

Abstract
Uric acid is the end product of purine metabolism in humans, and its excessive accumulation leads to hyperuricemia and urate crystal deposition in tissues including joints and kidneys. Hyperuricemia is considered an independent risk factor for cardiovascular and renal diseases. Although the symptoms of hyperuricemia-induced renal injury have long been known, the pathophysiological molecular mechanisms are not completely understood. In this review, we focus on the research advances in the mechanisms of hyperuricemia-caused renal injury, primarily on oxidative stress, endothelial dysfunction, renal fibrosis, and inflammation. Furthermore, we discuss the progress in hyperuricemia management.
1. Introduction
Uric acid (2,6,8-trioxypurine, molecular formula C5H4N4O3; UA), the final metabolite of endogenous and exogenous purine, is generated in the liver [1]. Owing to the evolutionary loss of urate oxidase, UA cannot be catabolized to allantoin in humans and primates, while most of other animals can catabolize UA [1]. As a major antioxidant [2], UA is beneficial to remove superoxide and oxygen free radicals in primates, which is unable to self-generate vitamin C [3, 4]. Hyperuricemia is clinically defined as the serum UA level ≥ 7 mg/dL in men and postmenopausal women and ≥6 mg/dL in premenopausal women, causing various diseases, such as gout and urinary stones [3–5]. An elevated UA level is also tightly associated with diabetes and cardiovascular and kidney diseases [6]. Recently, because of the changes in lifestyle and the increasing population of older people, the incidence of hyperuricemia in China has risen from merely 1.4% in the early 1980s to 10% in the early 21st century [7]. In fact, the morbidity from hyperuricemia has risen up to 20% in some coastal areas and developed cities in China, nearly reaching the level of developed countries [8, 9].
Numerous studies have shown that hyperuricemia is closely associated with kidney diseases [2, 10–13]. In a 6-year cohort study of 10,677 Chinese individuals with a normal estimated glomerular filtration rate (eGFR) and without proteinuria, a higher UA level was found to contribute to the onset of kidney disease and a rapid decline of eGFR [14]. A long-term follow-up cohort study of 13,338 volunteers with normal kidney function in two communities showed a significant relationship between the baseline UA level and the risk of kidney disease, with the risk of developing kidney disease rising by 7% to 11% per 1 mg/dL serum UA [15]. Another cohort study, which lasted for more than 25 years and enrolled 177,570 patients, showed an independent association between high UA levels and end-stage renal diseases (ESRDs) [16]. Hyperuricemia is now considered an independent risk factor for the occurrence and development of diabetic nephropathy (DN) [17, 18], acute kidney injury (AKI) [2, 12, 13], chronic kidney disease (CKD) [10, 11], and ESRD [16]. However, inconsistent results had been reported regarding the role of UA in the progression of CKD and there were insufficient evidences to suggest lowering UA therapy to prevent the progression of CKD [19–23]. Thus, the causal relationship between hyperuricemia and CKD remains controversial, and the pathophysiological mechanisms of hyperuricemia-induced renal injury are not entirely clear. In this review, we attempt to elucidate the recent advances in the mechanisms of hyperuricemia-induced renal injury.
2. Pathogenesis of Hyperuricemia
The cause of hyperuricemia is the imbalance between UA production and excretion. Purines are mainly degraded by xanthine oxidase (XO) in the liver, and targeting this enzyme, e.g., with allopurinol or febuxostat, is an effective therapeutic method for lowering serum UA levels [24]. A high-purine diet, increased purine metabolism, and excessive alcohol consumption contribute to the increased production of UA. Tumor lysis syndrome, in which a large number of cells are damaged, and the metabolism of nucleic acids is promoted, leads to an increase in UA production [25]. Other rare causes inducing acute hyperuricemia include seizures, rhabdomyolysis, and excessive exercise.
Most of UA is excreted by the kidneys (65–75%) and intestines (25–35%). The renal handling of UA consists of glomerular filtration, tubular reabsorption, tubular secretion, and reabsorption after secretion. A decrease in UA excretion and an increase in UA reabsorption cause hyperuricemia. UA transporters are required for the renal handling of UA and can be roughly divided into reabsorption-related and secretion-related proteins. Reabsorption-related proteins mainly include urate anion transporter 1 (URAT1), organic anion transporter 4 (OAT4), and glucose transporter 9 (GLUT9), while secretion-related transporters mainly consist of OAT1, OAT3, multidrug resistance protein 4 (MRP4/ABCC4), and breast cancer resistance protein (BCRP/ABCG2) [26]. For example, the function of URAT1 is to reabsorb UA at the apical membrane of proximal tubule epithelial cells (TECs) [27, 28]. GLUT9 acts as a transporter that reabsorbs both UA and glucose into tubular cells [26]. ABCG2, which was first found to be involved in the development of multidrug resistance in cancer cells, also takes part in the secretion of UA from proximal TECs through an ion pump [29]. Genetic defects or mutations in secretion-related transporters also contribute to hyperuricemia. In addition, some drugs, such as cyclosporine and diuretics, cause hyperuricemia by decreasing renal urate clearance [30].
3. Mechanisms of Hyperuricemia-Induced Renal Injury
3.1. Monosodium Urate (MSU) Crystal Deposition-Induced Renal Damage
UA has the characteristic of a weak organic acid, and most of it is ionized to MSU crystal at pH 7.4 and a temperature of 37°C [31, 32]. A solubility study showed that serum was supersaturated for MSU crystal when the concentration of UA exceeded 6.5 mg/dL [31]. As a consequence, UA and urate crystals may deposit in the joints, kidneys, and other tissues, inducing tissue damage. A reduced expression of BCRP/ABCG2 in TECs induced by hyperuricemia may promote MSU crystal deposition in TECs and the renal interstitium, resulting in substantial renal damage [33, 34]. Under long-term low pH and high UA concentration conditions in crude urine, urate crystal deposition in the renal tubular lumen and ureters contributes to cast formation and obstructive nephropathy [35–37]. Upon obstruction, a series of complications occur, such as local damage, infection, bleeding, and hydronephrosis [38]. In particular, insulin resistance leads to impaired UA excretion at a low urinary pH, contributing to the formation of urate stones [39].
3.2. Hyperuricemia-Induced Oxidative Stress
Once transported into the cell, UA becomes a prooxidant, which increases the production of reactive oxygen species (ROS), including the superoxide anion (O2−), H2O2, and 8-isoprostane [40, 41]. Many studies have shown that hyperuricemia-induced oxidative stress affects multiple organs and systems, including the kidneys [42–44]. Pathologically, hyperuricemia-associated oxidative stress gives rise to DNA damage, oxidation and inactivation of enzymes, inflammatory cytokine production, and cell apoptosis [45].
Mitochondria are the center of intracellular energy metabolism and the main site of oxidative phosphorylation, in which ROS are produced by the transfer of electrons from electron transport chain complexes to O2. It has been reported that long-term hyperuricemia could induce renal mitochondria dysfunction associated with renal cortex oxidative stress and tubular damage in rats [46]. Hyperuricemia was shown to mediate mitochondrial calcium overload and eventually cause endothelial dysfunction through mitochondrial Na+/Ca2+ exchange, which increases the production of ROS [47]. The mechanism of UA-induced endothelial dysfunction is closely associated with reduced mitochondria mass and ATP production [48]. Although mitochondria in TECs may undergo substantial damage under oxidative stress, glutathione (GSH) treatment shows an effective resistance as an antioxidant [46]. Another important source of ROS is NADPH oxidase. UA has been found to stimulate the synthesis of ROS by NADPH oxidase in various cells, such as adipocytes, vascular smooth muscle cells, and vascular endothelial cells [49]. Using stable isotope labeling with amino acids in cell culture and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to analyze differentially expressed proteins and the functional status of UA-stimulated human umbilical vein endothelial cells (HUVECs), Zhang et al. [50] found a significant relationship between hyperuricemia-induced endothelial dysfunction and aldose reductase- (AR-) mediated oxidative stress. Meanwhile, it has been reported that hyperuricemia induced endothelial dysfunction via regulation of AR, while inhibition of AR or degradation of ROS could restore endothelial function [51]. Similarly, antioxidant therapies, such as tempol and reduced GSH, may be beneficial for the recovery of endothelial function [45, 46]. Taken together, hyperuricemia-mediated oxidative stress directly damages the kidney, thus being a biotherapeutic target for UA-induced renal damage.
3.3. Hyperuricemia-Induced Endothelial Dysfunction
The renin-angiotensin system (RAS) mainly regulates the cardiovascular function and maintains the body fluid balance in cooperation with other compensatory mechanisms. Evidence from animal and patient studies showed that UA-mediated RAS activation is closely related to diabetic complications, such as cardiovascular and kidney diseases [52]. Upon UA stimulation, the expression of angiotensinogen, angiotensin-converting enzyme (ACE), and angiotensin II receptors was notably upregulated in vitro, resulting in the inhibition of proliferation and promotion of senescence, inflammation, and apoptosis of endothelial cells [53]. Moreover, UA-induced senescence and apoptosis of HUVECs were blocked by enalaprilat (an ACE inhibitor) or telmisartan (an angiotensin II receptor antagonist). These findings indicated that RAS activation was a novel mechanism of UA-induced endothelial dysfunction [40].
