

Abstract
There is a strong rationale to consider future cell therapeutic approaches for cystic fibrosis (CF) in which autologous proximal airway basal stem cells, corrected for CFTR mutations, are transplanted into the patient’s lungs. We assessed the possibility of editing the CFTR locus in these cells using zinc-finger nucleases and have pursued two approaches. The first, mutation-specific correction, is a footprint-free method replacing the CFTR mutation with corrected sequences. We have applied this approach for correction of ΔF508, demonstrating restoration of mature CFTR protein and function in air-liquid interface cultures established from bulk edited basal cells. The second is targeting integration of a partial CFTR cDNA within an intron of the endogenous CFTR gene, providing correction for all CFTR mutations downstream of the integration and exploiting the native CFTR promoter and chromatin architecture for physiologically relevant expression. Without selection, we observed highly efficient, site-specific targeted integration in basal cells carrying various CFTR mutations and demonstrated restored CFTR function at therapeutically relevant levels. Significantly, Omni-ATAC-seq analysis revealed minimal impact on the positions of open chromatin within the native CFTR locus. These results demonstrate efficient functional correction of CFTR and provide a platform for further ex vivo and in vivo editing.
Keywords: cystic fibrosis, gene editing, basal stem cell
Graphical Abstract
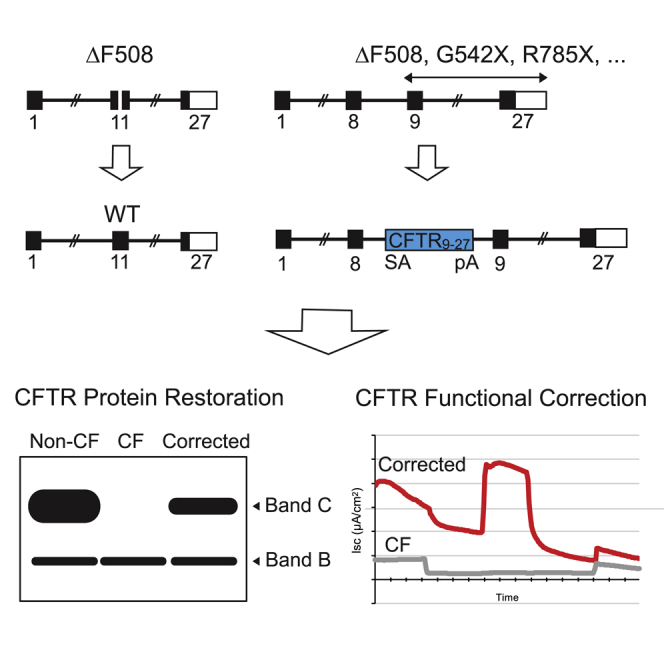
Suzuki et al. report correction of the CFTR defect in cystic fibrosis airway basal stem cells. They utilized gene-editing strategies either specific for the ΔF508 CFTR mutation or applicable to most CFTR mutations. Both approaches yielded highly efficient correction without selection, restoring CFTR function to therapeutically relevant levels.
Introduction
There is still much to be learned about turnover of cells in the human airway, as well as the identity of potential stem/progenitor cells responsible for overall pulmonary architecture and maintenance. There is an emerging consensus, however, that pseudo-stratified epithelial tissue of the human proximal airway contains basal cells capable both of self-renewing cell division, as well as differentiation to other specialized cells, including ciliated and secretory cells. As such, proximal airway basal cells serve as a major class of stem/progenitor cells within the proximal airway.1, 2, 3, 4 Thus, there is strong rationale to consider future cell therapeutic approaches for CF in which autologous proximal airway basal cells, corrected for CFTR gene mutations, are transplanted into the lungs of affected CF patients. To that end, our objective has been to prepare a population of CFTR corrected, patient-specific airway basal cells, which retain the ability to develop pseudo-stratified airway epithelium with restored CFTR function. Several groups, including our own, have utilized various methods to directly correct or compensate for CFTR mutations in relevant cell types via editing or gene transfer.5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21
There is currently uncertainty about the relative contribution of different cell types to overall CFTR activity in the airway epithelium. Does the rare ionocyte population expressing high levels of CFTR dominate22,23 or do other more abundant luminal cell types (secretory or ciliated), which apparently express CFTR at lower levels per cell, also contribute?24 Importantly, the aforementioned cell types are derived from airway basal stem cells, which justifies our editing focus.2,3,22
It is important to consider frequencies at which cells in the CF airway would need to be corrected, either as a consequence of transplantation of edited airway basal cells or direct in vivo editing, for therapeutic benefit. Johnson et al.25 reported that as few as 6%–10% corrected cells was sufficient to restore CF chloride ion transport levels to normal. A second study showed that 5% of pseudostratified epithelial cells expressing CFTR corrected the CF chloride transport defect.26 However, these two studies used highly efficient viral vector promoters to express CFTR and therefore it cannot be assured that comparable results would be obtained in differentiated human airway epithelia where CFTR is under the control of its endogenous promoter. In mixing experiments of human airway cells in air-liquid interface (ALI) cultures, it was reported that 20% of non-CF cells mixed into a background of 80% ΔF508/ΔF508 cells yielded CFTR chloride current at levels 70% of wild-type levels.27 These data supported the concept that even a small fraction of cells expressing CFTR from the endogenous CFTR locus would be sufficient to correct the chloride transport defect in CF cells.27
The two approaches for CFTR gene editing in CF airway basal cells pursued by this study are shown in Figure 1. Our rationale for performing gene editing specifically of the endogenous CFTR locus reflects an intent to achieve expression of the corrected CFTR gene at close to physiologic levels. The first approach is site-specific correction of CFTR mutations; namely, a footprint-free method replacing mutant CFTR sequences with the corrected sequences (Figure 1A). Because CF is a recessive disease, correction of only one CFTR allele per cell would be sufficient for rescue of CFTR function. Site-specific correction of CFTR mutations in the endogenous gene is expected to result in physiologically appropriate levels of CFTR protein and function. However, site-specific correction typically requires a distinct set of sequence-directed nuclease and oligonucleotide donor reagents for each CFTR mutation. Because approximately 2,000 different CF-associated mutations have been reported, the sequence-specific approach would only seem justified for the most frequently occurring variants such as ΔF508. The second approach is homologous recombination-mediated targeted integration (TI) of a codon optimized partial CFTR cDNA (also denoted as a “super-exon”)5,7 preceded by a splice acceptor (SA) and followed by a polyadenylation (pA) sequence, within an intron of the endogenous CFTR gene (Figure 1B). This approach is capable of providing correction for all CFTR mutations downstream of the targeted partial CFTR cDNA integration site. Furthermore, by targeting cleavage of the genomic DNA within intronic versus exonic sequences, the possibility that nuclease-induced indels will adversely impact uncorrected alleles is minimized. With the objective of achieving physiologically regulated levels of CFTR expression and function, this approach would also benefit, in principle, from endogenous CFTR promoter activity and native chromatin architecture, provided that TI of the partial CFTR cDNA does not disrupt chromatin architecture in edited cells.
Figure 1.

Site-Specific Editing of CF Airway Basal Cells
(A) Site-specific correction of ΔF508 mediated by targeted nuclease cleavage with wild-type DNA template serving as donor. (B) Integration of CFTR9-27 cDNA (encoding exons 9–27), preceded by a splice acceptor (SA), followed by polyadenylation (pA) sequences, and flanked by homology sequences, into CFTR intron 8; the spliced mRNA joins endogenous exon 8 with the exon 9–27 transgene.
