

Abstract
Developmental and epileptic encephalopathy (DEE) associated with de novo variants in the gene encoding dynamin-1 (DNM1) is a severe debilitating disease with no pharmacological remedy. Like most genetic DEEs, the majority of DNM1 patients suffer from therapy-resistant seizures and comorbidities such as intellectual disability, developmental delay, and hypotonia. We tested RNAi gene therapy in the Dnm1 fitful mouse model of DEE using a Dnm1-targeted therapeutic microRNA delivered by a self-complementary adeno-associated virus vector. Untreated or control-injected fitful mice have growth delay, severe ataxia, and lethal tonic-clonic seizures by 3 weeks of age. These major impairments are mitigated following a single treatment in newborn mice, along with key underlying cellular features including gliosis, cell death, and aberrant neuronal metabolic activity typically associated with recurrent seizures. Our results underscore the potential for RNAi gene therapy to treat DNM1 disease and other genetic DEEs where treatment would require inhibition of the pathogenic gene product.
Keywords: epilepsy, developmental and epileptic encephalopathy, DEE, dynamin-1, DNM1, gene therapy, RNA interferance, adeno-associated virus 9, AAV9, infantile spasms, Lennox-Gastaut syndrome
Graphical Abstract
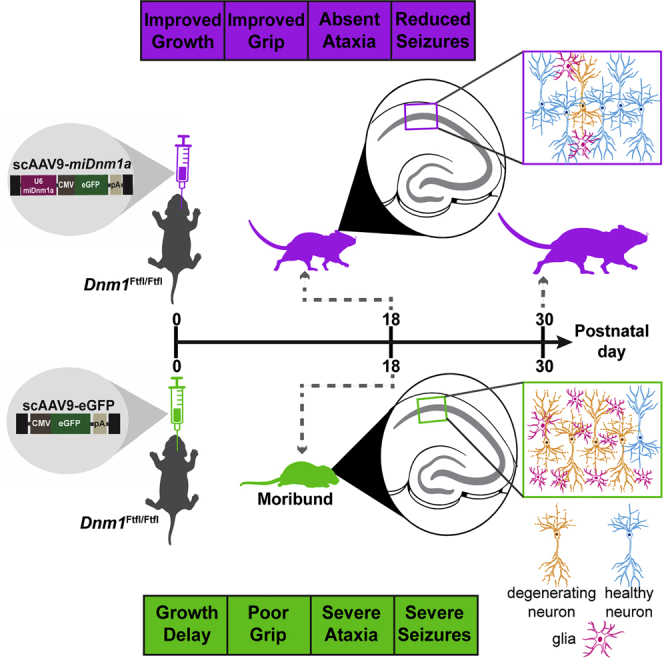
Toxic mutations in DNM1, a gene involved in the proper functioning of brain cells, cause developmental and epileptic encephalopathy—a severe class of epilepsy with no cure. Aimiuwu and colleagues show that a single gene therapy treatment via RNA interference eliminated the toxic allele sufficiently to curb severe lethal seizures, eliminate motor and developmental deficits, and extend survival of treated mice.
Introduction
DNM1 encodes a critical multimeric brain-specific GTPase, dynamin-1, that localizes to the presynapse, where it mediates endocytosis.1, 2, 3, 4 Individuals with pathogenic DNM1 variants suffer from two of the most severe developmental and epileptic encephalopathy (DEE) syndromes, Lennox-Gastaut syndrome and infantile spasms, with at least 20 heterozygous de novo variants identified in 33 patients predominantly in the critical GTPase and the middle domains of the protein.5, 6, 7, 8, 9, 10 The identification of affected individuals is likely to increase as DNM1 is now included on screening panels for severe childhood epilepsy. Children with DNM1 mutations suffer from intractable conditions manifesting as early-onset seizures, global developmental delay, profound intellectual disability, lack of speech, muscular hypotonia, dystonia, and spasticity.7, 8, 9 Affected individuals do not respond well to anti-epileptic drugs, leaving > 80% of patients with seizures,7,8 as is the case with many DEEs.
Prior to the identification of pathogenic human variants, the first direct link between DNM1 and severe epilepsy was a spontaneous missense mutation in the mouse ortholog, termed “fitful” (gene symbol, Dnm1Ftfl).11 This mutation occurs in a mutually exclusive alternate exon in the middle domain of Dnm1 that defines Dnm1a—which along with Dnm1b comprises two functionally semi-redundant isoforms of Dnm1. Peptides encoded by these very highly conserved exons form part of the assembly domain that is critical for oligomerization of dynamin monomers into ring structures that carry out endocytosis.7,11
Whereas Dnm1Ftfl/+ heterozygous mice show only mild spontaneous and handling-induced seizures from 2 to 3 months of age and have a normal lifespan,11 Dnm1Ftfl/Ftfl homozygotes show a DEE-like phenotype with severe ataxia, developmental delay, and fully penetrant lethal seizures by the end of the third postnatal week.4,6,11,12 While Dnm1b is expressed predominantly during gestation and expression wanes during early postnatal development, Dnm1a expression increases during early postnatal development and peaks during the second postnatal week, becoming the predominant isoform of adulthood.11 However, neither Dnm1a nor Dnm1b isoform-specific homozygous knockout mice (Dnm1Δa/Δa or Dnm1Δb/Δb) show seizures or other overt phenotypic characteristics associated with the Dnm1Ftfl allele.6,12 These and other in vivo and in vitro studies suggest that Dnm1Ftfl exerts a dominant-negative effect on protein function, as was modeled or predicted for all DNM1 pathogenic variants.4,6,7 The emerging successes of gene silencing therapies directed at specific variants or isoforms in animal models embolden the application of genetic-based therapies for this class of genetic diseases.13, 14, 15
Similar to the approach taken recently in a mouse model of peripheral neuropathy, where a virally delivered RNAi successfully prevented deleterious phenotypes associated with Charcot-Marie-Tooth type 2D (CMT2D),14 we developed a therapeutic microRNA (miRNA) that targets Dnm1a, the isoform that houses Dnm1Ftfl, and cloned it into self-complementary adeno-associated virus 9 (scAAV9) for in vivo delivery to neonatal mice. While the efficacy of AAV-mediated RNAi treatments has been established in other toxic genetic disease models,14, 15, 16 such treatments have not been previously applied to intractable DEE.
Here, we determined that a one-time bilateral intracerebroventricular injection of neonatal Dnm1Ftfl/Ftfl mouse pups reduced seizure severity, extended lifespan, improved growth, and abolished various developmental impairments. Treatment also greatly reduced or eliminated underlying cellular pathology. This study provides proof of principle for postnatal gene silencing to curb fundamental features of DNM1 DEE, with implied application to other early neurodevelopmental diseases caused by gain-of-function or dominant-negative genetic mechanisms.
