

Abstract
Low success rates during drug development are due, in part, to the difficulty of defining drug mechanism‐of‐action and molecular markers of therapeutic activity. Here, we integrated 199,219 drug sensitivity measurements for 397 unique anti‐cancer drugs with genome‐wide CRISPR loss‐of‐function screens in 484 cell lines to systematically investigate cellular drug mechanism‐of‐action. We observed an enrichment for positive associations between the profile of drug sensitivity and knockout of a drug's nominal target, and by leveraging protein–protein networks, we identified pathways underpinning drug sensitivity. This revealed an unappreciated positive association between mitochondrial E3 ubiquitin–protein ligase MARCH5 dependency and sensitivity to MCL1 inhibitors in breast cancer cell lines. We also estimated drug on‐target and off‐target activity, informing on specificity, potency and toxicity. Linking drug and gene dependency together with genomic data sets uncovered contexts in which molecular networks when perturbed mediate cancer cell loss‐of‐fitness and thereby provide independent and orthogonal evidence of biomarkers for drug development. This study illustrates how integrating cell line drug sensitivity with CRISPR loss‐of‐function screens can elucidate mechanism‐of‐action to advance drug development.
Keywords: CRISPR‐Cas9, drug mechanism‐of‐action, protein networks
Subject Categories: Chemical Biology
This study integrates pharmacological and CRISPR screens in 484 cancer cell lines to systematically investigate anticancer drug mechanism of action, yielding insights into the genetic contexts and cellular networks underpinning drug response.
Introduction
Understanding drug mechanism‐of‐action and evaluating in cellular activity is challenging (Santos et al, 2017), and widespread target promiscuity contributes to low success rates during drug development (Klaeger et al, 2017). For target‐based drug development, a detailed understanding of drug mechanism‐of‐action provides information about specificity and undesirable off‐target activity which could lead to toxicity and a reduced therapeutic window (Lin et al, 2019). Furthermore, molecular biomarkers can be used to monitor drug activity for patient stratification during clinical development.
The cellular activity of a drug is influenced by multiple factors including the affinity and selectivity of the compound for its target(s) and the penetrance of target engagement on cellular phenotypes. An array of biochemical, biophysical, computational and cellular assays are used to investigate drug mechanism‐of‐action (Schenone et al, 2013). For example, protein kinase inhibitors are profiled in vitro for their specificity and potency against panels of purified recombinant protein kinases. While informative, this approach fails to recapitulate the native context of the full‐length protein in cells which could influence true drug activity, nor does it identify potential non‐kinase off‐target effects. Cellular‐based approaches to investigate mechanism‐of‐action include transcriptional profiling following drug treatment of cells, chemical proteomics approaches such as kinobeads (Bantscheff et al, 2007; Médard et al, 2015) and cellular thermal shift assay (Savitski et al, 2014) to measure drug–protein interactions, and multiplexed imaging or flow‐cytometry to measure multiple cellular parameters upon drug treatment (Li et al, 2017; Subramanian et al, 2017; Reinecke et al, 2019). Despite the utility of these different approaches, gaining a full picture of drug mechanism‐of‐action, particularly in cells, remains a challenge and new approaches would be beneficial.
Pharmacological screens (Barretina et al, 2012; Garnett et al, 2012; Iorio et al, 2016; Subramanian et al, 2017; Lee et al, 2018) have been used to profile the activity of hundreds of compounds in highly annotated collections of cancer cell lines with the aim of identifying molecular markers of drug sensitivity to guide clinical development (Cook et al, 2014; Nelson et al, 2015). More recently, CRISPR‐based gene‐editing has enabled the evaluation of highly specific and penetrant gene knockout effects on cell fitness genome‐wide in hundreds of cancer cell lines (Jinek et al, 2012; Shalem et al, 2014; Hart et al, 2015; Meyers et al, 2017; Behan et al, 2019). This has provided rich functional resources to explore cancer vulnerabilities and to identify candidate drug targets (Marcotte et al, 2016; Meyers et al, 2017; Tsherniak et al, 2017; Behan et al, 2019). Parallel integration of gene loss‐of‐function screens with drug response can be used to investigate drug mechanism‐of‐action (Deans et al, 2016; Subramanian et al, 2017; Jost & Weissman, 2018; Wang et al, 2018; Zimmermann et al, 2018; Hustedt et al, 2019a,b).
Here, we integrate recent genome‐wide CRISPR‐Cas9 loss‐of‐function screens with pharmacological data for 397 unique anti‐cancer compounds in 484 cancer cell lines. This provided an unsupervised and comprehensive characterisation of drug mechanism‐of‐action using CRISPR‐Cas9 screens and recapitulated nominal drug targets, gave insights into drug potency and selectivity, and defined cellular networks underpinning drug sensitivity. Illustrating the utility of our approach, we identified a link between mitochondrial ubiquitin ligase MARCH5 in the response to MCL1 inhibitors, especially in breast cancer cell lines. Furthermore, we defined robust pharmacogenomic associations, represented by genetic biomarkers independently supported by drug response and gene fitness measurements. These identify genetic contexts associated with drug‐pathway dependency and provide a more refined set of biomarkers. Collectively, we present an approach to leverage pharmacological and CRISPR screening data to inform on drug in cellular mechanism‐of‐action and thus guide drug development.
Results
Cancer cell line drug sensitivity and gene fitness effects
We analysed data sets from a highly annotated collection of 484 histologically diverse human cancer cell lines (Dataset EV1). Cell line information is available through the Cell Model Passports web portal (http://cellmodelpassports.sanger.ac.uk/) (van der Meer et al, 2019). These have been extensively genetically characterised and utilised for both large‐scale drug sensitivity testing and CRISPR‐Cas9 whole‐genome loss‐of‐function screens (Garnett et al, 2012; Iorio et al, 2016; Meyers et al, 2017; van der Meer et al, 2019; Picco et al, 2019) as part of the Cancer Dependency Map initiative (https://depmap.sanger.ac.uk/). We expanded on published single agent drug sensitivity data (Garnett et al, 2012; Iorio et al, 2016; Lynch et al, 2016; Picco et al, 2019) to consider 199,219 IC50 values for 397 unique cancer drugs (480 drugs including duplicates, Dataset EV2). These encompassed FDA‐approved cancer drugs, drugs in clinical development and investigational compounds, with multiple modes of action, including 24 chemotherapeutic agents and 367 small molecule inhibitors. Drugs considered in this study had a response in at least three cell lines (IC50 lower than half of the maximum screened concentration) and 86% of all possible drug/cell line IC50 measurements have been experimentally evaluated (Fig EV1A, Dataset EV3). Two experimental protocols were used to generate drug sensitivity measurements, named GDSC1 (Iorio et al, 2016) and GDSC2 (Picco et al, 2019) (Fig EV1B). A principal component analysis (PCA) of IC50 values identified a screen specific batch effect associated with principal component (PC) 2 which explained 2.8% of the total variance (Fig EV1C). For this reason, despite the fact that compounds screened with both technologies showed good agreement (n = 66, mean Pearson's R = 0.50), we analysed the measurements of the screens separately. Analysis of the drug response variation across cell lines revealed that PC 1 (28.7% variance captured) was significantly and negatively correlated with cell line growth rate (Pearson's R = −0.51, P‐value = 1.2e‐28), particularly for chemotherapy agents and cell cycle inhibitors (Figs EV1D and E).
Figure EV1. Overview of the drug sensitivity data sets.
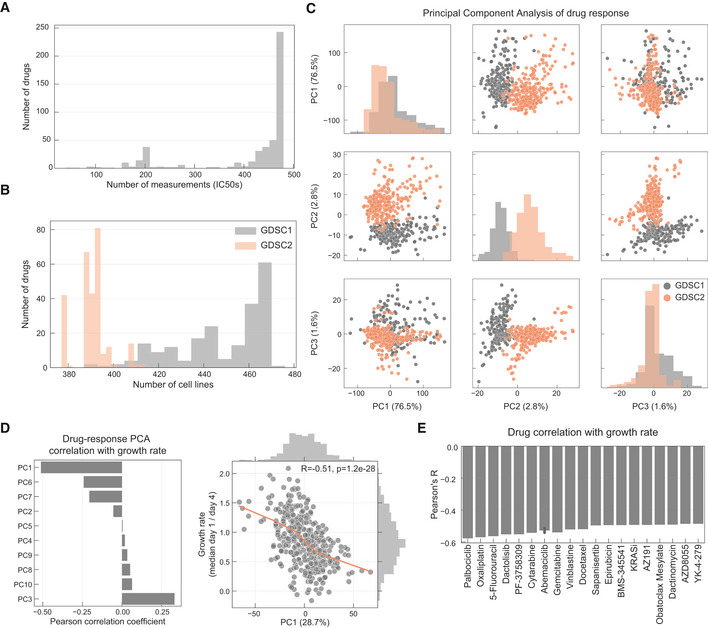
- Histogram of the number of IC50 values measured per drug.
- Number of drugs measured per cell line in each pharmacological data set.
- PCA analysis of the drug response measurements separated by GDSC1 and GDSC2.
- Pearson correlation coefficient between each principal component (PC) and cell lines growth rate.
- Top absolutely correlated drugs with growth rate.
