

Abstract
Chimeric antigen receptor (CAR)-modified T cells are endowed with novel antigen specificity and are most often administered to patients without an engineered mechanism to control the CAR T cells once infused. “Suicide switches” such as the small molecule-controlled, inducible caspase-9 (iCas9) system afford the ability to selectively eliminate engineered T cells; however, these approaches are designed for all-or-none, irreversible termination of an ongoing immune response. In order to permit reversible and adjustable modulation, we have created a CAR that is capable of on-demand downregulation by fusing the CAR to a previously developed ligand-induced degradation (LID) domain. Addition of a small molecule ligand triggers exposure of a cryptic degron within the LID domain, resulting in proteasomal degradation of the CAR-LID fusion protein and loss of CAR on the surface of T cells. This fusion construct allowed for reversible and “tunable” inhibition of CAR T cell activity in vitro. Delivery of the triggering molecule in CAR-LID-treated tumor-bearing mice temporarily reduced CAR activity through modulation of CAR surface expression. The ability to more flexibly modulate CAR T cell expression through a small molecule provides a platform for controlling possible adverse side effects, as well as preclinical investigations of CAR T cell biology.
Keywords: engineered cell therapy, CAR T cells, proteasomal degradation, synthetic biology
Graphical Abstract
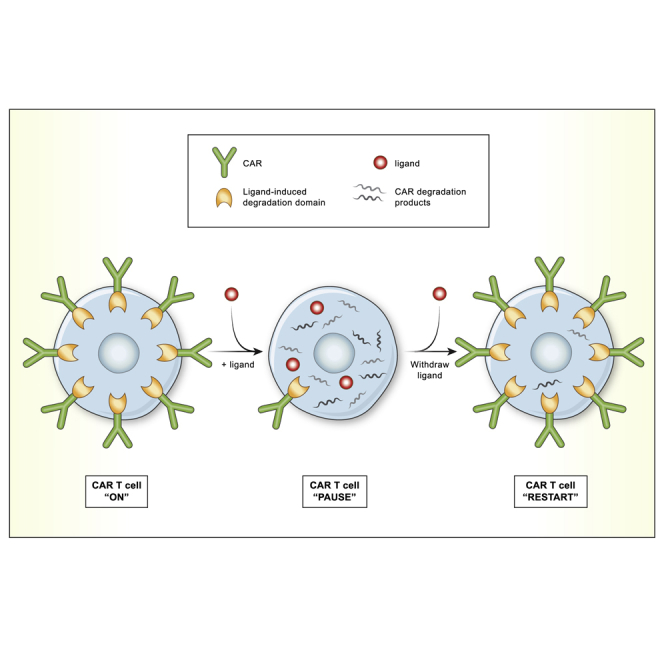
To enable remote control over CAR T cell activity, Richman et al. incorporated a ligand-induced degradation (LID) domain into the CAR. This system created a reversible off switch, depending on the presence or absence of a small molecule ligand, permitting CAR T cell regulation in vitro and in vivo.
Introduction
Engineered T cells are living drugs that have transformed the management of many cancers,1 and their use is currently expanding to address other maladies such as autoimmune disorders2 and fibrosis.3 These products can be very effective in patients whose disease is refractory to conventional therapies. However, unlike traditional drugs, once the engineered T cells have been administered to the patient, their fate is largely uncontrolled. Incorporating a control “knob” to modulate the strength of the T cells’ activity, thereby allowing the clinician to adjust the “dose” of the drug based on the patient’s response or toxicity, is desired, particularly as these therapies expand beyond CART19.4
Since current US Food and Drug Administration (FDA)-approved chimeric antigen receptor (CAR) T cell products do not incorporate methods to control the T cells once they have been administered, the clinical measures used to respond to CAR T cell-mediated toxicity have been largely restricted to either cytokine antagonists (e.g., tocilizumab), non-specific anti-inflammatory agents (glucocorticoids), or in rare cases cytotoxic agents such as cyclophosphamide.5,6 In cases where concern for CAR T cell-mediated toxicity is particularly heightened, investigational T cell products have been engineered to co-express safety switches that allow on-demand depletion of T cells. Examples of these incorporated in clinical products are a co-expressed inducible caspase-97,8 and antibody-mediated depletion targeting a co-expressed truncated epidermal growth factor receptor (EGFRt) via cetixumab9 or targeting co-expressed CD20 via rituximab.10,11 Other strategies being developed have provided reversibility and tunability to CAR T cell regulation by either splitting the CAR12, 13, 14 or globally suppressing T cells using a kinase inhibitor.15,16 In this study, we have developed an approach to CAR T cell regulation that utilizes an intact CAR and specifically targets the expression level of the CAR on the infused T cells. We surmised that direct regulation of the intact CAR could not only ultimately provide a mode of regulation in clinical product, but it could also serve as a useful tool for studying CAR T cell biology in vitro and in vivo.
For these studies, we chose to regulate the CAR post-translationally rather than at the transcriptional level so as to impart more direct and immediate control over the CAR. Several different technologies to allow post-translational modulation of a protein of interest have utilized a “degron,” or degradation signal. Most degron-based strategies have required expression of non-native proteins.17,18 A different degron-based system, developed at Stanford University by Bonger et al.,19 incorporates a cryptic degron to selectively regulate protein levels that requires only 19 amino acids (aa) of non-native peptide. This system, similar to many protein regulation technologies, leverages a “bumped” ligand-accommodating mutant of the human protein FKBP12 (a widely expressed FK506 binding protein). This mutant contains a F36V substitution, creating a “hole” that allows selective binding by a set of synthetic bump-containing ligands relative to the native protein.20,21 Added chemical groups serve as bumps that sterically hinder ligand binding to the endogenous target but can be accommodated by engineered mutants of the target protein. One of these bump-containing ligands, shield-1, binds the F36V mutant of FKBP12 with an affinity ~1,000-fold higher than the endogenous FKBP12.21, 22, 23 Bonger et al.19 thus engineered a 19-aa peptide that, when attached to the C terminus of the FKBP12 F36V, functions as a cryptic degron. At baseline, the degron is inert, tucked into the active site of the FKBP12 F36V. However, shield-1 binding to FKBP12 F36V displaces the degron, allowing it to target the mutant FKBP12, along with an N-terminally fused protein of interest, for degradation. The FKBP12 F36V fused to the cryptic degron was termed the ligand-induced degradation (LID) domain, and those investigators created a yellow fluorescent protein (YFP)-LID fusion expressed in a murine cell line for their proof-of-concept study. In this study, we have expanded this approach to primary human CAR T cells by fusing the CAR to the LID domain to create a CAR-LID construct (Figure 1A). We found that this fusion allowed on-demand downregulation of CAR surface expression. CAR downregulation was associated with inhibition of function that was reversible and permitted remote regulation of CAR activity in vitro and in vivo. These findings establish a platform for further study of CAR T cell biology and add to the tools for CAR regulation that may be combined for better control of CAR T cells in patients.
