

Abstract
Drug induced liver injury (DILI) is a common cause of acute liver injury. Paracetamol, also known as acetaminophen, is a widely used anti-pyretic that has long been established to cause liver toxicity once above therapeutic levels. Hepatotoxicity from paracetamol overdose, whether intentional or non-intentional, is the most common cause of DILI in the United States and remains a global issue. Given the increased prevalence of combination medications in the form of pain relievers and antihistamines, paracetamol can be difficult to identify and remains a significant cause of acute hepatotoxicity, as evidenced by its contribution to over half of all acute liver failure cases in the United States. This is especially concerning given that, when co-ingested with other medications, the rise in serum paracetamol levels may be delayed past the 4-hour post-ingestion mark that is currently used to determine patients that require medical therapy. This review serves to describe the clinical and pathophysiologic features of hepatotoxicity secondary to paracetamol and provide an update on current available knowledge and treatment options.
Keywords: Paracetamol, Drug-induced liver injury, Hepatotoxicity, Acute liver failure
Core tip: Paracetamol is a widely used anti-pyretic that has long been established to cause liver toxicity once above therapeutic levels. Given the increased prevalence of combination medications in the form of pain relievers and antihistamines, paracetamol can be difficult to identify and remains a significant cause of acute hepatotoxicity globally. This is especially concerning given that, when co-ingested with other medications, the rise in serum paracetamol levels may be delayed and alter medical management. This review serves to describe the clinical and pathophysiologic features of hepatotoxicity secondary to paracetamol and provide an update on current available knowledge and treatment options.
INTRODUCTION
Acute liver failure consists of severe liver dysfunction, as evidenced by coagulopathy, jaundice, and encephalopathy, usually in the absence of underlying liver disease[1]. The incidence of acute liver failure (also termed fulminant hepatic failure) is roughly 10 per one million people annually in developed countries[1] with over 2000 cases in the United States diagnosed each year[2]. While viral hepatitis is among the most common cause of acute liver failure worldwide[1], drug-induced liver injury (DILI) is another culprit of liver damage. Half of all cases of acute liver injury in the United States result from DILI[3]. Paracetamol, also known as acetaminophen, is a widely used anti-pyretic that has known liver toxicity once above therapeutic levels in the blood[4]. In fact, paracetamol is the most common cause of DILI in the United States[5]. Given its ease of access as an over-the-counter medication, the United States Food and Drug Administration had stated it is safe to consume up to a maximum dose of 4000 mg within 24 h[6,7] while experts recommend a dose of 2000 mg or less in patients with existing liver disease or with chronic alcohol use[8-10]. Alternatively, dosing guidelines from drug inserts in European countries recommends maximum of a 3000 mg of paracetamol in older adults either < 50 kg or in those > 50 kg with additional risk factors for hepatotoxicity[11,12]. However, toxicity from paracetamol has recently become more challenging to rapidly identify given the increased use of combination medications, such as over-the-counter cold medicine or prescription pain relievers, that also contain paracetamol. In addition, toxic ingestions with these medications or in combination with alcohol may have a delayed presentation of hepatotoxicity[13-15]. Previously, only limited data on the mechanism and outlook of patients with acute liver injury existed. Because acute liver failure was poorly studied and understood, centralized data registries, such as the United States Acute Liver Failure Study Group, were formed to improve detection and patient outcomes. Additionally, the United States Drug-Induced Liver Injury Network was formed with the goal of creating a centralized registry for all acute liver failure cases that result from the use of prescriptions, over-the-counters, and herbal medications[16]. Our aim is to describe the clinical and pathophysiologic features of hepatotoxicity secondary to paracetamol and provide an update on current available knowledge and treatment options.
PATHOPHYSIOLOGY
Paracetamol was first developed in 1878 from phenacetin and became widespread in the 1950s as an over-the-counter antipyretic and analgesic. Since that time, there have been numerous studies connecting paracetamol ingestion with liver injury in a dose-dependent fashion. These effects are compounded in the setting of concomitant alcohol abuse, starvation ketosis or concurrent infections. Hepatocytes metabolize paracetamol via microsomal cytochrome P450 (CYP450) into non-toxic byproducts. This metabolism pathway via CYP450, specifically cytochrome P450 2E1 (CYP2E1), produces reactive oxygen species[17], originally thought to be the ultimate cause of liver injury in paracetamol overdose. After recent debunking[18-20] of that long-standing belief, mitochondrial dysfunction has instead been attributed as the main source of free radicals and oxidative stress in paracetamol hepatotoxicity[21]. Mitochondrial dysfunction begins with the formation of drug-protein adducts between the reactive paracetamol metabolite, N-acetyl-p-benzoquinone imine (NAPQI), and mitochondrial proteins involved in the electron transport chain[22,23]. Additionally, increased activity of mitochondrial complex I, a known site of free radical generation[24], occurs with paracetamol overdose, and the level of activity was found to correlate with the degree of liver injury[23]. Oxidative stress induced by paracetamol overdose is mainly attributed to mitochondrial superoxide and peroxynitrite[24]. The superoxide reacts with nitric oxide to form the highly reactive peroxynitrite species that is main source of oxidative and nitrosative stress[24].