Results
Feeder-free Expansion of Primary Airway Basal Cells with Retention of CFTR Function
Starting with early passage primary human airway epithelial cells from explanted CF or non-CF lungs, we first compared feeder-free culture methodologies for their ability to expand airway basal cells for numerous passages while retaining functional capability. Both the previously reported dual SMAD inhibition medium28 and Pneumacult Ex-Plus medium allowed for continuous proliferation to at least passage 12 (p12; corresponding to 35–37 population doublings [PDs]; Figure S1A) while retaining markers characteristic of basal cells (e.g., CD49f, NGFR; Figure S1B). In order to assay functionality, expanded cells at various passage numbers were plated onto porous membranes and subject to ALI culture to assess development of well-differentiated airway epithelium. With increasing passage number, particularly for the dual SMAD inhibition culture, we noted progressive loss of ciliated cells, as measured by acetylated tubulin (Figure S1C). In order to measure CFTR ion transport function, ALI cultures were subjected to Ussing chamber assays. Airway basal cells expanded under Pneumacult Ex-Plus culture conditions exhibited robust levels of CFTR function through at least p8, while basal cells expanded in dual SMAD cultures exhibited lower levels of activity (Figures S1D and S1E). Under either culture condition, further passaging beyond p8 resulted in decreased levels of CFTR-dependent transepithelial transport (Figure S1E). Based on the above, we chose to perform our gene-editing manipulations on airway basal cells maintained maximally until p6 to p8 (∼18–25 PDs) in the Pneumacult Ex-Plus medium.
Sequence-Specific Correction of ΔF508 Mutation
Zinc-finger nucleases (ZFNs), targeted to recognize and cleave CFTR ΔF508 sequences in exon 11 (ZFN11; Figure S2A), were delivered as mRNA to ΔF508/ΔF508 airway basal cells via electroporation. For correction of the ΔF508 mutation, we evaluated two types of donor sequences encoding wild-type CFTR sequence. First, we co-delivered single-stranded (ss) 200-mer oligo DNAs together with the ZFN mRNAs via electroporation (Figure 2A). The ss oligo, centered on the ΔF508 mutation, included the wild-type restoring “CTT” bases (Figure S2A) spanned by approximately 100 bases of homology sequence on either side. In order to achieve maximal levels of correction while retaining cell viability, optimization was performed with respect to amount of ZFN mRNA (data not shown) and ssDNA donor oligo (e.g., Figures S2B and S2C). Next generation sequencing (NGS) was utilized to quantify the frequency of ΔF508 CFTR alleles exhibiting either no modification, indels, or correction (Figures 2A and 2B, Table S1). Delivery of ZFN mRNA alone resulted in 44.6% ± 2.4% of CFTR alleles exhibiting indels. When ssDNA donor was co-delivered with ZFN mRNA, the frequency of ΔF508 correction was 10.6% ± 2.6% (all editing frequencies are expressed on a per CFTR allele basis) (Figure 2B; Table S1). We sought to modify our methodology in order to achieve significantly higher efficiencies of correction. To this end, we utilized a longer donor (∼2.0 kb, again centered on the correcting “CTT” bases), but in this case delivered via adeno-associated virus type 6 (AAV-6) (Figure 2A); AAV-6 was selected due to its favorable tropism for the lung.29,30 Optimization was again performed, in this case focused on the amount of ZFN and the AAV-6 dose (Figure S2D). Delivery of ZFN mRNA via electroporation followed immediately thereafter by AAV-6 donor transduction resulted in a correction efficiency of 31.0% ± 4.0% (Figure 2B; Table S1).
Figure 2.

Sequence-Specific ΔF508 Correction with Restoration of Mature CFTR Protein Expression and Function
(A) Schematic of sequence-specific ΔF508 correction strategy. ZFNs were designed to site-specifically recognize and cleave CFTR ΔF508 sequences in exon 11 (ZFN11). Two donors carrying correcting CFTR sequence are shown: a single stranded 200-mer oligo DNA donor (ssDNA) and 2 kb AAV-6 donor (containing 1,963 bp of wild-type CFTR sequence spanning exon 11). Three anticipated outcomes are represented as unmodified, indels, and corrected. Primers f1 and r2, located outside the 1,963 bp donor sequence, were used to amplify the targeted region in order to assess genome modification efficiency. (B) Efficiency of genome modification analyzed by NGS. Z, ZFN11 alone; Z+D, ZFN11 plus donor DNA (mean ± SD, n = 3 biological replicates; see also Table S1). (C) H&E staining and immunofluorescence detection of airway epithelium markers in ALI culture. Major epithelial cell types were identified with markers: p63 or keratin 5 (CK5) for basal cells; mucin 5AC (MUC5AC) for secretory cells; acetylated tubulin (ACT) for ciliated cells. Representative 40× images in transverse section staining. Scale bar, 50 μm (panels). (D) Western blotting of CFTR. Sequence-specific ΔF508 correction restores expression of mature, fully glycosylated CFTR protein (band C). Calnexin, loading control. (E) Representative trace of short circuit current (Isc) from 4 samples tested: DMSO-treated, VX-809/VX-770 pre-treated and Z+D-treated CF (ΔF508/ΔF508), and non-CF evaluated by Ussing chamber analysis. (F) Summary of CFTR chloride current stimulated by forskolin and inhibited by CFTR inhibitor 172 (CFTR inh-172). Difference of Isc (Δlsc) (μA/cm2) before and after treatments are calculated from traces represented by (E). Values of Δlsc from samples listed in Table S2 are summarized and shown (mean ± SD, n = 3 experiments). (G) Restored CFTR activity as function of gene-editing frequency. The blue symbols reflect results from individual ΔF508/ΔF508 cell experiments with sequence-specific correction of ΔF508 utilizing either ssDNA or AAV-6 donor. CFTR activity (forskolin-induced) for each experiment is expressed as % of non-CF (e.g., see Table S2A). Frequency of genome modification for each experiment is from Table S1. Shown in open and filled blue circles are results of ΔF508 correction for ΔF508/ΔF508 cells with ssDNA and AAV-6 donors, respectively; the black filled circle is from ΔF508/ΔF508 cells. Linear regression for ΔF508 sequence-specific correction experiments resulted in an R2 = 0.92.
Sequence-Specific ΔF508 Correction Restores Mature CFTR Protein Expression and Function
In order to assess the functional consequences of ΔF508 correction, cells edited with the 2 kb AAV-6 donor were transferred onto porous supports and subjected to ALI culture conditions to generate well-differentiated airway epithelium containing basal cells (p63, keratin 5), secretory cells (mucin 5AC), ciliated cells (acetylated tubulin, FOXJ1), and ionocytes (FOXI1; Figures 2C and S3). Importantly, the manipulations required for editing did not alter the development of the pseudostratified epithelium. For example, cellular composition of the derived epithelium (i.e., % basal cells, % secretory cells, % ciliated cells, % ionocytes) was not affected by the manipulations (Figure S3).
