

Abstract
Gastrointestinal polyps are mucosal overgrowths that, if unchecked, can undergo malignant transformation. Although relatively uncommon in the pediatric age group, they can be the harbingers of multiorgan cancer risk and require close management and follow-up. Additionally, as many polyposis syndromes are inherited, appropriate genetic testing and management of relatives is vital for the health of the entire family. In this review, we discuss both common and uncommon childhood gastrointestinal polyposis syndromes in terms of clinical presentation, management, and surveillance. We also detail any additional malignancy risk and surveillance required in the pediatric age group (<21 years old). Through this review, we provide a framework for gastroenterologists to manage the multifaceted nature of pediatric polyposis syndromes.
Keywords: familial adenomatous polyposis, juvenile polyposis syndrome, pediatric polyp, pediatric polyposis, Peutz-Jeghers syndrome
Gastrointestinal polyposis is overall a rare finding in pediatric patients, but it can be a harbinger of cancer risk, and it always requires a complete evaluation. In the majority of polyposis syndromes, the lifetime cancer risk is over 50%, and in some cases approaches 100% (1). This risk can be assessed through careful coordination of genetic testing and institution of surveillance for both gastrointestinal (GI) and non-GI malignancies. It is important to recognize the need for surveillance in these patients to identify early-onset cancer in adolescence and adulthood.
Cancer risk in pediatric polyposis syndromes extends beyond the gastrointestinal tract, and it requires the astute provider to recommend and conduct surveillance for cancer and other medical complications. Specifically, surveillance for thyroid and germ cell malignancies are important components of care in some syndromes (2). Additionally, other high-risk clinical findings, such as vascular malformations, are commonly found in association with certain pediatric polyposis syndromes (3).
Polyposis syndromes are usually diagnosed by a gastroenterologist during upper endoscopic or colonoscopic evaluation. Next steps depend upon pathologic findings, including planning for genetic testing for the individual and their family members. In many cases, the gastroenterologist acts to manage what is often a multi-organ cancer predisposition. Our aim in this review is to explore pediatric polyposis syndromes—both common and rare—and define surveillance and management guidelines. We will specifically define guidelines for genetic testing, a workflow for evaluation and surveillance, and mention the important non-GI manifestations of each. We hope to provide a guide for gastroenterologists managing patients with pediatric polyposis syndromes. Additionally, involvement of a genetic counselor with expertise in cancer predisposition and/or polyposis, can be beneficial in managing these patients and organizing patient and family testing.
In approaching the care of pediatric polyposis syndromes, it is also important to recognize that guidelines listed herein should not outweigh clinical findings; should a patient develop clinical symptoms earlier than the recommended start of surveillance, the provider should act accordingly to initiate earlier clinical management.
COMMON POLYPOSIS SYNDROMES
Familial Adenomatous Polyposis
Familial Adenomatous Polyposis (FAP), previously called Gardner syndrome, carries the highest lifelong cancer risk of the common pediatric polyposis syndromes and requires aggressive management to prevent cancer and other complications. Of note, Gardner syndrome and Turcot syndrome (referring to both FAP and Lynch syndrome) were previously accepted eponymous disease names. Although most patients are diagnosed based on family history, patients may also present with other syndromic features that the astute clinician should recognize, as described below. Additionally, approximately 25% of cases are de novo, meaning there is no family history of FAP (4,5). Patients develop adenomatous polyps in childhood or adolescence, with progressively increasing polyp burden (Fig. 1A, B, and D). Noncolonic features of FAP can include osteomas of the mandible and skull, supernumerary and impacted/missing teeth, retinal pigmented epithelium (RPE) hamartomas, and skin fibromas, epidermoid cysts, lipomas, and pilomatricomas (6–9). Of note, congenital hypertrophy of the RPE (CHRPE) was initially reported to be associated with FAP, but more recently FAP-associated retinal lesions have been differentiated from CHRPE, and instead they are defined as RPE hamartomas associated with FAP (‘RPEH-FAP’) or pigmented ocular fundus lesions (‘POFL’) (8). However, as the 2 are difficult to distinguish by even experienced clinicians, a patient with a diagnosis of CHRPE should still be considered for FAP testing. The presence of any of the extracolonic findings listed here should indicate the need for genetic counseling and APC genetic testing, which should be conducted before initiation of GI surveillance program.
FIGURE 1.