Paracetamol has high bioavailability, with almost 80% of the drug being absorbed when taken orally[25]. In individuals without liver injury, the half-life of paracetamol is roughly 2-3 h[26]. At therapeutic levels in the blood, approximately 90% of paracetamol is broken down into non-toxic metabolites through sulfidation and glucuronidation pathways and then renally excreted[27]. However, at overdose levels, these pathways become saturated, resulting in large amounts of paracetamol being converted by CYP450 into its toxic metabolite, NAPQI[28]. NAPQI is subsequently excreted after glutathione conjugation renders it to harmless metabolites, as shown in Figure 1. Glutathione peroxidase activity is reduced by 60% in the setting of paracetamol[29]. This is dose-dependent, with larger amounts of paracetamol resulting in prolonged depletion of glutathione[21]. This reduction of glutathione in the mitochondria and cell cytosol results in decreased excretion of reactive oxygen species and peroxynitrite[30]. Additionally, without glutathione, oxidative stress activates the opening of mitochondrial permeability transition pores that results in the destruction of the membrane potential and halts ATP synthesis[21,30]. Ultimately, this results in the breakdown of DNA and cell membranes and the induction of apoptosis, resulting in cell death and acute inflammation[30].
Figure 1.
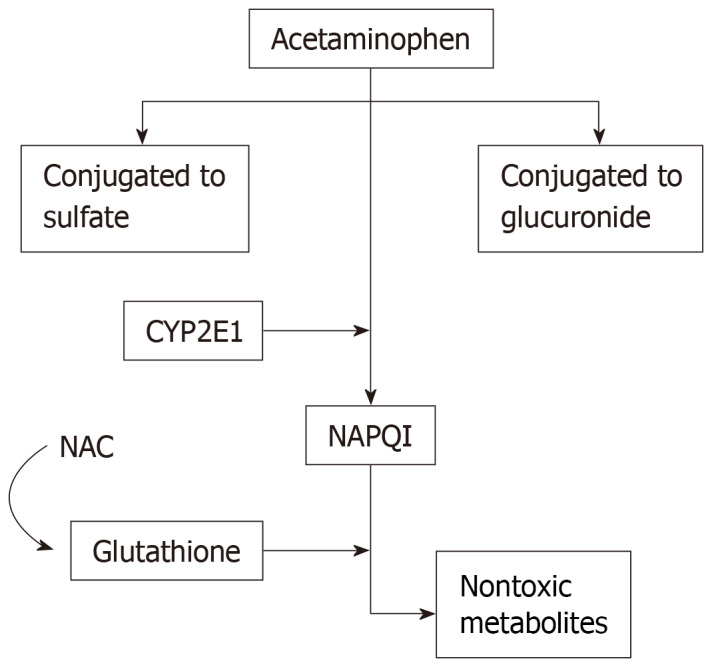
Paracetamol metabolism pathways and breakdown into N-acetyl-p-benzoquinone imine, by cytochrome P450 2E1. N-acetyl-p-benzoquinone imine is the toxic metabolite produced from paracetamol metabolism when the other two conjugation pathways become over-saturated. The resultant toxic N-acetyl-p-benzoquinone imine byproduct is converted into nontoxic metabolites by glutathione, which is regenerated by N-acetylcysteine. NAPQI: N-acetyl-p-benzoquinone imine; CYP2E1: Cytochrome P450 2E1; NAC: N-acetylcysteine.
CLINICAL PRESENTATION
In the United States, paracetamol is the one of the most widely used over-the-counter analgesics. According to the Third National Health and Nutrition Examination Survey from 1988-1994, 36% of Americans reported using acetaminophen within a month timeframe[31]. Due to its ease of access and availability in combination with other medications, such as opioids and antihistamines, paracetamol overdose can be accidental or in a suicide attempt. A thorough history and early recognition is key since long delays to the administration of treatment can result in worse outcomes for paracetamol-related hepatotoxicity. Depending on the severity of the liver damage, patients can present with a range of symptoms, from being initially asymptomatic to having overt signs of liver failure, including jaundice, confusion, ascites, nausea and vomiting.
Acute liver failure is defined by the presence of hepatic encephalopathy, jaundice and coagulopathy in individuals without any history of cirrhosis occurring for less than 26 wk[32]. This includes individuals with Wilson disease, autoimmune hepatitis or viral hepatitis as long as the disease was diagnosed within the last 26 wk[32]. An exclusion is severe alcoholic hepatitis, as this is likely accompanied with a history of prolonged alcohol abuse, resulting in chronicity of the liver injury.