Figure 1.

Fusion of the LID Domain to the CAR Allows for Dose-Dependent Shield-1-Mediated CAR Downregulation
(A) Diagram of shield-1-induced degradation of CAR-LID polypeptide. (B) Schematic of CAR constructs used in this study. SS, signal sequence; TM, transmembrane. See Figure S1A for nucleotide and amino acid sequence of the CAR-LID. (C) CAR-LID surface expression. Primary human T cells were activated ex vivo and transduced with lentivirus encoding either GD2-LID or the standard GD2 CAR. At day 7 of expansion, T cells were evaluated for CAR expression by staining with anti-mouse antibody (which binds to the murine scFvs) and analyzed by flow cytometry. Representative histograms shown from n = 6 independent experiments. (D) CAR expression across a range of shield-1 concentrations. GD2-LID T cells were incubated with shield-1 at the indicated concentrations for 24 h and evaluated for CAR expression as in (C). Values shown are percentage of GD2-LID CAR expression in a vehicle control sample that did not receive shield-1 (considered to represent “maximum” CAR expression) stained in parallel. Percent maximum expression, that is, (mean fluorescence intensity [MFI] of cells with shield-1/MFI of cells without shield-1) × 100, is plotted. The left panel depicts the dose response over a wide range of shield-1 concentrations. The non-linear curve was fit with GraphPad Prism software using one-phase exponential decay. The right panel highlights the dose response at a narrow range of shield-1 concentrations around the inflection point seen in the left panel (demarcated with a red bracket in the left panel). Values plotted are the mean, and error bars show the standard deviation (SD) from n = 3 different donors. (E) The LID system functions in additional CAR constructs. Incorporation of the LID domain into additional CAR constructs, m3F8-LID and VHH-LID, permits shield-1-mediated downregulation. The GD2 scFv was replaced by either the m3F8 scFv or VHH nanobody in the CAR-LID construct. Primary human T cells were activated ex vivo and transduced with either m3F8-LID or VHH-LID. On day 7, T cells were incubated with shield-1 (1 μM) or vehicle alone for 24 h. Surface CAR was then detected by flow cytometry. Representative histograms are shown from n ≥ 3 different T cell donors.
Results
Fusion of the LID Domain to the CAR Allows for Shield-1 Ligand-Mediated CAR Downregulation
In order to generate a CAR construct capable of regulation at the protein level, we first fused the LID domain to the C terminus of the anti-GD2-4-1BB-CD3ζ CAR to create the GD2 CAR-LID or “GD2-LID” construct (Figures 1B and S1A). Using lentiviral transduction of ex vivo-activated, primary human T cells to deliver either the GD2-LID or a control GD2 CAR transgene identical to the GD2-LID construct minus the LID domain, we observed that the GD2-LID can be expressed on T cells with surface expression that is comparable to the control standard GD2 CAR (Figure 1C). In order to compare the efficiency of expression of the GD2-LID construct to that of the standard GD2 CAR, we determined viral integrant copy number by qPCR (data not shown). This analysis revealed that the GD2-LID CAR T cells contained an average of 2.5-fold more transgene copies than did the standard GD2 CAR T cells, suggesting that there is a per transgene baseline impairment of expression efficiency attributable to the LID domain necessitating higher transduction to achieve similar CAR expression.
We next evaluated the effect of adding the degradation-inducing ligand, shield-1, on GD2-LID expression. GD2-LID T cells were incubated with a range of shield-1 concentrations for 24 h starting on day 7 of ex vivo expansion. A control aliquot of GD2-LID T cells was incubated with vehicle alone and analyzed in parallel. At the end of the 24-h incubation, CAR expression was measured by flow cytometry as for Figure 1C. GD2-LID T cells demonstrated a shield-1 concentration-dependent reduction in surface expression, with partially reduced surface expression noted at 10 nM, ~50% expression noted at ~50 nM, and maximal downregulation of 80% achieved between 100 and 1,000 nM (Figure 1D, left panel). Shield-1-mediated CAR downregulation was dependent on the presence of the LID domain, as expression of standard GD2 CAR control T cells was not altered by shield-1 (Figure S1B). To further delineate the concentration response in the region near the inflection point, we repeated the same analysis using shield-1 concentrations between 10 and 100 nM (Figure 1D, right panel). These data show a reduction in CAR expression that is linearly proportional to shield-1 concentration, demonstrating the tunability of this CAR system through control of shield-1 concentration.
To evaluate the versatility of this system across different CARs, we incorporated the LID domain into CARs containing an alternative GD2-specific single-chain variable fragment (scFv), m3F8 (derived from a completely different antibody clone), a camelid single VHH domain-containing CAR (termed VHH-LID) targeting EGFR, and two anti-fibroblast activation protein (FAP) scFv-containing CARs (one mouse, one human). Similar to the GD2-LID, these LID-containing CARs also demonstrated shield-1-mediated reductions in CAR expression (Figures 1E, S2A, and S2B). However, a sixth LID-based CAR containing the FMC63 anti-CD19 scFv exhibited almost complete abrogation of CAR surface expression in the absence of shield-1, suggesting that although most CAR-LID constructs retain activity, the applicability of this system to CARs is not completely universal and may depend on features of the CAR structure, such as the scFv, for optimal expression and function. We also incorporated the LID domain into a FAP CAR construct containing a different cytoplasmic domain structure based on the natural killer (NK) cell signaling molecule KIR2DS2 and its partner DAP12, (FAPKIRCAR-LID).24 Similar to the traditional CARs, we observed decreased expression and activity of the FAPKIRCAR-LID T cells after addition of shield-1 (Figure S3A).
We also determined that, in addition to applicability across constructs, this system is useful across different ligands. In addition to binding the selective synthetic ligands, shield-1 and its water-soluble counterpart aquashield-1 (AS-1), the LID domain is expected to retain its binding to the canonical FKBP ligand rapamycin and its 40-O-(2-hydroxyethyl) derivative, everolimus, that are widely used in clinical practice. We therefore incubated GD2-LID T cells with rapamycin and everolimus in addition to shield-1 and AS-1 and found comparable downregulation of GD2-LID CAR expression across ligands (Figure S1C). These data suggest that rapamycin or everolimus, anti-cancer and immunosuppressive drugs with well-established pharmacokinetics and long clinical track records, could be used in place of the selective shield-1/AS-1 ligands in scenarios where their concomitant anti-tumor and/or immunosuppressive effect would be desirable.
In addition to the drug concentration-response relationship of the LID system, the kinetics of CAR downregulation and its reversibility are key parameters for its use as a biological tool or potential component of a CAR T cell clinical product. The developers of the LID/shield-1 system, in evaluating it using a YFP fused to the LID domain, found a steep decline in YFP-LID fluorescence during the first 2 h following shield-1 addition, with a nadir of fluorescence by 4 h following shield-1 addition.19 By measuring CAR expression using flow cytometry at several time points following shield-1 addition, we found that the CAR-LID surface expression declined more slowly than that reported for YFP-LID, with the decrease not noted until 8 h and maximal decrease achieved >24 h after drug addition for the GD2-LID CAR (Figure 2), m3F8-LID, and VHH-LID CARs (Figure S4). To evaluate the reversibility of shield-1-mediated CAR-LID downregulation, we continued to culture the T cells for another 48 h following wash-out of the shield-1 and replacement with shield-1-free media. CAR-LID surface expression began to recover by 24 h following wash-out and was returning to near baseline by 48 h (Figures 2 and S4).