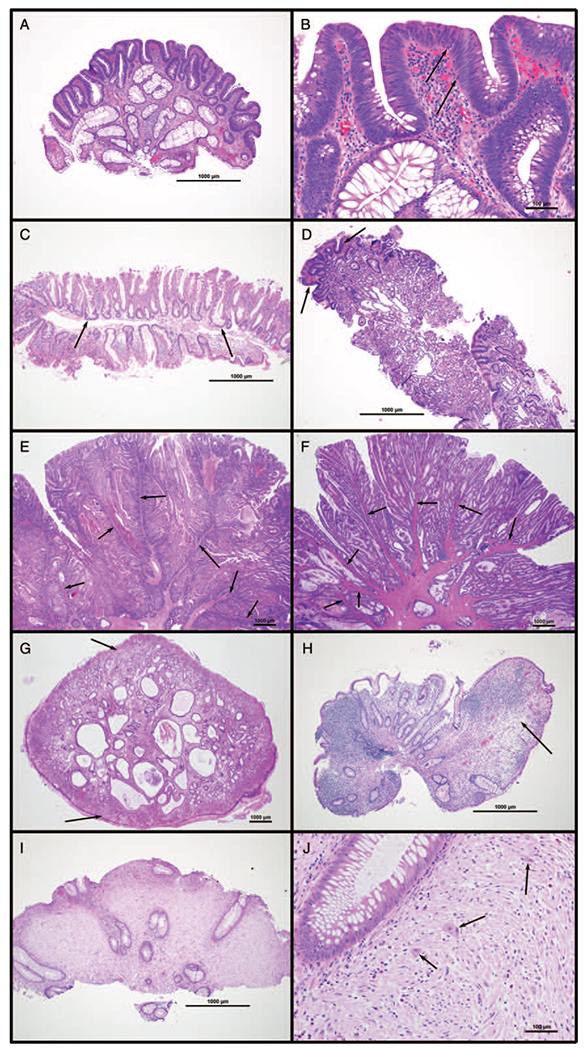
Histology of common gastrointestinal polyps. (A and B) Tubular adenoma; colon of 16-year-old girl with FAP. Surface hyperchromasia is caused by epithelial proliferation (adenomatous change), characterized by nuclear crowding and elongation, loss of cytoplasmic mucin, apical mitoses, and basal apoptotic bodies (arrows in B). (C) Sessile serrated adenoma; cecum of 17-y-old boy with Crohn colitis. Crypts have irregular, serrated luminal borders and lateral extension at the base along the muscularis mucosa (arrows). (D) Fundic gland polyp; gastric body of 16-year-old girl with FAP. Oxyntic mucosa with dilated central glands lined by a combination of parietal and mucous cells. Focal adenomatous change (arrows) is a rare finding. (E and F) Peutz-Jeghers polyps; (E) jejunum of 19-year-old woman with Peutz-Jeghers syndrome, (F) cecum of 8-y-old girl with Peutz-Jeghers syndrome. Polyps have an arborizing architecture, with smooth muscle cores (arrows) surrounded by benign mucosa. (G) Juvenile polyp; rectum of 2-year-old boy with rectal prolapse. Central dilated and mucin-filled crypts are set in an inflamed lamina propria; the surface is often ulcerated and has the appearance of granulation tissue (arrows). (H) Inflammatory pseudopolyp; cecum of 19-year-old woman with Crohn colitis. In contrast to juvenile polyps, inflammatory pseudopolyps lack central crypts, often contain only granulation tissue-like stroma (arrow), and are usually seen in a background of inflammatory bowel disease. (I and J) Polypoid ganglioneuroma; colon of 9-year-old boy with juvenile polyposis syndrome. Lamina propria is replaced by proliferation of schwannian stroma with interspersed mature ganglion cells (arrows in J).
The majority of patients with FAP have germline APC mutations, and the specific mutation can indicate disease severity (10). In particular, mutations between codons 1250 and 1464 (particularly codon 1309) can indicate more severe GI disease; alternately, mutations in the C-terminal domain can present with more attenuated disease (10). Mutations between codons 543 to 713 and 1310 to 2011 have been associated with desmoid tumors, and mutations between codons 311 and 1444 have been associated with RPE hamartomas/POFL (9–11). Regardless of genotype, patients will require surveillance, but patients with a family history of attenuated disease may start surveillance later in life. If a clinician is suspicious of FAP despite negative APC mutational testing, a cancer predisposition evaluation is warranted as other rare genetic variants can present with a similar phenotype, including somatic APC mosaicism, biallelic MSH3 deficiency, and biallelic NTHL1 deficiency (12,13).
Surveillance for FAP with colonoscopy should start between the ages of 10 and 12 years, and colectomy should occur in adolescence or young adulthood (Table 1) (2). In patients with hematochezia or other symptoms, surveillance may start sooner (14). Surveillance with upper endoscopy should usually start by age 20, or earlier for more severe phenotypes, as upper GI polyps usually occur 7 to 10 years after colon polyps (15). Surveillance in this case should include histologic evaluation of resected polyps for signs of dysplasia and malignant transformation. Endoscopy should include side-viewing endoscopy of the duodenum, given the risk of a dysplastic lesion at the papilla, which could be missed on standard endoscopy view (16). Surveillance may begin slightly later in patients with mutations known to cause attenuated disease, but should still start before age 18 years (2,10).
TABLE 1.