Endothelial cells secrete various vasoactive substances to regulate the relaxation and contraction of blood vessels, including the potent vasoconstrictor endothelin 1 (ET-1) and the effective vasodilator nitric oxide (NO) [54]. An imbalance between ET-1 and NO drives endothelial dysfunction, which plays an essential role in the pathophysiology of cardiovascular and renal diseases. Under physiological conditions, ET-1 stimulates the synthesis of NO by endothelial cells, while NO exerts a negative feedback effect on ET-1 [55]. Overexpression of ET-1 may elevate blood pressure and cause vascular and kidney injuries, such as reduced renal artery flow and small artery stiffening [56]. Accumulating evidence indicates that UA impacts endothelial function through downregulation of NO production and endothelial nitric oxide synthase (eNOS) activity, which subsequently decreases NO bioavailability. UA was shown to affect the activity of eNOS and production of NO in a dose- and time-dependent manner [57]. L-arginine is the substrate of eNOS and is converted to NO in mammalian endothelial cells [58]. However, it was reported that UA could not only stimulate arginase, an enzyme degrading L-arginine, but also enhance the affinity of L-arginine to arginase, which reduced the availability of the substrate for NO synthesis [58]. In addition, recent studies have demonstrated that UA markedly reduced the binding between eNOS and calmodulin (CaM), an eNOS activator, in both HUVECs and bovine aortic endothelial cells [57, 59]. Zhang et al. [60] suggested that high-level UA can induce endothelial dysfunction through miR-155-mediated eNOS suppression. In addition, UA regulated the PKC/eNOS pathway and endoplasmic reticulum (ER) stress, leading to endothelial dysfunction and apoptosis in HUVECs [57]. Although it was clearly demonstrated that UA inhibited eNOS activity and the interaction between eNOS and CaM, it did not influence the expression of eNOS and the intracellular amount of CaM [59]. The reason for the latter is not fully understood and may be related to posttranslational modifications and the activation of eNOS.
UA induces endothelial dysfunction via various pathways, while targeted therapy may ameliorate the endothelial dysfunction and alleviate kidney damage. It has been reported that iptakalim, an ATP-sensitive potassium channel opener, could improve endothelial dysfunction and defend against hypertension and hyperuricemia [60]. Additionally, XO inhibitors, such as allopurinol and febuxostat, exhibited protective effects on endothelial dysfunction in clinical therapy and animal models [24, 61, 62].
3.4. Hyperuricemia-Induced Renal Fibrosis
Renal fibrosis, characterized by glomerulosclerosis and tubulointerstitial fibrosis, is a common pathological process in all patients with CKD, leading to the loss of effective nephrons and a progressive decline in renal function, resulting in ESRD [63]. A clinical study of 1,700 biopsy-confirmed patients demonstrated that patients with high levels of plasma UA displayed not only more serious clinical renal dysfunction but also more severe renal pathology, particularly segmental glomerulosclerosis and tubular atrophy/interstitial fibrosis [64]. Recently, a line of evidence has indicated that hyperuricemia may directly cause glomerulosclerosis and tubulointerstitial fibrosis.
3.4.1. Hyperuricemia-Induced Glomerulosclerosis
Mild hyperuricemia causes renal arteriolosclerosis and glomerular hypertension and disrupts renal autoregulation, ultimately resulting in glomerulosclerosis [65]. Recently, the deleterious effects of hyperuricemia on glomerular intrinsic cells were explored, which may uncover the potential mechanisms of hyperuricemia-induced glomerulosclerosis.
As mentioned in the previous section, multiple pathways are involved in hyperuricemia-induced endothelial cell injury; thus, mild hyperuricemia may impair glomerular endothelial cells via similar mechanisms. In addition, similar to epithelial-to-mesenchymal transition (EMT), endothelial-to-mesenchymal transition (EndoMT) greatly contributes to the activation of fibroblasts and myofibroblasts and subsequent renal fibrosis [66]. EndoMT contributed to renal fibrosis in three mouse models of CKD, including unilateral ureteral obstructive nephropathy, streptozotocin-induced DN, and Alport kidney disease models [67–69]. These studies suggested that endothelial-origin myofibroblasts possibly contribute to the progression of glomerulosclerosis [68, 70]. Recent research has demonstrated that UA induced the phenotype transition in HUVECs via induction of oxidative stress and glycocalyx shedding [71]. However, direct evidence is still lacking for the contribution of UA-induced EndoMT to glomerulosclerosis. Therefore, further studies are needed to identify the role of UA-induced EndoMT in glomerulosclerosis by utilizing a conditionally immortalized human glomerular cell line [72] in vitro and an endothelial lineage-traceable mouse line in vivo [68].
Abnormal proliferation of glomerular mesangial cells (MCs) and overproduction of extracellular matrix (ECM) contribute to glomerulosclerosis. UA-mediated activation of the COX-2/mPGES-1/PGE2 inflammatory cascade not only has a direct proinflammatory effect but also induced the proliferation of MCs [73, 74]. Moreover, UA time-dependently induced MC proliferation through the NADPH/ROS/extracellular signal-regulated kinase (ERK)1/2 signaling pathway [75]. Albertoni et al. [76] proved that soluble UA stimulated the proliferation and contraction of human MCs via an angiotensin II-dependent mechanism and the production of ET-1 in vivo, which might have a long-term effect on glomerular function. In addition, soluble UA induced ER stress by upregulating the expression of α-smooth muscle actin (α-SMA), fibronectin (FN), and transforming growth factor-β 1 (TGF-β1) in a time- and concentration-dependent manner, which resulted in a phenotypic change in rat glomerular MCs [77]. Thus, UA-induced proliferation of glomerular MCs and production of extracellular matrix may lead to glomerular hypertrophy and sclerosis. These novel mechanisms suggest some potential targets for the treatment of hyperuricemia-induced glomerulosclerosis.
Notably, hyperuricemia also influences the function of glomerular podocytes, a key player in maintaining the glomerular filtration barrier, leading to albuminuria. Electron microscopy of kidney biopsies from patients with gout revealed varying degrees of podocyte proliferation and damage [78]. Signs of significant albuminuria were found in hyperuricemic model rats, accompanied by upregulation of desmin, a podocyte injury marker, and downregulation of podocin, a key component of the podocyte slit diaphragm [79, 80].
3.4.2. Hyperuricemia-Induced Renal Interstitial Fibrosis
Myofibroblasts act as collagen-producing cells in various pathologies, including renal interstitial fibrosis. During renal interstitial fibrosis, half of the myofibroblasts are derived from renal resident fibroblasts [81]. Under the stimulation of cytokines and growth factors, fibroblasts undergo activation and proliferation, achieve myofibroblast phenotype, and synthesize ECMs, including structural scaffolds, fibronectin, and various types of collagens [82]. UA promotes the renal fibroblast–myofibroblast transition mainly through the activation of the TGF-β/Smad3, epidermal growth factor receptor (EGFR), and ERK1/2 pathways [83, 84]. However, after treatment with 3-deazaneplanocin A, a selective inhibitor of the enhancer of zeste homolog 2, the above-activated pathways were inhibited, and the proliferation of renal fibroblasts was suppressed, alleviating renal interstitial fibrosis [83].
EMT is a physiological or pathophysiological process, leading to the phenotype transformation of renal tubular cells, which lose their epithelial phenotype and acquire that of mesenchymal cells [63, 85, 86]. It has been reported that almost one-third of myofibroblasts originate from EMT rather than from preexisting local fibroblasts [87]. EMT plays a primary role in the accumulation of myofibroblasts and the resulting production of ECM, which are the key steps in the progression of renal interstitial fibrosis [86, 88]. EMT is often accompanied by a decreased expression of the epithelial cell marker E-cadherin via upregulation of Snail and Slug and an increased expression of mesenchymal cell markers, such as α-SMA, vimentin, fibronectin, and smooth muscle 22 (SM22) [86]. Substantial evidence indicates that the TGF-β/Smad3 pathway plays a dominant role in the progression of EMT [84, 89, 90]. It has also been shown that UA activates the TGF-β/Smad3 signaling pathway in type 2 DN, promoting EMT and profibrogenic progression [18]. Setyaningsih et al. [91] reported that hyperuricemia induced EMT and kidney tubular injury in mice via regulation of the Wnt5a/Ror2 signaling pathway. Zhou et al. [63] also found that UA caused EMT through the stimulation of the toll-like receptor 4 (TLR4)/nuclear factor-kappa B (NF-κB) pathway. Furthermore, hyperuricemia may induce EMT through the PI3K/Akt signaling pathway [92]. EMT has already been deemed a new therapeutic target due to its reversibility [91, 93, 94]. Studies have demonstrated that UA-induced EMT could be inhibited by probenecid, an organic anion transport inhibitor [66]. In addition, in a hyperuricemia nephropathy rat model, a traditional Chinese medicine decreased the UA level and relieved renal interstitial fibrosis via inhibition of the EMT process [95]. Tao et al. [96] demonstrated that UA-induced EMT was prevented after blocking ERK1/2 with the specific inhibitor U0126.