There are four phases of acute paracetamol toxicity: Preclinical, hepatic injury, hepatic failure, and recovery[33]. Phase one, or the preclinical phase, occurs shortly after ingestion of toxic levels of paracetamol and can last 12-24 h. Non-specific symptoms such as nausea, vomiting, diaphoresis or lethargy may be seen. One to two days following the ingestion, the second phase begins, as evidenced by hepatotoxicity in laboratory values [elevation in hepatic enzymes, lactate, international normalized ratio (INR)][33,34]. Clinically, right upper quadrant abdominal pain may be present. In some cases, liver injury will progress to phase three, typically occurring at days three to five. Here, nausea and vomiting may recur or worsen and are accompanied with fatigue, jaundice, and central nervous system depression, varying from confusion to a coma[33]. Elevation in liver aminotransferases as high as 10000 IU/L can be seen[35]. The resultant hepatic necrosis and failure can be fatal and associated with multiorgan failure[33,34]. Lastly, phase four is recovery with normalization of laboratory values; roughly 70% of patients will fully recover, while 1%-2% will die from hepatic failure[33]. Death from untreated paracetamol toxicity occurs 4 to 18 days later[33].
Laboratory findings
Initial laboratory testing in acute liver failure will reveal prolonged prothrombin time, INR greater than 1.5, elevated aminotransferase and bilirubin levels, thrombocytopenia, electrolyte abnormalities, elevated ammonia levels and acid-base disturbances. Typically, aminotransferase levels are in the thousands in cases of paracetamol-induced acute liver failure. Paracetamol levels should always be drawn in acute liver injury cases. Since the time of ingestion is not always known, such as in accidental overdose, absent or low levels of paracetamol should not exclude paracetamol-induced liver injury in those cases where it is suspected. Laboratory variables most indicative of patient outcome were the peak bilirubin and prothrombin time levels with bilirubin directly correlated to survival[36].
Liver biopsy is not routinely performed in the diagnosis of acute liver failure as it is associated with a high risk of bleeding and mortality. In addition, in prior studies assessing the role and accuracy of liver biopsy in acute liver failure, the diagnosis changed in 18% of cases; there was no report on whether biopsy information altered the clinical course or treatment[37]. In fact, the American Gastroenterological Association “suggests against routine use of liver biopsy” in the workup of acute liver failure[37].
HEPATOTOXICITY RISK FACTORS
The dose of ingestion as well as the time span between ingestion of paracetamol and of the treatment drug N-acetylcysteine (NAC) are the most influential factors in the manifestation and severity of paracetamol hepatotoxicity[4,38-40]. While acute liver injury can occur when used at or below the recommended daily maximum dose (4000 mg)[4], paracetamol toxicity is often the result of ingestion of paracetamol over this maximum dose. In fact, the maximum daily dosage has been a topic of controversy, with some manufacturers voluntarily lowering this recommended threshold on their products in order to increase the safety of patients[41,42].
Beyond exceeding the recommended daily dose, the risk for liver injury increases when paracetamol is used in combination with other drugs and substances, such as alcohol. The interplay between paracetamol and alcohol is an interesting one, because these compounds are competitive substrates for CYP2E1, which reduces the production of the reactive NAPQI species generated in paracetamol metabolism; as a result, acute alcohol ingestion may in fact act as a protective mechanism against paracetamol hepatotoxicity[43-46]. On the other hand, paracetamol hepatotoxicity is augmented with chronic alcohol consumption through the up-regulation and increased synthesis and activity of CYP2E1 as well as the decreased production of glutathione; these activities result in enhanced liver necrosis and an exacerbated prognosis[43,46]. While the risk of liver failure may be increased in the case of chronic alcoholism in combination with paracetamol overdose, alcoholism does not necessarily increase the risk of paracetamol hepatotoxicity when in combination with therapeutic doses[43]. Beyond alcohol, there are various prescribed and over-the-counter medications that can predispose a patient to paracetamol hepatotoxicity, including opioids, anti-tuberculosis drugs[47], and anti-epileptic drugs as well as herbs and dietary supplements, such as St. John’s wort, garlic and germander, through their effects on CYP450 metabolism (Table 1)[7].
Table 1.
Drugs and substances that affect cytochrome P450 2E1 activity and can interfere with paracetamol metabolism
| Cytochrome P450 | Inducers | Inhibitors |
| CYP2E1 | Ethanol | Disulfram |
| Isoniazid | ||
| St. John’s wort | ||
| Garlic, Germander |
CYP2E1: Cytochrome P450 2E1.