Figure 2.

Kinetics of CAR Downregulation and Its Reversal
GD2-LID T cells were incubated with 1 μM shield-1 or vehicle, and CAR expression was evaluated at serial time points by flow cytometry. At 24 h after shield-1 addition, T cells were washed several times to remove shield-1 or vehicle, returned to culture, and stained for CAR at 24 and 48 h after wash-out. Percent maximum CAR expression is plotted as for Figure 1C. The connecting line is the average of three to four different T cell donors, and each data point represents a different donor, with the same symbol representing a particular donor across time points.
The CAR-LID Fusion Permits Shield-1-Dependent Regulation of CAR T Cell Effector Function In Vitro
Having observed shield-1-dependent, LID-mediated, reversible surface CAR downregulation in a variety of CAR-LID T cells, we next determined the impact of CAR downregulation on effector function in vitro. In this study, we assessed in vitro cytotoxicity, antigen-induced proliferation, and cytokine release. In order to monitor cytotoxicity, we co-incubated either GD2 CAR or GD2-LID CAR T cells that had been pre-incubated for 24 h with shield-1 with SY5Y GD2+ tumor cells. Shield-1 did not appear to interfere with the assay or impact standard GD2 CAR killing. GD2-LID-mediated specific cytotoxicity was nearly completely abrogated in the presence of shield-1 (Figure 3A, top panel). Similarly, an affinity-enhanced mutant of the GD2-LID CAR, E101K-LID, as well as m3F8-LID CAR T cells and FAPCAR-LID CAR T cells showed almost complete abrogation of their cytotoxicity following incubation with shield-1 (Figures 3A, lower panel, S5A, and S5B).
Figure 3.

Shield-1-Mediated CAR Downregulation Abrogates In Vitro Functional Activity
(A) 51Cr-release assay of in vitro cytotoxicity mediated by the indicated T cells pre-incubated for 24 h with 1 μM shield-1 or vehicle alone using a range of effector/target (E:T) ratios. Data are mean ± SD from n = 3 different donors. (B) In vitro expansion of the indicated T cells pre-incubated for 24 h with 1 μM shield-1 or vehicle following encounter with irradiated GD2+ SY5Y target cells. Viable cells were counted every 2–3 days using flow cytometric bead-based counting. Representative curves are shown from n = 4 (left panel) or n = 3 (right panel) different T cell donors (top row). Fold expansion on the last day of the experiment was normalized to maximum expansion for each donor (bottom row), and the mean fold expansion levels from the indicated groups are shown. Error bars are SD. Groups were compared using a one-way ANOVA with multiple (three) comparisons. ∗∗p < 0.01, ∗∗∗∗p < 0.0001. NS, not significant.
We also evaluated the impact of shield-1-mediated CAR downregulation on antigen-induced proliferation in vitro and observed a similar pattern. Whereas shield-1 did not impact proliferation of standard GD2 or m3F8 CAR T cells upon exposure to irradiated GD2+ tumor cells, it completely blocked proliferation of GD2-LID and m3F8-LID CAR T cells that correlated with the reduction in CAR expression in the presence of shield-1 (Figure 3B). Incorporation of the LID domain did not significantly alter the ratio of CD4+ to CD8+ T cells during antigen-induced proliferation (Figure S6A). Similar to cytotoxicity and antigen-induced proliferation, interferon (IFN)γ secretion was also substantially limited in GD2-LID, FAP-LID, and FAPKIRCAR-LID T cells exposed to shield-1. Shield-1 did not affect IFNγ secretion in standard CAR T cells (Figures S3A, S7A, and S7B).
Given the degree of functional inhibition in the CAR-LID T cells incubated with shield, we next determined whether function could be restored following removal of shield-1. We thus established an in vitro proliferation model as described for Figure 3B. However, 3 days into the assay, we separated aliquots of cells in shield-1-containing media, washed and resuspended the cells in shield-1-free media, and returned the washed T cells to the plate containing targets. We found that within 3 days following shield-1 washout, the CAR-LID cells had begun to proliferate, and within 10 days they had achieved as many-fold expansion as the CAR-LID cells that were never exposed to shield-1 (Figure 4). Following washout of the ligand, the CD4/CD8 ratio was not significantly altered in the CAR-LID cells exposed to shield-1 versus those exposed to vehicle alone (Figure S6B).
Figure 4.

In Vitro Antigen-Driven CAR T Cell Proliferation Can Be Restored Following Removal of Shield-1 from Media
The indicated CAR T cells were exposed to irradiated SY5Y target cells, and viable T cells were counted as described for Figure 3B. On day 3 after plating, shield-1 was washed from an aliquot of CAR-LID T cell culture containing shield-1, and washed T cells were returned to irradiated SY5Y target cells. Given normal donor-to-donor variability in T cell expansion kinetics, data are plotted as percent maximum expansion relative to CAR-LID T cells not exposed to shield-1 and grown in parallel from the same donor. Data points are mean ± SD from n = 3 different donors. Groups were compared using a one-sided t test. ∗p < 0.05.
In addition to reversibility, the ability to tune the CAR-LID system to display an intermediate degree of activity, rather than being limited to a toggle between either on or off, is another key aspect of this system’s utility, both as a biological tool as well as a potential component of a clinical product. As we had observed in our initial concentration-response studies that 25 nM shield-1 yielded intermediate CAR expression (~60% of maximum), we evaluated whether that degree of CAR expression was associated with an intermediate degree of T cell activation. To do so, we pre-incubated E101K-LID CAR T cells with an intermediate (25 nM) or fully suppressive concentration (1,000 nM) of shield-1 ligand and determined the E101K-LID CAR T cell cytotoxic activity toward GD2+ target cells relative to that of E101K-LID T cells pre-incubated with vehicle alone. T cells incubated with 25 nM shield-1 demonstrated partial cytotoxicity, an average of 30% of what was achieved by T cells without shield (Figure 5A). We also observed an intermediate degree of antigen-driven CAR-LID T cell expansion in the presence of 25 nM shield-1 (Figures 5B and S8). These data suggest that the LID system can be tuned such that, in addition to a reversible off-switch, it may also provide an adjustable system to finely control the degree of cytotoxic activity and proliferation of CAR T cells, provided the ligand concentration can be similarly controlled.
Figure 5.