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases involved in the degradation of extracellular and basement membranes [97]. It was shown that MMPs could promote EMT and establish a profibrotic environment, which may contribute to renal interstitial fibrosis [98–100]. Reports have shown that MMP2 and MMP9 were significantly activated in renal tissue of hyperuricemic rats [96]. Moreover, inhibition of the NF-κB/MMP9 signaling pathway by chloride channel 5 (CIC-5) overexpression suppressed TGF-β1-induced EMT [101]. ER stress is a cellular physiological or pathological response to the accumulation of misfolded and mismatched proteins in ER [102]. ER stress is closely associated with renal fibrosis [102, 103]. Recently, He et al. [104] found that a marker of ER stress (RTN1A) was markedly upregulated in hyperuricemic nephropathy; however, febuxostat suppressed ER stress, thereby improving kidney injury and interstitial fibrosis [105]. Thus, MMPs and ER stress may be additional hallmarks and therapeutic targets for hyperuricemia-induced renal interstitial fibrosis.
3.5. Hyperuricemia-Induced Renal Inflammation
During necrosis, the dying cell releases amount of danger signals, such as ATP, high-mobility group box protein 1 (HMGB1), heat shock proteins, and UA, to activate immune response. UA may crystallize into MSU crystal in the extracellular fluid and can be recognized by pattern recognition receptors (e.g., TLRs) expressed on antigen-presenting cells (APCs, such as macrophages and TECs) as one of the danger-associated molecular patterns (DAMPs), which ultimately activates immune and inflammatory responses. Notably, hyperuricemia may induce renal inflammation via crystal-dependent and crystal-independent pathways [106].
3.5.1. MSU Crystal-Induced Renal Inflammation
It is generally accepted that hyperuricemia induces renal inflammation in a crystal-dependent manner. Macrophages are considered key mediators and have been studied most in MSU crystal-induced renal inflammation. MSU crystals deposited in the tubular lumen or interstitial space can be recognized and engulfed into renal resident or infiltrated macrophages [63, 107]. Upon stimulation with MSU crystal, the production of chemokines, such as CXCL-12, which induce directional chemotaxis in nearby inflammatory cells, was significantly enhanced in tubular cells possibly leading to the accelerated renal recruitment of macrophages [108]. Moreover, MSU crystals strongly activated human primary macrophages to secrete the lysosomal protease cathepsin, proinflammatory cytokines, such as interleukin- (IL-) 1β, IL-18, and interferon through the Src/Pyk2/PI3K signaling pathway [109]. Nod-like receptor pyrin domain-containing protein 3 (NLRP3), an important member of NLRs, senses danger signals, including pathogen-associated molecular patterns (PAMPs) and DAMPs, in the cytosol and activates sterile inflammation [108, 110]. NLRP3 then assembles the functional NLRP3 inflammasome, which subsequently leads to the transformation of immature pro-IL-1β and pro-IL-18 into mature, bioactive IL-1β and IL-18, respectively, ultimately activating the entire cascade and amplifying downstream inflammatory signals [111]. Through endocytosis into macrophages, lysosomes capture MSU crystal for degradation; however, MSU crystal cannot be degraded but instead ruptures the lysosomal membrane and releases lysosomal cathepsins into the cytoplasm, leading to the activation of the inflammasome [33]. Active caspase-1 may cleave gasdermin D (GSDMD) into GSDMD-N, triggering cell pyroptosis [112, 113]. Interestingly, MSU crystal-induced macrophages not only secrete proinflammatory cytokines at the inflammatory activation stage but also produce anti-inflammatory cytokines during the resolution phase of inflammation. In particular, it was reported that MSU crystals promoted macrophages to secrete TGF-β1 through mediation of the metastatic tumor antigen 1 (MTA1)/transglutaminase 2 (TG2) signaling pathway, which contributed to self-limitation of inflammation [114]. TGF-β1 acts as a strong profibrotic cytokine, and aberrant TGF-β1 derived from MSU crystal-induced macrophages may promote renal fibrosis. Besides macrophages, infiltrated T cells not only could phagocytize MSU crystals but also could be directly activated and stimulated to proliferate by MSU crystals in the absence of APC [115].
Injured TECs also produce a lot of cytokines and chemokines to promote renal inflammation. Urate crystals can adhere to renal TECs through hydrogen bonding and hydrophobic interactions to induce TEC injury [116]. In addition, damaged TECs rapidly secrete migration inhibitory factor (MIF), a mediator of delayed-type hypersensitivity, to recruit macrophages and other immune cells [117]. MSU crystal also induces NLRP3 inflammasome activation in TECs triggered by lysosomal rupture. Released lysosomal cathepsins can initiate RIP3/MLKL-dependent necroptosis, which was confirmed by a study that showed that RIP3 deficiency attenuated hyperuricemia-caused tubular injury and renal inflammation in mice [118]. Necroptosis of TECs leads to the release of danger signals, further promoting renal inflammatory response in a positive feedback loop.
3.5.2. Soluble UA-Induced Renal Inflammation
Recent studies suggest that soluble UA may also have proinflammatory effects, independent of crystal formation. Braga et al. [119] found that soluble UA could also stimulate the activation of the NLRP3 inflammasome and the synthesis of IL-1β in vivo and in vitro. Moreover, soluble UA activated NLRP3 inflammasome to secrete IL-1β in macrophages and stimulated the release of CXCL12 and HMGB1 in TECs, while interaction between macrophages and TECs promoting the progression of DN [108]. Soluble UA significantly enhanced NLRP3, tumor necrosis factor- (TNF-) α as well as IL-1β in TECs, while AMP-activated protein kinase (AMPK) exerted a protective effect on UA-induced inflammatory response [120]. In the absence of lysosomal ruptures, mitochondria-derived ROS may mediate soluble UA-activated NLRP3 inflammasome [119]. In a rat model, hyperuricemia induced renal inflammation and promoted the progression of renal disease via a monocyte chemoattractant protein-1- (MCP-1-) related mechanism [121]. In cultured TECs (NRK-52E) and a hyperuricemia mouse model, UA induced the infiltration of inflammatory cells (T cells and macrophages) in tubular interstitial spaces and upregulated the production of the inflammatory cytokine tumor necrosis factor-α (TNF-α) and MCP-1 and regulated upon activation normal T cell expressed and secreted factor (RANTES) expression via the NF-κB signaling pathway [63]. In another rat model, soluble UA stimulated the production of MCP-1 in vascular smooth muscle cells (VSMCs), subsequently promoting the infiltration of inflammatory cells in kidney, causing profound renal vasoconstriction and chronic renal injury [122–124]. Soluble UA was also shown to directly stimulate proinflammatory cytokine production in human peripheral blood mononuclear cells (PBMCs) through breaking the IL-1β/IL-1 receptor antagonist (IL-1Ra) balance [125]. Plasma UA was found to induce endothelial dysfunction and inflammation in renal allograft recipients, which might lead to chronic renal allograft damage [126]. Therefore, the reduction in UA levels may bring many benefits. For example, after urate-lowering therapy (ULT) with benzbromarone in healthy volunteers for two weeks, the serum inflammatory cytokine IL-18 was significantly decreased [127]. Moreover, lowering plasma UA levels markedly decreased renal damage, the expression of MCP-1, and the macrophage M1/M2 ratio in a hyperuricemic mouse model [128].
High levels of UA significantly upregulate the expression of HMGB1 through activation of TLR4 and the MEK/ERK pathway [129]. HMGB1 amplifies inflammatory responses via various pathways, including the promotion of mononuclear cells to secrete proinflammatory cytokines, such as IL-1β and TNF-α, the expression of adhesion molecules, and inflammatory cell infiltration [130]. In particular, HMGB1 promotes its own release from endothelial cells by a positive-feedback mechanism. After binding to the receptor of advanced glycation end products (RAGE), UA-induced HMGB1 activates the NF-κB signaling pathway and promotes the production of cytokines, such as TNF-α and IL-6, leading to oxidative stress and inflammatory responses [131].
Recent evidence has demonstrated another inflammatory mechanism of renal damage, which involves hyperuricemia-induced Na+/K+-ATPase (NKA) degradation in lysosomes, whereas AMPK was shown to alleviate NKA downstream inflammation and maintain renal function. AMPK activators, such as metformin and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), significantly relieved hyperuricemia-induced renal damage and NKA signaling impairment in a rat model, although these compounds did not lower the serum UA levels in rats [132]. Recent studies have confirmed that autophagy also plays a functional role in hyperuricemia-induced inflammation [33, 133, 134]. Activation of autophagy may limit inflammasome activity induced by hyperuricemia through targeting ubiquitinated inflammasomes for degradation [135] and decreasing the production of ROS [136] and downstream inflammatory responses [137]. However, Bao et al. [138] demonstrated that the inhibition of autophagy with 3-methyladenine (3-MA) not only delayed the progression of renal fibrosis but also suppressed the infiltration of immune cells and the secretion of various inflammatory cytokines. More evidence is needed to explore the protective or deleterious role of autophagy in hyperuricemia-induced renal inflammation.