The risk for paracetamol hepatotoxicity is increased in patients with malnutrition, as glutathione stores are depleted and no longer available for conjugation with the reactive NAPQI species[48]. Individuals at a particular risk for poor nutritional status include those with chronic alcoholism[48], and while patients with anorexia nervosa are malnourished and have low glutathione reserves, they also have reduced CYP2E1 activity, which in fact does not exacerbate the risk of paracetamol toxicity in this subset of malnourished patients[49]. Age also impacts hepatotoxicity risk, with advanced age (over 40 years old) being associated with a higher risk of acute liver failure, liver transplantation, and death from paracetamol overdose[50]. The metabolism of paracetamol appears to be dependent on age[51], and paracetamol use alone and in combination with opiates is widespread among advanced-age adults for treatment of chronic pain or cancer. Chronic liver disease patients are also at increased risk for hepatotoxicity, as paracetamol metabolism is decreased in patients with cirrhotic livers. While there is no evidence suggesting pregnancy as a predisposing risk factor for paracetamol toxicity[7], the use of paracetamol during pregnancy should be carefully monitored, since paracetamol is the most common overdose during pregnancy, and toxicity in such cases can results in significant morbidity and mortality for both the fetus and mother[52].
The aforementioned confounding factors that influence the development and acuteness of liver injury are summarized in the flowchart in Figure 2.
Figure 2.

Factors that pre-dispose patients to increased paracetamol toxicity.
PROGNOSTICATION
While the extent of liver injury has been found to be dose dependent, there are a few possible risk factors for DILI. One study has found that men and younger age was associated with an increased risk in hepatocellular damage[53]. Traditionally, there are a few scoring systems available to prognosticate those with acute liver failure though none are considered gold standard criteria. The King’s College liver failure criteria[36] uses serum laboratory values to determine the prognosis of DILI and tested these prognostications by retrospectively analyzing those patients that had to undergo liver transplantation. The Roussel Uclaf Causality Assessment Method[54] is a sensitive test but difficult to perform based on its complicated system. The Roussel Uclaf Causality Assessment Method score is based off of seven measures that include the time of DILI onset, concomitant risk factors or drug use, non-drug related liver injury, the patient’s clinical course, prior liver injury toxicity and the response to re-challenge of the drug[54]. A modification to this is the Digestive Disease Week-Japan scale[55], which adds the lymphocyte stimulation test. Prior to transplantation, the finding of jaundice in DILI patients was associated with a poor prognosis with over 10% mortality prior to liver transplantation for paracetamol-induced liver injury[56]. This prognostic finding of hepatocellular injury significant enough to alter bilirubin excretion (with elevations greater than two times the upper limit of normal) is referred to as “Hy’s Law Cases”[56].
Patient outcomes are dependent upon what phase of paracetamol poisoning that treatment is initiated in. If the antidote is given during phase one (in cases where medical history reveals a suspicion of paracetamol overdose), patients are expected to fully recover with only a transient period of liver injury[57,58]. In fact, the administration of N-acetylcysteine will prevent most patients from progressing past phase two of hepatic injury[34]. Additionally, the presence of other organ involvement, such as altered mental status or acute renal failure portends a worse prognosis and is often an indication for monitoring the patient in a critical care setting[59].
TREATMENT
Early initiation of treatment is critical immediately following recognition of DILI. The Rumack-Matthew nomogram is a tool that uses serum paracetamol levels at a specific time point in the overdose, typically measured between 4- and 24-hours post-ingestion, to predict the risk of hepatotoxicity and guide medical management[60,61]. If the paracetamol level is above a certain cutoff, also called the “treatment line” that typically starts at 150 µg/mL at 4 h and extends to 4.7 µg/mL at 24 h, then treatment is indicated[62,63] (outlined in Figure 3). If the time of ingestion is unknown but within 24 h, the earliest possible time of ingestion should be estimated and plotted on the nomogram to see if treatment with NAC should be initiated (i.e. if above the treatment line). The use of the nomogram should be avoided until 4 h or more after ingestion as the levels may be misleading during this timeframe from the point of acute ingestion and not be an accurate predictor of toxicity[64,65]. Classically, the nomogram is used in conjunction with the patient’s history and laboratory findings to determine medical management. If medication review reveals co-ingestion with opioids or anticholingeric medications, the post-ingestion level should be checked at 4 h and repeated at 6 h post-ingestion if the initial level falls below the treatment line to account for possible delay in maximum serum concentrations of paracetamol[13]. However, other aspects of the history such as the reported dose in paracetamol toxicity, can be used to predict patient outcomes. A prospective study demonstrated that individuals who had a reported overdose with 50 g of paracetamol had a 90% probability of being over the treatment value cutoff, suggesting that dosing can help rapidly identify individuals that need treatment immediately initiated[66].
Figure 3.

Flowchart depicting the management pathway for acute paracetamol overdose/ toxicity. ALT: Aminotransferase; AST: Aspartate aminotransferase.