Partial CAR-LID T Cell Activity Achieved Using an Intermediate Concentration of Shield-1
(A) E101K-LID CAR T cells were pre-incubated for 24 h with either 1,000 nM shield-1, 25 nM shield-1, or vehicle alone and then co-incubated for 14–17 h with 51Cr-loaded SY5Y GD2+ cells at an effector/target (E:T) ratio of 20:1. Percent specific cytotoxicity was determined as described for Figure 3A. The percent specific cytotoxicity achieved by T cells incubated with vehicle alone was then set as maximum cytotoxicity, and the percent maximum cytotoxicity for T cells incubated with an intermediate dose of 25 or 1,000 nM shield-1 is shown for n = 3 different T cell donors. These two groups were compared using a two-sided t test. ∗p < 0.05. (B) m3F8-LID CAR T cells were pre-incubated for 24 h with 25–50 nM shield-1, 1,000 nM shield-1, or vehicle alone and added to irradiated SY5Y target cells at a ratio of 1:1. Viable T cells were then counted during the ensuing 10–13 days depending on the T cell donor as described for Figure 3B. Curves from a representative donor are shown here from n = 3 different donors. See Figure S8 for the other two donors. Bottom panels: paired data from three to four donors (each line connects an individual donor) were used to compare groups at the last time point via a two-sided paired t test. ∗p < 0.05.
Our findings of reversible, tunable CAR-LID regulation largely recapitulated those of Bonger et al.19 using their YFP-LID fusion. In investigating the mechanism of downregulation of the LID-fused proteins, Bonger et al.19 demonstrated the key role of the proteasome in YFP-LID degradation, suggesting that the exposed degron promotes downregulation by targeting the polypeptide to proteasomal degradation, possibly through ubiquitination. However, unlike the intracellular proteins evaluated in those studies, the CAR-LID is potentially subject to a different pathway of shield-1-induced degradation given its distinct location within the plasma membrane. An alternative degradation pathway for ubiquitinated plasma membrane proteins traffics them from the cell surface to endosomes and ultimately lysosomes, the site of polypeptide degradation by hydrolysis (reviewed in Clague and Urbé,25 Mukhopadhyay and Riezman,26 and Tai and Schuman27). In order to elucidate the dominant degradative pathway for CAR-LID, we determined the impact of inhibiting either the proteasome using inhibitor MG132 or the endosome-lysosome pathway using inhibitors bafilomycin and NH4Cl. Whereas inhibition of the endosome-lysosome pathway did not appreciably impact shield-1-mediated CAR-LID downregulation, inhibition of the proteasome pathway impaired the degradation leading to increased CAR-LID expression (Figures 6 and S9). These data suggest that the dominant mode of shield-1-mediated CAR-LID degradation is via targeting to the proteasome similar to YFP-LID fusions described by Bonger et al.19
Figure 6.

The Relative Contributions of Degradation Pathways in Shield-1-Mediated CAR Downregulation
(A) GD2-LID T cells were incubated for 24 h with 1 μM shield-1. During the last 6 h of shield-1 incubation, cultures were split and incubated with either the proteasome inhibitor MG132, endosome-lysosome inhibitor bafilomycin, or vehicle alone. Cells were then stained for CAR and analyzed by flow cytometry. (B) Standard GD2 CAR T cells were incubated for 6 h with or without the indicated inhibitors, and cells were stained for CAR and analyzed by flow cytometry.
The CAR-LID Fusion Permits Shield-1-Dependent Regulation of CAR T Cell Activity In Vivo
Finally, we extended these in vitro findings to xenograft models to assess the impact of in vivo ligand delivery on tumor control by CAR-LID T cells. In our first set of studies, we tested the effects of AS-1 on control of the FAP+ mesothelioma, EMMESO, by T cells expressing a variant of the FAPCAR that contains the killer inhibitory receptor (KIR) signaling domain to which the LID is fused (FAPKIRCAR-LID, Figure 1B). Our previous work in this relatively resistant tumor model showed that the FAPKIRCAR variant demonstrated improved anti-tumor activity over standard CAR.24 In the current study, AS-1 was administered intraperitoneally every 3 days beginning 18 days after injection of FAPKIRCAR-LID T cells in EMMESO tumor-bearing NOD (non-obese diabetic)-SCID (severe combined immunodeficiency)-Il2rg−/− (NSG) mice (Figure 7A). We chose to use AS-1 (Cheminpharma, Woodbridge, CT, USA), the water-soluble variant of shield-1, for these experiments due to its equivalent activity compared with shield-1 (Figure S1C), but improved solubility facilitating formulation for injection. AS-1 alone had no significant effect on tumor growth. Whereas the tumors in the FAPKIRCAR-LID group on day 31 were significantly smaller than the PBS- or AS-1-treated control tumors, the tumors in the mice that had received FAPKIRCAR-LID T cells and AS-1 were significantly larger than in those receiving FAPKIRCAR-LID T cells alone (Figure 7B). These data support a reduction in FAPKIRCAR-LID T cell function in the presence of AS-1.
Figure 7.

The Impact of AS-1 on CAR-LID Tumor Control In Vivo
(A) Experimental design. EMMESO cells were injected subcutaneously into the flanks of NSG mice. 14 days later, FAPKIRCAR-LID T cells (107 total T cells) or PBS alone was injected via tail vein. AS-1 was injected intraperitoneally on the indicated days at a dose of 10 mg/kg. (B) Left panel: tumor volume over time in mice receiving the indicated T cells and AS-1 where indicated. Right panel: tumor volume on day 31. Means ± SEM of five mice per group are shown. Groups were compared using a one-way ANOVA with Tukey’s multiple comparison test. ∗p = 0.01. (C) Experimental design. NSG mice were injected via tail vein on day 0 with 0.5 × 106 SY5Y human tumor cells (engineered to express luciferase to allow for tumor burden assessment using bioluminescent imaging [BLI], as described previously).28 Seven days later, 3 × 106 CAR+ T cells were injected. Five days after T cell injection, half of the mice underwent subcutaneous implantation of an osmotic pump filled with AS-1 such that 1.3 mg/day of AS-1 was released as a continuous infusion for 7 days. (D) Left panel: bioluminescence (total flux) over time of mice receiving E101K-LID CAR T cells, with or without AS-1-loaded pumps, or NTD control cells (without pumps). The duration of AS-1 delivery is marked with a red line. Data points shown are the means ± SEM of six mice per group. Right panel: BLI values from the two E101K-LID groups on the last evaluable day were compared using a Mann-Whitney comparison. ∗∗p < 0.01. (E) Survival curves of E101K-LID mice with and without AS-1 administration were compared using a log rank (Mantel-Cox) test. ∗∗p < 0.01. (F) Bioluminescence from mice receiving standard E101K CAR T cells with or without AS-1. Data points shown are the mean ± SEM of six mice per group.
