

Abstract
Respiratory motor neuron survival is critical for maintenance of adequate ventilation and airway clearance, preventing dependence to mechanical ventilation and respiratory tract infections. Phrenic motor neurons are highly vulnerable in rodent models of motor neuron disease versus accessory inspiratory motor pools (e.g. intercostals, scalenus). Thus, strategies that promote phrenic motor neuron survival when faced with disease and/or toxic insults are needed to help preserve breathing ability, airway defense and ventilator independence. Adenosine 2A receptors (A2a) are emerging as a potential target to promote neuroprotection, although their activation can have both beneficial and pathogenic effects. Since the role of A2a receptors in the phrenic motor neuron survival/death is not known, we tested the hypothesis that A2a receptor antagonism promotes phrenic motor neuron survival and preserves diaphragm function when faced with toxic, neurodegenerative insults that lead to phrenic motor neuron death. We utilized a novel neurotoxic model of respiratory motor neuron death recently developed in our laboratory: intrapleural injections of cholera toxin B subunit (CtB) conjugated to the ribosomal toxin, saporin (CtB-Saporin). We demonstrate that intrapleural CtB-Saporin causes: 1) profound phrenic motor neuron death (~5% survival); 2) ~7-fold increase in phrenic motor neuron A2a receptor expression prior to cell death; and 3) diaphragm muscle paralysis (inactive in most rats; ~7% residual diaphragm EMG amplitude during room air breathing). The A2a receptor antagonist istradefylline given after CtB-Saporin: 1) reduced phrenic motor neuron death (~20% survival) and 2) preserved diaphragm EMG activity (~46%). Thus, A2a receptors contribute to neurotoxic phrenic motor neuron death, an effect mitigated by A2a receptor antagonism.
Keywords: Adenosine, ADORA2A, A2a receptor, Neuroprotection, Phrenic motor neuron survival, Apoptosis, p38
Introduction
Respiratory motor neuron loss contributes to respiratory insufficiency, ventilator dependence, and death in clinical disorders such as ALS, cervical spinal cord injury, and infectious or toxic neuropathies (Nogués and Benarroch, 2008; Johnson and Mitchell, 2013; Seven and Mitchell, 2019). Since the diaphragm is the major inspiratory muscle, improved understanding of factors exacerbating/ameliorating phrenic motor neuron death is essential in our effort to design new therapies that promote phrenic motor neuron survival to preserve breathing function.
Mechanisms of neuroprotection are often studied on neuronal cultures and/or in vivo rodent models (Pedata et al., 2005; Melani et al., 2006; Mojsilovic-Petrovic et al., 2006; Jeanneteau et al., 2008; Paterniti et al., 2011). However, differential susceptibilities of discrete motor pools are seldom considered in these investigations (Lladó et al., 2006; Nichols et al., 2013; Seven et al., 2018c), despite the fact that motor neurons are diverse in their susceptibility to insults such as trauma, inflammation, infection and/or neurotoxins. For example, in the SOD1G93A rat model of ALS, phrenic motor neuron death (~80%) is more pronounced than intercostal (~50%) or hypoglossal (~0%) motor pools (Nichols et al., 2013; Seven et al., 2018c). Unknown differences in protein expression or signaling cascades likely determine differential motor neuron susceptibility to insults between motor pools. In particular, phrenic motor neurons present unique gene expression patterns versus motor neurons innervating limbs (Machado et al., 2014), and differ in cell signaling versus motor neurons innervating the intercostals (Navarrete-Opazo and Mitchell, 2014; Navarrete-Opazo et al., 2014). These differences may play a role in differential phrenic motor neuron susceptibility to death. Thus, neuroprotective therapies specialized to phrenic motor neuron survival should be investigated.
Adenosine receptors are emerging as potential targets for therapeutic interventions to promote neuroprotection. For example, adenosine 2A (A2a) receptor antagonism slows excitotoxic death of embryonic motor neurons in vitro (Mojsilovic-Petrovic et al., 2006), and elicits neuroprotective and anti-inflammatory effects in the face of certain challenges in vivo (Melani et al., 2006; Yu et al., 2008; Orr et al., 2018).
Although A2a receptors may contribute to death in some populations of CNS neurons, their role on phrenic motor neuron cell death is not known. Thus, we tested the hypothesis that A2a receptor antagonism promotes phrenic motor neuron survival and improves diaphragm function following a neurotoxic insult that causes phrenic motor neuron death. We utilized a novel and selective neuro-toxicological model of protein translation inhibition-induced neuronal loss via intrapleural injections of cholera toxin B subunit (CtB) conjugated to the ribosomal toxin saporin (CtB-Saporin). Intrapleural CtB-Saporin injections cause dose-dependent phrenic motor neuron death in the mid-cervical spinal cord and intercostal motor neuron death in the thoracic spinal cord (Nichols et al., 2015). Here, we utilized immunohistochemistry to assess phrenic motor neuron survival, A2a receptor expression and phosphorylation state of candidate downstream signaling proteins, as well as electrophysiology to assess diaphragm activity. We found that: 1) intrapleural CtB-Saporin injections upregulate A2a receptor expression in phrenic motor neurons prior to their death, and 2) A2a receptor antagonist administration reduces phrenic motor neuron loss, at least partially preserving diaphragm EMG activity.
Methods
Animals
Experiments were conducted on 32 adult, male, Sprague-Dawley rats weighing ~350g (Envigo, Indianapolis, IN, USA). Experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee and were conducted in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and the NIH Guide for the Care and Use of Laboratory Animals (2011). Rats were fed ad libitum, monitored daily for their body weights and behavior, and were exposed to 12-hour light-dark cycle. Intrapleural injections and terminal procedures were conducted under anesthesia, whereas intraperitoneal injections were performed while awake. Toe pinch and corneal reflexes were absent during anesthesia; blood pressure and heart rate responses to the toe pinch and were also absent during terminal EMG studies. All rats were sacrificed via transcardial perfusion.
Experimental Design
A total of 32 rats were studied. Different sets of rats were used for electrophysiology and phrenic motor neuron count (Day 8, n = 21), and protein analysis experiments to quantify protein expression prior to total phrenic motor neuron loss (Day 5, n = 11, see Figure 1). In electrophysiological experiments, we tested the hypothesis that A2a receptor antagonist enhances phrenic motor neuron survival and preserves diaphragm activity following CtB-Saporin injections. In immunohistochemistry experiments, we tested the hypothesis that A2a receptor expression is upregulated in CtB-identified phrenic motor neurons following CtB-Saporin injections prior to complete cell death.
