tumor homing in wild-type NSCs.7,8,34, 35, 36, 37, 38 Lastly, WWC1 encodes kidney/brain protein (KIBRA), which positively modulates the directional migration of podocytes and regulates the polarization of actin in brain-derived cell types.39 Further studies will be required to define fully the specific role that each of these pathways has in each relevant function; however, they should provide important guidance on initial starting points for exploration, as well as potential biomarkers to identify iNSCs that could impart the greatest tumor kill in human patient testing.
After the investigation of expression of these markers and the recording of TRAIL output, we hypothesized that iNSC-24-Ts would be less efficacious than the previously tested iNSC-7-Ts. Indeed, we discovered that when dosed at 4.0 × 105 iNSC-T per mouse, only iNSC-7-Ts significantly inhibited tumor progression, with a marked increase in survival compared to mice treated with iNSC-24-Ts or iNSC-GFPs. By increasing the dose to 1.0 × 106 cells per mouse, tumor inhibition and survival were significantly increased in both groups; however, mice treated with high-dose iNSC-24-Ts still underperformed compared to low-dose iNSC-7-Ts. This indicates a need for further characterization of potential CQAs affecting therapeutic cell half-life postimplantation. It is also likely that the differences in migratory capacity could account for this difference, as invasive tumor cells may escape initial suppression and become established far from the iNSC implant, where iNSC-24-Ts would be less likely to home than iNSC-7-Ts. Still, these data together provide evidence that autologous iNSCs can be generated from malignant glioma patient skin biopsies and engineered to inhibit disease progression in mouse models that reflect clinical GBM and provide an early methodology for CQA identification and QTPP establishment for therapeutic early-stage iNSCs. Although the approach described here may not be representative of the total population, it provides a framework for quality-by-design-based manufacturing for synthetic anticancer iNSCs.
Our ultimate goal is to move iNSC therapy for cancer into the clinical setting. As cell therapy continues to be one of the fastest growing areas in cancer treatment and beyond, this has driven a number of advancements in manufacturing. Led, in large part, by the chimeric antigen receptor T cell (CAR-T) market, protocols, as well as reagents, to generate cell therapies are becoming more standardized from culture media to plasmids for viral vector generation and cryopreservation media and beyond. Additionally, the pathway for approval of cell therapies is becoming clearer as the US Food and Drug Administration (FDA) becomes more familiar with cell therapies entering human patient testing. However, significant challenges do still remain for advancing treatments, such as iNSCs, into the clinical setting. Specific to manufacturing, variability in starting material between patients and resulting variability in the final iNSC product could be a challenge. The cost of transitioning preclinical media, vectors, and materials to GMP grade and scale is costly. Additionally, there are often numerous manual steps in the manufacturing process that can introduce additional variability, cost, and potential contamination. Lastly, queues for generating GMP virus and GMP cell products are long, commonly more than 12 months. Despite these challenges, highly promising results in preclinical testing and the continual entrance of new cell therapies into human patient testing speak to the power, promise, and ability of cell therapies to enter the clinic. Speed of production is an important aspect of iNSC therapy, as the aggressive nature of diseases, such as GBM, demands that therapies be delivered into patients within 2–3 weeks. Our current preclinical manufacturing utilizes a process that is close to this time frame. Additionally, new CAR-T manufacturing has shown the ability to generate human-scale doses of within 3 weeks, suggesting it is possible to generate cell therapy from patient to product within this rapid time frame.
