

Abstract
Adenine base editor (ABE) is a new generation of genome-editing technology through fusion of Cas9 nickase with an evolved E. coli TadA (TadA∗) and holds great promise as novel genome-editing therapeutics for treating genetic disorders. ABEs can directly convert A-T to G-C in specific genomic DNA targets without introducing double-strand breaks (DSBs). We recently showed that computer program-assisted analysis of Sanger sequencing traces can be used as a low-cost and rapid alternative of deep sequencing to assess base-editing outcomes. Here we developed a rapid fluorescence-based reporter assay (Base Editing ON [BEON]) to quantify ABE efficiency. The assay relies on the restoration of the downstream green fluorescent protein (GFP) in ABE-mediated editing of a stop codon located within the guide RNA (gRNA). We showed that this assay can be used to screen for effective ABE variants, characterize the protospacer adjacent motif (PAM) requirement of a novel NNG-targeting ABE based on ScCas9, and enrich for edited cells. Finally, we demonstrated that the reporter assay allowed us to assess the feasibility of ABE editing to correct point mutations associated with dysferlinopathy. Taken together, the BEON assay would facilitate and simplify the studies with ABEs.
Keywords: adenine base editor, ABE, base editing, CRISPR, genome editing, reporter
Graphical Abstract
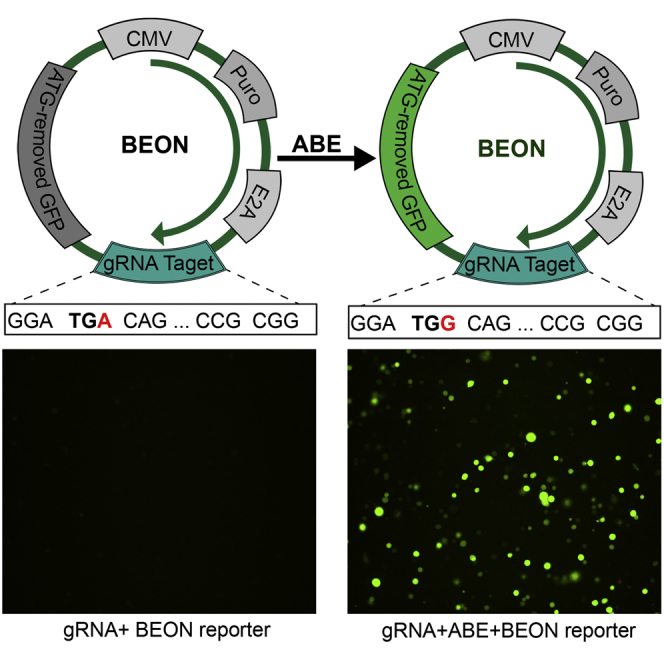
Han and colleagues developed a fluorescence-based reporter system (BEON) to rapidly quantify ABE activity. This system can be used to compare ABE variants, characterize the PAM requirements, enrich for edited cells, and assess the feasibility of ABE correction of disease mutations. BEON would facilitate the studies with ABEs.
Introduction
Recent advancements in genome-editing technologies have opened a new era of therapeutic development for genetic diseases. Analysis of the ClinVar database showed that about 48% of the pathogenic human single-nucleotide polymorphisms (SNPs) are caused by G-C to A-T conversion.1 Thus, the ability to efficiently convert the A-T base pair to G-C base pair would facilitate the correction of G-C to A-T pathogenic SNPs. Although homology-directed repair (HDR) of CRISPR-induced double-strand breaks (DSBs) can introduce precise base conversion at the target site,2 HDR is highly inefficient because repair of DSBs is predominantly achieved through non-homologous end joining (NHEJ), which generates undesired stochastic insertions and deletions (indels).3, 4, 5, 6, 7
Recently, a new generation of genome-editing technology, namely, base editors (BEs), was developed through fusion of the RNA-programmable Cas9 nickase with nucleobase deaminase enzymes.8, 9, 10, 11, 12, 13 By fusion with different DNA deaminases, two classes of DNA BEs have been developed: cytosine BEs (CBEs) converting a C-G base pair into a T-A base pair9,11, 12, 13 and adenine BEs (ABEs) converting an A-T base pair into a G-C base pair.10 These BEs can directly install point mutations into target DNA without making DSBs, requiring a DNA donor template, or relying on cellular HDR. It has been shown that the BEs are highly efficient in mammalian cells and introduce fewer indels as compared with HDR-based approaches.