4. Perspective and Conclusion
Currently, the standard treatment for patients with hyperuricemia is ULT, mainly including the XO inhibitors allopurinol and febuxostat, the UA reabsorption inhibitor benzbromarone and urate oxidase (rasburicase). In 2012, the American College of Rheumatology recommended using either allopurinol or febuxostat for first-line ULT [139]. In contrast to XO inhibitors, rasburicase lowers hyperuricemia quickly but does not induce the accumulation of xanthine, which is usually used in the prevention and treatment of tumor lysis symptoms. XO inhibitors may exert renoprotective effects beyond lowering UA. XO produces ROS, and inhibition of XO by allopurinol or febuxostat attenuates ROS-mediated kidney injury [140–142]. Some drugs targeting URAT1, such as SHR4640 and RDEA3170, are still under clinical trials, which may raise new hope for the treatment of hyperuricemia.
Apart from classical ULT drugs, losartan, as an angiotensin II receptor blocker, has been proven to reduce serum UA via inhibiting UA reabsorption mediated by URAT1 in TECs [143, 144]. Sodium-glucose cotransporter 2 inhibitors, which are approved antidiabetic drugs, promote UA excretion by suppressing UA reabsorption via commandeering UA transporter GLUT9 [145, 146].
Traditional medicines were also reported to reduce the level of serum UA and attenuate hyperuricemia-induced kidney injury. Extracts from Urtica hyperborea Jacq. Ex Wedd. significantly reduced the renal expression of URAT1 and increased that of OAT1, thereby lowering the serum UA level and improving renal injury [147]. Epigallocatechin gallate exerted hypouricemic effects by suppressing XO activity and GLUT9 expression and promoting OAT1 expression in vivo and in vitro [148]. Tu-Teng-Cao extract remarkably decreased the concentration of serum UA in potassium oxonate-induced hyperuricemia rats [149].
However, there is a long way to translate the identified novel mechanisms of hyperuricemia-induced renal injury based on experimental studies into clinical applications. Research methods used for hyperuricemia-induced renal injury have certain limitations. For instance, most of the in vivo and in vitro studies related to hyperuricemia-induced renal injury involve simple experimental models; however, clinical patients are more complex, and most of them either have a different underlying disease or various accompanying complications. Hence, complex models, such as models of CKD accompanied by hyperuricemia or hyperuricemia models with an AKI attack, should be considered because they might be more in line with the actual situation in clinical patients. Meanwhile, with the rapid development of modern computers and gene-related technologies, more emphasis should be placed on novel medical approaches, such as gene and metabolic pathway analyses, as well as on the combination of the modern information technology and clinical cases, which may become a new direction in the research of hyperuricemia-induced renal injury.
In conclusion, with a progressively higher incidence, hyperuricemia not only increases the risks but also affects the prognosis of renal diseases. The mechanisms of hyperuricemia-induced renal injury mainly include oxidative stress, endothelial dysfunction, renal fibrosis, and inflammation (Figure 1). However, the whole mechanisms of hyperuricemia-caused renal injury are complex and not fully understood, thus requiring further research. The novel underlying mechanisms may contribute to the development of clinical therapies with the potential to improve the treatment of hyperuricemia and hyperuricemia-caused renal injury.
Figure 1.
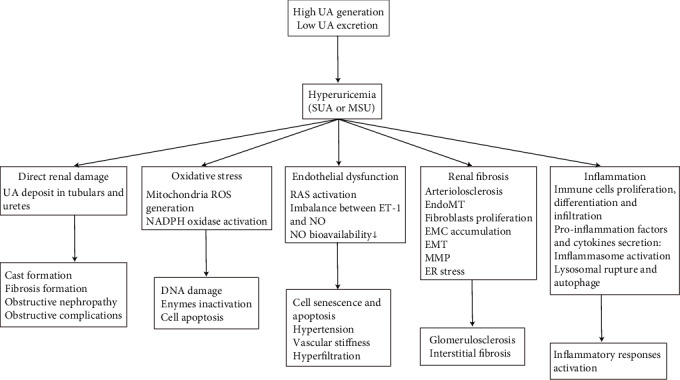
Mechanisms of hyperuricemia-induced renal injury. UA: uric acid; SUA: soluble uric acid; MSU: monosodium urate; ROS: reactive oxygen species; RAS; renin-angiotensin system; ET-1: endothelin 1; NO: nitric oxide; EndoMT: endothelial-to-mesenchymal transition; EMT: epithelial-to-mesenchymal transition; MMP: matrix metalloproteinase; ER stress: endoplasmic reticulum stress.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers: 81700627, 81670654, and 81974095), the Funds for Science and Technology Innovation Strategy of Guangdong Province (grant numbers: 2019A1515010678 and 2018A030313231), and the Guangdong Provincial Medical Research Fund (grant number: A2018072).
Abbreviations
- ACE:
Angiotensin-converting enzyme
- AICAR:
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside
- AKI:
Acute kidney injury
- AMPK:
AMP-activated protein kinase
- APC:
Antigen-presenting cell
- AR:
Aldose reductase
- BCRP/ABCG2:
Breast cancer resistance protein
- CaM:
Calmodulin
- CIC-5:
Chloride channel 5
- CKD:
Chronic kidney disease
- DAMP:
Danger-associated molecular pattern
- DN:
Diabetic nephropathy
- ECM:
Extracellular matrix
- eGFR:
Estimated glomerular filtration rate
- EGFR:
Epidermal growth factor receptor
- EMT:
Epithelial-to-mesenchymal transition
- EndoMT:
Endothelial-to-mesenchymal transition
- eNOS:
Endothelial nitric oxide synthase
- ER:
Endoplasmic reticulum
- ERK:
Extracellular signal-regulated kinase
- ESRD:
End-stage renal disease
- ET-1:
Endothelin 1
- FN:
Fibronectin
- GLUT9:
Glucose transporter 9
- GSH:
Glutathione
- GSDMD:
Gasdermin D
- HMGB1:
High-mobility group box protein 1
- HUVEC:
Human umbilical vein endothelial cell
- IL:
Interleukin
- LC-MS/MS:
Liquid chromatography-tandem mass spectrometry
- 3-MA:
3-methyladenine
- MCs:
Mesangial cells
- MCP-1:
Monocyte chemoattractant protein-1
- MMP:
Matrix metalloproteinase
- MRP4/ABCC4:
Multidrug resistance protein 4
- MSU:
Monosodium urate
- MTA1:
Metastatic tumor antigen 1
- NF-κB:
Nuclear factor-kappa B
- NKA:
Na+/K+-ATPase
- NLRP3:
Nod-like receptor pyrin domain-containing protein 3
- NO:
Nitric oxide
- OAT:
Organic anion transporter
- PAMP:
Pathogen-associated molecular pattern
- PBMC:
Peripheral blood mononuclear cells
- RAS:
Renin–angiotensin system
- ROS:
Reactive oxygen species
- α-SMA:
α-Smooth muscle actin
- SM22:
Smooth muscle 22
- TECs:
Tubule epithelial cells
- TG2:
Transglutaminase 2
- TGF-β:
Transforming growth factor-β
- TLR:
Toll-like receptor
- TNF-α:
Tumor necrosis factor-α
- UA:
Uric acid
- ULT:
Urate-lowering therapy
- URAT1:
Urate anion transporter 1
- VSMC:
Vascular smooth muscle cell
- XO:
Xanthine oxidase.
Conflicts of Interest
The authors declare that they have no conflicts of interest to disclose.
Authors' Contributions
Hong-yong Su and Chen Yang contributed equally to this work.