Figure 1:
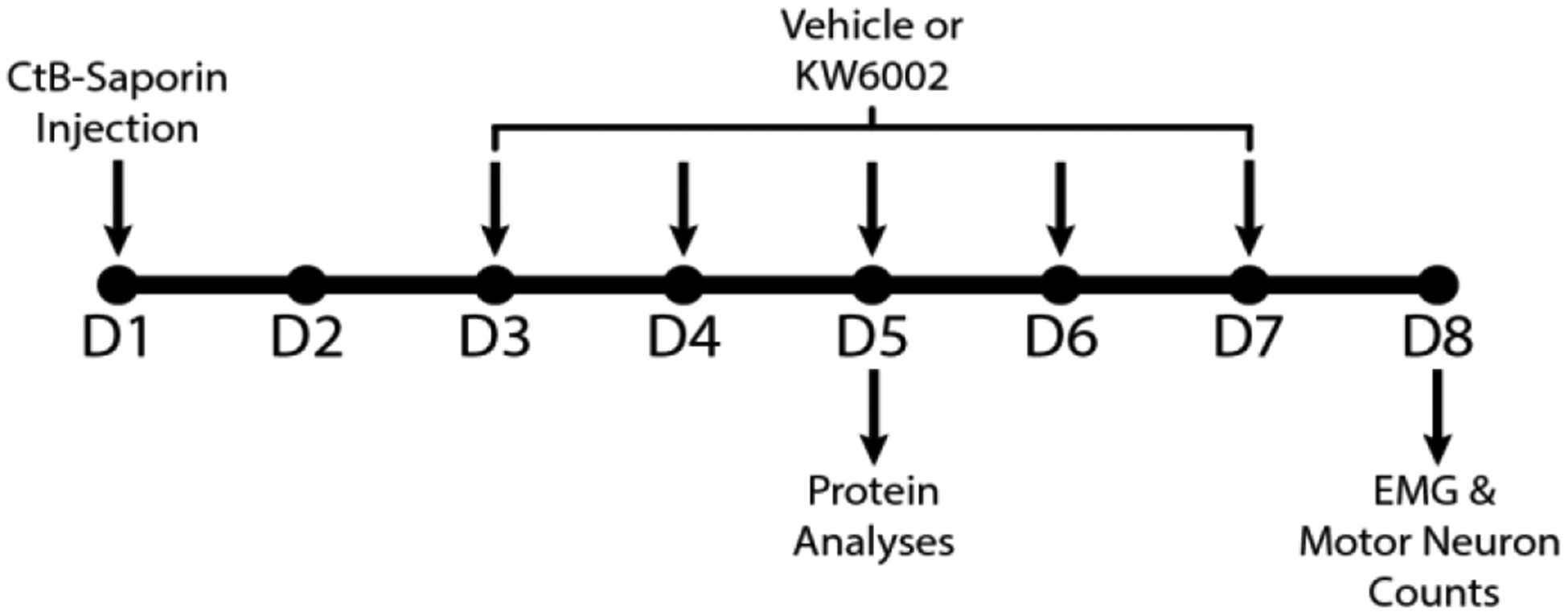
Timeline for experimental procedures: 36 hours after bilateral intrapleural CtB-Saporin injections (Total dose: 50 μg/rat), rats were treated with A2a receptor antagonist (KW6002; also known as Istradefylline) twice daily, attaining a total dose of 1 mg/kg/day. Terminal EMG measurements and phrenic motor neuron counts were performed on day 8 (24 h after the last A2a receptor antagonist injection). Because almost all phrenic motor neurons died by day 8, an earlier time-point when phrenic motor neurons were still viable was selected for immunohistochemical evaluation of protein expression and localization. Thus, for molecular analyses, rats were sacrificed and perfused at Day 5 post-CtB-Saporin injection.
Drugs
CtB-Saporin (50 μg, Catalog No: IT-14, Advanced Targeting Systems, San Diego, CA) was dissolved in 80 μl phosphate buffered saline (PBS) and administered intrapleurally to target phrenic motor neurons (25 μg per side) using 50 μl Hamilton syringes having semi-blunt needles of 23-gauge and 6 mm length through the fifth intercostal space anterior axillary line (Mantilla et al., 2009). Additionally, 50 μg CtB (Catalog No: 227039, EMD Millipore, Billerica, MA, USA) was dissolved in 25 μl sterile water and injected intrapleurally (12.5 μl on each side) to label the spared phrenic motor neurons.
Rats were administered with A2a receptor antagonist starting 36 hours after CtB-Saporin injections, until 6 hours before perfusion at day 5 or 24 hours before EMG recordings (to minimize acute effects of the drug on recordings) and subsequent perfusion. KW6002 (Istradefylline, Sigma-Aldrich) is a selective, blood-brain-barrier-penetrable A2a receptor antagonist (Yang et al., 2007). KW6002 was dissolved in DMSO (37°C) at 9.3 mg/ml concentration, sonicated, aliquoted and stored at 4°C in dark. Prior to use, KW6002 was re-warmed and administered intraperitoneally twice daily at a dose of 0.5 mg/kg (1.0 mg/kg per day total). The volume of DMSO administered to the vehicle rats was also normalized to body weight (~17–20 μl DMSO per injection, ~50 μl/kg).
EMG Recordings
Rats were anesthesized with isoflurane (3% induction, 2–2.3% maintenance), then converted to urethane anesthesia (1.5–1.6 g/kg) via tail vein intravenous fluid delivery (R-99, Razel Scientific Instruments, Saint Albans, Vermont). To maintain fluid and acid-base balance, Lactated Ringer’s Solution was administered via tail vein as necessary. Right femoral artery was catheterized to measure arterial pressure and to collect arterial blood sample to measure PO2, PCO2, pH, and standard base excess (ABL90 Flex, Radiometer, Copenhagen, Denmark). Body temperature was maintained at 37.5 ± 0.5 °C with a custom-built heating table and rectal thermistor (Physitemp, model 700 1H).
Teflon-coated stainless-steel multistrand electrodes (AS631, Cooner Wire, Chatsworth, CA) were uninsulated for a length of ~2 mm under a dissecting scope on graph paper. A pair of electrodes were implanted in parallel to the muscle fibers to the mid-costal region of diaphragm muscle following laparotomy. To eliminate potential influences of upper airway muscles, the trachea was cannulated with tubing matching upper airway volume. Experiments were performed during spontaneous breathing with an inspired gas composition regulated to match desired blood-gas values (Table 1). Mechanical ventilation was not performed at any point during the experimental protocols. Following surgical procedures, isoflurane was cleared from the tissues by waiting at least 1 hour or the total isoflurane exposure period, whichever is longer.
Table 1:
Physiological variables at baseline and maximal chemoreceptor stimulation in control, vehicle-treated CtB-Saporin injected and istradefylline-treated CtB-Saporin animals 24 h following the last treatment
| Condition | Control | CtB-Saporin + Vehicle | CtB-Saporin + KW6002 | |
|---|---|---|---|---|
| PaCO2 (mmHg) | Baseline | 43.9±0.5 | 44.7±0.6 | 44.7±1.3 |
| MCS | 58.6±1.2 | 59.8±1.3 | 57.0±1.5 | |
| sBE (m Eq/L) | Baseline | 2.4±1.0 | 2.5±1.2 | 2.0±1.1 |
| MCS | 3.5±0.9 | 2.3±1.4 | 4.3±1.0 | |
| PaO2 (mmHg) | Baseline | 92.1 ±1.6 | 93.7±1.3 | 97.9±3.3 |
| MCS | 46.3±1.8 | 41.4±2.0 | 45.3±2.2 | |
| Temperature | Baseline | 37.3±0.2 | 37.4±0.2 | 37.5±0.2 |
| MCS | 36.9±0.1 | 36.8±0.1 | 37.5±0.1 | |
| MAP (mmHg) | Baseline | 94.8±5.2 | 96.2±7.3 | 96.6±5.8 |
EMG signals were differentially amplified (1000x) and bandpass filtered (10–1000 Hz) using a biopotential amplifier (Model 1800, A-M Systems, Carlsborg, WA, USA) prior to digitization at 10 kHz (Powerlab, AD Instruments, Colorado, United States). Since root-mean-squared (RMS) EMG correlates with force generated by various muscles including diaphragm muscle (Fuglevand et al., 1993; Lawrence and De Luca, 1983; Mantilla et al., 2010), it was used to estimate EMG amplitude using a 50-ms sliding window.
Spontaneous EMG signals were recorded in supine position at 4 different diaphragm activation levels: (1) Eupnea (i.e. PO2>85 mmHg and PCO2 ~45 mmHg). To attain these blood-gas values, inspired O2 was slightly supplemented (FiO2 = 21–26%) (2) Maximum chemoreceptor stimulation (MCS; FiO2 = 10.5%, FiCO2 = 7%). (3) Augmented breaths/sighs during MCS (RMS EMG amplitude > 2 x Eupneic value, presenting post-sigh apnea). Augmented breaths elicit near-maximal respiratory muscle activation (Seven et al., 2013; Seven et al., 2014; Seven et al., 2018c).