Despite the advantages of BEs, there are several challenges that need to be addressed. First, there is a lack of effective ways to identify and isolate cell populations that have been edited, in particular when engineered cells will be used in clinics and there is no readily detectable phenotype to distinguish edited from unedited cells. Some approaches have been taken to facilitate enriching edited cells, such as co-transfecting a fluorescent protein plasmid or fusing BEs to fluorescent proteins, and using flow cytometry to isolate reporter-positive cells.14 However, these techniques lack the capability to directly report on base-editing activity per se but rather on transfection. Recently, fluorescent reporters for editing enrichment were reported, which are based on CBE-mediated conversion of a blue fluorescent protein (BFP) to green fluorescent protein (GFP).15,16 However, these reporters are limited to report on a particular target (a guide sequence on BFP), lack the flexibility of testing other desired targets, or do not work for ABE. Second, cells with the pathogenic SNPs for therapeutic development may not be readily available. Engineering or obtaining cells with the target pathogenic mutations is laborious, time consuming, and expensive. Designing a reliable fluorescence reporter with the flexibility to test desired targets would greatly facilitate the development of ABE therapies.
In this work, we developed a fluorescence reporter assay (Base Editing ON [BEON]) based on the rescue of a downstream GFP upon ABE-mediated conversion of a stop codon located within the guide RNA (gRNA) target fused in frame with GFP. We demonstrated that the BEON assay can be used to screen for effective ABE variants, characterize the protospacer adjacent motif (PAM) requirement of a novel ScCas9-ABE, enrich for edited cells, and test the feasibility of base correction of disease-causing mutations.
Results
Engineering a Fluorescence Reporter for Measuring Base-Editing Activity
The ABEs convert A to G or T to C, thus allowing the conversion of stop codons (such as TAG or TGA) to other codons that encode for amino acids (e.g., CAG, CGA, or TGG). We reasoned that insertion of a gRNA target sequence carrying a stop codon in the suitable editing window of ABEs upstream of an mCherry or GFP cassette and downstream of a puromycin-resistant gene cassette (Figure 1A) would serve as a fluorescent reporter assay to evaluate the ABE base-editing activity. We selected a target sequence with an NGG PAM in the human site 1 (S1) as a testing system and generated the corresponding reporter and gRNA plasmids. The stop codon TGA in the S1 target was fused in frame with the downstream mCherry cassette and the upstream puromycin and self-cleaving E2A cassette (Puro-E2A-S1-mCherry). Conversion of the stop codon TGA to TGG (the target A is located at position 5 of the gRNA) would allow the expression of downstream fluorescent mCherry or GFP. Although the Puro-E2A-S1-mCherry reporter showed some background signal, co-transfection with the S1-gRNA and ABEmax showed substantially increased mCherry fluorescence (data not shown).
Figure 1.

Design of the BEON Assay for Quantification of ABE Editing
(A) A schematic diagram of the reporter construct, consisting of a puromycin-resistant gene cassette, fused in frame with a gRNA target sequence carrying a stop codon (TGA) in the suitable editing window of ABE, E2A peptide sequence, and ATG-removed GFP cassette. The reporter is under the control of the CMV promoter. The PAM sequence is shown in blue. (B) Representative fluorescence and bright-field (merged) images of HEK293 cells transfected without (left) or with the reporter alone (middle), or together with S1-gRNA, S1-reporter, and ABEmax (right). Scale bar: 100 μm. (C) Representative flow cytometry plots of HEK293 cells as treated in (B). (D) Quantification of GFP+ HEK293 cells by flow cytometry. (E) Representative Sanger sequencing chromatogram of the RT-PCR amplicons from HEK293 cells as treated in (B). (F) Quantification of base-editing efficiency of HEK293 cells as treated in (B). ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
We reasoned that the baseline fluorescence from the Puro-E2A-S1-mCherry reporter is likely due to the presence of multiple in-frame start codons in the mCherry open reading frame, and that replacing the mCherry with start codon-deleted GFP would minimize the background fluorescence. We thus constructed the Puro-E2A-S1-GFP reporter (for simplicity, it is referred to as S1-reporter hereafter). Indeed, transfection of HEK293 cells with this reporter alone yielded little GFP fluorescence (Figure 1B, middle panel), similar to the non-transfection control (Figure 1B, left panel), as examined by fluorescence microscopy. Co-transfection of the cells with S1-gRNA, S1-reporter, and ABEmax evoked substantial GFP fluorescence (Figure 1B, right panel). Flow cytometry detected a low level of background fluorescence in the reporter control group as compared with non-transfected cells (Figures 1C and 1D), indicating that there is some leaky expression of GFP. However, the co-transfection group with the three plasmids showed the highest GFP-positive population (Figures 1C and 1D). To further determine whether the restoration of GFP fluorescence corresponded to conversion of the premature stop codon, we extracted total RNA from the cells, performed RT-PCR, and sequenced the PCR products. Analysis of the Sanger sequencing traces using BEAT17 showed that a 20% A-to-G conversion occurred at position 5 of the S1-gRNA target in the co-transfection group (Figures 1E and 1F). These results confirmed that the restoration of GFP fluorescence resulted from the conversion of the stop codon TGA to TGG at the targeted base.