In conclusion, these studies provide the first evidence that cytotoxic iNSCs can be generated from human cancer patients and are tumor-homing drug carriers that significantly inhibit GBM progression in clinically relevant mouse models of GBM resection/recurrence. We also demonstrate that interpatient variability is present in the iNSC lines, yet low-performing therapies can likely be rescued by elevating the dose that is delivered into the surgical cavity. Together with the genomic analysis that begins to define key pathways, these findings should serve as a guide for future clinical trials, where this same process is used to dose cytotoxic iNSC therapies back into the same patient to maximize tumor killing. This could have broad clinical impact, as cytotoxic NSC therapy is being explored for treatment of metastatic, pediatric, and peripheral cancers.2
Materials and Methods
Experimental Model and Subject Details
Cell Lines
U87 glioma cells were purchased from the American Type Culture Collection and cultured according to the manufacturer’s specifications. Human fibroblasts (NHF1) were provided by W. Kauffman (The University of North Carolina School of Medicine). Both were cultured at 37°C, 5% CO2, in Dulbecco’s modified Eagle’s medium (DMEM; 10% fetal bovine serum [FBS], 1% penicillin-streptomycin; Gibco). GBM8 glioma cells were a gift from H. Wakimoto (Massachusetts General Hospital) and were cultured at 37°C, 5% CO2, in EF medium (neurobasal media [Gibco], 1% L-glutamine [Gibco], 1× B27 supplement [Gibco], 0.5× N-2 supplement [Gibco], 2 μg/mL heparin [Sigma], 20 ng/mL recombinant human epidermal growth factor [EGF], 20 ng/mL recombinant human fibroblast growth factor 2 [FGF-2; R&D Systems]). iNSCs were generated as described below and cultured at 37°C, 5% CO2, in STEMdiff neural induction medium (STEMCELL Technologies) with 2 μg/mL doxycycline. Rat brain tissue explants inoculated with iNSCs/GBM8 were cultured in neurobasal medium (15% heat-inactivated horse serum, 10 mM KCl, 10 mM HEPES, penicillin-streptomycin, 1 mM sodium pyruvate, 1 mM L-glutamine, 0.5% reagent-grade agarose) under 5% CO2 at 32°C for up to 2 weeks. ReNcell VM (ventral mesencephalon) was also used throughout as a cortical-derived NSC control (Sigma-Aldrich). ReNcell was cultured in ReNcell maintenance medium with 20 ng/mL FGF-2 and 20 ng/mL EGF on laminin-coated cultureware under 5% CO2 at 37°C.
Primary Cell Cultures
Primary fibroblasts were isolated from skin biopsy samples taken from the incision border of glioblastoma patients. Samples were minced into 1.5 × 0.5 cm pieces using a scalpel blade in a laminar flow hood. Minced tissue was digested overnight in digestion media (high glucose DMEM, 20% FBS, 0.25% collagenase I, 0.05% DNase I, 1% penicillin-streptomycin [Gibco]), filtered through a 100-μm cell strainer, and seeded in a tissue-culture flask containing fibroblast growth media (high-glucose DMEM, 20% FBS, 1% penicillin-streptomycin [Gibco]). Cells were incubated at 37°C, 5% CO2, with media changed as needed to enable expansion of fibroblasts from the tissue. After 1 passage, FBS concentration of fibroblast growth media was reduced to 10%. Fibroblasts were expanded and banked at passage 4 for further use. Patient-derived iNSCs were generated as described below and cultured at 37°C, 5% CO2, in STEMdiff neural induction medium (STEMCELL Technologies) with 2 μg/mL doxycycline.
Lentiviral Vectors
The following lentiviral vectors (LVs) were used in this study and have been described previously: mCherry fused to firefly luciferase (LV-mCFL), a secreted variant of TRAIL (LV-TRAIL-IRES-GFP), a tetracycline-inducible SOX2 (LV-tetO-SOX2), and a reverse tetracycline-controlled transactivator (LV-rTTA).40 LV-TRAIL has IRES-GFP elements in the backbone, as well as a cytomegalovirus-driven puromycin element. All LV constructs were packaged as LV vectors in 293T cells using a helper virus-free packaging system, as described previously.7,8 Fibroblasts and GBM cells were transduced with LVs at varying multiplicities of infection by incubating virions in a culture medium containing hexadimethrine bromide (8 μg/mL; Sigma), and cells were visualized for fluorescent protein expression by fluorescence microscopy.
Animal Models
All animal studies were performed on female athymic nude mice, aged 6–12 weeks, and approved by the Institutional Animal Care and Use Committee at The University of North Carolina at Chapel Hill. Postnatal day 10 Sprague-Dawley rat pups of either sex (Charles River) were used to generate the brain-tissue explants; animals were sacrificed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee at Duke University.
