

Abstract
Translocase of outer mitochondrial membrane 34 (TOMM34) orchestrates heat shock protein 70 (HSP70)/HSP90–mediated transport of mitochondrial precursor proteins. Here, using in vitro phosphorylation and refolding assays, analytical size-exclusion chromatography, and hydrogen/deuterium exchange MS, we found that TOMM34 associates with 14-3-3 proteins after its phosphorylation by protein kinase A (PKA). PKA preferentially targeted two serine residues in TOMM34: Ser93 and Ser160, located in the tetratricopeptide repeat 1 (TPR1) domain and the interdomain linker, respectively. Both of these residues were necessary for efficient 14-3-3 protein binding. We determined that phosphorylation-induced structural changes in TOMM34 are further augmented by binding to 14-3-3, leading to destabilization of TOMM34's secondary structure. We also observed that this interaction with 14-3-3 occludes the TOMM34 interaction interface with ATP-bound HSP70 dimers, which leaves them intact and thereby eliminates an inhibitory effect of TOMM34 on HSP70-mediated refolding in vitro. In contrast, we noted that TOMM34 in complex with 14-3-3 could bind HSP90. Both TOMM34 and 14-3-3 participated in cytosolic precursor protein transport mediated by the coordinated activities of HSP70 and HSP90. Our results provide important insights into how PKA-mediated phosphorylation and 14-3-3 binding regulate the availability of TOMM34 for its interaction with HSP70.
Keywords: HSP70, molecular chaperone, dimerization, phosphorylation, protein–protein interaction, hydrogen-deuterium exchange, protein folding, protein import, protein kinase A (PKA), translocase of outer mitochondrial membrane 34 (TOMM34), 70-kDa heat shock protein (Hsp70), protein-nucleic acid interaction, 14-3-3 protein, Hsp70, Tomm34
Introduction
Two highly conserved families of molecular chaperones, HSP70 and HSP90, represent core machineries surveying folding and conformational status of cellular proteome (1, 2). Some client proteins of HSP70/HSP90 are either terminally folded or directed for degradation when misfolded to prevent their aggregation (3, 4). A working model for a subset of clients destined to particular cellular compartments, namely mitochondria, suggests that these precursors are kept in a semifolded transfer-competent state by cycling between HSP70 and HSP90 organized in large multiprotein complexes (5–8). To be imported, mitochondrial precursors processed by HSP70/HSP90 complexes are delivered to the translocase of outer mitochondrial membrane (TOM) complex, which serves as the main gate for diverse precursors transferred into mitochondria (5, 7, 9, 10). TOM is a multiprotein complex consisting of receptors and transmembrane channels. TOM20 and TOM22 receptors of TOM facilitate the transport of precursors containing cleavable presequences, as well as β-barrel proteins (11–13). The transfer of noncleavable hydrophobic precursors, such as the carrier precursors, is mediated by TOM70 receptor (5, 8). The precursors delivered via different receptors are then translocated through the transmembrane channel formed by TOM40 β-barrel protein (14). Recent studies in yeast demonstrated that the metabolic switch from respiration to fermentation is accompanied by a decrease of mitochondrial import capacity, which is induced by protein kinase A (PKA)–mediated phosphorylation of TOM components (15–18). These data indicate that mitochondrial import can be tuned by post-translational modification of the TOM import machinery.
In the multiprotein HSP70/HSP90 complexes transferring mitochondrial precursors, a group of co-chaperone proteins containing two or more tetratricopeptide repeat (TPR) domains enable recurrent precursor shuttling by physically bridging HSP70 to HSP90 (6, 7). This process is mediated through accommodation of the C-terminal HSP70/HSP90 EEVD motifs by conserved charged residues in the TPR domains, forming a so-called two-carboxylate clamp (19, 20). The function of TPR domain co-chaperones goes beyond mere scaffolding because they were reported to regulate the ATPase and folding activities of HSP70/HSP90 (6, 21, 22). In higher eukaryotes, the TOMM34 (34-kDa translocase of the outer mitochondrial membrane) co-chaperone, bearing two TPR domains joined by a flexible interdomain linker, participates with HSP70/HSP90 in mitochondrial precursor protein transport in the cytosol (7, 22, 23). At the molecular level, the TPR2 domain of TOMM34 accommodates the EEVD motif of HSP90 (23). The interaction between HSP70 and TOMM34 is more extensive and requires contacts between the ATP-bound conformation of HSP70 and determinants in the TPR1 domain and the interdomain linker (21). Formation of the HSP70·TOMM34 complex leads to rapid disassembly of ATP-bound dimers of human HSP70 (HSPA1A) and inhibition of HSP70/HSP40-mediated refolding (24). Nevertheless, the physiological role of TOMM34 in precursor protein transport is largely unknown.
14-3-3 dimeric adaptor proteins recognizing phosphorylated targets are involved in a plethora of cellular processes such as cell-cycle control, growth factor signaling, apoptosis, and protein trafficking, including precursor protein transport into mitochondria (25–31). The α-helical structure of 14-3-3 monomers is highly similar to TPR domains and contains residues that electrostatically interact with phosphorylated segments of their partner proteins (19, 32). Mode I RXX(pS/pT)X(P/G), mode II RX(F/Y)X(pS/pT)X(P/G), and mode III (pS/pT)X1–2–COOH were identified as optimal binding sites for 14-3-3 proteins; however, noncanonical modes of binding also exist (33–35). Motifs recognized by 14-3-3 proteins are predominantly found in structurally disordered regions of the partner proteins (36). Binding of 14-3-3 adaptors has a variety of effects on the bound phosphorylated proteins, including induction of structural changes and steric blockage of interaction interfaces with other proteins or with DNA (34, 37, 38).
In this study, we identified PKA-modified TOMM34 as a novel interaction partner of 14-3-3 proteins. TOMM34 phosphorylation induces structural changes in its TPR domains that are augmented by 14-3-3 protein binding and modulate the interaction of TOMM34 with HSP70. TOMM34 complexed with 14-3-3γ is largely excluded from interaction with HSP70, leaving HSP70/HSP40-mediated refolding intact. Our data suggest a role of both TOMM34 and 14-3-3 proteins in chaperone-assisted transport of precursors to mitochondria and show that post-translational modification of cytosolic, as well as TOM components of the mitochondrial import cascade, are involved in context-dependent regulation of the mitochondrial proteome.
Results
TOMM34 phosphorylation by PKA induces its binding to 14-3-3 proteins
We have shown previously that the TOMM34 TPR1 domain and the highly flexible interdomain linker (amino acids ∼140–190) play a role in ATP-dependent TOMM34·HSP70 complex formation (21). Because a number of serine and threonine residues are present in the TPR1 domain and the linker, post-translational modifications of these residues might regulate TOMM34 interaction with HSP70. Therefore, we performed a prediction analysis of potential kinase target sites in TOMM34 (Table S2) (39). This analysis revealed two high-scoring PKA motifs containing serine residues: Ser93 localized in the TPR1 domain and Ser160 present in the interdomain linker (Fig. 1a). Both of these sites were shown to be phosphorylated in vivo (40). To test whether PKA mediates phosphorylation of these sites, we performed in vitro phosphorylation of TOMM34 by PKA and analyzed the resulting phosphosites by MS (Fig. 1b and Table S3). We observed that PKA phosphorylates Ser93 and Ser160 with 94 and 65% efficiency, respectively. The higher modification level of Ser93 likely reflects the preference of PKA for the 91RRASA95 motif compared with 157RWNSLP162 (41). Other TOMM34 residues were phosphorylated to considerably lower degrees, with the exception of Ser280 exhibiting 24% modification (Fig. 1b and Table S3). Interestingly, both Ser93 and Ser160 are localized in motifs predicted to be recognized by 14-3-3 adaptor proteins, in contrast to Ser280 (42) (Fig. 1a and Table S4). Moreover, Ser160 resides in a highly flexible region of TOMM34, a structural feature facilitating binding of 14-3-3 proteins (Fig. 1a) (23, 43, 44). To test whether TOMM34 binds 14-3-3 proteins, we performed size-exclusion chromatography (SEC) analysis of PKA-phosphorylated pTOMM34 alone and preincubated with six 14-3-3 isoforms: β, γ, ε, σ, τ, and ζ (Fig. 2 and Fig. S1). Phosphorylation of TOMM34 does not change its SEC mobility, and nonphosphorylated TOMM34 protein does not interact with 14-3-3γ (Fig. 2 and Fig. S1b). The experiments showed that pTOMM34 forms early-eluting complexes with all tested 14-3-3 isoforms but with differing efficiency and peak distribution. Together, these experiments revealed that TOMM34 is readily phosphorylated by PKA at Ser93, Ser160, and Ser280 residues, and this modification enables 14-3-3 isoforms binding.
Figure 1.

Unstructured and structured regions of TOMM34 contain PKA-targeted serine residues. a, prediction of TOMM34 disordered regions (43). The red dashed line denotes the prediction threshold. TOMM34 domain structure with the indicated location and sequence context of the preferentially PKA-targeted serine residues is depicted. The regions of TOMM34 implicated in the interaction with HSP70 (TPR1 domain and interdomain linker) are marked. b, summed extracted ion chromatograms for selected TOMM34 peptides covering PKA phosphorylation sites Ser93 (peptides 79–95), Ser160 (peptides 145–170), and Ser280 (peptides 265–281). Traces for nonphosphorylated (blue) and phosphorylated (red) versions of the peptides derived from nonphosphorylated TOMM34 and PKA-phosphorylated TOMM34 (pTOMM34) are shown. The level of the targeted sites phosphorylation is indicated as percentages.
Figure 2.

PKA-phosphorylated TOMM34 interacts with 14-3-3 proteins. PKA-phosphorylated TOMM34 (T34, 35 μm) was preincubated with various 14-3-3 isoforms (70 μm) for 30 min at 21 °C before separation by analytical SEC.