As expected, ALI cultures derived from unmanipulated ΔF508/ΔF508 airway basal cells only expressed the core glycosylated CFTR protein (band B; Figure 2D), whereas non-CF ALI cultures primarily exhibited the mature, fully-glycosylated, membrane-bound form of CFTR (band C; Figure 2D). Importantly, the AAV-6 edited ΔF508/ΔF508 cultures demonstrated the emergence of band C, corresponding to the presence of corrected CFTR alleles in the treated cells (Figures 2D and S4). Electrophysiological measurement of CFTR channel function was performed via Ussing chamber analysis. In Figure 2E, tracings for a representative experiment are presented; results of several experiments are presented in Table S2A and summarized in Figure 2F and Table S2B. As expected, non-treated ΔF508/ΔF508 cultures exhibit negligible forskolin-activated CFTR current (Figure 2F). ALI cultures derived from ΔF508/ΔF508 cells treated with ZFNs plus AAV-6 donor exhibited CFTR-dependent current (20.2 ± 3.5 μA/cm2 (Figure 2F), robustly blocked by the CFTR channel inhibitor (CFTRinh-172; Figures 2E and 2F). This level of restored CFTR activity in edited cultures is significant in two respects. First, this level of rescue in bulk-treated ΔF508/ΔF508 cultures (with a mean correction efficiency of 31.0%; Figure 2B) is 40.2% of that seen in the non-CF culture (50.2 ± 4.7 μA/cm2; Figure 2F). Second, this activity is 152% of that resulting from exposure of non-edited ΔF508/ΔF508 cells to the clinically approved CFTR modulators VX-809 and VX-770 (13.3 ± 0.9 μA/cm2; Figure 2F). This particular combination of modulators has been shown to be of therapeutic benefit in ΔF508/ΔF508 CF patients31 and thus provides a metric against which the CFTR activity in the edited ΔF508/ΔF508 cells can be compared. Thus, by both measures (% of non-CF, % of VX-809/VX-770 treated ΔF508/ΔF508), CFTR function resulting from this efficiency of ΔF508 correction is meaningful (see Discussion).
Similar analysis of ALI cultures derived from the 200-mer ssDNA donor-corrected airway basal cells (mean correction frequency of 10.6%; Figure 2B; Table S1) also demonstrated restored expression of mature CFTR protein (Figure S4), albeit at lower levels. For ssDNA donor edited cells, the level of CFTR current was 13.2% of the non-CF control (Tables S2A and S2B). However, rescue in this case did not reach the level of the VX-809/VX-770 treated ΔF508/ΔF508 control (21.3% of the non-CF control; Tables S2A and S2B). For sequence-specific ΔF508 correction, we observed an approximately linear correlation (R2 = 0.92) between the frequency of correction and level of restored CFTR current (Figure 2G). Taken together, these data, comparing the ssDNA and AAV-6 donors, support the importance of achieving significant levels of editing.
Efficient TI of SA-CFTR9-27-pA into CFTR Intron 8 of ΔF508/ΔF508 Airway Basal Cells
We selected CFTR intron 8 to demonstrate proof of principle for the homologous recombination-mediated TI approach. ZFNs targeting CFTR intron 8 were designed and verified in K562 cells (data not shown). In order to facilitate simultaneous quantification of both the TI and indel frequencies in the same population of edited cells, an intron 8 specific primer, followed by a random sequence and an EcoRI site (to permit a rapid assessment of TI), was incorporated into the donor immediately downstream of the SA-CFTR9-27-pA cassette (Figure 3A); PCR amplification would yield a PCR product of identical size as that from unmodified cells and NGS would permit an accurate determination of TI and indel rates. We note the downstream primer was positioned outside of donor sequences to only amplify from transgene sequences integrated at the targeted site (Figure 3A). Electroporation-mediated delivery of intron 8 ZFN mRNA to ΔF508/ΔF508 airway basal cells was followed almost immediately by AAV-6 transduction of SA-CFTR9-27-pA. Initial evidence for sequence-specific TI of SA-CFTR9-27-pA into intron 8 came from upstream inside-outside PCR amplification (Figure S5A) and from EcoRI digestion of downstream PCR amplicons (Figure S5B). These results were quantified by NGS, with TI rates of 56.5% ± 7.4% observed 4 days post editing (Figure 3B; Table S3A). Efficient intron 8 TI was confirmed by Southern blotting of genomic DNA isolated from the edited ΔF508/ΔF508 cells (Figure 3C).
Figure 3.

Efficient SA-CFTR9-27-pA TI into CFTR Intron 8 of ΔF508/ΔF508 Airway Basal Cells
(A) Schematic of site-specific targeted editing of CFTR intron 8. Unmodified and indel diagrams highlight CFTR genomic sequences between exons 8 and 9 (black boxes; not to scale). TI-8 diagram shows intron 8 TI of human codon optimized CFTR9-27 cDNA preceded by a splice acceptor, followed by bovine growth hormone (bGH) pA sequence, and flanked by 313 bp homology left (HL) and 351 bp homology right (HR) intron 8 sequences. Arrows indicate oligos amplifying unmodified, indel, or TI-8 events and used to quantify frequency of each by NGS. (B) The percentage of genome modification (mean ± SD) determined by NGS (Table S3A) from five independent TI-8 experiments (utilizing the AAV-6 donor at either 2 × 106 or 6 × 106 viral genomes [vg]/cell). There was no enhancement of TI efficiency as one increased the amount of donor. Efficiency was measured 4 days after the delivery of ZFNs targeting intron 8 (ZFN8) followed immediately by AAV-6 CFTR9-27 cDNA donor. Z, ZFN8 alone; Z+D, ZFN8 and AAV-6 donor. (C) Southern blot analysis of TI-8. The schematic shows the expected genomic organization of non-targeted (no TI-8) and targeted (TI-8) alleles with the expected sizes resulting from EcoRI digestion. The non-targeted allele yields a 9.8 kb band while the TI-8 allele yields a 6.9 kb band due to the new EcoRI site in the CFTR9-27 cDNA donor construct. (D) H&E staining and immunostaining of well-differentiated airway epithelium in ALI culture. Representative images of cross sections from ΔF508/ΔF508 or TI-8 ΔF508/ΔF508. Immunostaining shows major epithelial cell types: basal cell (CK5 and P63), secretory cells (MUC5AC), and ciliated cells (ACT). 40× images, scale bar, 50 μm. (E) Detection of transgene CFTR9-27 mRNA. Schematic of endogenous and transgene CFTR mRNA is shown here. Primer T9 recognizes codon-optimized transgene exon 9 sequence only (blue) while Primer E9 recognizes endogenous exon 9. A 250 bp E8-T9 RT-PCR amplicon, evidence of the chimeric endogenous-transgene CFTR mRNA, was present only in the TI-8 ΔF508/ΔF508 sample. (F) Restoration of fully glycosylated CFTR protein via TI-8. Western blots of protein lysates harvested at 4 weeks of ALI culture. Band C, representing the mature, fully glycosylated form of CFTR, is present in non-CF, absent in ΔF508/ΔF508 cells, and restored in TI-8 ΔF508/ΔF508. Calnexin, loading control. (G) CFTR functional assay in ALI cultures at 4 weeks. TI-8 ΔF508/ΔF508 cells show the restoration of CFTR function measured as Δlsc (μA/cm2) ± SD and compared with ΔF508/ΔF508 treated with VX-809/VX-770. (H) Restored CFTR activity as function of gene-editing frequency. The blue and black symbols and blue linear regression line are from individual ΔF508/ΔF508 cell experiments with sequence-specific correction of ΔF508 (from Figure 2G). Shown in filled square and open square orange symbols are the results of ΔF508/ΔF508 cell TI-8 and ΔF508/ΔF508 cell TI-7 experiments, respectively.
