

Abstract
Rickets is a common bone disease worldwide that is associated with disturbances in calcium and phosphate homeostasis and can lead to short stature and joint deformities. Rickets can be diagnosed based on history and physical examination, radiological features, and biochemical tests. It can be classified into 2 major groups based on phosphate or calcium levels: phosphopenic and calcipenic. Knowledge of categorization of the type of rickets is essential for prompt diagnosis and proper management. Nutritional rickets is a preventable disease through adequate intake of vitamin D through both dietary and sunlight exposure. There are other subtypes of rickets, such as vitamin D–dependent type 1 rickets and vitamin D–dependent type 2 rickets (due to defects in vitamin D metabolism), renal rickets (due to poor kidney function), and hypophosphatemic rickets (vitamin D–resistant rickets secondary to renal phosphate wasting wherein fibroblast growth factor-23 (FGF-23) often plays a major role), which requires closer monitoring and supplementation with activated vitamin D with or without phosphate supplements. An important development has been the introduction of burosumab, a human monoclonal antibody to FGF-23, which is approved for the treatment of X-linked hypophosphatemia among children 1 year and older.
Keywords: chronic kidney disease, hypocalcemia, hypophosphatemia, phosphorus, vitamin D
Rickets, a common disease worldwide,1,2 substantially affects the health, growth, and development of children and adolescents. It results from abnormalities of the growth plate cartilage predominantly affecting longer bones and leads to poor bone growth, defective mineralization, and bony deformities, such as bow-legs and knock-knees.3 This is usually secondary to deficiencies of calcium or phosphorus because they are essential for normal bone growth and mineralization.4,5 This review article delves and analyzes different types of rickets and their appropriate management plan.
Pathogenesis and Types of Rickets
Bones consist of cells that have various specific roles during the bone formation process. Osteoblasts are bone-forming cells that secrete the extracellular matrix and mineralize the osteoid, whereas osteoclasts break down the bone matrix during the stage of remodeling, disease conditions, or aging. For bone maturation, the organic component of the bone matrix, the osteoid, must be mineralized by calcium salts. In rickets, this process is hampered and results in amassing of osteoid beneath the growth plate leading to softness in the bone over a gradual period of time.4 Rickets can be classified into 2 major groups: phosphopenic and calcipenic3,6 (Figure 1).
Figure 1.

Different types of rickets. ADHR, autosomal dominant hypophosphatemic rickets; ARHR, autosomal recessive hypophosphatemic rickets; CKD, chronic kidney disease; VDDR, vitamin D–dependent type 1 rickets; XLHR, X-linked hypophosphatemic rickets.
Abundant in all tissues of the body, phosphorus is a vital structural component for mineralization of bone. Both calcium and phosphorus keep the bone in a healthy, functional state.7 In phosphopenic/hypophosphatemic rickets, the defect usually results from increased renal excretion of phosphate.3 Urinary loss of phosphate can be either as a part of generalized tubular dysfunction as seen in Fanconi syndrome, or secondary to either increased synthesis/ reduced catabolism of the FGF-23, or inactivating mutations in genes encoding for sodium-dependent phosphate transporters in the proximal renal tubule (Figure 1).8
Calcipenic rickets, as the name suggests, happens primarily because of a lack of calcium, which is most commonly due to a low availability or defective functioning of vitamin D in the body (Figure 1). Hence, calcipenic rickets can occur due to severe vitamin D deficiency (nutritional), inability to form either 25-hydroxyvitamin D (as in liver failure/drug intoxication; e.g., phenytoin) or 1,25-dihydroxy vitamin D (as in chronic kidney disease), or due to end-organ resistance to 1,25-dihydroxy vitamin D3. As a result, calcium absorption in the gut is decreased, which in turn increases parathyroid hormone (PTH) secretion by the parathyroid gland. PTH aims to preserve blood calcium levels by (i) activating bone resorption mediated by increasing RANKL by osteoblasts, (ii) decreasing renal calcium loss, and (iii) increasing renal phosphate loss by internalization and subsequent degradation of sodium-dependent phosphate cotransporter protein (NaPi-2a and NaPi-2c), which decreases tubular phosphate reabsorption.8 The common pathway in the development of rickets in both calcipenic and phosphopenic forms is reduced phosphate concentration.2,9
Calcipenic Rickets
Sources and Metabolism of Vitamin D
Vitamin D plays an essential role in skeletal health by regulating normal blood levels of calcium and phosphorus (Figure 2).10,11 There are 2 main forms of vitamin D: vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol). Vitamin D2 is primarily derived from plant sources. In addition to being present in foods such as fish, eggs, milk, and cod liver oil, the synthesis of vitamin D3 occurs naturally through the conversion of dehydrocholesterol to cholecalciferol in the skin by sunlight (ultraviolet B in the 290–315-nm range). Vitamin D binds to the vitamin D binding protein and is transported to the liver for hydroxylation, and converted by 25-hydroxylase (encoded by CYP2R1, Cytochrome P450 Family 2 Subfamily R Member 1) into calcidiol (also known as hydroxyl-cholecalciferol, 25-hydroxyvitamin, calcifediol) which is then absorbed in the proximal tubule of the kidney through the endocytic receptors megalin and cubilin12 and hydroxylated by the enzyme 1 alpha-hydroxylase (encoded by CYP27B1, Cytochrome P450 Family 27 Subfamily B Member 1) to form the active metabolite of vitamin D, calcitriol (also known as 1,25-dihydroxy vitamin D).13 1,25-dihydroxy vitamin D acts on the vitamin D receptor in intestinal cells to increase the gut absorption of calcium by upregulating the calcium channel, TRPV6 (Transient receptor potential cation channel subfamily V member 6).14 As shown in Figure 3, there is a complex interaction between the hormones produced by the kidneys (1,25 dihydroxy vitamin D), bone (FGF-23), and PTH. Understanding these interactions is essential for proper management of rickets.
Figure 2.

Sources and metabolism of vitamin D. PTH, parathyroid hormone.
Figure 3.

Parathyroid/bone/kidney axis. 1. Parathyroid hormone (PTH) increases 1,25 dihydroxy vitamin (vit) D synthesis in the kidney. 2. Fibroblast growth factor 23 (FGF-23) is produced by bone and it acts on the kidney. 3. FGF-23 decreases PTH and 1,25 dihydroxy vitamin D. 4. Both PTH and 1,25 dihydroxy vitamin D increase FGF-23 syntheses.