Common pediatric polyposis syndromes and malignancy risk*
| Syndrome | Gene (s) | Cancer risk | Screening | Onset | Interval | Other |
|---|---|---|---|---|---|---|
| Familial adenomatous polyposis | APC | Colon | Colonoscopy | 10 years | 1 year | Colectomy recommended by 20 to 25 years |
| Gastric | Endoscopy | 18 to 20 years | 1 to 4 years | |||
| Thyroid | Examination or ultrasound | 18 years | 1 year | |||
| Brain (medulloblastoma) | Neurologic examination | Infancy | 1 year | |||
| Liver (hepatoblastoma) | Alpha-fetoprotein, abdominal ultrasound | Infancy | 3 to 6 months | Until ages 5 years | ||
| Desmoid tumor | Abdominal examination, Abdomen/pelvis MRI | Postcolectomy | 1 to 3 years | Can lengthen to 5 to 10 years if first interval normal | ||
| Juvenile polyposis syndrome | SMAD4 and BMPR1A | Gastric | Endoscopy | 15 years | 1 to 3 years | Evaluate for HHT with SMAD4 mutation |
| Colon | Colonoscopy | 15 years | 1 to 3 years | |||
| Peutz-Jeghers syndrome | STK11 | Gastric | Endoscopy | 8 to 10 years | 2 to 3 years | Defer second study to 18 years ofage if no polyps |
| Colon | Colonoscopy | 8 to 10 years | 2 to 3 years | Defer second study to 18 years of age if no polyps | ||
| Small intestine | MRE or video capsule study | 8 to 10 years | 2 to 3 years | Defer second study to 18 years of age if no polyps | ||
| Reproductive—F (ovarian/cerv/uterine) | Pap smear, pelvic examination | 18 years | 1 year | |||
| Reproductive—M (testes) | Testicular examination | 10 years | 1 year | |||
| Pancreas | MRCP or endoscopic US | 30 to 35 years | 1 to 2 years | |||
| Breast | Mammogram/breast MRI Breast self-examination | 25 years | 1 year 6 months | |||
| PTEN hamartoma tumor syndrome | PTEN | Colon | Colonoscopy | 35 years | 1 to 5 years | Earlier if symptomatic |
| Thyroid | Examination and ultrasound | 18 years | 1 year |
Modified from Achatz et al, Clinical Cancer Research, 2016. F = female; M = male; MRI = magnetic resonance imaging; MRE = magnetic resonance enterography; MRCP = magnetic resonance cholangiopancreatography; US = ultrasound.
Decisions about colectomy are highly patient-specific, but a general guideline is to undergo a single-stage procedure when polyp burden outweighs ability for removal, with preference for total abdominal colectomy with ileopouch anal anastomosis (usually with a J-pouch), versus ileorectal anastamosis (2,17). Choice of surgery should be based on patient phenotype, and it is important that the patient understand the different needs for ongoing surveillance with each procedure. This is especially important to consider given the risk of desmoid tumors in patients with FAP, which increases significantly postoperatively; risk can be decreased by minimizing abdominal instrumentation (18,19). Ongoing surveillance of ileal pouch and/or rectal cuff is necessary postsurgery, and risk of ileal pouch adenoma increases in the years postcolectomy (75% risk at 15 years postcolectomy, with higher risk in patients with higher polyp burden) (20,21). Additionally, ongoing surveillance with upper endoscopy on an every 1 to 4-year basis is necessary, based on the upper GI polyp burden (17).
Patients with FAP also require surveillance for thyroid cancer, as they are at increased risk of papillary thyroid cancer (particularly cribriform-morular variant), with an increased risk in female patients, thus thyroid examination or ultrasound should start at age 15 to 18 years (17,22–24). FAP patients should also have an annual neurologic examination, given risk of medulloblastoma, which can be done by their gastroenterologist or other provider, and does not need neurology subspecialty referral or imaging unless there are abnormalities uncovered (2,17). There is some debate as to whether these patients should undergo surveillance for hepatoblastoma, given the slight increase in prevalence in patients with an APC mutation; generally, this requires discussion with the family regarding risk and is not routinely recommended (14,25,26). Patients with a family history of desmoid tumor should also undergo routine abdominal MRI every 3 to 5 years postoperatively from colectomy (2,17). Recommendations for family testing are discussed further below; FAP is autosomal-dominant and parents and siblings should be offered testing when a mutation is identified.
Juvenile Polyposis Syndrome
Juvenile polyposis syndrome (JPS) is associated with a 10% to 50% increased risk of colorectal cancer, with median age of onset of 35 years and occasional gastrointestinal cancer in childhood (27). Patients present childhood or adolescence in the setting of hematochezia and anemia, but presentation can take a variety of forms. Rarely, patients will present with severe anemia and/or signs of obstruction from polyposis. On histology, juvenile polyps have specific pathologic features that aid in diagnosis of the syndrome (Fig. 1G), but they can be confused with inflammatory pseudopolyps (Fig. 1H). The presence of 5 or more juvenile polyps in the colon, any number of juvenile polyps present on both upper and lower endoscopy, or any number of juvenile polyps with a positive family history are all sufficient to make the clinical diagnosis of JPS (17,28,29).
Patients should be offered testing for SMAD4 and BMPR1A mutations, as these are the 2 known germline alterations to cause JPS and can occur de novo (30). SMAD4 and BMPR1A are both members of the TGF-β/BMP pathway and are involved in intestinal development and maintenance of the intestinal epithelium. Genotype-phenotype correlation has been described in SMAD4, associated with more severe gastric disease and also associated more severe and earlier onset disease occurring in exons 4 to 8 (31). In many patients (40%–60%), an aberration in either gene will not be present, and the underlying germline cause will remain unknown (27). Given the significant number of patients without a known mutation, GI surveillance should be considered in all first-degree relatives starting at age 15 years, even if a mutation is not identified (Table 2) (30–32).
TABLE 2.