References
- 1.Skinner K. A., White C. R., Patel R., et al. Nitrosation of uric acid by peroxynitrite. Formation of a vasoactive nitric oxide donor. Journal of Biological Chemistry. 1998;273(38):24491–24497. doi: 10.1074/jbc.273.38.24491. [DOI] [PubMed] [Google Scholar]
- 2.Ejaz A. A., Johnson R. J., Shimada M., et al. The role of uric acid in acute kidney injury. NEPHRON. 2019;142(4):275–283. doi: 10.1159/000499939. [DOI] [PubMed] [Google Scholar]
- 3.Wang H., Zhang H., Sun L., Guo W. Roles of hyperuricemia in metabolic syndrome and cardiac-kidney-vascular system diseases. American Journal of Translational Research. 2018;10(9):2749–2763. [PMC free article] [PubMed] [Google Scholar]
- 4.Komori H., Yamada K., Tamai I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2018;1860(5):973–980. doi: 10.1016/j.bbamem.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Mallat S. G., Al Kattar S., Tanios B. Y., Jurjus A. Hyperuricemia, Hypertension, and Chronic Kidney Disease: an Emerging Association. Current Hypertension Reports. 2016;18(10) doi: 10.1007/s11906-016-0684-z. [DOI] [PubMed] [Google Scholar]
- 6.Kumagai T., Ota T., Tamura Y., Chang W. X., Shibata S., Uchida S. Time to target uric acid to retard CKD progression. Clinical and Experimental Nephrology. 2017;21(2):182–192. doi: 10.1007/s10157-016-1288-2. [DOI] [PubMed] [Google Scholar]
- 7.Lai S. W., Tan C. K., Ng K. C. Epidemiology of hyperglycemia in elderly persons. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2000;55(5):M257–M259. doi: 10.1093/gerona/55.5.m257. [DOI] [PubMed] [Google Scholar]
- 8.Luk A. J., Simkin P. A. Epidemiology of hyperuricemia and gout. American Journal of Managed Care. 2005;11(15, Supplement) [PubMed] [Google Scholar]
- 9.Zhou Z., Dong Y., Zhou H., Liu J., Zhao W. MiR-143-3p directly targets GLUT9 to reduce uric acid reabsorption and inflammatory response of renal tubular epithelial cells. Biochemical and Biophysical Research Communications. 2019;517(3):413–420. doi: 10.1016/j.bbrc.2019.07.114. [DOI] [PubMed] [Google Scholar]
- 10.Liu X., Zhai T., Ma R., Luo C., Wang H., Liu L. Effects of uric acid-lowering therapy on the progression of chronic kidney disease: a systematic review and meta-analysis. Renal Failure. 2018;40(1):289–297. doi: 10.1080/0886022x.2018.1456463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng X. X., Tang Y., Hu K., et al. Efficacy of febuxostat in hyperuricemic patients with mild-to-moderate chronic kidney disease. Medicine. 2018;97(13):p. e0161. doi: 10.1097/md.0000000000010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X., Hu J., Song N., Chen R., Zhang T., Ding X. Hyperuricemia increases the risk of acute kidney injury: a systematic review and meta-analysis. BMC Nephrology. 2017;18(1):p. 27. doi: 10.1186/s12882-016-0433-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai C. W., Lin S. Y., Kuo C. C., Huang C. C. Serum uric acid and progression of kidney disease: a longitudinal analysis and mini-review. PLOS ONE. 2017;12(1):p. e0170393. doi: 10.1371/journal.pone.0170393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou F., Yu G., Wang G., et al. Association of serum uric acid levels with the incident of kidney disease and rapid eGFR decline in Chinese individuals with eGFR > 60 mL/min/1.73 m2 and negative proteinuria. Clinical and Experimental Nephrology. 2019;23(7):871–879. doi: 10.1007/s10157-019-01705-w. [DOI] [PubMed] [Google Scholar]
- 15.Weiner D. E., Tighiouart H., Elsayed E. F., Griffith J. L., Salem D. N., Levey A. S. Uric Acid and Incident Kidney Disease in the Community. Journal of the American Society of Nephrology. 2008;19(6):1204–1211. doi: 10.1681/asn.2007101075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu C.-y., Iribarren C., McCulloch C. E., Darbinian J., Go A. S. Risk Factors for End-Stage Renal Disease. Archives of Internal Medicine. 2009;169(4):342–350. doi: 10.1001/archinternmed.2008.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kosugi T., Nakayama T., Heinig M., et al. Effect of lowering uric acid on renal disease in the type 2 diabetic db/db mice. American Journal of Physiology-Renal Physiology. 2009;297(2):F481–F488. doi: 10.1152/ajprenal.00092.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S.-M., Choi Y. W., Seok H. Y., et al. Reducing Serum Uric Acid Attenuates TGF-β1-Induced Profibrogenic Progression in Type 2 Diabetic Nephropathy. Nephron Experimental Nephrology. 2013;121(3-4):e109–e121. doi: 10.1159/000343567. [DOI] [PubMed] [Google Scholar]
- 19.Chini L. S. N., Assis L. I. S., Lugon J. R. Relationship between uric acid levels and risk of chronic kidney disease in a retrospective cohort of Brazilian workers. Brazilian Journal of Medical and Biological Research. 2017;50(9):p. e6048. doi: 10.1590/1414-431x20176048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharaf El Din U. A. A., Salem M. M., Abdulazim D. O. Uric acid in the pathogenesis of metabolic, renal, and cardiovascular diseases: A review. Journal of Advanced Research. 2017;8(5):537–548. doi: 10.1016/j.jare.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez-Sandoval J. C., Madero M. Treatment of Hyperuricemia in Chronic Kidney Disease. Contributions to Nephrology. 2018;192:135–146. doi: 10.1159/000484288. [DOI] [PubMed] [Google Scholar]
- 22.Campion E. W., Glynn R. J., Delabry L. O. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. The American Journal of Medicine. 1987;82(3):421–426. doi: 10.1016/0002-9343(87)90441-4. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh Y. P., Chang C. C., Yang Y., Wen Y. K., Chiu P. F., Lin C. C. The role of uric acid in chronic kidney disease patients. Nephrology. 2017;22(6):441–448. doi: 10.1111/nep.12679. [DOI] [PubMed] [Google Scholar]
- 24.Maruhashi T., Hisatome I., Kihara Y., Higashi Y. Hyperuricemia and endothelial function: from molecular background to clinical perspectives. Atherosclerosis. 2018;278:226–231. doi: 10.1016/j.atherosclerosis.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Ngo J. S., Ho M. H. M. Evaluation of rasburicase use in the Fraser Health Authority: a retrospective review. The Canadian Journal of Hospital Pharmacy. 2019;72(4):311–319. doi: 10.4212/cjhp.v72i4.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu L., Shi Y., Zhuang S., Liu N. Recent advances on uric acid transporters. Oncotarget. 2017;8(59):100852–100862. doi: 10.18632/oncotarget.20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enomoto A., Kimura H., Chairoungdua A., et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature. 2002;417(6887):447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- 28.Hagos Y., Stein D., Ugele B., Burckhardt G., Bahn A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. Journal of the American Society of Nephrology. 2007;18(2):430–439. doi: 10.1681/ASN.2006040415. [DOI] [PubMed] [Google Scholar]
- 29.Lu X., Chen M., Shen J., Xu Y., Wu H. IL-1β functionally attenuates ABCG2 and PDZK1 expression in HK-2 cells partially through NF-ĸB activation. Cell Biology International. 2019;43(3):279–289. doi: 10.1002/cbin.11100. [DOI] [PubMed] [Google Scholar]
- 30.Scott J. T. Drug-induced gout. Baillieres Clinical Rheumatology. 1991;5(1):39–60. doi: 10.1016/S0950-3579(05)80295-X. [DOI] [PubMed] [Google Scholar]
- 31.Ruilope L. M., Garcia-Puig J. Hyperuricemia and renal function. Current Hypertension Reports. 2001;3(3):197–202. doi: 10.1007/s11906-001-0038-2. [DOI] [PubMed] [Google Scholar]
- 32.Marangella M. Uric acid elimination in the urine. Pathophysiological implications. Contributions to Nephrology. 2005;147:132–148. doi: 10.1159/000082551. [DOI] [PubMed] [Google Scholar]
- 33.Maejima I., Takahashi A., Omori H., et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. The EMBO Journal. 2013;32(17):2336–2347. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasegawa J., Maejima I., Iwamoto R., Yoshimori T. Selective autophagy: Lysophagy. Methods. 2015;75:128–132. doi: 10.1016/j.ymeth.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 35.Schlee S., Bollheimer L. C., Bertsch T., Sieber C. C., Harle P. Crystal arthritides - gout and calcium pyrophosphate arthritis. Zeitschrift Fur Gerontologie Und Geriatrie. 2018;51(5):579–584. doi: 10.1007/s00391-017-1198-2. [DOI] [PubMed] [Google Scholar]
- 36.Moe O. W., Abate N., Sakhaee K. Pathophysiology of uric acid nephrolithiasis. Endocrinology and Metabolism Clinics of North America. 2002;31(4):895–914. doi: 10.1016/S0889-8529(02)00032-4. [DOI] [PubMed] [Google Scholar]
- 37.Maahs D. M., on behalf of the PERL Consortium, Caramori L., et al. Uric acid lowering to prevent kidney function loss in diabetes: the preventing early renal function loss (PERL) allopurinol study. Current Diabetes Reports. 2013;13(4):550–559. doi: 10.1007/s11892-013-0381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafsson D., Unwin R. The pathophysiology of hyperuricaemia and its possible relationship to cardiovascular disease, morbidity and mortality. BMC Nephrology. 2013;14(1):p. 164. doi: 10.1186/1471-2369-14-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spatola L., Angelini C., Badalamenti S., Maringhini S., Gambaro G. Kidney stones diseases and glycaemic statuses: focus on the latest clinical evidences. Urolithiasis. 2017;45(5):457–460. doi: 10.1007/s00240-016-0956-8. [DOI] [PubMed] [Google Scholar]