Immunohistochemistry and Image Acquisition
Immunohistochemistry was performed to count the phrenic motor neurons at day 8 and to evaluate protein expression and phosphorylation at day 5. Protein analyses were performed at day 5, because vehicle-treated CtB-Saporin group had nearly no surviving phrenic motor neurons by the 8th day. All rats were perfused transcardially using 0.01 M PBS followed by 4% paraformaldehyde (PFA) in 0.01 M PBS at the pH of 7.4 and 4°C. Rats perfused at day 8, had received their last A2a antagonist injection ~24 h ago. On the other hand, rats perfused at day 5, had received their last A2a antagonist injection ~4 h ago. Spinal cords were harvested, post-fixed at 4°C in 4% PFA overnight, and cryo-protected in 20% sucrose for 1 day and 30% sucrose for 3 days until sinking. C3, C4, and C5 spinal segments were sectioned at the transverse plane at 40 μm thickness using a freezing microtome (SM2010R, Leica, Buffalo Grove, IL, USA). Sections were stored in antifreeze solution (30% glycerol + 30% ethylene glycol in 0.01 M PBS at the pH of 7.4). All sections were uniformly sampled throughout C3–5 spinal segments for each protein of interest, washed (3x) in 0.01 M PBS, and blocked in the blocking solution (5% normal donkey serum, 0.5% bovine serum albumin, and 0.1% Triton X-100 in PBS) for 1 h. Sections were incubated in primary antibodies and 2.5% normal donkey serum, 0.25% bovine serum albumin, and 0.1% Triton X-100 in PBS at 4°C for 2 days. Then, sections were washed in PBS and incubated with their respective secondary antibodies at room temperature for 2 h. After the final washes, sections were mounted on charged slides with hard-set anti-fade solution (Vector Labs, Burlingame, CA, USA). Slides were imaged at 20x magnification (Numerical aperture: 0.75) with a fluorescence microscope (BZ-X710, Keyence Co., Osaka, Japan). The same exposure times and excitation intensities were used for all groups.
Phrenic Motor Neuron Counts.
Twelve sections per rat were systematically sampled from the C3-C5 spinal segments. CtB-labeled phrenic motor neurons were immunostained using goat anti-CtB primary antibody (1:2500, catalogue no: 227040, Millipore, Billerica, MA, USA) and anti-goat secondary antibody (1:1000, Alexa Fluor 488; Thermo Fisher Scientific, Waltham, MA). Imaging was performed with a GFP filter (BZ-X, model no: OP-87763) with exposure time of 1/35 s. CtB-positive motor neurons with intact nuclei were counted; phrenic motor neuron number was averaged per slice/ side.
Protein Expression, Phosphorylation and Localization.
Six sections per animal were systematically sampled from C3-C5 spinal segment and double-labeled for CtB and the protein of interest. CtB-labeled phrenic motor neurons were immunostained using goat anti-CtB primary antibody (1:2500, catalogue no: 227040, Millipore, Billerica, MA, USA) and anti-goat secondary antibody (1:1000, Alexa Fluor 488, Thermo Fisher). Other proteins were immunostained with the following primary antibodies: mouse anti-A2a receptor (1:500, 05–717, Millipore, Billerica, MA, USA), rabbit anti-phospho-p38 MAPK (1:500, 4370, Cell Signaling, Danvers, MA, USA), rabbit anti-phospho-ERK1/2 (1:500, 4370, Cell Signaling, Danvers, MA, USA), rabbit anti-phospho-JNK (1:250, 4668, Cell Signaling, Danvers, MA, USA); and the anti-mouse and anti-rabbit secondary antibodies (1:500, Alexa Fluor 594, Thermo Fisher). The exposure time for A 2A receptor labeling was 1/5 s. For the other proteins, sectioning module of the Keyence Imaging Software was used with the exposure times of 1/5, 1/4, and 1/3 s for phospho -ERK1/2, phospho-p38 MAPK, and phospho-JNK, respectively.
Image Analysis
Fluorescence images for A2a receptors, p-p38 MAPK, and pJNK were analyzed based on colocalization with the CtB-labelled phrenic motor neuron somata. A histogram was created for each image channel containing CtB immunostaining (green). For each CtB image, a threshold was set at a binarized pixel intensity value corresponding to a fixed percentile (P99.2) for all rats and images to account for changes in signal and background intensities. Using binarized images, CtB+ areas greater than 200 μm2 were used to mask the other channel to assess immunofluorescence intensity of the protein of interest.
Statistics
EMG recordings were averaged within each experimental condition to represent the value for each condition for each rat. Statistical analyses were performed using ANOVA. When the outcome of ANOVA was significant, Tukey-Kramer honestly significant difference test was used to assess pairwise differences post hoc. Data was presented as mean ± standard error of the mean. JMP 14.0 software was used for statistical calculations.
Results
Increased A2a receptor expression following CtB-Saporin injections
A2a receptors were observed in CtB-labelled phrenic motor neurons and other neurons in the spinal cord (Figure 2A–I) in agreement with our earlier report (Seven et al., 2018a). Extra-neuronal A2a receptor expression was uncommon. Five days after intrapleural CtB-Saporin injections, A2a receptors were upregulated by ~7-fold within identified phrenic motor neurons (3). This increase temporally precedes the 8-day time-point when most phrenic motor neurons die. Furthermore, A2a receptors were translocated to the nucleus following CtB-Saporin injections (the mechanism is currently under investigation). With A2a antagonist administration, no changes were observed in A2a receptor expression or localization within phrenic motor neurons.