Suggested familial screening in pediatric polyposis syndromes
| Syndrome | Familial targeted testing | Familial screening | Familial screening interval (if first negative) |
|---|---|---|---|
| Familial adenomatous polyposis | APC | Genetic testing only* | |
| Juvenile polyposis syndrome | SMAD4 and BMPR1A | If genetic testing negative in proband, screening colonoscopy/endoscopy at 15 years in first degree family members | 5 years |
| Peutz-Jeghers syndrome | STK11 | If genetic testing negative in proband, screening colonoscopy/endoscopy at 15 years in first degree family members | 5 years |
| PTEN hamartomas tumor syndrome | PTEN | Genetic testing only* | |
| Constitutional mismatch repair Deficiency | PMS2, MLH1, MSH2, MSH6 | Genetic testing only* | |
| 10p23 deletion | 10p23 deletion | Genetic testing only* | |
| Hereditary mixed polyposis | GREM1 | Genetic testing only* | |
| Serrated polyposis | None | First degree relatives: (earliest of following) ages 40, 10 years before earliest CRC diagnosis, age of earliest diagnosis of serrated polyposis | Every 5 years |
In those listed as “genetic testing only,” the presumption is that the grand majority of affected individuals have a mutation in a known gene; should amutation not be identified despite clear clinical phenotype, we recommend evaluation of first-degree family members at 10 years before phenotype onset in proband.
Surveillance timing and intensity depends significantly on the polyp burden in an individual patient, but should occur at least every 1 to 3 years in affected patients, with earlier interval follow-up if polyp burden is higher (Table 1) (2,17). As with FAP, surveillance should include histologic evaluation of resected polyps for signs of dysplasia and malignant transformation. Patients with JPS because of a SMAD4 mutation should also be evaluated for hereditary hemorrhagic telangiectasia (HHT), particularly with certain hotspot mutations; for this reason, genetic testing earlier in life (at birth) is recommended. Patients with a known SMAD4 mutation should undergo a physical examination (including neurologic examination to evaluate for evidence of any deficits indicating possible vascular malformation) and echocardiogram evaluation, and are best managed by a provider with experience in HHT as bleeding can be life-threatening. Additionally, although there is a slightly increased risk of pancreatic and small intestinal cancer in adulthood, there are no current recommendations for surveillance of these organs (17,28,29).
Peutz-Jeghers Syndrome
Patients with Peutz-Jeghers syndrome (PJS) are often clinically evaluated for other syndromic features, particularly oral freckling, before diagnosis of polyposis (2). Similarly, they can present with signs of precocious puberty before manifestations of any gastrointestinal symptoms (33). PJS can be recognized in infancy because of freckling around the mouth, nostrils, perianal area, dorsal aspects of the hands and feet, and fingers and toes. Oral freckling is not pathognomonic for PJS, but should raise clinical suspicion and requires evaluation, as lesions can fade with age (9,33). Polyps in PJS (Fig. 1E and F) are of a specific arborizing histologic subtype, and can grow significantly in childhood and cause symptomatic GI obstruction/intussusception, particularly, in the small bowel. Over 90% of patients with clinical PJS will have a germline mutation in the STK11 tumor suppressor gene, which can be de novo, and genotype-phenotype correlations are not well-defined (34). Diagnosis can also be made clinically when an individual has 2 or more of the following: 2 or more PJS-type polyps, mucocutaneous hyperpigmentation (lips, mouth, nose, eyes, genitalia, fingers), and family history of PJS (17).
The purpose of GI surveillance in childhood is to prevent complications from polyp obstruction, including intussusception and bleeding, as cancer risk starts mainly in adulthood; additionally, it is controversial whether CRC risk results from malignant transformation of PJS polyps or is a separate process (33). For this reason, all PJS patients should undergo surveillance early in life for a polyposis phenotype, and if present should undergo continued surveillance. This should start with colonoscopy, endoscopy, and small bowel imaging (magnetic resonance enterography or video capsule study) at the age of 8 to 10 years (2,17,33,35). If polyps are found on initial study, evaluation should be repeated every 2 to 3 years; if the initial study is negative, next GI imaging can be deferred to 18 years of age, and then repeated at 2- to 3-year intervals (Table 1) (17). Additionally, if a patient presents with symptomatic intussusception, the recommendation would be for surgical resection rather than radiologic/endoscopic reduction (35).
Patients with PJS should also be evaluated for signs of precocious puberty, and are at risk for sex cord/stromal tumors, particularly large-cell calcifying Sertoli cell tumors (of note, precocious puberty may be present without presence of a tumor). In male patients, gynecomastia can be an indicator of precocious puberty and can be reversed with medical management (36). If signs of precocious puberty are present, however, patients should be evaluated by an oncologist or endocrinologist with ultrasound and testosterone levels as indicators of an underlying malignancy. In the absence of these signs, patients with PJS should still be evaluated with annual physical examination (33). In adulthood, risk of breast, pancreatic, and lung cancer increases and screening will commence (Table 1), although it is important to counsel teenage patients not to smoke, or to stop smoking if they have started (17).
PTEN Hamartoma Tumor Syndrome (Also Known as Cowden Syndrome, or Bannayan-Riley-Ruvalcaba Syndrome)
PTEN Hamartoma Tumor Syndrome (PHTS) can present in childhood in a variety of ways, and occasionally polyposis may occur in the pediatric age group, most commonly presenting with hematochezia. Other indicators of this syndrome include macrocephaly and autism spectrum disorder. The term PHTS refers to any patients with an underlying germline PTEN mutation, and can encompass patients meeting clinical criteria for Cowden syndrome, Bannayan-Ruvalcaba-Riley syndrome, Proteus syndrome, and Proteus-like syndrome. Although recommendations do not specify the need for surveillance colonoscopy/endoscopy in the pediatric age group, providers should be aware of the possibility if patients present with symptoms, as the majority of patients with this syndrome will develop polyposis (37). PHTS is considered a mixed polyposis syndrome, with multiple polyp types including juvenile, hamartomatous, inflammatory, ganglioneuromatous, lipomatous, and others (Fig. 1) (38). Patients will require thyroid screening starting at the age of 7 years (39).