TI of SA-CFTR9-27-pA into CFTR Intron 8 of ΔF508/ΔF508 Airway Basal Cells Restores Mature CFTR Protein Expression and Function
Edited cells were plated in ALI cultures to develop well differentiated epithelium as described previously. In the one experiment for which we performed the analysis, we observed roughly comparable frequencies of intron 8 TI in basal cells prior to plating in the ALI cultures (50.1%) versus frequency of correction in mature, well differentiated airway epithelium in ALI cultures (43.9%) (Table S3B). The TI editing and minimal expansion required for early passage airway basal cell cultures also did not adversely affect the ability to establish well-differentiated airway epithelium comprising basal, secretory, and ciliated cells (Figure 3D). Successful TI-mediated correction of bulk cultures was confirmed by detection of CFTR transgene mRNA (Figure 3E) and by restoration of CFTR band C protein (Figure 3F). Ussing chamber assays from two independent experiments demonstrated restoration of forskolin-activated CFTR currents in intron 8 TI cultures (Figure 3G; sample trace in Figure S5C). CFTR current levels in edited cells from two independent experiments were 34.4% to 42.7% of the level seen in non-CF cultures (Figure 3G). Furthermore, the mean level of restored CFTR channel activity was 157% of the level seen for ΔF508/ΔF508 cultures exposed to VX-809/VX-770 (Figure 3G). Thus, with our current TI efficiency, we are in a therapeutically relevant range for a mixed population of corrected and uncorrected ΔF508/ΔF508 cells.
We also evaluated, in a limited number of experiments, the TI strategy for intron 7 by delivery of intron 7 specific ZFNs followed shortly thereafter by AAV-6 delivery of a donor construct now including CFTR exons 8 to 27 (SA-CFTR8-27-pA). Evidence for intron 7 TI, first suggested via EcoRI digestion (Figure S6A), was confirmed by NGS (Figure S6B). Successful targeting was confirmed by expression of the transgenic mRNA (Figure S6C), restored CFTR band C protein (Figure S6D), and restored CFTR current (Figure S6E; sample trace in Figure S5C). This demonstrates that potential effectiveness of the TI approach is not limited to intron 8.
The level of TI-8 or TI-7 restored CFTR function in ΔF508/ΔF508 cells, as a function of editing efficiency, is shown in Figure 3H. These results confirm significant CFTR functional restoration with the TI approach. Given the limited number of experimental data points for either the sequence-specific correction or TI approaches, it is premature to draw any firm conclusions, However, the tendency of the TI data points to perhaps lie slightly below the sequence-specific trendline suggests that further improvements may be possible (see Discussion).
CFTR Intron 8 TI Preserves the Open Chromatin Structure of the ΔF508/ΔF508 CFTR Locus
Our rationale for directly targeting integration of the partial CFTR cDNA at the endogenous CFTR locus was to exploit the native CFTR promoter and chromatin architecture, with the goal of achieving physiologically relevant levels of restored CFTR protein and function. The presence in ALI cultures of transgene-derived CFTR mRNA (Figure 3E), protein (Figure 3F), and function (Figure 3G) from bulk TI-8 edited cells provides support for this targeting strategy. We further wished to confirm that neither the integrated SA-CFTR9-27-pA transgene nor indels at the target site significantly disrupted the sites of open chromatin normally present in the native CFTR locus. To this end, we mapped, via Omni-ATAC-seq, the sites of open chromatin in ALI cultures established from TI-8 edited ΔF508/ΔF508 cells (Exp. 2; Table S3B; Figure 3G). At this time point, 43.9% and 47.3% of the CFTR alleles exhibited TI and indels, respectively (Table S3B). ALI cultures derived from unmanipulated ΔF508/ΔF508 cells or non-CF cells were used as controls. Significantly, Omni-ATAC-seq analysis of intron 8 targeted ALI cultures revealed minimal impact on the positions of open chromatin within the native CFTR locus (Figure 4). Unaffected sites of open chromatin included the CFTR topological domain boundaries at −80.1 kb and +48.9 kb, as well as the CFTR promoter and the previously identified −44 kb and −35 kb cis-regulatory elements (CREs) shown to play a critical role in directing CFTR expression in airway cells (Figure 4). Of note, other dominant peaks of open chromatin observed across the locus in HBE-ALI cells (all of which correspond to known CREs32) are also unaffected by the insertion. Upon closer examination, we did observe a new site of accessible chromatin near the start of the integrated SA-CFTR9-27-pA transgene (Figure 4, lower panel). The significance of this particular finding is unclear. In summary, we find that intron 8 TI of the transgene has minimal impact on the open chromatin profile within the native CFTR locus.
Figure 4.

Targeted Integration of a Partial cDNA at CFTR Intron 8 Does Not Disrupt the Open Chromatin Profile of the ΔF508/ΔF508 CFTR Locus
Integrative Genomics Viewer (IGV) browser graphics of CFTR TAD regions showing Omni-ATAC profiles of non-CF, ΔF508/ΔF508, and TI-8 ΔF508/ΔF508 ALI cells. The number of sequencing reads is shown in Table S7. Key cis-regulatory elements are marked above the ATAC profiles by arrows. DHS denotes all DNase I-hypersensitive sites across the locus. Below is a magnified panel of the intron 8 region showing appearance of a peak of open chromatin at the TI-8 cDNA insertion site.
Efficient TI Restores Mature CFTR Protein Expression and Function in Airway Basal Cells Carrying CFTR Premature Termination Codon Mutations
TI strategies as described here are highly relevant to CFTR mutations resulting in little-or-no CFTR protein (for example, due to premature termination codons [PTCs] or splicing defects); i.e., variants not otherwise responsive to CFTR modulators. Evidence for sequence-specific TI of SA-CFTR9-27-pA into intron 8 of ΔF508/R553X or G542X/R785X airway basal cells was obtained from 5′ inside-outside PCR amplification (Figure S7A). Amplification using 3′ PCR followed by EcoRI digestion was consistent with efficient TI (Figure S7B). NGS confirmed highly efficient TI in both the ΔF508/R553X (50.0% ± 6.0%) and G542X/R785X (61.8% ± 6.0%) airway basal cells (Tables S3C and S3D; Figure 5A), and efficient intron 8 TI was confirmed by Southern blot analysis (Figure 5B). Successful TI-mediated correction in the edited cultures was further established by restoration of CFTR band C protein (Figure 5C). Ussing chamber assays demonstrated restoration of forskolin-activated CFTR current in intron 8 TI ALI cultures at levels 55.8% of non-CF cultures for ΔF508/R553X and 33.8% of non-CF for G542X/R785X cultures (Figure 5D; sample traces in Figures S7C and S7D). These results demonstrate that targeted correction of PTC containing airway cells results in significant restoration of CFTR channel activity, either when the PTC occurs in combination with ΔF508 or with a second PTC.
Figure 5.

Efficient TI-8 Correction in CF Airway Basal Cells Carrying CFTR PTC Mutations
(A) Frequency of genome modification determined by NGS in CF basal cells carrying ΔF508/R553X (mean ± SD, n = 3 experiments) or G542X/R785X (mean ± SD, n = 4 experiments) variants (from Tables S3C and S3D). Highly efficient TI-8 was confirmed in CF basal cells treated with ZFN8 and AAV-6 CFTR9-27 cDNA donor (Z+D) 4 days after genome editing. (B) Southern blot analysis of TI-8. A 6.9 kb fragment confirmed the expected TI-8 genomic organization in CF basal cells of either genotype. (C) Restoration of fully glycosylated CFTR protein via TI-8. Western blot of protein lysates harvested at 4 weeks of ALI culture shows the presence of band C in TI-8 and the absence in non-targeted cells. Calnexin, loading control. (D) CFTR functional assay in ALI cultures. Shown are short-circuit current measurements for bulk TI-8 ΔF508/R553X and TI-8 G542X/R785X cells. Restored CFTR currents are shown as Δlsc (μA/cm2) ± SD and are in excess of the VX-809/VX-770 treated cells. No VX-809/VX-770 response is expected for G542X/R785X.