Results
scAAV9-miDnm1a Selectively Inhibits Dnm1a
To knock down Dnm1Ftfl, we identified a miRNA that specifically and efficiently targets the Dnm1a isoform that harbors the fitful mutation.6,11,12 We first designed four different miRNA constructs (miDnm1a-1 through miDnm1a-4) and tested their efficacy in vitro using a dual-luciferase reporter assay (Figure 1A). miDnm1a-4 was the most effective at reducing the amount of Dnm1a mRNA (Figure 1A). We then packaged miDnm1a-4 into a scAAV9 virus vector (scAAV9-miDnm1a) for in vivo validation (Figure 1B). Mice were either administered the treatment (scAAV9-miDnm1a) or control (scAAV9-EGFP) via a one-time bilateral intracerebroventricular (i.c.v.) injection at postnatal day 0 (PND 0; see the Materials and Methods; Figures 1B and 1C). miDnm1a-treated mice showed significant reduction of Dnm1a mRNA 2 weeks postdelivery compared to scAAV9-EGFP mice (p < 0.0001), assessed by using qRT-PCR (Figure 1C). Expression of the other Dnm1 isoform, Dnm1b, was unaltered, reflecting the specificity of miDnm1a for the Dnm1a isoform and fitful silencing (p = 0.783; Figure 1C). Because all viral constructs express GFP, viral expression was verified histologically using an anti-GFP antibody (see the Materials and Methods). GFP expression was detected in the brains of scAAV9-miDnm1a- and scAAV9-EGFP-injected mice (Figure 1D).
Figure 1.

scAAV9-miDnm1a Selectively Inhibits Dnm1a
(A) Knockdown efficacy of Dnm1a in vitro by four different miRNA constructs. miDnm1a-4 was the most effective with 95% knockdown. (B) Experimental and control constructs delivered via i.c.v. injection at PND 0. The black boxes indicate the viral inverted terminal repeats, and pA indicates an SV40 polyadenylation signal. (C) Validation of Dnm1a knockdown efficacy in vivo by scAAV9-miDnm1a (n = 8) compared to scAAV9-EGFP control (n = 6) shows significant decrease of Dnm1a but not Dnm1b in whole-brain extracts from scAAV9-miDnm1a-treated mice (p < 0.0001 and p > 0.05 respectively; two-way ANOVA with Sidak’s correction for multiple comparisons). (D) Broad viral transduction of both Dnm1Ftfl/Ftfl and Dnm1+/+ scAAV9-miDnm1a-treated mice 30 days after i.c.v. injection. Images were taken at 10× magnification. Data reported as mean ± SEM. Scale bar on whole brain represents 500 μm, and scale bar of region of interest (ROI) represents 20 μm. n.s - not significant; ∗∗p < 0.0001.
scAAV9-miDnm1a Treatment Improves Developmental and Seizure Behaviors
To assess the efficacy of scAAV9-miDnm1a in extending survival and to identify and address any logistical constraints in the use of the construct, including viral delivery, we undertook a pilot study in the C57BL/6J (B6J) mouse strain background. Dnm1Ftfl/Ftfl mice experience severe and fully penetrant tonic-clonic seizures and comorbidities resulting in lethality by the third postnatal week, irrespective of mouse strain background.11 B6J-Dnm1Ftfl/Ftfl mice were treated with three doses of miDnm1a: 1 × 1010, 1.85 × 1011, 3.25 × 1011 vector genomes (vg). Treatment of Dnm1Ftfl/Ftfl mutants extended survival in a dose-dependent manner: 30% (p = 0.0001) and 50% (p = 0.01) of mice treated with the latter two doses, respectively, survived to PND 30 and showed growth improvements (p = 0.003; Figures S1B and S1C). The highest dose at 3.25 × 1011 vg did not produce overt adverse effects, suggesting that similar or higher doses could be used in a full experiment.
To further evaluate the effectiveness of miDnm1a, we employed (B6J × FVB/NJ)F2 hybrid background mice because of their large litter size, animal size, and good maternal care. Dnm1Ftfl/Ftfl and Dnm1+/+ F2 hybrids were either treated (scAAV9-miDnm1a) or control injected (i.e., scAAV9-EGFP or saline; see Materials and Methods; Figure 2A) at PND 0. Mice were observed for survival, seizure activity, and weight until PND 30, the chosen endpoint for this study (Figure 2A). While control-injected Dnm1Ftfl/Ftfl mice, similar to prior observation,11 did not survive past PND 19 (n = 27; Figure 2B), 88% of treated mice lived past PND 20, and 75% (n = 25) survived until PND 30 (p = 7.5 × 10−10; Figure 2B). These data show the effectiveness of miDnm1a at extending survival of Dnm1Ftfl/Ftfl mice considerably.
Figure 2.

scAAV9-miDnm1a Treatment Improves Survival, Growth, and Seizure Outcomes
(A) Experimental design with i.c.v. injection administered at PND 0, developmental phenotyping executed between PND 4 and PND 11, survival, seizure, and growth measurement assessed from PND 4–PND 30 (the endpoint of the study) and cellular phenotyping performed at PND 18 and PND 30. (B) Treatment with miDnm1a led to 75% survival of Dnm1Ftfl/Ftfl mice (n = 25) to PND 30 compared to control-injected mice (EGFP or saline, n = 27), which were 100% lethal before PND 20 (p < 0.0001, log-rank Mantel-Cox test). Treated Dnm1Ftfl/Ftfl mice differed from treated Dnm1+/+ (n = 24) or control-injected Dnm1+/+ (n = 19) mice (p = 0.0239, p = 0.0097, respectively; log-rank Mantel-Cox test). (C) Although treated and control-injected Dnm1Ftfl/Ftfl mice were notably smaller as early as PND 8, miDnm1a-treated Dnm1Ftfl/Ftfl showed growth improvement beginning at PND 12. Repeated-measures ANOVA was performed until PND 18 when control-injected Dnm1Ftfl/Ftfl mice exited the study, including genotype-treatment effects (combining the two control treatments, EGFP and saline), plus other independent variables including sex, virus dose, and litter size. For treated versus control-injected Dnm1Ftfl/Ftfl, the effect of treatment was highly significant (p = 3.3 × 10−13), despite a significant effect of litter size (p = 1.4 × 10−7) but no significant impact of virus dose or sex. Growth differences at the PND 30 study endpoint between treated Dnm1Ftfl/Ftfl and treated wild-type were significant (p = 0.004), with a modest effect of litter size (p = 0.048). Using similar analysis, treated wild-type mice also showed growth delay compared to control wild-type (p = 0.004), with a modest effect of sex (p = 0.01) and litter size (p = 0.033). (D) Both miDnm1a-treated and control-injected Dnm1Ftfl/Ftfl mice show seizure-like behavior; however, control-injected Dnm1Ftfl/Ftfl mice had significantly more seizures at PND 14 and PND 18. Seizure behaviors of treated mice decreased over time. See Table 1 for sample numbers and analysis. Data reported as mean ± SEM. See also Figure S1.