Hypocalcemia, hypophosphatemia, and PTH stimulate the synthesis of 1,25-dihydroxy vitamin D. FGF-23, a hormone produced by osteocytes, plays an important role in bone metabolism. It inhibits the synthesis of 1,25-dihydroxy vitamin D and binds with the FGF receptors with the help of klotho, a membrane-bound protein, and increases the renal excretion of phosphate by reducing the number of the main renal phosphate transporters, sodium-dependent phosphate transport proteins, NaPi-2a and NaPi-2c, on the apical surface of proximal renal tubular cells.15 FGF-23 is regulated by 2 major proteins in bone: phosphate-regulating neutral endopeptidase homolog (PHEX) and dentin matrix acidic phosphoprotein 1 (DMP1). Both of which are made primarily in bone, specifically by osteocytes. Dysregulation of these proteins results in osteomalacia, suggesting that osteocytes play a role in the regulation of skeletal mineralization.
Nutritional Rickets/Vitamin D Deficiency Rickets
Nutritional rickets is the most common form of bone disease, primarily affecting infants and young children. Although primarily caused by vitamin D deficiency, calcium and phosphate deficiencies also play a significant role. Vitamin D regulates calcium and phosphorus in the blood and deficiency of vitamin D does result in inadequate mineralization of osteoid produced by osteoblasts.16 The primary cause of vitamin D deficiency usually involves interplay of nutritional inadequacy and lack of sunlight exposure with overlapping contributions by cultural, environmental, and genetic factors.17 Table 1 shows the classification of the severity of vitamin D deficiency.18 There is no clear consensus on the definition of normal vitamin D concentration in healthy children and guidelines differ in their target levels for optimal vitamin D status.19 The Global Consensus meeting on the prevention and management of nutritional rickets defined deficiency as vitamin D level <30 ng/ml.18
Table 1.
Severity of 25 (OH) vitamin D deficiency
| Vitamin D status | ng/ml |
|---|---|
| Deficiency | <30 |
| Insufficiency | 30–50 |
| Adequate | >50 |
| Toxicity | >250 |
Both skeletal and extraskeletal manifestations are present in patients with nutritional rickets. Skeletal symptoms include swollen wrists and ankles, delayed tooth eruption, leg deformity, rachitic rosary, frontal bossing, craniotabes, and bone pain.8,16 On the other hand, extraskeletal findings include muscle weakness and hypocalcemic seizures.4,18 Nutritional/medical history, biochemical testing, and radiographs are used to diagnose nutritional rickets. Along with low level of 25 (OH) vitamin D, laboratory findings are also helpful in its diagnosis as well as in differentiating it from other causes of rickets (Table 2).4,18,20,21
Table 2.
Salient features of different types of rickets
| Type | Calcium | Phosphorus | Alkaline phosphatase | PTH | 25 (OH)D | 1,25 (OH)2 D |
|---|---|---|---|---|---|---|
| Calcipenic rickets | ||||||
| Vitamin D deficiency | ↓ or N | ↓ or N | ↑ or ↑↑ | ↑ | ↓ | Variable |
| Vitamin D–dependent rickets type I | ↓ | ↓ or N | ↑↑ | ↑ | N | ↓ |
| Vitamin D–dependent rickets type II | ↓ | ↓ or N | ↑↑ | ↑ | N | N or ↓ |
| Phosphenic rickets | ||||||
| Nutritional phosphate deficiency | ↑ or N | ↓ | ↑ or ↑↑ | ↓ or N | N | ↑ |
| X-linked hypophosphatemic rickets | N | ↓ | ↑ | N or slightly ↑ | N | N or ↓ |
| Autosomal dominant hypophosphatemic rickets | N | ↓ | ↑ | N | N | ↓ |
| Autosomal recessive hypophosphatemic rickets | N | ↓ | ↑ | N | N | ↓ |
| Hereditary hypophosphatemic rickets with hypercalciuria | N | ↓ | ↑ | N or ↓ | N | ↑ |
25 (OH)D, 25-hydroxy vitamin D; 1,25 (OH)2 D, 1,25 dihydroxy vitamin D; N, normal levels; PTH, parathyroid hormone; ↑, increased levels; ↓, decreased levels.
The treatment of nutritional vitamin D deficiency with cholecalciferol consists of an intensive phase followed by a maintenance phase. The National Osteoporosis Society in the United Kingdom recommends 3000 IU (in infants <6 months old), 6000 IU (6 months–12 years old), and 10,000 IU (12–18 years old) of cholecalciferol per day in the intensive phase followed by 400 to 600 IU/d in the maintenance phase.22 The U.S. Endocrine Society recommends 2000 IU/d cholecalciferol for 6 weeks for all age groups in the intensive phase followed by 400 to 1000 IU/d in the maintenance phase.23 Also, evaluation of nutritional calcium intake of the child is important. The recommended daily dietary allowance of calcium is 200 mg for infants 0 to 6 months, 260 mg for infants 7 to 12 months, 700 mg for children 1 to 3 years old, 1000 mg for 4 to 8 years old, and 1300 mg for children 9 to 18 years old.24 Table 3 shows age-based normal values of serum calcium and phosphorus in children.25 In children in whom calcium deficiency is the primary cause of nutritional rickets, calcium supplements (≥1000 mg/d) is recommended.2
Table 3.
Normal age-based serum calcium and phosphorus levels in children25
| Age | Age-based serum calcium (mg/dl) | Age-based serum phosphorus (mg/dl) |
|---|---|---|
| 0–3 mo | 8.8–11.3 | 4.8–7.4 |
| 1–5 yr | 9.4–10.8 | 4.5–6.5 |
| 6–12 yr | 9.4–10.3 | 3.6–5.8 |
| 13–20 yr | 8.8–10.2 | 2.3–4.5 |
To convert units: calcium 1 mg/dl = 0.25 mmol/l; phosphorus 1 mg/dl = 0.32 mmol/l.