- 40.Yu M. A., Sanchez-Lozada L. G., Johnson R. J., Kang D. H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. Journal of Hypertension. 2010;28(6):1234–1242. [PubMed] [Google Scholar]
- 41.Roumeliotis S., Roumeliotis A., Dounousi E., Eleftheriadis T., Liakopoulos V. Dietary antioxidant supplements and uric acid in chronic kidney disease: a review. Nutrients. 2019;11(8):p. 1911. doi: 10.3390/nu11081911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Yamamoto T., Hisatome I., et al. Uric acid induces oxidative stress and growth inhibition by activating adenosine monophosphate-activated protein kinase and extracellular signal- regulated kinase signal pathways in pancreatic β cells. Molecular and Cellular Endocrinology. 2013;375(1-2):89–96. doi: 10.1016/j.mce.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 43.Doehner W., Schoene N., Rauchhaus M., et al. Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation. 2002;105(22):2619–2624. doi: 10.1161/01.CIR.0000017502.58595.ED. [DOI] [PubMed] [Google Scholar]
- 44.Lytvyn Y., Perkins B. A., Cherney D. Z. Uric acid as a biomarker and a therapeutic target in diabetes. Canadian Journal of Diabetes. 2015;39(3):239–246. doi: 10.1016/j.jcjd.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 45.Yang L., Chang B., Guo Y., Wu X., Liu L. The role of oxidative stress-mediated apoptosis in the pathogenesis of uric acid nephropathy. Renal Failure. 2019;41(1):616–622. doi: 10.1080/0886022X.2019.1633350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cristóbal-García M., García-Arroyo F. E., Tapia E., et al. Renal oxidative stress induced by long-term hyperuricemia alters mitochondrial function and maintains systemic hypertension. Oxidative Medicine and Cellular Longevity. 2015;2015:8. doi: 10.1155/2015/535686.535686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong Q., Qi K., Feng Z., et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium. 2012;51(5):402–410. doi: 10.1016/j.ceca.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Sánchez-Lozada L. G., Lanaspa M. A., Cristóbal-García M., et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Experimental Nephrology. 2013;121(3-4):e71–e78. doi: 10.1159/000345509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadowaki D., Sakaguchi S., Miyamoto Y., et al. Direct radical scavenging activity of benzbromarone provides beneficial antioxidant properties for hyperuricemia treatment. Biological & Pharmaceutical Bulletin. 2015;38(3):487–492. doi: 10.1248/bpb.b14-00514. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y., Hong Q., Huang Z., et al. ALDR enhanced endothelial injury in hyperuricemia screened using SILAC. Cellular Physiology and Biochemistry. 2014;33(2):479–490. doi: 10.1159/000358628. [DOI] [PubMed] [Google Scholar]
- 51.Huang Z., Hong Q., Zhang X., et al. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Communication and Signaling. 2017;15(1):p. 3. doi: 10.1186/s12964-016-0158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lytvyn Y., Bjornstad P., Lovshin J. A., et al. Association between uric acid, renal haemodynamics and arterial stiffness over the natural history of type 1 diabetes. Diabetes Obesity & Metabolism. 2019;21(6):1388–1398. doi: 10.1111/dom.13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang X., Gu J., Lv H., et al. Uric acid induced inflammatory responses in endothelial cells via up- regulating(pro)renin receptor. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019;109:1163–1170. doi: 10.1016/j.biopha.2018.10.129. [DOI] [PubMed] [Google Scholar]
- 54.Song X., Sun Z., Chen G., et al. Matrix stiffening induces endothelial dysfunction via the TRPV4/microRNA-6740/endothelin-1 mechanotransduction pathway. Acta Biomaterialia. 2019;100:52–60. doi: 10.1016/j.actbio.2019.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Tsuprykov O., Chaykovska L., Kretschmer A., et al. Endothelin-1 overexpression improves renal function in eNOS knockout mice. Cellular Physiology and Biochemistry. 2015;37(4):1474–1490. doi: 10.1159/000438516. [DOI] [PubMed] [Google Scholar]
- 56.Coelho S. C., Berillo O., Caillon A., et al. Three-month endothelial human endothelin-1 overexpression causes blood pressure elevation and vascular and kidney injury. Hypertension. 2018;71(1):208–216. doi: 10.1161/HYPERTENSIONAHA.117.09925. [DOI] [PubMed] [Google Scholar]
- 57.Li P., Zhang L., Zhang M., Zhou C., Lin N. Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: a mechanism for uric acid-induced endothelial dysfunction. International Journal of Molecular Medicine. 2016;37(4):989–997. doi: 10.3892/ijmm.2016.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan J., Shi M., Ma L., Fu P. Mechanistic insights of soluble uric acid-related kidney disease. Current Medicinal Chemistry. 2018;26 doi: 10.2174/0929867326666181211094421. [DOI] [PubMed] [Google Scholar]
- 59.Park J. H., Jin Y. M., Hwang S., Cho D. H., Kang D. H., Jo I. Uric acid attenuates nitric oxide production by decreasing the interaction between endothelial nitric oxide synthase and calmodulin in human umbilical vein endothelial cells: a mechanism for uric acid-induced cardiovascular disease development. Nitric Oxide. 2013;32:36–42. doi: 10.1016/j.niox.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 60.Long C. L., Qin X. C., Pan Z. Y., et al. Activation of ATP-sensitive potassium channels protects vascular endothelial cells from hypertension and renal injury induced by hyperuricemia. Journal of Hypertension. 2008;26(12):2326–2338. doi: 10.1097/HJH.0b013e328312c8c1. [DOI] [PubMed] [Google Scholar]
- 61.George J., Carr E., Davies J., Belch J. J. F., Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation. 2006;114(23):2508–2516. doi: 10.1161/CIRCULATIONAHA.106.651117. [DOI] [PubMed] [Google Scholar]
- 62.Khosla U. M., Zharikov S., Finch J. L., et al. Hyperuricemia induces endothelial dysfunction. Kidney International. 2005;67(5):1739–1742. doi: 10.1111/j.1523-1755.2005.00273.x. [DOI] [PubMed] [Google Scholar]
- 63.Zhou Y., Fang L., Jiang L., et al. Uric acid induces renal inflammation via activating tubular NF-κB signaling pathway. PLoS One. 2012;7(6, article e39738) doi: 10.1371/journal.pone.0039738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan S., Zhang P., Wang A. Y., et al. Hyperuricemia and its related histopathological features on renal biopsy. BMC Nephrology. 2019;20(1):p. 95. doi: 10.1186/s12882-019-1275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson R. J., Nakagawa T., Jalal D., Sanchez-Lozada L. G., Kang D.-H., Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrology, dialysis, transplantation. 2013;28(9):2221–2228. doi: 10.1093/ndt/gft029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang D. H. Hyperuricemia and progression of chronic kidney disease: role of phenotype transition of renal tubular and endothelial cells. Contributions to Nephrology. 2018;192:48–55. doi: 10.1159/000484278. [DOI] [PubMed] [Google Scholar]
- 67.Xavier S., Vasko R., Matsumoto K., et al. Curtailing endothelial TGF-βSignaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. Journal of the American Society of Nephrology. 2015;26(4):817–829. doi: 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J., Qu X., Bertram J. F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. American Journal of Pathology. 2009;175(4):1380–1388. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zeisberg E. M., Potenta S. E., Sugimoto H., Zeisberg M., Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. Journal of the American Society of Nephrology. 2008;19(12):2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kato T., Mizuno S., Ito A. A decrease in glomerular endothelial cells and endothelial-mesenchymal transition during glomerulosclerosis in the tensin2-deficient mice (ICGN strain) Acta Histochemica et Cytochemica. 2014;47(6):265–271. doi: 10.1267/ahc.14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ko J., Kang H. J., Kim D. A., et al. Uric acid induced the phenotype transition of vascular endothelial cellsviainduction of oxidative stress and glycocalyx shedding. The FASEB Journal. 2019;33(12, article j201901148R):13334–13345. doi: 10.1096/fj.201901148r. [DOI] [PubMed] [Google Scholar]
- 72.Satchell S. C., Tasman C. H., Singh A., et al. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney International. 2006;69(9):1633–1640. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 73.Li S., Sun Z., Zhang Y., et al. COX-2/mPGES-1/PGE2 cascade activation mediates uric acid-induced mesangial cell proliferation. Oncotarget. 2017;8(6):10185–10198. doi: 10.18632/oncotarget.14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Convento M. S., Pessoa E., Dalboni M. A., Borges F. T., Schor N. Pro-inflammatory and oxidative effects of noncrystalline uric acid in human mesangial cells: contribution to hyperuricemic glomerular damage. Urological Research. 2011;39(1):21–27. doi: 10.1007/s00240-010-0282-5. [DOI] [PubMed] [Google Scholar]
- 75.Zhuang Y., Feng Q., Ding G., et al. Activation of ERK1/2 by NADPH oxidase-originated reactive oxygen species mediates uric acid-induced mesangial cell proliferation. American Journal of Physiology-Renal Physiology. 2014;307(4):F396–F406. doi: 10.1152/ajprenal.00565.2013. [DOI] [PubMed] [Google Scholar]