Ser160 phosphorylation by PKA is crucial for 14-3-3 dimer binding
To define the contribution of individual serine residue phosphorylation on the interactions between pTOMM34 and 14-3-3, we analyzed the binding of in vitro phosphorylated WT, S93A, S160A, and S93A/S160A TOMM34 variants to 14-3-3γ protein using SEC (Fig. 3). WT, pWT TOMM34 and 14-3-3γ proteins eluted as single peaks with elution volumes of 14.3 and 13.7 ml, respectively. Complex pWT·14-3-3γ eluted in a main peak with elution volume of 12.5 ml and a minor peak (shoulder of 14-3-3γ elution peak) eluting at ∼13.1 ml. The elution profile of pS93A/14-3-3γ protein mixture revealed largely intact complex formation; however, the main elution peak shifted to elution volume of 12.8 ml. Importantly, only a small fraction of pS160A/14-3-3γ protein mixture formed a complex with elution volume 12.5 ml, and mixture of p(S93A/S160A) with 14-3-3γ eluted as noninteracting single proteins. Coomassie staining of selected fractions separated in Phos-tag gels (45), together with MS analysis of phosphosites (% of phosphorylated peptides in the selected fractions), enabled the distinction of individual phosphorylated forms (Fig. 3c and Table S3b). We observed that the main elution peak of the pWT·14-3-3γ complex (12.8 ml) contains TOMM34 protein phosphorylated simultaneously at both Ser93 (98%) and Ser160 (99%) residues. Noninteracting pWT fraction (14.3 ml) contains residual nonphosphorylated protein, pSer93 (93%), and only 7% pSer160 protein. Further, 98% of pSer160 in the pS93A/14-3-3γ sample is complexed with 14-3-3γ (elution volume 12.8 ml). Interestingly, because the bulk pWT and pS93A proteins are phosphorylated at Ser160 to 65 and 58%, respectively (Fig. 1 and Table S3a), the 14-3-3γ–interacting fractions are highly enriched for pSer160 modification. Conversely, pSer93 species in pS160A/14-3-3γ protein mixture remain largely in the unbound TOMM34 fraction. These results clearly show that Ser160 phosphorylation by PKA is both necessary and sufficient for the interaction between pTOMM34 and 14-3-3γ. However, the simultaneous presence of a phosphate group on Ser93 and Ser160 accelerates pTOMM34·14-3-3γ complex mobility in SEC, suggesting that the architecture of Ser93/pSer160·14-3-3γ and pSer93/pSer160·14-3-3γ differ. The phosphorylation of Ser280 does not contribute to the interaction between pTOMM34 and 14-3-3γ. To determine the stoichiometry of pTOMM34·14-3-3γ assembly, we performed native electrospray ionization MS with TOMM34/14-3-3γ mixture (Fig. S2). We detected ions corresponding selectively to pTOMM34 complexed with 14-3-3γ dimer, excluding the possibility of 14-3-3γ monomer binding to TOMM34 protein (46).
Figure 3.

Ser160 phosphorylation by PKA is crucial for 14-3-3 dimers binding. a, PKA-phosphorylated TOMM34 variants (T34, 35 μm) were preincubated with 14-3-3γ protein (70 μm) for 30 min at 21 °C before separation by analytical SEC. b, indicated fractions from (a) and from SEC separations of nonphosphorylated TOMM34/14-3-3γ (see Fig. 2 and Fig. S1) were analyzed by gel electrophoresis and Coomassie staining. c, indicated fractions from (a) and (b) as well as nonfractioned proteins were separated in Phos-tag gels retarding the migration of phosphorylated proteins and stained by Coomassie (45). MS-identified phosphoforms of TOMM34 in 14-3-3γ-bound (elution volume, 12.8 ml) and unbound (elution volume, 14.3 ml) fractions are indicated.
Phosphorylation induces destabilization of TOMM34 secondary structure strengthened by 14-3-3γ binding
To gain insight into the structural consequences of TOMM34 phosphorylation by PKA and subsequent 14-3-3γ binding, we performed hydrogen/deuterium exchange (HDX)–MS measurement of nonphosphorylated/phosphorylated WT, S93A, S160A, and S93A/S160A TOMM34 in the presence and absence of 14-3-3γ (Fig. 4). This method measures the level of peptide bond hydrogens exchange for deuterium present in the reaction buffer, reflecting the changes in hydrogen bonding and in solvent accessibility of the corresponding protein regions (47). The deuteration levels were followed on peptides/phosphopeptides derived from TOMM34, pTOMM34 and 14-3-3γ proteins reflecting site-specific structural changes introduced by phosphorylation. Introduction of alanine substitutions (S93A, S160A, and S93A/S160A) had virtually no effect on the overall TOMM34 deuteration, indicating that these mutations do not affect TOMM34 secondary structure (Fig. S3). PKA-mediated phosphorylation of WT TOMM34, targeting preferentially Ser93, Ser160, and Ser280 residues (Fig. 1 and Table S3), led to destabilization of the TPR2 domain detectable at early deuteration intervals (20 s, 2 min) followed by TPR1 domain destabilization at longer incubation times (20 min, 2 h) (Fig. 4a). The analyses of pS93A, pS160A, and p(S93A/S160A) variants revealed that pSer160 has a minor role in phosphorylation-induced changes in TOMM34 (Fig. 4a). Conversely, mutants lacking Ser93 phosphorylation (pS93A and p(S93A/S160A)) exhibited similar deuteration profiles that differ from the pWT protein (Fig. 4a). These differences include slightly reduced destabilization of TPR2 domain opening and particularly the absence of TPR1 destabilization. These findings support the role of Ser93 phosphorylation for the stability of the TPR1 domain. The rapid opening of the TPR2 domain region, localized in proximity to Ser280, is present in all phosphorylated TOMM34 variants, demonstrating that Ser280 phosphorylation regulates local structural features of the TPR2 domain.
Figure 4.

Phosphorylation induces destabilization of TOMM34 secondary structure augmented by 14-3-3γ binding. Time resolved deuteration level differences between selected experimental conditions (indicated on the left side) presented in the form of heat maps. Horizontal gray lines separate individual experimental conditions. Within each experimental condition, the four rows correspond to exchange times (20 s, 2 min, 20 min, and 2 h from top to bottom), indicated on the right side. The vertical gray lines mark sequence positions (spaced by 50 amino acids). a, difference between phosphorylated (p) TOMM34 WT, S93A, S160A, and/or S93A/S160A proteins and their nonphosphorylated forms. TOMM34 domain structure is depicted above the heat map with PKA-modified sites indicated by arrowheads (see Fig. 1). Regions in gray boxes were not covered. b, differences between nonphosphorylated TOMM34 WT and phosphorylated (p) TOMM34 WT, S93A, S160A, and/or S93A/S160A proteins in the presence and absence of 14-3-3γ protein. c, differences between 14-3-3γ protein incubated in the presence or absence of nonphosphorylated TOMM34 WT and PKA-phosphorylated (p) TOMM34 WT, S93A, S160A, and/or S93A/S160A. 14-3-3γ domain structure is depicted above the heat map. d, deuteration level differences between 14-3-3γ protein incubated with and without PKA-phosphorylated TOMM34 protein after 2 h of deuteration (indicated in c by an arrowhead) mapped on the structure of 14-3-3γ dimer in complex with phosphopeptide (in red) (PBD code 6A5S; Ref. 50). Residues involved in phosphoserine interaction (Lys50, Arg57, Arg132, Tyr133, Glu136, Leu177, and Glu185) and α-helices are highlighted in one of 14-3-3γ protomers.
To test the structural effects of 14-3-3γ association with TOMM34, we measured HDX of PKA-phosphorylated WT and phosphoablative variants in the presence or absence of 14-3-3γ (Fig. 4b). We recapitulated that the pSer160 modification is crucial for pTOMM34·14-3-3γ interaction (Fig. 3), because both pS160A and p(S93A/S160A) variants showed lower HDX changes in the presence of 14-3-3γ compared with pWT and pS93A proteins (Fig. 4b). Interestingly, pWT association with 14-3-3γ leads to simultaneous destabilization of both TPR domains (Fig. 4b). Comparable structural loosening in the TPR2 domain is detectable also in the pS93A·14-3-3γ complex, but in contrast, TOMM34 lacking Ser93 phosphorylation has compromised TPR1 destabilization. These observations are in agreement with our SEC analyses (Fig. 3), suggesting that Ser160 phosphorylation is both necessary and sufficient for 14-3-3γ binding to TOMM34, whereas Ser93 phosphorylation is required for additional positioning of bound 14-3-3γ. A subtle structural loosening of the TPR2 domain in the presence of 14-3-3γ is observed also in nonphosphorylated TOMM34, indicating that 14-3-3γ transiently contacts this part of the TOMM34 protein.
HDX levels monitored in 14-3-3γ peptides were largely unchanged by TOMM34 or its variants, mirroring the structural rigidity of dimeric 14-3-3 (48, 49). However, several regions showed differential deuteration (Fig. 4, c and d). We observed phosphorylation-independent opening of the C-terminal end of helix H9 in 14-3-3γ, which was slightly enhanced in the presence of phosphorylated TOMM34 proteins. This region might be responsible for the above-mentioned contacts with the TPR2 domain of nonphosphorylated TOMM34. 14-3-3γ helices 3, 5, and 7, which map to the phosphopeptide-binding groove, were protected from deuterium exchange selectively by phosphorylated TOMM34 (Fig. 4d) (50). Conversely, the protection of helix 8 cannot be ascribed directly to phosphopeptide accommodation and implies the presence of an additional protein–protein interface between phosphorylated TOMM34 and 14-3-3γ as has been shown previously for other 14-3-3 interaction partners (38, 51).
Together, using HDX-MS, we have determined that phosphorylation of TOMM34 Ser93 by PKA is responsible for overall structural opening of the TPR1 domain and has a role in positioning of 14-3-3γ during pTOMM34·14-3-3γ complex formation mediated through phosphorylation of the TOMM34 Ser160 residue. 14-3-3γ is likely to interact with TOMM34 through accommodating its phosphosites in the ligand-binding groove and also by an additional interface localized at helix 8 of 14-3-3γ (38, 50, 51).
Phosphorylation of Ser160 modulates TOMM34 interaction with HSP70
The HDX-MS (Fig. 4, a and b) analyses suggested that TOMM34 phosphorylation by PKA at Ser93 and Ser280 residues destabilizes its TPR1 and TPR2 domains, respectively, and destabilization is further strengthened by pSer160–dependent 14-3-3γ recruitment. Because the TPR1 and TPR2 domains interact with the C termini of HSP70 and HSP90, respectively, to enable efficient binding of the molecular chaperones (21, 23), we tested the interaction of phosphorylated TOMM34 variants with HSP70 and HSP90 C-terminal EEVD peptides in the presence of 14-3-3γ by fluorescence polarization (Fig. 5a). We observed that all nonphosphorylated proteins, as well as phosphorylated TOMM34 variants bearing a S93A mutation, bound HSP70 EEVD peptide with comparable affinity as nonphosphorylated WT protein. Conversely, proteins phosphorylated at Ser93 residue exhibited reduced association with HSP70 EEVD peptide independently of 14-3-3γ. Experiments with HSP90 C-terminal EEVD peptide did not show a phosphorylation/14-3-3γ–dependent decrease of TOMM34 interaction with the peptide (Fig. 5a). 14-3-3γ did not exhibit binding to EEVD peptides. To further delineate the structural consequences of HSP70/HSP90 EEVD peptides binding to phosphorylated TOMM34 variants in the presence of 14-3-3γ, we performed HDX-MS (Fig. 5b and Fig. S4). Differences in deuteration of peptides covering the two-carboxylate clamp residues accommodating HSP70/HSP90 C-terminal motifs revealed an unchanged ability of HSP90 EEVD peptide to induce protection of the TOMM34 TPR2 domain (23). On the contrary, protection of the TPR1 domain, the main binding site for HSP70 EEVD peptide, was reduced (23). Again, the effect of 14-3-3γ on the peptide-induced deuteration changes was low. These results revealed that although destabilization of the TPR1 domain by Ser93 phosphorylation (Fig. 4a) decreases affinity to the HSP70 C terminus (Fig. 5a), the loosening of TPR2 by Ser280 modification is not accompanied by diminished binding of HSP90 EEVD peptide. The role of 14-3-3γ in reducing the affinity of pTOMM34 for HSP70/HSP90 EEVD motifs is negligible.