We next examined the effects of varying doses of the reporter vector and the ABE/gRNA plasmids on GFP fluorescence restoration. We observed that the GFP+ populations in both the reporter alone and the co-transfection group were increased with the increasing amount of the reporter plasmid transfected (Figures 2A and 2C). We chose 0.5:0.75:0.75 (reporter:ABE:gRNA, in micrograms per well for a six-well plate) for the following experiments, because this ratio yielded good rescue of GFP fluorescence relative to the reporter alone (Figures 2A and 2C).
Figure 2.

The Effects of Reporter Dosage and the Transfection Time on the BEON Assay
(A) Representative fluorescence and bright-field (merged) images of HEK293 cells transfected with different amounts of the S1-reporter (0.125, 0.25, 0.5, 0.75, 1.0 μg) and 1:1 of ABEmax and S1-gRNA with a total of 2 μg DNA per well in a six-well plate. The images were taken at day 3 after transfection. Scale bar: 100 μm. (B) Representative fluorescence and bright-field (merged) images of HEK293 cells transfected with S1-reporter (0.5 μg), ABEmax (0.75 μg), and S1-gRNA (0.75 μg), imaged at different time points after transfection. (C) FACS quantification of GFP+ cells for conditions described in (A). (D) FACS quantification of GFP+ cells for conditions described in (B). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
We also examined the time course of base-editing-induced restoration of GFP. HEK293 cells were co-transfected with S1-reporter, ABEmax, and S1-gRNA, and the cells were examined at different time points using fluorescence microscopy and flow cytometry analysis. We observed that GFP started to show up at 24 h post-transfection, gradually increased over time in the cells co-transfected with the three plasmids, and peaked at around 72 h post-transfection (Figures 2B and 2D) (notably, the higher GFP+ reading at 120 h was inaccurate due to the high cell density-induced cell detachment). In the reporter alone group, GFP+ cells peaked at 48 h (Figure 2D). Thus, in all of the following experiments, the GFP reporter assays were analyzed at 72 h unless noted otherwise.
Screening for Relative Activities of Different ABEs
Previous studies showed that codon optimization improves the base-editing efficiency.14,18 To examine the relative activities of ABEs with or without codon optimization or with modified PAM targeting,19,20 we compared ABE7.10, ABEmax, and xABE [ABE7.10 with xCas(3.7)] using the BEON assay. Fluorescence microscopy examination showed that all of these ABEs can restore GFP fluorescence of the S1-reporter; however, substantially higher GFP-positive cells were induced by ABEmax editing as compared with ABE7.10 (Figure 3A). Fluorescence-activated cell sorting (FACS) analysis further confirmed that ABEmax had higher editing activity than ABE7.10 (31.57% ± 1.08% versus 18.03% ± 0.90%) (Figures 3B and 3C). As compared with ABE7.10, the xABE (31.67% ± 0.94%) also had higher activity (Figures 3B and 3C). Thus, the BEON assay can be used to screen for relative efficacy of BEs at desired target sites.
Figure 3.