In an effort to evaluate whether the LID approach can modulate CAR-induced toxicity in addition to anti-tumor function, we extended the FAPKIRCAR-LID studies to a different, 4-1BB-based standard CAR that bears the 14G2a-derived scFv with a single point mutation in the VH domain (E101K CAR) that binds GD2 with increased affinity over the parental CAR. Our laboratory has shown that this E101K CAR exhibits both enhanced anti-tumor function in a rapidly growing SY5Y neuroblastoma xenograft model, but with associated severe neurotoxicity.28 In vitro analysis of shield-1-mediated E101K-LID CAR downregulation and subsequent impact on cytotoxicity are shown in Figures 3B and S3B. The introduction of the LID domain into the E101K CAR partially reduced the anti-tumor tumor activity (Figure S10B); however, in the absence of ligand, E101K-LID CAR T cells still induced tumor regression with associated neurotoxicity as seen in the standard E101K CAR T cells.
Although our first study and a prior published study used intermittent doses of shield-1/AS-1,29 intermittent dosing may be insufficient to consistently achieve the necessary plasma concentration (1 μM) required to fully induce CAR-LID downregulation, owing to the ligands’ short in vivo half-life (t1/2). (The t1/2 of AS-1 in mice is 58 min [data not shown].) In order to achieve consistent exposure to AS-1 over time, we chose to conduct our second study using a continuous infusion of AS-1 via an implanted osmotic pump. AS-1 was thus delivered to mice via subcutaneously implanted osmotic pumps for 7 days to achieve a predicted concentration of approximately 4,325 ng/mL based on the measured t1/2 (Figure 7C). No difference in tumor control between E101K-LID CAR-treated mice with or without AS-1 treatment was observed at day 3 of AS-1 infusion, but by day 6 tumor growth in the E101K-LID-treated mice receiving AS-1 exceeded that of control mice not receiving ligand, which maintained tumor control (Figures 7D and S10A, left panel). At this later time point (AS-1 infusion day 6), whereas all control E101K-LID CAR-treated mice had developed signs of severe toxicity necessitating euthanasia, mice receiving AS-1 still appeared healthy. Demonstrating the reversibility of the LID control, 2 days following the end of AS-1 infusion, tumor growth slowed in the mice exposed to AS-1 (Figures 7D and S10A, right panel), which was associated with subsequent toxicity requiring euthanasia. Toxicity was delayed by a median of 5 days in mice receiving the 7-day AS-1 infusion (Figure 7E). Due to the confounding phenomenon of antigen-induced CAR downregulation, AS-1-induced CAR downregulation was not measured in the in vivo studies in the presence of tumor antigen. The effect of AS-1 was dependent on the LID domain, as mice receiving standard CAR and AS-1 did not demonstrate a difference in tumor control (Figure 7F) or onset of toxicity (data not shown) compared to mice receiving standard CAR alone.
Discussion
As the clinical applications of CAR T cell therapy expand beyond the groundbreaking efforts targeting CD19+ leukemia/lymphoma, it may become necessary to introduce regulatory elements, largely to promote safety.7,9,11,13 As noted previously, thus far, most of the domains that permit “remote control” of T cells have incorporated permanent off-switches. However, a system that permits a temporary turn-off or turn-down of CAR T cell activity may provide a useful alternative in scenarios where a less drastic approach is appropriate, for example, in the setting of cytokine release syndrome, where it would be an advantage to “dial down” the CAR activity temporarily rather than eliminate the CAR T cells needed to remove tumor cells. An additional potential of this system stems from a recent report evaluating the preclinical activity of CAR T cells whose activation was intermittently switched off compared to those left on, which showed that the CAR T cells allowed “rest” periods achieved superior activity and persistence.30 The CAR-LID system described herein could thus be integrated into a rest paradigm to investigate the impact of activating through a second CAR while the other CAR is resting.
The CAR-LID system that we have developed in this study permits reversible inhibition of CAR T cell activity in vitro that translated to a “pause button” in vivo. We noted a rapid effect on T cell activity of the ligand in vivo: within 6 days from the initiation of AS-1 delivery, T cell activity had been sufficiently inhibited to allow tumor to already appreciably re-grow. In the subsequent 2 days following ligand shut-off, tumor control had resumed. Our in vitro data suggest that, in addition to being reversible, the system can be tuned to yield an intermediate level of activity by lowering the dose of ligand used, indicating the potential of the LID system to ultimately provide a translatable method for achieving an additional layer of control over infused CAR T cells. Note that the inducible caspase-9 system can be titrated,7 and it will be interesting to compare the impact of partial removal of CAR T cells (inducible caspase-9) versus partial pausing of CAR T cells (CAR-LID system) on toxicity and anti-tumor effect.
Although we chose to use the selective ligands shield-1 and AS-1 that lack immunosuppressive activity to evaluate the CAR-LID system in the animal model, the LID system is also regulatable using rapamycin or everolimus, both of which bind the FKBP12 domain variant upon which the LID system was designed. Both compounds are pharmaceuticals with established pharmacokinetics, regulatory approval, and clinical track records. Although neither rapamycin nor its analogs are specific to the LID domain and would also exert their immune-suppressive effects based on binding to native FKBP, in circumstances where T cell activity is meant to be temporarily dampened, their use would be expected to be constructive. Their intrinsic anti-cancer effect may also be helpful and their incorporation into this system warrants further exploration.31
In addition to its ligand versatility, another feature of the LID system is its potential to be used in combination with other regulation strategies. For example, the developers of the LID system have also created a shield-1 controlled destabilizing domain (DD) fused to a protein of interest that is degraded at baseline but can be stabilized in the presence of shield-1.23,32 These investigators used two different fluorescent reporters incorporating either the LID or DD domains to demonstrate that two different proteins could be inversely titrated using the same shield-1 ligand.19 These systems could also be combined with an orthogonally regulated approach, such as tetracycline (Tet)-on-regulated transgene expression to further increase the flexibility of regulation.33
There are a number of potential limitations to the CAR-LID system that should be considered. It is possible that the incorporation of the LID domain sequences in the CAR could lead to enhanced immunogenicity in patients with certain histocompatibility leukocyte antigen (HLA) haplotypes, although the small size of the degron (19 aa) would reduce this risk. Our data suggest that CAR downregulation with shield-1 administration will require 12–24 h. Therefore, in the context of toxicity mitigation, LID/shield-1 is a tool one would want to utilize earlier in the course of toxicity, before a patient becomes critically ill. However, the reversible (and titratable) nature of the LID/shield system can permit a lower threshold for providers to engage the LID system given that, once the patient has stabilized, the drug can be partially or completely withdrawn in order to resume anti-tumor activity. Prior pharmacokinetic data in mice revealed a short half-life for shield-1 and AS-1, thus allowing rapid recovery of CAR T cell activity when needed. In contrast, the threshold to engage an irreversible system such as the inducible caspase-97,8 would likely be higher, as the treatment would irreversibly deplete active CAR T cells, necessitating reinfusion (if possible) to resume an anti-tumor response. An alternative approach using a T cell inhibitor such as dasatinib permits rapid and reversible inhibition of T cell activity.15,16 However, unlike the LID/shield-1 or inducible caspase 9-system, dasatinib is not selective for the engineered T cells.