Figure 2:
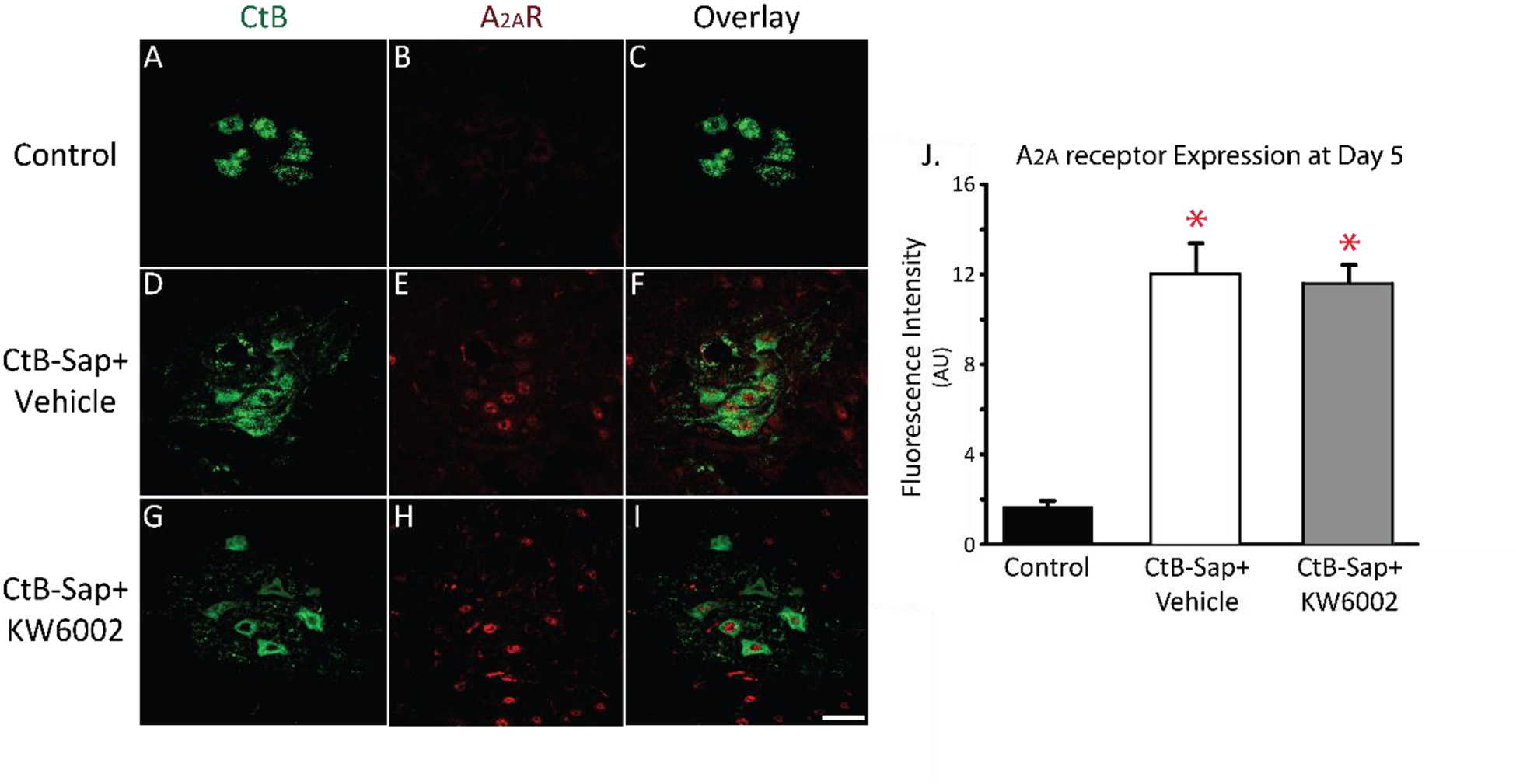
Adenosine 2A (A2a) receptor expression in phrenic motor neurons 5 days after intrapleural cholera toxin B (CtB)-Saporin injections (i.e. before phrenic motor neuron loss). A-I: Representative images (20x) for CtB (green) and A2a receptors (red) at Day 5. J: A2a receptor fluorescence intensity is increased following intrapleural CtB-Saporin injections versus control (p<0.01; red asterisks). Mean ± 1 SEM. Scale bar = 40 μm.
A2a receptor Inhibition Protects Phrenic Motor Neurons from Death
Only 5.4±2.9% of CtB-labelled phrenic motor neurons survived one week after intrapleural injection of 50 μg CtB-Saporin; no CtB-labelling was observed in 4 of 7 rats treated with CtB-Saporin. Punctate CtB staining is sometimes observed in phagocytic bodies at 1-week. These results were confirmed by the absence of neuronal nucleus (and peri-nucleus) marker (NeuN) staining and neuron-specific membrane marker (KCC2: Chloride potassium symporter 5) in the region of the phrenic motor nucleus in CtB-Saporin injected rats (unpublished observations). Selective A2a receptor antagonism protected phrenic motor neurons from toxic cell death; phrenic motor neuron survival was significantly increased (19.6±3.3%, p<0.0001).
A2a receptor inhibition preserves diaphragm activity after toxic insult
Viability of motor neuron soma may not imply phrenic motor neuron functionality. Thus, we evaluated diaphragm muscle activity (EMG) during different levels of diaphragm muscle effort. Representative diaphragm EMG traces (Figure 5) show diaphragm EMG activation profiles comparable to our previous report (Seven et al., 2018c). Rat diaphragm muscle utilizes approximately a fifth of its functional reserve during normal breathing. With hypoxia and hypercapnia, diaphragm EMG amplitude is augmented, although this activation is well below maximal activation. Near-maximal diaphragm effort is observed during airway protective reflexes, such as augmented breaths, sneezing and coughing.
Figure 5:
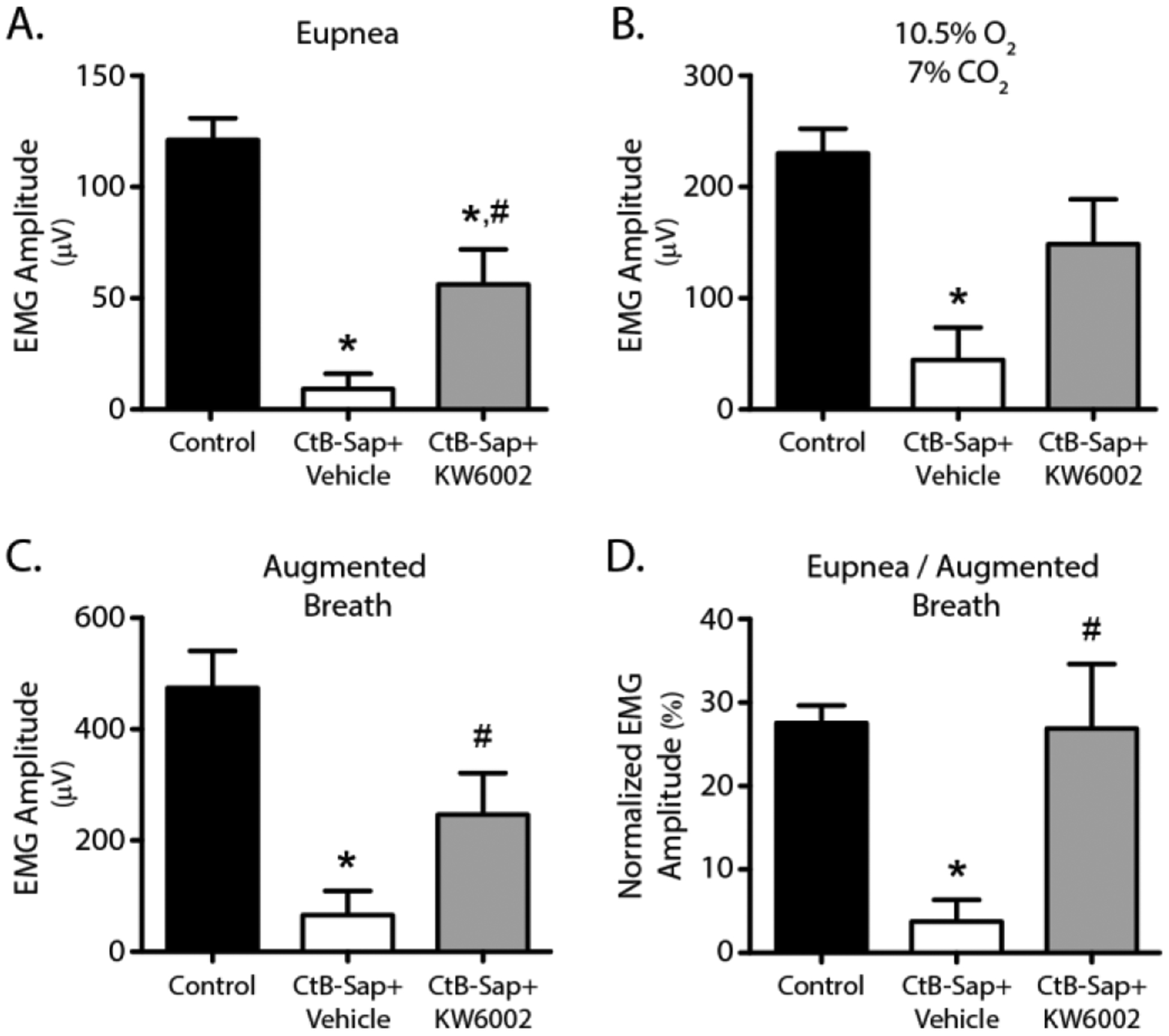
Diaphragm EMG activity 8 days after intrapleural cholera toxin B-Saporin (CtB-Saporin) injections (50 μg). Diaphragm EMG amplitude is estimated via root-mean-squared (RMS) calculation over a 50 -ms window. Diaphragm EMG was nearly abolished following CtB-Saporin injections during (A) eupnea, (B) hypoxia (10.5%)-hypercapnia (7%), and (C) augmented breaths (p<0.01). Adenosine 2A (A2a) receptor antagonist (KW6002) following intrapleural CtB-Saporin injections increases preserved diaphragm EMG activity compared to vehicle treatment (p<0.05 during eupnea and augmented breath). (D) EMG activity was normalized to augmented breaths since this normalization reveals the functional reserve of the diaphragm muscle. Normalized EMG value was assumed to be zero in the rats with no EMG activity across all conditions tested, since there was no functional reserve in these rats. Diaphragmatic functional reserve was obliterated following intrapleural CtB-Saporin injections (p<0.001). A2a antagonist treatment restored normalized EMG value (p=0.82 compared to control, p<0.05 compared to CtB-Saporin + Vehicle), suggesting restored functional reserve. Mean ± SEM.