Cell fitness effects for 16,643 gene knockouts have been measured using genome‐wide CRISPR‐Cas9 screens at the Sanger and Broad Institutes (Meyers et al, 2017; Behan et al, 2019; DepMap Broad, 2019) (Dataset EV4). The first PC across the cell lines (6.8% variance explained) separated the two institutes of origin (Fig EV2A), consistent with a comparative analysis performed on an overlapping set of cell lines (Dempster et al, 2019). Growth rate was less significantly associated with CRISPR knockout response (Figs EV2B and C).
Figure EV2. Overview of the CRISPR‐Cas9 data sets.
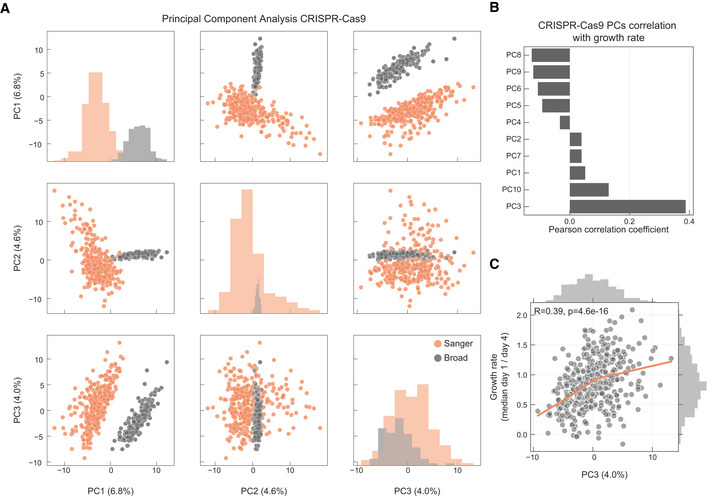
- PCA analysis of the samples in the CRISPR‐Cas9 screens, samples institute of origin is highlighted.
- Correlation coefficients between all top 10 PCs and growth rate.
- Correlation between cell lines growth rate and PC3 (Pearson correlation coefficient reported in the top left).
Gene knockout fitness effects correspond with drug targets
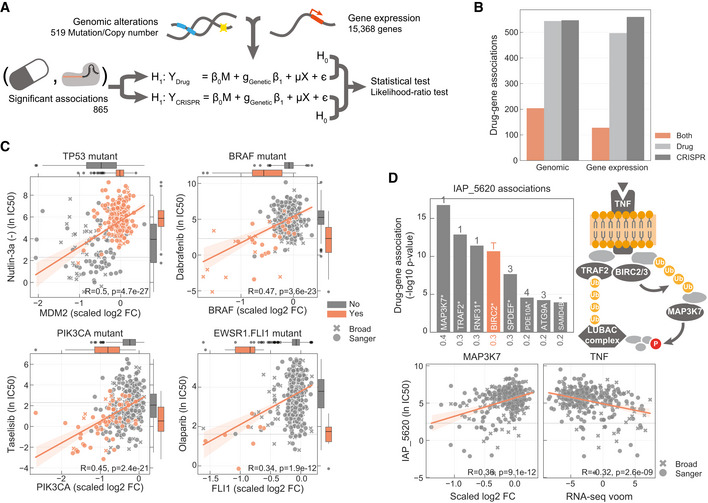
We began by investigating the extent to which drug sensitivity corresponded to CRISPR knockout of drug targets. In an unsupervised way, we systematically searched for all possible associations between the profile of drug sensitivity (n = 480) and gene fitness effects (n = 16,643) across the 484 cell lines (Fig 1A). We expect this to capture a variety of relationships ranging from direct drug–target interactions to more complex associations arising from interactions with regulators of the drug target(s). We tested a total of 7,988,640 single‐feature gene–drug associations using linear mixed regression models. Potential confounding effects such as growth rate, culture conditions, data source and sample structure were considered in the models. We identified 865 significant associations (FDR‐adjusted P‐value < 10%, Dataset EV5) between drug response and gene fitness profiles (Fig 1B), termed hereafter as significant drug–gene pairs. For this analysis, we were able to manually curate the nominal therapeutic target(s) for 94.7% (n = 376) of the anti‐cancer drugs screened (Fig EV3A and Dataset EV1).
Figure 1. Integration of drug and CRISPR gene dependencies in cancer cell lines.
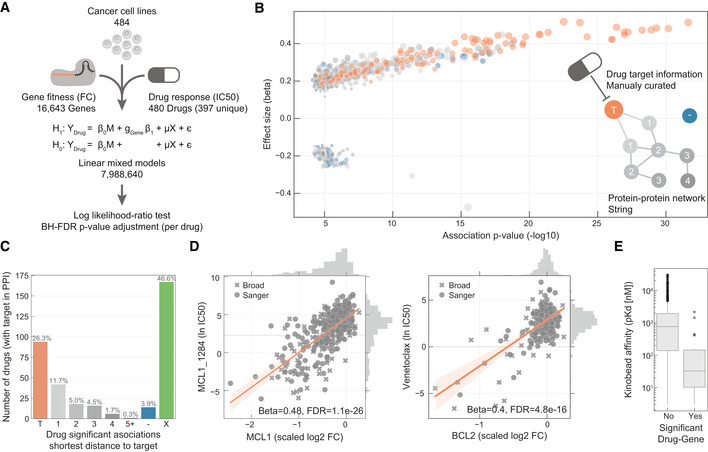
- Linear models were used to integrate drug sensitivity (IC50 values) and gene fitness measurements.
- Volcano plot showing the effect sizes and the P‐value for statistically significant associations, Benjamini–Hochberg false discovery rate (FDR)‐adjusted likelihood‐ratio test P‐value < 10%. Drug–gene associated pairs are coloured according to their shortest distance in a protein–protein interaction network of the gene to any of the nominal target of the drug.
- Percentage of the 358 drugs with significant associations and their shortest distance in the PPI network to the drug nominal targets. T represents drugs that have a significant association with at least one of their canonical targets, “−” represents no link was found, and X are those which have no significant association.
- Examples of the top drug response correlations with target gene fitness. Each point represents an individual cell line. MCL1_1284 and venetoclax are MCL1 and BCL2 selective inhibitors, respectively. Gene fitness log2 fold changes (FC) are scaled by using previously defined sets of essential (median scaled log2 FC = −1) and non‐essential (median scaled log2 FC = 0) genes. Drug response IC50 measurements are represented using the natural log (ln IC50).
- Kinobead affinity is significantly higher (lower pK d) for compounds with a significant association with their target (n = 20, Mann–Whitney P‐value = 3.1e‐07). Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
Figure EV3. Drug response and gene fitness associations.
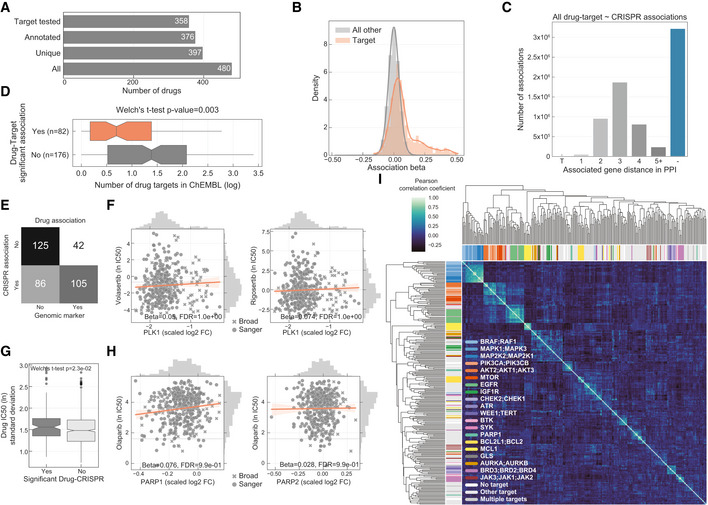
- Total number of drugs utilised in the study and the different levels of information available: “All” represents all the drugs including replicates screened with different technologies (GDSC1 and GDSC2); “Unique” counts the number of unique drug names; “Annotated” shows the number of unique drugs with manual annotation of nominal targets; and “Target tested” represents the number of unique drugs, with target information, for which the target has been knocked‐out in the CRISPR‐Cas9 screens.
- Histogram of the drug–gene associations effect sizes (beta) highlighting drug–target associations.
- Distribution of the shortest path lengths between all the tested drug–gene pairs. For drugs with multiple targets the smallest shortest path of all the targets was taken. T represents the drug target and “−” represents no link was found.
- Distribution of number of drug targets found in an unsupervised way using the ChEMBL database. Drugs are grouped by having significant drug–target associations. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
- Contingency matrix of significant drug associations with CRISPR fold changes and binarised event matrix of genomic features, i.e. mutations and copy number gain or loss.
- PLK1 inhibitors drug response correlation with PLK1 knockout log2 fold change (FC) gene fitness effects. The dashed grey line indicates the dose response highest drug concentration.
- Drug–target associations split by significance (FDR‐adjusted likelihood‐ratio test P‐value < 10%) plotted against the standard deviation of the drug IC50 (ln) measurements of the respective pair (significant “Yes” n = 129, significant “No” n = 684). Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
- Similar to (F), correlation of olaparib drug response and both targets PARP1 and PARP2 gene fitness effects.
- Correlation heatmap of the drug–gene effect size across all the genes. Drugs are coloured according to their targets.