The clear disadvantage to the short half-life of the shield-1/AS-1 ligands is that it necessitates continuous infusion, which introduces challenges for both preclinical and potential clinical applications. We are therefore investigating modifications to increase the half-life in order to expand upon our in vivo preclinical studies and design experiments with longer dosing schedules and repeated dosing, and further biodistribution studies are planned. Although AS-1 has not been tested in humans, as it has 1,000-fold selectivity for the F36V mutant of FKBP over endogenous FKBP, it is expected to be relatively biologically inert (aside from its effects on the engineered LID domain) and not pose a safety issue. However, this would need to be tested.
Additionally, one potential disadvantage to the CAR-LID system described herein is the apparent lower per-integrant expression of CAR-LID relative to standard CAR and the partial, yet statistically significant decrease in tumor control relative to standard CAR observed in these experiments. One approach to overcome this decreased surface expression is simply to increase the multiplicity of infection (MOI). However, with a higher MOI comes a theoretically greater chance of an insertional mutagenesis, although this has not been observed in T cells with either retroviral or lentiviral vectors. Alternatively, future protein engineering efforts could be directed at selecting for improved LID activity to augment baseline CAR-LID expression levels. Likewise, alternative degradation strategies, such as the dTAG system that targets the protein of interest for degradation via direct ubiquitination by a small molecule ligand, rather than via displaced degron, are worth exploring.34
In addition to laying the foundation for future development of CAR-LID as a potential component of clinical T cell products, our findings raised questions of receptor dynamics that warrant further investigation. When reviewing the degree of functional inhibition of CAR-LID T cells in the presence of shield-1, we were intrigued that functional inhibition was out of proportion to the degree of CAR downregulation. For example, at fully suppressive shield-1 concentrations, CAR-LID T cell activity was virtually eliminated while CAR-LID expression remained at 30% of maximum (rather than close to 0% as would be expected based on activity). Likewise, at a concentration of 25 nM, CAR-LID expression was still 75% of maximum but function was less than 50%. Considered with our data implicating the proteasome in CAR-LID degradation (rather than removal of surface CAR-LID via the endosome-lysosome pathway), these findings lead us to speculate that shield-1 induces proteasomal degradation of the nascent CAR-LID polypeptide forming within the endoplasmic reticulum,35 thereby preventing freshly translated CAR-LID from arriving at the plasma membrane in the presence of shield-1. Perhaps the trafficking of newly synthesized CAR molecules to the cell surface is essential to allow antigen-mediated CAR T cell activation to proceed at detectable levels. This finding is a focus of our future studies.
In conclusion, our data suggest that a CAR fusion protein that incorporates a peptide that targets the molecule for proteasome degradation allows for reversible and tunable inhibition of CAR T cell activity in vitro and in vivo through modulation of the levels of CAR protein expression. The ability to more flexibly and reversibly modulate CAR T cell expression through a small molecule provides a platform for controlling possible adverse side effects, as well as preclinical investigations of CAR T cell biology.
Materials and Methods
CAR Constructs
The LID domain19 was incorporated into the GD2 and FAP-KIR CAR24 constructs using gene synthesis (Genewiz, South Plainfield, NJ, USA). The GD2-LID and FAPKIRCAR-LID inserts were then subcloned into the pTRPE lentiviral plasmid. The LID domain was also subcloned into lentiviral vectors containing the GD2-E101K mutant and m3F8 CARS (previously described in Richman et al.28), as well the VHH anti-EGFR CAR. All CARs incorporated CD8A (CD8α) hinge and transmembrane domains as well as TNFRSF9 (4-1BB) co-stimulatory domains. The nucleotide and amino acid sequence of the CAR-LID construct (excluding the antigen binding domain) is shown for reference in Figure S1A.
Ligands
Shield-1 and AS-1 were obtained from Cheminpharma (Woodbridge, CT, USA). Shield-1 was resuspended in 100% ethyl alcohol to a concentration of 1 mM, aliquoted, and stored at −20°C. AS-1 was resuspended in sterile de-ionized water to a concentration of 1 mM for in vitro use and to a concentration of 78 mM for in vivo studies, and stored at 4°C. Rapamycin (Calbiochem, San Diego, CA, USA) was resuspended in 100% ethyl alcohol to a concentration of 10 mg/mL, aliquoted, stored at −20°C, and used at a final concentration of 100 nM. Everolimus (RAD001, Novartis, Cambridge, MA, USA) was resuspended in 100% ethyl alcohol to a concentration of 10 mg/mL, aliquoted, stored at −20°C, and used at a final concentration of 100 nM.
Cell Lines
Human neuroblastoma cell lines SY5Y and NB16 were a generous gift of Dr. John Maris (Children’s Hospital of Philadelphia). EMMESO cells were derived from malignant mesothelioma pleural effusion and used as described in Wang et al.24 and Moon et al.36 The SY5Y-click beetle green (CBG) cell line was created as described previously.28 Cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer, 100 U/mL penicillin, and 100 g/mL streptomycin sulfate.
Primary Human CAR T Cell Generation
Primary human T cells were isolated from normal donors by the Human Immunology Core at the University of Pennsylvania. All specimens were collected under a University Institutional Review Board-approved protocol, after written informed consent was obtained. Cells were expanded by adding anti-CD3/anti-CD28 antibody-coated beads (Dynabeads, Thermo Fisher Scientific, Waltham, MA, USA) at a bead/cell ratio of 3:1. One day following the addition of beads, lentivirus (prepared as described previously37) was added to T cells at an MOI of 3–5, and culture was expanded until T cells had rested to a cellular volume of ~350–400 fL. Beads were then removed and T cells were washed in PBS and cryopreserved in FBS + 5% DMSO.
Flow Cytometry
T cells were washed in PBS, pelleted, and incubated for 15 min at room temperature with Biotin-SP-AffiniPure goat anti-mouse immunoglobulin G (IgG) F(ab′)2 (fragment specific) (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at approximately 0.075 mg/mL (for GD2 and m3F8-containing CAR) or biotinylated protein A (Pierce, Waltham, MA, USA) for VHH-LID CAR, washed, and stained with R-phycoerythrin (R-PE)-conjugated streptavidin (SA) (BD Biosciences, Franklin Lakes, NJ, USA) at approximately 0.08 mg/mL for 5 min at room temperature. Stained cells were analyzed on an LSR II flow cytometer. Flow cytometry data were analyzed using FlowJo software.
Cytotoxicity Assays
T cells were thawed, washed, and, following a 24-h pre-incubation with shield-1 (or vehicle alone), were co-incubated with SY5Y target cells that had been loaded with 100 μCu 51Cr per million cells for 14–17 h at a range of E:T ratios from 2.5:1 to 20:1. Shield-1 concentration was maintained throughout the co-incubation. Supernatant was removed at the end of the co-incubation and counts per minute (cpm) were measured using a MicroBeta2 Lumijet (PerkinElmer, Waltham, MA, USA). Percent specific cytotoxicity was calculated using the following formula: [(experimental cpm − spontaneous cpm)/(maximum cpm − spontaneous cpm)] × 100, where spontaneous cpm was obtained from wells containing targets and media alone, and maximum cpm was obtained from wells containing targets and 5% sodium dodecyl sulfate (SDS).
