

Abstract
The urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) catalyzes the initial step of the urea cycle; bi-allelic mutations typically present with hyperammonemia, vomiting, ataxia, lethargy progressing into coma, and death due to brain edema if ineffectively treated. The enzyme deficiency is particularly difficult to treat; early recognition is essential to minimize injury to the brain. Even under optimal conditions, therapeutic interventions are of limited scope and efficacy, with most patients developing long-term neurologic sequelae. One significant encumberment to gene therapeutic development is the size of the CPS1 cDNA, which, at 4.5 kb, nears the packaging capacity of adeno-associated virus (AAV). Herein we developed a split AAV (sAAV)-based approach, packaging the large transgene and its regulatory cassette into two separate vectors, thereby delivering therapeutic CPS1 by a dual vector system with testing in a murine model of the disorder. Cps1-deficient mice treated with sAAVs survive long-term with markedly improved ammonia levels, diminished dysregulation of circulating amino acids, and increased hepatic CPS1 expression and activity. In response to acute ammonia challenging, sAAV-treated female mice rapidly incorporated nitrogen into urea. This study demonstrates the first proof-of-principle that sAAV-mediated therapy is a viable, potentially clinically translatable approach to CPS1 deficiency, a devastating urea cycle disorder.
Keywords: carbamoyl phosphate synthetase deficiency, ureagenesis, hyperammonemia, gene therapy, adeno-associated virus, urea cycle disorder, split AAV
Graphical Abstract
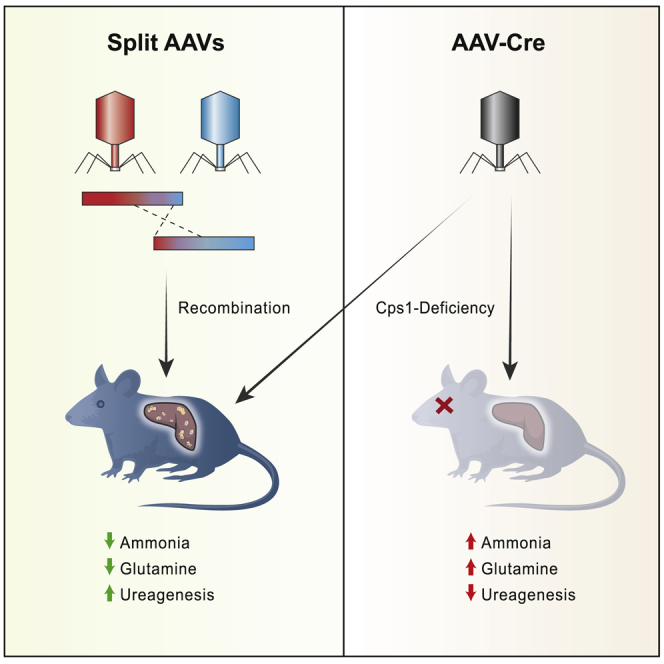
Present-day therapy for the urea cycle disorder carbamoyl phosphate synthetase deficiency is incomplete, resulting in CNS injury or neonatal death. Here, Lipshutz and colleagues demonstrate a dual AAV approach to overcome the cDNA size restriction, resulting in enzyme expression with improved ammonia control and ureagenesis restoration, both supporting long-term survival.
Introduction
Carbamoyl phosphate synthetase 1 (CPS1; E.C. 6.3.4.16) catalyzes the ATP-dependent condensation of ammonia and bicarbonate into carbamoyl phosphate,1 which subsequently proceeds through the urea cycle in hepatocytes or the arginine biosynthesis pathway in intestinal epithelium.2 CPS1 resides in the mitochondria, where its function is dependent on the allosteric activator N-acetylglutamate (NAG). CPS1 deficiency (OMIM #237300) is a rare autosomal recessive disorder caused by mutations in the CPS1 gene leading to diminished or abolished CPS1 protein function. The estimated incidence is 1 in 1.3 million in the United States and Europe,3 though estimates vary widely4 and this is likely an underestimate due to the difficulty in making an accurate early diagnosis.
Patients afflicted with CPS1 deficiency often present in the neonatal period with vomiting, lethargy, and coma due to elevated plasma ammonia. With acute life-threatening episodes of hyperammonemia, hemodialysis may be necessary. Timely and aggressive treatment is essential for reducing plasma ammonia to prevent mortality; however, these treatments are typically unable to prevent marked hyperammonemic episodes and cannot reverse neurological injury that has been acquired during these crises. Even in the best of circumstances, patients often remain severely nitrogen vulnerable. Current chronic treatment strategies rely primarily on dietary protein restriction and ammonia scavenger administration in combination with L-arginine and/or L-citrulline supplementation.5
Recent advances in gene therapy have led to the concept of treating CPS1 deficiency using adeno-associated virus (AAV)-based technology; as a monogenic disorder that is a well-defined enzyme deficiency with no completely effective medical treatment except liver transplantation (which has accompanying inherent risks, both short- and long-term), it is an appropriate disorder to target for therapeutic advances. While AAV-mediated gene transfer has proven effective in animal models for other urea cycle disorders including deficiencies of ornithine transcarbamylase,6 argininosuccinate synthetase,7 argininosuccinate lyase,8 and arginase 1,9 no gene therapy strategies using AAV have yet been established for CPS1 deficiency for multiple reasons. First, until recently there were no viable animal models of CPS1 deficiency. While a neonatal murine model was developed 20 years ago,10 this was not submitted to a repository and was lost for future investigation.11 In that model, neonatal mice perished within the first 24 h after birth; developing therapies in a newborn mouse with the early intake of milk and consequent early rise in plasma ammonia poses significant challenges. As an alternative to that neonatal model, an adult model of CPS1 deficiency was recently established utilizing primary human hepatocytes from patients transplanted into immune compromised mice and expanded in vivo.12 However, this model suffers from being technically challenging and time consuming, in addition to requiring access to a relatively rare cell population. Second, CPS1, being the most abundant mitochondrial protein in human liver, must be expressed at high levels in hepatocytes. The enzyme itself accounts for 15%–20% of the total mitochondrial protein as a 165,000 molecular weight polypeptide;1 its expression is pan-hepatic,13,14 not exclusively periportal like some of the other urea cycle enzymes, further leading to challenges of obtaining adequate expression. This is in contrast to gene therapy targets such as hemophilia that require as little as 1%–2% protein levels to achieve therapeutic benefit.15 Finally, the size of the CPS1 cDNA at 4.5 kb, and limitations of the transgene expression cassette to ∼5 kb to maximize packaging efficiency, may be the greatest hindrance for a clinically translatable vector that can be produced at high titer such as AAV; with inverted terminal repeats and regulatory elements, the genome size easily exceeds the canonical limit of AAV.
Vector systems exist to package and deliver large transgenes, though they have significant drawbacks. Adenoviral vectors are highly immunogenic,16 while lentiviral vectors suffer from low titer and random, widespread genomic integration.17 Plasmids can be difficult to deliver and are largely silenced due to the presence of bacterial genomic elements.18 To overcome these limitations, an approach was developed to capitalize on the recombinogenic nature of AAV vectors. This is achieved by generating two separate AAVs that contain complementary halves of a transgene cassette that includes a region of homology between them. These so-called dual or split AAVs (sAAVs) can potentiate homologous recombination between the vectors upon co-transduction of the same cell,19,20 resulting in expression of the full-length therapeutic transgene. Variations on this system include relying on inverted terminal repeat (ITR)-mediated concatemerization and subsequent removal by splicing, known as trans-splicing19 and the hybrid approach, which combines elements from both the overlapping and trans-splicing approaches.21 sAAV approaches have been implemented successfully in, among others, the retina for Alström syndrome,22 skeletal muscle for dysferlin deficiency,23,24 and the liver for glycogen storage deficiency type III.25
Here we describe the design and implementation of a sAAV dual vector approach to treat CPS1 deficiency. Specifically, using our recently established conditional knockout murine model of CPS1 deficiency,26 we demonstrate that sAAV-mediated gene transfer is able to restore CPS1 hepatic expression, control plasma ammonia, and re-establish ureagenesis. These studies lay the proof-of-concept groundwork for a clinically translatable gene therapy approach to treat CPS1 deficiency.
Results
In Vitro Transduction with sAAVs Leads to RNA and Protein Expression of hcoCPS1
A schematic of the sAAV strategy is depicted in Figure 1A. Two separate viral vectors were prepared: the first (5′ [or left] sAAV) contains the constitutively active CMV enhancer/chicken β-actin promoter (CAG) and the 5′ half of the human codon-optimized CPS1 transgene (hcoCPS1) (cDNA base pairs 1–2,181); the second virus (3′ sAAV [or right]) contains the 3′ half of hcoCPS1 (cDNA base pairs 1,714–4,521) and the rabbit β-globin polyadenylation sequence. A 467 bp region of homology within the transgene (overlap) potentiates homologous recombination upon co-transduction of the same cell, resulting in the reconstituted full-length transgene with no intervening viral ITR (combined sAAV).
Figure 1.