For 26% (n = 94) of the 358 drugs with target annotation and for which the target was knocked‐out with the CRISPR‐Cas9 library, we identified significant drug–gene pairs with their putative targets (Fig 1C). For example, there were strong associations between MCL1 and BCL2 inhibitor sensitivity and their respective gene knockouts (Fig 1D). Notably, drug–gene associations with the drug target had a skewed distribution towards positive effect sizes (Mann–Whitney U‐test P‐value < 1.36e‐105, Fig EV3B) and were amongst the strongest associations (Fig 1B). To investigate this further, we utilised independently acquired kinobead drug–protein affinity measurements for an overlapping set of 64 protein kinase inhibitors which were profiled for their specificity against 202 kinases (Klaeger et al, 2017). Drugs with significant associations with knockout of their target also had stronger affinity to their target in the kinobead assay, providing independent evidence that the strongest drug–gene associations are enriched for targets of the drugs (Fig 1E). Overall, we identified the nominal target of approximately one‐quarter of the drugs tested using orthogonal CRISPR gene fitness screens, and drug targets were amongst the most significant gene–drug associations.
Cellular networks underpinning drug response
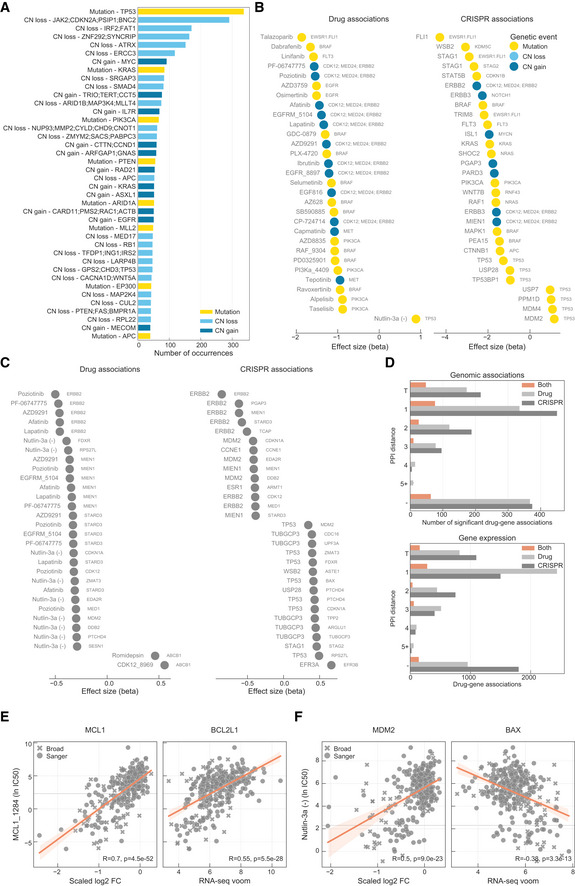
For the remaining drugs (n = 264) which were not significantly associated with the CRISPR loss‐of‐function measurements of their nominal targets (Fig 1C), we reasoned that superimposing the significant drug–gene pairs onto a protein interaction network may shed further insights into the relationship of the associations. We used a protein–protein interaction (PPI) network assembled from the STRING database (Szklarczyk et al, 2017) (10,587 nodes and 205,251 interactions), and for the significant drug–gene pairs calculated the minimal distance between the drug nominal target(s) and the associated gene. Out of the 264 drugs, 76 drugs had a significant association with their target's first neighbour or a protein closely related in the network (1, 2 or 3 PPI distance from drug targets; Fig 1B and C). Thus, despite these drugs not showing significant associations with their nominal targets, CRISPR associations revealed potential mechanisms‐of‐actions which are functionally related to their targets. Taken together, for the 358 drugs with target annotation and covered by the CRISPR‐Cas9 screens, 47.5% (n = 170) had an association with either the target (26.3%) or a functionally related protein (21.2%; Fig 1C).
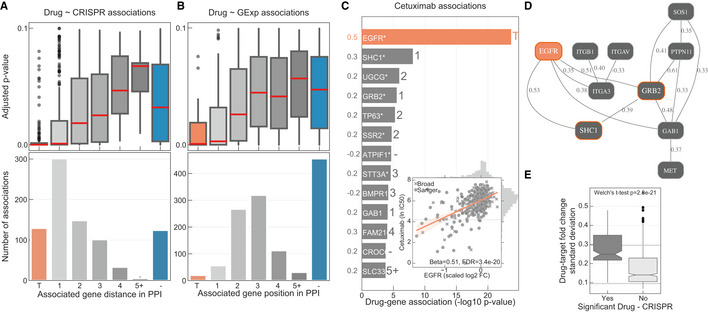
The strongest drug–gene pair associations were between a drug and its canonical target(s) rather than components of the PPI network, and significance decreased (along with the number of associations) as the interaction distance increased (Fig 2A). To exclude the possibility that this observation is biased by the topology of the network, we calculated the length of all the shortest paths between the drug target(s) and their associated genes and confirmed the enrichment of first and second neighbours in significant drug–gene associations (Fig EV3C). In comparison, cell line gene expression identified considerably fewer associations with the PPI neighbours of the drug target (Fig 2B; Dataset EV6). In particular, the number of drugs significantly associated with their targets substantially decreased (n = 17), and significant associations were predominantly found with proteins further away in the PPI network, close to the average length of all paths (l G = 3.9). As an example, MIEN1 gene expression is positively correlated with multiple EGFR and ERBB2 inhibitors, which can be explained, not by a functional relationship, but more likely by genomic co‐localisation with ERBB2 on chromosome 17. Hence, CRISPR‐Cas9 screens are powered to discriminate genes which are likely to be functionally linked to drug response.
Figure 2. Drug response protein–protein networks.
-
ADistribution of the FDR‐adjusted P‐values (top) and count (bottom) of the significant (FDR‐adjusted likelihood‐ratio test P‐value < 10%) drug–gene (CRISPR) associations according to their distance between the gene and corresponding drug targets in the protein–protein interaction network. T represents drugs that have a significant association with at least one of their canonical targets and “−” represents no link was found. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
-
BSimilar to (A), but instead gene expression (GExp) was tested to identify associations with drug response. T represents drugs that have a significant association with at least one of their canonical targets and “−” represents no link was found. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
-
C, D(C), Representative example, i.e. cetuximab—EGFR inhibitor, of the associations and (D), networks that can be obtained from the integrative analysis. Edges in the network are weighted with the Pearson correlation coefficient obtained between the fitness profiles of interacting nodes. For representation purposes only edges with the highest correlation coefficient were represented, R 2 > 0.3. Nodes with orange borders represent significant associations with drug response, cetuximab.
-
EDrug–target associations grouped by statistical significance (FDR‐adjusted likelihood‐ratio test P‐value < 10%) and plotted against the standard deviation of the drug–target CRISPR fold changes (significant “Yes” n = 129, significant “No” n = 684). Upper and lower dashed lines represent the standard deviations of essential and non‐essential genes, respectively. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
To investigate putative regulatory networks for drugs, we weighted the PPI network edges with the correlation between the fitness profiles of the two connected nodes and integrated the resulting weighted network with drug response associations. EGFR inhibitors are the most abundant drug class in our set, and we observed that multiple inhibitors (e.g. cetuximab) showed significant associations with EGFR and known pathway members, for example SHC1 and GRB2 (Scaltriti & Baselga, 2006; Zheng et al, 2013) (Fig 2C). Additionally, the weighted network shows pathway members that have strongly correlated fitness profiles, which are likely functionally related (Wang et al, 2017a,b; Boyle et al, 2018; Pan et al, 2018; Rauscher et al, 2018; Kim et al, 2019). For EGFR inhibitors, these included receptor tyrosine kinase MET and the protein phosphatase PTPN11 (Wang et al, 2017a; Pan et al, 2018) (Fig 2D). Drug–target tailored networks can be used to understand drug mechanism‐of‐action and have the potential to identify resistance mechanisms and thus can be used to identify new alternative drug targets. Collectively, our network analysis demonstrates that CRISPR screens can provide functional insights into drug in cellular activity, extending beyond the direct drug target, into the associated functional network.
Despite our finding that we can illuminate drug functional networks, 46.6% (n = 167) of the tested drugs had no significant drug–gene associations. A number of possible technical and biological factors may underpin this observation. In support of a link between drug selectivity and significant gene–drug associations (Fig 1B and E), drugs with no significant association with their target had in general a higher number of putative targets based on ChEMBL bioactivity profiles (two‐sided Welch's t‐test P‐value = 0.003; Fig EV3D), and drugs with no significant associations were approximately three times less likely to be associated with a genomic biomarker linked to sensitivity (Fig EV3E). Alternative explanations include lower variability in CRISPR fold change measurements for the target of these drugs (Fig 2E). For example, where genetic knockout of an essential gene induces a strong loss‐of‐fitness effect uniformly across all cell lines, whereas a drug has more variable effects likely due to incomplete target inhibition (Fig EV3F). The lack of variability was much less pronounced in the drug sensitivity measurements since we only considered drugs which showed a minimal level of activity, i.e. IC50 lower than half of the maximum screened concentration (Fig EV3G). Inhibition of a protein is also intrinsically different than a knockout, as observed for PARP inhibitors whose activity is mediated through formation of cytotoxic PARP‐DNA complexes, whereas PARP knockout has little or no effect on cell fitness (Gill et al, 2015; Murai & Pommier, 2015; Antolin et al, 2020) (Fig EV3H). Similarly, redundancy of gene paralogs when using single‐gene CRISPR knockout may confound comparisons with drugs that target multiple paralogs (preprint: Dede et al, 2020). Thus, although the reasons may vary for each drug, the absence of an association between drug sensitivity and CRISPR loss‐of‐function effects could warrant further investigation into drug mechanism‐of‐action to understand possible underlying factors, such as low potency, alternative molecular mechanisms, or polypharmacology.