Figure 5.

Phosphorylation of Ser93 decreases TOMM34 interaction with HSP70 C-terminal EEVD motif. a, equilibrium binding curves of fluorescein-labeled C-terminal HSP70 (GGSGSGPTIEEVD) or HSP90 (GDDDTSRMEEVD) peptides binding to nonphosphorylated and PKA-phosphorylated (p) TOMM34 WT, S93A, S160A, and/or S93A/S160A proteins in the presence or absence of 14-3-3γ measured by fluorescence polarization. Error bars represent S.D.; n = 3 independent experiments. b, deuteration level differences (percentage of deuteration) between selected peptides covering the respective two-carboxylate clamps of TPR1 and TPR2 domains in nonphosphorylated and PKA-phosphorylated (p) TOMM34 WT and/or S93A/S160A proteins detected in the presence of HSP70 (70) and HSP90 (90) C-terminal peptides and in the peptides' absence. The effect of 14-3-3γ presence on peptide-induced deuteration changes is also shown.
Having investigated the binding of HSP70/HSP90 C-terminal peptides, we proceeded with analyses of full-length protein interactions by pulldown with streptavidin-binding peptide (SBP)-tagged proteins (Fig. 6). Because the TOMM34·HSP70 interaction is ATP-dependent (21, 24), the corresponding buffers were supplemented with ATP. All the nonphosphorylated TOMM34 variants interacted with SBP-HSP70 to the same level in the presence of ATP, indicating that the introduced mutations do not affect binding (Fig. 6a). Expectedly, no interaction was detected between SBP-HSP70 and phosphorylated TOMM34 variants in the absence of ATP. Surprisingly, PKA treatment decreased the association of TOMM34 pWT and pS93A proteins with SBP-HSP70, whereas pS160A and p(S93A/S160A) proteins exhibited unaffected binding to SBP-HSP70. These results show that phosphorylation of Ser160, rather than pSer93–mediated structural changes of the TPR1 domain (Figs. 4a), decreases TOMM34 binding to full-length HSP70. SBP-HSP90 pulldown showed that all phosphorylated TOMM34 variants are able to bind HSP90, recapitulating that the opening of the TPR2 domain by Ser280 phosphorylation does not preclude HSP90 binding (Figs. 4a and 5).
Figure 6.

Phosphorylation of Ser160 modulates TOMM34 interaction with HSP70 protein. a, SBP pulldown analysis of the interaction between SBP–HSP70/SBP–HSP90 and nonphosphorylated (Non-phos) and PKA-phosphorylated (Phos) TOMM34 protein variants. Because the interaction of TOMM34 with HSP70 is ATP-dependent (21), these experiments were performed in the presence or absence of ATP. b, SBP pulldown analysis of SBP–HSP70/SBP–HSP90 interaction with nonphosphorylated and PKA-phosphorylated TOMM34 and/or S93A/S160A proteins in the presence and the absence of 14-3-3γ. C+ denotes purified 14-3-3γ protein used as a positive control for Western blotting.
To delineate the influence of 14-3-3γ binding on the interactions between TOMM34 and HSP70, we performed SBP-pulldown assay using the S93A/S160A variant that is unable to bind 14-3-3γ (Fig. 3). We detected a phosphorylation-dependent and 14-3-3γ–independent decrease of TOMM34 binding to SBP-HSP70 (Fig. 6b). 14-3-3γ protein was not co-precipitated with residual WT TOMM34 interacting with SBP-HSP70. This result indicates that the pWT·14-3-3γ complex is excluded from interaction with SBP-HSP70. Conversely, SBP-HSP90·pWT·14-3-3γ assembly is effectively pulled down using the same experimental setup (Fig. 6b). Collectively, the data show that although phosphorylation of Ser93 decreases binding of the TOMM34 TPR1 domain to HSP70 EEVD peptide, the interaction of full-length HSP70 with phosphorylated TOMM34 is precluded mainly by phosphorylation at Ser160.
pTOMM34 complexed with 14-3-3γ dimer does not disrupt ATP-dependent HSP70 dimer
We next used SEC to further study interactions between HSP70, TOMM34, and 14-3-3γ, because this method does not require immobilization (Fig. 7, a and b). All analyses were performed in the presence of ATP. First, we evaluated HSP70 and TOMM34 protein samples in the absence of 14-3-3γ. HSP70 eluted as a major peak with elution volume 11.9 ml, which corresponds to the ATP-bound HSP70 dimer (24). The HSP70 dimer was disrupted by interaction with nonphosphorylated TOMM34, forming a complex with 1:1 stoichiometry (24) eluting at ∼12.4 ml. Interestingly, the presence of phosphorylated TOMM34 also led to HSP70 dimer disruption, although the pTOMM34·HSP70 complex was less abundant, and a significant portion of HSP70 redistributed into early-eluting oligomeric fractions. This observation suggests that the decreased level of pTOMM34 binding to HSP70 detected by pulldown analysis (Fig. 6a) is caused by modulated mechanism/kinetics of pTOMM34/HSP70 interaction compared with nonphosphorylated TOMM34 protein. To unravel the role of 14-3-3γ in TOMM34·HSP70 complex formation, we first analyzed HSP70/14-3-3γ protein mixture, showing that 14-3-3γ does not interact with the ATP-bound HSP70 dimer and leaves it intact (Fig. 7a). Nonphosphorylated TOMM34 interacts with HSP70, disrupting its dimeric structure independently of the presence of 14-3-3γ. Strikingly, pTOMM34/HSP70/14-3-3γ mixtures eluted as two main peaks with elution volumes 12.5 and 13.1 ml, respectively. Although the peak at 13.1 ml corresponds to pTOMM34·14-3-3γ late eluting assembly enriched for HSP70 monomers, the 12.5-ml peak contains early-eluting pTOMM34·14-3-3γ complex together with HSP70, as shown by Coomassie staining (Fig. 7, a and b). That the elution profile of pTOMM34·14-3-3γ complexes is unchanged by the presence of HSP70 (Fig. 7b) strongly suggests that pTOMM34 complexed with 14-3-3γ is largely excluded from interaction with HSP70. This is further demonstrated by the presence of a 12.5-ml peak shoulder corresponding to ATP-bound HSP70 dimer (elution volume of ∼11.9 ml).
Figure 7.

Binding of pTOMM34 to 14-3-3γ dimer prevents disruption of ATP-dependent HSP70 dimer. a, phosphorylated/nonphosphorylated TOMM34 WT preincubated with/without 14-3-3γ (30 min, 21 °C) was mixed with HSP70 in the presence of 0.2 mm ATP and incubated 20 min at 21 °C before separation by analytical SEC. The final concentrations of TOMM34, 14-3-3γ, and HSP70 in the protein mixtures were 35, 70, and 35 μm, respectively. b, indicated fractions from (a) were analyzed by gel electrophoresis and Coomassie staining. c, intact or PKA-phosphorylated WT and/or S93A/S160A (final concentration, 60 μm) proteins were mixed with 14-3-3γ (final concentration, 120 μm) or buffer. HSP70 protein (60 μm) preincubated with or without ATP (0.4 mm) was added to TOMM34/14-3-3γ samples in 1:1 ratio, and the mixture was chemically cross-linked by glutaraldehyde addition. The reactions were stopped after 10 min with Tris, pH 8, and separated by SDS-PAGE, blotted, and probed with anti-TOMM34 and HSP70 antibodies. Molecular mass markers and captured protein assemblies are indicated by numbers and dots, respectively.
To further test whether pTOMM34 complexed with 14-3-3γ loses its interaction with ATP-bound HSP70 dimers, we performed chemical cross-linking with glutaraldehyde and analyzed the resulting complexes by SDS-PAGE/Western blotting using antibodies recognizing HSP70, TOMM34, and 14-3-3γ proteins (Fig. 7c and Fig. S5). ATP-induced HSP70 dimer migrated at ∼150 kDa. The addition of nonphosphorylated WT TOMM34 and S93A/S160A proteins led to HSP70 dimer disruption, and comparable levels of HSP70·WT and HSP70·S93A/S160A complexes appeared migrating at 110 kDa. In contrast, both pWT and p(S93A/S160A) proteins disrupted HSP70 dimers; however, the level of pWT·HSP70 assembly was lower compared with the p(S93A/S160A)·HSP70 complex. This observation corresponds with our SEC analysis of pWT/HSP70 samples (Fig. 7a) showing that pTOMM34 contacts HSP70 dimer inducing its disassembly, but the resulting pTOMM34·HSP70 complex is less stable. Importantly, the simultaneous presence of p(S93A/S160A) and 14-3-3γ led to HSP70 dimer disassembly, and a p(S93A/S160A)·HSP70 complex was formed. Conversely, a significant fraction of HSP70 dimer is preserved in the presence of the pTOMM34·14-3-3γ complex, and the level of pTOMM34·HSP70 assembly is strongly decreased. Surprisingly, pTOMM34·14-3-3γ assembly was detected migrating at ∼70 kDa by TOMM34 antibody, indicating 1:1 stoichiometry (Fig. 7b), which is in contrast to our native ESI analysis of pTOMM34·14-3-3γ complex stoichiometry (1:2, Fig. S2). The corresponding signal for p(S93A/S160A)·14-3-3γ complex is less populated. Detection using 14-3-3γ antibody showed the presence of 14-3-3γ monomers/dimers and only a weak signal for corresponding pTOMM34·14-3-3γ dimer tripartite complex (Fig. S5). Coomassie staining of pWT and p(S93A/S160A) proteins cross-linked to 14-3-3γ revealed clear separation of pWT, p(S93A/S160A) and 14-3-3γ monomers/dimers (Fig. S5). These observations suggest that the pTOMM34 complex with 14-3-3γ dimer is inefficiently cross-linked and decays in the presence of SDS leaving only traces of pTOMM34·14-3-3γ assembly with 1:1 stoichiometry detectable by Western blotting. Alternatively, the epitopes of antibodies raised against 14-3-3γ and TOMM34 are not readily accessible in the cross-linked tripartite complex. Taken together, the SEC and chemical cross-linking analyses revealed that the modulated interaction of phosphorylated TOMM34 protein with HSP70 is largely prevented by interaction of pTOMM34 with 14-3-3γ dimers, leaving the ATP-bound HSP70 dimer intact.