As shown from prior studies,11 control-injected Dnm1Ftfl/Ftfl mice started showing growth deficits from PND 8 until moribundity or a terminal seizure event by PND 18–19 (Figures 2B and 2C). In contrast, treated Dnm1Ftfl/Ftfl mice grew steadily until PND 30, although they lagged behind treated and control-injected Dnm1+/+ mice starting at PND 8 and PND 18, respectively (Figure 2C). Overall, there was a significant growth improvement in treated Dnm1Ftfl/Ftfl mice compared to control-injected Dnm1Ftfl/Ftfl by PND 18 (repeated-measures ANOVA; p = 3.3 × 10−13). We also note that even treated Dnm1+/+ mice showed some growth delay starting at PND 8 compared to control-injected Dnm1+/+ (p = 0.004; Figure 2C). Although germline Dnm1a null mice were previously reported to lack any overt impairments,6,12 it is plausible that modest growth delay as seen in treated Dnm1+/+ is a feature of postnatal Dnm1a elimination. Alternatively, the growth deficits observed in treated Dnm1+/+ mice could be due to unpredictable off-target effects of miDnm1a. Regardless, these data reinforce the effectiveness of miDnm1a in improving growth outcomes of Dnm1Ftfl/Ftfl mice.
To determine the effect of miDnm1a on seizure phenotypes, treated and control-injected Dnm1Ftfl/Ftfl homozygotes were assessed for overt seizure and seizure-associated activity, including wild runs, Straub tail, continuous vertical jumping lasting for more than 10 s with subsequent facial grooming, continuous jerking of the limbs, and full-blown tonic-clonic seizures. These assessments were done on alternate days, starting at PND 14 during weight examination sessions. While both treated and control-injected Dnm1Ftfl/Ftfl mice exhibited seizure behaviors, treated mice had fewer overall observed events between PND 14 and 18 (Figure 2D; Table 1). By PND 18, all control-injected Dnm1Ftfl/Ftfl mice were moribund and incapable of staying upright. Treated Dnm1Ftfl/Ftfl mice showed handling seizures between PND 14 and 24 (Table 1). After this period, the number and intensity of their observed seizure and seizure-like events decreased (Figure 2D; Table 1). These data suggest that miDnm1a treatment decreased seizures and seizure-associated activity in Dnm1Ftfl/Ftfl mice.
Table 1.
Observed Seizure or Seizure-Associated Behaviors in Treated or Control-Injected Dnm1Ftfl/Ftfl Mice
| Postnatal Day | Group | Positive | Negative | Total | % Positive | p Valuea |
|---|---|---|---|---|---|---|
| 14 | control | 25 | 19 | 44 | 56.8% | 0.017 |
| miDnm1a | 8 | 21 | 29 | 27.6% | ||
| 16 | control | 24 | 19 | 43 | 55.8% | 0.156 |
| miDnm1a | 11 | 18 | 29 | 37.9% | ||
| 18 | control | 39 | 3 | 42 | 92.9% | 2 × 10−6 |
| miDnm1a | 12 | 17 | 29 | 41.4% | ||
| 20 | miDnm1a | 5 | 19 | 24 | 20.8% | n.a. |
| 22 | miDnm1a | 9 | 12 | 21 | 20.8% | n.a. |
| 24 | miDnm1a | 7 | 13 | 20 | 35.0% | n.a. |
| 26 | miDnm1a | 3 | 17 | 20 | 15.0% | n.a. |
| 28 | miDnm1a | 1 | 19 | 20 | 5.0% | n.a. |
| 30 | miDnm1a | 1 | 19 | 20 | 5.0% | n.a. |
Fisher exact test, two-tail, 2 × 2 contingency analysis. Treated Dnm1Ftfl/Ftfl mice show fewer seizures compared to control-injected Dnm1Ftfl/Ftfl mice, and this is statistically significant at PND 14 and PND 18. At PND 18, four treated mice were removed from the study for histological analysis. n.a. - not applicable.
To determine the outcome of miDnm1a treatment on neurodevelopmental phenotypes, strength, sensorimotor development, and gait were assayed (see the Materials and Methods). Treated and control-injected Dnm1Ftfl/Ftfl homozygotes were tested for grip strength, an assay whereby mouse pups at PND 9 and PND 11 were placed on a vertical screen mesh and latency to fall recorded. Treated Dnm1Ftfl/Ftfl mice (n = 30) showed improved grip strength compared to control-injected Dnm1Ftfl/Ftfl mice (n = 28) at PND 11 (p = 0.0009; Figure 3A). However, treated Dnm1Ftfl/Ftfl mice did not differ in grip strength from treated (n = 24) or control-injected (n = 19) Dnm1+/+ at PND 9 and PND 11 (p > 0.05; Figure 3A). We then evaluated possible sensorimotor impairment using the negative geotaxis assay, which challenges the innate behavior in mice to utilize vestibular cues for motor coordination.17,18 Mice were placed on a 45° incline with heads pointing downward, and latencies to turn 90° and 180° were recorded. Unlike control-injected Dnm1Ftfl/Ftfl mice, treated Dnm1Ftfl/Ftfl mice, like wild-type controls, tended to exhibit a shorter latency to turn 90° and 180° at PND 11, but only the more challenging 180° turn was statistically significant compared with control-injected Dnm1Ftfl/Ftfl mice (p = 0.0004; Figures 3B and 3C). We further assessed gait impairments since ataxia is a prominent phenotype of Dnm1Ftfl/Ftfl mice.11 We recorded PND 14 treated and control-injected Dnm1Ftfl/Ftfl and Dnm1+/+ mice for 10-min intervals using automated software and counted the number of falls and wobbles (see the Materials and Methods). As expected, control-injected Dnm1Ftfl/Ftfl mice had numerous ataxic events (overtly wobbly gait and falls), but miDnm1a treatment completely abolished this ataxic phenotype observed in the control-injected Dnm1Ftfl/Ftfl mice (p < 0.0001; Figure 3D; Video S1). We also quantified ambulation during this time but found no difference in either distance traveled or velocity between groups (p > 0.05; Figures 3E and 3F). These results show that miDnm1a treatment improved the developmental outcomes of Dnm1Ftfl/Ftfl mice.
Figure 3.

scAAV9-miDnm1a Treatment Improves Developmental Outcomes
(A) Treatment significantly improved the grip strength at PND 11 of Dnm1Ftfl/Ftfl mice (n = 30) compared to control-injected (n = 28) mice (p = 0.0009, least-squares regression using rank- and normal-quantile transformed data), with no effect of litter size, sex, or virus dose. Treated Dnm1Ftfl/Ftfl mice did not differ from treated (n = 24) or control-injected (n = 19) Dnm1+/+ mice (same test as above with Tukey’s HSD post hoc test, p > 0.05). (B and C) In an assay for sensorimotor development, at PND 9 and PND 11, control-injected Dnm1Ftfl/Ftfl mice had a higher latency, albeit not significant for the easier 90° turn (B) (p > 0.05, least-squares regression using rank- and normal-quantile transformed data, Dunnett’s post hoc test). However, for the more difficult 180° turn (C), control-injected Dnm1Ftfl/Ftfl mice showed a significantly higher latency at PND 11 compared to the other three groups (p < 0.001, Dunnett’s post hoc test). (D) Control-injected Dnm1Ftfl/Ftfl mice (n = 24) show severe ataxia; importantly, treatment with miDnm1a (n = 17) eliminates this phenotype (p = 6.9 × 10−17, log-Poisson test) and restores Dnm1Ftfl/Ftfl motor coordination back to the level of treated (n = 16) and control-injected (n = 23) Dnm1+/+ mice (p = 0.19 mixed model log-Poisson test). (E and F) Using locomotion (E) and velocity (F) as a proxy for possible hyperactivity, we observed that there was no significant difference between all groups (p > 0.05, one-way ANOVA). ∗p < 0.01; ∗∗p < 0.001; ∗∗∗∗p < 0.00001.