In Vitro Characterization of sAAVs
(A) Schematic of overlapping sAAVs. A 467 bp overlapping region of homology (overlap) mediates homologous recombination between the 5′ (left) and 3′ (right) sAAVs, resulting in the full-length combined sAAV containing hcoCPS1 (green-blue rectangles) and removing the intervening viral ITR. (B) DNA: PCR amplification of left (top) and right (middle) sAAVs and the combined sAAV (bottom) with increasing viral genomes/cell in transduced HEK293T cells. Amplicons are generated by primers indicated by black half-arrows in A (LF+LR amplify left sAAV; RF+RR amplify right sAAV); combined sAAV product is generated by amplifying with the outer primer of each pair, i.e., primers LF+RR. (C) RNA: hcoCPS1 mRNA is detectable by qPCR in transduced HeLa cells. Primers used to amplify either half are indicated by red arrows (left sAAV) and blue arrows (right sAAV), and expression was normalized to GAPDH. Data presented are mean ± SEM of triplicate experiments. (D) Protein: western blot for CPS1 in HEK293T cells either non-transduced (-, left lane) or transduced with both sAAVs (+, right lane); β-actin, loading control. (Note: increased density of CPS1 band from sAAV transduced cells.) Abbreviations: CAG, CMV enhancer/chicken β-actin promoter; sAAV, split adeno-associated viral vector; hcoCPS1, human codon-optimized carbamoyl phosphate synthetase 1; HEK, human embryonic kidney; ITR, inverted terminal repeat. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To determine whether sAAVs containing hcoCPS1 have the same propensity for homology-directed concatemerization as previously tested transgenes by others,20,27 we transduced HEK293T cells with sAAVs followed by genomic DNA extraction. There was a dose-dependent increase (log increments from 102 to 105 viral genomes/cell) in detectable concatemerized product when amplified using primers to specifically recognize either the 5′ (primers LF and LR), 3′ (primers RF and RR), or combined sAAV (primers LF and RR) (Figure 1B; primers shown as black half-arrows in Figure 1A). To show that concatemerization leads to gene and protein expression, we transduced HeLa and 293T cells and extracted RNA and protein, respectively. Gene-expression analysis by qPCR demonstrated that the individual halves of hcoCPS1 are only detectable in cells transduced with the relevant sAAV (relative to non-transduced cells) (Figure 1C; primers shown as red/blue arrows in Figure 1A). CPS1 protein levels were also increased over endogenous levels in 293T cells transduced with both sAAVs (2.7-fold increase relative to non-transduced cells; Figure 1D).
In Vivo sAAV Treatment Extends Lifespan of Cps1-Deficient Mice
Following in vitro testing, serotype 8 sAAVs were tested for therapeutic efficacy in vivo. A diagram of the general workflow is depicted in Figure 2A. To determine the optimal viral dose for subsequent in vivo studies, we injected adult female Cps1 conditional knockout mice (Cps1flox/flox) with AAV8-Cre (as described previously26) and increasing doses of sAAVs ranging from 1 × 1013 genome copies (GC)/kg to 3 × 1014 GC/kg of each virus (Figure 2B; n = 3 per dose). The minimum dose required to extend lifespan to 30 days in all mice in a group was 3 × 1014 GC/kg. Because the mice at the highest dose still showed a slight decline in body weight (data not shown), a surrogate for overall health, in addition to our previous work demonstrating that therapeutic CPS1 level requirements are relatively high,26 all subsequent studies were carried out with 5 × 1014 GC/kg/virus. No acute or chronic side effects were seen at this dose, and plasma alanine aminotransferase levels (ALT, collected on day 36) were not significantly different from control mice (sAAV-treated: 31.8 ± 3.0 U/L, n = 4; wild-type: 37.8 ± 12.7 U/L, n = 5; p = 0.69; Figure S1A).
Figure 2.

sAAVs Promote Lifespan Extension and Weight Maintenance in Mice In Vivo
(A) Workflow of in vivo sAAV administration. Adult Cps1flox/flox mice were injected with AAV8-Cre alone (mice depicted in brown) or in combination with sAAVs (mice depicted in gray) at time 0 (top left). AAV8-Cre results in hepatic disruption of Cps1 expression resulting in loss of functional protein. All mice were weighed daily to assess general health (bottom left), after which only the surviving sAAV-treated mice (gray) were subjected to an exogenous ammonia challenge after day 120 (bottom right). Mice were subsequently euthanized and the livers removed for molecular and biochemical analysis (top right). (B) Dose escalation of sAAVs in female AAV8-Cre-treated Cps1flox/flox mice. The minimum dose required to ensure survival to 30 days was determined to be 3 × 1014 GC/kg/virus; n = 3 mice per group. (C) Survival of 5 × 1014 GC/kg sAAV-treated (treated [red and blue lines]) mice and AAV8-Cre-only Cps1flox/flox (untreated [black and orange]) controls. (D) Weight maintenance in sAAV-treated mice. AAV8-Cre-only mice (black, orange) rapidly lost weight beginning around day 14 after AAV8-Cre administration, while sAAV-treated mice (red, blue) did not. Red F-series arrows correspond to the red line of treated females; blue M-series arrow corresponds to the blue line of treated males. Red arrows F1 and F3 indicate administration of NCA and dextrose in female sAAV-treated mice (red line); red arrow F2 indicates removal of NCA and dextrose from sAAV-treated females; red arrow F4 indicates removal of dextrose only from NCA water for sAAV-treated female mice; note stability of weight without dextrose. Blue arrow M1 represents addition of NCA and dextrose to male mice. (E and F) Plasma ammonia levels were measured in female (E) and male (F) mice. Legend is shared between (E) and (F); day 21 data points for untreated mice represent values obtained at time of death or euthanasia and are grouped together for clarity. Data represent mean ± SEM with n = 5 mice per group; ∗p < 0.05; ∗∗p < 0.01. Abbreviations: NCA, N-carglumic acid; sAAV, split AAV; SEM, standard error of the mean.
To investigate the ability of sAAVs to treat CPS1 deficiency, we injected adult Cps1flox/flox mice with AAV8-Cre alone (referred to as untreated) or in combination with sAAVs at 5 × 1014 GC/kg (referred to as treated; n = 5 each for both males and females, all groups). At this dose, both female and male mice survived beyond 120 days (Figure 2C), significantly longer (p < 0.0001) than untreated controls that perished by day 22 (females: range 19–22 days) or 21 (males: range 17–21 days).
The studies with the female mice began first. All untreated Cps1−/− mice (Figure 2D, black line) had significant weight loss and died or were euthanized due to humane endpoints by day 22. By day 38, treated female Cps1−/− mice began to slowly lose weight (10% of their starting weights) (Figure 2D, red line). As an additional intervention in the treated Cps1−/− mice, N-carglumic acid (NCA), a structural analog of the Cps1 allosteric activator NAG (and the active ingredient in CarbaGlu), was added to the drinking water to increase CPS1 activity (Figure 2D, red arrow F1); dextrose (5%) was also included in the water to encourage consumption. Dextrose/NCA-treated female mice subsequently regained the lost weight and continued to gain over time (red line). These mice were dependent on this supplementation as its withdrawal (red arrow F2) resulted in weight loss prior to re-administration (beginning at red arrow F3). In addition, NCA treatment alone, without dextrose, was sufficient to maintain stable weights (Figure 2D, red arrow F4) and plasma ammonia levels (Figure S1B, p = 0.8). With these findings in the female mice, male mice (blue line) were subsequently administered water with dextrose/NCA beginning at day 14 (blue arrow M1).
sAAV Treatment Improves Biochemical Profiles of Cps1-Deficient Mice
To understand the extent of the therapeutic response, we measured plasma ammonia in AAV8-Cre-only treated Cps1−/− (i.e., untreated), sAAV-treated Cps1−/− (i.e., treated), and wild-type mice (n = 5 per group for both males and females). At day 21, plasma ammonia levels in treated mice (red line) were significantly lower than untreated control mice (black line) in both females (339.0 ± 42.3 μM versus 1,244.6 ± 180.9 μM [treated versus untreated, respectively], p < 0.01) and males (581.3 ± 60.9 μM versus 1,090.4 ± 166.7 μM, p = 0.021) (Figures 2E and 2F). At 120 days, compared to baseline (day 0) levels, females (138.9 ± 38.7 μM [day 0] versus 464.3 ± 65.0 μM [day 120], p = 0.026) and males (231.8 ± 41.9 μM [day 0] versus 601.0 ± 53.7 μM [day 120], p = 0.033) demonstrated controlled plasma ammonia, albeit females with superior control.
To further investigate metabolic health, untreated Cps1−/−, sAAV-treated Cps1−/−, and wild-type mice underwent urea cycle-related plasma amino acid profiling to determine the extent of residual metabolic derangement (Figure 3). In untreated female Cps1−/− mice (black line) compared to wild-type controls (blue line), though not statistically significantly, arginine trended toward reduction (p = 0.067) (Figure 3A), while citrulline (p = 0.70) and ornithine were not significantly reduced (Figures 3B and 3C, black lines). In contrast, glutamine was significantly elevated in Cps1 deficiency at the time of death relative to wild-type controls (853.4 ± 74.9 μM versus 630.6 ± 18.9 μM; p = 0.03; Figure 3D). Alanine, a non-toxic molecular carrier of nitrogen, was reduced at baseline (269 ± 9.5 μM versus 356.2 ± 8.0 μM, p < 0.01) but increased slightly by day 21 in Cps1−/− mice when they perished (p = 0.5), while glutamate did not significantly increase (Figure 3F). Conversely, sAAV-treated females (red line) showed no significant differences compared to wild-type mice in plasma arginine, citrulline, glutamate, and glutamine (Figures 3A, 3B, 3D, and 3F [red lines], respectively). Ornithine was significantly reduced in treated mice at days 21 (38.1 ± 5.8 μM; p = 0.03) and 60 (36.5 ± 4.1 μM; p = 0.02) compared to wild-type controls (64.2 ± 3.9 μM), though not at days 30 and 120 (p = 0.8 and 0.13, respectively) (Figure 3C). Alanine levels were not significantly elevated compared to wild-type controls (Figure 3E).