Discussion
In this study we have demonstrated highly efficient editing of the mutant CFTR locus in primary CF airway basal cells. High efficiency was achieved utilizing either a mutation-specific approach, demonstrated for correction of ΔF508, or a mutation-agnostic approach, requiring only that the CFTR mutation lies downstream of the targeted integration. Our objective was to achieve an efficiency of gene editing such that neither selection (e.g., via drug selection or sorting) nor single-cell derived cloning would be required to obtain a pool of cells corrected at a sufficiently high level to be directly transplantable. We are not aware of prior reports demonstrating efficient TI in CF primary airway basal stem cells; these levels of correction resulted in restored CFTR function for the basal cell-derived airway epithelium. Future studies will be required to determine how to prepare the CF airways for receipt and integration of the edited airway basal cells and how best to maximize the frequency of corrected cells in the resulting engrafted airway tissue.33, 34, 35
For ΔF508-specific correction utilizing AAV-6 donor delivery, we achieved correction to wild-type in approximately 30% of CFTR alleles. These editing results are generally similar to those recently reported for correction of ΔF508 in nasal and bronchial primary epithelial cells mediated by CRISPR/Cas9 together with AAV-6.11 This 30% correction frequency corresponds to between 30% (if both ΔF508 alleles per cell are restored) and 60% (if only one ΔF508 allele is corrected per cell) of target cells demonstrating correction. In principle, molecular characterization of individual basal cells or single-cell-derived basal cell clones could provide a more accurate quantitative assessment of correction (or indels) on a per cell basis;36 this will be addressed in future studies. Importantly, neither the manipulations required for editing (i.e., electroporation, AAV-6 transduction) nor the editing itself (i.e., correction, indels) altered the development or cellular composition of the derived pseudostratified epithelium. Studies of various CFTR mutations suggest that even very modest levels of CFTR gene correction may provide therapeutic benefit. For example, as little as 4.7% of the normal level of wild-type CFTR mRNA is sufficient to result in a milder form of CF.37 In another study, 8% of wild-type CFTR transcripts was suggested to be sufficient for normal lung function.38 According to this measure, even our modest 10.6% ± 2.6% ΔF508 correction efficiency with the ssDNA oligo donor, resulting in 13.2% ± 3.2% of wild-type current levels, might offer partial therapeutic benefit. The restoration of CFTR activity to levels 40.2% ± 4.7% of non-CF, resulting from AAV-6 delivery of the 2 kb donor approximates a therapeutically beneficial range, and exceeds the CFTR activity resulting from VX-809/VX-770 treatment (26.7% ± 3.6% of non-CF). Whereas the restoration of CFTR activity in edited cultures is presumably due only to the fraction of cells with correction, it is possible that modulators act more generally. Administration of the VX-809/VX-770 drug combination to homozygous ΔF508 patients resulted in improvements in FEV1, reduced rate of pulmonary exacerbations, reduced events leading to hospitalization, and reduced use of intravenous antibiotics.31
For sequence-specific ΔF508 correction, we observed an approximately linear correlation (R2 = 0.92) between the frequency of ΔF508 correction and level of restored CFTR current (Figure 2G). In mixing experiments of primary human airway cells, 10%:90%, 20%:80%, and 60%:40% mixtures of non-CF:ΔF508/ΔF508 cells yielded CFTR chloride current at levels ∼35%, 70%, and 100% that of non-CF cells;27 a linear relationship between CFTR current and % non-CF cells was maintained up to approximately 20% non-CF cell percentages.27 On first examination, our levels of CFTR activity, as a function of gene-corrected cells, might appear to be somewhat lower than would be predicted by the mixing experiments. However, it is difficult to make direct comparisons of this type since it is currently unknown precisely what percentage of our cells were corrected at both CFTR alleles (corresponding to the non-CF cells utilized in the mixing experiment described above) versus at only a single CFTR allele per cell (corresponding to a carrier state). Although it might be assumed that carrier state cells would have CFTR activity similar to that of non-CF cells, it has been suggested that presence of the ΔF508 protein can adversely affect the processing and chloride channel function of the wild-type CFTR protein.39
While the mutation-specific editing approach achieved efficient correction and restored CFTR activity, there are at least two potential drawbacks to this strategy. First is the potential requirement for a new set of editing reagents (e.g., sequence specific nuclease and donor) for each mutation or small region spanning several mutations. Although this may be warranted for a common mutation such as ΔF508, development of editing materials and performance of clinical trials for each individual variant would be difficult. Second, even in the presence of donor, a significant frequency of CFTR alleles with nuclease-induced indels was observed; based on other editing studies,36 as well as our own experience (data not shown), introduction of indels at both alleles is expected to occur with some frequency. For CFTR mutations occurring within exon sequences (e.g., ΔF508), such residual indels would need to be examined to ensure there are not unintended consequences, for example the potential loss of response to current or future modulators.
As an alternative strategy capable of treating airway basal cells carrying various CFTR mutations, we demonstrated efficient TI of CFTR partial cDNA sequences into CFTR introns 7 and 8. We first showed successful intron 8 TI in ΔF508/ΔF508 cells at efficiencies 56.5% ± 7.4% as measured 4 days after editing. Again, the manipulations required for highly efficient editing (mean allele modification of 56.5% for TI and 33.0% for indels) did not adversely affect the ability to establish well-differentiated airway epithelium. Intron 8 TI of ΔF508/ΔF508 cells restored CFTR function to levels 34.4%–42.7% of non-CF.
By targeting integration of a CFTR partial cDNA to the endogenous CFTR locus, our intent was to retain, to the greatest extent possible, the regulated level and pattern of expression of the native gene. Importantly, Omni-ATAC-seq analysis of the ΔF508/ΔF508 TI-8 ALI culture revealed no apparent alteration in the pattern of open chromatin at previously identified regulatory sites within the CFTR locus. This result is encouraging with respect to maintaining physiologically regulated transgene expression. We also demonstrated successful TI and restoration of CFTR activity for TI-7. It is likely that the overall reduced TI-7 efficiency (with respect to TI-8) is due to the lower activity of the intron 7 ZFNs (with respect to the intron 8 ZFNs; Figures 3B and S6B)
Recently, there have been important advances in development of CF modulators (e.g., the recently available triple drug combination) potentially providing significant therapeutic benefit to the vast majority of CF patients.40,41 Consequently, increased focus is now being devoted to developing novel therapeutic approaches for the 7% of CF patients who are unable to benefit from modulators, due to an insufficient amount of CFTR protein (e.g., due to PTCs or splicing mutations). We thus applied the TI methodology to restore CFTR function in CF airway basal cells carrying PTCs. Again, we demonstrated highly efficient TI-8 at frequencies 50.0% ± 6.0% and 61.8% ± 6.0% in ΔF508/R553X and G542X/R785X cells, respectively (Figures 5A and 5B). After differentiation in ALI culture, restoration of both CFTR protein (Figure 5C) and function (Figure 5D) were observed.