Genotype-phenotype correlations have been shown to correlate with multiple adult-onset cancers, but this will not be discussed in detail in this review. In brief, promoter mutations have been shown to correlate with breast and colon cancer risk, and variants with partial loss of PTEN function have been associated with autism risk (40,41). There has not been any genotype-phenotype correlation shown to relate to other features of disease, such as macrocephaly and polyposis (40), and it is not currently recommended that mutation type should guide surveillance recommendations.
RARE POLYPOSIS SYNDROMES
Constitutional Mismatch Repair Deficiency
Constitutional mismatch repair deficiency (CMMRD) is a newly recognized cancer predisposition syndrome that can present in infancy with polyposis (42). Patients may present with early-onset malignancy, a significant familial cancer history, and have other clinical features, such as café-au-lait hyperpigmented lesions, agenesis of the corpus callosum, and immunoglobulin deficiency (43). Although a heterozygous DNA-mismatch repair defect can cause Lynch syndrome (an adult-onset colorectal cancer syndrome), a homozygous or compound heterozygous defect in DNA repair leads to significant anticipation of symptoms and early onset of polyposis. The cancer risk in this syndrome also extends to other tumors, including brain tumors and leukemia/lymphoma (44). Thus, the proposed guidelines for surveillance start in infancy for brain tumors, and onset of colonoscopy and endoscopy should start no later than 6 years of age (43,45). Should there be concern for CMMRD, the patient should be referred to a cancer predisposition program to initiate intensive screening protocols, such as those recommended by the American Association for Cancer Research guidelines (43).
As CMMRD is a relatively new cancer predisposition syndrome, it is important that should a patient present with concerning features, such as early-onset polyposis, café-au-lait macules, and/or early onset malignancy, they should be evaluated by a cancer predisposition specialist. Certain rare variants in DNA repair genes (such as the V411L variant of POLE) may confer a similar phenotype (46). The proofreading associated polyposis syndrome associated with POLE and POLD1 is usually an adult onset syndrome, but mutations in the exonuclease domain of these genes may lead to childhood onset disease (47).
10q23 Deletion
10q23 deletion including BMPR1A and PTEN can be a compound syndrome leading to extremely early onset of polyposis and high likelihood of needing a colectomy. Although rare, case reports describe onset of polyposis before the age of 6 years, which can occur anywhere in the GI tract and can present with bleeding and/or obstruction (48–50). Early colectomy is also likely to be required in this syndrome based on these reports, but there are no established guidelines for surveillance. On the basis of the available data, we propose the start of fecal occult blood surveillance at time of diagnosis, followed by annual colonoscopy with onset of positive fecal occult blood testing. However, given the rarity of this syndrome and lack of evidence to support this approach, surveillance should be timed as deemed appropriate by the provider based upon clinical findings.
Serrated Polyposis Syndrome
Serrated polyps are often found in adult patients (21% of asymptomatic individuals), but the finding of several such pathologically defined polyps should raise concern for serrated polyposis syndrome (51). Serrated polyposis syndrome is defined by the presence of at least 5 serrated polyps in the proximal colon (at least 2 of which are >10 mm), and/or at least 1 serrated polyp proximal to the sigmoid colon with a family history of SPS, and/or more than 20 serrated polyps throughout the colon (Fig. 1C) (17,52). Of note, the pathologic term “serrated polyp” has undergone many changes over the years and can include hyperplastic polyps, sessile serrated adenomas, and traditional serrated adenomas, so involvement of an experienced pathologist in diagnosis is recommended. Although germline RNF43 mutation was recently identified as a rare cause of serrated polyposis (53), a germline driver mutation may not be found in individual patients.
The risk of colorectal and extracolonic cancer in patients with serrated polyposis is high (incidence ratio 18.72 for CRC and 31.20 for extracolonic cancer); however, surveillance for extracolonic cancer is not currently recommended (52,54). Recommended surveillance for patients with serrated polyposis syndrome is annual colonoscopy, with possibility to space to every 3 years if the polyp burden is low. There is currently no standard genetic testing that is done in these patients, although if other nonserrated adenomas are present, an argument can be made for multigene polyposis panel testing. For family members, first degree relatives should have surveillance starting 10 years earlier than first family CRC diagnosis, age 40, or at the age of onset of serrated polyposis in unaffected family members, whichever is earlier (17).
Hereditary Mixed Polyposis Syndrome
Hereditary mixed polyposis syndrome is an autosomal-dominant polyposis syndrome with increased risk of CRC, almost exclusively seen in the Ashkenazi Jewish population (55). Patients can present with many different morphologies including adenomatous polyps, juvenile polyps, serrated polyps, Peutz-Jegher type polyps, and occasionally polyps with mixed elements (56). Additionally, these patients do not have extracolonic features of disease (56). This syndrome is because of increased GREM1 expression because of duplication of the GREM1 promoter (57). Of note, targeted testing should be done, as these duplications can be missed in panel/exome sequencing (58). Phenotype is varied and heterogeneous among families, with a trend towards rapid progression from polyposis to advanced/dysplastic adenomatous lesions when compared with other polyposis syndromes (55,59). Although surveillance does not start until adulthood in asymptomatic family members (ages 25 years), if polyps are identified in the pediatric age group, the patient should then be followed with annual colonoscopy (59).