Binding of pTOMM34 to 14-3-3γ eliminates TOMM34's inhibitory role in HSP70-mediated refolding
TOMM34 protein at high concentrations inhibits HSP70/HSP40-mediated luciferase refolding, likely through HSP70 dimer disruption (21, 24). Therefore, we tested the influence of TOMM34 phosphorylation by PKA on HSP70/HSP40-mediated luciferase refolding (Fig. 8a). We detected that both WT and S93A/S160A proteins either phosphorylated or nonphosphorylated completely inhibited luciferase refolding. This observation reflects the finding that HSP70 dimers are disrupted by both modified and unmodified TOMM34 proteins (Fig. 7). Next, we monitored HSP70/HSP40-mediated luciferase refolding in the presence of constant pTOMM34 concentration and increasing concentrations of 14-3-3γ (Fig. 8b and Fig. S6). pTOMM34-inhibited luciferase refolding was recovered by 14-3-3γ in a concentration-dependent mode. At equimolar concentrations of pTOMM34 and 14-3-3γ dimers (5 μm), the refolding efficiency reached the level of TOMM34-free refolding mixtures. Together, the refolding assays suggest that complex formation between pTOMM34 and 14-3-3γ prevents TOMM34-mediated decomposition of ATP-bound HSP70 dimers and allows for productive luciferase refolding.
Figure 8.

Sequestration of pTOMM34 by 14-3-3γ eliminates TOMM34's inhibitory role in HSP70-mediated refolding. a, firefly luciferase incubated with HSP70 (1 μm), HSP40 (2 μm), and BAG1 (0.5 μm) proteins was thermally denatured at 42 °C for 30 min in the presence of 5 μm nonphosphorylated/PKA-phosphorylated TOMM34 WT and S93A/S160A proteins. The kinetics of luciferase reactivation was measured after shifting the reaction temperature to 37 °C. b, firefly luciferase denatured as in (a) was refolded in the presence of 5 μm PKA-phosphorylated TOMM34 WT protein and increasing concentrations of 14-3-3γ dimer (0.3, 1.25, and 5 μm). The signal from samples with native luciferase was set as 100%. As negative controls, we measured the luciferase activity of denatured luciferase only. Error bars represent S.D.; n = 3 independent experiments.
14-3-3 Isoforms interact with TOMM34 ex vivo
To verify whether the interactions between 14-3-3 isoforms and pTOMM34 detected in vitro (Fig. 2) are physiologically relevant, we performed ex vivo pulldown of HA-tagged TOMM34 WT and S93A/S160A proteins reintroduced into TOMM34−/− MCF-7 cells (52) using biotinylated 14-3-3 proteins (Fig. 9). To induce cellular signaling, the cells were treated with 12-O-tetradecanoylphorbol-13-acetate (PMA) (53–55). Because PKA kinase is encoded by several genes in the human genome (56), we decided to use H89 PKA kinase inhibitor with relaxed specificity (57, 58) to ensure robust inhibition of all kinases sharing PKA-like activity. This allowed us to distinguish phosphorylation/PKA-dependent pTOMM34·14-3-3 complexes. We observed that all the analyzed 14-3-3 isoforms (β, γ, ε, σ, τ, and ζ) interacted with TOMM34 in a PMA-dependent manner, and the interaction was decreased in PMA/H89 co-treated samples. S93A/S160A variant also exhibited PMA-induced binding to 14-3-3 isoforms that was sensitive to H89; however, the levels of pulled down S93A/S160A protein were lower. These results indicate that TOMM34 interaction with 14-3-3 isoforms takes place in cells and is heavily dependent on Ser93/Ser160 phosphorylation by PKA or possibly other kinases inhibited by H89 inhibitor (57). Nevertheless, the residual PMA-induced S93A/S160A interaction with 14-3-3 isoforms suggests that an pSer93– and pSer160–independent mechanism of TOMM34/14-3-3 binding also exists in vivo.
Figure 9.
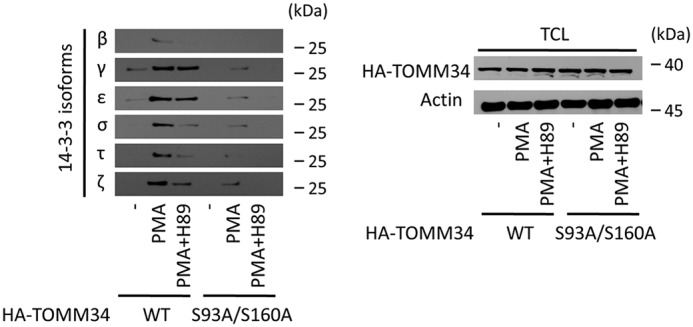
14-3-3 isoforms interact with TOMM34 ex vivo in a phosphorylation-dependent manner. TOMM34−/− MCF-7 cells transfected with pCMV-N-HA-TOMM34 WT and pCMV-N-HA-TOMM34-S93A/S160A constructs were treated with PMA (0.3 μm), PKA inhibitor H89 (20 μm), and vehicle control (DMSO, -) for 12 h, collected, and lysed. Next, biotinylated 14-3-3 isoforms prebound to streptavidin–agarose beads were incubated with the cell lysates at 4 °C for 1 h. The eluted complexes were analyzed by gel electrophoresis and Western blotting using anti-HA antibody.
Discussion
TOMM34·HSP70 complex formation requires both accommodation of the HSP70 EEVD C-terminal motif by the TOMM34 TPR1 domain and docking of so-far-unknown determinants in the TOMM34 interdomain linker to HSP70 in the ATP-bound conformation (21, 23). The TPR1 domain and the interdomain linker of TOMM34 contain several serine/threonine residues, suggesting that their modification might regulate interaction between TOMM34 and HSP70.
Here, we have shown that Ser93 (TPR1 domain) and Ser160 (interdomain linker) TOMM34 residues are effectively phosphorylated by PKA (Fig. 1 and Tables S2 and S3). The phosphorylation of Ser93 induces destabilization of the TPR1 domain (Fig. 4a). Opening of the TPR1 domain can be ascribed to repulsive electrostatic interactions introduced by the phosphate group into a structurally stable region of TPR1 domain (Fig. 1a) (23, 59). These structural rearrangements are a likely cause of diminished HSP70 EEVD peptide binding because the two-carboxylate clamp of TPR1 domain preferentially accommodates the C terminus of HSP70 (Fig. 5b) (23). However, the interaction of the phosphorylated S160A TOMM34 variant (bearing pSer93) with full-length HSP70 is unaffected (Fig. 6a). This indicates that the reduced binding of the HSP70 EEVD motif into a destabilized TPR1 domain does not preclude the formation of a high-affinity interaction interface between ATP-bound HSP70 and TOMM34 in vitro (21). PKA modification of residue Ser93 might, however, regulate the TOMM34/HSP70 interaction in vivo, where the TOMM34 TPR1 domain competes with other TPR co-chaperones for binding to the HSP70 C terminus (60, 61). Analogously, phosphorylation of Ser280 residue interferes with TPR2 stability (Fig. 4a), however, without reducing the affinity of HSP90 C-terminal peptide toward this domain in vitro (Fig. 5).
The phosphorylation of Ser160 does not have an effect on TOMM34 structure (Fig. 4a). This observation is in line with that fact that Ser160 is located in the flexible solvent-exposed interdomain linker (23) (Fig. 1a). Importantly, Ser160 phosphorylation modulated the TOMM34/HSP70 interaction (Figs. 6–8). Although the ATP-bound HSP70 dimer (24) is effectively disrupted by phosphorylated TOMM34, the level of pTOMM34·HSP70 assembly is substantially reduced compared with TOMM34·HSP70 (Fig. 7, a and b) unless stabilized by chemical cross-linking (Fig. 7c and Fig. S5). Because the TOMM34 interdomain linker is involved in the entropically driven TOMM34 interactions with HSP70, the presence of the phosphate group at Ser160 is likely to preclude the formation of a stable protein–protein interface (21). The exact topology of this interface is unknown. These observations also indicate that dissociation of ATP-bound HSP70 dimers is independent of the stable TOMM34·HSP70 complex formation. Accordingly, pTOMM34 exhibits an inhibitory effect on HSP70/HSP40-mediated refolding (Fig. 8), which requires the presence of ATP-dependent HSP70 dimers (24). It is of note that disassembly of HSP70 dimers selectively by pTOMM34 is accompanied by HSP70 oligomerization (Fig. 7, a and b). We speculate that the released HSP70 monomers lacking stabilization through interaction with TOMM34 oligomerize to form a mixed population of molecules in different nucleotide states (21, 62). However, detailed analysis of these oligomers is yet to be performed. Taken together, the mechanism of PKA phosphorylation-induced reduction of TOMM34 binding to HSP70 is 2-fold: pSer93 diminishes TPR1-EEVD interactions, and pSer160 perturbs the interaction interface formed between the TOMM34 interdomain linker and HSP70.
Structural changes triggered in proteins phosphorylated at particular motifs are often secured by subsequent binding of 14-3-3 proteins (63–65). All tested 14-3-3 isoforms (β, γ, ε, σ, τ, and ζ) bound phosphorylated TOMM34 in vitro and ex vivo (Figs. 2 and 9), and pTOMM34·14-3-3γ assembly was analyzed in detail. Phosphorylation of Ser160 in the context of the 152RXX(pS/pT)X(P/R)WNSLP162 consensus binding motif localized in the flexible interdomain region is both necessary and sufficient for pTOMM34·14-3-3γ interaction (Figs. 3 and 4) (36, 65). However, the distinct SEC profiles of pWT and pS93A proteins in complex with 14-3-3γ (Fig. 3) indicate that the architecture of these assemblies differ. The structural analysis supports this notion by showing that pS93A retains 14-3-3γ–induced TPR2 destabilization but lacks the opening of TPR1 domain observed in pWT protein (Fig. 4b). 14-3-3γ forms a structurally stable dimer (48) that interacts with TOMM34 monomer (Figs. 4 and 7b and Figs. S2 and S5). We suggest that the phosphorylated Ser160 residue serves as a “gatekeeper” enabling the initial contact of TOMM34 with one of the two amphipathic grooves in the 14-3-3γ dimer (66, 67). Subsequently, the phosphorylated Ser93 residue can be accommodated by the second amphipathic groove. Although the 90RRASAY95 motif deviates from the consensus mainly by lacking proline residue at +2 positon (65), it was shown that the amphipathic grooves of 14-3-3 proteins can coordinate noncanonical sequences (34). Simultaneous binding of two distinct phosphosites in one peptide/protein to 14-3-3 dimers has been previously described (34, 68–70). Moreover, a bidentate 14-3-3 binding was shown to induce conformational changes in Nth11–751 enzyme regulating its activity (34). The presence of a flexible solvent-exposed interdomain linker allows the TPR1 and TPR2 domains of TOMM34 to be mutually oriented with a certain degree of freedom (23, 71). Thus, simultaneous accommodation of phosphorylated Ser93 and Ser160 residues by structurally rigid 14-3-3γ (Fig. 4, c and d) (32, 48, 66) might force TOMM34 TPR domains to assume a more defined orientation. As TOMM34 becomes generally more solvent-accessible in the presence of 14-3-3γ (Fig. 4b), the bidentate binding mode of 14-3-3γ might impose constraints on TOMM34 structure, leading to its structural loosening. This mode of interaction is rather unusual because studies dealing with the structural consequences of 14-3-3 binding mostly report 14-3-3–induced stabilization of the partner protein (38, 49, 51). However, the fast deuteration kinetics of the flexible protein regions may abrogate the detection of the stabilizing interaction event, as seen with other 14-3-3–interacting proteins (38). Similarly, the protein–protein contacts mediated mainly by electrostatic and side-chain interactions may also show minimal or no detectable changes in peptide bond hydrogen-exchange rates (23, 38, 49). As the gatekeeper pSer160 modification resides in a flexible part of TOMM34 protein (Fig. 1), the putative local stabilizing effects of 14-3-3 binding, therefore, could be undetectable by HDX-MS. Moreover, our data support the existence of an additional interaction interface between pTOMM34 and 14-3-3γ involving the surface of helix 8 (Fig. 4, b–d). Similar interfaces encompassing helix 8 of 14-3-3 proteins have been reported previously (38, 51). Conversely, phosphorylation-independent transient contacts are likely to occur between the TOMM34 TPR2 domain and helix 9 of 14-3-3γ (Fig. 4, b–d) (72, 73). Helix 9 is commonly a part of phosphopeptide-binding grooves and becomes stabilized by the presence of phosphorylated protein partner (38, 51). In contrast, helix 9 was also shown to exhibit intrinsic flexibility in the presence of nonphosphorylated interaction partner that was increased in the presence of phosphorylated interactor, allowing the assembly to reach a more compact conformation (67). This model is supported by our HDX data (Fig. 4, c and d), although more detailed structural data are needed to fully delineate pTOMM34·14-3-3γ complex architecture and the role of helix 9.