Figure 3.

sAAV Treatment Improves Some Amino Acid Imbalances in Mice with Cps1 Deficiency
(A–L) Plasma amino acids show improvement in females (A)–(F) and moderate correction in males (G)–(L); note sustained elevation of glutamine in sAAV-treated male mice. Data represent mean ± SEM, n = 5 per group except in treated females at day 0, where n = 4. Asterisks denote significant deviation from wild-type values, where solid and dashed lines are mean and ± SEM, respectively; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (Female plasma results are depicted in A-F while male plasma results are depicted in G-L.) (A,G: arginine; B,H: Citrulline; C,I: Ornithine; D, J: Glutamine; E, K: Alanine; F, L: Glutamate)
Like the female mice, untreated Cps1−/− males (black line) showed a trend toward reduced arginine levels compared to wild-types (50.6 ± 5.2 μM versus 64.6 ± 4.1 μM; p = 0.066) (Figure 3G) while citrulline was also reduced, however not significantly different from wild-type controls (Figure 3H). Ornithine was significantly reduced at time of death compared to wild-type mice (30.6 ± 5.9 μM versus 45.2 ± 2 μM, p = 0.047) (Figure 3I) while plasma glutamine was significantly elevated compared to wild-type mice (932.9 ± 73.1 μM versus 487.3 ± 46 μM; p < 0.001) (Figure 3J), as expected; however, alanine and glutamate showed no significant differences in untreated Cps1−/− mice compared to wild-type controls (Figures 3K and 3L). In contrast, treated male mice (red line) had significantly elevated arginine at days 21, 30, and 60 (peak at day 30: 109.7 ± 12.9 μM, p = 0.01) compared to wild-type mice (Figure 3G). Citrulline was also significantly elevated at days 60 and 120 compared to wild-type mice (peak at day 60: 104.9 ± 10 μM versus 67.2 ± 8.3 μM, p = 0.019) (Figure 3H) while ornithine was significantly reduced at days 21 and 60 (trough at day 60: 33 ± 0.9 μM, p < 0.001), though not at days 30 and 120 (Figure 3I). Similar to untreated controls, glutamine was significantly elevated in treated mice from day 21 to the end of the study at day 120 (peak at day 60: 1,090 ± 96.9 μM, p < 0.001) compared to wild-type control mice (Figure 3J), while alanine was significantly elevated at days 30 and 60 relative to wild-type mice (peak at day 30: 497.2 ± 59.8 μM versus 293.9 ± 21.3 μM, p = 0.012) (Figure 3K). Glutamate showed no significant differences from wild-type mice (Figure 3L).
Acutely Elevated Ammonia Is Metabolized by Female but Not Male sAAV-Treated Mice
To measure the ability of sAAV-treated mice to metabolize ammonia into urea, we subjected sAAV-treated and wild-type mice (male and female, n = 5 per gender) to an acute ammonia challenge by injection with 4 mmol/kg of 15N-labeled ammonium chloride (Figure 4A). Plasma samples were taken before (time 0) and at 10, 20, 30, 60, and 90 min after injection to measure the plasma ammonia and ureagenesis capacity. In sAAV-treated females (red line), plasma ammonia peaked relative to baseline at 20 min (535.4 ± 40.4 μM versus 2,471.0 ± 317.4 μM; p < 0.01) and subsequently decreased over time, with a trajectory toward baseline levels by 90 min after injection (937.0 ± 167.0 μM; p = 0.16; Figure 4A, red line). Wild-type control female mice had a reduced peak ammonia and a more rapid correction to baseline ammonia levels (blue line). Mice were scored at 15 min post-injection to examine for any behavioral abnormalities. Treated females averaged a score of 6 (range: 4–7; 7 being no signs of abnormal behavior, i.e., no noticeable differences from wild-types) with no seizure activity while all wild-type mice were scored at 7 (Table S1).
Figure 4.

Ammonia Metabolism after Exogenous Ammonia Loading
(A) After a marked increase with exogenous administration of 15NH4Cl, plasma ammonia levels decrease toward baseline over 90 min in female sAAV-treated mice (red line), albeit less rapidly than in wild-type mice (blue line). In male sAAV-treated mice plasma ammonia levels do not decline (orange line). Asterisks denote significant deviation from baseline values within a gender. (Treated male mice data at 60 and 90 min was n = 2 per time point due to animal deaths, along with large value variation accounts for statistical non-significance.) (B) 15N-labeled urea is produced in mice treated with 15NH4Cl, with enrichment peaking faster in wild-type mice (blue, black) than in sAAV-treated mice (red, orange) of both genders. Legend is also shared with (A). (A and B: for females at all time points and experimental groups: n = 5 per group. For males: n = 5 at time 0, n = 4 at time 20, and n = 2 at times 60 and 90 in the treated group due to animal deaths from seizure activity; n = 5 at all times for the wild-type males.) (C) Instantaneous rate of ureagenesis in female (n = 5, both groups) and male mice (n = 4 sAAV-treated and n = 5 wild-type), based on best-fit curves from (B). Data represent mean values ± SEM. Asterisks denote significant deviation from baseline values in the ammonia curves and wild-type values in the urea curves; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
The results were strikingly different with the male mice. While all female mice completed the challenge, only 2 treated male mice (orange line) survived until the 90-min time point; the others perished during the challenge (i.e., seizure activity and death). Ammonia levels in treated males, which were higher than females at time 0, reached greater levels above baseline at 20 min (n = 4 mice; 3,389 ± 348.2 μM; p < 0.05) and continued to climb during the challenge without returning toward baseline levels (90 min: n = 2 mice; 5,292.5 ± 841.5 μM; Figure 4A, orange line). Similar to the females, male mice were also scored for behavioral abnormalities at 15 min post-injection: treated males scored substantially worse however, averaging only a score of 4 (range: 3–6; p = 0.067; Table S1); 2 male mice died shortly after with seizure activity. All wild-type males scored either 6 or 7 (n = 3 scored 6, n = 2 scored 7).
In female mice, concomitant with decreases in plasma ammonia, plasma [15N]-urea increased over time before reaching equilibrium at 30 min in wild-type (blue line) and 60 min in sAAV-treated mice (red line; Figure 4B, p = 0.29 at 60 min). To compare the maximum ureagenic output, we calculated the instantaneous rate of ureagenesis from the slope of the best-fit curve of percent enrichment at time 0, which subsequently demonstrated that sAAV-treated females have 60.3% of wild-type capacity (0.53% ± 0.04% enrichment/min versus 0.88% ± 0.04% enrichment/min [sAAV-treated versus wild-type, respectively], p < 0.001; Figure 4C). Male sAAV-treated mice (orange line) also showed gradual incorporation of 15NH4Cl into urea, though at a significantly reduced rate; [15N]-urea enrichment did not reach equilibrium in these mice (Figure 4B, p < 0.01 at 60 min relative to wild-type males [black line]). Males produced [15N]-urea at a reduced instantaneous rate compared to wild-types (0.27% ± 0.03% enrichment/min versus 0.86% ± 0.08% enrichment/min, respectively, p < 0.001), representing 31.4% wild-type capacity (Figure 4C).
hcoCPS1 Transgene and Protein Expression is Upregulated in sAAV-Treated Mice
To measure the amount of hcoCPS1 transgene expression, we euthanized sAAV-treated mice (n = 5 per group per gender) after 120 days post-injection of AAV8-Cre and sAAVs, and livers were removed for molecular analysis. Total DNA was extracted, and vector copy number was determined (Figure 5A). There was no significant difference seen in the amount of 5′ and 3′ sAAVs as copy number per diploid genome in females (5′ [left]: 90.08 ± 37.64, 3′ [right]: 78.46 ± 32.57; p = 0.82). However, in the male mice, there was near statistical significance in the difference in the amount of 5′ and 3′ sAAVs (left: 132.48 ± 28.67, right: 62.08 ± 10.76; p = 0.051).
Figure 5.

Molecular Analysis of Livers from Treated Mice
(A) Vector copy number of both the left (5′) and right (3′) sAAVs in males (n = 5) and females (n = 5). (B) Total CPS1 mRNA were measured by qPCR in AAV8-Cre-only (untreated), sAAV-treated (treated), and wild-type mice for both females and males (n = 5 per group per gender). (C) Western blot for CPS1 protein. (D) Immunofluorescent staining of liver tissue sections for CPS1 (red) and glutamine synthetase (GS) (green); nuclear stained with DAPI (blue). (E) CPS1 enzyme functional assay on liver tissue lysates in both females and males (n = 5 per group per gender). Western blot and immunofluorescence data are from representative samples; quantitative data represent mean ± SEM. ∗∗∗∗p < 0.0001.