Causes of Vitamin D Deficiency
Along with nutritional factors, vitamin D deficiency can be compounded by a multitude of non-nutritional factors. Differences among racial groups, including skin pigmentation, substantially affect vitamin D status. From 2008 to 2012, 110 children younger than 5 in the Norwegian Patient Registry were diagnosed with active rickets, of whom 42 children were classified as having nutritional rickets (vitamin D deficiency). Although the number was substantially low in comparison to years before 2008 to 2012, 93% of the patients had a non-Caucasian background.26
A cohort study conducted among the South Asian and white population suggested the effect of both seasonal variation and skin pigmentation. During the summer, the median 25-hydroxyvitamin level in the South Asian group was 9.1 ng/ml, which decreased to 5.8 ng/ml in the winter. In comparison, the Caucasians also had a decrease in winter vitamin D levels but from a median of 26.2 ng/ml during the summer to 18.9 ng/ml in the winter.27 Apart from skin pigmentation, sociocultural issues also play an important role. As discussed, ultraviolet radiation is essential for the production of vitamin D,4,28 but despite having plentiful sunlight and average summer seasonal temperature of 45 °C, 100% of young healthy Saudi female individuals suffer from vitamin D deficiency. In a study of the 465 female participants, all participants had low vitamin D levels (25-hydroxyvitamin <75 nmol/l) with a mean level of 18.34 ± 8.2 nmol/l.29 This paradox might be secondary to the traditional requirement of Muslim women in Saudi Arabia to be covered from head to toe, thus hindering sun exposure.
Ultraviolet radiation, although important, is not necessarily the only important cause for vitamin D deficiency. Despite India’s geographic location giving its population excessive amounts of sunlight exposure, vitamin D deficiency has been reported significantly in the range of 70% to 100% in the adult population, which may be due to the widespread practice of a vegetarian-based diet.4,30
Infancy is another age group quite vulnerable to 25-hydroxy vitamin D deficiencies. During pregnancy, there is a transfer of vitamin D between the mother and fetus and hence if the mother has low vitamin D level storage then so does the fetus.31 Even if the mother’s vitamin D level is adequate, infants get a maximum of approximately 40 IU of vitamin D per 750 ml of the breast milk. On the other hand, formula-fed infants receive plentiful amounts of vitamin D because of the fortification of minerals within the milk. Hence, vitamin D deficiency is uncommon in formula-fed infants. A cohort study was undertaken in northwest England with breast-fed infants reporting a rate of 67% of vitamin D deficiency, whereas formula-fed infants had a 2% rate of deficiency.4,32 Preterm infants, especially those with birth weight <1000 g are also at a high risk for rickets primarily because of calcium and phosphate deficiency.2
Prevention of vitamin D deficiency can be practiced through adequate sun exposure, vitamin D supplementation, fortification of dietary intake, and meeting the normal calcium intake. Regular vitamin D supplementation is recommended for infants from birth until 12 months of age (400 IU/d). For those older, 600 IU/d of vitamin D through diet or supplementation is advised.4,11,18,33, 34, 35, 36
Vitamin D–Dependent Type 1 Rickets
Occurring generally during the first year of life, vitamin D–dependent type 1 rickets (VDDR I) is a rare autosomal recessive disease of vitamin D metabolism that occurs due to homozygous inactivating mutations in the CYP27B1 gene leading to impaired production of the enzyme 1 alpha-hydroxylase, which in turn leads to low serum levels of the active metabolite calcitriol.37, 38, 39 Specific clinical manifestations include typical features of rickets, such as growth failure, hypotonia, rachitic rosary, genu valgum, and increased susceptibility to fractures, despite the patient getting sufficient vitamin D intake.37 The laboratory findings (Table 2) would typically show low calcium, low phosphate, elevated PTH, and high alkaline phosphatase, but in contrast to nutritional rickets they will have normal or high 25-hydroxyvitamin levels and low calcitriol levels.37,38
As expected, these children will not respond to high doses of cholecalciferol but respond to physiologic doses of calcitriol or 1α-hydroxyvitamin D (1–2 μg daily). Adequate intake of dietary calcium (30–75 mg/kg per day of elemental calcium) should be maintained.33 Typically, radiological healing occurs within 6 to 8 weeks of therapy. These children should be monitored for potential side effects of hypercalcemia, hypercalciuria, and nephrocalcinosis secondary to calcitriol therapy.33 Regular blood work (serum creatinine and calcium, phosphate), urine examination for urine calcium and creatinine ratio, and kidney ultrasound examination should be performed.
Vitamin D–Dependent Type 2 Rickets
Also occurring during infancy, VDDR type 2 (VDDR II) or hereditary vitamin D–resistant rickets, is a rare autosomal recessive disease caused by a defect in the calcitriol vitamin D receptor.38 The defect causes the body to be irresponsive to calcitriol.38 Children with VDDR II present early in life and may have hypocalcemia, rickets, growth failure, seizures, enamel hypoplasia, and dental caries. Alopecia also occurs in two-thirds of cases due to a lack of vitamin D receptor activity within keratinocytes and is a marker of disease severity.33,40 The laboratory tests show high levels of calcitriol, hypocalcemia, hypophosphatemia, high serum levels of alkaline phosphatase, and PTH (Table 2).37,41 Low levels of 1,25 dihydroxy vitamin D differentiates VDDR II from VDDR I, among which the levels are usually high.