Cancer drug mechanism‐of‐action
Next, we set out to investigate in detail some of the strongest drug sensitivity and CRISPR gene fitness associations (Dataset EV5). Strikingly, 46 of the top 50 strongly associated drugs had significant associations with their nominal target and with known functionally related genes. Some of the strongest associations were between MCL1 inhibitors and their target (Fig 3—MCL1 and BLC2 inhibitors panel), including AZD5991 which is currently in clinical trials for treatment of haematologic cancers (Hird et al, 2017). Additionally, for several insulin‐like growth factor 1 receptor (IGFR1) inhibitors the association with the target was recapitulated. Moreover, significant associations with proprotein convertase furin were observed, supporting the genetic association that IGFR1 is a furin substrate. Moreover, increased levels of furin are associated with increased levels of processed IGFR1 and worse prognosis in several cancers (Thomas, 2002).
Figure 3. Representative examples of top most significantly associated drug classes.
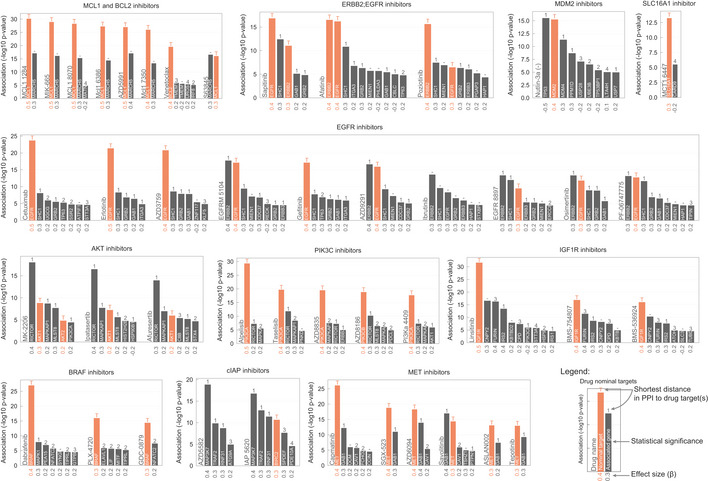
Each bar plot group represents a unique drug where genes are ranked by statistical significance (likelihood‐ratio test P‐value) of their association. Effect sizes of the associations are reported under the bars along the x axis. Shortest distance (number of interactions) in a protein–protein interaction network between the gene and the drug nominal target(s) is represented on the top of the bars, where T and orange bar represent the target and ““−” represents no link was found.
The clinical development of kinase inhibitors is hampered by poor selectivity because of the conserved structural features of the commonly targeted kinase domain, which could lead to undesirable off‐target activity (Klaeger et al, 2017). Furthermore, some kinases have multiple isoforms with non‐redundant roles. For example, isoform‐selective PI3K inhibitors have been developed in part to reduce toxicity and increase the therapeutic window (Thorpe et al, 2015). Interestingly, several PI3K inhibitors had strong associations with only one gene encoding a single isoform (Fig 3—PIK3C inhibitors panel). This together with the increased kinobead binding affinity of significant associations (Fig 1E) suggests these are isoform‐selective compounds. For example, alpelisib was associated with PIK3CA, consistent with its development as an alpha‐isoform‐selective compound (Thorpe et al, 2015), whereas AZD8186 was only associated with PIK3CB confirming its beta‐selectivity. Conversely, two pan‐PI3K inhibitors (buparlisib and omipalisib) displayed no significant association with any PI3K isoform (Dataset EV5), consistent with its lack of isoform specificity and potential polypharmacology. Interestingly, MTOR and pan‐PI3K inhibitor, dactolisib, had significant associations with RPTOR and MTOR but none with PI3K isoforms (Dataset EV5), consistent with recently reported greater specificity for inhibition of the MTOR complex (Reinecke et al, 2019). Similarly, we observed that selective EGFR inhibitors cetuximab, erlotinib and gefitinib (Fig 3—EGFR inhibitors panel) were associated with EGFR but not ERBB2, whereas sapatinib, afatinib and poziotinib (Fig 3—ERBB2; EGFR inhibitors panel) were all associated with both EGFR and ERBB2.
Our analysis can also provide insights into possible off‐target activity of drugs. Unsupervised clustering of the drug–gene associations effect sizes (betas) revealed classes of inhibitors with similar targets and mechanism‐of‐action (Fig EV3I). Of note, BTK inhibitor, ibrutinib, clustered with EGFR inhibitors and displayed significant associations with EGFR and ERBB2 gene fitness (Fig 3—EGFR inhibitors panel). This is consistent with recent findings that ibrutinib covalently binds to and inhibits EGFR (Lee et al, 2018) and is also supported by kinobead measurements (Klaeger et al, 2017). Additionally, 24 compounds have significant associations with genes identified as essential core fitness (Behan et al, 2019) across multiple cancer types, indicating an increased risk of cellular toxicity. Out of these, two compounds, PD0166285 and CCT244747, have significant associations with their nominal target (PKMYT1 and CHEK1/WEE1) and the remaining compounds (n = 22) are correlated with core fitness proteins closely connected in the PPI network.
A functional link between MARCH5 and MCL1 inhibitor sensitivity
Seven out of nine unique inhibitors of the anti‐apoptotic BCL2 family member myeloid cell leukaemia 1 (MCL1) were strongly and nearly exclusively associated with their putative target, suggesting these are potent and specific compounds in cells (Fig 4A). MCL1 is frequently amplified in human cancers (Beroukhim et al, 2010) and associated with chemotherapeutic resistance and relapse (Wuillème‐Toumi et al, 2005; Wei et al, 2006). MCL1 is a negative regulator of the mitochondrial apoptotic pathway, regulating BAX/BAK1 which co‐localise with Drp1/Fis1 in the mitochondria outer membrane and control mitochondrial fragmentation and cytochrome c release, both of which are important for inducing apoptosis (Youle & Karbowski, 2005; Mojsa et al, 2014; Morciano et al, 2016). Interestingly, knockout of a key regulator of mitochondrial fission, mitochondrial E3 ubiquitin‐protein ligase MARCH5 (Karbowski et al, 2007 ), is significantly associated with MCL1 inhibitors sensitivity (Fig EV4A) and positively correlated with MCL1 gene fitness, suggesting a functional relationship (Fig 4B). Correlation between MCL1 and MARCH5 fitness profiles shows that cell lines dependent on MARCH5 are also dependent on MCL1, while the inverse is not necessarily true with a subgroup of cell lines dependent on MCL1 but not on MARCH5 (Fig 4B). Cell lines independently dependent on both gene products have increased sensitivity to MCL1 inhibitors (Fig EV4B). This is particularly marked in breast carcinoma cancer cell lines, with MCL1‐ and MARCH5‐dependent cells having similar sensitivity to haematologic cancer cell lines (acute myeloid leukaemia), where MCL1 inhibitors are in clinical development (Fig 4C).
Figure 4. MCL1 inhibitors associations.
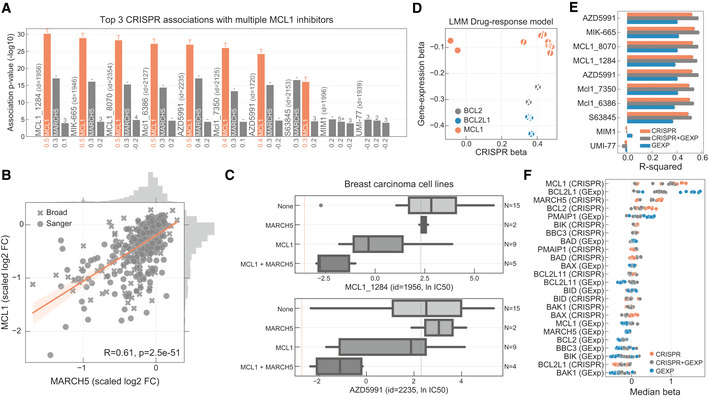
- Top three CRISPR associations with all MCL1 inhibitors screened. Each bar represents the likelihood‐ratio test P‐value of each drug–gene (CRISPR) association and effect sizes reported under the bars. Shortest distance (number of interactions) in a protein–protein interaction network between the gene and the drug nominal target(s) is represented on the top of the bars, where T and orange bar represent the target and “−” represents no link was found.
- Association between the gene fitness profiles of MCL1 and MARCH5.
- Stratification of the MCL1 inhibitor sensitivity according to the essentiality profile of MCL1 and MARCH5, where MCL1 + MARCH5 represents a cell line that is independently dependent on both genes. Dashed orange line (left) represents the mean IC50 in acute myeloid leukaemia cell lines. Grey dashed line (right) represents the maximum concentration used in the dosage response curve. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
- BCL2, BCL2L1 and MCL1 inhibitors and the respective association with their targets, on the x axis with CRISPR gene fitness and on the y axis with gene expression. The statistical significance (FDR‐adjusted likelihood‐ratio test P‐value < 10%) of the association is represented with a backward slash for CRISPR and forward slash for GEXP.
- Regularised multilinear regression to predict drug response of all MCL1 inhibitors using gene expression, fitness or both of known regulators of the BCL2 family and MARCH5. Predictive performance is estimated using R 2 metric represented in the x axis.