Screening for Optimal Base Editors Using the BEON Assay
(A) Representative bright-field (upper) and GFP fluorescence (lower) images of HEK293 cells without transfection (control), transfected with the reporter alone, or co-transfected with S1-gRNA, S1-reporter, and one of the base editors: ABE7.10, xABE7.10, or ABEmax. Scale bar: 100 μm. (B) Representative flow cytometry plots of HEK293 cells as treated in (A). (C) Quantification of GFP+ population by FACS analysis for the HEK293 cells as treated in (A). ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Determination of the PAM Requirement of a Novel ScCas9-ABE Using the BEON Assay
Recently, a new Cas9 variant from Streptococcus canis (ScCas9) was reported to recognize a minimal 5'-NNG-3' PAM sequence.21 We constructed ScCas9-ABE by fusing the TadA-TadA∗ with codon-optimized ScCas9-D10A nickase. We also constructed 11 S1-reporter plasmids that carry the same S1-gRNA target but vary in the PAM sequences (S1-NNG1 to S1-NNG11, excluding TAG and four NGG PAMs) (Figure 4A). The two adjacent positions following NNG were fixed as TT. HEK293 cells were transfected with the S1-gRNA and ScCas9-ABE with 1 of the 11 reporter plasmids. After 72 h of transfection, the cells were imaged by fluorescence microscopy and analyzed by flow cytometry. We observed that ScCas9-ABE activated GFP expression in all of these 11 S1-reporters (Figure 4B; Figures S1–S9). Flow cytometry demonstrated that all of the reporters except ATG and GAG PAM had low baseline GFP+ cells, and that ScCAs9-ABE was efficient to rescue the GFP fluorescence in these reporters (Figure 4C). The high baseline GFP expression in ATG and GAG reporters is most likely due to a combination of the leaky scanning mechanism22 (allowing the initiation from a downstream start codon) and usage of AUG and non-AUG start codons23 carried by the PAM sequences. Both of these mechanisms are well illustrated in adeno-associated virus capsid genes, in which a single open reading frame allows the expression of VP1, VP2, and VP3 proteins.24 Quantification of the fluorescence intensity in the ATG reporters showed that the average baseline fluorescence intensity was substantially lower than that with ScCas9-ABE editing (Figure S2C), indicating that the ScCas9 editing further increased the expression of the downstream GFP. Taken together, these results confirmed that ScCas9 recognizes a minimal NNG PAM.
Figure 4.

Determination of the PAM Requirement of a Novel ScCas9-ABE Using the BEON Assay
(A) Samples of the reporter sequences consisting of the same S1-gRNA target but different PAM sequences (in blue). The target “A” base is highlighted in red in the bolded stop codon. (B) Representative fluorescence and bright-field (merged) images of HEK293 cells transfected with S1-gRNA, ScCas9-ABE, and S1NNG1 (AAG) or S1NNG2 (CAG) reporter plasmids, or the corresponding reporter alone. The PAM sequences are listed in yellow. Scale bars: 100 μm. (C) FACS quantification of GFP+ cells for various reporters with or without ScCas9-ABE editing. The PAM sequence is listed on each graph. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Enrichment of Base-Edited Cells Using the BEON System
We reasoned that successful editing of the reporter DNA would reflect the efficiency of editing in the endogenous DNA and thus envisioned that the reporter assay could allow us to enrich the base-edited cell population. At 72 h after transfection, the cells were sorted into two populations with either low (GFP-low) or high (GFP-high) GFP fluorescence intensity (Figure 5A). These two cell populations and cells without sorting were analyzed for base editing of the endogenous S1 by Sanger sequencing and the BEAT software.17 As compared with the cells before sorting, the GFP-high population showed a significantly increased overall editing at position 5 (after sorting versus before sorting: 70.61% ± 7.27% versus 29.54% ± 4.86%) (Figures 5B and 5C), a 2.4-fold enrichment. In contrast, the GFP-low population showed only 12.26% ± 1.42% editing (Figures 5B and 5C). As a comparison, we replaced the BEON reporter with a simple GFP-expressing construct and sorted cells into GFP-high and GFP-low populations. We observed that the GFP-high population still showed higher editing compared with the GFP-low population. However, the editing detected in the GFP-high population was significantly lower than the GFP-high population resulting from the BEON reporter (Figure 5C), suggesting that the BEON reporter can yield higher enrichment than a simple GFP-expressing construct.
Figure 5.