In conclusion, treated Dnm1Ftfl/Ftfl mice showed extended survival, improved growth, decreased lethal seizures, improved developmental outcomes, and an absence of ataxia. These data together show the effectiveness of miDnm1a at curbing or eliminating the most severe fitful behavioral phenotypes including seizures, growth, and ataxia, culminating in an overall improvement in their quality of life and survival to the endpoint.
miDnm1a Treatment Improves Cellular Pathology
The significant mitigating effect of Dnm1Ftfl mRNA silencing on severe seizures and impaired neurodevelopment prompted us to examine the extent to which treatment impacts underlying cellular pathology. Gliosis and neuronal cell death are hallmarks of neuronal insults and recurrent seizure activity,19,20 features not previously examined in the published studies on the Dnm1Ftfl mouse model. Here, we investigated the presence of gliosis and cell degeneration via immunolabeling using Glial Fibrillary Acidic Protein (GFAP) and Fluoro-Jade C (FJC). At PND 18, control-injected Dnm1Ftfl/Ftfl mice showed intense fibrillary gliosis (GFAP) in the hippocampus, specifically around CA1, compared to treated Dnm1Ftfl/Ftfl mice (p = 0.018), indicative of a hippocampus under stress (Figure 4A). In contrast, treated Dnm1Ftfl/Ftfl mice did not show gliosis, and their hippocampi did not differ from wild-type controls (p > 0.05; Figure 4A; Figure S2A). By the study endpoint at PND 30, surviving treated Dnm1Ftfl/Ftfl mice showed an increase in GFAP intensity in the hippocampus compared to treated and control-injected Dnm1+/+ mice (p = 0.0059 and p = 0.0072, respectively; Figure 4B; Figure S2A). FJC labeling of control-injected Dnm1Ftfl/Ftfl mice revealed striking cellular degeneration in the hippocampus and more so in the CA1 region compared to treated Dnm1Ftfl/Ftfl mice (p = 0.015; Figure 4C; Figure S2B). This cell death phenotype was significantly diminished in treated Dnm1Ftfl/Ftfl mice that did not differ from control-injected Dnm1Ftfl/Ftfl mice (Figure 4C). By the PND 30 endpoint, treated Dnm1Ftfl/Ftfl mice showed some cell death, however, not significantly more compared to treated and control-injected Dnm1+/+ mice (Figure 4D; Figure S2B). These results revealed a susceptibility of the hippocampal neurons to Dnm1Ftfl. Additionally, these results showed that miDnm1a treatment was successful at curbing or delaying gliosis and cellular degeneration in the hippocampus of Dnm1Ftfl/Ftfl mice at PND 18 and PND 30. (Figures 4A–4D; Figures S2A and S2B).
Figure 4.

miDnm1a Treatment Diminishes Gliosis and Cellular Degeneration at PND 18 until PND 30
(A) Control-injected Dnm1Ftfl/Ftfl mice showed strikingly increased gliosis specifically in the hippocampal CA1, as identified with the marker GFAP compared to treated Dnm1Ftfl/Ftfl mice (p = 0.018). Treated Dnm1Ftfl/Ftfl mice did not differ from treated and control-injected Dnm1+/+ mice (p > 0.05) at PND 18. (B) By PND 30, treated Dnm1Ftfl/Ftfl showed significantly more GFAP intensity compared to treated and control-injected Dnm1+/+ mice (p = 0.0052 and p = 0.0072, respectively). Images (A and B) were taken at 10× magnification, and analysis was done using an ordinary one-way ANOVA followed by Tukey’s multiple comparisons test. Scale bar of entire hippocampus represents 200 μm, and region of interest (ROI) scale bar represents 20 μm. (C and D) FJC labeling at PND 18 (C) showed significant cell death in the hippocampus of control-injected Dnm1Ftfl/Ftfl mice, specifically along the CA1, unlike treated Dnm1Ftfl/Ftfl mice (p = 0.015). Treated Dnm1Ftfl/Ftfl mice did not differ from treated and control-injected Dnm1+/+ mice (p > 0.05). By PND 30 (D), treated Dnm1Ftfl/Ftfl mice had little apparent cell death as identified by FJC labeling. However, they did not differ from treated and control-injected Dnm1+/+ mice (p > 0.05). Images were taken at 10× magnification, and scale bars correspond to 100 μm. Analyses were executed using the Poisson overdispersion option in the GMLJ module of Jamovi’s software. 3–5 mice were used in these analysis and data are reported as mean ± SEM. ∗p < 0.05; ∗∗p < 0.01. See also Figure S2.
Given the increased gliosis and cellular degeneration observed in the hippocampus of control-injected Dnm1Ftfl/Ftfl mice, we examined hippocampal involvement further by using cellular footprints of metabolic activity typically associated with recurrent limbic seizure behavior. We examined Neuropeptide Y (NPY), as upregulation of NPY in the hippocampus is a known method of evidencing hyperexcitability in rodent models of epilepsy.21,22 We observed aberrant cellular NPY expression in the hippocampal cornu ammonis areas, specifically in CA3, of control-injected Dnm1Ftfl/Ftfl mice compared to treated Dnm1Ftfl/Ftfl mice at PND 18 (p = 0.0012; Figure 5A; Figure S3A). Treated Dnm1Ftfl/Ftfl mice, however, showed NPY expression similar to treated and control-injected Dnm1+/+ mice at PND 18 (p > 0.05; Figure 5A; Figure S3A). By PND 30, treated Dnm1Ftfl/Ftfl mice still did not differ from treated and control-injected Dnm1+/+ mice, although treated Dnm1Ftfl/Ftfl mice tended to have more aberrant NPY+ cells in the CA3 (p > 0.05; Figure 5B; Figure S3A). Additionally, we immunostained for the immediate-early gene marker of neuronal activity, c-Fos.23 We observed a significant increase in c-Fos+ cells in the hippocampus, specifically in the CA3 of control-injected Dnm1Ftfl/Ftfl mice (Figure 5C; Figure S3B). In contrast, this increased neuronal activity was abated in treated Dnm1Ftfl/Ftfl mice at PND 18 (p < 0.00001) and persisted until PND 30 (Figures 5C and 5D; Figure S3B). Moreover, treated Dnm1Ftfl/Ftfl mice did not differ from treated and control-injected Dnm1+/+ mice at PND 18 or PND 30 (p > 0.05; Figures 5C and 5D; Figure S3B). These results suggest that miDnm1a treatment decreased the abnormal cellular metabolic activity of Dnm1Ftfl/Ftfl mice.