- 76.Albertoni G., Maquigussa E., Pessoa E., Barreto J. A., Borges F., Schor N. Soluble uric acid increases intracellular calcium through an angiotensin II-dependent mechanism in immortalized human mesangial cells. Experimental Biology and Medicine. 2010;235(7):825–832. doi: 10.1258/ebm.2010.010007. [DOI] [PubMed] [Google Scholar]
- 77.Li S., Zhao F., Cheng S., Wang X., Hao Y. Uric acid-induced endoplasmic reticulum stress triggers phenotypic change in rat glomerular mesangial cells. Nephrology. 2013;18(10):682–689. doi: 10.1111/nep.12127. [DOI] [PubMed] [Google Scholar]
- 78.Dikshtein E. A., Vasilenko I. V., Siniachenko O. V., Diadyk A. I., Shevchenko N. I. Morphological changes in the kidney glomeruli in gout. Arkhiv patologii. 1986;48(9):54–58. [PubMed] [Google Scholar]
- 79.Asakawa S., Shibata S., Morimoto C., et al. Podocyte injury and albuminuria in experimental hyperuricemic model rats. Oxidative Medicine and Cellular Longevity. 2017;2017:14. doi: 10.1155/2017/3759153.3759153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kawamorita Y., Shiraishi T., Tamura Y., et al. Renoprotective effect of topiroxostat via antioxidant activity in puromycin aminonucleoside nephrosis rats. Physiological Reports. 2017;5(15):p. e13358. doi: 10.14814/phy2.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mack M., Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney International. 2015;87(2):297–307. doi: 10.1038/ki.2014.287. [DOI] [PubMed] [Google Scholar]
- 82.Sutariya B., Jhonsa D., Saraf M. N. TGF-β: the connecting link between nephropathy and fibrosis. Immunopharmacology and Immunotoxicology. 2016;38(1):39–49. doi: 10.3109/08923973.2015.1127382. [DOI] [PubMed] [Google Scholar]
- 83.Shi Y., Xu L., Tao M., et al. Blockade of enhancer of zeste homolog 2 alleviates renal injury associated with hyperuricemia. American Journal of Physiology-Renal Physiology. 2019;316(3):F488–F505. doi: 10.1152/ajprenal.00234.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zavadil J., Bottinger E. P. TGF- β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 85.Zha D., Wu S., Gao P., Wu X. Telmisartan attenuates uric acid-induced epithelial-mesenchymal transition in renal tubular cells. Biomed Research International. 2019;2019:12. doi: 10.1155/2019/3851718.3851718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature Reviews Molecular Cell Biology. 2014;15(3):178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zaza G., Masola V., Granata S., et al. Sulodexide alone or in combination with low doses of everolimus inhibits the hypoxia-mediated epithelial to mesenchymal transition in human renal proximal tubular cells. Journal of Nephrology. 2015;28(4):431–440. doi: 10.1007/s40620-015-0216-y. [DOI] [PubMed] [Google Scholar]
- 88.Meran S., Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. International Journal of Experimental Pathology. 2011;92(3):158–167. doi: 10.1111/j.1365-2613.2011.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sader F., Denis J. F., Laref H., Roy S. Epithelial to mesenchymal transition is mediated by both TGF-β canonical and non-canonical signaling during axolotl limb regeneration. Science Reports. 2019;9(1):p. 1144. doi: 10.1038/s41598-018-38171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zavadil J., Cermak L., Soto-Nieves N., Bottinger E. P. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. Embo Journal. 2004;23(5):1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Setyaningsih W., Arfian N., Suryadi E., Romi M. M., Tranggono U., Sari D. Hyperuricemia induces Wnt5a/Ror2 gene expression, epithelial-mesenchymal transition, and kidney tubular injury in mice. Iranian journal of medical sciences. 2018;43(2):164–173. [PMC free article] [PubMed] [Google Scholar]
- 92.Xiong X. Y., Bai L., Bai S. J., Wang Y. K., Ji T. Uric acid induced epithelial-mesenchymal transition of renal tubular cells through PI3K/p-Akt signaling pathway. Journal of Cellular Physiology. 2019;234(9):15563–15569. doi: 10.1002/jcp.28203. [DOI] [PubMed] [Google Scholar]
- 93.Grande M. T., Sánchez-Laorden B., López-Blau C., et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nature Medicine. 2015;21(9):989–997. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- 94.Ryu E. S., Kim M. J., Shin H. S., et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. American Journal of Physiology-Renal Physiology. 2013;304(5):F471–F480. doi: 10.1152/ajprenal.00560.2012. [DOI] [PubMed] [Google Scholar]
- 95.Huijuan W., Xiaoxu C., Rui S., Xinghui L., Beibei T., Jianchun M. Qi-Zhu-Xie-Zhuo-Fang reduces serum uric acid levels and ameliorates renal fibrosis in hyperuricemic nephropathy rats. Biomedicine & Pharmacotherapy. 2017;91:358–365. doi: 10.1016/j.biopha.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 96.Tao M., Shi Y., Tang L., et al. Blockade of ERK1/2 by U0126 alleviates uric acid-induced EMT and tubular cell injury in rats with hyperuricemic nephropathy. American journal of physiology-Renal physiology. 2019;316(4):F660–F673. doi: 10.1152/ajprenal.00480.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mehde A. A., Mehdi W. A., Yusof F., et al. Association of MMP-9 gene polymorphisms with nephrolithiasis patients. Journal of Clinical Laboratory Analysis. 2018;32(1):p. e22173. doi: 10.1002/jcla.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Du X., Shimizu A., Masuda Y., et al. Involvement of matrix metalloproteinase-2 in the development of renal interstitial fibrosis in mouse obstructive nephropathy. Laboratory Investigation. 2012;92(8):1149–1160. doi: 10.1038/labinvest.2012.68. [DOI] [PubMed] [Google Scholar]
- 99.Zheng G., Lyons J. G., Tan T. K., et al. Disruption of E-Cadherin by Matrix Metalloproteinase Directly Mediates Epithelial-Mesenchymal Transition Downstream of Transforming Growth Factor-β1 in Renal Tubular Epithelial Cells. American Journal of Pathology. 2009;175(2):580–591. doi: 10.2353/ajpath.2009.080983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tan T. K., Zheng G., Hsu T. T., et al. Macrophage Matrix Metalloproteinase-9 Mediates Epithelial-Mesenchymal Transition _in Vitro_ in Murine Renal Tubular Cells. American Journal of Pathology. 2010;176(3):1256–1270. doi: 10.2353/ajpath.2010.090188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang S. X., Zhang Z. C., Bai H. L. ClC-5 alleviates renal fibrosis in unilateral ureteral obstruction mice. Human Cell. 2019;32(3):297–305. doi: 10.1007/s13577-019-00253-5. [DOI] [PubMed] [Google Scholar]
- 102.Cybulsky A. V. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature Reviews Nephrology. 2017;13(11):681–696. doi: 10.1038/nrneph.2017.129. [DOI] [PubMed] [Google Scholar]
- 103.Kropski J. A., Blackwell T. S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. Journal of Clinical Investigation. 2018;128(1):64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.He L., Fan Y., Xiao W., et al. Febuxostat attenuates ER stress mediated kidney injury in a rat model of hyperuricemic nephropathy. Oncotarget. 2017;8(67):111295–111308. doi: 10.18632/oncotarget.22784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim H., Baek C. H., Chang J. W., Yang W. S., Lee S. K. Febuxostat, a novel inhibitor of xanthine oxidase, reduces ER stress through upregulation of SIRT1-AMPK-HO-1/thioredoxin expression. Clinical and Experimental Nephrology. 2020;24(3):205–215. doi: 10.1007/s10157-019-01804-8. [DOI] [PubMed] [Google Scholar]
- 106.Braga T. T., Foresto-Neto O., Camara N. O. S. The role of uric acid in inflammasome-mediated kidney injury. Current Opinion in Nephrology and Hypertension. 2020;29(4):423–431. doi: 10.1097/MNH.0000000000000619. [DOI] [PubMed] [Google Scholar]
- 107.Liu N., Wang L., Yang T., et al. EGF receptor inhibition alleviates hyperuricemic nephropathy. Journal of the American Society of Nephrology. 2015;26(11):2716–2729. doi: 10.1681/ASN.2014080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim S. M., Lee S. H., Kim Y. G., et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. American Journal of Physiology-Renal Physiology. 2015;308(9):F993–F1003. doi: 10.1152/ajprenal.00637.2014. [DOI] [PubMed] [Google Scholar]
- 109.Valimaki E., Miettinen J. J., Lietzen N., Matikainen S., Nyman T. A. Monosodium urate activates Src/Pyk2/PI3 kinase and cathepsin dependent unconventional protein secretion from human primary macrophages. Molecular & Cellular Proteomics. 2013;12(3):749–763. doi: 10.1074/mcp.M112.024661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen J., Chen Z. J. PtdIns4P on dispersed _trans_ -Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–76. doi: 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Conforti-Andreoni C., Spreafico R., Qian H. L., et al. Uric acid-driven Th17 differentiation requires inflammasome-derived IL-1 and IL-18. Journal of Immunology. 2011;187(11):5842–5850. doi: 10.4049/jimmunol.1101408. [DOI] [PubMed] [Google Scholar]
- 112.Xia S., Hollingsworth L. R., Wu H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harbor Perspectives in Biology. 2020;12(3) doi: 10.1101/cshperspect.a036400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang G., Zuo T., Li R. The mechanism of arhalofenate in alleviating hyperuricemia―activating PPARγ thereby reducing caspase-1 activity. Drug Development Research. 2020 doi: 10.1002/ddr.21699. [DOI] [PubMed] [Google Scholar]