In this project we evaluated TI for both introns 7 and 8. There are several considerations relevant to determining which CFTR intron is optimal for TI. One consideration would be to maximize the number of CF patients (or CFTR mutations) that could, in principle, benefit from site-specific TI. We note that intron 8 TI, for example, would provide correction for 89.1% of the CF-causing CFTR alleles listed in the CFTR2 database: https://www.cftr2.org/ (March 2019 version). Notably, this includes ΔF508, several of the most common PTCs such as G542X, R785X, and W1282X, and common splicing mutations such as 3,849 + 10 kb C > T. A second consideration is the size of the requisite CFTR partial cDNA and whether this can be appropriately delivered with a chosen vector. Although intron 1 targeting would appear to be preferred (by providing correction for all downstream mutations), limitations on packaging size for AAVs (∼4.7 kb) potentially precludes incorporating SA-CFTR2-27-pA together with homology arms of sufficient size for efficient homology directed repair. A third aspect comprises the need to achieve integration without disrupting activity of critical CREs and the chromatin architecture of the CFTR.
A further consideration in the selection of which intron to target involves the relative strength(s) of the transgene splice acceptor versus the native splice acceptor in the immediate downstream CFTR exon. If the targeted intron were immediately upstream of a strong splice acceptor sequence, then it is possible that a significant amount of splicing could jump across the corrective partial CFTR cDNA. We previously noted that the level of CFTR activity, as a function of editing frequency for either TI-7 or TI-8 (Figure 3H), was perhaps suggestive of a reduction relative to that resulting from the same frequency of site-specific correction. To quantitatively assess what fraction of CFTR mRNA transcripts from targeted alleles were spliced from the endogenous exon 8 to the transgenic exon 9 (thus including transgenic corrective sequences) versus alternative splicing downstream to the endogenous exon 9 (including only endogenous mutant sequences), we examined several single-cell-derived clones exhibiting homozygous intron 8 TI (Figures S8A–S8C). Quantitative RT-PCR demonstrated that the majority (58.0%–89.9%; depending on the clone) of CFTR transcripts exhibited the desired splicing from the endogenous exon 8 into the corrective human codon optimized exon 9 sequences; however, 10.1%–42.0% of transcripts did, in fact, reflect splicing across the corrective exon sequences directly into endogenous mutant exon 9 sequences (Figure S8D). A failure to capture only corrective cDNA sequences in CFTR mRNA transcripts from successfully targeted alleles would potentially result in reduced levels of CFTR protein and activity as a function of editing frequency. This finding perhaps suggests room for improvement in the TI strategy (e.g., strengthening either the splice acceptor sequence or, alternatively the poly-adenylation sequence to maximize termination of transcription). In addition, it is possible that our use of non-native amino acid-encoding transgene sequences (i.e., human codon optimized) may influence the efficiency of generating the appropriately folded CFTR protein necessary for functional activity.42
For any gene-editing approach, it is important to assess the nature and incidence of off-target effects that could potentially affect therapeutic safety. We experimentally identified via unbiased genome-wide oligonucleotide capture, in K562 cells, potential off-target sites (Table S4) for the intron 8-specific ZFNs employed in the editing experiments reported in this study;43 we then quantitatively assessed the incidence of off-target indels in ZFN-treated primary ΔF508/ΔF508 airway basal cells (Figure S9A). This validated the occurrence of indels at varying frequency at 10 of the previously identified off-target sites. Although all of these sites lie in intergenic or intronic sequences (Figure S9A), we chose to optimize the ZFN design, in order to reduce the frequency of such off-target indels, while still retaining recognition of the same intended target sequences.44 The improved intron 8 ZFNs maintained efficient on-target cleavage while exhibiting a significantly reduced incidence of off-target indels (Figure S9A). Importantly, the optimized ZFNs facilitated intron 8 TI of ΔF508/ΔF508 airway basal cells (Figures S9B and S9C) at levels similar to the original ZFNs.
Although we utilized airway epithelial cells from explanted lungs in these experiments, recently developed methodologies are capable of significantly expanding (ex vivo) the number of airway basal cells present in accessible primary tissues of CF patients, e.g., bronchial brushings (Figure S1A).28,45 These developments make possible the consideration of autologous, tissue-resident basal cells from CF patients, and their editing for therapeutic use. We have readily expanded basal cells obtained from airway brushings and do not anticipate any difficulty in obtaining greater than the 6 × 107 cells estimated to be required for engraftment in a human lung.33,46 There is compelling clinical evidence that small airways are a critical site of early obstruction and disease.47 We expect that our studies using basal cells from the large airways (bronchi) will be representative of basal cells from bronchioles—an assertion that will require formal testing in the future.
With the objective of generating CFTR-corrected patient-specific airway basal stem cells, we have performed gene editing directly in minimally expanded primary airway basal cells. It would, in principle, be possible to first correct for CFTR mutations in CF patient induced pluripotent stem cells (iPSCs) and then derive airway basal cells, provided that robust differentiation protocols can be developed. Whereas earlier work by our group and others utilized sequence-specific editing for correction of CF iPSCs,6,9,16 the TI of partial CFTR cDNAs, as reported here, would represent a more generalizable approach.5,7 Finally, we note that although the CFTR gene-editing strategies pursued in the present studies were exclusively evaluated on airway basal cells ex vivo, it is possible that these tools, appropriately vectored, may eventually be applied for in vivo correction.
Materials and Methods
Complete methods can be found in the Supplemental Information online. All primary/secondary antibodies and oligos used in this report are listed in Tables S5 and S6, respectively. Human subject review was performed by University of Texas Health Science Center institutional review committee.
Culture of Airway Basal Cells
Passage 1 CF and non-CF airway epithelial cells in this study were obtained from the Tissue Procurement and Cell Culture core facility at the University of North Carolina (Chapel Hill, NC, USA). Lungs were obtained following informed consent under UNC Office of Human Research Ethics/Institutional Review Board Study #03-1396 and cells were isolated as previously described.48 Basal cells were cultured with Pneumacult-Ex Plus medium (StemCell Technologies, Vancouver, BC, Canada) on plates pre-coated with 804G cell-conditioned medium (CM) and maintained at 37°C in humidified air with 5% CO2. At 50%–70% confluence, cells were dissociated by trypsinization and either re-plated at a 1:10 ratio or directly utilized for gene editing.
Gene Editing
Gene editing of basal cells was performed by electroporation-mediated delivery of ZFN mRNA (2–3 μg per reaction) followed immediately by AAV-6 donor transduction. Electroporations in this study were performed with the BTX ECM 830 electroporation generator (Harvard Apparatus, Holliston, MA, USA). The ZFN mRNA-electroporated cells were immediately transduced with AAV-6 donor at a multiplicity of infection (MOI) of 2 − 6 × 106 viral genomes per cell (vg/cell) overnight. Alternatively, for single-strand oligo DNA donors (ssDNA), basal cells were co-electroporated with ZFN mRNA and ssDNA and directly plated. All donor DNA sequences are shown in the Supplemental Information (donor DNA sequences).
Assessment of Genome Modification
The efficiency of induction of on-target indels, sequence-specific correction, and targeted integration was assessed quantitatively through NGS deep sequencing on the Illumina platform. Examples of sequencing data are uploaded in supplemental data files (Data S1 and S2). Additionally, targeted integration was confirmed by Southern blot analysis.
In Vitro Differentiation of Basal Cells at Air Liquid Interface (ALI)
200,000 airway basal cells were seeded on a 6.5 mm Transwell with 0.4 μm pore polyester membrane inserts (Corning, Corning, NY, USA) pre-coated with 804G-CM and subject to air liquid interface culture with Pneumacult-ALI medium (StemCell Technologies) for up to 4 weeks. Fully differentiated airway epithelia were confirmed by hematoxylin and eosin (H&E) staining and immunofluorescence analysis.