Currently, the mainstay therapy is NAC, given intravenously as soon as the diagnosis of paracetamol hepatotoxicity is made. NAC acts by restoring glutathione levels that then allow for the removal of NAPQI from the body[67]. Specifically, NAC is hydrolyzed to cysteine, which in turns restores glutathione as well as provides thiol groups that react directly with NAPQI in the hepatocytes[57,68]. Administration of NAC is the mainstay treatment and standard of care in paracetamol overdose, with the most benefit seen if initiated within the first 8 h from the time of paracetamol overdose[57,58]. However, it has been shown that mortality is significantly decreased by the administration of NAC even up to 36 h after the toxic ingestion and that this cohort of patients is less likely to progress to grade III/IV hepatic coma after receiving treatment with NAC[69]. NAC is typically administered intravenously over three weight-based doses: The initial 150 mg/kg dose in the first 15-60 minutes, followed by 50 mg/kg over 4 h, and then 100 mg/kg over 16 h[60,70]. In cases where NAC is administered orally, the typical dosing regimen is a loading dose of 140 mg/kg, subsequently followed by 70 mg/kg every 4 h until 18 doses are administered[60,68]. After NAC dosing is complete, re-evaluation of the paracetamol level and liver function tests should be done to assess if repeat dosing is indicated. Dosing of NAC can be continued if the serum paracetamol level is above 10 µg/mL or if alanine aminotransferase (ALT) elevation persists, especially in the setting of acidosis, coagulopathy, acute kidney injury and hyperbilirubinemia as these patients have worse outcomes[71].
With opioid use being more prevalent, medication interactions that slow gut motility have important implications in paracetamol toxicity. Recent studies have shown that there are limitations to the Rumack-Matthew nomogram in predicting the risk of hepatotoxicity in the setting of combination medications due to a delay in the onset of symptoms and laboratory abnormalities[13-15]. In particular, paracetamol combined with antihistamines or opioids have been shown to have serum paracetamol levels below 150 µg at the 4-hour post-ingestion mark but would cross above the treatment threshold when levels were rechecked within the 24-hour period[13]. In fact, a United States prospective cohort study[14] showed that 6% of patients with an acute combination medication overdose of paracetamol with antihistamines or opioids had paracetamol levels that were initially low at the 4-hour time mark but were later found to be above the 150 µg/mL treatment threshold.
In conjunction with NAC therapy, activated charcoal has been proven to be beneficial in reducing the number of patients that develop toxic serum paracetamol levels[72] and has been shown to decrease the extent of liver injury, as evidenced through a reduction in serum transaminase levels and prothrombin time[72,73]. Since the majority of paracetamol absorption in the gastrointestinal tract occurs within the first 4 h[40], activated charcoal is generally believed to be most beneficial if administered within that time period, as its mechanism of action is to interfere with paracetamol absorption. However, activated charcoal has been shown to provide some benefit even with later administration, suggesting an additional mechanism for improvement in hepatotoxicity[72].
Treatment updates and alternatives
While the mechanism of toxicity in paracetamol overdose is thought to be due to glutathione depletion and subsequent buildup of harmful metabolites as previously mentioned, studies on the repletion of glutathione as a therapy option are few and in early stages but yielded positive results. In one animal study[74], both free and niosomal (or encapsulated) glutathione administered intravenously had been shown to reduce hepatotoxicity in paracetamol overdose with serum concentration at 150 mg/kg. This promising finding may represent an avenue for treatment in the future after further investigations are performed. An additional novel therapy is N-acetylcysteine amide (NACA), which is a variant of NAC with an amide in place of a carboxyl group, which in turn increases the compound’s lipophilicity[75]. This allows NACA to more easily transverse cell membranes, meaning it is effective at lower doses when compared to NAC and potentially avoid some adverse side effects. NACA’s therapeutic benefit is multifactorial: It acts as a precursor to glutathione, promotes intracellular metabolism of toxic compounds and is a free radical scavenger. In this study, NACA was dosed at 106 mg/kg every 12 h for a maximum of up to 72 h. With this dosing regimen, NACA was found to have increased survival in mice as well as improved ability to decrease damage from oxidation and paracetamol[75].
Additionally, recent research has explored the effectiveness of lower doses of NAC in the treatment of hepatotoxicity from paracetamol overdose. The study by Shen et al[76] demonstrated that NAC was still effective at an initial lower infusion rate (200 mg/kg over 9 h, or 23 mg/kg/h) followed by the third dose of the conventional treatment regimen. The lower initial infusion would allow for immediate treatment in suspected acute liver failure from overdose cases to prevent delay while awaiting serum paracetamol levels and liver function tests. This is beneficial as serious adverse events including hypersensitivity reactions, such as rashes to even anaphylaxis, can occur following high dose NAC infusions[58,77,78]. Furthermore, gastric lavage and molecular adsorbent recirculating system (MARS) are two other treatment options for paracetamol overdose. While gastric lavage is used for numerous types of drug overdose, its use for the treatment of paracetamol toxicity has fallen out of favor as there are more effective conventional treatments with better outcomes[79]. A study looking at the use of MARS in acute liver failure patients showed that this system could increase the removal of paracetamol and was associated with improved survival times when compared to current standard therapy alone[80].