Figure 5.

miDnm1a Treatment Improves Metabolic Cellular Activity at PND 18 until PND 30
(A) Aberrant NPY expression was observed in the hippocampus and specifically in the CA3 of control-injected Dnm1Ftfl/Ftfl mice at PND 18; treatment with miDnm1a reverted this phenotype (p = 0.0012). Treated Dnm1Ftfl/Ftfl mice did not differ from treated and control-injected Dnm1+/+ mice (p > 0.05). (B) NPY expression varied among treated Dnm1Ftfl/Ftfl mice at PND 30. However, treated Dnm1Ftfl/Ftfl mice trended toward significance compared to treated Dnm1+/+ mice (p = 0.057) and control-injected Dnm1+/+ mice (p = 0.074). (C) At PND18 c-Fos staining showed increased neuronal activation in the hippocampal CA3 of control-injected Dnm1Ftfl/Ftfl mice, which was significantly diminished in treated Dnm1Ftfl/Ftfl mice (p < 0.00001). Treated Dnm1Ftfl/Ftfl mice did not differ from Dnm1+/+ controls (p > 0.05). (D) By PND 30, there was a modest increase in neuronal activation in the hippocampus, specifically in the CA3 region, of treated Dnm1Ftfl/Ftfl mice compared to Dnm1+/+controls, but this increase was not significant (p > 0.05). All images (A–D) were taken at 10× magnification. Scale bar of entire hippocampus represents 200 μm and ROI scale bar represents 20 μm. Analyses were executed using the Poisson overdispersion option in the GMLJ module of Jamovi’s software. 3–5 mice were used in these analysis, and data are reported as mean ± SEM. ∗∗p < 0.01; ∗∗∗p < 0.0001. See also Figure S3.
Overall, unlike control-injected Dnm1Ftfl/Ftfl mice, treated Dnm1Ftfl/Ftfl mice showed a decrease in gliosis, cell death, and aberrant neuronal activity at PND 18, which persisted and led to their survival until the PND 30 endpoint. These results together show that miDnm1a treatment diminished the cellular pathology associated with the Dnm1Ftfl mutation.
Discussion
Here, we examined an RNAi-based gene therapy approach in an animal model of severe childhood epilepsy. The preclinical features of Dnm1 fitful mice are representative of many genetic gain-of-function or dominant-negative DEE, which represent the most severe forms.24 Similar to a recently described approach in the Gars model of a genetic peripheral neuropathy,14 we used a miRNA that specifically targets the pathogenic Dnm1Ftfl isoform, packaged it into a self-complementary AAV9 vector, and delivered it via intracerebroventricular injection of neonatal mice. We determined that a single treatment eliminated ataxia, improved growth and development, and decreased lethal seizures, thus improving survival at least to the endpoint of our study at postnatal day 30. We also observed significant accompanying improvements in cellular pathology.
Although treatment rendered the seizures survivable, eliminated the ataxia, greatly reduced underlying cellular pathology, and improved animal growth, miDnm1a treatment did not completely eliminate seizures, as mutants still had observable seizures prior to weaning age, nor was 100% survival achieved. Nevertheless, compared with most other DEE models, it is critical to note that the Dnm1 model is exceptionally debilitating, culminating in 100% moribundity or lethality before the third postnatal week, and yet the RNAi approach led to 75% survival until at least PND 30. Though a 30-day study endpoint was set to mark the end of the developmental stage and without expectations of the kind of success observed, the apparent vigor of treated mutants suggests even longer survival—a focus of future experiments. Regardless, our results highlight the significant potential of RNAi therapy for rescuing the developmental phenotypes and decreasing the early-onset seizure occurrence associated with DEE. It is possible that a higher viral dose might have led to a more complete rescue, but experimental logistics precluded this. However, our hypothesis is that AAV9 delivery, even as early as the first postnatal day for mice, was unable to fully compete against disease pathogenesis already in rapid progress. That is, gene expression from self-complementary AAV9 is not instantaneous—gene expression begins 4 days after transduction and peaks at 14 days,25 lagging behind Dnm1a peak expression—but Dnm1Ftfl/Ftfl-associated symptoms are already evident by the end of the first postnatal week. Overall, despite limitations intrinsic to an initial study of this type, the results demonstrate the potential effectiveness of virally delivered RNAi as a strategy for treating DEE where it is necessary to block or eliminate, rather than replace, the abnormal gene product.
RNAi therapy presents a promising and perhaps necessary avenue for treating such debilitating diseases. Indeed, this approach is currently being used to treat various other diseases caused by dominant-negative or gain-of-function mutations in both mouse models and humans with success.13, 14, 15 Very recently, the RNAi therapy Patisiran was approved for treating hereditary transthyretin amyloidosis, and more similar therapies are likely to follow.13 Such advances together with our results and recent success in using AAV as a delivery system for treating various neurological diseases14, 15, 16 suggest that RNAi therapy for the treatment of DEEs caused by dominant-negative or gain-of-function mutations is directly translationally feasible. Although the lag in AAV9 gene expression may be a limitation for treating this early severe DEE in mice, one would expect that the larger therapeutic window of human postnatal development compared to mouse would facilitate a more complete therapeutic response. Furthermore, given that human mutations are heterozygous, for some mutations, RNAi would allow for allele-specific silencing, leaving the wild-type copy intact, as was the case in the Gars model.14 There are also alternative albeit more transient therapy vehicles, such as antisense oligos (ASOs)26 that may be more suited for instantaneous results, better control of dosage, and the potential for stopping treatment if side effects manifest. This, however, means that patients will require multiple “booster” applications, whereas AAV9 is expected to be more robust and longer-term, as vector expression is persistent.26,27
In summary, this study serves as a strong proof-of-principle of the efficacy of RNAi therapy for the treatment of DNM1 DEE with potential broad application to other dominant-negative or gain-of-function DEEs. Indeed, even conversion of a severely debilitating, untreatable disease to a more manageable form of epilepsy would immensely improve the quality of life and be transformative for patients and their families.
Materials and Methods
Study Design
Experiments were performed blinded to genotype and treatment and randomized as appropriate. PND 30 was selected prospectively as the adult endpoint because pups are typically weaned between PND 22 and PND 28, marking the end of the developmental period. To conform to institutional compliance, data collection from live Dnm1Ftfl/Ftfl mice was stopped upon moribundity. For 95% confidence in detecting improvement from inhibition of mutant Dnm1a, at least 12 animals per group (genotype × treatment, sexes combined) were necessary to detect an effect with 80% power. For the primary study, to assess a possible dose effect, two experiments were executed in the F2 hybrid background, representing two viral doses: ∼5.2 × 1011 vg and ∼6.0 × 1011 vg. Mice were randomly assigned to either miDnm1a treatment or control condition, which included EGFP or saline administration; in analysis for simplicity, the two control conditions were combined since they did not differ in effect. Thus, for analysis when modeling treated versus control-injected groups, we combined as covariates the two dose experiments and sexes (using categorical indicator variables for each) and litter size. Cellular analysis was executed at PND 18, because that is the time point by which almost all untreated or control-injected mice become moribund, and PND 30, the endpoint of the study. For this study, we show three representative replicates for the cellular characterization of the effect of miDnm1a treatment.