Adult Polyposis Syndromes
Although a full discussion of adult-onset polyposis syndromes is outside the scope of this article, rarely some patients with these syndromes can present with symptomatic polyposis in the pediatric age group, especially those syndromes discussed previously. Lynch syndrome, polymerase-proofreading-associated polyposis, and MUTYH-associated polyposis are not expected to have a pediatric presentation, and families should be counseled that screening is not indicated in asymptomatic children unless there is family history of very onset CRC (less than 25 years). Additionally, conditions that are mainly adult-onset and are very rare, including MSH3-associated polyposis and NTLH1-associated polyposis, will not be discussed in this context, but should be considered in phenotypically concerning individuals with otherwise negative genetic testing, as noted above.
MANAGEMENT OF INDETERMINATE POLYPOSIS
The current guidelines recommend evaluation based on known clinical and genetic diagnostic criteria, but in many cases, pediatric patients will present without diagnostic features of a specific syndrome. For example, the presence of juvenile polyps in inadequate numbers to reach a clinical diagnosis of JPS could be followed clinically and with further surveillance if symptomatic, as there is a 17% chance that additional juvenile polyps will develop (30,60). Evaluation for fecal occult blood can be considered in absence of symptoms. Similar guidelines could be followed in patients with a single PJS-type polyp or serrated polyp, which is inadequate to qualify as diagnostic for either syndrome, or other single indeterminate polyps.
Adenomatous polyps are an uncommon finding in children, and the presence of 1 to 2 adenomatous polyps in children should be concerning enough to warrant further evaluation with colonoscopy/ endoscopy at a 5-year interval (or sooner with symptoms), similar to adult-screening guidelines (61). Additionally, even if family history is negative, we would recommend evaluation for underlying APC aberration in patients with the presence of more than 1 to 2 adenomatous polyps in childhood.
MANAGEMENT OF FAMILY MEMBERS
When a patient is diagnosed with a familial polyposis syndrome, it is imperative to also consider the screening needs of other family members. This is detailed in Table 2 for each of the syndromes discussed above. Of note, even if a parent has not had symptoms of a syndrome, it is still important to discuss screening. Additionally, even if parents test negative for a genetic syndrome, testing of siblings should still be considered, given the risk of gonadal mosaicism.
For several of the syndromes discussed above, especially JPS, there is a chance that no mutation will be detected in the pediatric patient. In those cases, where genetic testing is not informative, it is important to offer screening to parents and siblings with colonoscopy/endoscopy at the age indicated in Table 2. Involvement of a genetic counselor and/or a childhood cancer predisposition program can be helpful in deciding on next steps for family member evaluation.
CONCLUSIONS
Polyposis in childhood can be a harbinger of cancer predisposition requiring lifelong surveillance and involvement of other subspecialties. By paying attention to other pertinent clinical features and conducting appropriate genetic testing and routine surveillance, major complications can be avoided in these patients. The involvement of a multidisciplinary polyposis team—including gastroenterology, oncology, genetics, and surgery—can assist in access to appropriate surveillance and other psychosocial supports for affected families.
What Is Known
Familial adenomatous polyposis, Peutz-Jegher syndrome, juvenile polyposis syndrome, and other polyposis syndromes can present with hematochezia and obstruction in childhood or adolescence.
Pediatric polyposis syndromes may be harbingers of cancer risk and require close follow-up with gastrointestinal surveillance.
What Is New
Pediatric Cancer Predisposition guidelines have been recently formalized and require close interval followup, including gastrointestinal and nongastrointestinal screening
Involvement of a genetic counselor or cancer risk team can help to facilitate surveillance of affected patients and genetic testing of family members.
Recent guidelines published by the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition recently addressed the 3 most common of these syndromes; we expand on these recommendations to provide guidance on nongastrointestinal complications and surveillance.
Acknowledgments
This work is supported by the National Institutes of Health (K12CA076931 SPM, K08 DK099379 BJW, K08DK106489 BWK), the Precious Jules Foundation (SPM) the Audrey E. Evans Endowed Chair in Molecular Oncology (GMB), and The Children’s Hospital of Philadelphia Chair’s Initiative (GMB).
Footnotes
B.W.K. for Janssen Pharmaceuticals (travel) and Exact Sciences (consulting); no other potential conflicts of interest to declare.