Importantly, formation of a complex of pTOMM34 with 14-3-3γ dimer eliminates its ability to disrupt ATP-bound HSP70 dimers and interact with HSP70 (Fig. 7), allowing HSP70/HSP40-mediated refolding in the presence of pTOMM34 (Fig. 8b). We propose that 14-3-3γ sterically occludes the TOMM34 interdomain linker, thereby preventing its interaction with ATP-bound HSP70 (21). Conversely, destabilization of the TPR2 domain in pTOMM34·14-3-3γ assembly (Fig. 4b) does not inhibit HSP90·pTOMM34·14-3-3γ complex formation (Fig. 6). Thus, Ser160 phosphorylation and subsequent 14-3-3 protein binding might serve as a specific mechanism for suspending HSP70 from TOMM34 alone or as a part of HSP70·TOMM34·HSP90 tripartite complex (Fig. 10) (21, 23). Whether this mode of TOMM34·14-3-3 interaction is generic for other 14-3-3 isoforms awaits further investigation; however, peak distribution heterogeneity of TOMM34 complexes with different 14-3-3 isoforms (Fig. 2) suggests possible structural/functional diversification of TOMM34·14-3-3 assemblies (32, 48).
Figure 10.

TOMM34 interaction with HSP70 dimers is regulated by its PKA-mediated phosphorylation and 14-3-3 binding. ATP-bound HSP70 antiparallel dimers cooperate with HSP40 during protein substrate (dotted red line) processing (24). a, nonphosphorylated TOMM34 stably interacts with HSP70 in the ATP-bound state through TPR1-EEVD (c) contacts and an interface encompassing the TOMM34 interdomain linker (21, 24). This interaction leads to HSP70 dimer disruption, preventing substrate processing. b, PKA mediates TOMM34 phosphorylation (pTOMM34) on Ser93 and Ser160 residues (red circles), leading to structural loosening of the TPR1 domain and perturbation of the interaction interface formed between the TOMM34 interdomain linker and HSP70. pTOMM34 transiently interacts with HSP70, disrupting its dimeric structure and substrate processing activity, but a stable pTOMM34·HSP70 complex is not formed. c, 14-3-3 binding to pTOMM34 sequesters TOMM34 from HSP70 dimers leaving their substrate processing activity intact.
What is the physiological outcome of PKA-mediated TOMM34 interaction with 14-3-3 proteins? From a systemic perspective, TOMM34 (74) was reported to be transactivated by NRF1 and NRF2, transcription factors governing the expression of mitochondrial respiratory subunits (75) and Tom20/Tom70 translocases (76, 77). At the protein level, TOMM34 participates on preprotein transport in the cytosol as a component of large multichaperone complexes in which it coordinates HSP70 and HSP90 (7, 23). Elevated levels of TOMM34 inhibited import of mitochondrial precursor proteins containing N-terminal presequences translocated by the Tom20 pathway (ornithine transcarbamylase), as well as of carrier proteins (phosphate carrier, PiC; ADP/ATP carrier protein, ACC) characterized by multiple integral targeting elements imported via a Tom70-dependent mechanism (7, 8, 11, 78). In contrast, an antibody against TOMM34 blocked mitochondrial import of preproteins bearing presequences (79). These data suggest that tightly regulated levels of TOMM34 are necessary for effective transport of a variety of import-competent precursors from ribosomes to mitochondria. In recent years, PKA has been shown to suppress mitochondrial protein import by phosphorylating Tom70 (15), Tom40 (16), and Tom22 (17) translocases upon metabolic switch from respiratory to glycolytic metabolism. Interestingly, PKA can be sequestered to the proximity of the outer mitochondrial membrane by protein kinase A anchor proteins (AKAPs) (80, 81) to perform a plethora of functions including regulation of apoptosis by Bad protein phosphorylation (18, 82). Local accumulation of PKA at the surface of TOM might therefore integrate signaling and metabolic demands of the cell by tuning of mitochondrial import pathways, with TOMM34 as a newly identified PKA target. In mammals, 14-3-3 proteins were first described to participate in mitochondrial import by recognizing presequences of preprotein; however, later studies showed that they also facilitate the import of β-barrel proteins lacking presequences (26–31). These observations reveal a more general role of 14-3-3 proteins in mitochondrial import, although the exact mechanism of their action remains elusive. A recurrent bidirectional transfer of precursor proteins between HSP70 and HSP90 seems to be vital for maintaining their import-competent conformation while preventing aggregation (5, 6). We suggest that the action of TOMM34 and 14-3-3 proteins coordinated by PKA phosphorylation of TOMM34 helps to fine-tune shuttling of various transport-competent precursors between HSP70 and HSP90 by timely destabilization of TOMM34 binding to HSP70. This process would be further regulated by the level of PKA activation under different signaling and metabolic circumstances (15–17).
Collectively, we have described TOMM34 protein phosphorylated by PKA as a novel interaction partner of 14-3-3 adaptor proteins. This interaction, established via phosphorylation of residues Ser160 and Ser93, leads to large structural changes in TOMM34 and prevents TOMM34 binding to HSP70 eliminating its inhibitory role on HSP70/HSP40-mediated refolding. Because the mechanism of HSP70/HSP90–regulated precursor protein transport to mitochondria is largely unknown, our data suggest novel testable hypotheses about the role of TOMM34 and 14-3-3 proteins in this process.
Experimental procedures
Cloning and protein preparation
All coding sequences were cloned by Gateway recombination technology (Invitrogen). The full coding sequences of the human TOMM34 (NM_006809.4), DNAJB1 (HSP40, NM_006145.2), and human BAG1 (BAG1s, NM_001172415.1) genes, sequences coding for TOMM34 point mutants (S93A, S160A, S93A/S160A), were cloned into a vector containing an N-terminal His6-GST tag cleavable by tobacco etch virus protease. The sequences coding for TOMM34 WT and S93A/S160A point mutant were furthermore cloned into a vector containing an N-terminal SBP tag. The full coding sequences of the human HSPA1A (HSP70, NM_005345.5) and HSP90AA1 (HSP90α, NM_001017963.2) genes were cloned into a vector containing an N-terminal His6 or SBP tag cleavable by tobacco etch virus protease. All cloned genes were expressed in BL21(DE3) RIPL cells and purified as described previously (21). 14-3-3 protein isoforms (β, γ, ε, σ, τ, and ζ) were cloned into a vectors containing either N- or C-terminal His6 tag (see Table S1) and expressed in BL21(DE3) RIPL. Bacteria expressing 14-3-3 proteins were grown in LB medium at 37 °C with an A600 of up to 0.5. Induction of gene expression was achieved by adding isopropyl β-d-thiogalactopyranoside to the culture (final concentration, 1 mm). The bacterial culture was grown at 30 °C for another 3-4 h and then pelleted by centrifugation. Next, the cells were resuspended in His-binding buffer (50 mm Hepes, pH 7.6, 0.3 m KCl, 5 mm imidazole, 5% glycerol). Cell suspensions were enriched with lysozyme (1 mg/ml) and phenylmethylsulfonyl fluoride (1 mm) and then sonicated. Bacterial lysates were obtained by centrifugation for 30 min at 12,000 × g. His6-tagged 14-3-3 proteins were captured on a HisTrap column and eluted with 250 mm imidazole. The eluted fractions were subsequently subjected to buffer exchange into His-binding buffer to remove imidazole. All proteins were finally exchanged into final assay buffers using 7-kDa molecular mass cutoff Zeba spin desalting columns (Thermo Fisher Scientific).
Cell culture, transfection, and treatment
TOMM34−/− MCF-7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 300 mg/liter l-glutamine in a humidified atmosphere of 5% CO2 at 37 °C. Transient transfections of cells (grown to 70% confluency on 15-mm-diameter dishes) with the plasmids pCMV-N-HA-TOMM34 WT and pCMV-N-HA-TOMM34-S93A/S160A (10 μg of DNA) were performed by Lipofectamine 3000 according to the manufacturer's manual (Thermo Fisher Scientific). 12 h after transfection, the cells were split to three 15-mm-diameter dishes and cultured for another 6 h before treatment with PMA (0.3 μm), PKA inhibitor H89 (Selleckchem, Munich, Germany, 20 μm), and vehicle control (DMSO) for 12 h. Next, the cells were collected and lysed in 50 mm Hepes, pH 7.5, 150 mm KCl, 2 mm MgCl2, 5 mm NaF, 1% Nonidet P-40 supplemented with proteinase and phosphatase inhibitors mixture (Sigma–Aldrich).
Site-directed mutagenesis
The full-length TOMM34 coding sequences were mutated to encode desired mutations using QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) and mutagenic oligonucleotides according to the manufacturer's instructions.
In vitro phosphorylation assay
TOMM34 and all its variants (40 μm) were incubated with PKA (New England Biolabs, Ipswich, MA, USA; 62,500 units/mg TOMM34) and ATP (2 mm) in PKA assay buffer at 30 °C for 24 h. After incubation, the phosphorylation reaction was stopped by exchanging the buffer to 50 mm Hepes, pH 7.5, 150 mm KAc, and 2 mm MgCl2 (HKM buffer).