CLINICAL OUTCOMES
Outcomes of paracetamol overdose have been reported from numerous countries. A study from Australia reported over 440 deaths from paracetamol in combination with codeine from accidental overdose, with roughly 25% of these cases also involving other sedating medications, such as antihistamines[81]. While paracetamol has been the main cause of DILI in the United States and the England, it is less common in other European countries, such as Portugal and Germany, only making up roughly 10% of ALF cases according to the European Liver Transplant Registry (ELTR) database[82]. In part, this could be from the increased usage of paracetamol intake in the United States in comparison to European countries. A summary of clinical outcomes from paracetamol-induced acute liver failure can be found in Table 2.
Table 2.
Outcomes of acute liver failure from paracetamol among selected countries
| Country | Acute liver failure from paracetamol | Hepatic failure resulting in death or transplant | Concomitant medication use/ suicide attempts |
| Australia[81] | Not disclosed | Death in 8.8% of cases (39 deaths total) | 79% of cases with co-ingestion of opioids or benzodiazepines |
| United Kingdom[83] | 2163 cases (65% of total ALF cases) | Death in 36% of cases (778 deaths total), 147 transplant cases | Not disclosed |
| United States[5] | 120 cases (39% of total ALF cases) | Transplantation in 6% of cases, mortality in 27% of cases | 44 cases (37%) were suicide attempts |
| Portugal[84] | 5 cases over 3 years (11% of total ALF cases) | 1 liver transplant case | Not disclosed |
| Germany[85] | 10 cases (9.2% of total ALF cases) | 3 liver transplant cases, 1 death | Not disclosed |
Recently, a multinational study, the Study of Acute Liver Transplantation (SALT)[86], identified cases where drug exposure, specifically nonsteroidal anti-inflammatory drugs (NSAIDs) and paracetamol, had led to acute liver failure and resultant registration for liver transplantation. Among all the individuals in the study, there was no significant difference in the development of acute liver failure in the setting of exposure to various NSAIDs with incidence of patients registered for liver transplantation in these cases being rare. In comparison, paracetamol was associated with a three-fold higher risk of being registered for liver transplantation from associated acute liver injury in non-overdose levels and a seven-fold higher in overdose levels of paracetamol exposure[86]. In a follow up study, Gulmez et al[87] analyzed all the cases of individuals on the transplant registry and identified those that had drug overdose with resultant liver failure. Of those, paracetamol was responsible for one-sixth of all cases, and paracetamol overdose was responsible for 97% of all drug overdoses associated with acute liver failure. In response to the findings of the SALT study, the EPIHAM study[88] was conducted to compare the risk of non-overdose levels of paracetamol versus NSAIDs resulting in admission for acute liver injury. However, the three-fold risk associated with paracetamol in transplantation registry was not seen for acute liver injury in this study[88].
Acute liver failure from paracetamol toxicity has a high mortality rate of 30% if there is no liver transplantation available[89,90]. Among intentional and accidental overdose, the liver transplant-free survival rate was not found to significantly differ. Prognostic criteria, such as the King’s College criteria, can be used to determine appropriate candidates for liver transplantation referral based on those with expected high mortality of over 80%[36,91]. As previously mentioned, those with phase three liver injury and signs of progressive organ dysfunction, severe acidosis or multiorgan failure have a poor prognosis and should be referred for possible liver transplantation. Those patients that undergo liver transplantation due to paracetamol liver failure have good clinical outcomes, with a 5-year survival of over 70%[59].
CONCLUSION
Paracetamol toxicity, albeit accidental or intentional overdose, is an ongoing global problem that continues to result in cases of hepatotoxicity, acute liver failure, and even irreversible liver injury necessitating liver transplantation. Given the increased prevalence of combination medications in the form of pain relievers and antihistamines, paracetamol remains a significant cause of acute hepatotoxicity, as evidenced by paracetamol contributing to over half of acute liver failure cases in the United States. This is especially concerning given that when co-ingested with other medications, the rise in serum paracetamol levels may be delayed past the 4-hour post-ingestion mark that is currently used to determine patients that require medical therapy. Current research is exploring the outcomes of paracetamol-related DILI cases and its relationship with liver transplantation as well as other treatment modalities.
Footnotes
Conflict-of-interest statement: Dr. Pyrsopoulos reports grants from Allergan, grants from Bayer, grants from Beigene, grants from Bristol Myers, grants from Confirm, grants from Conatus, grants from Intercept, grants from Mallinckrodt, grants from Novartis, grants from Resusix, grants from Saro, grants from Valeant, grants from Gilead, grants from Exelixis, grants from Hologic, grants from Shire, grants from Genfit, grants from Prometheus, outside the submitted work. Dr. Rotundo certifies that she has no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
Manuscript source: Unsolicited manuscript
Peer-review started: October 11, 2019
First decision: November 2, 2019
Article in press: February 17, 2020
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: United States
Peer-review report´s scientific quality classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Cure E, De Ponti F, Lei YC, Qadir MI S-Editor: Wang YQ L-Editor: A E-Editor: Wu YXJ
Contributor Information
Laura Rotundo, Department of Medicine, Rutgers New Jersey Medical School, Newark, NJ 07103, United States.