The authors report no conflicts of interest
REFERENCES
- 1.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 2007;21:2525–38. [DOI] [PubMed] [Google Scholar]
- 2.Achatz MI, Porter CC, Brugieres L, et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res 2017;23:e107–14. [DOI] [PubMed] [Google Scholar]
- 3.van Hattem WA, Langeveld D, de Leng WW, et al. Histologic variations in juvenile polyp phenotype correlate with genetic defect underlying juvenile polyposis. Am J Surg Pathol 2011;35:530–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis 2009;4:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisgaard ML, Fenger K, Bulow S, et al. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Human Mutation 1994;3:121–5. [DOI] [PubMed] [Google Scholar]
- 6.Gardner EJ. Follow-up study of a family group exhibiting dominant inheritance ofa syndrome including intestinal polyps, osteomas, fibromas, and epidermal cysts. Am J Hum Genet 1962;14:376–90. [PMC free article] [PubMed] [Google Scholar]
- 7.Aihara H, Kumar N, Thompson CC. Diagnosis, surveillance, and treatment strategies for familial adenomatous polyposis: rationale and update. Eur J Gastroenterol Hepatol 2014;26:255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shields JA, Shields CL. Tumors and related lesions of the pigmented epithelium. Asia Pac J Ophthalmol (Phila) 2017;6:215–23. [DOI] [PubMed] [Google Scholar]
- 9.Winship IM, Dudding TE. Lessons from the skin—cutaneous features of familial cancer. Lancet Oncol 2008;9:462–72. [DOI] [PubMed] [Google Scholar]
- 10.Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007;61:153–61. [DOI] [PubMed] [Google Scholar]
- 11.Slowik V, Attard T, Dai H, et al. Desmoid tumors complicating familial adenomatous polyposis: a meta-analysis mutation spectrum of affected individuals. BMC Gastroenterol 2015;15:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adam R, Spier I, Zhao B, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet 2016;99:337–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciavarella M, Miccoli S, Prossomariti A, et al. Somatic APC mosaicism and oligogenic inheritance in genetically unsolved colorectal adenomatous polyposis patients. Eur J Hum Genet 2018;26:387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyer W, Cohen S, Attard T, et al. Management of familial adenomatous polyposis in children and adolescents: position paper from the ESP-GHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2018;68:428–41. [DOI] [PubMed] [Google Scholar]
- 15.Singh A, Steinhagen E, Katona BW. Approach to upper gastrointestinal tract lesions in familial adenomatous polyposis. Semin Colon Rectal Surg 2018;29:102–7. [Google Scholar]
- 16.Gluck N, Strul H, Rozner G, et al. Endoscopy and EUS are key for effective surveillance and management of duodenal adenomas in familial adenomatous polyposis. Gastrointest Endosc 2015;81:960–6. [DOI] [PubMed] [Google Scholar]
- 17.Genetic/familial high-riskassessment: colorectal. NCCN Clinical Practice Guidelines in Oncology 2018;V1.2018.
- 18.Lefevre JH, Parc Y, Kerneis S, et al. Risk factors for development of desmoid tumours in familial adenomatous polyposis. Br J Surg 2008;95:1136–9. [DOI] [PubMed] [Google Scholar]
- 19.Vitellaro M, Sala P, Signoroni S, et al. Risk of desmoid tumours after open and laparoscopic colectomy in patients with familial adenomatous polyposis. Br J Surg 2014;101:558–65. [DOI] [PubMed] [Google Scholar]
- 20.Parc YR, Olchwang S, Desaint B, et al. Familial adenomatous polyposis: prevalence of adenomas in the ileal pouch after restorative proctocolectomy. Ann Surg 2001;233:360–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tonelli F, Ficari F, Bargellini T, et al. Ileal pouch adenomas and carcinomas after restorative proctocolectomy for familial adenomatous polyposis. Dis Colon Rectum 2012;55:322–9. [DOI] [PubMed] [Google Scholar]
- 22.Ito Y, Miyauchi A, Ishikawa H, et al. Our experience of treatment of cribiform morular viarant of papillary thyroid carcinoma; difference in clinicopathological features of FAP-associated and sporadic patients. EndocrJ 2011;58:685–6889. [DOI] [PubMed] [Google Scholar]
- 23.Uchino S, Ishikawa H, Miyauchi A, et al. Age- and gender-specific risk of thyroid cancer in patients with familial adenomatous polyposis. J Clin Endocrinol Metab 2016;101:4611–7. [DOI] [PubMed] [Google Scholar]
- 24.Sada H, Hinoi T, Ueno H, et al. Prevalence of and risk factors for thyroid carcinoma in patients with familial adenomatous polyposis: results of a multicenter study in Japan and a systematic review. Surg Today 2018;49:72–81. [DOI] [PubMed] [Google Scholar]
- 25.Aretz S, Koch A, Uhlhaas S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 2006;47:811–8. [DOI] [PubMed] [Google Scholar]
- 26.Trobaugh-Lotrario AD, Lopez-Terrada D, Li P, et al. Hepatoblastoma in patients with molecularly proven familial adenomatous polyposis: clinical characteristics and rationale for surveillance screening. Pediatr Blood Cancer 2018;65:e27103. [DOI] [PubMed] [Google Scholar]
- 27.Cichy W, Klincewicz B, Plawski A. Juvenile polyposis syndrome. Arch Med Sci 2014;10:570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howe JR. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet 2004;41:484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latchford AR, Neale KF, Phillips RK, et al. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum 2012;55:1038–43. [DOI] [PubMed] [Google Scholar]
- 30.Cohen S, Hyer W, Mas E, et al. Management of juvenile polyposis syndromes in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019;68:453–62. [DOI] [PubMed] [Google Scholar]
- 31.Howe JR, Shellnut J, Wagner B, et al. Common deletion of SMAD4 in juvenile polyposis is a mutational hotspot. Am J Hum Genet 2002;70:1357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howe JR, Ringold JC, Hughes JH, et al. Direct genetic testing for Smad4 mutations in patients at risk for juvenile polyposis. Surgery 1999;126:162–70. [PubMed] [Google Scholar]
- 33.Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975–86. [DOI] [PubMed] [Google Scholar]
- 34.Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat 2005;26: 513–9. [DOI] [PubMed] [Google Scholar]
- 35.Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndromes in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019;68:442–52. [DOI] [PubMed] [Google Scholar]
- 36.Di Grezia G, Romano T, De Francesco F, et al. Breast ultrasound in the management of gynecomastia in Peutz-Jeghers syndrome in monozygotic twins: two case reports. J Med Case Rep 2014;8:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genetic/Familial HIgh-Risk Assessment: Breast and Ovarian NCCN Clinical Practice Guidelines in Oncology 2018.