Enrichment of Base-Edited Cells Using the BEON Assay
(A) Representative fluorescence and bright-field (merged) images of HEK293 cells transfected with the S1-reporter, S1-gRNA, and ABEmax before and after sorting. Scale bar: 100 μm. (B) BEAT analyses of the PCR amplicons of the endogenous site 1 for the cells before and 24 h (24h) after sorting. The red arrows point to the target “A” base at position 5. (C) Quantification of the A-to-G conversion efficiency of site 1 target for the sorted GFP-high and GFP-low HEK293 cells with the BEON reporter or a simple GFP-expressing construct. (D) Quantification of the A-to-G conversion efficiency of site 1 target for the sorted GFP-high and GFP-low HEK293 cells cultured for 1 more week. (E) Quantification of the A-to-G conversion efficiency of CAR target for the sorted GFP-high and GFP-low HEK293 cells using the BEON reporter. (F) Quantification of the A-to-G conversion efficiency of mouse Tmem5 target for the sorted GFP-high and GFP-low Neuro2A cells using the BEON reporter. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
To examine whether the editing at the endogenous S1 of HEK293 cells will be preserved after prolonged culture, the sorted cells were maintained in culture for another week and analyzed by Sanger sequencing. Despite the GFP fluorescence loss (due to several cell doublings and gradual loss of the transfected plasmids), the editing events at the target S1 could still be detected with high efficiency in the GFP-high group similar to the cells analyzed at 24 h after sorting (Figure 5D), indicating that the editing is long-lasting.
Previous studies showed that the target DNA sequence features per se,25,26 nucleosome occupation of the target DNA,27, 28, 29, 30, 31 and chromatin accessibility26,32, 33, 34, 35 may affect the editing efficiency by Cas9 endonuclease. We tested whether the BEON reporter could allow enrichment of endogenous editing at a previously identified refractory site: CAR (nuclear receptor subfamily 1 group I member 3). The GFP-high population showed 37.54% ± 1.27% editing, significantly higher than the editing activity in GFP-low population (3.11% ± 2.15%) (Figure 5E), suggesting that the BEON reporter system can facilitate the enrichment of edited cells even for refractory targets.
Finally, to test whether the BEON system could be used in other cell types, we designed a reporter and gRNA for mouse Tmem5. Again, the GFP-high population sorted by FACS showed substantially higher editing activity than the GFP-low population (Figure 5F).
Exploring the Feasibility of ABE-Mediated Correction of Nonsense Mutations Causing Dysferlinopathy
Finally, we applied the BEON assay to determine whether disease-causing nonsense point mutations can be corrected using ABE editing. We chose the DYSF gene because its mutations have been identified in patients with muscular dystrophy manifested as distal anterior compartment myopathy, limb girdle muscular dystrophy type 2B and Miyoshi myopathy, proximodistal dysferlinopathy, and isolated/long-lasting hyperCKemia, together termed dysferlinopathy.36 Specifically, the nonsense mutations of R377X (GenBank: NM_003494.3, c.1129C>T), Q605X (c.1813C>T), and Q1278X (c.3832C>T) were selected (Figure 6A). Because the patient cells with these mutations are not readily available in the laboratory, we tested whether these mutations could be corrected by ABE editing using the reporter assay. As shown in Figure 6B, the R377X and Q1278X reporters showed robust GFP expression after co-transfection with ABEmax and their corresponding gRNAs. However, the Q605X reporter showed only background signal with ABEmax and gRNA treatment, indicating that ABEmax was inefficient for this target, obviously due to the non-compatible PAM (AGT) for ABEmax. We speculated that xABE or the ABE based on the NG variant of SpCas9 (ABE-NG)37 may be able to work on the Q605X target. Indeed, co-transfection with ABE-NG and Q605X-gRNA was effective in turning on GFP expression in the Q605X reporter, whereas xABE was still ineffective (Figure 6C). Quantification with flow cytometry showed that ABEmax editing induced GFP expression in 47.9% and 50.3% of cells for R377X and Q1278X reporters, respectively (Figure 6D), and that ABE-NG editing evoked 28.2% GFP-positivity for the Q605X reporter (Figure 6E), suggesting that these nonsense mutations are targetable by ABE-mediated editing.
Figure 6.

Feasibility Evaluation of ABE Editing to Correct Nonsense Mutations Causing Dysferlinopathy
(A) The target sequences and gRNAs of three nonsense mutations causing dysferlinopathy. The mutations are shown in red, and the PAM sequences are shown in blue. (B) Representative merged fluorescence and bright-field images of HEK293 cells transfected with R377X and Q1278X reporters plus or minus their corresponding gRNAs and ABEmax. Scale bar: 100 μm. (C) Representative merged fluorescence and bright-field images of HEK293 cells transfected with the Q605X reporter, Q605X gRNA and ABEmax, xABE, or ABE-NG. Scale bar: 100 μm. (D) Quantification of GFP+ cells for the conditions described in (B). (E) Quantification of GFP+ cells for the conditions described in (C). ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
Discussion
In this study, we report a fluorescence reporter system BEON that enables rapid and easy quantification, enrichment, and preclinical evaluation of ABE activity for targeted correction of disease-causing point mutations.