Method Details
iNSC Generation
iNSC generation was performed following our previously defined protocol.8 Briefly, PFs between passages 4 and 15 were seeded in six-well plates at 2.0 × 105 cells/well and incubated with LV-SOX2/rTTA at varying MOIs (1–4, with 2 selected as optimal) in DMEM (10% FBS) containing hexadimethrine bromide (8 μg/mL; Sigma) for 24 h. Medium was replaced with fresh DMEM (10% FBS, 1% penicillin-streptomycin) following this 24-h period. Next, the medium was changed to STEMdiff neural induction medium containing 2 μg/mL doxycycline. Medium was changed every other day for 5 days. Adherent cells were lifted with Accutase (STEMCELL Technologies) for 5 min at room temperature and then diluted in PBS and centrifuged for 5 min at 1,000 rpm to obtain a cell pellet before use.
RNA-seq Analyses
RNA was extracted from NHF1, PF, iNSC, and wild-type NSC cellular pellets using the Invitrogen Purelink RNA Micro Kit (Thermo Fisher Scientific). Three biologic replicates for each cell type were randomized, and automated library preparation was performed with the Ion Chef System using the Ion AmpliSeq Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Automated template preparation on Ion 540 chips was also performed using the Ion Chef system. The chips were sequenced using the Ion S5 system and the Ion 540 Kit (Thermo Fisher Scientific). PCA, hierarchical cluster analysis, differential expression testing with DESeq2, and plotting of these results were performed both in the Transcriptome Analysis Console and in R 3.6.0.
Transcriptome Analysis Console v.4.0 (Applied Biosystems, Foster City, CA, USA) was used to visualize the results of the AmpliSeq chips. Differential expression was calculated using DESeq2, version 1.16.1. Statistics were run with the R package “statmod,” version 1.4.30, built under R, version 3.4.0. Linear regression analysis was performed using Limma, version 3.32.10.
Cell Viability Assay
To assess the proliferation of PFs and iNSCs, cells were seeded in 96-well plates at 5.0 × 103 cells/well. Cell viability was assessed 1 to 7 days after seeding using the CellTiter-Glo luminescent cell viability kit (Promega) and quantified on a Synergy HTX multi-mode microplate reader (BioTek).
Brain Slices
Living rat-tissue slices were generated as described previously.20,41 Briefly, 250 μm-thick coronal rat-brain tissue explants were sliced while immersed in ice-cold artificial cerebrospinal fluid, then transferred to six-well plates, and embedded on top of semi-solidified culture medium. To assess the potency of iNSC-Ts, GBM8s transduced with LV-mCherry, mixed with iNSC-Ts at varying ratios (1:1, 1:2, 1:4, 1:10, 1:50, and 1:100 iNSC:GBM), and seeded in droplets onto the brain-slice surface at approximately 2.5 × 104 total cells/droplet. Two droplets were added to each brain slice for a total of 4 replicates/ratio. Fluorescence of each droplet was measured by quantifying radiant efficiency, 3 h after seeding and 24 h after initial imaging, using an IVIS Kinetic imaging system (PerkinElmer), and tumor volume was normalized to day 0 and expressed as percentage growth.
Flow Cytometric Analysis
Blank or GFP-expressing fibroblasts or iNSCs were washed in PBS and resuspended at 1 × 106 cells/mL in ice-cold PBS, supplemented with 1% BSA, 0.1% sodium azide (Gibco). Single-cell suspensions were seeded in either a 96-well plate on ice for acquisition conducted on an iQue Screener PLUS (Intellicyt) or in FACS tubes on ice for purification based on GFP expression conducted using the BD FACSAria II. FCS (Flow Cytometry Software) Express was used to perform analysis after data collection. Forward-scatter (FSC) and side-scatter gates were set to exclude debris, and double-discrimination gates (FSC-area versus FSC-height) were set to exclude multimers. Negative cell gates were set on nontransduced, blank cells. Cells positive for GFP were quantified using the ratio of cells that fell above the negative cell gates.
ELISA
TRAIL secretion from iNSCs was determined by measuring TRAIL levels in cell-culture supernatant, 6 h after incubation with iNSC-Ts. A Human TRAIL ELISA Kit (Abcam) was used, according to the manufacturer’s instructions.