- Effect size of each feature used in each MCL1 inhibitor model.
Figure EV4. MCL1 inhibitors.
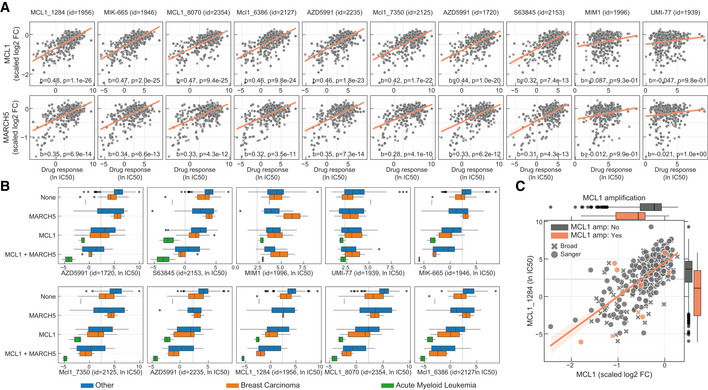
- Correlation of all MCL1 inhibitor IC50 values against MCL1 and MARCH5 gene fitness profiles. Effect sizes (b) and FDR (P) of the association are reported on the bottom.
- Stratification of the MCL1 inhibitors drug response measurements according to the cell line dependency on MARCH5 and/or MCL1. Gene vulnerabilities are independent from each other, meaning knockouts were introduced independently and not at the same time. Responses are then split according to the cancer type of the cell lines. Number of measurements per boxplot are: acute myeloid leukaemia (MCL1 = 3, MCL1 + MARCH5 = 2, None = 1), breast carcinoma (MARCH5 = 2, MCL1 = 9, MCL1 + MARCH5 = 4, None = 14) and Other (MARCH5 = 9, MCL1 = 52, MCL1 + MARCH5 = 21, None = 236). Vulnerable cell lines to MARCH5 and MCL1 knockout were defined as those with a depletion of at least 50% of that visible for essential genes on the particular cell line (scaled log2 fold change < −0.5). Grey dashed line represents the maximum concentration used in the dosage response curve of the respective drug. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
- Representative example of a MCL1 inhibitor and their relation with MCL1 gene fitness, with cell lines containing copy number amplification of MCL1 highlighted in orange. Copy number amplified cells were defined taking into consideration their ploidy status, cells with (ploidy ≤ 2.7 and copy number ≥ 5) or (ploidy > 2.7 and copy number ≥ 9) were considered as having MCL1 amplified. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
We investigated the molecular mechanisms underlying the responses of MCL1 inhibitors. MCL1 copy number and gene expression alone are not a good predictor of MCL1 inhibitors sensitivity (Figs 4D and EV4C). This is in contrast to BCL2 and BCL2L1 inhibitors, where their target gene expression is significantly correlated with drug sensitivity (Fig 4D). Next, we used multilinear regression models to predict sensitivity to each MCL1 inhibitor using gene fitness and/or gene expression of known regulators of MCL1 (e.g. BCL2, BCL2L1, BAX) (Czabotar et al, 2014) and MARCH5. For two MCL1 inhibitors, MIM1 and UMI‐77, the trained models performed poorly, likely due to lack of in cellular activity of these compounds as suggested by their drug response poor correlation with MCL1 gene essentiality profiles (Figs 4A and EV4A). For the remaining seven MCL1 inhibitors, drug response was well predicted (CRISPR + GEXP mean R 2 = 0.55). Models trained with only CRISPR displayed overall better predictions compared to models only trained with gene expression, and models trained with both data types out‐performed all others (Fig 4E). As expected, MCL1 fitness effect was the most predictive feature, followed by decreased BCL2L1 expression and increased MARCH5 essentiality (Fig 4F). No genomic feature, mutation or copy number alterations correlated significantly with MCL1 inhibitors response, including MCL1 amplifications (Fig EV4C), likely a consequence of the strong post‐transcriptional regulation and short half‐life of the MCL1 protein.
Altogether, these results highlight a functional link between MARCH5 and sensitivity to MCL1 inhibitors. This is consistent with recent reports of a synthetic‐lethal interaction between MARCH5 and the MCL1 negative regulator BCL2L1 (DeWeirdt et al, 2020), and MARCH5‐dependent degradation of the MCL1/NOXA complex (Djajawi et al, 2020; Haschka et al, 2020). With further investigation, the link between MCL1 and MARCH5 could shed light on the mechanism‐of‐action of MCL1 inhibitors and the development of stratification approaches in solid tumours, such as breast carcinomas.
Robust molecular markers of drug sensitivity networks
The identification of molecular biomarkers of drug sensitivity is fundamental to guide drug development. We hypothesised that molecular biomarkers independently linked with both drug response and gene fitness would be of particularly high value—termed robust pharmacogenomic biomarkers. To identify these, we used the set of significant drug–gene pairs (n = 865) and we searched independently for significant associations between each measurement type (drug response or gene fitness) and 519 genomic (mutations and copy number alterations) and 15,368 gene expression features (Figs 5A and EV5A) (Garnett et al, 2012; Iorio et al, 2016; Garcia‐Alonso et al, 2018). This analysis recapitulated established genomic and expression biomarkers of either drug sensitivity or gene fitness effects in cancer cells (Figs EV5B and C). A total of 224 and 679 robust pharmacogenomic associations (FDR < 10%) were identified with genomic (Dataset EV7) and gene expression features (Dataset EV8), respectively. Overall, 30.6% (265 of 865) of drug–gene pairs have at least one robust molecular marker that correlated significantly with both drug response and gene fitness (Fig 5B). The number of robust pharmacogenomic biomarkers was smaller than the number of biomarkers associated with only one type of measurement, likely due to the stringent requirement for an independent association with both drug sensitivity and gene fitness effects.
Figure 5. Robust pharmacological associations.
- Diagram representing how genomic and gene expression data sets are integrated to identify significant associations with drug–gene pairs that were previously found to be significantly correlated.
- Number of drug–gene pairs with at least one significant association with drug response, gene fitness or both, considering either genomic or gene expression profiles.
- Canonical examples of robust pharmacological associations. Box‐and‐whisker plots show 1.5× interquartile ranges and 5–95th percentiles, centres indicate medians.
- Representative example of a BIRC2/BIRC3 inhibitor, IAP_5620, showing the significant associations (FDR‐adjusted likelihood‐ratio test P‐value < 10%) with CRISPR gene fitness profiles and their location in a representation of the TNF pathway. Bar plot is defined similarly to Fig 3.
Figure EV5. Robust pharmacological associations.
-
AMost frequent genomic alterations across the cancer cell lines.
-
B, CMost significant associations between (B), genomic features and (C), gene expression profiles with drug response and gene fitness.
-
DNumber of significant drug–gene pairs across the different types of interactions. Drug–gene pairs were categorised considering the shortest path length between the drug target(s) and the associated gene. T represents the drug target and “−” represents no link was found.
-
ERobust pharmacological association between the expression of BCL2L1 and the significantly correlated pair of MCL1_1284 drug and MCL1 gene fitness profile.
-
FSimilarly to (E), but instead it represents a robust pharmacological association between BAX and MDM2 and Nutlin‐3a.
From the subset of 129 drug–gene pair associations that were linked by the drug target, 50.4% (n = 65) had one or more robust pharmacogenomic associations (Fig EV5D). Most of these were established dependencies of cancer cells, including Nutlin‐3a sensitivity and MDM2 gene fitness independently associated with TP53 mutation status; BRAF and PIK3CA mutation induced drug and CRISPR dependency; olaparib sensitivity mediated by the presence of EWSR1‐FLI1 fusion, also recapitulated by FLI1 essentiality profile; MCL1 inhibitor and gene fitness associations with BCL2L1 expression, and Nutlin‐3a sensitivity and MDM2 gene fitness associated with BAX expression (Figs 5C and EV5E and F). Similarly, of the 413 significant gene–drug pairs closely related within the PPI network (≤ 3 interactions from the drug target), we identified robust pharmacogenomic associations for 29.5% (n = 122; Fig EV5D), enabling the discovery of cellular contexts where drug response networks are important. For example, we identified increased tumour necrosis factor (TNF) expression as a robust pharmacogenomic marker for drugs targeting the downstream cellular inhibitor of apoptosis (cIAP) proteins BIRC2 and BIRC3 (e.g. IAP_5620), and based on CRISPR dependency data, for multiple members of the cIAP pathway, including BIRC2, MAP3K7 and RNF31 (Beug et al, 2012) (Fig 5D).
Discussion
Understanding drug mechanism‐of‐action and the biological pathways underpinning drug response is an important step in preclinical studies. Here, we demonstrate how the integration of drug sensitivity and CRISPR‐Cas9 gene fitness data can be used to inform on multiple aspects of drug mechanism in cells, including drug specificity, isoform selectivity and potency. Our analysis recapitulated drug targets for approximately a quarter of the drugs tested and for approximately another quarter revealed associations enriched for proteins closely related with the drug target(s). Critically, the strength of these associations reflects specificity and polypharmacology of the cancer drugs, with highly selective and potent drugs showing the strongest associations with their nominal target. Significant drug–gene associations define networks of protein interactions that are functionally related with drug targets and underpin drug response. This revealed a previously unappreciated interaction between MARCH5 and MCL1 inhibitors, with potential utility to derive predictive models of MCL1 inhibitor response across multiple cancer types, and particularly in solid tumours such as breast carcinomas. Robust pharmacogenomic biomarkers leveraged both data sets to provide refined biomarkers that are correlated with both drug response and biological networks. Interestingly, the networks we have defined can provide alternative targets that are functionally related with the drug target and mediate similar effects on cell fitness, potentially providing strategies for combination therapies to limit therapy resistance.