We first demonstrated the flexibility and quantitative nature of BEON to profile different ABEs with codon optimization or PAM modification. Our experiments revealed the robustness of ScCas9-ABE in editing NNG-PAM target sites. Although we studied only a relatively small panel of ABEs, BEON could be implemented in characterizing and comparing features of newly generated enzymes in various mammalian cell systems.
Several recent studies have described base-editing reporters that evoke or suppress GFP expression. For instance, BE-FLARE16 and TREE15 employed a fluorescence shift from BFP to GFP following cytidine deamination of codon 66. Moreover, Martin et al.38 established a panel of GFP reporters for CBEs based on the findings that a T-to-C mutation ablates fluorescence in three GFP codons, and thus CBE-mediated C-to-T conversion switches on GFP. However, these reporters lack the flexibility to test different gRNA targets and also are limited for detecting CBE activities. During writing up this manuscript, we realized that Katti et al.39 reported a similar reporter system named GO as our BEON system, which utilized the BE-mediated conversion of an upstream stop codon or the translation initiation codon.
We demonstrated that the BEON system enabled over 2-fold enrichment of base-edited cells following a single FACS sorting step. Although we used only GFP and mCherry, our system can be easily adapted to use other non-fluorescent marker genes, such as antibiotic resistance genes, to facilitate enrichment of endogenous base-editing events without using cell sorting.
One major advantage of these reporters is their flexibility to test different targets. This is particularly important for modeling or correcting human disease variants, where patient-derived cells may not be readily available. As a proof-of-concept test, we showed that BEON enabled rapid validation of ABE base editing as a potential therapeutic approach to correct three disease-causing mutations in dysferlinopathy.
BEON is a rapid and robust functional reporter system that can be used to detect, quantify, and enrich base-editing activity. It can be easily modified to adopt a range of functional reporters and thus expand the applicability of base-editing technologies and potentially the most recent prime editing technology40 as well.
Materials and Methods
Plasmids Construction
The ABE plasmids, including pCMV-ABE7.10 (#102919; Addgene), xCas9(3.7)-ABE(7.10) (#108382; Addgene), and pCMV-ABEmax (#112095; Addgene), were obtained from Addgene (Watertown, MA, USA). The codon-optimized ScCas9 was synthesized by GenScript and subcloned into pCMV-ABEmax to generate pCMV-ABEmaxSc. The pCMV-ABEmaxNG was constructed by site-directed mutagenesis of pCMV-ABEmax. The gRNAs were designed to target human S1 and human dysferlin gene. The annealed gRNA oligos were cloned into pLenti-OgRNA-Zeo plasmid, which was modified from pLenti-sgRNA(MS2)_zeo (#61427; Addgene). The target reporter plasmid backbone pLKO- puro-E2A-GFP was constructed by cloning the puro-E2A-GFP (with ATG removed from GFP) into pLKO-GFP-E2A-SpCas9.41 The annealed reporter oligos were then cloned into EcoRI/BamHI linearized pLKO-puro-E2A-GFP. All oligos for gRNAs, target reporters, and PCR are listed in Table S1. All plasmids are listed in Table S2.
Cell Culture and Transfection
HEK293 cells and Neuro2A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma, St. Louis, MO, USA) containing 10% fetal bovine serum (FBS) and 1% 100× penicillin-streptomycin (Invitrogen). Cells were plated in six-well plates and transfected with the 2 μg plasmids (0.5 μg reporter, 0.75 μg gRNA, and 0.75 μg ABE) per well unless specified otherwise by polyethylenimine (PEI) as previously described.42
Fluorescence Microscopy
After 3 days post-transfection or as stated otherwise, HEK293 cells transfected with the BEON reporter, gRNA, and ABE plasmids were collected from six-well plates and re-seeded in poly-l-lysine-coated 35-mm glass dishes. Fluorescence images were collected on a Nikon Ti-E inverted fluorescence microscope equipped with an Andor Zyla sCMOS camera and Nikon Super Fluor 20× 0.75 NA objective lens. Images were recorded using NIS-Elements Advanced Research software package (Nikon) and processed using Adobe Photoshop CC software.