In Vivo Bioluminescence Imaging
To track tumor progression, iNSC homing, and therapeutic efficacy, serial bioluminescence was performed, as described previously.25 Briefly, mice were given an intraperitoneal injection of D-luciferin (4.5 mg/mouse in 150 μL of saline), and photon emission was determined 5 min later using an IVIS Kinetic Optical System (PerkinElmer) with a 5-min acquisition time. Images were processed and photon emission quantified using Living Image software (PerkinElmer). Additionally, mice were followed for survival over time.
iNSCMG Migration In Vivo
To determine the migration of iNSCs to solid GBM, nude mice were stereotactically implanted with U87 cells expressing mCFL (50,000 cells/mouse). 3 days later, iNSCs from 4 different patients were labeled with Invitrogen Qtracker 655 (Thermo Fisher Scientific) and implanted 1.5 mm apart in the right frontal lobe. Mice were sacrificed 8 days after iNSC implantation, and brains were processed as described below. Fluorescent microscopy of postmortem tissue sections was used to visualize colocalization of iNSCs with GBM (EVOS FL Auto system).
Assessing the Efficacy of GBM Patient iNSC Therapy In Vivo
Patient-derived GBM8-mCFL was harvested at 80% confluency and implanted stereotactically (5 × 105 cells) in the right frontal lobe: 2 mm lateral to the bregma and 0.5 mm from the dura. 10 days later, mice were immobilized on a stereotactic frame and placed under an Olympus MVX-10 microscope. A midline incision was made in the skin above the skull exposing the cranium of the mouse. The intracranial xenograft was identified using mCherry fluorescence. A small portion of the skull covering the tumor was surgically removed using a bone drill and forceps, and the overlying dura was gently peeled back from the cortical surface to expose the tumor. Under mCherry fluorescence, the GBM8-mCFL tumor was surgically excised using a combination of surgical dissection and aspiration. Following tumor removal, the resulting resection cavity was thoroughly irrigated. iNSC-Ts or control iNSCs (4 × 105 or 1 × 106 cells) were generated on biocompatible gelatin matrices (approximately 2 × 2 mm in size) and transplanted into the postoperative GBM cavity before skin was closed using surgical glue. No procedure-related mortality was observed. GBM recurrence was visualized by Fluc imaging, as described above, and mice were monitored for survival.
Tissue Processing
Mice were perfused with 10% formalin immediately following sacrifice. Brains were then extracted and immediately immersed in 10% formalin overnight. Following complete fixation, tissue was transferred to 30% sucrose in PBS overnight and then embedded in optimal cutting temperature (OCT) solution for frozen sectioning. 40 μm-thick coronal sections were cut, and OCT was dissolved in PBS. Sections were added to slides, and coverslips were mounted using FluoroGel. Fluorescence was imaged using an EVOS FL Auto system.
Quantification and Statistical Analysis
Data were analyzed by one-way ANOVA or two-way ANOVA with Bonferroni post hoc analysis when comparing 2 or more groups or by the log-rank test when comparing Kaplan-Meier survival curves. Data were expressed as mean ± SEM, and significance between groups is denoted by ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. p > 0.05 is considered not significant. Replicate number is defined by n in figure legends. Data falling greater than 2 standard deviations apart from the mean were excluded as outliers.
Data and Software Availability
Transcriptome Analysis Console v.4.0 (Applied Biosystems, Foster City, CA) was used to visualize the results of the AmpliSeq chips. Differential expression was calculated using DESeq2, version 1.16.1. Statistics were run with the R package statmod, version 1.4.30, built under R, version 3.4.0. Linear regression analysis was performed using Limma, version 3.32.10.
Author Contributions
Conceptualization, S.B.H., C.R.M., and S.D.H.; Methodology, S.B.H., V.L., S.K., J.R.B., and S.D.H.; Investigation, S.B.H. and S.M.M.; Writing – Original Draft, S.B.H.; Writing – Review & Editing, S.B.H., C.R.M., and S.D.H; Resources, Supervision, and Funding Acquisition, S.D.H.
Conflicts of Interest
S.D.H. and M.G.E. have an equity interest in Falcon Therapeutics, which has licensed aspects of the iNSC technology.