Nikolaos Pyrsopoulos, Department of Gastroenterology and Hepatology, Rutgers New Jersey Medical School, Newark, NJ 07103, United States. pyrsopni@njms.rutgers.edu.
References
- 1.Bernal W, Wendon J. Acute liver failure. N Engl J Med. 2013;369:2525–2534. doi: 10.1056/NEJMra1208937. [DOI] [PubMed] [Google Scholar]
- 2.Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102:2459–2463. doi: 10.1111/j.1572-0241.2007.01388.x. [DOI] [PubMed] [Google Scholar]
- 3.Reuben A, Koch DG, Lee WM Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology. 2010;52:2065–2076. doi: 10.1002/hep.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, Harris SC. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296:87–93. doi: 10.1001/jama.296.1.87. [DOI] [PubMed] [Google Scholar]
- 5.Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, Crippin JS, Blei AT, Samuel G, Reisch J, Lee WM U. S. Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 6.Herndon CM, Dankenbring DM. Patient perception and knowledge of acetaminophen in a large family medicine service. J Pain Palliat Care Pharmacother. 2014;28:109–116. doi: 10.3109/15360288.2014.908993. [DOI] [PubMed] [Google Scholar]
- 7.Bunchorntavakul C, Reddy KR. Acetaminophen-related hepatotoxicity. Clin Liver Dis. 2013;17:587–607, viii. doi: 10.1016/j.cld.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Bosilkovska M, Walder B, Besson M, Daali Y, Desmeules J. Analgesics in patients with hepatic impairment: pharmacology and clinical implications. Drugs. 2012;72:1645–1669. doi: 10.2165/11635500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Chandok N, Watt KD. Pain management in the cirrhotic patient: the clinical challenge. Mayo Clin Proc. 2010;85:451–458. doi: 10.4065/mcp.2009.0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delcò F, Tchambaz L, Schlienger R, Drewe J, Krähenbühl S. Dose adjustment in patients with liver disease. Drug Saf. 2005;28:529–545. doi: 10.2165/00002018-200528060-00005. [DOI] [PubMed] [Google Scholar]
- 11.Perfalgan 10 mg/ml, solution for infusion. Middlesex: BMSP. Available from: https://www.medicines.org.uk/emc/files/pil.60.pdf. [Google Scholar]
- 12.Mian P, Allegaert K, Spriet I, Tibboel D, Petrovic M. Paracetamol in Older People: Towards Evidence-Based Dosing? Drugs Aging. 2018;35:603–624. doi: 10.1007/s40266-018-0559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dougherty PP, Klein-Schwartz W. Unexpected late rise in plasma acetaminophen concentrations with change in risk stratification in acute acetaminophen overdoses. J Emerg Med. 2012;43:58–63. doi: 10.1016/j.jemermed.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 14.Kirschner RI, Rozier CM, Smith LM, Jacobitz KL. Nomogram line crossing after acetaminophen combination product overdose. Clin Toxicol (Phila) 2016;54:40–46. doi: 10.3109/15563650.2015.1110591. [DOI] [PubMed] [Google Scholar]
- 15.Graudins A. Overdose with modified-release paracetamol (Panadol Osteo®) presenting to a metropolitan emergency medicine network: a case series. Emerg Med Australas. 2014;26:398–402. doi: 10.1111/1742-6723.12249. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi PH. Drug-Induced Liver Injury Network Causality Assessment: Criteria and Experience in the United States. Int J Mol Sci. 2016;17:201. doi: 10.3390/ijms17020201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wendel A, Feuerstein S. Drug-induced lipid peroxidation in mice--I. Modulation by monooxygenase activity, glutathione and selenium status. Biochem Pharmacol. 1981;30:2513–2520. doi: 10.1016/0006-2952(81)90576-1. [DOI] [PubMed] [Google Scholar]
- 18.Smith CV, Jaeschke H. Effect of acetaminophen on hepatic content and biliary efflux of glutathione disulfide in mice. Chem Biol Interact. 1989;70:241–248. doi: 10.1016/0009-2797(89)90047-1. [DOI] [PubMed] [Google Scholar]
- 19.Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80:343–349. doi: 10.1093/toxsci/kfh151. [DOI] [PubMed] [Google Scholar]
- 20.Lauterburg BH, Smith CV, Hughes H, Mitchell JR. Biliary excretion of glutathione and glutathione disulfide in the rat. Regulation and response to oxidative stress. J Clin Invest. 1984;73:124–133. doi: 10.1172/JCI111182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev. 2012;44:88–106. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem. 1998;273:17940–17953. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 23.Du K, Ramachandran A, Weemhoff JL, Chavan H, Xie Y, Krishnamurthy P, Jaeschke H. Editor's Highlight: Metformin Protects Against Acetaminophen Hepatotoxicity by Attenuation of Mitochondrial Oxidant Stress and Dysfunction. Toxicol Sci. 2016;154:214–226. doi: 10.1093/toxsci/kfw158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016;10:148–156. doi: 10.1016/j.redox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ameer B, Divoll M, Abernethy DR, Greenblatt DJ, Shargel L. Absolute and relative bioavailability of oral acetaminophen preparations. J Pharm Sci. 1983;72:955–958. doi: 10.1002/jps.2600720832. [DOI] [PubMed] [Google Scholar]