Because VDDR II is a hereditary disease resistant to 1,25-dihydroxyvitamin D, no completely proven treatment is available.38,42 Despite its difficulty, the most plausible way is to saturate the normal receptors through mega-doses of calcitriol and calcium. Without treatment, the disorder further leads to severe skeletal deformity, respiratory infections, and most likely death by age 8.37 Therapy consists of high doses of calcitriol, starting at low dose (0.05 μg/kg per day), which may be increased up to 0.2 μg/kg per day along with calcium and phosphate supplementation. Some patients also may require high-dose i.v. calcium infusion for many months.37,41
Renal Rickets
The term renal rickets is usually restricted to those with chronic kidney disease. Chronic kidney disease results in the deficiency of the enzyme 1 alpha-hydroxylase, which decreases the production of 1,25 hydroxy vitamin D (calcitriol).33 A history of renal failure will be evident that excludes the disorder from other bone diseases. Laboratory findings (Table 2) usually show low calcitriol levels, but 25-hydroxyvitamin D levels may even be normal. The most characteristic finding is the elevated phosphate level secondary to poor renal function of chronic kidney disease.16 Because patients with chronic kidney disease cannot convert the calcidiol into the active form calcitriol, vitamin D supplementation alone is therefore ineffective for renal rickets. Instead, a low-phosphate diet, dietary phosphate binders, and oral administration of 1 alfacalcidol or calcitriol is advised, along with maintaining normal 25-hydroxyvitamin D level.33,43,44
Hypophosphatemic Rickets
The main defect in these forms of rickets is the increased loss of phosphate through the urine. Hereditary causes occur due to genetic mutations involving phosphate-regulating neutral endopeptidase (PHEX), or dentin matrix acidic phosphoprotein 1 (DMP1), or FGF-23 resulting in X-linked dominant, autosomal recessive (ARHR), or autosomal dominant hypophosphatemic rickets, respectively.45
Hypophosphatemic rickets should not be confused with hypophosphatesia, which is a rare inborn error of metabolism due to the dysfunction of the tissue nonspecific alkaline phosphatase enzyme.46 Childhood hypophosphatesia has onset of symptoms between 6 months and 18 years and can present as rickets, reduced mobility, fractures, and poor growth.46 It is characterized by low alkaline phosphatase levels, which is paradoxical because other forms of rickets are associated with high alkaline phosphatase levels.46
X-Linked Dominant Hypophosphatemic Rickets
It is the most common genetic form of hypophosphatemia, with an incidence of 1:20,000.47 The disease results from mutations of the phosphate-regulating gene on the X chromosome (PHEX gene), which impairs the inactivation of FGF-23 by an ill-understood mechanism.48 Increased FGF-23 level results in renal wasting of phosphorus at the proximal tubule level, which results in hypophosphatemia.49,50 Elevated FGF-23 also results in low calcitriol levels. Unlike vitamin D deficiency, craniotabes and rachitic rosary are not common. One of the initial clinical findings is frontal bossing, which may appear as early as 6 months of age. As the child starts walking, progressive limb deformities become evident, leading to disproportionate short stature with shorter limbs. Lower limbs are more affected, leading to coxa vara, genu valgum, or genu varum. Dental abnormalities are common and may often be the presenting complaints.51 These abnormalities include abscessed noncarious teeth, enamel defects, and enlarged pulp chambers. Inadequate dentin mineralization results in cracks that allow penetration of systemic bacteria, leading to dental abscess and severe infections, like facial cellulitis from dental focus in some patients.52 In addition, recent studies have shown higher incidence of craniovertebral and cranial vault anomalies, especially early closure of the cranial sutures (craniosynostosis) and Chiari type 1 malformation.53 A retrospective study of head and skull computed tomography scans involving 44 children with X-linked hypophosphatemic rickets found incidence of craniosynostosis (complete or partial fusion of the sagittal sutures) to be 59%, and 25% of children showed cerebellar tonsillar protrusion (Chiari type 1 malformation).53 Further, craniosynostosis was found to be associated with a history of dental abscesses.53 Neurologic symptoms were observed in only 2 children and 9% (4 of 44) of children had neurosurgery.53
Autosomal Dominant Hypophosphatemic Rickets
Autosomal dominant hypophosphatemic rickets has a variable age of onset and an incomplete penetrance. The defect being an activating mutation in FGF-23 leading to phosphaturia.54 In addition, studies have shown that iron deficiency increases expression of the FGF-23 gene.55 Elevated levels of FGF-23 and hypophosphatemia were observed with iron deficiency in humans and mice with autosomal dominant hypophosphatemic rickets mutation.55 Based on the age of presentation, there are 2 subgroups. One presents during childhood and mimics X-linked dominant hypophosphatemic rickets. The other subgroup presents during adolescence or adulthood with bone pain, weakness, and pseudo fractures but no deformity.
Autosomal Recessive Hypophosphatemic Rickets
ARHR type 1 occurs due to loss of function mutations in DMP 1, a noncollagenous bone matrix protein expressed in osteoblasts and osteocytes.45,56 DMP1 has a role in osteocyte proliferation and in the downregulation of FGF-23 and hence loss of its function will augment FGF-23 activity, explaining renal phosphate loss and hypophosphatemia. Another form of ARHR (ARHR 2) has been recently described. It occurs due to loss of function mutations in ectonucleotide pyrophosphatase/phosphodiesterase 1, which is an enzyme that generates pyrophosphate from ATP, and pyrophosphate is a mineralization inhibitor.57 Loss of function mutations in this protein can lead to generalized arterial calcification of infancy. Clinical manifestations and biochemical findings of patients with ARHR are similar to those with X-linked dominant hypophosphatemic rickets.