Consistent with the motor neuron numbers, diaphragm EMG activity was abolished at all levels of effort in most CtB-Saporin injected rats, and was blunted in the rest. A2a receptor antagonist partially rescued diaphragm EMG activity across all effort levels (p<0.01). During normoxic breathing, diaphragm EMG activity was ~7% of control following CtB-Saporin (p<0.001). However, ~46% of diaphragm activity during normoxic breathing was observed in rats that had received the A2a antagonist (p<0.01 vs. CtB-Saporin + Vehicle). Similar effects were observed across all effort levels. During maximum chemoreceptor stimulation, diaphragm EMG activity was ~19% vs. ~65% of control in CtB-Saporin + vehicle (p<0.001) and CtB-Saporin + A2a antagonist treated rats, respectively (p<0.01). Augmented breaths during maximum chemoreceptor stimulation generated diaphragm EMG activity ~14% vs. ~51% of control in rats that had received CtB-Saporin + vehicle (p<0.001) and CtB-Saporin + A2a antagonist, respectively (p<0.01). Lastly, normoxic EMG activity was normalized to augmented breath amplitude since this normalization reveals the diaphragm functional reserve. In this analysis, normalized EMG values were assumed to be zero in rats with no detectable EMG activity across all conditions tested (i.e. there was no functional reserve in these rats). Normalized eupneic amplitude dropped from 27% (Control) to 4% following CtB-Saporin injections (p<0.001), suggesting the diaphragmatic functional reserve was eliminated. CtB-Saporin + A2a antagonist treatment restored the normalized EMG value (p=0.82 vs. control, p<0.05 vs. CtB-Saporin + Vehicle), demonstrating improved functional reserve.
A2a Receptor Inhibition Reduces CtB-Saporin-Induced p38 MAPK Phosphorylation
Levels of phospho-p38 MAPK were significantly higher in phrenic motor neurons 5 days post-CtB-Saporin injections versus control (p<0.05). With A2a receptor antagonism, phospho-p38 MAPK levels were reduced versus CtB-Saporin+Vehicle treated rats (p<0.05), although it was still higher than in controls (p<0.05).
Phosphorylated extracellular signal-regulated kinase (phospho-ERK) is observed as boutons surrounding phrenic motor neurons (Dale et al., 2012). Following CtB-Saporin injections, phospho-ERK immunoreactivity is significantly increased (p<0.05). A2a receptor antagonism did not change phospho-ERK immunoreactivity (p>0.05).
Phosphorylated c-Jun N-terminal kinase (phospho-JNK) was not observed in phrenic motor neurons until terminal stages of cell death (i.e. phagocytic clearance).
Discussion
Given the vital role of phrenic motor neurons in maintaining proper breathing, understanding mechanisms of phrenic motor neuron death with injury, disease or toxic insults is of considerable importance. Our central hypothesis is that, with disease or injury, increased A2a receptor expression and signaling accelerates phrenic motor neuron death, diminishing diaphragm function. Thus, following a neurotoxic insult that normally causes phrenic motor neuron death, A2a receptor inhibition would preserve both phrenic motor neuron survival and diaphragm function. Consistent with our hypothesis, following intrapleural CtB-Saporin injections, we observed: 1) A2a receptors are significantly upregulated prior to phrenic motor neuron cell death; 2) A2a receptor signaling hastens CtB-Saporin-induced phrenic motor neuron death; and 3) A2a receptor antagonism improves phrenic motor neuron survival and diaphragm function. These findings indicate that A2a receptors play a key role in early processes initiating phrenic motor neuron dysfunction and death.
Adenosine is present in both intracellular and extracellular CNS compartments at low concentrations under physiological conditions (Zetterström et al., 1982; Dale and Frenguelli, 2009). With challenges such as increased activity, hypoxia, injury and inflammation, adenosine rapidly accumulates in the extracellular space, maintaining homeostasis by modulating synaptic plasticity, regulating energy balance, reducing oxygen demand, inducing vasodilation, orchestrating inflammatory responses and other functions mostly due to its actions on adenosine receptors (Winn et al., 1980; Fredholm et al., 1999; Fredholm and Lindström, 1999; Berman et al., 2000; McAdoo et al., 2000; Dale and Frenguelli, 2009). However, when the challenge becomes too severe to be contained, adenosine can switch from protective to detrimental actions (Gomes et al., 2011), likely depending on receptor expression levels, the specific cell-type expressing adenosine receptors, and the severity and type of insult.
A2a receptors are implicated in phrenic motor neuron plasticity, increasing phrenic activity at a constant level of chemoreflex activation (Golder et al., 2008; Seven et al., 2018b) or undermining other forms (e.g. serotonin-dependent) of phrenic motor plasticity (Hoffman et al., 2010). Thus, adenosine receptors play a key role in regulating important forms of respiratory motor plasticity that are presumably beneficial in mounting a response to compensate for injury or disease.
Conversely, A2a receptors are suspected to play key roles in excitotoxic toxicity (e.g. glutamate and kainate), leading to or accelerating neuronal death (Mojsilovic-Petrovic et al., 2006; Gomes et al., 2011). Thus, A2a receptor activation can trigger neurodegeneration when extracellular glutamate levels are high (Mojsilovic-Petrovic et al., 2006). This is particularly important since increased excitatory glutamatergic drive to phrenic motor neurons is a compensatory strategy of the respiratory control system in protecting against respiratory pathology (Johnson and Mitchell, 2013; Seven and Mitchell, 2019). Thus, A2a receptors are integral regulators of both compensatory and pathogenic responses when neurons are challenged by injury and/or disease.
Earlier work concerning A2a receptor involvement in neuronal death/survival utilized neuronal cultures to study the relevant signaling mechanisms and in vivo rodent studies to test efficacies of potential therapeutic approaches. Unfortunately, there is little information concerning susceptibilities of discrete motor neuron pools to toxic or other challenges. Differences are often reported in motor neuron death in rodent models of ALS, such as SOD1G93A over-expressing rodents. Using this same ALS rat model, we reported differential susceptibility of respiratory motor neurons with phrenic > intercostal >hypoglossal motor neurons (Nichols et al., 2013; Seven et al., 2018c). Furthermore, there are differences in response to A2a receptor antagonism in the ability to elicit respiratory motor plasticity by exposing normal rats to modest intermittent hypoxia (Navarette-Opazo et al., 2014). Here, we demonstrate that A2a receptors have a neuroprotective effect on phrenic motor neurons under toxic insult from CtB-Saporin. Our results parallel previous work in primary motor neuron cultures showing A2a receptors accelerate motor neuron death from excitotoxic insult (Mojsilovic-Petrovic et al., 2006). The similarity in experimental outcomes using two different toxic insults suggests a common mechanism whereby A2a receptors promote motor neuron loss under stress from, for example, toxins.