Acknowledgments
We thank H. Wakimoto (Massachusetts General Hospital) for providing the GBM8 cell line, W. Kauffman (UNC School of Medicine) for providing the normal human fibroblast cell line, and T. Kafri (UNC Gene Therapy Center) and S. Magness (UNC Department of Medicine) for providing lentiviral backbones. pLV-tetO-SOX2 and LV-rTTA were a gift from Konrad Hochedlinger (Addgene). We thank Scott Floyd and Denise Dunn (Duke University) for generation of rat brain tissue explants. We thank the Small Animal Imaging Facility at the UNC Biomedical Imaging Research Center for providing the IVIS kinetic imaging service, and the imaging core is supported, in part, by an NCI Cancer Core Grant (P30-CA016086-40). Research reported in this publication was primarily supported by the Eshelman Institute for Innovation (RX03512417). Research reported in this publication was also supported, in part, by the North Carolina Biotech Center Institutional Support Grant (2015-IDG-1001). We also thank the UNC Flow Cytometry Core Facility, which is supported, in part, by the Cancer Center Core Support Grant (P30 CA016086)to the UNC Lineberger Comprehensive Cancer Center, as well as the Lineberger Cancer Center Bioinformatics Core Facility, particularly Dr. Yi-Hsuan Tsai, who assisted with bioinformatics analyses.
References
- 1.Ostrom Q.T., Gittleman H., Truitt G., Boscia A., Kruchko C., Barnholtz-Sloan J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro-oncol. 2018;20(Suppl 4):iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lettry V., Hagler S.B., Khagi S., Hingtgen S.D. Tumor-homing Stem Cell Therapy for Brain Cancer. Curr. Surg. Rep. 2017;5:28. [Google Scholar]
- 3.Burri S.H., Gondi V., Brown P.D., Mehta M.P. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma: Guide for Oncologists. Am. J. Clin. Oncol. 2018;41:191–196. doi: 10.1097/COC.0000000000000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adamson C., Kanu O.O., Mehta A.I., Di C., Lin N., Mattox A.K., Bigner D.D. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin. Investig. Drugs. 2009;18:1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R., Taillibert S., Kanner A.A., Kesari S., Steinberg D.M., Toms S.A., Taylor L.P., Lieberman F., Silvani A., Fink K.L. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA. 2015;314:2535–2543. doi: 10.1001/jama.2015.16669. [DOI] [PubMed] [Google Scholar]
- 6.Erpolat O.P., Akmansu M., Goksel F., Bora H., Yaman E., Büyükberber S. Outcome of newly diagnosed glioblastoma patients treated by radiotherapy plus concomitant and adjuvant temozolomide: a long-term analysis. Tumori. 2009;95:191–197. doi: 10.1177/030089160909500210. [DOI] [PubMed] [Google Scholar]
- 7.Bagó J.R., Alfonso-Pecchio A., Okolie O., Dumitru R., Rinkenbaugh A., Baldwin A.S., Miller C.R., Magness S.T., Hingtgen S.D. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat. Commun. 2016;7:10593. doi: 10.1038/ncomms10593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagó J.R., Okolie O., Dumitru R., Ewend M.G., Parker J.S., Vander Werff R., Underhill T.M., Schmid R.S., Miller C.R., Hingtgen S.D. Tumor-homing cytotoxic human induced neural stem cells for cancer therapy. Sci. Transl. Med. 2017;9:eaah6510. doi: 10.1126/scitranslmed.aah6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hingtgen S.D., Kasmieh R., van de Water J., Weissleder R., Shah K. A novel molecule integrating therapeutic and diagnostic activities reveals multiple aspects of stem cell-based therapy. Stem Cells. 2010;28:832–841. doi: 10.1002/stem.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aboody K.S., Najbauer J., Danks M.K. Stem and progenitor cell-mediated tumor selective gene therapy. Gene Ther. 2008;15:739–752. doi: 10.1038/gt.2008.41. [DOI] [PubMed] [Google Scholar]