Hereditary Hypophosphatemic Rickets With Hypercalciuria
This particular variant of inherited hypophosphatemic rickets results from a genetic defect wherein there is loss of function mutation in the gene SLC34A3 that encodes sodium-phosphate cotransporter (NaPi-2c), disrupting its key role in maintaining phosphate homeostasis.58, 59, 60 Bone pain, muscle weakness, and pseudo fractures are the common presenting complaints, but dental abnormalities are not usually reported. In contrast to other variants of inherited hypohosphatemic rickets, FGF-23 level is normal and 1,25-dihydroxy vitamin D levels are elevated for low phosphorus levels. These patients also exhibit hypercalciuria, which predisposes them to nephrolithiasis.50
Secondary or Idiopathic Hypophosphatemic Rickets
Idiopathic hypophosphatemic rickets has been described recently in children who are fed with an amino acid–based elemental formula, especially Neocate formula products.61 A retrospective study of 51 children who were evaluated for fractures or rickets were found to have unexplained hypophosphatemia.61 Most of the children were suffering from complex illnesses, and the common finding was that they were solely fed with Neocate formula products.61 Elevated alkaline phosphatase activity was noted along with preserved renal phosphate conservative function.61 In spite of sufficient elemental formula composition, biochemistries pointed to dietary deficiency or severe phosphate malabsorption.61 Furthermore, improvement in biochemistry with eventual improvement in skeletal abnormalities was observed following transition to a different elemental formula, indicating decreased bioavailability of formula phosphorus in some clinical settings.61 Close monitoring of micronutrients in children fed with amino acid–based elemental formula is very important, and prompt phosphate supplementation or switching to alternate formula is recommended if hypophosphatemia is present.61 Another less common cause of secondary hypophosphatemia is excessive use of phosphate binding agents, such as antacid abuse that results in reduced intestinal absorption of phosphate.61 Also, elevated gastric pH due to gastric acid-modifying agents such as proton pump inhibitors affect calcium and phosphate solubility, resulting in decreased intestinal absorption.61 However, correction of phosphate levels may be done without changing acid-modifying agents.61
Management of Hypophosphatemic Rickets
Figure 4 gives an algorithmic approach to a child with rickets. As per European evidence-based guideline for the diagnosis and management, diagnosis of X-linked hypophosphatemic rickets is based on clinical signs of rickets and/or osteomalacia along with hypophosphatemia and renal phosphate loss without deficiency of vitamin D or calcium.62 The diagnosis of X-linked hypophophatemic rickets should be confirmed where possible by genetic testing or measuring FGF-23 levels before initiating treatment.62 Phosphate supplementation (elemental phosphorus in 3 to 5 doses of 20 to 60 mg/kg per day) remains the cornerstone of management. Most of them also benefit from calcitriol supplementation (20–30 ng/kg per day) or alfacalcidol 30 to 50 ng/kg per day, as the FGF-23 suppresses formation of 1,25 dihydroxy cholecalciferol.7,48,62,63 If supplemented by calcitriol, urinary calcium should be monitored closely to avoid nephrocalcinosis.62 Of note, the management of patients with hereditary hypophosphatemic rickets with hypercalciuria differs because the former does not require calcitriol, which may worsen hypercalciuria and increases the predisposition for nephrocalcinosis. Supplementation with phosphate forms the mainstay of its treatment.64,65 In addition to the preceding standard therapy, evaluation and treatment of iron deficiency is important in children with autosomal dominant hypophosphatemic rickets, as iron deficiency causes increased expression of the FGF-23 gene.55 In a prospective open-label clinical trial involving 6 adults with autosomal dominant hypophosphatemic rickets with iron deficiency, oral iron supplementation normalized FGF-23 and serum phosphorus levels.55 Further, marked symptomatic improvement was noted with correction of hypophosphatemia, and calcitriol and phosphate supplements were discontinued.55 In the management of X-linked hypophosphatemia, secondary hyperparathyroidism results from the persistent stimulation of parathyroid cells by FGF-23 and phosphate supplements, and also from the suppression of 1,25 dihydroxy vitamin D by FGF-23 in children not managed with active vitamin D.62 This secondary hyperparathyroidism leads to increased phosphaturia and bone resorption.62 In contrast, excessive vitamin D treatment and/or inadequate phosphate intake suppresses PTH levels and results in reduced bone turnover, jeopardizing the healing process of rickets and growth.62 This necessitates the adjustment of therapies and monitoring of PTH levels to keep within normal limits (10–65 pg/ml).62 Evidence-based European guidelines recommend treatment with calcimimetic agents, such as cinacalcet, for persistent secondary hyperparathyroidism if higher dosage of active vitamin D and/or lower dosage of oral phosphate fail to normalize PTH levels.62 However, extreme caution should be exercised while using cinacalcet because of severe side effects, such as hypocalcemia and prolonged QT interval.62 Further, guidelines recommend parathyroidectomy for persistent hypercalcemic hyperparathyroidism despite optimized active vitamin D and cinacalcet treatment.62 Also, a multidisciplinary approach led by a metabolic bone disease expert is recommended, given the multisystemic nature of the disease.62
Figure 4.
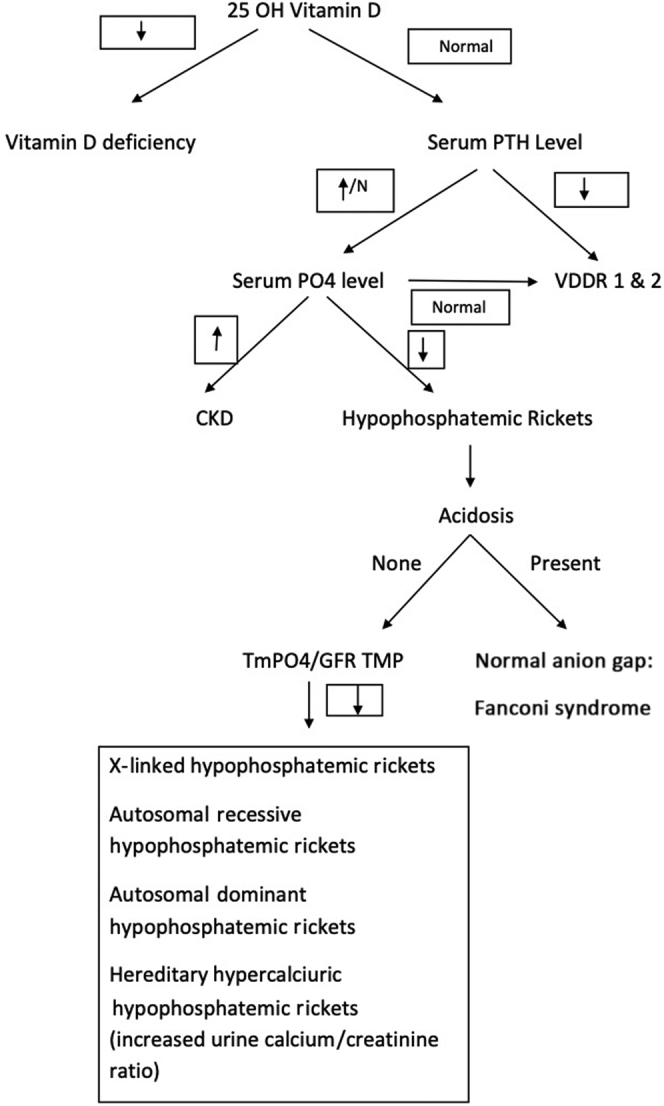
Algorithmic approach to a child with rickets. CKD, chronic kidney disease; GFR, glomerular filtration rate; N, normal; PTH, parathyroid hormone; TMP, renal threshold phosphate concentration; TmPO4, renal tubular reabsorption of phosphate; VDDR, vitamin D–dependent type 1 rickets.