Total RNA was isolated and used to generate cDNA for qPCR analysis. Using primers that amplify hcoCPS1 and endogenous Cps1 mRNA (located on the left sAAV), both AAV8-Cre-only (untreated Cps1−/− [n = 5]: 0.02 ± 0.02 fold (2% of wild-type), p < 0.0001; black bar) and sAAV-treated Cps1−/− (treated [n = 5]: 0.24 ± 0.07 fold [24% of wild-type], p < 0.001; red bar) females had reduced total CPS1 mRNA relative to wild-type Cps1 gene expression (blue bar; Figure 5B, left). In male mice, total CPS1 mRNA levels were significantly reduced compared to wild-types in both untreated [n = 5; black bar at x axis] and sAAV-treated [n = 5; red bar] mice (0.0009 ± 0.0005 fold [< 1% of wild-type] and 0.03 ± 0.008 fold [3% of wild-type], respectively; p < 0.0001 for both groups; Figure 5B).
Liver tissue was used to assess protein levels. In females, sAAV-treated mice showed reduced but substantial CPS1 protein expression compared to the Cps1 protein of wild-type controls by western blot (41.7% ± 11.5%, p < 0.05 [n = 5]), and trending toward significantly more than untreated controls (17.3% ± 4.9%, p = 0.08 [n = 5]; Figure 5C, left); however, CPS1 protein in males was substantially less than wild-type Cps1 protein (10.2% ± 3.5%, p < 0.0001 [n = 5]; Figure 5C, right), and lower than untreated controls (27.2% ± 2.3%, p < 0.01 [n = 5]), likely only achieving sAAV-based expression just at the level to provide survival with persistent nitrogen vulnerability. Similar differences between untreated and treated mice are seen in tissue sections stained for CPS1 (red). Particularly in females, treated mice show CPS1 protein broadly distributed in the parenchyma with little or no residual endogenous expression visible in untreated mice (Untreated: 85.0 ± 27.3 CPS1+ cells/field; sAAV-treated: 446.5 ± 92.9 CPS1+ cells/field, p < 0.01; Figure 5D, top images). Male sAAV-treated mice also had increased quantities of CPS1+ cells relative to untreated mice (sAAV-treated: 417.4 cells ± 80.1; untreated: 34.0 cells ± 20.8; p < 0.01) (Figure 5D, top images). An enzymatic functional assay was performed on liver lysates to examine the amount of CPS1 activity in sAAV-treated mice (n = 5 per gender per group). Female treated mice analyzed after day 120 showed enzyme function of 31.3% ± 6.6% relative to wild-type controls (p < 0.0001; Figure 5E, left). In males, treated mice had 15.0% ± 1.6% of wild-type activity (Figure 5E, right; p < 0.0001). Taken together, these results demonstrate that sAAV-derived hcoCPS1 expression leads to substantial gene and protein expression in the livers of treated mice, albeit to different extents in females and males.
Discussion
CPS1 deficiency has two clinical presentations: (1) neonatal onset, which is typically severe with high morbidity and mortality, and despite treatment, disease progression is difficult to prevent;28 and (2) late onset, which has milder symptoms brought on by protein-rich meals, infection, pregnancy, and the postpartum period, or other situations of high metabolic demand.29 Biochemically, neonatal onset CPS1 deficiency results in markedly elevated plasma ammonia, leading to encephalopathy with growth and neurological deficits, along with an increase in plasma glutamine and a decrease of plasma citrulline and arginine.5 In many cases, unrelenting hyperammonemia results in patient deaths. Orthotopic liver transplantation, while curative,30 is limited due to an inadequate supply of available healthy donor livers and has risks of substantial complications;31 no other curative treatment currently exists. CarbaGlu, an FDA-approved treatment for N-acetylglutamate synthase (NAGS) deficiency, has been shown to be effective in treating a small subset of CPS1 patients,32 though this is dependent on the nature of the mutation and is not broadly applicable to patients with complete protein loss. The development of novel and efficacious therapeutics is therefore essential to improve patients’ quality of life and prevent the morbidity and mortality that accompany this disorder.
To have clinical benefit for patients afflicted with CPS1 deficiency, an effective therapy would need to maintain plasma ammonia in an acceptable range and resolve or prevent hyperammonemic episodes to avoid repeated neurological injury. However, the development of a gene therapy approach for CPS1 deficiency is challenged not only in part by (1) the large size of the cDNA, and (2), until recently, the lack of a clinically relevant animal model of the disorder, but also the expectation of a need for high-level mitochondrial protein expression based on its abundant quantity in normal hepatocytes.1 A recent manuscript employed mathematical modeling to study ureagenesis of the individual urea cycle enzymes,33 confirming this expectation. A progressive decline in ureagenesis was predicted using a one compartment model when liver-wide CPS1 activity was reduced below 50%. In contrast, ureagenesis was not predicted to decline until enzyme activity was below 3% for ornithine transcarbamylase, 20% for argininosuccinate synthetase, 3% argininosuccinate lyase, and 0.5% for arginase activity.
While our group redeveloped a constitutive Cps1 knockout murine model,34 we chose to test the AAV vector approach in our recently developed conditional Cps1 knockout mouse,26 avoiding investigating this treatment in mice that perish within the first 24 h after delivery, are very small (∼1 g in size), and develop hyperammonemia shortly after birth with early intake of milk.34 Use of the conditional model allowed for proof-of-concept studies avoiding these further model complexities. Recent work from our laboratory developing and characterizing the conditional Cps1 knockout mouse model also had demonstrated both the model’s amenability to gene therapy strategies26 and as a representative model of CPS1 deficiency. The use of an AAV encoding Cre recombinase does introduce a small amount of mouse-to-mouse variability in this model; however, at the dose used here, no appreciable differences were seen in the efficiency of knockout.
While AAV is an efficient gene therapy vehicle, its limited ability to package large DNA sequences with the cis regulatory elements necessary for expression is a significant limitation in its application to certain disorders. To address the large size of the CPS1 cDNA, we took an approach taking advantage of the ability of AAVs to undergo intermolecular concatemerization by developing dual vectors with a region of homology, splitting the large transgene into two separate AAV vectors. However, there is inherent inefficiency of transgene expression by this approach; hepatocytes must be transduced by both vectors, intracellular head-to-head and tail-to-tail concatemerization can occur, limiting successful reconstitution, and homologous recombination is required. Together these issues would typically be expected to result in expression that is lower than that achieved by single AAV vectors.22,35,36 While it would likely be more efficacious if a single hcoCPS1 cDNA vector could be made, previous unpublished attempts by our lab to generate single AAV vectors expressing CPS1 were unsuccessful, despite repeated efforts. This is possibly due to excessive shearing and genome truncation caused by the oversized AAV genome; encapsidated oversized DNA tends not to be a pure population of large genomes but rather a heterogeneous mixture of mostly genomes truncated to 5 kb or less in size.37, 38, 39
To address the need for high level expression, utilization of the dual vector approach allowed for incorporation of a large and strong promoter (the CMV enhancer/chicken β-actin promoter); together with codon optimization of the human CPS1 cDNA, therapeutically relevant expression levels of successfully recombined genomes to control plasma ammonia were achieved. The successful employment of these approaches has allowed us to overcome these major hurdles and permitted the AAV-based delivery and expression of CPS1, avoiding use of less ideal viral vectors that may have limited clinical translatability.26 Despite previous reports of limitations in CAG-based vectors, such as transgene silencing, immune response to therapeutic protein, and tumorigenesis, no evidence for these effects were seen, similar to previously published work by others.40
The studies described herein demonstrate the following results for a dual AAV gene therapy approach for CPS1 deficiency: (1) high doses of sAAV vectors with a strong constitutive promoter can lead to expression of CPS1 and result in long-term survival in both male and female mice; however, there is a dependence on N-carglumic acid to allosterically activate the enzyme to increase the level of enzyme activity; (2) chronic control of plasma ammonia and glutamine (along with alanine, another intermolecular carrier of nitrogen) is achievable, albeit better in females; and (3) acute control of elevated ammonia and restoration of high-level ureagenesis is achievable in female mice. Untreated control mice perished early and exhibited many of the circulating amino acid perturbations typical of CPS1 deficiency,5 which were notably reversed or mitigated by sAAV treatment.
However, this approach was met with an unexpected result. While female Cps1−/− mice had near-equal transduction of both the 5′and 3′ sAAV vectors and long-term ammonia control with avoidance of marked hyperglutaminemia, similar control was not present in the male mice. In contrast to females, male sAAV-treated mice showed a marked increase in glutamine, which remained elevated for most of the duration of the study. Elevated glutamine is a typical presentation in proximal urea cycle disorders, and this result supports the moderately elevated plasma ammonia levels and reduced ureagenesis capacity found in the male mice. N-carglumic acid (NCA) with dextrose supplementation was efficacious in contributing to improved nitrogen metabolism; as the active ingredient in CarbaGlu, treatment was found to be effective to further assist in control of plasma ammonia long-term. However, NCA and dextrose alone were not sufficient to extend lifespan in mice that did not receive sAAV treatment (data not shown).