Animals and Genotyping
C57BL/6J-Dnm1Ftfl-flox mice used for these studies (hereafter termed B6J-Dnm1Ftfl) were generated in Dnm1Δ1a/Δ1a mice by The Jackson Laboratory’s Genetic Engineering Technology core, introducing the Dnm1Ftfl mutation using CRISPR/Cas9. To generate interstrain F2 hybrid mice, B6J-Dnm1Ftfl/+ mice were mated to FVB/NJ females (JAX, Bar Harbor, ME, USA; stock 004624), and F1 hybrid mice mated inter se. Mice were genotyped with a PCR protocol designed to detect the presence of loxP sites in the fitful allele. The genotyping primers (5′-CCTCTCTGTCCACTTGTAGCCATT-3′ and 5′-ACTGGGTGATGCTCACTAGAACCT -3′) produce a 321-bp mutant allele and a 215-bp wild-type allele. Mice for this study were between 0 and 30 days old. To identify individual pups, they were tattooed at PND 0 according to the AIMS pup tattoo identification system (Budd Lake, NJ, USA) using Ketchum animal tattoo ink (cat. #329AA). Mice were also ear notched at PND 10. Postweaning at PND 21, mice had access to food and water in their home cages ad libitum. Pups were never separated from their home cage for more than 10 min at a time. Lights were maintained on a 12-h light/dark cycle with behavioral testing occurring during the light portion of the cycle. All procedures were approved by Columbia University’s Institutional Animal Care and Use Committee and were performed in accordance with the National Institute of Health guide for the care and use of laboratory animals.
Dnm1a miRNA Production and Viral Packaging
Four miRNA sequences potentially targeting Dnm1a were designed and cloned into a mir-30-based construct with expression driven by the U6 promoter, as previously described.15 Qualifying miRNAs had to be 22 nt long, with the first four and last four nt being at least 75% GC rich and AU rich, respectively. Additionally, the string of 22 nt had to be 40% AU rich. miRNA-specific targeting of Dnm1a capitalizes on the 42-nt difference between Dnm1a and Dnm1b. The full miRNA sequence is 5′-cucgagugagcgaaccaucagaaaguguagugaacu guaaagccacagauggguucacuacacuuucugauggugugccuacuaga-3′. The antisense strand of the mature miRNA (red) binds Dnm1a mRNA. After in vitro testing, the lead candidate U6 miRNA was cloned into a self-complementary adeno-associated viral vector and packaged as serotype 9 (scAAV9). Vectors contained a cytomegalovirus (CMV) promoter-driven EGFP reporter. scAAV9 vectors were generated and titered by the Harper lab and the Viral Vector Core at Nationwide Children’s Hospital.
i.c.v. Injection of scAAV9-miDnm1a
For i.c.v. delivery of scAAV9-miDnm1a, PND 0 mice were anesthetized using hypothermia by being placed on a chilled metal block until properly anesthetized.28 The injection site was approximately 2/5 the distance from the lambda suture to each eye.28 All injections were executed free hand using a point style 4, 33G needle and a 10 μL or 25 μL Hamilton syringe (cat. #65460-06 and cat. #65460-10) for escalating doses of scAAV9-miDnm1a. F2 mice were treated with ∼5.2 × 1011 vg to 6.0 × 1011 vg per mouse based on the titers acquired. B6J mice were treated with varying doses (1 × 1010, 1.85 × 1011, and 3.25 × 1011 vg) that correspond to volumes of ∼6–13 μLs in an effort to establish possible dosage effects. Control scAAV9-EGFP injections were matched to miDnm1a dosage, and saline controls were matched to the volume of virus injected.
In Vivo Quantification of Transduction and Dnm1a Knockdown
Dnm1+/+ mice were treated with miDnm1a or EGFP at PND 0. Whole brain was isolated from six EGFP- and eight miDnm1a-injected mice at PND 14. The tissues were flash frozen with 2-methylbutane and stored at −80°C. Samples were homogenized using a dounce, and RNA was isolated using TRIzol Reagent (Thermo Fisher, Waltham, MA, USA; cat. #15596018). RNA was converted to cDNA using Invitrogen SuperScript III first-strand synthesis system (Carlsbad, CA, USA; cat. #18080051). Dnm1a knockdown was assessed using the primers (5′-CTCGCTTTTGAAGCCACAGT-3′ and 5′-GAGTGCAGGTGGTAGTCTTT -3′). Dnm1b expression was evaluated using the primers (5′-GGCCTTTGAAACCATTGTGA-3′, and 5′-AGTCGTGCCAATCTGTCACG -3′). SYBR select master mix (Applied Biosystems, Waltham, MA, USA; cat. #4472903) was used for qPCR, which was run using Applied Biosystems-Quant Studio 5.
Pup Developmental Milestones and Behaviors
Developmental milestones in pups are important readouts of developmental delays. To begin the test, each pup was gently removed from the nest and placed on a clean piece of bench protector. The cage lid was immediately and gently placed back to reduce agitation in the nest. All assessment was completed by a deft experimenter within 3 min. At the end of the session, the pup was quickly returned to the nest. For all behaviors, mice from both groups (genotype × treatment) were handled every day from PND 4–12. On all even days (PND 4 to PND 12) they were weighed, and on odd days (PND 5 to PND 11), they were assessed for strength and sensorimotor development. From PND 14 to PND 30, mice were weighed on every other day on even days. Testing was executed blind.
Negative Geotaxis
Mice are placed head down on a mesh screen set at a 45° angle. The latency for the pups to turn 90° (sideways) and 180° (heads up) from a downward facing start position was recorded.17,18,29 Mice were given 30 s to perform the task and were allowed two attempts. Both attempts were averaged for the analysis.
Vertical Screen Hold
To evaluate strength, mice were placed on a vertical mesh screen, and their latency to fall off the screen was recorded. Mice were observed for 30 s and were allowed two attempts to complete the task. The scores from both trials were averaged. Mice were tested at PND 9 and PND 11 because the average age of a rodent to perform this task is PND 8.17
Open Field
To quantify the ataxic phenotype, we observed PND 14 pups in the open field using the EthoVision XT video tracking software (Noldus). Mice were tested in a 28.5-cm arena with luminescence of 100 lux. Mice were recorded for 10 min. Videos were subsequently scored blind by counting the number of times each mouse wobbled or fell during movement over a 10-min window. Mice that moved less than 100 cm were excluded. Additionally, distance traveled and velocity of each mouse were quantified.
Growth and Survival Monitoring
Between PND 4 and PND 30, mice were weighed every other day. In addition, general health and survival were monitored every day from PND 0 to PND 30 (study endpoint).