An important recent development is the introduction of burosumab (KRN23), a human monoclonal antibody against FGF-23 that has been shown to be effective in children with X-linked hypophosphatemia.66 A randomized, active-controlled, open-label, phase 3 trial involving 61 children with X-linked hypophosphatemia from 16 clinical sites compared the efficacy and safety of burosumab versus conventional treatment with oral phosphate and active vitamin D supplements.67 This study found the burosumab group showed significant improvement in severity of rickets, growth, and serum biochemistries compared with the conventional therapy group.67 In another open-label phase 2 trial, 52 children with X-linked hypophosphatemia were randomly assigned, in a 1:1 ratio, to receive subcutaneous burosumab either every 2 weeks or every 4 weeks, and the dose was adjusted to achieve a low normal serum phosphorus level.66 Treatment with burosumab every 2 weeks provided a more sustained increase in the phosphorus levels and healing of rickets compared with the every 4 weeks regimen. The authors concluded that burosumab was effective in improving the renal tubular phosphate reabsorption, serum phosphorus levels, linear growth, and physical function, and reduced pain and the severity of rickets in children with X-linked hypophosphatemia.66 Burosumab therapy is expensive, and its cost-effectiveness and long-term outcome data are awaited.62 The most common side effects of burosumab therapy are extremities pain, headaches, and injection site reactions.62 It was recently approved by the U.S. Food and Drug Administration for its intended purpose, in patients aged 1 year and older in 2018.68 Also, the European Medicines Agency granted conditional marketing authorization to burosumab in February 2018.62 The evidence-based European guidelines recommend starting burosumab therapy at a dose of 0.4 mg/kg subcutaneously every 2 weeks in X-linked hypophosphatemic children aged 1 year or older with growing skeletons and presence of overt bone disease on radiographs, or refractoriness or complications or noncompliance to conventional treatment.62 The recommended dosage adjustments of burosumab should not be more often than every 4 weeks, and are in increments of 0.4 mg/kg to a maximum of 2 mg/kg to maintain fasting serum phosphate levels within the lower end of normal limit for the age.62
Fanconi Syndrome
Fanconi syndrome, in which underlying proximal tubulopathy results in glycosuria, hypokalemia, proximal renal tubular acidosis, hyperuricosuria, and generalized aminoaciduria also can have significant urinary phosphate loss resulting in hypophosphatemic rickets. It may be primary or secondary to conditions like cystinosis, Lowe's syndrome, Fanconi-Bickel syndrome, or drugs. Management includes any specific therapy for the underlying disorder if identified along with phosphate supplementation and acidosis correction.69
Supplementary Table S1 describes the various animal models, including detailed descriptions of different types of rickets.56,70, 71, 72, 73, 74, 75, 76, 77
Conclusions
Rickets is a disorder of growing children that arises from defective mineralization of the growth plate. Nutritional rickets is a preventable disease by maintaining an adequate intake of vitamin D through both dietary and sunlight exposure. Vitamin D supplementation will work in nutritional rickets secondary to vitamin D deficiency but not in most of the non-nutritional variants of rickets. Figure 4 gives an algorithmic approach to a child with rickets and Table 2 summarizes the major differentiating points between the various types of rickets. Knowledge of these conditions is essential for prompt diagnosis and proper management.
Disclosure
All the authors declared no competing interests.
Footnotes
Table S1. Description of various animal models, including different types of rickets.
Supplementary Material
References
- 1.Craviari T., Pettifor J.M., Thacher T.D. Rickets: an overview and future directions, with special reference to Bangladesh. A summary of the Rickets Convergence Group meeting, Dhaka, 26–27 January, 2006. J Health Popul Nutr. 2008;26:112–121. [PMC free article] [PubMed] [Google Scholar]
- 2.Carpenter T.O., Shaw N.J., Portale A.A. Rickets. Nat Rev Dis Primers. 2017;3:17101. doi: 10.1038/nrdp.2017.101. [DOI] [PubMed] [Google Scholar]
- 3.Jagtap V.S., Sarathi V., Lila A.R. Hypophosphatemic rickets. Indian J Endocrinol Metab. 2012;16:177–182. doi: 10.4103/2230-8210.93733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahay M., Sahay R. Rickets-vitamin D deficiency and dependency. Indian J Endocrinol Metab. 2012;16:164–176. doi: 10.4103/2230-8210.93732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitt M.J. Rickets and osteomalacia are still around. Radiol Clin North Am. 1991;29:97–118. [PubMed] [Google Scholar]
- 6.Tiosano D., Hochberg Z. Hypophosphatemia:the common denominator of all rickets. J Bone Miner Metab. 2009;27:392–401. doi: 10.1007/s00774-009-0079-1. [DOI] [PubMed] [Google Scholar]
- 7.Goldsweig B.K., Carpenter T.O. Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep. 2015;13:88–97. doi: 10.1007/s11914-015-0259-y. [DOI] [PubMed] [Google Scholar]
- 8.Mughal M.Z. Rickets. Curr Osteoporos Rep. 2011;9:291–299. doi: 10.1007/s11914-011-0081-0. [DOI] [PubMed] [Google Scholar]
- 9.Sabbagh Y., Carpenter T.O., Demay M.B. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102:9637–9642. doi: 10.1073/pnas.0502249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas M.K., Lloyd-Jones D.M., Thadhani R.I. Hypovitaminosis D in medical inpatients. N Engl J Med. 1998;338:777–783. doi: 10.1056/NEJM199803193381201. [DOI] [PubMed] [Google Scholar]
- 11.Yan X., Han X., Zhang H.F. [Interpretation for the global consensus recommendations on prevention and management of nutritional rickets] Zhonghua er ke za zhi. 2016;54:891–895. doi: 10.3760/cma.j.issn.0578-1310.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Kaseda R., Hosojima M., Sato H. Role of megalin and cubilin in the metabolism of vitamin D(3) Ther Apher Dial. 2011;15(Suppl 1):14–17. doi: 10.1111/j.1744-9987.2011.00920.x. [DOI] [PubMed] [Google Scholar]
- 13.Mozos I., Marginean O. Links between vitamin D deficiency and cardiovascular diseases. Biomed Res Int. 2015;2015:109275. doi: 10.1155/2015/109275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christakos S., Dhawan P., Benn B. Vitamin D. Ann N Y Acad Sci. 2007;1116:340–348. doi: 10.1196/annals.1402.070. [DOI] [PubMed] [Google Scholar]