Vector dosing in this study was based on weight and, not unexpectedly, male mice were larger. While the circulating blood volume of male compared to female mice is larger,41 this is not as great as the weight difference between genders; thus effectively an overall greater number of AAV genomes was administered to male mice. This is reflected in the viral DNA copy number per diploid genome of the 5′ sAAV: murine hepatocytes from male mice demonstrated nearly 50% more genomes (90.08 versus 132.48 vector genomes per diploid nucleus, female versus male, respectively) than hepatocytes from female mice. However, unexpectedly, the number of 3′ AAV genomes in male hepatocytes was ∼50% of the 5′ AAV and was only ∼80% of the female 3′ AAV genomes per diploid nucleus. This reduced 3′ sAAV genome number in hepatocytes from male mice resulted in less full-length hcoCPS1 cDNA, CPS1 protein and enzymatic activity, and a reduced ability to handle an exogenous ammonia challenge with a reduction in ureagenesis. The mechanism responsible for this finding is unclear. As both genders received the same dose per kilogram of sAAVs, and male cells have been demonstrated to be transduced more efficiently,42 male mice were expected to have more robust CPS1 enzymatic activity and to perform better than females in the exogenous ammonia challenge and ureagenesis experiments. Stochastic differences in vector copy number and sAAV concatemerization, sexually dimorphic AAV expression, and viral retention43 may have contributed to the differences seen between male and female mice in terms of mRNA and protein expression, enzyme activity, and tolerance to exogenous ammonia loading. Although previous studies have demonstrated that a doubling of vector dose does not reduce viral transduction,35 it is possible that saturation from the larger number of virions (i.e., greater weight in males and thus a greater number of total virions administered) may occur at differential viral loads for females and males. In addition, to our knowledge only two studies using sAAVs have been conducted in both male and female animals,44,45 raising the possibility of sexually dimorphic responses to sAAV administration, concatemerization, expression, or retention that warrant further investigation.
In this study, we injected mice with 5 × 1014 GC/kg/virus, which is higher than other reports for gene therapies. A recent study in nonhuman primates demonstrated vector-related toxicity at a dose of 2 × 1014 GC/kg,46 indicating that the high viral load could be a barrier to translation. Despite this, high vector dosing has been used for a clinical trial for Duchenne Muscular Dystrophy (2 × 1014 vg/kg; NCT03375164) and X-linked myotubular myopathy (3 × 1014 vg/kg; NCT03199469). Though no negative effects have been detected at this dose in our studies, such high vector dosing may pose a barrier in terms of production and cost. However, despite this dose, there was no evidence of acute toxicity or chronic liver injury in mice used in this study. In addition, we found that in a small cohort of females, a reduced dose of 2 × 1014 GC/kg/virus was still effective for long-term survival (data not shown). Further vector optimization is required and may be achieved by transitioning to a trans-splicing19 or hybrid21 approach in contrast to the overlapping one used here, which we pursued first due to encouraging early results. Both alternative strategies warrant further investigation for their potential capacity to reduce the viral load necessary for therapeutic benefit.
In summary, we have shown for the first time that CPS1 deficiency may be treated using a split AAV-based gene therapy approach. This work bridges the gap between previously described pre-clinical efforts and more translatable tools to deliver therapeutic CPS1 protein safely and effectively. To date, no other potentially clinically translatable nucleic acid or protein replacement strategies have been successfully implemented to treat CPS1 deficiency.
Materials and Methods
Animal Care and Mouse Procedures
All mice were kept according to the National Institutes of Health guidelines and all experimental procedures were approved by and conducted according to the guidelines of the Institutional Animal Care and Use Committee. Unless otherwise stated, mice had ad lib access to standard chow and water and were maintained on a 12-h light-dark cycle. Cps1flox/flox mice on the C57BL/6 background had been developed as previously described26 and were used for all of the studies. Genomic DNA was prepared from ear clip by standard methods and genotyping was performed by PCR amplification as previously described.26
Mice were randomly assigned to either the control or the experimental group. Male and female mice were equally represented throughout the study. For injections, AAV was diluted in sterile pharmaceutical grade 0.9% saline (Medline, Northfield, IL, 5T-NFSF-10011) and injected intravenously by a retro-orbital approach under isoflurane anesthesia. AAV8 TBG-Cre recombinase (University of Pennsylvania Vector Core, Philadelphia, PA, USA) was administered (2 × 1011 GC/mouse, a dose previously demonstrated to result in Cps1 deficiency in this model26) simultaneously with the sAAVs. All mice were weighed daily and closely monitored for any signs of deteriorating health. Ill-appearing mice (e.g., disheveled fur, hunched appearance, lethargy) were euthanized at a humane endpoint prior to expiration. Mice receiving N-carglumic acid (NCA)-supplemented water were administered a dose of 200 mg/L NCA (Sigma-Aldrich, St. Louis, MO, USA, C4375-10G) with 5% (w/v) dextrose (Sigma-Aldrich, G8270); water was replaced daily for females and every 2–3 days for males (on an as-needed basis). Scheduled blood sampling was obtained from the retro-orbital plexus under isoflurane anesthesia with plasma frozen immediately and stored at −80°C until analysis. Samples of liver were either snap frozen at the time of euthanasia and stored at −80°C until analyzed or were or prepared for immunohistochemistry as described below.
Molecular Cloning
Full-length codon-optimized human CPS1 (hcoCPS1) was synthesized by Blue Heron Biotech (Bothell, WA). A 467-bp region encompassing base pairs 1,714–2,181 of the hcoCPS1 transgene was used as the region of homology between the left and right sAAVs. The size of the overlapping region was chosen based on previous data demonstrating that increasing overlap length does not significantly improve recombination efficiency.47 Molecular cloning of viral vectors was performed according to standard practices. 5′ sAAV: The viral backbone (p1044, provided by the University of Pennsylvania Vector Core) containing AAV2 ITRs was digested with KpnI and XhoI (all restrictions enzymes purchased from New England Biolabs [Ipswich, MA, USA]); the CAG promoter was digested with KpnI and SalI; hcoCPS1 was then PCR-amplified (cDNA base pairs 1–2,181) and subsequently digested with KpnI and SalI. Inserts were ligated with Quick Ligase (New England Biolabs, M2200S) into the backbone at a 3:1 molar ratio and transformed into Stbl3 E. coli (Invitrogen, Carlsbad, CA, USA, C737303). 