Seizure Quantification
Each litter of pups was monitored for approximately 5 min during weighing. Seizure activity was characterized by wild runs, Straub tail, vertical jumps that lasted more than 10 s with subsequent facial grooming, continuous jerking of the forelimbs and hindlimbs, and tonic-clonic seizures. All seizures within the observation window were counted as one event.
Histology
At least 3–5 mice from each group (genotype, Dnm1Ftfl/Ftfl and Dnm1+/+, × treatment, miDnm1a and EGFP) were perfused with 4% paraformaldehyde (PFA) for immunohistochemical assessment at PND 18. We also evaluated Dnm1Ftfl/Ftfl miDnm1a mice surviving until PND 30 and Dnm1+/+ controls. All animals were handled in the same way prior to euthanasia. Brains were dissected from the skull and postfixed in 4% PFA overnight at 4°C. Brains were transferred to gradient concentrations of sucrose (15% and 30%) overnight at 4°C. Once saturated, the brains were embedded in Tissue-Tek® (Fisher Healthcare, cat. #4585) and frozen. Free-floating 40-μm sections were collected using a cryostat (Leica CM3050S). The sections were permeabilized with 0.1% Triton X in PBS for 30 min and blocked with 5% normal goat serum in PBS (blocking buffer) for 1 h at room temperature. Slices were incubated with either anti-Neuropeptide Y (NPY) (1:500, ImmunoStar, Hudson, WI, USA; cat. #22940), anti-c-Fos (1:250, Abcam, Cambridge, UK; cat. #ab190289), anti-GFP (1:250, Invitrogen, Carlsbad, CA, USA; cat. #A11120), or anti-glial fibrilary acidic protein (GFAP) (1:750, Sigma, St. Louis, MO, USA; cat. #M4403) in blocking buffer overnight at 4°C. Afterward, sections were washed in PBST (PBS with 0.1% Triton X) for 10 min and incubated in Alexa Fluor secondary 555 (1:1,000, Thermo Fisher Scientific, Waltham, MA, USA; ref. #A31428 and ref. #A32727) for 2 h at room temperature. Sections were incubated in DAPI for 5 min before washing with PBS. Sections were finally mounted on slides and coverslipped with fluoromount-G (Southern Biotech, Birmingham, AL, USA; cat. #0100-01).
For FJC labeling, 3–5 PND 18 and PND 30 mice (genotype × treatment) were euthanized, and their brains were dissected and flash frozen. 20-μm-thick sections were collected with a cryostat and mounted on gelatin-coated slides (FD NeuroTechnologies, Columbia, MD, USA; cat. #PO101). Sections were postfixed with 2% PFA and washed in PBS. Slides were air-dried on a 50°C heating block for 20–30 min and subsequently placed in 80% ethanol solution consisting of 1% NAOH for 5 min. Slides were moved to 70% ethanol for 2 min, rinsed in deionized water for 2 min, incubated in 0.06% potassium permanganate in deionized water for 10 min, rinsed for 2 min in deionized water, and finally incubated in FJC working solution consisting of 0.0001% FJC in 0.1% acetic acid for 10 min. Finally, slides were washed three times for 2 min each in deionized water, air-dried completely, and cleared in xylene for at least 1 min before they were coverslipped with mounting media (DPX, St. Louis, MO, USA; cat. #06522).30 Slides were imaged using a Zeiss LSM-800 confocal microscope and Zen v2.3. Tiled images of 10× magnification were acquired of the hippocampus for each section keeping the laser and gain settings constant. For FJC, the Axio X-Cite series 120 Q epifluorescence microscope was used, and images of 10× magnification were acquired. Postprocessing of images was carried out with Adobe Photoshop. All image processing was kept consistent.
Quantification
IHC images were quantified blind using ImageJ (Fiji; www.nih.gov). Images were converted to 8-bit and brightness and contrast keep constant. The cell counter plugin was used to count aberrant NPY and c-Fos-positive cells in the whole hippocampal CA3 and to count FJC-positive cells in the hippocampal CA1. GFAP fluorescence intensity was determined using the measure tool with the region of interest (ROI) in the CA1. Measurements were set to include area, min and max gray value, and integrated density. For fluorescence intensity measurement, background intensity was collected. Relative fluorescence was calculated by first subtracting the integrated intensity of the background from the integrated intensity of the CA1 ROI to yield a background-corrected intensity. Area was kept constant across all intensity measures. The corrected intensities of the wild-type control group were then averaged. Each corrected intensity was divided by the average of the wild-type control corrected intensity and multiplied by 100.
Statistical Analysis
Statistical analysis was done using either Prism 8 software (GraphPad), JMP 14 software (JMP), depending on the test. qPCR data (Figure 1) were analyzed using two-way ANOVA (Prism 8). Survival analysis for the pilot study (Figure S1) was analyzed using the log-rank Mantel-Cox test (Prism 8), and for the primary study (Figure 2), the proportional hazards test with risk ratios was used to accommodate covariates (JMP 14). Growth (Figure 2; Figure S1) was analyzed using repeated-measures MANOVA (JMP). Seizure-like behaviors (Figure 2) were analyzed using 2 × 2 Fisher exact test (JMP 14). Grip strength and negative geotaxis (Figure 3) were analyzed using least-squares regression after rank- and normal-quantile transformation of the data (JMP). Ataxia (counts of falls and wobbles) was analyzed using the log-Poisson test and contrast modeling to compare specific groups (JMP 14). IHC quantifications were analyzed using the Poisson overdispersion option in the generalized analysis for linear models module (gamlj) in Jamovi.31 p values were Bonferroni adjusted.
Author Contributions
W.N.F., S.Q.H., and O.V.A. contributed to experimental design, data interpretation, data analysis, and manuscript preparation. N.K.P. made the miDnm1 constructs and performed the initial screening, and A.M.F. prepared subsequent miDnm1a constructs for virus packaging. i.c.v. injections were performed by O.V.A. as well as RNA isolation and cDNA synthesis from mouse tissue. Behavioral phenotyping was performed by O.V.A. and A.K. under the supervision of M.Y. Counting of ataxic events was performed by A.K. and C.R.-W. Perfusions were performed by O.V.A. and S.P. Histology was performed by O.V.A., M.S., and J.J.T.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank the Viral Vector Core at Nationwide Children’s Hospital (NCH-VVC) for producing the AAV9 virus used in this study. We would also like to extend our gratitude to the Mouse Neurobehavior Core (MNBC) at CUIMC and Elizabeth Rafikian for setting up the EthoVision software and exporting the acquired data. Graphical abstract was created using BioRender. W.N.F. is funded by the NIH (R37 NS031348). O.V.A. is supported by a NIH predoctoral fellowship (F31 NS111808). S.Q.H. is funded by NIH (U54 OD020351).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.007.