- 15.Martin A., David V., Quarles L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92:131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wharton B., Bishop N. Rickets. Lancet. 2003;362:1389–1400. doi: 10.1016/S0140-6736(03)14636-3. [DOI] [PubMed] [Google Scholar]
- 17.Konradsen S., Ag H., Lindberg F. Serum 1,25-dihydroxy vitamin D is inversely associated with body mass index. Eur J Nutr. 2008;47:87–91. doi: 10.1007/s00394-008-0700-4. [DOI] [PubMed] [Google Scholar]
- 18.Munns C.F., Shaw N., Kiely M. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016;101:394–415. doi: 10.1210/jc.2015-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shroff R., Wan M., Nagler E.V. Clinical practice recommendations for native vitamin D therapy in children with chronic kidney disease Stages 2–5 and on dialysis. Nephrol Dial Transplant. 2017;32:1098–1113. doi: 10.1093/ndt/gfx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ross A.C., Taylor C.L., Yaktine A.L., Del Valle H.B., editors. Dietary Reference Intakes for Calcium and Vitamin D. National Academies Press, National Academy of Sciences; Washington, DC: 2011. Institute of Medicine Committee to Review Dietary Reference Intakes for Vitamin D, Calcium. The National Academies Collection: Reports funded by National Institutes of Health; pp. 345–402. [Google Scholar]
- 21.Ariganjoye R. Pediatric hypovitaminosis D. Glob Pediatr Health. 2017;4 doi: 10.1177/2333794X16685504. 2333794X16685504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aspray T.J., Bowring C., Fraser W., Gittoes N. National Osteoporosis Society vitamin D guideline summary. Age Ageing. 2014;43:592–595. doi: 10.1093/ageing/afu093. [DOI] [PubMed] [Google Scholar]
- 23.Holick M.F., Binkley N.C., Bischoff-Ferrari H.A. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911–1930. doi: 10.1210/jc.2011-0385. [DOI] [PubMed] [Google Scholar]
- 24.Graf L., Reidy K., Kaskel F.J. Nutrition management in childhood kidney disease: an integrative and lifecourse approach. In: Avner E.D., Harmon W.E., Niaudet P., editors. Pediatric Nephrology. Springer; Berlin: 2016. pp. 341–360. [Google Scholar]
- 25.National Kidney Foundation K/DOQI clinical practice guidelines for bone metabolism and disease in children with chronic kidney disease. Am J Kidney Dis. 2005;46:S1–S122. [PubMed] [Google Scholar]
- 26.Meyer H.E., Skram K., Berge I.A. Nutritional rickets in Norway: a nationwide register-based cohort study. BMJ Open. 2017;7 doi: 10.1136/bmjopen-2016-015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kift R., Berry J.L., Vail A. Lifestyle factors including less cutaneous sun exposure contribute to starkly lower vitamin D levels in U.K. South Asians compared with the white population. Br J Dermatol. 2013;169:1272–1278. doi: 10.1111/bjd.12518. [DOI] [PubMed] [Google Scholar]
- 28.Lo C.W., Paris P.W., Holick M.F. Indian and Pakistani immigrants have the same capacity as Caucasians to produce vitamin D in response to ultraviolet irradiation. Am J Clin Nutr. 1986;44:683–685. doi: 10.1093/ajcn/44.5.683. [DOI] [PubMed] [Google Scholar]
- 29.Al-Mogbel E.S. Vitamin D status among adult Saudi females visiting primary health care clinics. Int J Health Sci. 2012;6:116–126. doi: 10.12816/0005987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhas Y., Mishra N., Banerjee J. Vitamin D deficiency and oxidative stress in type 2 diabetic population of India. Cardiovasc Hematol Agents Med Chem. 2017;14:82–89. doi: 10.2174/1871525714666160426150233. [DOI] [PubMed] [Google Scholar]
- 31.Thandrayen K., Pettifor J.M. Maternal vitamin D status: implications for the development of infantile nutritional rickets. Endocrinol Metab Clin North Am. 2010;39:303–320. doi: 10.1016/j.ecl.2010.02.006. table of contents. [DOI] [PubMed] [Google Scholar]
- 32.Emmerson A.J.B., Dockery K., Mughal M.Z. Vitamin D status of white pregnant women and infants at birth and 4 months in North West England: a cohort study. Matern Child Nutr. 2018;14(1) doi: 10.1111/mcn.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahay M., Sahay R. Renal rickets-practical approach. Indian J Endocrinol Metab. 2013;17:S35–S44. doi: 10.4103/2230-8210.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson P.D., Hogler W., Craig M.E. The re-emerging burden of rickets: a decade of experience from Sydney. Arch Dis Child. 2006;91:564–568. doi: 10.1136/adc.2004.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prentice A. Nutritional rickets around the world. J Steroid Biochem Mol Biol. 2013;136:201–206. doi: 10.1016/j.jsbmb.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 36.Gartner L.M., Greer F.R. Prevention of rickets and vitamin D deficiency: new guidelines for vitamin D intake. Pediatrics. 2003;111:908–910. doi: 10.1542/peds.111.4.908. [DOI] [PubMed] [Google Scholar]
- 37.Takeda E., Yamamoto H., Taketani Y. Vitamin D-dependent rickets type I and type II. Acta Paediatr Jpn. 1997;39:508–513. doi: 10.1111/j.1442-200x.1997.tb03629.x. [DOI] [PubMed] [Google Scholar]
- 38.Holick M.F. Resurrection of vitamin D deficiency and rickets. J Clin Invest. 2006;116:2062–2072. doi: 10.1172/JCI29449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ting T.H., Devnani A.S. Vitamin-D-deficiency rickets even with abundant sunlight -A case to highlight emerging problem. Med J Malaysia. 2016;71:354–356. [PubMed] [Google Scholar]
- 40.Forghani N., Lum C., Krishnan S. Two new unrelated cases of hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from the same novel nonsense mutation in the vitamin D receptor gene. J Pediatr Endocrinol Metab. 2010;23:843–850. doi: 10.1515/jpem.2010.136. [DOI] [PubMed] [Google Scholar]
- 41.Choudhury S., Jebasingh K.F., Ranabir S. Familial vitamin D resistant rickets: end-organ resistance to 1,25-dihydroxyvitamin D. Indian J Endocrinol Metab. 2013;17:S224–S227. doi: 10.4103/2230-8210.119579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraser D., Scriver C.R. Familial forms of vitamin D-resistant rickets revisited. X-linked hypophosphatemia and autosomal recessive vitamin D dependency. Am J Clin Nutr. 1976;29:1315–1329. doi: 10.1093/ajcn/29.11.1315. [DOI] [PubMed] [Google Scholar]
- 43.Reddy P. Clinical approach to renal tubular acidosis in adult patients. Int J Clin Pract. 2011;65:350–360. doi: 10.1111/j.1742-1241.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- 44.Walsh S.B., Shirley D.G., Wrong O.M. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment:an alternative to ammonium chloride. Kidney Int. 2007;71:1310–1316. doi: 10.1038/sj.ki.5002220. [DOI] [PubMed] [Google Scholar]