3′ sAAV: The same viral backbone as the 5′ sAAV was digested with BamHI; hcoCPS1 was PCR-amplified (cDNA base pairs 1,714–4,521) and subsequently digested with BglII. hcoCPS1 was ligated and transformed similar to the 5′ sAAV. The sequences for all primers used here are listed in Table S2. PCR conditions for amplifications using Platinum SuperFi DNA polymerase (Thermo Fisher Scientific, Boston, MA, USA, 12351010) were: 95°C for 30 s; 67°C for 30 s; 72°C for 90 s; repeat for 35 cycles. Both constructs were verified by restriction digest and subsequently with sequencing. Viral amplification and purification of serotype 8 vectors were completed at the University of Pennsylvania Vector Core. The complete hcoCPS1 sequence: 5′−ATGCCTCAGATCATAAAGATGACCCGGATTCTTACCGCATTCAAGGTTGTAAGGACCCTTAAAACCGGCTTCGGCTTTACTAACGTGACCGCACACCAAAAGTGGAAGTTTAGCAGGCCCGGAATTCGCCTCCTTAGTGTGAAAGCCCAGACCGCTCATATAGTCCTTGAAGACGGCACAAAAATGAAAGGGTACTCATTCGGCCATCCATCATCTGTAGCCGGTGAGGTCGTGTTCAATACTGGATTGGGGGGTTATCCCGAGGCCATAACAGACCCAGCTTATAAGGGCCAGATCCTGACCATGGCCAACCCAATCATCGGGAACGGAGGTGCGCCGGATACAACTGCGTTGGATGAGCTGGGACTGTCCAAGTACTTGGAGAGCAATGGAATTAAAGTTTCTGGACTGCTGGTACTGGACTACTCAAAGGACTACAATCATTGGCTGGCCACCAAAAGTCTGGGGCAATGGCTGCAGGAGGAGAAGGTGCCAGCTATATACGGAGTTGACACTAGAATGCTTACCAAAATTATAAGAGACAAAGGTACTATGCTGGGAAAAATTGAGTTTGAAGGACAGCCCGTGGATTTCGTAGACCCTAATAAGCAGAATCTTATCGCCGAGGTGAGCACAAAGGACGTTAAGGTCTACGGAAAAGGAAATCCAACTAAGGTGGTGGCTGTTGATTGTGGCATTAAGAACAACGTGATCAGACTGCTGGTGAAACGCGGAGCTGAAGTCCATCTTGTCCCATGGAATCATGATTTTACGAAAATGGAGTATGATGGAATTCTCATCGCCGGCGGACCAGGGAACCCAGCCTTGGCTGAACCCCTTATCCAAAACGTTAGAAAAATACTCGAATCTGATAGGAAAGAGCCCCTTTTTGGTATATCCACCGGAAACTTGATTACAGGCCTTGCTGCAGGGGCCAAGACATATAAGATGAGCATGGCAAACCGCGGACAGAATCAGCCCGTACTGAACATTACTAATAAGCAGGCTTTTATCACCGCACAGAATCACGGTTACGCTCTCGATAATACGCTCCCTGCCGGCTGGAAGCCGCTCTTCGTTAACGTAAATGATCAGACAAACGAGGGAATAATGCACGAATCCAAACCCTTCTTCGCCGTCCAGTTCCACCCTGAAGTCACTCCAGGCCCTATTGACACAGAATATCTCTTTGACTCCTTCTTTAGCCTGATAAAAAAGGGGAAGGCCACCACCATAACGTCCGTCCTGCCTAAGCCAGCTCTCGTGGCATCAAGAGTAGAGGTCTCCAAAGTGCTCATACTTGGTAGCGGGGGACTGTCAATCGGCCAAGCAGGCGAGTTCGATTACTCCGGAAGCCAAGCAGTTAAGGCTATGAAAGAAGAGAACGTTAAAACTGTGCTGATGAATCCAAATATAGCCTCCGTGCAGACCAATGAGGTGGGTCTCAAGCAAGCAGATACTGTTTACTTTCTTCCAATTACCCCCCAATTCGTAACCGAAGTCATTAAGGCCGAGCAGCCTGATGGATTGATCCTGGGTATGGGCGGACAGACTGCACTGAATTGCGGAGTGGAGTTGTTCAAAAGGGGTGTGTTGAAGGAATATGGAGTTAAGGTACTCGGCACCTCCGTTGAGAGCATCATGGCGACCGAGGATAGACAGTTGTTCTCTGATAAACTGAACGAGATTAATGAGAAGATCGCCCCCTCATTCGCCGTGGAGTCTATCGAAGATGCACTGAAAGCCGCTGATACGATTGGCTATCCTGTAATGATAAGAAGCGCCTACGCCCTGGGTGGCCTGGGGTCTGGCATCTGCCCTAACCGAGAGACGCTGATGGACCTCTCCACAAAAGCCTTCGCCATGACTAACCAGATTCTGGTAGAAAAATCCGTCACCGGCTGGAAGGAAATTGAATACGAAGTAGTAAGAGACGCTGATGACAATTGCGTCACAGTCTGCAACATGGAAAACGTCGATGCGATGGGCGTGCACACCGGAGATTCCGTCGTTGTGGCGCCAGCACAAACACTCTCCAATGCTGAGTTCCAGATGCTCAGAAGAACAAGCATTAACGTTGTGCGACATCTTGGGATAGTTGGCGAATGTAACATCCAATTTGCACTGCACCCAACTAGCATGGAATACTGCATTATCGAAGTGAATGCGCGGCTGAGCCGAAGCAGCGCTCTCGCCAGCAAAGCCACAGGCTACCCACTTGCCTTCATTGCCGCAAAGATTGCACTGGGCATTCCACTGCCTGAGATTAAGAATGTCGTAAGCGGGAAGACAAGCGCCTGTTTTGAACCTTCCCTGGACTATATGGTGACTAAGATTCCTCGGTGGGACCTTGATAGGTTCCATGGGACCTCATCTaGAATAGGATCATCAATGAAGTCTGTGGGTGAAGTGATGGCTATCGGGCGGACCTTcGAaGAGAGTTTTCAGAAAGCACTTCGGATGTGTCACCCCTCAATTGAGGGCTTCACCCCCCGGTTGCCAATGAACAAGGAGTGGCCATCAAACCTGGACCTGAGAAAAGAGCTCAGCGAGCCTAGCTCAACTAGAATCTACGCAATCGCCAAGGCAATCGACGATAACATGTCATTGGATGAGATAGAGAAGTTGACATACATAGACAAATGGTTCCTCTACAAAATGCGAGACATTCTGAATATGGAGAAAACACTGAAGGGACTGAATTCTGAGAGCATGACGGAGGAGACACTTAAGAGAGCAAAAGAGATTGGGTTCAGCGATAAGCAAATTTCAAAGTGCCTTGGACTGACCGAAGCCCAGACACGGGAGCTGAGACTGAAGAAAAATATACACCCATGGGTGAAGCAGATCGACACCCTGGCGGCCGAATATCCCAGCGTTACTAATTACCTGTATGTTACATATAACGGCCAAGAGCATGACGTAAATTTTGACGATCATGGAATGATGGTTTTGGGATGCGGTCCCTACCACATTGGCTCTTCAGTGGAGTTTGATTGGTGCGCAGTGAGCTCCATTCGGACCCTCAGACAGCTTGGAAAAAAAACAGTGGTGGTAAATTGTAACCCGGAGACTGTGTCAACCGACTTCGACGAATGCGACAAGTTGTATTTTGAGGAATTGAGTCTTGAAAGGATTCTTGATATCTACCATCAGGAAGCATGCGGAGGCTGTATTATCTCAGTGGGCGGGCAGATACCCAACAACCTTGCTGTACCTCTCTATAAAAACGGTGTAAAGATCATGGGCACCTCTCCCCTCCAGATTGACAGGGCCGAGGACCGCTCAATTTTCAGTGCTGTGCTGGACGAACTCAAAGTCGCTCAAGCTCCTTGGAAAGCTGTTAATACTCTTAACGAGGCCCTcGAgTTCGCCAAGTCTGTGGATTACCCATGTCTTCTTCGGCCCTCCTACGTGCTGTCAGGaTCcGCAATGAACGTCGTGTTCAGCGAGGATGAAATGAAGAAATTTCTGGAGGAGGCTACACGGGTGAGTCAAGAGCATCCTGTGGTTTTGACTAAGTTCGTTGAGGGCGCCCGGGAAGTCGAGATGGATGCAGTCGGTAAAGATGGACGGGTAATTAGCCACGCAATTAGTGAACACGTGGAAGATGCCGGGGTCCATTCTGGCGACGCCACTCTCATGCTGCCAACACAGACAATTAGTCAGGGTGCTATAGAGAAAGTGAAAGATGCGACTAGGAAGATCGCAAAAGCCTTCGCAATATCTGGCCCATTTAACGTGCAGTTTCTCGTGAAAGGTAACGACGTCCTGGTGATCGAGTGTAATCTCCGAGCGTCACGATCCTTCCCTTTCGTAAGCAAGACCCTCGGCGTAGACTTTATTGACGTGGCCACGAAAGTTATGATTGGAGAGAATGTAGACGAGAAACACCTCCCCACTCTTGACCATCCGATCATCCCCGCGGATTATGTTGCCATCAAGGCCCCAATGTTCTCTTGGCCGCGCCTGCGAGACGCTGATCCCATCTTGCGCTGTGAAATGGCAAGCACAGGCGAAGTAGCATGCTTCGGCGAAGGTATTCATACCGCATTTCTGAAGGCCATGCTGAGCACCGGCTTCAAGATCCCCCAGAAGGGTATCCTCATCGGCATCCAGCAGTCTTTCCGCCCAAGATTCCTGGGGGTAGCAGAACAACTTCATAACGAAGGCTTCAAGCTGTTTGCAACAGAAGCAACCTCTGATTGGCTGAACGCTAATAATGTTCCTGCGACTCCAGTCGCCTGGCCCAGCCAGGAAGGACAAAATCCCAGCCTGTCTAGCATCAGAAAACTCATACGAGATGGCTCTATCGACCTTGTTATCAACCTGCCTAATAACAACACCAAATTTGTCCACGACAACTACGTCATCAGAAGAACTGCCGTGGATAGCGGTATCCCCCTGCTGACCAATTTCCAGGTTACCAAGCTCTTTGCAGAAGCTGTTCAGAAATCTCGCAAGGTGGATAGCAAGTCACTGTTTCACTATCGACAATATTCAGCGGGGAAGGCTGCATAG-3′.
In Vitro Transduction
HEK293T and HeLa cells were grown in Dulbecco’s modified Eagle’s medium (11885-084) supplemented with 10% fetal bovine serum (10437028), 1% GlutaMAX (35050061; all from Invitrogen), and 0.1 mg/mL primocin (Invivogen, San Diego, CA, USA, ant-pm-1). Prior to transduction, all cells were passaged and plated at a density of 5.3 × 104 cells/cm2. The following day, cells were transduced with increasing sAAV serotype 8 viral genomes/cell in log increments from 102 to 105 (DNA from 293T cells) or 106 (RNA and protein from HeLa and 293T cells, respectively) in growth media supplemented with 5 μg/mL polybrene (Vector Builder, Chicago, IL, USA) and incubated overnight (approximately 16 h). Virus was removed from cells in the morning following transduction by changing to fresh growth media, and cells were grown one additional day in growth media. Cells were then harvested, and total DNA extracted (QIAGEN, Germantown, MD, USA, 69504). PCR amplification was performed with Taq DNA Polymerase (QIAGEN, 201443). PCR conditions: 95°C for 30 s, 60°C for 30 s, and 72°C for 60 s for 35 cycles. RNA was extracted, cDNA synthesized, and qPCR performed for hcoCPS1 as described below. Protein was extracted and analyzed by western blot as described below.
Immunohistochemistry
Whole livers were collected and analyzed as described previously.26 Briefly, tissues were prepared using standard procedures by fixing in 10% buffered formalin and storing in 70% ethanol until embedding in paraffin. Blocks were sectioned at 4 μm thickness onto glass slides. Sections were baked at 60°C for 1 h before being deparaffinized in xylene and rehydrated in serial ethanol washes. Slides were boiled in 10 mM sodium citrate pH 6.0 to retrieve antigens before permeabilization in 0.1% Triton X-100. Sections were blocked with 10% normal goat serum and incubated with primary antibodies overnight at 4°C. After washing, secondary antibodies were incubated for 1 h at RT, followed by washing and mounting with VectaShield containing DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Burlingame, CA, USA, H-1200). Primary antibodies used were: CPS1 (Abcam, Cambridge, MA, USA, ab45956, used at 1:1,000); GS (Abcam, ab64613, used at 1:1,000). Secondary antibodies used were as follows: goat anti-mouse FITC (against GS primary antibody; Invitrogen, M30101, used at 1:200), and goat anti-rabbit AF594 (against CPS1 primary antibody; Invitrogen, A-11012, used at 1:200). Quantitation of CPS1+ cells was performed on random fields of the same size in all mice.
qPCR
qPCR was performed as described.26 Briefly, total RNA was extracted and used to generate cDNA for the PCR reaction according to the manufacturer’s instructions (Roche, Indianapolis, IN, USA, 11828665001 and 4897030001, respectively). qPCR was carried out using BioRad ssoAdvanced Universal SYBRGreen (Hercules, CA, USA, 1725271). Primers used are listed in Table S2.