Supplemental Information
References
- 1.Ferguson S.M., Brasnjo G., Hayashi M., Wölfel M., Collesi C., Giovedi S., Raimondi A., Gong L.W., Ariel P., Paradise S. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- 2.Marks B., Stowell M.H., Vallis Y., Mills I.G., Gibson A., Hopkins C.R., McMahon H.T. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature. 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- 3.Powell K.A., Robinson P.J. Dephosphin/dynamin is a neuronal phosphoprotein concentrated in nerve terminals: evidence from rat cerebellum. Neuroscience. 1995;64:821–833. doi: 10.1016/0306-4522(94)00337-5. [DOI] [PubMed] [Google Scholar]
- 4.Dhindsa R.S., Bradrick S.S., Yao X., Heinzen E.L., Petrovski S., Krueger B.J., Johnson M.R., Frankel W.N., Petrou S., Boumil R.M., Goldstein D.B. Epileptic encephalopathy-causing mutations in DNM1 impair synaptic vesicle endocytosis. Neurol. Genet. 2015;1:e4. doi: 10.1212/01.NXG.0000464295.65736.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.EuroEPINOMICS-RES Consortium. Epilepsy Phenome/Genome Project, Epi4K Consortium De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. Am. J. Hum. Genet. 2017;100:179. doi: 10.1016/j.ajhg.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asinof S.K., Sukoff Rizzo S.J., Buckley A.R., Beyer B.J., Letts V.A., Frankel W.N., Boumil R.M. Independent Neuronal Origin of Seizures and Behavioral Comorbidities in an Animal Model of a Severe Childhood Genetic Epileptic Encephalopathy. PLoS Genet. 2015;11:e1005347. doi: 10.1371/journal.pgen.1005347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Spiczak S., Helbig K.L., Shinde D.N., Huether R., Pendziwiat M., Lourenço C., Nunes M.E., Sarco D.P., Kaplan R.A., Dlugos D.J., Epi4K Consortium. EuroEPINOMICS-RES NLES Working Group DNM1 encephalopathy: A new disease of vesicle fission. Neurology. 2017;89:385–394. doi: 10.1212/WNL.0000000000004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brereton E., Fassi E., Araujo G.C., Dodd J., Telegrafi A., Pathak S.J., Shinawi M. Mutations in the PH Domain of DNM1 are associated with a nonepileptic phenotype characterized by developmental delay and neurobehavioral abnormalities. Mol. Genet. Genomic Med. 2018;6:294–300. doi: 10.1002/mgg3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolnikova M., Skopkova M., Ilencikova D., Foltan T., Payerova J., Danis D., Klimes I., Stanik J., Gasperikova D. DNM1 encephalopathy - atypical phenotype with hypomyelination due to a novel de novo variant in the DNM1 gene. Seizure. 2018;56:31–33. doi: 10.1016/j.seizure.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Li H., Fang F., Xu M., Liu Z., Zhou J., Wang X., Wang X., Han T. Clinical Assessments and EEG Analyses of Encephalopathies Associated With Dynamin-1 Mutation. Front. Pharmacol. 2019;10:1454. doi: 10.3389/fphar.2019.01454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boumil R.M., Letts V.A., Roberts M.C., Lenz C., Mahaffey C.L., Zhang Z.-W., Moser T., Frankel W.N. A missense mutation in a highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice. PLoS Genet. 2010;6:e1001046. doi: 10.1371/journal.pgen.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asinof S., Mahaffey C., Beyer B., Frankel W.N., Boumil R. Dynamin 1 isoform roles in a mouse model of severe childhood epileptic encephalopathy. Neurobiol. Dis. 2016;95:1–11. doi: 10.1016/j.nbd.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.-C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 14.Morelli K.H., Griffin L.B., Pyne N.K., Wallace L.M., Fowler A.M., Oprescu S.N., Takase R., Wei N., Meyer-Schuman R., Mellacheruvu D. Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J. Clin. Invest. 2019;129:5568–5583. doi: 10.1172/JCI130600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace L.M., Liu J., Domire J.S., Garwick-Coppens S.E., Guckes S.M., Mendell J.R., Flanigan K.M., Harper S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 2012;20:1417–1423. doi: 10.1038/mt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daya S., Berns K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008;21:583–593. doi: 10.1128/CMR.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feather-Schussler D.N., Ferguson T.S. A Battery of Motor Tests in a Neonatal Mouse Model of Cerebral Palsy. J. Vis. Exp. 2016;2016 doi: 10.3791/53569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang M., Bozdagi O., Scattoni M.L., Wöhr M., Roullet F.I., Katz A.M., Abrams D.N., Kalikhman D., Simon H., Woldeyohannes L. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J. Neurosci. 2012;32:6525–6541. doi: 10.1523/JNEUROSCI.6107-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pollen D.A., Trachtenberg M.C. Neuroglia: gliosis and focal epilepsy. Science. 1970;167:1252–1253. doi: 10.1126/science.167.3922.1252. [DOI] [PubMed] [Google Scholar]
- 20.Southard E.E. On the Mechanism of Gliosis in Acquired Epilepsy. Am. J. Ment. Sci. 1908;64:607–641-22. [Google Scholar]
- 21.Manno I., Macchi F., Caleo M., Bozzi Y. Environmental enrichment reduces spontaneous seizures in the Q54 transgenic mouse model of temporal lobe epilepsy. Epilepsia. 2011;52:e113–e117. doi: 10.1111/j.1528-1167.2011.03166.x. [DOI] [PubMed] [Google Scholar]
- 22.Sørensen A.T., Nikitidou L., Ledri M., Lin E.-J.D., During M.J., Kanter-Schlifke I., Kokaia M. Hippocampal NPY gene transfer attenuates seizures without affecting epilepsy-induced impairment of LTP. Exp. Neurol. 2009;215:328–333. doi: 10.1016/j.expneurol.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kadiyala S.B., Papandrea D., Tuz K., Anderson T.M., Jayakumar S., Herron B.J., Ferland R.J. Spatiotemporal differences in the c-fos pathway between C57BL/6J and DBA/2J mice following flurothyl-induced seizures: A dissociation of hippocampal Fos from seizure activity. Epilepsy Res. 2015;109:183–196. doi: 10.1016/j.eplepsyres.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McTague A., Howell K.B., Cross J.H., Kurian M.A., Scheffer I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–316. doi: 10.1016/S1474-4422(15)00250-1. [DOI] [PubMed] [Google Scholar]
- 25.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 26.Rinaldi C., Wood M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018;14:9–21. doi: 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- 27.Gray S.J. Gene therapy and neurodevelopmental disorders. Neuropharmacology. 2013;68:136–142. doi: 10.1016/j.neuropharm.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 28.Kim J.-Y., Grunke S.D., Levites Y., Golde T.E., Jankowsky J.L. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J. Vis. Exp. 2014;214:51863. doi: 10.3791/51863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill J.M., Lim M.A., Stone M.M. Developmental Milestones in the Newborn Mouse. In: Gozes I., editor. Neuropeptide Techniques. Humana Press; 2008. pp. 131–149. [Google Scholar]
- 30.Schmued L.C., Stowers C.C., Scallet A.C., Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 31.The Jamovi Project . 2020. jamovi (Version 1.2) [Computer Software]. Retrieved from https://www.jamovi.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.