Analytical size-exclusion chromatography
Separations by SEC were carried out using Superdex 200 Increase 10/300 GL (GE Healthcare) pre-equilibrated with HKM buffer. 75 μl of phosphorylated/nonphosphorylated TOMM34 proteins (35 μm) preincubated with/without different 14-3-3 isoforms (70 μm) for 30 min at 21 °C was injected and isocratically eluted at 0.3 ml/min. In experiments testing the influence of TOMM34·14-3-3γ complex formation on HSP70 dimers, phosphorylated/nonphosphorylated TOMM34 preincubated with/without 14-3-3γ (30 min, 21 °C) was mixed with HSP70 in the presence of 0.2 mm ATP and incubated for 20 min at 21 °C. The final concentrations of TOMM34, 14-3-3γ, and HSP70 in the protein mixtures were 35, 70, and 35 μm, respectively. Injection volume and flow rate was as described above. From each run, 300-μl fractions corresponding to protein peaks were collected and analyzed using standard or Phos-tag (Wako Pure Chemical Industries, Osaka, Japan) gel electrophoresis followed by Coomassie staining.
Pulldown assay for biotinylated proteins ex vivo
10 μg of biotinylated 14-3-3 isoforms (EZ-LinkTM Thermo Fisher Scientific) per sample were prebound to streptavidin–agarose beads and incubated at 4 °C for 1 h with TOMM34−/− MCF-7 cell lysates prepared after cell transfection and treatment as described above. After washing with assay buffer (50 mm Hepes, pH 7.5, 150 mm KCl, 2 mm MgCl2, 5 mm NaF, 1% Nonidet P-40), the bound complexes were eluted by boiling the beads in × loading sample buffer for 10 min and analyzed by SDS-PAGE and Western blotting.
SBP pulldown assays
Before performing pulldown experiments, all proteins were exchanged to HKM buffer. Next, 60 pmol of SBP-tagged HSP70 or HSP90 was incubated with streptavidin–agarose beads at 4 °C for 30 min After washing with assay buffer, 60 pmol of phosphorylated/nonphosphorylated TOMM34 variants (WT or mutant proteins) preincubated with/without 120 pmol of 14-3-3γ (30 min, 21 °C) were added to the beads. Mixtures containing SBP–HSP70 were supplemented with/without 0.2 mm ATP. After 1 h of incubation at 4 °C, the beads were washed with ATP-containing/ATP-free buffers. Finally, the proteins were eluted with 2 mm biotin in assay buffer and analyzed by Coomassie staining or Western blotting.
Antibodies
Monoclonal anti-TOMM34 and anti-HA tag antibodies used in this study were prepared in-house. Monoclonal anti-14-3-3γ was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). For SBP-tagged protein detection, we used peroxidase-conjugated streptavidin (Sigma–Aldrich) diluted in 5% milk in the presence of avidin (10 ng/µl). Avidin was added to prevent streptavidin binding to biotinylated proteins present in the milk. The blots were developed with polyclonal anti-mouse/rabbit IgG secondary antibodies conjugated with horseradish peroxidase (Dako, Santa Clara, CA, USA).
Luciferase refolding assay
Firefly luciferase (50 nm, Promega) incubated with HSP70 (1 μm), HSP40 (2 μm), and BAG1 (0.5 μm) proteins in HKM buffer supplemented with 5 mm DTT, 5 mm ATP was thermally denatured at 42 °C for 30 min in the presence of 5 μm phosphorylated/nonphosphorylated TOMM34 WT or S93A/S160A and increasing concentrations of 14-3-3γ dimer (0.3, 1.25, and 5 μm). The refolding reaction was started by shifting the temperature to 37 °C. At given time points, 2 µl of the reaction were mixed with d-luciferin (100 μm in 100 µl of 50 mm glycylglycine, pH 7.8, 30 mm MgSO4, 4 mm DTT), and the luciferase activity was measured at 21 °C using an Infinite M1000 Pro (Tecan, Männedorf, Switzerland) at emission wavelengths of 560 nm and 500/100 ms settle/integration time. The signal from samples with native luciferase was set as 100%. As negative controls, we measured the luciferase activity of denatured luciferase only.
Peptide binding
Phosphorylated/nonphosphorylated TOMM34 WT, S93A, S160A, and S93A/S160A in various concentrations were titrated against 30 nm fluorescein-labeled C-terminal HSP70 (GGSGSGPTIEEVD) or HSP90 (GDDDTSRMEEVD) peptide in HKM buffer supplemented with 0.01% Tween 20. In titrations containing 14-3-3γ protein, the molar ratio between 14-3-3γ and TOMM34 was 2:1. All reactions were carried out in a total volume of 12 µl in a 384-well black Nunc Plate (Thermo Fisher Scientific). The plate was incubated for 1 h at room temperature with shaking. Fluorescence polarization was measured at 21 °C using an Infinite M1000 Pro (Tecan) with excitation and emission wavelengths of 470 and 520 nm, respectively. The analysis was performed using GraphPad Prism, version 5.03, for Windows (GraphPad Software, San Diego, CA, USA).
Chemical cross-linking
Intact or PKA-phosphorylated TOMM34 WT or S93A/S160A proteins (final concentration, 60 μm) were mixed with 14-3-3γ (final concentration, 120 μm) or buffer in HKM buffer supplemented with 0.4 mm ATP and incubated for 30 min at 21 °C. Next, HSP70 protein (60 μm) preincubated with or without ATP (0.4 mm) for 30 min at 21 °C was added to TOMM34/14-3-3γ samples in 1:1 ratio, and the mixture was incubated for 1 min at 21 °C. Glutaraldehyde was added (final concentration, 1 mm), and the reactions stopped after 10 and 20 min with Tris, pH 8 (final concentration, 80 mm). The samples were diluted 100× in 2× CSB loading buffer, and 5 µl was separated by SDS-PAGE, blotted, and probed with TOMM34, HSP70, and 14-3-3γ antibodies.
MS and hydrogen/deuterium exchange
Native ESI-MS analysis of TOMM34 and 14-3-3γ was performed on proteins alone or mixed in 1:2 molar ratio (TOMM34:14-3-3γ, 10 μm:20 μm). After preincubation in HKM buffer, the proteins were buffer exchanged into 200 mm ammonium acetate, pH 7.5, using Zeba Spin columns (0.5 ml, 7-kDa cutoff). The samples were loaded to homemade quartz tips and electrosprayed to Waters Synapt G2Si. MS settings was a follows: spray voltage, 1.2 kV; trap collision energy, 70 V; sampling cone voltage, 20 V; source offset, 10 V; trap gas, 4 ml/min; and source temperature, 30 °C.
HDX was followed for all TOMM34 variants (WT, S93A, S160A, S93A/S160A) in phosphorylated and nonphosphorylated state. Nonphosphorylated and phosphorylated WT and phosphorylated mutant forms of TOMM34 were also studied in the presence of 14-3-3γ. Finally, nonphosphorylated and phosphorylated WT and phosphorylated S93A/S160A mutant were analyzed in complex with HSP70 or HSP90 C-terminal peptides or with the peptides and 14-3-3γ. TOMM34 concentration during HDX (initiated by a 5-fold dilution into D2O based HKM buffer) was 10 μm. 14-3-3γ was used in 2-molar excess and C-terminal peptides in 5-molar excess to TOMM34. Exchange was followed for 20 s, 2 min, 20 min, and 2 h and then quenched by 1:9 dilution into 0.5 m glycine (pH 2.3) and subjected to rapid freezing in liquid nitrogen. Each sample was thawed, online-digested on immobilized pepsin column (66-μl bed volume), desalted, and separated using water–acetonitrile gradient (10–45% B in 7 min) on a setup consisting of a trap cartridge (ACQUITY UPLC BEH C18, 130 Å, 1.7 μm, 2.1 mm × 5 mm, Waters) and an analytical column (ACQUITY UPLC BEH C18, 130 Å, 1.7 μm, 1 mm × 100 mm, Waters). Digestion and desalting by 0.4% formic acid (FA) in water was driven by the 1260 Infinity II quaternary pump (Agilent Technologies) pumping at 100 µl/min and lasted 3 min. Separation was done using the 1290 Infinity II LC system (Agilent Technologies) pumping at 40 µl/min and using the following solvents: A: 0.1% FA, 2% acetonitrile in water; and B: 0.1% FA, 2% water in acetonitrile. All steps were performed at pH 2.5 and 0 °C to minimize back-exchange. The LC system was interfaced to an ESI FT-ICR MS (15T Solarix XR, Bruker Daltonics) operating in positive MS mode. Hydrogen/deuterium data processing used the in-house developed program Deutex as described previously (24, 83). Identification of pepsin generated peptides was achieved by separate LC–MS/MS analyses and database searching (MASCOT, MatrixScience) against a custom database containing sequences of the studied protein forms and proteases. Phosphorylation of Ser and Thr was included as a variable modification. Estimation of the phosphorylation extent on the identified sites via extracted ion chromatograms was done in DataAnalysis 5.0 (Bruker Daltonics).
Data availability
All data are contained within the article and the accompanying supporting information.
Supplementary Material
Acknowledgments
We thank Dr. P. J. Coates for critical reading of the manuscript.
This article contains supporting information.
Author contributions—F. T., P. Muller, and P. Man conceptualization; F. T., M. D., P. V., D. K., and P. Man formal analysis; F. T., M. D., P. V., and D. K. validation; F. T., M. D., P. V., V. V., O. S., D. K., and P. Man investigation; F. T., M. D., P. V., V. V., D. K., and P. Man visualization; F. T., M. D., P. V., V. V., O. S., D. K., P. Muller, and P. Man methodology; F. T. writing-original draft; F. T., M. D., P. V., V. V., O. S., D. K., B. V., P. Muller, and P. Man writing-review and editing; P. V., D. K., and P. Man data curation; P. V., D. K., and P. Man software; V. V. and O. S. resources; B. V., P. Muller, and P. Man supervision; B. V., P. Muller, and P. Man funding acquisition; B. V., P. Muller, and P. Man project administration.
Funding and additional information—This work was mainly supported by Czech Science Foundation Grant 16-20860S. Additional experiments were funded by Czech Science Foundation Grant 19-03796S. Further institutional and instrumental support from the Ministry of Education, Youth and Sports of the Czech Republic through Grant LQ1604, Czech Infrastructure for Integrative Structural Biology (CIISB) through Grant LM2015043, and the European Union through Grant CZ.1.05/1.1.00/02.0109, the Ministry of Health of the Czech Republic Conceptual Development of Research Organization through Grant 00209805 is gratefully acknowledged.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- HSP
- heat shock protein
- ESI
- electrospray ionization
- HDX
- hydrogen/deuterium exchange
- PKA
- protein kinase A
- SBP
- streptavidin-binding peptide
- SEC
- size-exclusion chromatography
- TOM
- translocase of outer mitochondrial membrane
- TPR
- tetratricopeptide repeat
- -P
- PKA-phosphorylated
- PMA
- 12-O-tetradecanoylphorbol-13-acetate
- FA
- formic acid.