Western Blot
Unfixed frozen liver was homogenized, and soluble protein was isolated in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, 89900) and Halt protease inhibitor (Thermo Fisher Scientific, 78430). Protein was quantified using BioRad protein assay dye (BioRad, 5000006). Protein was loaded into a 12% gel (BioRad, 4561045) and transferred to polyvinylidene fluoride (PVDF) membranes using the TransBlot Turbo system according to the manufacturer’s instructions (BioRad, 1704156). Membranes were blocked in 5% milk and incubated with primary antibodies overnight at 4°C. Secondary antibodies were incubated for 1 h at RT before detection with SuperSignal West Pico PLUS (Thermo Fisher Scientific, PI34579). Primary antibodies: anti-CPS1 (Abcam, ab45956, used at 1:1,000), and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA, SC47778, used at 1:1,000). Secondary antibody: goat anti-rabbit-HRP (against CPS1 primary; Santa Cruz Biotechnology, SC2004, used at 1:5,000). No secondary antibody was used for β-actin as the primary antibody is HRP-conjugated. Semi-quantitation was completed using NIH ImageJ version 1.51, normalizing to its own β-actin band to give a ratio of CPS1:β-actin. The average of 5 mice per group was obtained for comparisons.
Ammonia Assay
Whole blood was collected by retro-orbital bleeding under isoflurane anesthesia into capillary tubes coated with sodium heparin (Spectrum Chemical, New Brunswick, NJ, USA, 631-11095) and immediately centrifuged at 2,000 × g for 10 min at 4°C. Plasma was collected and stored at −80°C until analysis. Ammonia levels were detected using a colorimetric ammonia assay (Abcam, ab83360) according to the manufacturer’s instructions.
Ammonia Challenge and Ureagenesis
Mice were fasted for 3–4 h at the same time of the day. To simulate a nitrogen challenge, we injected mice by intraperitoneal method with a 4 mmol/kg solution of 15NH4Cl (Cambridge Isotope Laboratories, Andover, MA, USA, NLM-467-1). At 15 min after injection, the mice were evaluated behaviorally by scoring from one observer using the scale outlined in Ye et al.48 Blood was collected at the indicated time points after injection. Plasma was isolated and stored at −80°C until analyzed as described above for the ammonia assay. At each bleed, 5 μL of blood was dispensed in duplicate onto a filter paper card (Perkin Elmer, Waltham, MA, USA, GR2261005) for the ureagenesis assay. 15N-labeled urea enrichment was determined on these samples by gas chromatography-mass spectrometry as previously described.49
CPS1 Enzyme Function Assay
The activity of CPS1 was determined colorimetrically as previously described50 by assaying at 37°C in an ornithine transcarbamylase coupled reaction, where carbamoyl phosphate was immediately converted to citrulline.
Amino Acid Analysis
The concentration of amino acids in plasma and liver was determined by high-performance liquid chromatography as previously described.26
Statistical Analysis
Collected data was analyzed with the Prism8 (GraphPad Software, San Diego, CA, USA) statistical package. Results were expressed as mean ± standard error of the mean (SEM) and p values were determined using one-way ANOVA with Tukey’s multiple comparison’s test or unpaired t test when applicable. Error bars represent SEM p < 0.05 was considered significant.
Author Contributions
Experimental design, data acquisition, interpretation, analysis, and manuscript preparation: M.N. and G.S.L. Data acquisition, analysis, and critical review of the manuscript: G.A., S.K., B.T., J.H., and G.M.
Conflicts of Interest
G.S.L. serves as a consultant to Audentes Therapeutics in an area unrelated to the work described in this manuscript. All authors declare no competing interests.
Acknowledgments
Amino acid profiles were performed in the Metabolomics Core facility, The Children’s Hospital of Philadelphia (https://metabolomic.research.chop.edu/). Liver tissue for immunohistochemistry was prepared and sectioned by UCLA Translational Pathology Core Laboratory. Mouse and syringe drawings in Figure 2A were developed by Sarah Pyle at Sarah Pyle Design (San Francisco, CA, USA). These studies were funded by grant R21NS091654 (G.S.L.) from the United States NIH/National Institute of Neurological Disorders and Stroke (NINDS), the Philip Whitcome Pre-Doctoral Fellowship from the University of California, Los Angeles Molecular Biology Institute (M.N.), and the Swiss National Science Foundation grant 320030_176088 (J.H.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.011.
Supplemental Information
References
- 1.Clarke S. A major polypeptide component of rat liver mitochondria: carbamyl phosphate synthetase. J. Biol. Chem. 1976;251:950–961. [PubMed] [Google Scholar]
- 2.Shi D., Caldovic L., Tuchman M. Sources and Fates of Carbamyl Phosphate: A Labile Energy-Rich Molecule with Multiple Facets. Biology (Basel) 2018;7:E34. doi: 10.3390/biology7020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Summar, M.L., Koelker, S., Freedenberg, D., Le Mons, C., Haberle, J., Lee, H.S., and Kirmse, B.; European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD). Electronic address: http://www.e-imd.org/en/index.phtml; Members of the Urea Cycle Disorders Consortium (UCDC). Electronic address: http://rarediseasesnetwork.epi.usf.edu/ucdc/ (2013). The incidence of urea cycle disorders. Mol. Genet. Metab. 110, 179–180. [DOI] [PMC free article] [PubMed]
- 4.Nettesheim S., Kölker S., Karall D., Häberle J., Posset R., Hoffmann G.F., Heinrich B., Gleich F., Garbade S.F., Arbeitsgemeinschaft für Pädiatrische Stoffwechselstörungen (APS); European registry and network for Intoxication type Metabolic Diseases (E-IMD); Erhebungseinheit für Seltene Pädiatrische Erkrankungen in Deutschland (ESPED); Austrian Metabolic Group; Swiss Paediatric Surveillance Unit (SPSU) Incidence, disease onset and short-term outcome in urea cycle disorders -cross-border surveillance in Germany, Austria and Switzerland. Orphanet J. Rare Dis. 2017;12:111. doi: 10.1186/s13023-017-0661-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diez-Fernandez C., Häberle J. Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin. Ther. Targets. 2017;21:391–399. doi: 10.1080/14728222.2017.1294685. [DOI] [PubMed] [Google Scholar]
- 6.Wang L., Morizono H., Lin J., Bell P., Jones D., McMenamin D., Yu H., Batshaw M.L., Wilson J.M. Preclinical evaluation of a clinical candidate AAV8 vector for ornithine transcarbamylase (OTC) deficiency reveals functional enzyme from each persisting vector genome. Mol. Genet. Metab. 2012;105:203–211. doi: 10.1016/j.ymgme.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kok C.Y., Cunningham S.C., Carpenter K.H., Dane A.P., Siew S.M., Logan G.J., Kuchel P.W., Alexander I.E. Adeno-associated virus-mediated rescue of neonatal lethality in argininosuccinate synthetase-deficient mice. Mol. Ther. 2013;21:1823–1831. doi: 10.1038/mt.2013.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baruteau J., Perocheau D.P., Hanley J., Lorvellec M., Rocha-Ferreira E., Karda R., Ng J., Suff N., Diaz J.A., Rahim A.A. Argininosuccinic aciduria fosters neuronal nitrosative stress reversed by Asl gene transfer. Nat. Commun. 2018;9:3505. doi: 10.1038/s41467-018-05972-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee E.K., Hu C., Bhargava R., Rozengurt N., Stout D., Grody W.W., Cederbaum S.D., Lipshutz G.S. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol. Ther. 2012;20:1844–1851. doi: 10.1038/mt.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schofield J.P., Cox T.M., Caskey C.T., Wakamiya M. Mice deficient in the urea-cycle enzyme, carbamoyl phosphate synthetase I, die during the early neonatal period from hyperammonemia. Hepatology. 1999;29:181–185. doi: 10.1002/hep.510290112. [DOI] [PubMed] [Google Scholar]
- 11.Deignan J.L., Cederbaum S.D., Grody W.W. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol. Genet. Metab. 2008;93:7–14. doi: 10.1016/j.ymgme.2007.08.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srinivasan R.C., Zabulica M., Hammarstedt C., Wu T., Gramignoli R., Kannisto K., Ellis E., Karadagi A., Fingerhut R., Allegri G. A liver-humanized mouse model of carbamoyl phosphate synthetase 1-deficiency. J. Inherit. Metab. Dis. 2019;42:1054–1063. doi: 10.1002/jimd.12067. [DOI] [PubMed] [Google Scholar]
- 13.Smith D.D., Jr., Campbell J.W. Distribution of glutamine synthetase and carbamoyl-phosphate synthetase I in vertebrate liver. Proc. Natl. Acad. Sci. USA. 1988;85:160–164. doi: 10.1073/pnas.85.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaasbeek Janzen J.W., Moorman A.F., Lamers W.H., Charles R. Development of the heterogeneous distribution of carbamoyl-phosphate synthetase (ammonia) in rat-liver parenchyma during postnatal development. J. Histochem. Cytochem. 1985;33:1205–1211. doi: 10.1177/33.12.4067274. [DOI] [PubMed] [Google Scholar]