Preclinical biomarker development is an important step in drug discovery and is associated with increased success rates during clinical development (Nelson et al, 2015). Traditionally, this has been performed by building predictive models of drug response using mutation, copy number and gene expression (Iorio et al, 2016; Tsherniak et al, 2017). Here we extended this approach and propose what we term as robust pharmacogenomic association—a drug response and gene fitness pair that are significantly correlated and are also both significantly related to the same molecular biomarker. This approach gives greater confidence in molecular biomarkers identified, since they are recapitulated using data from two orthogonal assays and provides markers at the level of the network. In addition, by focusing only on drugs involved in significant gene–drug pairs, we enrich for drugs most likely to have greater specificity, and thereby better enabling biomarker discovery.
Nearly half of the drugs did not have a significant association with gene fitness effects and may warrant further investigation. Possible explanations for this include the following: (i) drug polypharmacology which is difficult to deconvolute using single‐gene knockout data; (ii) intrinsic difference between protein inhibition and knockout; (iii) a dosage‐dependent response leading to incomplete inhibition of the drug target; (iv) functional redundancy between protein isoforms resulting in less penetrant effects with gene knockout; and (v) technical limitations of CRISPR‐Cas9 screens such as the introduction of DNA double‐strand breaks and sgRNA on‐target and off‐target effects variability across cancer cell lines. We expect that some of these issues can be addressed by expanding this analysis to integrate other types of functional genomic screens, such as CRISPR inhibition, which might mimic drug inhibition more closely and does not introduce double‐strand breaks.
We used CRISPR loss‐of‐function data sets to comprehensively study drug mechanism‐of‐action in cancer cells. CRISPR screening data are now available for many cell lines, and the profiling of compounds across cell line panels is already regularly performed, and so this approach could become a routine step during drug development. In particular, it is likely to have utility during the hit‐to‐lead or lead optimisation stages of drug development to select compound series with optimal potency and selectivity. It could also be useful for novel and uncharacterised cell active compounds, particularly if integrated with orthogonal experimental (e.g. such as kinobead assays) or computational approaches (e.g. drug pocket binding) to find direct targets and interrogate mechanisms‐of‐action. The utility of this approach is likely to expand as the availability of CRISPR knockout screening data, and other data sets such as CRISPR activation and inhibition, increases across ever larger collections of highly annotated cancer cell models. In conclusion, this study illustrates a new approach for investigating cellular drug mechanism‐of‐action that can be applied to multiple critical aspects of drug development.
Materials and Methods
Reagents and Tools table
| Software | |
|---|---|
| Python v3.7.3 | https://www.python.org/downloads/release/python-370/ |
| Matplotlib v3.1.0 | https://matplotlib.org/ (Hunter, 2007) |
| Seaborn v0.10.0 | https://seaborn.pydata.org/ (Waskom et al, 2020) |
| Limix v3.0.3 | https://docs.limix.io/ (preprint: Lippert et al, 2014; Casale et al, 2017) |
| Numpy v1.18.1 | https://numpy.org/ (van der Walt et al, 2011) |
| Scipy v1.4.1 | https://www.scipy.org/ (Virtanen et al, 2020) |
| Pandas v1.0.1 | https://pandas.pydata.org/ (McKinney, 2010) |
| Scikit‐learn v0.21.2 | https://scikit-learn.org/ (Pedregosa et al, 2011) |
| Pydot v1.4.1 | https://pypi.org/project/pydot/ |
| Python‐igraph v0.7.1 | https://igraph.org/python/ (Csardi & Nepusz, 2006) |
| Crispy (Cy) v0.4.6 | https://pypi.org/project/cy/ (Gonçalves et al, 2019) |
Methods and Protocols
Cancer cell lines panel
The 484 cancer cell lines used in this manuscript have been compiled from publicly available repositories as well as private collections and maintained following the supplier guidelines. STR and SNP fingerprints were used to ensure cell lines selected were genetically unique and matched those in public repositories (http://cancer.sanger.ac.uk/cell_lines/download). Detailed cell line model information is available through Cell Model Passports database (https://cellmodelpassports.sanger.ac.uk/) (van der Meer et al, 2019). Cell lines growth rate is represented as the ratio between the mean of the untreated negative controls measured at day 1 (time of drug treatment) and the mean of the DMSO treated negative controls at day 4 (72 h post‐drug treatment).
High‐throughput drug sensitivity
Experimental details of both GDSC1 and GDSC2 screens can be found in the Genomics of Drug Sensitivity in Cancer (GDSC) project (www.cancerRxgene.org) (Yang et al, 2013). Cell viability and dose response curve fitting models were previously described in detail (Iorio et al, 2016; Vis et al, 2016). Maximum screened drug concentrations (μM) are provided in Dataset EV1. Each compound was measured on average across 393 cell lines rendering a nearly complete matrix with only 14.2% missing values. All considered compounds displayed an IC50 lower than half of the maximum screened concentration in at least three cell lines. This ensures the compounds display an informative profile in at least a subset of cell lines. Drug nominal oncology target(s) annotation was manually curated from literature (Dataset EV1).
Genome‐wide CRISPR‐Cas9 dropout screens
The CRISPR‐Cas9 screens for the 484 cancer cell lines considered in this study (Dataset EV4) were assembled from two distinct projects, 320 were generated as part of Sanger DepMap Project Score (Behan et al, 2019) and 164 from the Broad DepMap version 19Q3 (Meyers et al, 2017; DepMap, 2019). Only cell lines that passed quality control filtering similarly to Behan et al (2019) and with matched drug response measurements were considered. Different CRISPR‐Cas9 sgRNA libraries were used in each project (Koike‐Yusa et al, 2014; Doench et al, 2016; Tzelepis et al, 2016). Consequently, library‐specific effects were present (Dempster et al, 2019) (Fig EV2A) which hampers averaging of cell lines that were screened in both data sets. Thus, for the overlapping cell lines only data from Sanger DepMap Project Score was used. This also minimises potential cell line specific differences, for example due to genetic drift (Ben‐David et al, 2018), and thereby increasing concordance with the drug response data set also generated at the Wellcome Sanger Institute. Fold changes (log2) were estimated comparing samples with the respective control plasmid. Gene‐independent deleterious effects induced by copy number amplifications in CRISPR‐Cas9 screens (Aguirre et al, 2016; Munoz et al, 2016; Gonçalves et al, 2019) were corrected on a per sample basis using the unsupervised method CRISPRcleanR (Iorio et al, 2018). Replicates were mean averaged, and gene level fold changes were estimated by taking the mean of all the mapping sgRNAs. Gene level fold changes were quantile normalised per sample and then median scaled using previously defined lists of cancer cell lines essential and non‐essential genes (Hart et al, 2015); thus, essential genes have a median log2 fold change of −1 and non‐essential genes a median log2 fold change of 0. Only overlapping genes between the two libraries were considered, thus generating a full matrix of 16,643 genes across the 484 cell lines. A cell line was considered dependent on a gene if the knockout had a log2 fold change of at least 50% of that expected of essential genes (scaled log2 fold change < −0.5).
PCA of drug sensitivity and gene fitness
Principal component analysis was performed using scikit‐learn (Pedregosa et al, 2011) and the sklearn.decomposition.PCA class with default parameters and the number of components (n_components) set to 10. For the drug response data set, and only for the PCA analysis, missing values of each drug were imputed using the drug mean IC50 response across the rest of the cell lines. Imputation was not required for the CRISPR‐Cas9 data set since the matrix had no missing values.
Drug response linear mixed model associations
Associations between drug response and gene fitness scores were performed using an efficient implementation of mixed‐effect linear models available in the Limix Python module (preprint: Lippert et al, 2014; Casale et al, 2017). We considered the following covariates in the model: (i) binary variables indicating the institute of origin of the cell line CRISPR‐Cas9 screen; (ii) principal component 1 of the drug response data set which is a correlative of cell lines growth rate; and (iii) growing conditions (adherent, suspension or semi‐adherent) represented as binary variables. Additionally, gene fitness similarity matrix of the samples is considered as random effects in the model to account for potential sample structure. Taken together, we fitted the following mixed linear regression model for each drug–gene pair:
| (1) |
where d represents a vector of the drug response IC50 values across the cell lines; M is the matrix of covariates; β 0 is the vector of effect sizes; e is the vector of gene CRISPR‐Cas9 log2 fold changes and β 1 the effect size; X the similarity matrix based on the CRISPR‐Cas9 gene fitness measurements; μ is the random effects; ε is the general noise term. For each drug, cell lines with missing values were dropped from the fit.
We statistically assessed the significance of each association by performing likelihood‐ratio tests between the alternative model () and the null model which excludes the CRISPR gene fitness scores vector e and its parameter . The parameter inference is performed using maximum likelihood estimation:
| (2) |
And the P‐value of the association is defined by:
| (3) |
We tested all the single‐feature pairwise associations between the 480 compounds and the 16,643 genes, making a total of 7,988,640 tested associations. P‐value adjustment for multiple testing was performed per drug using the Benjamini–Hochberg false discovery rate (FDR). Contrary to performing the adjustment across all tests, per drug correction has the following benefits: (i) associations assembled from the different screening platforms (GDSC1 and GDSC2) are kept separate hence not biasing for measurement type; and (ii) drugs with responses across larger subsets of cancer cell lines, for example Nutlin‐3a response across TP53 wild‐type cell lines, display stronger associations than most drugs; thus, correcting across all drugs would retain more associations from these drugs at a specific error rate, i.e. 10%, compared to the rest.