- 38.Stanich PP, Pilarski R, Rock J, et al. Colonic manifestations of PTEN hamartoma tumor syndrome: case series and systematic review. World J Gastroenterol 2014;20:1833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med 2009;11:687–94. [DOI] [PubMed] [Google Scholar]
- 40.Tan MH, Mester JL, Ngeow J, et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012;18:400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spinelli L, Black FM, Berg JN, et al. Functionally distinct groups of inherited PTEN mutations in autism and tumor syndromes. J Med Genet 2015;52:128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durno CA, Sherman PM, Aronson M, et al. , International BMMRD Consortium. Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer 2015;51:977–83. [DOI] [PubMed] [Google Scholar]
- 43.Tabori U, Hansford JR, Achatz MI, et al. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res 2017;23:e32–7. [DOI] [PubMed] [Google Scholar]
- 44.Shlien A, Campbell BB, de Borja R, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 2015;47:257–62. [DOI] [PubMed] [Google Scholar]
- 45.Durno C, Boland CR, Cohen S, et al. Recommendations on surveillance and management of Biallelic Mismatch Repair Deficiency (BMMRD) Syndrome: a consensus statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2017;156:1605–14. [DOI] [PubMed] [Google Scholar]
- 46.Wimmer K, Beilken A, Nustede R, et al. A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency. Fam Cancer 2017;16: 67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elsayed FA, Kets CM, Ruano D, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet 2015;23:1080–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliveira PH, Cunha C, Almeida S, et al. Juvenile polyposis of infancy in a child with deletion of BMPR1A and PTEN genes: surgical approach. J Pediatr Surg 2013;48:e33–7. [DOI] [PubMed] [Google Scholar]
- 49.Alimi A, Weeth-Feinstein LA, Stettner A, et al. Overlap of Juvenile polyposis syndrome and Cowden syndrome due to de novo chromosome 10 deletion involving BMPR1A and PTEN: implications for treatment and surveillance. Am J Med Genet A 2015;167:1305–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hiljadnikova Bajro M, Sukarova-Angelovska E, Adelaide J, et al. A new case with 10q23 interstitial deletion encompassing both PTEN and BMPR1A narrows the genetic region deleted in juvenile polyposis syndrome. J Appl Genet 2013;54:43–7. [DOI] [PubMed] [Google Scholar]
- 51.Rosty C, Hewett DG, Brown IS, et al. Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis, and clinical management. J Gastroenterol 2013;48:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Alcalde D, Carballal S, Moreira L, et al. , Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. High incidence of advanced colorectal neoplasia during endoscopic surveillance in serrated polyposis syndrome. Endoscopy 2018;51:142–51. [DOI] [PubMed] [Google Scholar]
- 53.Quintana I, Mejias-Luque R, Terradas M, et al. Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 2018;67:2230–2. [DOI] [PubMed] [Google Scholar]
- 54.Edelstein DL, Cruz-Correa M, Soto-Salgado M, et al. Risk of colorectal and other cancers in patients with serrated polyposis. Clin Gastroenterol Hepatol 2015;13:1697–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lieberman S, Walsh T, Schechter M, et al. Features of patients with hereditary mixed polyposis syndrome caused by duplication of GREM1 and implications for screening and surveillance. Gastroenterology 2017;152:1876.e1-80.e1. [DOI] [PubMed] [Google Scholar]
- 56.Whitelaw SC, Murday VA, Tomlinson IP, et al. Clinical and molecular features of the Hereditary Mixed Polyposis Syndrome. Gastroenterology 1997;112:327–34. [DOI] [PubMed] [Google Scholar]
- 57.McKenna DB, Van Den Akker J, Zhou AY, et al. Identification of a novel GREM1 duplication in a patient with multiple colon polyps. Fam Cancer 2018;18:63–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jaeger E, Leedham S, Lewis A, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 2012;44:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plesec T, Brown K, Allen C, et al. Clinicopathological features of a kindred with SCG5-GREM1-associated hereditary mixed polyposis syndrome. Hum Pathol 2017;60:75–81. [DOI] [PubMed] [Google Scholar]
- 60.Fox VL, Perros S, Jiang H, et al. Juvenile polyps: recurrence in patients with multiple and solitary polyps. Clin Gastroenterol Hepatol 2010;8:795–9. [DOI] [PubMed] [Google Scholar]
- 61.Lieberman DA, Rex DK, Winawer SJ, et al. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012;143:844–57. [DOI] [PubMed] [Google Scholar]