Molecular and Functional Analysis
ALI-cultured cells were assayed for CFTR mRNA expression with RT-PCR, CFTR protein expression via western blotting, and the functional CFTR correction by Ussing chamber analysis as described previously with modification.16
Omni Assay for Transposase Accessible Chromatin and Deep Sequencing
Omni-ATAC-seq was performed on 50,000 cells as described previously with minor modifications.49 Omni-ATAC-seq libraries were purified with Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA) with a sample to bead ratio of 1:1.2, and eluted in Buffer EB (Hilden, Germany). Libraries were pooled and sequenced on a HiSeq4000 machine (Illumina, San Diego, CA, USA) with 100 bp paired end reads. Data were processed by the ENCODE-DCC/atac-seq-pipeline. A custom genome was constructed using the Reform package (ver. August 2019) (https://github.com/gencorefacility/reform) to insert the partial cDNA sequence into the hg19 genome build.
Author Contribution
Conceptualization: B.R.D., A.C., A.H.; Project Administration: B.R.D., A.C., E.J.S., A.H.; Supervision: S.S.; Funding Acquisition: B.R.D., A.C., E.J.S., A.H., S.H.R.; Methodology: S.S., A.M.C., N.M.; Investigation: S.S., A.M.C., V.A., N.M., C.B., A.R., J.L.K., S.Y., M.M., K.K.; ZFN Design: L.Z.; Resources: S.H.R.; Writing – Original Draft: B.R.D., A.C., S.S., A.M.C., A.H.; Writing – Review & Editing: B.R.D., A.C., S.S., A.M.C., C.B., A.H., S.H.R., J.L.K., E.J.S.
Conflicts of Interest
A.C., M.M., K.K., and L.Z. are employees of Sangamo Therapeutics.
Acknowledgments
We gratefully acknowledge funding agencies enabling this study: Cystic Fibrosis Foundation (CFF): SANGAM16X0 to A.C., DAVIS15XX0 and DAVIS17XX0 to B.R.D., RANDEL15XX0, RANDEL17XX0, and BOUCHE15RO to S.H.R., and HARRIS15/17XX0, HARRIS16G0, and HARRIS18P0 to A.H.; National Institutes of Health (NIH): R01 HL139876 to E.J.S. and B.R.D., DK065988 to S.H.R., and R01 HL094585 to AH; and UTHealth Pulmonary Center of Excellence to B.R.D. We also acknowledge histological sample preparation by Dr. Zhengmei Mao (UTHealth Microscopy Core Facility), Dr. Susan Reynolds for guidance and discussions, and Dr. Hongmei Mou for guidance on the dual SMAD inhibition expansion methodology and providing the 804G cell line.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.021.
Contributor Information
Anthony Conway, Email: aconway@sangamo.com.
Brian R. Davis, Email: brian.r.davis@uth.tmc.edu.
Supplemental Information
References
- 1.Hogan B.L., Barkauskas C.E., Chapman H.A., Epstein J.A., Jain R., Hsia C.C., Niklason L., Calle E., Le A., Randell S.H. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15:123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rock J.R., Onaitis M.W., Rawlins E.L., Lu Y., Clark C.P., Xue Y., Randell S.H., Hogan B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rock J.R., Randell S.H., Hogan B.L. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010;3:545–556. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong K.U., Reynolds S.D., Watkins S., Fuchs E., Stripp B.R. Basal cells are a multipotent progenitor capable of renewing the bronchial epithelium. Am. J. Pathol. 2004;164:577–588. doi: 10.1016/S0002-9440(10)63147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison P.T., Hoppe N., Martin U. Gene editing & stem cells. J. Cyst. Fibros. 2018;17:10–16. doi: 10.1016/j.jcf.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 6.Firth A.L., Menon T., Parker G.S., Qualls S.J., Lewis B.M., Ke E., Dargitz C.T., Wright R., Khanna A., Gage F.H., Verma I.M. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient ipscs. Cell Rep. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bednarski C., Tomczak K., Vom Hövel B., Weber W.M., Cathomen T. Targeted integration of a super-exon into the cftr locus leads to functional correction of a cystic fibrosis cell line model. PLoS ONE. 2016;11:e0161072. doi: 10.1371/journal.pone.0161072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merkert S., Bednarski C., Göhring G., Cathomen T., Martin U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Res. (Amst.) 2017;23:95–97. doi: 10.1016/j.scr.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki S., Sargent R.G., Illek B., Fischer H., Esmaeili-Shandiz A., Yezzi M.J., Lee A., Yang Y., Kim S., Renz P. Talens facilitate single-step seamless sdf correction of f508del cftr in airway epithelial submucosal gland cell-derived cf-ipscs. Mol. Ther. Nucleic Acids. 2016;5:e273. doi: 10.1038/mtna.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maule G., Casini A., Montagna C., Ramalho A.S., De Boeck K., Debyser Z., Carlon M.S., Petris G., Cereseto A. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat. Commun. 2019;10:3556. doi: 10.1038/s41467-019-11454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaidyanathan S., Salahudeen A.A., Sellers Z.M., Bravo D.T., Choi S.S., Batish A., Le W., Baik R., de la O.S., Kaushik M.P. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues cftr function in differentiated epithelia. Cell Stem Cell. 2020;26:161–171. doi: 10.1016/j.stem.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jennings S., Ng H.P., Wang G. Establishment of a ΔF508-CF promyelocytic cell line for cystic fibrosis research and drug screening. J. Cyst. Fibros. 2019;18:44–53. doi: 10.1016/j.jcf.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Ruan J., Hirai H., Yang D., Ma L., Hou X., Jiang H., Wei H., Rajagopalan C., Mou H., Wang G. Efficient gene editing at major cftr mutation loci. Mol. Ther. Nucleic Acids. 2019;16:73–81. doi: 10.1016/j.omtn.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramalingam S., London V., Kandavelou K., Cebotaru L., Guggino W., Civin C., Chandrasegaran S. Generation and genetic engineering of human induced pluripotent stem cells using designed zinc finger nucleases. Stem Cells Dev. 2013;22:595–610. doi: 10.1089/scd.2012.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valley H.C., Bukis K.M., Bell A., Cheng Y., Wong E., Jordan N.J., Allaire N.E., Sivachenko A., Liang F., Bihler H. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J. Cyst. Fibros. 2019;18:476–483. doi: 10.1016/j.jcf.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Crane A.M., Kramer P., Bui J.H., Chung W.J., Li X.S., Gonzalez-Garay M.L., Hawkins F., Liao W., Mora D., Choi S. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4:569–577. doi: 10.1016/j.stemcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinn P.L., Hwang B.Y., Li N., Ortiz J.L.S., Shirazi E., Parekh K.R., Cooney A.L., Schaffer D.V., McCray P.B., Jr. Novel GP64 envelope variants for improved delivery to human airway epithelial cells. Gene Ther. 2017;24:674–679. doi: 10.1038/gt.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooney A.L., Thornell I.M., Singh B.K., Shah V.S., Stoltz D.A., McCray P.B., Jr., Zabner J., Sinn P.L. Novel aav-mediated gene delivery system corrects cftr function in pigs. Am. J. Respir. Cell Mol. Biol. 2019;61:747–754. doi: 10.1165/rcmb.2019-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cmielewski P., Donnelley M., Parsons D.W. Long-term therapeutic and reporter gene expression in lentiviral vector treated cystic fibrosis mice. J. Gene Med. 2014;16:291–299. doi: 10.1002/jgm.2778. [DOI] [PubMed] [Google Scholar]