Two relevant factors in the process whereby A2a receptors potentially accelerate phrenic motor neuron death are increased extracellular adenosine concentration and increased A2a receptor expression by phrenic motor neurons. Here, we show that A2a receptor expression is upregulated by CtB-Saporin in phrenic motor neurons per se. A2a receptor expression is regulated by different mechanisms. For example, A2a receptors are selectively upregulated at high extracellular adenosine concentrations due to direct A2a receptor activation in mast cells (Sereda et al., 2011). Thus, increased extracellular adenosine levels may activate A2a receptors, thereby enhancing their expression. Greater understanding of factors regulating A2a receptor activation and expression in phrenic motor neurons is needed to understand their precise roles in neuroprotection and neuroplasticity.
After neurotoxic insults, A2a receptors can activate several intracellular cascades, including downstream signaling via p38, ERK and JNK MAP kinases. Activation of these kinases contributes to neuronal death with a variety of lethal triggers. For example, p38 plays a key role in neuronal cell death from excessive reactive oxygen species formation (Kralova et al., 2008). Increased phospho-p38 immunoreactivity with CtB- Saporin points to potential involvement of the MAP kinase in toxic death, paralleling earlier observations in rodent ALS models (Bhinge et al., 2017). The observed increase in phospho-ERK is similar to observations in a rat model of ALS (Satriotomo et al., 2012; Satriotomo et al., 2016), although it could either contribute to accelerated cell death or compensatory responses that promote neuroprotection. The lack of observed changes in phospho-JNK provides little evidence for any role in CtB-Saporin/A2a receptor enhanced phrenic motor neuron death. Although the roles of p38 and ERK MAP kinases require additional investigation, our finding that CtB-Saporin increases phosphorylation (and activation) of p38 MAP kinases prior to phrenic motor neuron death, and that A2a receptor antagonism reverses this effect with associated neuroprotection leads us to suspect the p38 MAP kinase pathway is critical in the mechanism of accelerated cell death. Further investigation are needed to confirm the causality of p38 MAP kinase activation-phrenic motor neuron cell death relationship.
Figure 3:
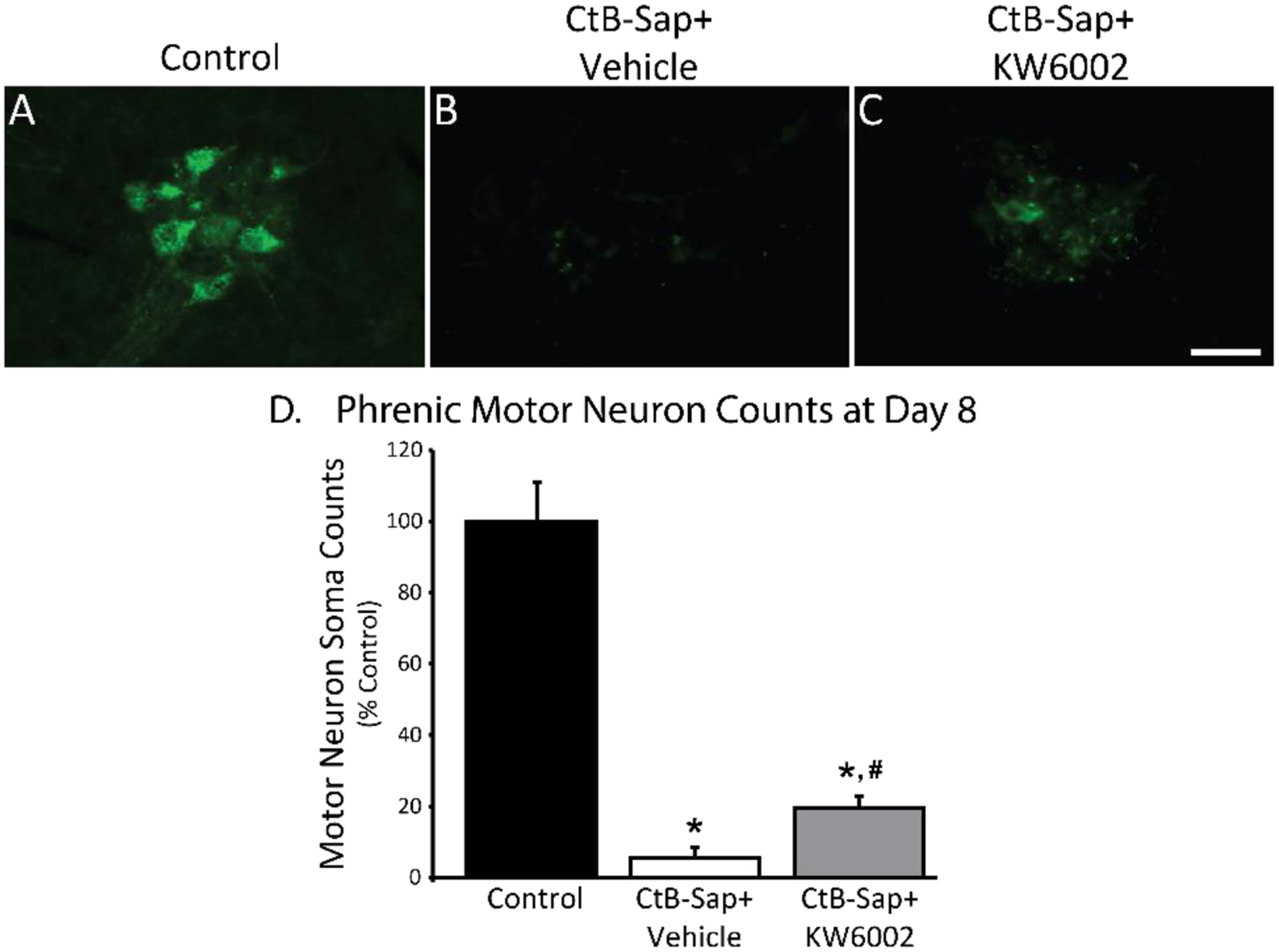
Phrenic motor neuron counts at Day 8 post-intrapleural cholera toxin (CtB-Saporin) injections (50 μg/rat total). A-C: Representative images (20x) for CtB-labelled phrenic motor neurons. D: CtB-Saporin causes almost complete loss of phrenic motor neurons (p<0.01 vs control; asterisk). Adenosine 2A (A2a) receptor antagonist (Istradefylline; KW6002) administration following intrapleural CtB-Saporin injections improves phrenic motor neuron survival versus CtB-Saporin injected + vehicle treated rats (p<0.05; #). Mean ± 1 SEM. Scale bar = 40 μm.