To measure activation and killing of FAPCAR and FAP-CAR-LID cells, T cells were mixed at various E:T ratios with mouse 3T3BALB/c cells that had been transfected with both murine FAP and with luciferase. After 24 h, supernatants were collected for IFNγ ELISA. Dead cells were washed away and the remaining cell number was determined using luciferase assays as previously described.38
Cytokine Release Assays
Supernatant was removed from overnight co-incubations of T cells and targets. IFNγ concentration in the supernatants was determined by ELISA (R&D Systems, Minneapolis, MN, USA).
Proliferation Assays
T cells were thawed, washed, and, following a 24-h pre-incubation with shield-1 (or vehicle alone), were added 1:1 to SY5Y cells that had been exposed to 100 Gy irradiation to prevent their outgrowth. Viable T cells were then counted by flow cytometry during the ensuing 10–14 days using CountBright absolute counting beads (Life Technologies, Carlsbad, CA, USA). Cells were co-stained with anti-CD4 and anti-CD8 antibodies (BD Biosciences, Franklin Lakes, NJ, USA). Shield-1 concentration was maintained throughout the experiment. For the shield-1 washout samples, an aliquot of cells cultured in the presence of ligand was removed, washed three times in 30-fold excess PBS and once in 30-fold excess shield-1-free media and returned to the plate of fresh irradiated targets.
Proteasome/Endosome-Lysosome Inhibitor Assays
MG132 (Sigma, St. Louis, MO, USA) was resuspended in DMSO and used at a final concentration of 10 μM. Bafilomycin (Sigma, St. Louis, MO, USA) was resuspended in DMSO and used at a final concentration of 100 nM. NH4Cl (Sigma, St. Louis, MO, USA) was resuspended in distilled H2O (dH2O) and used at a final concentration of 50 mM. T cells transduced with CAR as described above were incubated with 1 μM shield-1 or vehicle on day 7 of expansion. 18 h later, proteasome inhibitor (MG132) or endosome-lysosome inhibitors (bafilomycin or NH4Cl) were added for the last 6 h of the 24-h shield-1 incubation. Cells were then stained for CAR using biotin goat anti-mouse as described above with a live/dead violet viability co-stain (Thermo Fisher Scientific, Waltham, MA, USA). Stained cells were then analyzed by flow cytometry as described above. Activity of the lysosome inhibitors under these assay conditions was confirmed by inhibition of unquenching of DQ Green BSA (Molecular Probes/Thermo Fisher Scientific, Waltham, MA, USA) fluorescence following uptake into the endosome-lysosome compartment (Figure S9B).
In Vivo Studies
6- to 8-week-old female NSG mice were used. (Only females were included in the osmotic pump experiments in an effort to maximize AS-1 concentration given their smaller average body size.) Mice were housed in the Xenograft Core Facility at the University of Pennsylvania under pathogen-free conditions, and experimental protocols, including surgical procedure and post-procedure analgesia and monitoring, were approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). Mice were euthanized based on standard criteria of body condition score or tumor burden.
EMMESO cells were injected into the flanks of the NSG mice. After 14 days, when the tumors were ~180 mm3), four groups were designated as follows: control (injected with PBS), injected with AS-1 alone, injected with thawed cryopreserved FAPKIRCAR-LID T cells alone, or injected with FAPKIRCAR-LID T cells plus AS-1. 107 FAPKIRCAR-LID T cells were injected intravenously at this time point. Mice received four doses of AS-1 (10 mg/kg) or PBS intraperitoneally (i.p.) on days 18, 21, 24, and 28. Tumor volumes were determined by caliper measurements.
SY5Y-CBG tumor cells were injected via tail vein at a dose of 0.5 × 106 cells per mouse. Cryopreserved CAR T cells (or NTD control T cells) were thawed, washed, resuspended in PBS, and injected via tail vein at a dose of 3 × 106 CAR+ cells per mouse in 100 μL (~5 × 106 total T cells per mouse). Osmotic pumps designed to deliver 1 μL/h for 7 days (Alzet model 2001) were filled with 200 μL of sterile-filtered 78 mM AS-1 and pre-primed at 37°C in PBS for several hours prior to surgical implantation in the dorsal subcutaneous tissue. Tumor burden was monitored at multiple time points via bioluminescent imaging of anesthetized mice following i.p. injection of ~115 mg/kg D-luciferin (Caliper Life Sciences, Waltham, MA, USA). Images were obtained using an IVIS Spectrum imager and analyzed using Living Image software (PerkinElmer, Waltham, MA, USA).
Author Contributions
Conceptualization, S.A.R., L.-C.W., S.M.A., and M.C.M.; Investigation, S.A.R. and L.-C.W.; Resources, U.R.K.; Writing – Original Draft, S.A.R.; Writing –Review and Editing, L.-C.W., U.R.K., S.M.A., and M.C.M.
Conflicts of Interest
M.C.M. is an inventor on several granted and pending patents related to CAR T cells and their use in cancer therapy. He is also co-founder and co-chair of the Scientific Advisory Board of Cabaletta Bio. U.R.K. is the CEO of Cheminpharma LLC, a for-profit drug discovery services and reagents company. AS-1 is a proprietary ligand developed by Cheminpharma LLC. S.A.R. is a co-inventor on a pending patent related to chimeric autoantigen receptor-engineered T cells. L.-C.W. is an employee and stockholder of Incyte Corporation. S.M.A. declares no competing interests.
Acknowledgments
The authors would like to thank John Leferovich and Chune Zhang for technical assistance, as well as the staff of the University of Pennsylvania Stem Cell and Xenograft Core and Small Animal Imaging Facility for assistance with the in vivo studies. We thank Dr. Michael Marks in the Department of Pathology and Laboratory Medicine at CHOP for helpful discussion. We also acknowledge the Human Immunology Core as well as Dr. Irina Kulikovsaya and the Translational Correlative Studies Laboratory at the University of Pennsylvania. This work was supported in part through funding provided by Novartis Pharmaceuticals through a research alliance with the University of Pennsylvania, institutional funds from the University of Pennsylvania (to M.C.M.), and NCI P01-CA217805 (to S.M.A.). S.A.R. was supported by a St. Baldrick’s Foundation Scholar Award (524831), St. Baldrick’s Foundation Fellowship/Ben’s Green Drakkoman Fund, and CHOP Cancer Center K12 (CA076931, NIH/NCI).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.06.004.
Contributor Information
Sarah A. Richman, Email: richmans@email.chop.edu.
Michael C. Milone, Email: milone@pennmedicine.upenn.edu.