References
- 1. Morán Luengo T., Mayer M. P., and Rüdiger S. G. D. (2019) The Hsp70–Hsp90 chaperone cascade in protein folding. Trends Cell Biol. 29, 164–177 10.1016/j.tcb.2018.10.004 [DOI] [PubMed] [Google Scholar]
- 2. Mayer M. P. (2010) Gymnastics of molecular chaperones. Mol. Cell 39, 321–331 10.1016/j.molcel.2010.07.012 [DOI] [PubMed] [Google Scholar]
- 3. Davis A. K., Pratt W. B., Lieberman A. P., and Osawa Y. (2020) Targeting Hsp70 facilitated protein quality control for treatment of polyglutamine diseases. Cell. Mol. Life Sci. 77, 977–996 10.1007/s00018-019-03302-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morán Luengo T., Kityk R., Mayer M. P., and Rüdiger S. G. D. (2018) Hsp90 breaks the deadlock of the HSP70 chaperone system. Mol. Cell 70, 545–552.e9 10.1016/j.molcel.2018.03.028 [DOI] [PubMed] [Google Scholar]
- 5. Bhangoo M. K., Tzankov S., Fan A. C., Dejgaard K., Thomas D. Y., and Young J. C. (2007) Multiple 40-kDa heat-shock protein chaperones function in Tom70-dependent mitochondrial import. Mol. Biol. Cell 18, 3414–3428 10.1091/mbc.e07-01-0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brychzy A., Rein T., Winklhofer K. F., Hartl F. U., Young J. C., and Obermann W. M. (2003) Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. EMBO J. 22, 3613–3623 10.1093/emboj/cdg362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faou P., and Hoogenraad N. J. (2012) Tom34: a cytosolic cochaperone of the Hsp90/Hsp70 protein complex involved in mitochondrial protein import. Biochim. Biophys. Acta 1823, 348–357 10.1016/j.bbamcr.2011.12.001 [DOI] [PubMed] [Google Scholar]
- 8. Young J. C., Hoogenraad N. J., and Hartl F. U. (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112, 41–50 10.1016/S0092-8674(02)01250-3 [DOI] [PubMed] [Google Scholar]
- 9. Wiedemann N., and Pfanner N. (2017) Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 86, 685–714 10.1146/annurev-biochem-060815-014352 [DOI] [PubMed] [Google Scholar]
- 10. Jores T., Lawatscheck J., Beke V., Franz-Wachtel M., Yunoki K., Fitzgerald J. C., Macek B., Endo T., Kalbacher H., Buchner J., and Rapaport D. (2018) Cytosolic Hsp70 and Hsp40 chaperones enable the biogenesis of mitochondrial β-barrel proteins. J. Cell Biol. 217, 3091–3108 10.1083/jcb.201712029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abe Y., Shodai T., Muto T., Mihara K., Torii H., Nishikawa S., Endo T., and Kohda D. (2000) Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell 100, 551–560 10.1016/S0092-8674(00)80691-1 [DOI] [PubMed] [Google Scholar]
- 12. Jores T., Klinger A., Gross L. E., Kawano S., Flinner N., Duchardt-Ferner E., Wöhnert J., Kalbacher H., Endo T., Schleiff E., and Rapaport D. (2016) Characterization of the targeting signal in mitochondrial β-barrel proteins. Nat. Commun. 7, 12036 10.1038/ncomms12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moczko M., Bömer U., Kübrich M., Zufall N., Hönlinger A., and Pfanner N. (1997) The intermembrane space domain of mitochondrial Tom22 functions as a trans binding site for preproteins with N-terminal targeting sequences. Mol. Cell Biol. 17, 6574–6584 10.1128/MCB.17.11.6574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hill K., Model K., Ryan M. T., Dietmeier K., Martin F., Wagner R., and Pfanner N. (1998) Tom40 forms the hydrophilic channel of the mitochondrial import pore for preproteins. Nature 395, 516–521 10.1038/26780 [DOI] [PubMed] [Google Scholar]
- 15. Schmidt O., Harbauer A. B., Rao S., Eyrich B., Zahedi R. P., Stojanovski D., Schönfisch B., Guiard B., Sickmann A., Pfanner N., and Meisinger C. (2011) Regulation of mitochondrial protein import by cytosolic kinases. Cell 144, 227–239 10.1016/j.cell.2010.12.015 [DOI] [PubMed] [Google Scholar]
- 16. Rao S., Schmidt O., Harbauer A. B., Schönfisch B., Guiard B., Pfanner N., and Meisinger C. (2012) Biogenesis of the preprotein translocase of the outer mitochondrial membrane: protein kinase A phosphorylates the precursor of Tom40 and impairs its import. Mol. Biol. Cell 23, 1618–1627 10.1091/mbc.e11-11-0933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gerbeth C., Schmidt O., Rao S., Harbauer A. B., Mikropoulou D., Opalińska M., Guiard B., Pfanner N., and Meisinger C. (2013) Glucose-induced regulation of protein import receptor Tom22 by cytosolic and mitochondria-bound kinases. Cell Metab. 18, 578–587 10.1016/j.cmet.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 18. Dagda R. K., and Das Banerjee T. (2015) Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Rev. Neurosci. 26, 359–370 10.1515/revneuro-2014-0085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F. U., and Moarefi I. (2000) Structure of TPR domain–peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101, 199–210 10.1016/S0092-8674(00)80830-2 [DOI] [PubMed] [Google Scholar]
- 20. Brinker A., Scheufler C., Von Der Mulbe F., Fleckenstein B., Herrmann C., Jung G., Moarefi I., and Hartl F. U. (2002) Ligand discrimination by TPR domains: relevance and selectivity of EEVD-recognition in Hsp70·Hop·Hsp90 complexes. J. Biol. Chem. 277, 19265–19275 10.1074/jbc.M109002200 [DOI] [PubMed] [Google Scholar]
- 21. Durech M., Trcka F., Man P., Blackburn E. A., Hernychova L., Dvorakova P., Coufalova D., Kavan D., Vojtesek B., and Muller P. (2016) Novel entropically driven conformation-specific interactions with Tomm34 protein modulate Hsp70 protein folding and ATPase activities. Mol. Cell. Proteomics 15, 1710–1727 10.1074/mcp.M116.058131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee C. T., Graf C., Mayer F. J., Richter S. M., and Mayer M. P. (2012) Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 31, 1518–1528 10.1038/emboj.2012.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trcka F., Durech M., Man P., Hernychova L., Muller P., and Vojtesek B. (2014) The assembly and intermolecular properties of the Hsp70–Tomm34–Hsp90 molecular chaperone complex. J. Biol. Chem. 289, 9887–9901 10.1074/jbc.M113.526046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trcka F., Durech M., Vankova P., Chmelik J., Martinkova V., Hausner J., Kadek A., Marcoux J., Klumpler T., Vojtesek B., Muller P., and Man P. (2019) Human stress-inducible Hsp70 has a high propensity to form ATP-dependent antiparallel dimers that are differentially regulated by cochaperone binding. Mol. Cell. Proteomics 18, 320–337 10.1074/mcp.RA118.001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park J. H., Jang H. R., Lee I. Y., Oh H. K., Choi E. J., Rhim H., and Kang S. (2017) Amyotrophic lateral sclerosis-related mutant superoxide dismutase 1 aggregates inhibit 14-3-3–mediated cell survival by sequestration into the JUNQ compartment. Hum. Mol. Genet. 26, 3615–3629 10.1093/hmg/ddx250 [DOI] [PubMed] [Google Scholar]
- 26. Hachiya N., Alam R., Sakasegawa Y., Sakaguchi M., Mihara K., and Omura T. (1993) A mitochondrial import factor purified from rat liver cytosol is an ATP-dependent conformational modulator for precursor proteins. EMBO J. 12, 1579–1586 10.1002/j.1460-2075.1993.tb05802.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hachiya N., Komiya T., Alam R., Iwahashi J., Sakaguchi M., Omura T., and Mihara K. (1994) MSF, a novel cytoplasmic chaperone which functions in precursor targeting to mitochondria. EMBO J. 13, 5146–5154 10.1002/j.1460-2075.1994.tb06844.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hachiya N., Mihara K., Suda K., Horst M., Schatz G., and Lithgow T. (1995) Reconstitution of the initial steps of mitochondrial protein import. Nature 376, 705–709 10.1038/376705a0 [DOI] [PubMed] [Google Scholar]
- 29. Komiya T., Hachiya N., Sakaguchi M., Omura T., and Mihara K. (1994) Recognition of mitochondria-targeting signals by a cytosolic import stimulation factor, MSF. J. Biol. Chem. 269, 30893–30897 [PubMed] [Google Scholar]
- 30. Komiya T., and Mihara K. (1996) Protein import into mammalian mitochondria: characterization of the intermediates along the import pathway of the precursor into the matrix. J. Biol. Chem. 271, 22105–22110 10.1074/jbc.271.36.22105 [DOI] [PubMed] [Google Scholar]
- 31. Komiya T., Sakaguchi M., and Mihara K. (1996) Cytoplasmic chaperones determine the targeting pathway of precursor proteins to mitochondria. EMBO J. 15, 399–407 10.1002/j.1460-2075.1996.tb00370.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gardino A. K., Smerdon S. J., and Yaffe M. B. (2006) Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3–ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol. 16, 173–182 10.1016/j.semcancer.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 33. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., and Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 10.1016/S0092-8674(00)80487-0 [DOI] [PubMed] [Google Scholar]
- 34. Alblova M., Smidova A., Docekal V., Vesely J., Herman P., Obsilova V., and Obsil T. (2017) Molecular basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. Proc. Natl. Acad. Sci. U.S.A. 114, E9811–E9820 10.1073/pnas.1714491114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ottmann C., Marco S., Jaspert N., Marcon C., Schauer N., Weyand M., Vandermeeren C., Duby G., Boutry M., Wittinghofer A., Rigaud J. L., and Oecking C. (2007) Structure of a 14-3-3 coordinated hexamer of the plant plasma membrane H+-ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell 25, 427–440 10.1016/j.molcel.2006.12.017 [DOI] [PubMed] [Google Scholar]
- 36. Bustos D. M., and Iglesias A. A. (2006) Intrinsic disorder is a key characteristic in partners that bind 14-3-3 proteins. Proteins 63, 35–42 10.1002/prot.20888 [DOI] [PubMed] [Google Scholar]
- 37. Silhan J., Vacha P., Strnadova P., Vecer J., Herman P., Sulc M., Teisinger J., Obsilova V., and Obsil T. (2009) 14-3-3 protein masks the DNA binding interface of forkhead transcription factor FOXO4. J. Biol. Chem. 284, 19349–19360 10.1074/jbc.M109.002725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rezabkova L., Man P., Novak P., Herman P., Vecer J., Obsilova V., and Obsil T. (2011) Structural basis for the 14-3-3 protein-dependent inhibition of the regulator of G protein signaling 3 (RGS3) function. J. Biol. Chem. 286, 43527–43536 10.1074/jbc.M111.273573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blom N., Gammeltoft S., and Brunak S. (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294, 1351–1362 10.1006/jmbi.1999.3310 [DOI] [PubMed] [Google Scholar]
- 40. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hennrich M. L., Marino F., Groenewold V., Kops G. J., Mohammed S., and Heck A. J. (2013) Universal quantitative kinase assay based on diagonal SCX chromatography and stable isotope dimethyl labeling provides high-definition kinase consensus motifs for PKA and human Mps1. J. Proteome Res. 12, 2214–2224 10.1021/pr400074f [DOI] [PubMed] [Google Scholar]