- 11.Aboody K.S., Najbauer J., Metz M.Z., D’Apuzzo M., Gutova M., Annala A.J., Synold T.W., Couture L.A., Blanchard S., Moats R.A. Neural Stem Cell-Mediated Enzyme/Prodrug Therapy for Glioma: Preclinical Studies. Sci. Transl. Med. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hingtgen S., Kasmieh R., Elbayly E., Nesterenko I., Figueiredo J.-L., Dash R., Sarkar D., Hall D., Kozakov D., Vajda S. A First-Generation Multi-Functional Cytokine for Simultaneous Optical Tracking and Tumor Therapy. PLoS One. 2012;7:e40234. doi: 10.1371/journal.pone.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hingtgen S., Ren X., Terwilliger E., Classon M., Weissleder R., Shah K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and Temozolomide. Mol. Cancer Ther. 2008;7:3575–3585. doi: 10.1158/1535-7163.MCT-08-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehtesham M., Kabos P., Kabosova A., Neuman T., Black K.L., Yu J.S. Advances in Brief The Use of Interleukin 12-secreting Neural Stem Cells for the Treatment of. Stem Cells. 2002;62:5657–5663. [PubMed] [Google Scholar]
- 15.Tobias A.L., Thaci B., Auffinger B., Rincón E., Balyasnikova I.V., Kim C.K., Han Y., Zhang L., Aboody K.S., Ahmed A.U., Lesniak M.S. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl. Med. 2013;2:655–666. doi: 10.5966/sctm.2013-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed A.U., Alexiades N.G., Lesniak M.S. The use of neural stem cells in cancer gene therapy: Predicting the path to the clinic. Curr. Opin. Mol. Ther. 2010;12:546–552. [PMC free article] [PubMed] [Google Scholar]
- 18.Shah K., Hingtgen S., Kasmieh R., Figueiredo J.L., Garcia-Garcia E., Martinez-Serrano A., Breakefield X., Weissleder R. Bimodal viral vectors and in vivo imaging reveal the fate of human neural stem cells in experimental glioma model. J. Neurosci. 2008;28:4406–4413. doi: 10.1523/JNEUROSCI.0296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Portnow J., Synold T.W., Badie B., Tirughana R., Lacey S.F., D’Apuzzo M., Metz M.Z., Najbauer J., Bedell V., Vo T. Neural stem cell-based anticancer gene therapy: A first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 2017;23:2951–2960. doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satterlee A.B., Dunn D.E., Lo D.C., Khagi S., Hingtgen S.D. Tumoricidal Stem Cell Therapy Enables Killing in Novel Hybrid Models of Heterogeneous Glioblastoma. Nero. Oncol. 2019;21:1552–1564. doi: 10.1093/neuonc/noz138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okolie O., Bago J.R., Schmid R.S., Irvin D.M., Bash R.E., Miller C.R., Hingtgen S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-oncol. 2016;18:1622–1633. doi: 10.1093/neuonc/now117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose J.B., Pacelli S., Haj A.J.E., Dua H.S., Hopkinson A., White L.J., Rose F.R.A.J. Gelatin-based materials in ocular tissue engineering. Materials (Basel) 2014;7:3106–3135. doi: 10.3390/ma7043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Colli M., Massimi M., Barbetta A., Di Rosario B.L., Nardecchia S., Conti Devirgiliis L., Dentini M. A biomimetic porous hydrogel of gelatin and glycosaminoglycans cross-linked with transglutaminase and its application in the culture of hepatocytes. Biomed. Mater. 2012;7:055005. doi: 10.1088/1748-6041/7/5/055005. [DOI] [PubMed] [Google Scholar]
- 24.Kauer T.M., Figueiredo J.L., Hingtgen S., Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011;15:197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bagó J.R., Pegna G.J., Okolie O., Hingtgen S.D. Fibrin matrices enhance the transplant and efficacy of cytotoxic stem cell therapy for post-surgical cancer. Biomaterials. 2016;84:42–53. doi: 10.1016/j.biomaterials.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bagó J.R., Pegna G.J., Okolie O., Mohiti-Asli M., Loboa E.G., Hingtgen S.D. Electrospun nanofibrous scaffolds increase the efficacy of stem cell-mediated therapy of surgically resected glioblastoma. Biomaterials. 2016;90:116–125. doi: 10.1016/j.biomaterials.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirst T.C., Vesterinen H.M., Sena E.S., Egan K.J., Macleod M.R., Whittle I.R. Systematic review and meta-analysis of temozolomide in animal models of glioma: was clinical efficacy predicted? Br. J. Cancer. 2013;108:64–71. doi: 10.1038/bjc.2012.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang N.C., Song T.Y., Chang Y.Z., Chen M.Y., Hu M.L. Up-regulation of nicotinamide phosphoribosyltransferase and increase of NAD+ levels by glucose restriction extend replicative lifespan of human fibroblast Hs68 cells. Biogerontology. 2015;16:31–42. doi: 10.1007/s10522-014-9528-x. [DOI] [PubMed] [Google Scholar]