Protein–protein interaction network
We assembled from the STRING database (Szklarczyk et al, 2017) a high confidence undirected protein–protein interaction network. We only consider interactions with a combined confidence score higher than 900. STRING identifiers were converted to HUGO gene symbols, and those not mapping or with multiple mappings were removed. Using igraph Python wrapper (Csardi & Nepusz, 2006), the network was simplified by removing unconnected nodes, self‐loops and duplicated edges, leaving a total of 10,587 nodes and 205,251 interactions. A weighted version of the network was also assembled by correlating the gene fitness profiles of the connected nodes. Network nodes, and corresponding edges, that were not covered by the CRISPR‐Cas9 screens were removed, making a total of 9,595 nodes and 172,584 weighted interactions.
Robust pharmacogenomic associations
Robust pharmacological associations were estimated similarly to the previous associations, but in this case only drug–gene pairs that are significantly correlated were considered to test associations with the genomic features (binarised copy number and mutation status (Iorio et al, 2016)) and gene expression profiles (RNA‐seq voom (Law et al, 2014) transformed RPKMs (Garcia‐Alonso et al, 2018)). A robust pharmacogenomic association is defined as follows: (i) a drug–gene pair whose drug sensitivity and gene fitness is significantly correlated, and (ii) genomic alteration or gene expression profile is significantly correlated with both drug response and gene fitness. Log‐ratio test P‐values are independently estimated for drug response and gene fitness measurements and corrected per drug–gene. Drug–gene pairs where both are associated with a genomic or gene expression feature with an FDR lower than 10% are called robust pharmacogenomic associations ([Link], [Link]).
Predictive models of drug response of MCL1 inhibitors
L2‐regularised linear regression models to predict MCL1 inhibitors drug response were trained using gene fitness, gene expression measurements or both of canonical regulators of MCL1, namely MARCH5, MCL1, BCL2, BCL2L1, BCL2L11, PMAIP1, BAX, BAK1, BBC3, BID, BIK, BAD. For the 9 MCL1 inhibitors considered in this study, predictive models of drug response measurements were trained using Ridge regressions with an internal cross‐validation optimisation of the regularisation parameter, implemented in Sklearn with RidgeCV class (Pedregosa et al, 2011). Additionally, drug response measurements are split randomly 1,000 times, where 70% of the measurements are for training the model and 30% are left out as a test set. Model's performance is quantified using the R 2 metric on the test set, comparing the predicted vs the observed drug response measurements.
Unsupervised drug target annotation using ChEMBL bioactivity profiles
Curated therapeutic targets were extracted from the ChEMBL database (version 25) (Mendez et al, 2019) for each drug, where available. Additional targets (including potential off‐targets) were identified from the in vitro bioactivity data in ChEMBL where the “target type” description was one of the following: “single protein,” “protein complex,” “protein complex group,” “protein–protein interaction” or “protein family.” Only assays where the target organism corresponds to “Homo sapiens” were considered.
In order to define whether a drug is considered active in an in vitro assay, only those situations where a pChEMBL value was defined were selected for inclusion in the subsequent analysis. The pChEMBL value is aimed to harmonise all the comparable measures of half‐maximal responses (molar IC50, XC50, EC50, AC50, Ki, Kd, potency and ED50) on a negative logarithmic scale, and it is calculated only when the standard relation in an assay is known to be “=”. Activity thresholds were defined according to the target protein family, based on the Illuminating the Druggable Genome consortium (IDG https://druggablegenome.net/ProteinFam) as follows: Kinases: ≤ 30 nM; GPCRs: ≤ 100 nM; Nuclear Receptors: ≤ 100 nM; Ion Channels: ≤ 10 μM; Others: ≤ 1 μM.
Author contributions
Conceptualisation: EG and MJG; Formal analysis: EG; Data curation: EG, CP, DvdM, ABa, HL, JTL, BS, CC, FI, SF and MJG; Drug response acquisition and processing: DvdM, TM, ABa, LR, WY, EL, JH, CT, CH, IM, FT, JM, ABe and HL; Drug annotation: EG, AS‐C, GP, FMB, PJ, EAC, ARL, CC and MJG; Writing original draft preparation: EG and MJG; Writing, reviewing and editing: all authors; Visualisation: EG; Supervision: ABa, AL, JTL, BS, CC, FI, SF and MJG; Funding acquisition: SF and MJG.
Conflict of interest
This work was funded in part by AstraZeneca. M.J.G. receives funding from AstraZeneca. M.J.G., F.I. and A.R.L. receive funding from Open Targets, a public–private initiative involving academia and industry. F.I. performs consulting for the CRUK‐AstraZeneca Functional Genomics Centre. J.T.L., B.S., C.C. and S.F. are current employees of AstraZeneca and hold stock in AstraZeneca.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Review Process File
Acknowledgements
We would like to thank all members of the Translational Cancer Genomics team who provided useful insight, including Matthew Coelho for providing careful reading of the manuscript. We are grateful for the diligent contributions of Jon Winter‐Holt in terms of compound identification and data clearance, Pedro Beltrao for helpful discussions and Danilo Horta for support integrating the Limix package. Work in M.J.G laboratory was funded by Wellcome (206194) and AstraZeneca.
Mol Syst Biol. (2020) 16: e9405
Data availability
Source code, analysis reports and Jupyter notebooks are publicly available in GitHub https://github.com/EmanuelGoncalves/dtrace. Drug response and CRISPR‐Cas9 measurements are included in [Link], [Link], respectively, and available at FigShare https://doi.org/10.6084/m9.figshare.10338413.v1.
References
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C‐Z, Ben‐David U, Cook A, Ha G, Harrington WF, Doshi MB et al (2016) Genomic copy number dictates a gene‐independent cell response to CRISPR/Cas9 targeting. Cancer Discov 6: 914–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antolin AA, Ameratunga M, Banerji U, Clarke PA, Workman P, Al‐Lazikani B (2020) The kinase polypharmacology landscape of clinical PARP inhibitors. Sci Rep 10: 2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C et al (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol 25: 1035–1044 [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D et al (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M et al (2019) Prioritization of cancer therapeutic targets using CRISPR‐Cas9 screens. Nature 568: 511–516 [DOI] [PubMed] [Google Scholar]
- Ben‐David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R et al (2018) Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M et al (2010) The landscape of somatic copy‐number alteration across human cancers. Nature 463: 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beug ST, Cheung HH, LaCasse EC, Korneluk RG (2012) Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol 33: 535–545 [DOI] [PubMed] [Google Scholar]
- Boyle EA, Pritchard JK, Greenleaf WJ (2018) High‐resolution mapping of cancer cell networks using co‐functional interactions. Mol Syst Biol 14: e8594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale FP, Horta D, Rakitsch B, Stegle O (2017) Joint genetic analysis using variant sets reveals polygenic gene‐context interactions. PLoS Genet 13: e1006693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G, Pangalos MN (2014) Lessons learned from the fate of AstraZeneca's drug pipeline: a five‐dimensional framework. Nat Rev Drug Discov 13: 419–431 [DOI] [PubMed] [Google Scholar]
- Csardi G, Nepusz T (2006) The Igraph software package for complex network research. Int J 1696: 1–9 [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL‐2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15: 49–63 [DOI] [PubMed] [Google Scholar]
- Deans RM, Morgens DW, Ökesli A, Pillay S, Horlbeck MA, Kampmann M, Gilbert LA, Li A, Mateo R, Smith M et al (2016) Parallel shRNA and CRISPR‐Cas9 screens enable antiviral drug target identification. Nat Chem Biol 12: 361–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dede M, Kim E, Hart T (2020) Biases and blind‐spots in genome‐wide CRISPR knockout screens. bioRxiv 10.1101/2020.01.16.909606 [PREPRINT] [Accessed March 28, 2020] [DOI] [Google Scholar]
- Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill‐Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H et al (2019) Agreement between two large pan‐cancer CRISPR‐Cas9 gene dependency data sets. Nat Commun 10: 5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DepMap Broad (2019) DepMap 19Q3 Public. https://figshare.com/articles/DepMap_19Q3_Public/9201770
- DeWeirdt PC, Sangree AK, Hanna RE, Sanson KR, Hegde M, Strand C, Persky NS, Doench JG (2020) Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat Commun 11: 752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djajawi TM, Liu L, Gong J‐N, Huang AS, Luo M‐J, Xu Z, Okamoto T, Call MJ, Huang DCS, van Delft MF (2020) MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex. Cell Death Differ 10.1038/s41418-020-0517-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R et al (2016) Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol 34: 184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alonso L, Iorio F, Matchan A, Fonseca N, Jaaks P, Peat G, Pignatelli M, Falcone F, Benes CH, Dunham I et al (2018) Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res 78: 769–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J et al (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SJ, Travers J, Pshenichnaya I, Kogera FA, Barthorpe S, Mironenko T, Richardson L, Benes CH, Stratton MR, McDermott U et al (2015) Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in Ewing's sarcoma. PLoS ONE 10: e0140988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves E, Behan FM, Louzada S, Arnol D, Stronach EA, Yang F, Yusa K, Stegle O, Iorio F, Garnett MJ (2019) Structural rearrangements generate cell‐specific, gene‐independent CRISPR‐Cas9 loss of fitness effects. Genome Biol 20: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet‐Turcotte A, Sun S et al (2015) High‐resolution CRISPR screens reveal fitness genes and genotype‐specific cancer liabilities. Cell 163: 1515–1526 [DOI] [PubMed] [Google Scholar]