- 42. Madeira F., Tinti M., Murugesan G., Berrett E., Stafford M., Toth R., Cole C., MacKintosh C., and Barton G. J. (2015) 14-3-3-Pred: improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 31, 2276–2283 10.1093/bioinformatics/btv133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishida T., and Kinoshita K. (2007) PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res 35, W460–W464 10.1093/nar/gkm363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uhart M., and Bustos D. M. (2014) Protein intrinsic disorder and network connectivity: the case of 14-3-3 proteins. Front. Genet. 5, 10 10.3389/fgene.2014.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5, 749–757 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 46. Sluchanko N. N., and Gusev N. B. (2012) Oligomeric structure of 14-3-3 protein: what do we know about monomers?. FEBS Lett. 586, 4249–4256 10.1016/j.febslet.2012.10.048 [DOI] [PubMed] [Google Scholar]
- 47. Oganesyan I., Lento C., and Wilson D. J. (2018) Contemporary hydrogen deuterium exchange mass spectrometry. Methods 144, 27–42 10.1016/j.ymeth.2018.04.023 [DOI] [PubMed] [Google Scholar]
- 48. Yang X., Lee W. H., Sobott F., Papagrigoriou E., Robinson C. V., Grossmann J. G., Sundström M., Doyle D. A., and Elkins J. M. (2006) Structural basis for protein–protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 103, 17237–17242 10.1073/pnas.0605779103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kacirova M., Kosek D., Kadek A., Man P., Vecer J., Herman P., Obsilova V., and Obsil T. (2015) Structural characterization of phosducin and its complex with the 14-3-3 protein. J. Biol. Chem. 290, 16246–16260 10.1074/jbc.M115.636563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Y., Ren J., He X., Chen H., Wei T., and Feng W. (2019) YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization. Autophagy 15, 1017–1030 10.1080/15548627.2019.1569928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Macakova E., Kopecka M., Kukacka Z., Veisova D., Novak P., Man P., Obsil T., and Obsilova V. (2013) Structural basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. Biochim. Biophys. Acta 1830, 4491–4499 10.1016/j.bbagen.2013.05.025 [DOI] [PubMed] [Google Scholar]
- 52. Muller P., Coates P. J., Nenutil R., Trcka F., Hrstka R., Chovanec J., Brychtova V., and Vojtesek B. (2019) Tomm34 is commonly expressed in epithelial ovarian cancer and associates with tumour type and high FIGO stage. J. Ovarian Res. 12, 30 10.1186/s13048-019-0498-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Baillie G., MacKenzie S. J., and Houslay M. D. (2001) Phorbol 12-myristate 13-acetate triggers the protein kinase A–mediated phosphorylation and activation of the PDE4D5 cAMP phosphodiesterase in human aortic smooth muscle cells through a route involving extracellular signal regulated kinase (ERK). Mol. Pharmacol. 60, 1100–1111 10.1124/mol.60.5.1100 [DOI] [PubMed] [Google Scholar]
- 54. Wu J., Li J., Huang K. P., and Huang F. L. (2002) Attenuation of protein kinase C and cAMP-dependent protein kinase signal transduction in the neurogranin knockout mouse. J. Biol. Chem. 277, 19498–19505 10.1074/jbc.M109082200 [DOI] [PubMed] [Google Scholar]
- 55. Tai T. C., and Wong D. L. (2003) Protein kinase A and protein kinase C signaling pathway interaction in phenylethanolamine N-methyltransferase gene regulation. J. Neurochem. 85, 816–829 10.1046/j.1471-4159.2003.01728.x [DOI] [PubMed] [Google Scholar]
- 56. Søberg K., Moen L. V., Skålhegg B. S., and Laerdahl J. K. (2017) Evolution of the cAMP-dependent protein kinase (PKA) catalytic subunit isoforms. PLoS One 12, e0181091 10.1371/journal.pone.0181091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Limbutara K., Kelleher A., Yang C. R., Raghuram V., and Knepper M. A. (2019) Phosphorylation changes in response to kinase inhibitor H89 in PKA-null cells. Sci. Rep. 9, 2814 10.1038/s41598-019-39116-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. García-Bermúdez J., Sánchez-Aragó M., Soldevilla B., Del Arco A., Nuevo-Tapioles C., and Cuezva J. M. (2015) PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H+-ATP synthase. Cell Rep. 12, 2143–2155 10.1016/j.celrep.2015.08.052 [DOI] [PubMed] [Google Scholar]
- 59. Kumar A., Gopalswamy M., Wolf A., Brockwell D. J., Hatzfeld M., and Balbach J. (2018) Phosphorylation-induced unfolding regulates p19(INK4d) during the human cell cycle. Proc. Natl. Acad. Sci. U.S.A. 115, 3344–3349 10.1073/pnas.1719774115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Muller P., Ruckova E., Halada P., Coates P. J., Hrstka R., Lane D. P., and Vojtesek B. (2013) C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32, 3101–3110 10.1038/onc.2012.314 [DOI] [PubMed] [Google Scholar]
- 61. Assimon V. A., Southworth D. R., and Gestwicki J. E. (2015) Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry 54, 7120–7131 10.1021/acs.biochem.5b00801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aprile F. A., Dhulesia A., Stengel F., Roodveldt C., Benesch J. L., Tortora P., Robinson C. V., Salvatella X., Dobson C. M., and Cremades N. (2013) Hsp70 oligomerization is mediated by an interaction between the interdomain linker and the substrate-binding domain. PLoS One 8, e67961 10.1371/journal.pone.0067961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Muslin A. J., Tanner J. W., Allen P. M., and Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 10.1016/S0092-8674(00)81067-3 [DOI] [PubMed] [Google Scholar]
- 64. Oldfield C. J., Meng J., Yang J. Y., Yang M. Q., Uversky V. N., and Dunker A. K. (2008) Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9, S1 10.1186/1471-2164-9-S1-S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johnson C., Crowther S., Stafford M. J., Campbell D. G., Toth R., and MacKintosh C. (2010) Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 427, 69–78 10.1042/BJ20091834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yaffe M. B. (2002) How do 14-3-3 proteins work?: Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 513, 53–57 10.1016/S0014-5793(01)03288-4 [DOI] [PubMed] [Google Scholar]
- 67. Masone D., Uhart M., and Bustos D. M. (2017) On the role of residue phosphorylation in 14-3-3 partners: AANAT as a case study. Sci. Rep. 7, 46114 10.1038/srep46114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Molzan M., and Ottmann C. (2012) Synergistic binding of the phosphorylated S233- and S259-binding sites of C-RAF to one 14-3-3ζ dimer. J. Mol. Biol. 423, 486–495 10.1016/j.jmb.2012.08.009 [DOI] [PubMed] [Google Scholar]
- 69. Obsil T., Ghirlando R., Anderson D. E., Hickman A. B., and Dyda F. (2003) Two 14-3-3 binding motifs are required for stable association of Forkhead transcription factor FOXO4 with 14-3-3 proteins and inhibition of DNA binding. Biochemistry 42, 15264–15272 10.1021/bi0352724 [DOI] [PubMed] [Google Scholar]
- 70. Kostelecky B., Saurin A. T., Purkiss A., Parker P. J., and McDonald N. Q. (2009) Recognition of an intra-chain tandem 14-3-3 binding site within PKCε. EMBO Rep. 10, 983–989 10.1038/embor.2009.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bhaskara R. M., de Brevern A. G., and Srinivasan N. (2013) Understanding the role of domain-domain linkers in the spatial orientation of domains in multi-domain proteins. J. Biomol. Struct. Dynamics 31, 1467–1480 10.1080/07391102.2012.743438 [DOI] [PubMed] [Google Scholar]
- 72. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., and Hallberg B. (2007) Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26, 902–913 10.1038/sj.emboj.7601530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ito T., Nakata M., Fukazawa J., Ishida S., and Takahashi Y. (2014) Phosphorylation-independent binding of 14-3-3 to NtCDPK1 by a new mode. Plant Signal. Behavior 9, e977721 10.4161/15592324.2014.977721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Blesa J. R., Prieto-Ruiz J. A., Abraham B. A., Harrison B. L., Hegde A. A., and Hernández-Yago J., et al. (2008) NRF-1 is the major transcription factor regulating the expression of the human TOMM34 gene. Biochem. Cell Biol. 86, 46–56 10.1139/O07-151 [DOI] [PubMed] [Google Scholar]
- 75. Scarpulla R. C. (2011) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 10.1016/j.bbamcr.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blesa J. R., Prieto-Ruiz J. A., Hernández J. M., and Hernández-Yago J. (2007) NRF-2 transcription factor is required for human TOMM20 gene expression. Gene 391, 198–208 10.1016/j.gene.2006.12.024 [DOI] [PubMed] [Google Scholar]
- 77. Blesa J. R., Hernández J. M., and Hernández-Yago J. (2004) NRF-2 transcription factor is essential in promoting human Tomm70 gene expression. Mitochondrion 3, 251–259 10.1016/j.mito.2004.02.001 [DOI] [PubMed] [Google Scholar]
- 78. Brix J., Rüdiger S., Bukau B., Schneider-Mergener J., and Pfanner N. (1999) Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J. Biol. Chem. 274, 16522–16530 10.1074/jbc.274.23.16522 [DOI] [PubMed] [Google Scholar]
- 79. Chewawiwat N., Yano M., Terada K., Hoogenraad N. J., and Mori M. (1999) Characterization of the novel mitochondrial protein import component, Tom34, in mammalian cells. J. Biochem. 125, 721–727 10.1093/oxfordjournals.jbchem.a022342 [DOI] [PubMed] [Google Scholar]
- 80. Wang L., Sunahara R. K., Krumins A., Perkins G., Crochiere M. L., Mackey M., Bell S., Ellisman M. H., and Taylor S. S. (2001) Cloning and mitochondrial localization of full-length D-AKAP2, a protein kinase A anchoring protein. Proc. Natl. Acad. Sci. U.S.A. 98, 3220–3225 10.1073/pnas.051633398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cardone L., de Cristofaro T., Affaitati A., Garbi C., Ginsberg M. D., Saviano M., Varrone S., Rubin C. S., Gottesman M. E., Avvedimento E. V., and Feliciello A. (2002) A-kinase anchor protein 84/121 are targeted to mitochondria and mitotic spindles by overlapping amino-terminal motifs. J. Mol. Biol. 320, 663–675 10.1016/S0022-2836(02)00479-5 [DOI] [PubMed] [Google Scholar]
- 82. Affaitati A., Cardone L., de Cristofaro T., Carlucci A., Ginsberg M. D., Varrone S., Gottesman M. E., Avvedimento E. V., and Feliciello A. (2003) Essential role of A-kinase anchor protein 121 for cAMP signaling to mitochondria. J. Biol. Chem. 278, 4286–4294 10.1074/jbc.M209941200 [DOI] [PubMed] [Google Scholar]
- 83. Kavan D., and Man P. (2011) MSTools: web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrometry 302, 53–58 10.1016/j.ijms.2010.07.030 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article and the accompanying supporting information.