Flow Cytometry
At 72 h post-transfection, HEK293 cells transfected with ABE plasmids were collected from six-well plates and analyzed on Becton Dickinson LSR II (BD Biosciences) to determine GFP-positive cells. A total of 100,000 cell events were collected, and data analysis was performed using the FlowJo software (Tree Star, Ashland, OR, USA). For the cell sorting experiment, the transfected HEK293 cells were sorted on Aria III by the staff at the Analytical Cytometry Shared Resource of the Ohio State University Comprehensive Cancer Center.
PCR and Sanger Sequencing
Genomic DNA or total RNA was extracted from HEK293 cells 3 or 5 days after transfection. PCRs were carried out with 100 ng genomic DNA or cDNA in the GoTaq Master Mix (Promega) according to the manufacturer’s instruction. PCR conditions were 5 min at 95°C, followed by 32 cycles of 15 s at 95°C, 15 s at 60°C, and 30 s at 72°C. The PCR products were purified using the Wizard SV Gel and PCR Clean-up System (Promega). Purified PCR products (100 ng) were subjected to Sanger sequencing at the Genomics Shared Resource of the Ohio State University Comprehensive Cancer Center and analyzed by BEAT program.17
Statistical Analysis
The data are expressed as the mean ± SD unless otherwise stated. Statistical significance was determined using Student’s t test for two groups or one-way ANOVA followed by Bonferroni post hoc tests for multiple groups using GraphPad Prism v.8.0.1 (GraphPad Software). A p value <0.05 was considered as significant.
Author Contributions
R.H. conceived the study and wrote the manuscript. P.W. and L.X. performed the experiments, analyzed the data, and participated in drafting the manuscript. Y.G. helped plasmid construction. All co-authors have reviewed and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health (grant HL116546) and the Parent Project Muscular Dystrophy award. We thank members of the Han lab for their helpful discussion and suggestions.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.009.
Supplemental Information
References
- 1.Tang J., Lee T., Sun T. Single-nucleotide editing: From principle, optimization to application. Hum. Mutat. 2019;40:2171–2183. doi: 10.1002/humu.23819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brookhouser N., Raman S., Potts C., Brafman D.A. May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells. Cells. 2017;6:e5. doi: 10.3390/cells6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T., Tanaka M., Amano N., Watanabe A., Sakurai H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyaoka Y., Chan A.H., Judge L.M., Yoo J., Huang M., Nguyen T.D., Lizarraga P.P., So P.L., Conklin B.R. Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nat. Methods. 2014;11:291–293. doi: 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X., Wang Y., Yan W., Smith C., Ye Z., Wang J., Gao Y., Mendelsohn L., Cheng L. Production of Gene-Corrected Adult Beta Globin Protein in Human Erythrocytes Differentiated from Patient iPSCs After Genome Editing of the Sickle Point Mutation. Stem Cells. 2015;33:1470–1479. doi: 10.1002/stem.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reinhardt P., Schmid B., Burbulla L.F., Schöndorf D.C., Wagner L., Glatza M., Höing S., Hargus G., Heck S.A., Dhingra A. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Grobarczyk B., Franco B., Hanon K., Malgrange B. Generation of Isogenic Human iPS Cell Line Precisely Corrected by Genome Editing Using the CRISPR/Cas9 System. Stem Cell Rev. Rep. 2015;11:774–787. doi: 10.1007/s12015-015-9600-1. [DOI] [PubMed] [Google Scholar]
- 8.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida K., Arazoe T., Yachie N., Banno S., Kakimoto M., Tabata M., Mochizuki M., Miyabe A., Araki M., Hara K.Y. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y., Zhang J., Yin W., Zhang Z., Song Y., Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods. 2016;13:1029–1035. doi: 10.1038/nmeth.4027. [DOI] [PubMed] [Google Scholar]
- 13.Hess G.T., Frésard L., Han K., Lee C.H., Li A., Cimprich K.A., Montgomery S.B., Bassik M.C. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods. 2016;13:1036–1042. doi: 10.1038/nmeth.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–893. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Standage-Beier K., Tekel S.J., Brookhouser N., Schwarz G., Nguyen T., Wang X., Brafman D.A. A transient reporter for editing enrichment (TREE) in human cells. Nucleic Acids Res. 2019;47:e120. doi: 10.1093/nar/gkz713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coelho M.A., Li S., Pane L.S., Firth M., Ciotta G., Wrigley J.D., Cuomo M.E., Maresca M., Taylor B.J.M. BE-FLARE: a fluorescent reporter of base editing activity reveals editing characteristics of APOBEC3A and APOBEC3B. BMC Biol. 2018;16:150. doi: 10.1186/s12915-018-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L., Liu Y., Han R. BEAT: A Python Program to Quantify Base Editing from Sanger Sequencing. CRISPR J. 2019;2:223–229. doi: 10.1089/crispr.2019.