- 114.Yen J. H., Lin L. C., Chen M. C., et al. The metastatic tumor antigen 1-transglutaminase-2 pathway is involved in self-limitation of monosodium urate crystal-induced inflammation by upregulating TGF-β1. Arthritis Research & Therapy. 2015;17(1):p. 65. doi: 10.1186/s13075-015-0592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eleftheriadis T., Pissas G., Sounidaki M., et al. Urate crystals directly activate the T-cell receptor complex and induce T-cell proliferation. Biomedical Reports. 2017;7(4):365–369. doi: 10.3892/br.2017.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koka R. M., Huang E., Lieske J. C. Adhesion of uric acid crystals to the surface of renal epithelial cells. American Journal of Physiology-Renal Physiology. 2000;278(6):F989–F998. doi: 10.1152/ajprenal.2000.278.6.F989. [DOI] [PubMed] [Google Scholar]
- 117.Kim Y. G., Huang X. R., Suga S., et al. Involvement of macrophage migration inhibitory factor (MIF) in experimental uric acid nephropathy. Molecular Medicine. 2000;6(10):837–848. doi: 10.1007/BF03401822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang K., Hu L., Chen J. K. RIP3-deficience attenuates potassium oxonate-induced hyperuricemia and kidney injury. Biomedicine & Pharmacotherapy. 2018;101:617–626. doi: 10.1016/j.biopha.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 119.Braga T. T., Forni M. F., Correa-Costa M., et al. Soluble uric acid activates the NLRP3 inflammasome. Science Reports. 2017;7(1):p. 39884. doi: 10.1038/srep39884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang Q., Fu C., Zhang X., et al. Adiponectin protects against uric acid‑induced renal tubular epithelial inflammatory responses via the AdipoR1/AMPK signaling pathway. International Journal of Molecular Medicine. 2019;43(3):1542–1552. doi: 10.3892/ijmm.2019.4072. [DOI] [PubMed] [Google Scholar]
- 121.Yu S., Ren Q., Wu W. Effects of losartan on expression of monocyte chemoattractant protein-1 (MCP-1) in hyperuricemic nephropathy rats. Journal of Receptors and Signal Transduction. 2015;35(5):458–461. doi: 10.3109/10799893.2015.1006332. [DOI] [PubMed] [Google Scholar]
- 122.Kanellis J., Watanabe S., Li J. H., et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension. 2003;41(6):1287–1293. doi: 10.1161/01.HYP.0000072820.07472.3B. [DOI] [PubMed] [Google Scholar]
- 123.Shimada M., Dass B., Ejaz A. A. Paradigm shift in the role of uric acid in acute kidney injury. Seminars in Nephrology. 2011;31(5):453–458. doi: 10.1016/j.semnephrol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 124.Roncal C. A., Mu W., Croker B., et al. Effect of elevated serum uric acid on cisplatin-induced acute renal failure. American Journal of Physiology-Renal Physiology. 2007;292(1):F116–F122. doi: 10.1152/ajprenal.00160.2006. [DOI] [PubMed] [Google Scholar]
- 125.Crișan T. O., Cleophas M. C. P., Oosting M., et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Annals of the Rheumatic Diseases. 2016;75(4):755–762. doi: 10.1136/annrheumdis-2014-206564. [DOI] [PubMed] [Google Scholar]
- 126.Karbowska A., Boratynska M., Kusztal M., Klinger M. Hyperuricemia is a mediator of endothelial dysfunction and inflammation in renal allograft recipients. Transplantation Proceedings. 2009;41(8):3052–3055. doi: 10.1016/j.transproceed.2009.07.080. [DOI] [PubMed] [Google Scholar]
- 127.Kimura Y., Yanagida T., Onda A., Tsukui D., Hosoyamada M., Kono H. Soluble uric acid promotes atherosclerosis via AMPK (AMP-activated protein kinase)-mediated inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020;40(3):570–582. doi: 10.1161/ATVBAHA.119.313224. [DOI] [PubMed] [Google Scholar]
- 128.Haryono A., Nugrahaningsih D., Sari D., Romi M. M., Arfian N. Reduction of serum Uric Acid associated with attenuation of renal injury, Inflammation and macrophages M1/M2 Ratio in hyperuricemic mice model. The Kobe journal of medical sciences. 2018;64(3):E107–E114. [PMC free article] [PubMed] [Google Scholar]
- 129.Rabadi M. M., Kuo M. C., Ghaly T., et al. Interaction between uric acid and HMGB1 translocation and release from endothelial cells. American Journal of Physiology-Renal Physiology. 2012;302(6):F730–F741. doi: 10.1152/ajprenal.00520.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Choe J. Y., Choi C. H., Park K. Y., Kim S. K. High-mobility group box 1 is responsible for monosodium urate crystal-induced inflammation in human U937 macrophages. Biochemical and Biophysical Research Communications. 2018;503(4):3248–3255. doi: 10.1016/j.bbrc.2018.08.139. [DOI] [PubMed] [Google Scholar]
- 131.Cai W., Duan X. M., Liu Y., et al. Uric acid induces endothelial dysfunction by activating the HMGB1/RAGE signaling pathway. Biomed Research International. 2017;2017:11. doi: 10.1155/2017/4391920.4391920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiao J., Zhu S., Guan H., et al. AMPK alleviates high uric acid-induced Na+-K+-ATPase signaling impairment and cell injury in renal tubules. Experimental and Molecular Medicine. 2019;51(5):1–14. doi: 10.1038/s12276-019-0254-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saitoh T., Fujita N., Jang M. H., et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 134.Hu J., Wu H., Wang D., et al. Weicao capsule ameliorates renal injury through increasing autophagy and NLRP3 degradation in UAN rats. International Journal of Biochemistry and Cell Biology. 2018;96:1–8. doi: 10.1016/j.biocel.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 135.Shi C. S., Shenderov K., Huang N. N., et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology. 2012;13(3):255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Isaka Y., Takabatake Y., Takahashi A., Saitoh T., Yoshimori T. Hyperuricemia-induced inflammasome and kidney diseases. Nephrology, dialysis, transplantation. 2016;31(6):890–896. doi: 10.1093/ndt/gfv024. [DOI] [PubMed] [Google Scholar]
- 137.Choe J. Y., Jung H. Y., Park K. Y., Kim S. K. Enhanced p 62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology. 2014;53(6):1043–1053. doi: 10.1093/rheumatology/ket474. [DOI] [PubMed] [Google Scholar]
- 138.Bao J., Shi Y., Tao M., Liu N., Zhuang S., Yuan W. Pharmacological inhibition of autophagy by 3-MA attenuates hyperuricemic nephropathy. Clinical Science. 2018;132(21):2299–2322. doi: 10.1042/CS20180563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nuki G. An appraisal of the 2012 American College of Rheumatology Guidelines for the Management of Gout. Current Opinion in Rheumatology. 2014;26(2):152–161. doi: 10.1097/BOR.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 140.Foresto-Neto O., Ávila V. F., Arias S. C. A., et al. NLRP3 inflammasome inhibition ameliorates tubulointerstitial injury in the remnant kidney model. Laboratory Investigation. 2018;98(6):773–782. doi: 10.1038/s41374-018-0029-4. [DOI] [PubMed] [Google Scholar]
- 141.Yang K. J., Kim J. H., Chang Y. K., Park C. W., Kim S. Y., Hong Y. A. Inhibition of xanthine oxidoreductase protects against contrast-induced renal tubular injury by activating adenosine monophosphate-activated protein kinase. Free Radical Biology and Medicine. 2019;145:209–220. doi: 10.1016/j.freeradbiomed.2019.09.027. [DOI] [PubMed] [Google Scholar]
- 142.Tsuda H., Kawada N., Kaimori J. Y., et al. Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochemical and Biophysical Research Communications. 2012;427(2):266–272. doi: 10.1016/j.bbrc.2012.09.032. [DOI] [PubMed] [Google Scholar]
- 143.Daskalopoulou S. S., Tzovaras V., Mikhailidis D. P., Elisaf M. Effect on serum uric acid levels of drugs prescribed for indications other than treating hyperuricaemia. Current Pharmaceutical Design. 2005;11(32):4161–4175. doi: 10.2174/138161205774913309. [DOI] [PubMed] [Google Scholar]
- 144.Hamada T., Ichida K., Hosoyamada M., et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. American Journal of Hypertension. 2008;21(10):1157–1162. doi: 10.1038/ajh.2008.245. [DOI] [PubMed] [Google Scholar]
- 145.Zhao Y., Xu L., Tian D., et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obesity & Metabolism. 2018;20(2):458–462. doi: 10.1111/dom.13101. [DOI] [PubMed] [Google Scholar]
- 146.Lytvyn Y., Skrtic M., Yang G. K., Yip P. M., Perkins B. A., Cherney D. Z. Glycosuria-mediated urinary uric acid excretion in patients with uncomplicated type 1 diabetes mellitus. American Journal of Physiology-Renal Physiology. 2015;308(2):F77–F83. doi: 10.1152/ajprenal.00555.2014. [DOI] [PubMed] [Google Scholar]
- 147.Han S., Wei R., Han D., et al. Hypouricemic effects of extracts from Urtica hyperborea Jacq. ex Wedd. in hyperuricemia mice through XOD, URAT1, and OAT1. BioMed Research International. 2020;2020:8. doi: 10.1155/2020/2968135.2968135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li F., Liu Y., Xie Y., Liu Z., Zou G. Epigallocatechin gallate reduces uric acid levels by regulating xanthine oxidase activity and uric acid excretion in vitro and in vivo. Annals of Palliative Medicine. 2020;9(2):331–338. doi: 10.21037/apm.2019.11.28. [DOI] [PubMed] [Google Scholar]
- 149.Yao R., Geng Z., Mao X., et al. Tu-Teng-Cao extract alleviates monosodium urate-induced acute gouty arthritis in rats by inhibiting uric acid and inflammation. Evidence-Based Complementary and Alternative Medicine. 2020;2020:13. doi: 10.1155/2020/3095624.3095624 [DOI] [PMC free article] [PubMed] [Google Scholar]