Supplemental Information
References
- 1.Boyiadzis M.M., Dhodapkar M.V., Brentjens R.J., Kochenderfer J.N., Neelapu S.S., Maus M.V., Porter D.L., Maloney D.G., Grupp S.A., Mackall C.L. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J. Immunother. Cancer. 2018;6:137. doi: 10.1186/s40425-018-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maldini C.R., Ellis G.I., Riley J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018;18:605–616. doi: 10.1038/s41577-018-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aghajanian H., Kimura T., Rurik J.G., Hancock A.S., Leibowitz M.S., Li L., Scholler J., Monslow J., Lo A., Han W. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. doi: 10.1038/s41586-019-1546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe K., Kuramitsu S., Posey A.D., Jr., June C.H. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front. Immunol. 2018;9:2486. doi: 10.3389/fimmu.2018.02486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude S.L., Barrett D., Teachey D.T., Grupp S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Acharya U.H., Dhawale T., Yun S., Jacobson C.A., Chavez J.C., Ramos J.D., Appelbaum J., Maloney D.G. Management of cytokine release syndrome and neurotoxicity in chimeric antigen receptor (CAR) T cell therapy. Expert Rev. Hematol. 2019;12:195–205. doi: 10.1080/17474086.2019.1585238. [DOI] [PubMed] [Google Scholar]
- 7.Diaconu I., Ballard B., Zhang M., Chen Y., West J., Dotti G., Savoldo B. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 2017;25:580–592. doi: 10.1016/j.ymthe.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straathof K.C., Pulè M.A., Yotnda P., Dotti G., Vanin E.F., Brenner M.K., Heslop H.E., Spencer D.M., Rooney C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paszkiewicz P.J., Fräßle S.P., Srivastava S., Sommermeyer D., Hudecek M., Drexler I., Sadelain M., Liu L., Jensen M.C., Riddell S.R., Busch D.H. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016;126:4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serafini M., Manganini M., Borleri G., Bonamino M., Imberti L., Biondi A., Golay J., Rambaldi A., Introna M. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum. Gene Ther. 2004;15:63–76. doi: 10.1089/10430340460732463. [DOI] [PubMed] [Google Scholar]
- 11.Tasian S.K., Kenderian S.S., Shen F., Ruella M., Shestova O., Kozlowski M., Li Y., Schrank-Hacker A., Morrissette J.J.D., Carroll M. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood. 2017;129:2395–2407. doi: 10.1182/blood-2016-08-736041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung W.H., Gay J., Martin U., Garrett T.E., Horton H.M., Certo M.T., Blazar B.R., Morgan R.A., Gregory P.D., Jarjour J., Astrakhan A. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight. 2019;5 doi: 10.1172/jci.insight.124430. e124430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foster A.E., Mahendravada A., Shinners N.P., Chang W.C., Crisostomo J., Lu A., Khalil M., Morschl E., Shaw J.L., Saha S. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol. Ther. 2017;25:2176–2188. doi: 10.1016/j.ymthe.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu C.Y., Roybal K.T., Puchner E.M., Onuffer J., Lim W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mestermann K., Giavridis T., Weber J., Rydzek J., Frenz S., Nerreter T., Mades A., Sadelain M., Einsele H., Hudecek M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau5907. eaau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber E.W., Lynn R.C., Sotillo E., Lattin J., Xu P., Mackall C.L. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3:711–717. doi: 10.1182/bloodadvances.2018028720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung H.K., Jacobs C.L., Huo Y., Yang J., Krumm S.A., Plemper R.K., Tsien R.Y., Lin M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015;11:713–720. doi: 10.1038/nchembio.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 19.Bonger K.M., Chen L.C., Liu C.W., Wandless T.J. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat. Chem. Biol. 2011;7:531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spencer D.M., Wandless T.J., Schreiber S.L., Crabtree G.R. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 21.Clackson T., Yang W., Rozamus L.W., Hatada M., Amara J.F., Rollins C.T., Stevenson L.F., Magari S.R., Wood S.A., Courage N.L. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang W., Rozamus L.W., Narula S., Rollins C.T., Yuan R., Andrade L.J., Ram M.K., Phillips T.B., van Schravendijk M.R., Dalgarno D. Investigating protein-ligand interactions with a mutant FKBP possessing a designed specificity pocket. J. Med. Chem. 2000;43:1135–1142. doi: 10.1021/jm9904396. [DOI] [PubMed] [Google Scholar]
- 23.Maynard-Smith L.A., Chen L.C., Banaszynski L.A., Ooi A.G., Wandless T.J. A directed approach for engineering conditional protein stability using biologically silent small molecules. J. Biol. Chem. 2007;282:24866–24872. doi: 10.1074/jbc.M703902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang E., Wang L.C., Tsai C.Y., Bhoj V., Gershenson Z., Moon E., Newick K., Sun J., Lo A., Baradet T. Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol. Res. 2015;3:815–826. doi: 10.1158/2326-6066.CIR-15-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clague M.J., Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 26.Mukhopadhyay D., Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 27.Tai H.C., Schuman E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- 28.Richman S.A., Nunez-Cruz S., Moghimi B., Li L.Z., Gershenson Z.T., Mourelatos Z., Barrett D.M., Grupp S.A., Milone M.C. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res. 2018;6:36–46. doi: 10.1158/2326-6066.CIR-17-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banaszynski L.A., Sellmyer M.A., Contag C.H., Wandless T.J., Thorne S.H. Chemical control of protein stability and function in living mice. Nat. Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viaud S., Ma J.S.Y., Hardy I.R., Hampton E.N., Benish B., Sherwood L., Nunez V., Ackerman C.J., Khialeeva E., Weglarz M. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA. 2018;115:E10898–E10906. doi: 10.1073/pnas.1810060115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer I.A., Arteaga C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
- 32.Chu B.W., Banaszynski L.A., Chen L.C., Wandless T.J. Recent progress with FKBP-derived destabilizing domains. Bioorg. Med. Chem. Lett. 2008;18:5941–5944. doi: 10.1016/j.bmcl.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakemura R., Terakura S., Watanabe K., Julamanee J., Takagi E., Miyao K., Koyama D., Goto T., Hanajiri R., Nishida T. A Tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol. Res. 2016;4:658–668. doi: 10.1158/2326-6066.CIR-16-0043. [DOI] [PubMed] [Google Scholar]
- 34.Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Printsev I., Curiel D., Carraway K.L., 3rd Membrane protein quantity control at the endoplasmic reticulum. J. Membr. Biol. 2017;250:379–392. doi: 10.1007/s00232-016-9931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon E.K., Wang L.C., Dolfi D.V., Wilson C.B., Ranganathan R., Sun J., Kapoor V., Scholler J., Puré E., Milone M.C. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellebrecht C.T., Bhoj V.G., Nace A., Choi E.J., Mao X., Cho M.J., Di Zenzo G., Lanzavecchia A., Seykora J.T., Cotsarelis G. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moon E.K., Carpenito C., Sun J., Wang L.-C.S., Kapoor V., Predina J., Powell D.J., Jr., Riley J.L., June C.H., Albelda S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.