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang T.P., Zhao K.T., Miller S.M., Gaudelli N.M., Oakes B.L., Fellmann C., Savage D.F., Liu D.R. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 2019;37:626–631. doi: 10.1038/s41587-019-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee P., Jakimo N., Jacobson J.M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018;4:eaau0766. doi: 10.1126/sciadv.aau0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 23.Kearse M.G., Goldman D.H., Choi J., Nwaezeapu C., Liang D., Green K.M., Goldstrohm A.C., Todd P.K., Green R., Wilusz J.E. Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev. 2019;33:871–885. doi: 10.1101/gad.324715.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becerra S.P., Koczot F., Fabisch P., Rose J.A. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J. Virol. 1988;62:2745–2754. doi: 10.1128/jvi.62.8.2745-2754.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jensen K.T., Fløe L., Petersen T.S., Huang J., Xu F., Bolund L., Luo Y., Lin L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. doi: 10.1002/1873-3468.12707. [DOI] [PubMed] [Google Scholar]
- 27.Hinz J.M., Laughery M.F., Wyrick J.J. Nucleosomes Inhibit Cas9 Endonuclease Activity in Vitro. Biochemistry. 2015;54:7063–7066. doi: 10.1021/acs.biochem.5b01108. [DOI] [PubMed] [Google Scholar]
- 28.Hinz J.M., Laughery M.F., Wyrick J.J. Nucleosomes Selectively Inhibit Cas9 Off-target Activity at a Site Located at the Nucleosome Edge. J. Biol. Chem. 2016;291:24851–24856. doi: 10.1074/jbc.C116.758706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E., Torigoe S.E., Tjian R., Weissman J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife. 2016;5:e12677. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isaac R.S., Jiang F., Doudna J.A., Lim W.A., Narlikar G.J., Almeida R. Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. eLife. 2016;5:e13450. doi: 10.7554/eLife.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yarrington R.M., Verma S., Schwartz S., Trautman J.K., Carroll D. Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc. Natl. Acad. Sci. USA. 2018;115:9351–9358. doi: 10.1073/pnas.1810062115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knight S.C., Xie L., Deng W., Guglielmi B., Witkowsky L.B., Bosanac L., Zhang E.T., El Beheiry M., Masson J.B., Dahan M. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350:823–826. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- 33.Chen X., Rinsma M., Janssen J.M., Liu J., Maggio I., Gonçalves M.A. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016;44:6482–6492. doi: 10.1093/nar/gkw524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X., Liu J., Janssen J.M., Gonçalves M.A.F.V. The Chromatin Structure Differentially Impacts High-Specificity CRISPR-Cas9 Nuclease Strategies. Mol. Ther. Nucleic Acids. 2017;8:558–563. doi: 10.1016/j.omtn.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daer R.M., Cutts J.P., Brafman D.A., Haynes K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017;6:428–438. doi: 10.1021/acssynbio.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariano A., Henning A., Han R. Dysferlin-deficient muscular dystrophy and innate immune activation. FEBS J. 2013;280:4165–4176. doi: 10.1111/febs.12261. [DOI] [PubMed] [Google Scholar]
- 37.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin A.S., Salamango D.J., Serebrenik A.A., Shaban N.M., Brown W.L., Harris R.S. A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep. 2019;9:497. doi: 10.1038/s41598-018-36739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katti A., Foronda M., Zimmerman J., Diaz B., Zafra M.P., Goswami S., Dow L.E. GO: A functional reporter system to identify and enrich base editing activity. bioRxiv. 2019 doi: 10.1101/862458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L., Zhao L., Gao Y., Xu J., Han R. Empower multiplex cell and tissue-specific CRISPR-mediated gene manipulation with self-cleaving ribozymes and tRNA. Nucleic Acids Res. 2017;45:e28. doi: 10.1093/nar/gkw1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J., Xu L., Lau Y.S., Gao Y., Moore S.A., Han R. A novel ANO5 splicing variant in a LGMD2L patient leads to production of a truncated aggregation-prone Ano5 peptide. J. Pathol. Clin. Res. 2018;4:135–145. doi: 10.1002/cjp2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.