Figure 4:
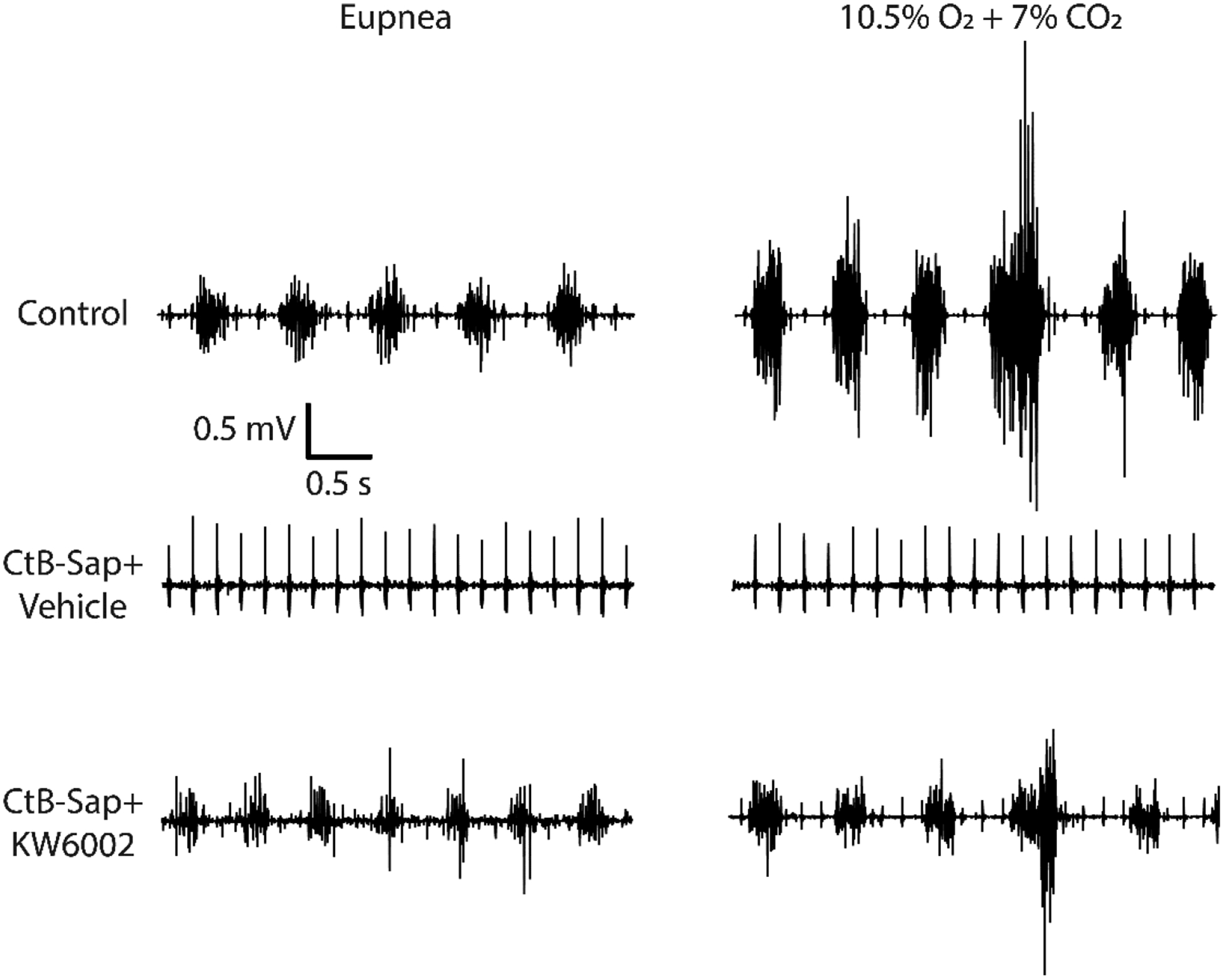
Representative diaphragm EMG recordings during eupnea, maximum chemoreceptor stimulation (10.5% O2 + 7% CO2), and spontaneous deep breaths (sighs) 7 days after CtB (control) and CtB-Saporin injections. Intrapleural CtB-Saporin injections abolished diaphragm EMG activity across all behaviors in 4 of 7 rats. Twice daily treatment of A2a receptor antagonist partially preserved the diaphragm EMG activity.
Figure 6:
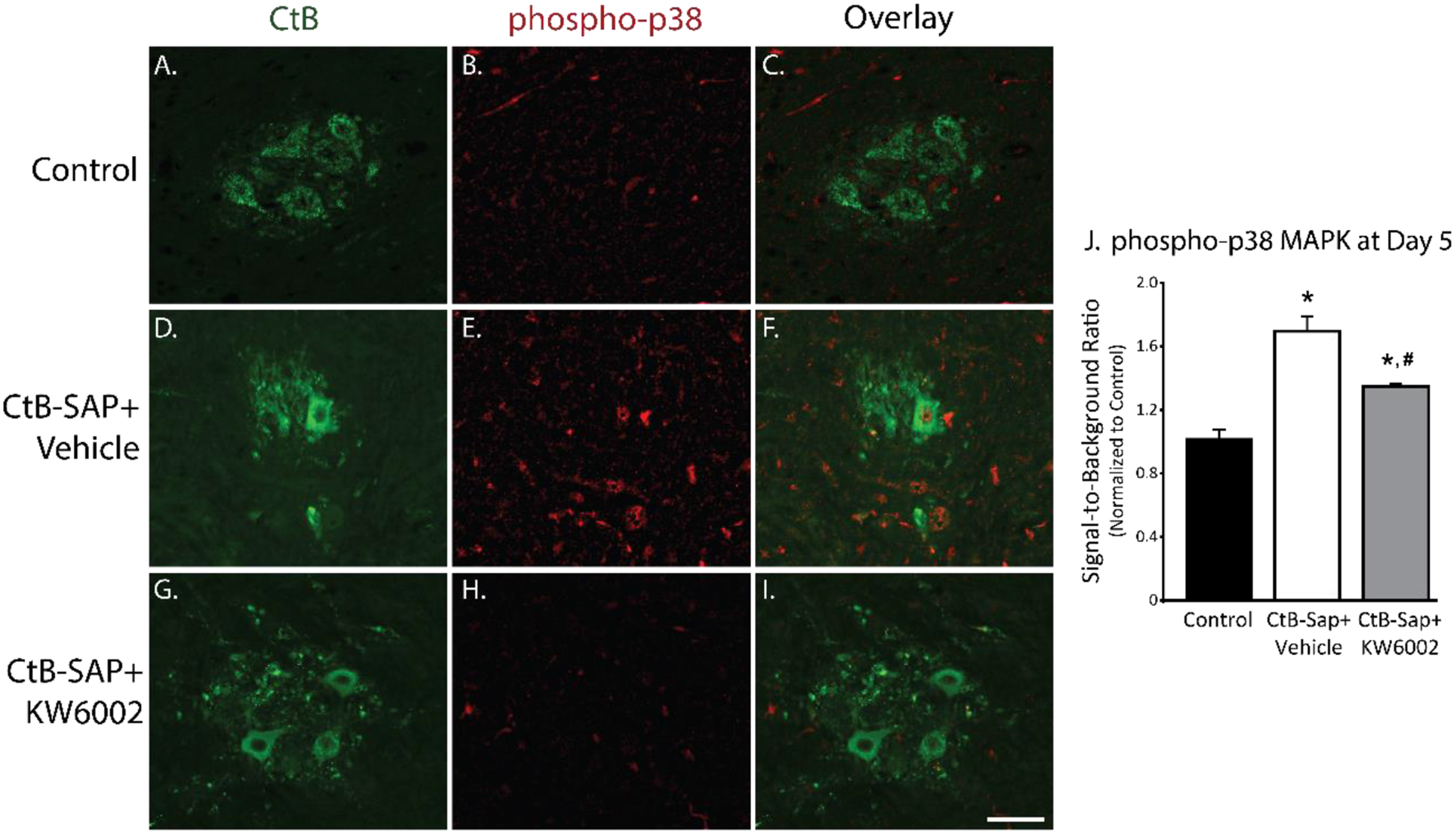
Phosphorylation of p38 MAPK within the phrenic motor neurons 5 days after intrapleural cholera toxin B-Saporin (CtB-Saporin) injections. A-I. Representative images (20x) for CtB (green) and phospho-p38 MAPK (red) at Day 5. J. Signal-to-background ratio of phospho-p38 fluorescence intensity is increased following intrapleural CtB-Saporin injections compared to control (p<0.001). A2a antagonist treatment reduced phosphor-p38 MAPK levels compared to CtB-Saporin+Vehicle group (p<0.05); however, was still higher than control group (p<0.05). Mean ± SEM. SEM in the control group represents the variability in the raw values normalized to averaged mean value. Scale bar = 40 μm.