- Haschka MD, Karbon G, Soratroi C, O'Neill KL, Luo X, Villunger A (2020) MARCH5‐dependent degradation of MCL1/NOXA complexes defines susceptibility to antimitotic drug treatment. Cell Death Differ 10.1038/s41418-020-0503-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird AW, Paul Secrist J, Adam A, Belmonte MA, Gangl E, Gibbons F, Hargreaves D, Johannes JW, Kazmirski SL, Kettle JG et al (2017) Abstract DDT01‐02: AZD5991: a potent and selective macrocyclic inhibitor of Mcl‐1 for treatment of hematologic cancers. Cancer Res 77: DDT01–02 [Google Scholar]
- Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9: 90–95 [Google Scholar]
- Hustedt N, Álvarez‐Quilón A, McEwan A, Yuan JY, Cho T, Koob L, Hart T, Durocher D (2019a) A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol 9: 190156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Saito Y, Zimmermann M, Álvarez‐Quilón A, Setiaputra D, Adam S, McEwan A, Yuan JY, Olivieri M, Zhao Y et al (2019b) Control of homologous recombination by the HROB‐MCM8‐MCM9 pathway. Genes Dev 33: 1397–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H et al (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166: 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Behan FM, Gonçalves E, Bhosle SG, Chen E, Shepherd R, Beaver C, Ansari R, Pooley R, Wilkinson P et al (2018) Unsupervised correction of gene‐independent cell responses to CRISPR‐Cas9 targeting. BMC Genom 19: 604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Weissman JS (2018) CRISPR approaches to small molecule target identification. ACS Chem Biol 13: 366–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Dede M, Lenoir WF, Wang G, Srinivasan S, Colic M, Hart T (2019) A network of human functional gene interactions from knockout fitness screens in cancer cells. Life Sci Alliance 2: e201800278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P‐A, Reinecke M, Ruprecht B, Petzoldt S, Meng C et al (2017) The target landscape of clinical kinase drugs. Science 358: eaan4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike‐Yusa H, Li Y, Tan E‐P, Velasco‐Herrera MDC, Yusa K (2014) Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol 32: 267–273 [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK (2014) voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol 15: R29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐K, Liu Z, Sa JK, Shin S, Wang J, Bordyuh M, Cho HJ, Elliott O, Chu T, Choi SW et al (2018) Pharmacogenomic landscape of patient‐derived tumor cells informs precision oncology therapy. Nat Genet 50: 1399–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK et al (2017) Characterization of human cancer cell lines by reverse‐phase protein arrays. Cancer Cell 31: 225–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, Sausville EL, Lukow DA, Liu L, Chait AR et al (2019) Off‐target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med 11: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert C, Casale FP, Rakitsch B, Stegle O (2014) LIMIX: genetic analysis of multiple traits. bioRxiv 10.1101/003905 [PREPRINT] [DOI] [Google Scholar]
- Lynch JT, McEwen R, Crafter C, McDermott U, Garnett MJ, Barry ST, Davies BR (2016) Identification of differential PI3K pathway target dependencies in T‐cell acute lymphoblastic leukemia through a large cancer cell panel screen. Oncotarget 7: 22128–22139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez‐Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB et al (2016) Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164: 293–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney W (2010) Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, pp 51–56. Austin, TX: SciPy 2010; [Google Scholar]
- Médard G, Pachl F, Ruprecht B, Klaeger S, Heinzlmeir S, Helm D, Qiao H, Ku X, Wilhelm M, Kuehne T et al (2015) Optimized chemical proteomics assay for kinase inhibitor profiling. J Proteome Res 14: 1574–1586 [DOI] [PubMed] [Google Scholar]
- van der Meer D, Barthorpe S, Yang W, Lightfoot H, Hall C, Gilbert J, Francies HE, Garnett MJ (2019) Cell Model Passports‐a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res 47: D923–D929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, Magariños MP, Mosquera JF, Mutowo P, Nowotka M et al (2019) ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res 47: D930–D940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S et al (2017) Computational correction of copy number effect improves specificity of CRISPR‐Cas9 essentiality screens in cancer cells. Nat Genet 49: 1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsa B, Lassot I, Desagher S (2014) Mcl‐1 ubiquitination: unique regulation of an essential survival protein. Cells 3: 418–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M, Pinton P (2016) Mcl‐1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell 27: 20–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G et al (2016) CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false‐positive hits for highly amplified genomic regions. Cancer Discov 6: 900–913 [DOI] [PubMed] [Google Scholar]
- Murai J, Pommier Y (2015) Classification of PARP inhibitors based on PARP trapping and catalytic inhibition, and rationale for combinations with topoisomerase I inhibitors and alkylating agents In PARP Inhibitors for Cancer Therapy, Curtin NJ, Sharma RA. (eds), pp 261–274. Cham: Springer International Publishing; [Google Scholar]
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J et al (2015) The support of human genetic evidence for approved drug indications. Nat Genet 47: 856–860 [DOI] [PubMed] [Google Scholar]
- Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC et al (2018) Interrogation of mammalian protein complex structure, function, and membership using genome‐scale fitness screens. Cell Syst 6: 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V et al (2011) Scikit‐learn: machine learning in Python. J Mach Learn Res 12: 2825–2830 [Google Scholar]
- Picco G, Chen ED, Alonso LG, Behan FM, Gonçalves E, Bignell G, Matchan A, Fu B, Banerjee R, Anderson E et al (2019) Functional linkage of gene fusions to cancer cell fitness assessed by pharmacological and CRISPR‐Cas9 screening. Nat Commun 10: 2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauscher B, Heigwer F, Henkel L, Hielscher T, Voloshanenko O, Boutros M (2018) Toward an integrated map of genetic interactions in cancer cells. Mol Syst Biol 14: e7656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke M, Ruprecht B, Poser S, Wiechmann S, Wilhelm M, Heinzlmeir S, Kuster B, Médard G (2019) Chemoproteomic selectivity profiling of PIKK and PI3K kinase inhibitors. ACS Chem Biol 14: 655–664 [DOI] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al‐Lazikani B, Hersey A, Oprea TI et al (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16: 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S et al (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346: 1255784 [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12: 5268–5272 [DOI] [PubMed] [Google Scholar]
- Schenone M, Dančík V, Wagner BK, Clemons PA (2013) Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 9: 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK et al (2017) A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171: 1437–1452.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P et al (2017) The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res 45: D362–D368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol 3: 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15: 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill‐Burger JM et al (2017) Defining a cancer dependency map. Cell 170: 564–576.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike‐Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M et al (2016) A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep 17: 1193–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J et al (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17: 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vis DJ, Bombardelli L, Lightfoot H, Iorio F, Garnett MJ, Wessels LF (2016) Multilevel models improve precision and speed of IC50 estimates. Pharmacogenomics 17: 691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt S, Colbert SC, Varoquaux G (2011) The NumPy Array: a structure for efficient numerical computation. Comput Sci Eng 13: 22–30 [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM (2017a) Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell 168: 890–903.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu ZZ, Costanzo M, Boone C, Lange CA, Myers CL (2017b) Pathway‐based discovery of genetic interactions in breast cancer. PLoS Genet 13: e1006973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song J‐Y, Zevenhoven J, Los‐de Vries GT et al (2018) An acquired vulnerability of drug‐resistant melanoma with therapeutic potential. Cell 173: 1413–1425.e14 [DOI] [PubMed] [Google Scholar]
- Waskom M, Botvinnik O, Ostblom J, Lukauskas S, Hobson P, MaozGelbart, Gemperline DC, Augspurger T, Halchenko Y, Cole JB et al (2020) mwaskom/seaborn: v0.10.0 (January 2020) ZENODO
- Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam RW, Opferman JT, Sallan SE, den Boer ML, Pieters R et al (2006) Gene expression‐based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 10: 331–342 [DOI] [PubMed] [Google Scholar]
- Wuillème‐Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet‐Loiseau H, Harousseau J‐L, Amiot M, Bataille R (2005) Mcl‐1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 19: 1248–1252 [DOI] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR et al (2013) Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41: D955–D961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Karbowski M (2005) Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 6: 657 [DOI] [PubMed] [Google Scholar]
- Zheng Y, Zhang C, Croucher DR, Soliman MA, St‐Denis N, Pasculescu A, Taylor L, Tate SA, Hardy WR, Colwill K et al (2013) Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 499: 166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskait≐ Ž, Muir M, Fluteau A, Aregger M, McEwan A et al (2018) CRISPR screens identify genomic ribonucleotides as a source of PARP‐trapping lesions. Nature 559: 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Review Process File
Data Availability Statement
Source code, analysis reports and Jupyter notebooks are publicly available in GitHub https://github.com/EmanuelGoncalves/dtrace. Drug response and CRISPR‐Cas9 measurements are included in [Link], [Link], respectively, and available at FigShare https://doi.org/10.6084/m9.figshare.10338413.v1.