- 15.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brunetti-Pierri N., Palmer D.J., Beaudet A.L., Carey K.D., Finegold M., Ng P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum. Gene Ther. 2004;15:35–46. doi: 10.1089/10430340460732445. [DOI] [PubMed] [Google Scholar]
- 17.Rothe M., Modlich U., Schambach A. Biosafety challenges for use of lentiviral vectors in gene therapy. Curr. Gene Ther. 2013;13:453–468. doi: 10.2174/15665232113136660006. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z.Y., He C.Y., Meuse L., Kay M.A. Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther. 2004;11:856–864. doi: 10.1038/sj.gt.3302231. [DOI] [PubMed] [Google Scholar]
- 19.Sun L., Li J., Xiao X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat. Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- 20.Duan D., Yue Y., Engelhardt J.F. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol. Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh A., Yue Y., Lai Y., Duan D. A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol. Ther. 2008;16:124–130. doi: 10.1038/sj.mt.6300322. [DOI] [PubMed] [Google Scholar]
- 22.Maddalena A., Tornabene P., Tiberi P., Minopoli R., Manfredi A., Mutarelli M., Rossi S., Simonelli F., Naggert J.K., Cacchiarelli D., Auricchio A. Triple Vectors Expand AAV Transfer Capacity in the Retina. Mol. Ther. 2018;26:524–541. doi: 10.1016/j.ymthe.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grose W.E., Clark K.R., Griffin D., Malik V., Shontz K.M., Montgomery C.L., Lewis S., Brown R.H., Jr., Janssen P.M., Mendell J.R., Rodino-Klapac L.R. Homologous recombination mediates functional recovery of dysferlin deficiency following AAV5 gene transfer. PLoS ONE. 2012;7:e39233. doi: 10.1371/journal.pone.0039233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sondergaard P.C., Griffin D.A., Pozsgai E.R., Johnson R.W., Grose W.E., Heller K.N., Shontz K.M., Montgomery C.L., Liu J., Clark K.R. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015;2:256–270. doi: 10.1002/acn3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vidal P., Pagliarani S., Colella P., Costa Verdera H., Jauze L., Gjorgjieva M., Puzzo F., Marmier S., Collaud F., Simon Sola M. Rescue of GSDIII Phenotype with Gene Transfer Requires Liver- and Muscle-Targeted GDE Expression. Mol. Ther. 2018;26:890–901. doi: 10.1016/j.ymthe.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khoja S., Nitzahn M., Hermann K., Truong B., Borzone R., Willis B., Rudd M., Palmer D.J., Ng P., Brunetti-Pierri N., Lipshutz G.S. Conditional disruption of hepatic carbamoyl phosphate synthetase 1 in mice results in hyperammonemia without orotic aciduria and can be corrected by liver-directed gene therapy. Mol. Genet. Metab. 2018;124:243–253. doi: 10.1016/j.ymgme.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dyka F.M., Molday L.L., Chiodo V.A., Molday R.S., Hauswirth W.W. Dual ABCA4-AAV Vector Treatment Reduces Pathogenic Retinal A2E Accumulation in a Mouse Model of Autosomal Recessive Stargardt Disease. Hum. Gene Ther. 2019;30:1361–1370. doi: 10.1089/hum.2019.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeoka M., Soman T.B., Shih V.E., Caviness V.S., Jr., Krishnamoorthy K.S. Carbamyl phosphate synthetase 1 deficiency: a destructive encephalopathy. Pediatr. Neurol. 2001;24:193–199. doi: 10.1016/s0887-8994(00)00259-9. [DOI] [PubMed] [Google Scholar]
- 29.Häberle J., Shchelochkov O.A., Wang J., Katsonis P., Hall L., Reiss S., Eeds A., Willis A., Yadav M., Summar S., Urea Cycle Disorders Consortium Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum. Mutat. 2011;32:579–589. doi: 10.1002/humu.21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bates T.R., Lewis B.D., Burnett J.R., So K., Mitchell A., Delriviere L., Jeffrey G.P. Late-onset carbamoyl phosphate synthetase 1 deficiency in an adult cured by liver transplantation. Liver Transpl. 2011;17:1481–1484. doi: 10.1002/lt.22407. [DOI] [PubMed] [Google Scholar]
- 31.Foschi F.G., Morelli M.C., Savini S., Dall’Aglio A.C., Lanzi A., Cescon M., Ercolani G., Cucchetti A., Pinna A.D., Stefanini G.F. Urea cycle disorders: a case report of a successful treatment with liver transplant and a literature review. World J. Gastroenterol. 2015;21:4063–4068. doi: 10.3748/wjg.v21.i13.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi D., Zhao G., Ah Mew N., Tuchman M. Precision medicine in rare disease: Mechanisms of disparate effects of N-carbamyl-l-glutamate on mutant CPS1 enzymes. Mol. Genet. Metab. 2017;120:198–206. doi: 10.1016/j.ymgme.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kok C.Y., Cunningham S.C., Kuchel P.W., Alexander I.E. Insights into Gene Therapy for Urea Cycle Defects by Mathematical Modeling. Hum. Gene Ther. 2019;30:1385–1394. doi: 10.1089/hum.2019.053. [DOI] [PubMed] [Google Scholar]
- 34.Khoja S., Nitzahn M., Truong B., Lambert J., Willis B., Allegri G., Rüfenacht V., Häberle J., Lipshutz G.S. A constitutive knockout of murine carbamoyl phosphate synthetase 1 results in death with marked hyperglutaminemia and hyperammonemia. J. Inherit. Metab. Dis. 2019;42:1044–1053. doi: 10.1002/jimd.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trapani I., Colella P., Sommella A., Iodice C., Cesi G., de Simone S., Marrocco E., Rossi S., Giunti M., Palfi A. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014;6:194–211. doi: 10.1002/emmm.201302948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colella P., Trapani I., Cesi G., Sommella A., Manfredi A., Puppo A., Iodice C., Rossi S., Simonelli F., Giunti M. Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 2014;21:450–456. doi: 10.1038/gt.2014.8. [DOI] [PubMed] [Google Scholar]
- 37.Dong B., Nakai H., Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol. Ther. 2010;18:87–92. doi: 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Z., Yang H., Colosi P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y., Ling C., Song L., Wang L., Aslanidi G.V., Tan M., Ling C., Srivastava A. Limitations of encapsidation of recombinant self-complementary adeno-associated viral genomes in different serotype capsids and their quantitation. Hum. Gene Ther. Methods. 2012;23:225–233. doi: 10.1089/hgtb.2012.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mochizuki S., Mizukami H., Ogura T., Kure S., Ichinohe A., Kojima K., Matsubara Y., Kobayahi E., Okada T., Hoshika A. Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 2004;11:1081–1086. doi: 10.1038/sj.gt.3302262. [DOI] [PubMed] [Google Scholar]
- 41.Sluiter W., Oomens L.W., Brand A., Van Furth R. Determination of blood volume in the mouse with 51chromium-labelled erythrocytes. J. Immunol. Methods. 1984;73:221–225. doi: 10.1016/0022-1759(84)90046-2. [DOI] [PubMed] [Google Scholar]
- 42.Davidoff A.M., Ng C.Y., Zhou J., Spence Y., Nathwani A.C. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 43.Dane A.P., Cunningham S.C., Graf N.S., Alexander I.E. Sexually dimorphic patterns of episomal rAAV genome persistence in the adult mouse liver and correlation with hepatocellular proliferation. Mol. Ther. 2009;17:1548–1554. doi: 10.1038/mt.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Rosenberg J.B., De B.P., Ferris B., Wang R., Rivella S., Kaminsky S.M., Crystal R.G. In vivo gene transfer strategies to achieve partial correction of von Willebrand disease. Hum. Gene Ther. 2012;23:576–588. doi: 10.1089/hum.2011.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Al-Moyed H., Cepeda A.P., Jung S., Moser T., Kügler S., Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol. Med. 2019;11:e9396. doi: 10.15252/emmm.201809396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Halbert C.L., Allen J.M., Miller A.D. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat. Biotechnol. 2002;20:697–701. doi: 10.1038/nbt0702-697. [DOI] [PubMed] [Google Scholar]
- 48.Ye X., Robinson M.B., Pabin C., Quinn T., Jawad A., Wilson J.M., Batshaw M.L. Adenovirus-mediated in vivo gene transfer rapidly protects ornithine transcarbamylase-deficient mice from an ammonium challenge. Pediatr. Res. 1997;41:527–534. doi: 10.1203/00006450-199704000-00012. [DOI] [PubMed] [Google Scholar]
- 49.Allegri G., Deplazes S., Grisch-Chan H.M., Mathis D., Fingerhut R., Häberle J., Thöny B. A simple dried blood spot-method for in vivo measurement of ureagenesis by gas chromatography-mass spectrometry using stable isotopes. Clin. Chim. Acta. 2017;464:236–243. doi: 10.1016/j.cca.2016.11.038. [DOI] [PubMed] [Google Scholar]
- 50.Diez-Fernandez C., Martínez A.I., Pekkala S., Barcelona B., Pérez-Arellano I., Guadalajara A.M., Summar M., Cervera J., Rubio V. Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Hum. Mutat. 2013;34:1149–1159. doi: 10.1002/humu.22349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.