- 20.Yan Z., McCray P.B., Jr., Engelhardt J.F. Advances in gene therapy for cystic fibrosis lung disease. Hum. Mol. Genet. 2019;28(R1):R88–R94. doi: 10.1093/hmg/ddz139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steines B., Dickey D.D., Bergen J., Excoffon K.J., Weinstein J.R., Li X., Yan Z., Abou Alaiwa M.H., Shah V.S., Bouzek D.C. CFTR gene transfer with AAV improves early cystic fibrosis pig phenotypes. JCI Insight. 2016;1:e88728. doi: 10.1172/jci.insight.88728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montoro D.T., Haber A.L., Biton M., Vinarsky V., Lin B., Birket S.E., Yuan F., Chen S., Leung H.M., Villoria J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 2018;560:319–324. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plasschaert L.W., Žilionis R., Choo-Wing R., Savova V., Knehr J., Roma G., Klein A.M., Jaffe A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature. 2018;560:377–381. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okuda K., Kobayashi Y., Dang H., Nakano S., Barbosa Cardenas S.M., On V.K., Kato T., Chen G., Gilmore R.C., Chua M. Vol. 54. 2019. (Regional regulation of cftr and ionocyte expression in normal human airways. 2019 North American CF Conference, Nashville TN, Pediatric Pulmonology). Abstract 48. [Google Scholar]
- 25.Johnson L.G., Olsen J.C., Sarkadi B., Moore K.L., Swanstrom R., Boucher R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 26.Goldman M.J., Yang Y., Wilson J.M. Gene therapy in a xenograft model of cystic fibrosis lung corrects chloride transport more effectively than the sodium defect. Nat. Genet. 1995;9:126–131. doi: 10.1038/ng0295-126. [DOI] [PubMed] [Google Scholar]
- 27.Farmen S.L., Karp P.H., Ng P., Palmer D.J., Koehler D.R., Hu J., Beaudet A.L., Zabner J., Welsh M.J. Gene transfer of CFTR to airway epithelia: low levels of expression are sufficient to correct Cl- transport and overexpression can generate basolateral CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:L1123–L1130. doi: 10.1152/ajplung.00049.2005. [DOI] [PubMed] [Google Scholar]
- 28.Mou H., Vinarsky V., Tata P.R., Brazauskas K., Choi S.H., Crooke A.K., Zhang B., Solomon G.M., Turner B., Bihler H. Dual smad signaling inhibition enables long-term expansion of diverse epithelial basal cells. Cell Stem Cell. 2016;19:217–231. doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Limberis M.P., Vandenberghe L.H., Zhang L., Pickles R.J., Wilson J.M. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol. Ther. 2009;17:294–301. doi: 10.1038/mt.2008.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan Z., Lei-Butters D.C., Keiser N.W., Engelhardt J.F. Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther. 2013;20:328–337. doi: 10.1038/gt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wainwright C.E., Elborn J.S., Ramsey B.W., Marigowda G., Huang X., Cipolli M., Colombo C., Davies J.C., De Boeck K., Flume P.A., TRAFFIC Study Group. TRANSPORT Study Group Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del cftr. N. Engl. J. Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swahn H., Harris A. Cell-selective regulation of cftr gene expression: Relevance to gene editing therapeutics. Genes (Basel) 2019;10:E235. doi: 10.3390/genes10030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh M., Ahmad S., White C.W., Reynolds S.D. Transplantation of airway epithelial stem/progenitor cells: A future for cell-based therapy. Am. J. Respir. Cell Mol. Biol. 2017;56:1–10. doi: 10.1165/rcmb.2016-0181MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milman Krentsis I., Rosen C., Shezen E., Aronovich A., Nathanson B., Bachar-Lustig E., Berkman N., Assayag M., Shakhar G., Feferman T. Lung injury repair by transplantation of adult lung cells following preconditioning of recipient mice. Stem Cells Transl. Med. 2018;7:68–77. doi: 10.1002/sctm.17-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farrow N., Cmielewski P., Donnelley M., Rout-Pitt N., Moodley Y., Bertoncello I., Parsons D. Epithelial disruption: a new paradigm enabling human airway stem cell transplantation. Stem Cell Res. Ther. 2018;9:153. doi: 10.1186/s13287-018-0911-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramalho A.S., Beck S., Meyer M., Penque D., Cutting G.R., Amaral M.D. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2002;27:619–627. doi: 10.1165/rcmb.2001-0004OC. [DOI] [PubMed] [Google Scholar]
- 38.Chu C.S., Trapnell B.C., Curristin S.M., Cutting G.R., Crystal R.G. Extensive posttranscriptional deletion of the coding sequences for part of nucleotide-binding fold 1 in respiratory epithelial mRNA transcripts of the cystic fibrosis transmembrane conductance regulator gene is not associated with the clinical manifestations of cystic fibrosis. J. Clin. Invest. 1992;90:785–790. doi: 10.1172/JCI115952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker T.A., Fortenberry J.A., Zsembery A., Schwiebert L.M., Schwiebert E.M. The ΔF508-CFTR mutation inhibits wild-type CFTR processing and function when co-expressed in human airway epithelia and in mouse nasal mucosa. BMC Physiol. 2012;12:12. doi: 10.1186/1472-6793-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keating D., Marigowda G., Burr L., Daines C., Mall M.A., McKone E.F., Ramsey B.W., Rowe S.M., Sass L.A., Tullis E., VX16-445-001 Study Group Vx-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two phe508del alleles. N. Engl. J. Med. 2018;379:1612–1620. doi: 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middleton P.G., Mall M.A., Dřevínek P., Lands L.C., McKone E.F., Polineni D., Ramsey B.W., Taylor-Cousar J.L., Tullis E., Vermeulen F., VX17-445-102 Study Group Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single phe508del allele. N. Engl. J. Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim S.J., Yoon J.S., Shishido H., Yang Z., Rooney L.A., Barral J.M., Skach W.R. Protein folding. Translational tuning optimizes nascent protein folding in cells. Science. 2015;348:444–448. doi: 10.1126/science.aaa3974. [DOI] [PubMed] [Google Scholar]
- 43.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller J.C., Patil D.P., Xia D.F., Paine C.B., Fauser F., Richards H.W., Shivak D.A., Bendaña Y.R., Hinkley S.J., Scarlott N.A. Enhancing gene editing specificity by attenuating DNA cleavage kinetics. Nat. Biotechnol. 2019;37:945–952. doi: 10.1038/s41587-019-0186-z. [DOI] [PubMed] [Google Scholar]
- 45.Butler C.R., Hynds R.E., Gowers K.H., Lee Ddo.H., Brown J.M., Crowley C., Teixeira V.H., Smith C.M., Urbani L., Hamilton N.J. Rapid expansion of human epithelial stem cells suitable for airway tissue engineering. Am. J. Respir. Crit. Care Med. 2016;194:156–168. doi: 10.1164/rccm.201507-1414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayes D., Jr., Kopp B.T., Hill C.L., Lallier S.W., Schwartz C.M., Tadesse M., Alsudayri A., Reynolds S.D. Cell therapy for cystic fibrosis lung disease: Regenerative basal cell amplification. Stem Cells Transl. Med. 2019;8:225–235. doi: 10.1002/sctm.18-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenow T., Ramsey K., Turkovic L., Murray C.P., Mok L.C., Hall G.L., Stick S.M., Arest C.F., AREST CF Air trapping in early cystic fibrosis lung disease-Does CT tell the full story? Pediatr. Pulmonol. 2017;52:1150–1156. doi: 10.1002/ppul.23754. [DOI] [PubMed] [Google Scholar]
- 48.Fulcher M.L., Randell S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013;945:109–121. doi: 10.1007/978-1-62703-125-7_8. [DOI] [PubMed] [Google Scholar]
- 49.Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.