- 29.Sanokawa-Akakura R., Akakura S., Tabibzadeh S. Replicative senescence in human fibroblasts is delayed by hydrogen sulfide in a NAMPT/SIRT1 dependent manner. PLoS ONE. 2016;11:e0164710. doi: 10.1371/journal.pone.0164710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maas-Szabowski N., Fusenig N.E. Interleukin-1-induced growth factor expression in postmitotic and resting fibroblasts. J. Invest. Dermatol. 1996;107:849–855. doi: 10.1111/1523-1747.ep12331158. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura F., Kalb R.G., Strittmatter S.M. Molecular basis of semaphorin-mediated axon guidance. J. Neurobiol. 2000;44:219–229. doi: 10.1002/1097-4695(200008)44:2<219::aid-neu11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 32.Falk J., Bechara A., Fiore R., Nawabi H., Zhou H., Hoyo-Becerra C., Bozon M., Rougon G., Grumet M., Püschel A.W. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 2005;48:63–75. doi: 10.1016/j.neuron.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 33.Nasarre P., Gemmill R.M., Drabkin H.A. The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. OncoTargets Ther. 2014;7:1663–1687. doi: 10.2147/OTT.S37744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S.Y., Kim J.M., Cho S.Y., Kim H.S., Shin H.S., Jeon J.Y., Kausar R., Jeong S.Y., Lee Y.S., Lee M.A. TIMP-1 modulates chemotaxis of human neural stem cells through CD63 and integrin signalling. Biochem. J. 2014;459:565–576. doi: 10.1042/BJ20131119. [DOI] [PubMed] [Google Scholar]
- 35.Niedermeier M., Hennessy B.T., Knight Z.A., Henneberg M., Hu J., Kurtova A.V., Wierda W.G., Keating M.J., Shokat K.M., Burger J.A. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113:5549–5557. doi: 10.1182/blood-2008-06-165068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagci-Onder T., Wakimoto H., Anderegg M., Cameron C., Shah K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2011;71:154–163. doi: 10.1158/0008-5472.CAN-10-1601. [DOI] [PubMed] [Google Scholar]
- 37.Koh S.H., Lo E.H. The role of the PI3K pathway in the regeneration of the damaged brain by neural stem cells after cerebral infarction. J. Clin. Neurol. 2015;11:297–304. doi: 10.3988/jcn.2015.11.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelouahab H., Zhang Y., Wittner M., Oishi S., Fujii N., Besancenot R., Plo I., Ribrag V., Solary E., Vainchenker W. CXCL12/CXCR4 pathway is activated by oncogenic JAK2 in a PI3K-dependent manner. Oncotarget. 2016;8:54082–54095. doi: 10.18632/oncotarget.10789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L., Yang S., Wennmann D.O., Chen Y., Kremerskothen J., Dong J. KIBRA: In the brain and beyond. Cell. Signal. 2014;26:1392–1399. doi: 10.1016/j.cellsig.2014.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maherali N., Ahfeldt T., Rigamonti A., Utikal J., Cowan C., Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reinhart P.H., Kaltenbach L.S., Essrich C., Dunn D.E., Eudailey J.A., DeMarco C.T., Turmel G.J., Whaley J.C., Wood A., Cho S., Lo D.C. Identification of anti-inflammatory targets for Huntington’s disease using a brain slice-based screening assay. Neurobiol. Dis. 2011;43:248–256. doi: 10.1016/j.nbd.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Transcriptome Analysis Console v.4.0 (Applied Biosystems, Foster City, CA) was used to visualize the results of the AmpliSeq chips. Differential expression was calculated using DESeq2, version 1.16.1. Statistics were run with the R package statmod, version 1.4.30, built under R, version 3.4.0. Linear regression analysis was performed using Limma, version 3.32.10.