- 45.Karaplis A.C., Bai X., Falet J.P. Mineralizing enthesopathy is a common feature of renal phosphate-wasting disorders attributed to FGF23 and is exacerbated by standard therapy in Hyp mice. Endocrinology. 2012;153:5906–5917. doi: 10.1210/en.2012-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whyte M.P. Hypophosphatasia: an overview. Bone. 2017;102:15–25. doi: 10.1016/j.bone.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 47.Endo I., Fukumoto S., Ozono K. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J. 2015;62:811–816. doi: 10.1507/endocrj.EJ15-0275. [DOI] [PubMed] [Google Scholar]
- 48.Imel E.A., Econs M.J. Approach to the hypophosphatemic patient. J Clin Endocrinol Metab. 2012;97:696–706. doi: 10.1210/jc.2011-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misra M., Pacaud D., Petryk A. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. 2008;122:398–417. doi: 10.1542/peds.2007-1894. [DOI] [PubMed] [Google Scholar]
- 50.Page K., Bergwitz C., Jaureguiberry G. A patient with hypophosphatemia, a femoral fracture, and recurrent kidney stones: report of a novel mutation in SLC34A3. Endocr Pract. 2008;14:869–874. doi: 10.4158/EP.14.7.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sattur A., Naikmasur V., Shrivastava R. Familial hypophosphatemic rickets. J Indian Soc Pedod Prev Dent. 2010;28:302–306. doi: 10.4103/0970-4388.76163. [DOI] [PubMed] [Google Scholar]
- 52.Lambert A.S., Zhukouskaya V., Rothenbuhler A. X-linked hypophosphatemia: management and treatment prospects. Joint Bone Spine. 2019;86:731–738. doi: 10.1016/j.jbspin.2019.01.012. [DOI] [PubMed] [Google Scholar]
- 53.Rothenbuhler A., Fadel N., Debza Y. High incidence of cranial synostosis and Chiari I malformation in children with x-linked hypophosphatemic rickets (XLHR) J Bone Miner Res. 2019;34:490–496. doi: 10.1002/jbmr.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.White K.E., Carn G., Lorenz-Depiereux B. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 55.Imel E.A., Liu Z., Coffman M. Oral iron replacement normalizes fibroblast growth factor 23 in iron-deficient patients with autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2020;35:231–238. doi: 10.1002/jbmr.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng J.Q., Ward L.M., Liu S. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levy-Litan V., Hershkovitz E., Avizov L. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86:273–278. doi: 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bergwitz C., Roslin N.M., Tieder M. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lorenz-Depiereux B., Benet-Pages A., Eckstein G. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ichikawa S., Sorenson A.H., Imel E.A. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027. doi: 10.1210/jc.2005-2840. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez Ballesteros L.F., Ma N.S., Gordon R.J. Unexpected widespread hypophosphatemia and bone disease associated with elemental formula use in infants and children. Bone. 2017;97:287–292. doi: 10.1016/j.bone.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haffner D., Emma F., Eastwood D.M. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15:435–455. doi: 10.1038/s41581-019-0152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruppe M.D. X-linked hypophosphatemia. In: Pagon R.A., Adam M.P., Ardinger H.H., editors. GeneReviews(R) University of Washington; Seattle: 1993. [Google Scholar]
- 64.Baroncelli G.I., Toschi B., Bertelloni S. Hypophosphatemic rickets. Curr Opin Endocrinol Diabetes Obes. 2012;19:460–467. doi: 10.1097/MED.0b013e328358be97. [DOI] [PubMed] [Google Scholar]
- 65.Pavone V., Testa G., Gioitta Iachino S. Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol. 2015;25:221–226. doi: 10.1007/s00590-014-1496-y. [DOI] [PubMed] [Google Scholar]
- 66.Carpenter T.O., Whyte M.P., Imel E.A. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378:1987–1998. doi: 10.1056/NEJMoa1714641. [DOI] [PubMed] [Google Scholar]
- 67.Imel E.A., Glorieux F.H., Whyte M.P. Burosumab versus conventional therapy in children with X-linked hypophosphataemia:a randomised, active-controlled, open-label, pase 3 trial. Lancet. 2019;393:2416–2427. doi: 10.1016/S0140-6736(19)30654-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.FDA. FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-rare-inherited-form-rickets-x-linked-hypophosphatemia. Accessed September 20, 2018. 2018.
- 69.Mohandas Nair K., Sakamoto O., Jagadeesh S. Fanconi-Bickel syndrome. Indian J Pediatr. 2012;79:112–114. doi: 10.1007/s12098-011-0373-5. [DOI] [PubMed] [Google Scholar]
- 70.Lorenz-Depiereux B., Guido V.E., Johnson K.R. New intragenic deletions in the PHEX gene clarify X-linked hypophosphatemia-related abnormalities in mice. Mamm Genome. 2004;15:151–161. doi: 10.1007/s00335-003-2310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li Y.C., Pirro A.E., Amling M. Targeted ablation of the vitamin D receptor:an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dardenne O., Prud’homme Je, Arabian A. Targeted Inactivation of the 25-hydroxyvitamin D3–1α-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin d-deficiency rickets∗. Endocrinology. 2001;142:3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 73.Shimada T., Kakitani M., Yamazaki Y. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao X., Dittmer K.E., Blair H.T. A novel nonsense mutation in the DMP1 gene identified by a genome-wide association study is responsible for inherited rickets in Corriedale sheep. PloS One. 2011;6 doi: 10.1371/journal.pone.0021739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garner S.C., Pi M., Tu Q. Rickets in cation-sensing receptor-deficient mice: an unexpected skeletal phenotype. Endocrinology. 2001;142:3996–4005. doi: 10.1210/endo.142.9.8364. [DOI] [PubMed] [Google Scholar]
- 76.Kaune R., Harmeyer J. Vitamin D3 metabolism in a pig strain with pseudo vitamin D-deficiency rickets, type I. Acta Endocrinol (Copenh) 1987;115:345–352. doi: 10.1530/acta.0.1150345. [DOI] [PubMed] [Google Scholar]
- 77.Safadi F.F., Thornton P., Magiera H. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. 1999;103:239–251. doi: 10.1172/JCI5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.