Highlights.
Toxic phrenic motor neuron death can be caused by selective protein synthesis inhibition induced by intrapleural cholera-toxin beta subunit conjugated saporin (CtB-Sap) administration.
Intrapleural CtB-Sap causes upregulation of neuronal A2a receptors prior to phrenic motor neuron death.
A2a receptor inhibition protects phrenic motor neurons from death and preserves diaphragm activity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berman RF, Fredholm BB, Aden U, O’Connor WT (2000) Evidence for increased dorsal hippocampal adenosine release and metabolism during pharmacologically induced seizures in rats. Brain Res 872:44–53. [DOI] [PubMed] [Google Scholar]
- Bhinge A, Namboori SC, Zhang X, VanDongen AMJ, Stanton LW (2017) Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Reports 8:856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale EA, Satriotomo I, Mitchell GS (2012) Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and Akt signaling. J Neurosci 32:5973–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Frenguelli BG (2009) Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol 7:160–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Lindström K (1999) Autoradiographic comparison of the potency of several structurally unrelated adenosine receptor antagonists at adenosine A1 and A(2A) receptors. Eur J Pharmacol 380:197–202. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51:83–133. [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, Mitchell GS (2008) Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation. J Neurosci 28:2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA (2011) Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta 1808:1380–1399. [DOI] [PubMed] [Google Scholar]
- Hoffman MS, Golder FJ, Mahamed S, Mitchell GS (2010) Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J Physiol 588:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, Garabedian MJ, Chao MV (2008) Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A 105:4862–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Mitchell GS (2013) Common mechanisms of compensatory respiratory plasticity in spinal neurological disorders. Respir Physiol Neurobiol 189:419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralova J, Dvorak M, Koc M, Kral V (2008) p38 MAPK plays an essential role in apoptosis induced by photoactivation of a novel ethylene glycol porphyrin derivative. Oncogene 27:3010–3020. [DOI] [PubMed] [Google Scholar]
- Lladó J, Haenggeli C, Pardo A, Wong V, Benson L, Coccia C, Rothstein JD, Shefner JM, Maragakis NJ (2006) Degeneration of respiratory motor neurons in the SOD1 G93A transgenic rat model of ALS. Neurobiol Dis 21:110–118. [DOI] [PubMed] [Google Scholar]
- Machado CB, Kanning KC, Kreis P, Stevenson D, Crossley M, Nowak M, Iacovino M, Kyba M, Chambers D, Blanc E, Lieberam I (2014) Reconstruction of phrenic neuron identity in embryonic stem cell-derived motor neurons. Development 141:784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC (2009) Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods 182:244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdoo DJ, Robak G, Xu GY, Hughes MG (2000) Adenosine release upon spinal cord injury. Brain Res 854:152–157. [DOI] [PubMed] [Google Scholar]
- Melani A, Gianfriddo M, Vannucchi MG, Cipriani S, Baraldi PG, Giovannini MG, Pedata F (2006) The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res 1073–1074:470–480. [DOI] [PubMed] [Google Scholar]
- Mojsilovic-Petrovic J, Jeong GB, Crocker A, Arneja A, David S, Russell DS, Russell D, Kalb RG (2006) Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci 26:9250–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Opazo A, Mitchell GS (2014) Recruitment and plasticity in diaphragm, intercostal, and abdominal muscles in unanesthetized rats. J Appl Physiol (1985) 117:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Opazo AA, Vinit S, Mitchell GS (2014) Adenosine 2A receptor inhibition enhances intermittent hypoxia-induced diaphragm but not intercostal long-term facilitation. J Neurotrauma 31:1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Vinit S, Bauernschmidt L, Mitchell GS (2015) Respiratory function after selective respiratory motor neuron death from intrapleural CTB-saporin injections. Exp Neurol 267:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS (2013) Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med 187:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogués MA, Benarroch E (2008) Abnormalities of respiratory control and the respiratory motor unit. Neurologist 14:273–288. [DOI] [PubMed] [Google Scholar]
- Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, Kim DH, Knox A, Saito T, Saido TC, Simms J, Toddes C, Wang X, Yu GQ, Mucke L (2018) Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis 110:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I, Melani A, Cipriani S, Corti F, Mello T, Mazzon E, Esposito E, Bramanti P, Cuzzocrea S, Pedata F (2011) Selective adenosine A2A receptor agonists and antagonists protect against spinal cord injury through peripheral and central effects. J Neuroinflammation 8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedata F, Gianfriddo M, Turchi D, Melani A (2005) The protective effect of adenosine A2A receptor antagonism in cerebral ischemia. Neurol Res 27:169–174. [DOI] [PubMed] [Google Scholar]
- Satriotomo I, Dale EA, Dahlberg JM, Mitchell GS (2012) Repetitive acute intermittent hypoxia increases expression of proteins associated with plasticity in the phrenic motor nucleus. Exp Neurol 237:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satriotomo I, Nichols NL, Dale EA, Emery AT, Dahlberg JM, Mitchell GS (2016) Repetitive acute intermittent hypoxia increases growth/neurotrophic factor expression in non-respiratory motor neurons. Neuroscience 322:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sereda MJ, Bradding P, Vial C (2011) Adenosine potentiates human lung mast cell tissue plasminogen activator activity. J Immunol 186:1209–1217. [DOI] [PubMed] [Google Scholar]
- Seven YB, Mitchell GS (2019) Mechanisms of compensatory plasticity for respiratory motor neuron death. Respir Physiol Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Mantilla CB, Sieck GC (2014) Recruitment of rat diaphragm motor units across motor behaviors with different levels of diaphragm activation. J Appl Physiol (1985) 117:1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Mantilla CB, Zhan WZ, Sieck GC (2013) Non-stationarity and power spectral shifts in EMG activity reflect motor unit recruitment in rat diaphragm muscle. Respir Physiol Neurobiol 185:400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Perim RR, Hobson OR, Simon AK, Tadjalli A, Mitchell GS (2018a) Phrenic motor neuron adenosine 2A receptors elicit phrenic motor facilitation. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Perim RR, Hobson OR, Simon AK, Tadjalli A, Mitchell GS (2018b) Phrenic motor neuron adenosine 2A receptors elicit phrenic motor facilitation. J Physiol 596:1501–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seven YB, Nichols NL, Kelly MN, Hobson OR, Satriotomo I, Mitchell GS (2018c) Compensatory plasticity in diaphragm and intercostal muscle utilization in a rat model of ALS. Exp Neurol 299:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn HR, Welsh JE, Rubio R, Berne RM (1980) Brain adenosine production in rat during sustained alteration in systemic blood pressure. Am J Physiol 239:H636–641. [DOI] [PubMed] [Google Scholar]
- Yang M, Soohoo D, Soelaiman S, Kalla R, Zablocki J, Chu N, Leung K, Yao L, Diamond I, Belardinelli L, Shryock JC (2007) Characterization of the potency, selectivity, and pharmacokinetic profile for six adenosine A2A receptor antagonists. Naunyn Schmiedebergs Arch Pharmacol 375:133–144. [DOI] [PubMed] [Google Scholar]
- Yu L, Shen HY, Coelho JE, Araújo IM, Huang QY, Day YJ, Rebola N, Canas PM, Rapp EK, Ferrara J, Taylor D, Müller CE, Linden J, Cunha RA, Chen JF (2008) Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol 63:338–346. [DOI] [PubMed] [Google Scholar]
- Zetterström T, Vernet L, Ungerstedt U, Tossman U, Jonzon B, Fredholm BB (1982) Purine levels in the intact rat brain. Studies with an implanted perfused hollow fibre. Neurosci Lett 29:111–115. [DOI] [PubMed] [Google Scholar]