- 26.Tylenol (acetaminophen) professional product information. In: McNeil Consumer Healthcare. Fort Washington: PA, 2010. [Google Scholar]
- 27.Hodgman MJ, Garrard AR. A review of acetaminophen poisoning. Crit Care Clin. 2012;28:499–516. doi: 10.1016/j.ccc.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Ramachandran A, Jaeschke H. Oxidative Stress and Acute Hepatic Injury. Curr Opin Toxicol. 2018;7:17–21. doi: 10.1016/j.cotox.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tirmenstein MA, Nelson SD. Acetaminophen-induced oxidation of protein thiols. Contribution of impaired thiol-metabolizing enzymes and the breakdown of adenine nucleotides. J Biol Chem. 1990;265:3059–3065. [PubMed] [Google Scholar]
- 30.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 31.NHANES III Second Laboratory Data. U.S. Department of Health and Human Services, 1988-1994; Centers for Disease Control and Prevention, 1998. In: Third National Health and Nutrition Examination Survey. [Google Scholar]
- 32.Lee WM, Stravitz RT, Larson AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011. Hepatology. 2012;55:965–967. doi: 10.1002/hep.25551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun LJ, Tong MJ, Busuttil RW, Hiatt JR. Acetaminophen hepatotoxicity and acute liver failure. J Clin Gastroenterol. 2009;43:342–349. doi: 10.1097/MCG.0b013e31818a3854. [DOI] [PubMed] [Google Scholar]
- 34.Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med. 2010;77:19–27. doi: 10.3949/ccjm.77a.09084. [DOI] [PubMed] [Google Scholar]
- 35.Kwo PY, Cohen SM, Lim JK. ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries. Am J Gastroenterol. 2017;112:18–35. doi: 10.1038/ajg.2016.517. [DOI] [PubMed] [Google Scholar]
- 36.O'Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology. 1989;97:439–445. doi: 10.1016/0016-5085(89)90081-4. [DOI] [PubMed] [Google Scholar]
- 37.Flamm SL, Yang YX, Singh S, Falck-Ytter YT AGA Institute Clinical Guidelines Committee. American Gastroenterological Association Institute Guidelines for the Diagnosis and Management of Acute Liver Failure. Gastroenterology. 2017;152:644–647. doi: 10.1053/j.gastro.2016.12.026. [DOI] [PubMed] [Google Scholar]
- 38.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127:1760–1774. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 39.Xie Y, McGill MR, Dorko K, Kumer SC, Schmitt TM, Forster J, Jaeschke H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol Appl Pharmacol. 2014;279:266–274. doi: 10.1016/j.taap.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Douglas DR, Sholar JB, Smilkstein MJ. A pharmacokinetic comparison of acetaminophen products (Tylenol Extended Relief vs regular Tylenol) Acad Emerg Med. 1996;3:740–744. doi: 10.1111/j.1553-2712.1996.tb03508.x. [DOI] [PubMed] [Google Scholar]
- 41.Krenzelok EP, Royal MA. Confusion: acetaminophen dosing changes based on NO evidence in adults. Drugs R D. 2012;12:45–48. doi: 10.2165/11633010-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Organ-specific warnings: internal analgesic, antipyretic, and antirheumatic drug products for over-the-counter human use-labeling for products that contain acetaminophen; guidance for industry; availability. Available from: https://www.fda.gov/media/83588/download. [Google Scholar]
- 43.Dart RC, Erdman AR, Olson KR, Christianson G, Manoguerra AS, Chyka PA, Caravati EM, Wax PM, Keyes DC, Woolf AD, Scharman EJ, Booze LL, Troutman WG American Association of Poison Control Centers. Acetaminophen poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila) 2006;44:1–18. doi: 10.1080/15563650500394571. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt LE, Dalhoff K, Poulsen HE. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology. 2002;35:876–882. doi: 10.1053/jhep.2002.32148. [DOI] [PubMed] [Google Scholar]
- 45.Waring WS, Stephen AF, Malkowska AM, Robinson OD. Acute ethanol coingestion confers a lower risk of hepatotoxicity after deliberate acetaminophen overdose. Acad Emerg Med. 2008;15:54–58. doi: 10.1111/j.1553-2712.2007.00019.x. [DOI] [PubMed] [Google Scholar]
- 46.McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30:2174–2187. doi: 10.1007/s11095-013-1007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang P, Pradhan K, Zhong XB, Ma X. Isoniazid metabolism and hepatotoxicity. Acta Pharm Sin B. 2016;6:384–392. doi: 10.1016/j.apsb.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA. 1994;272:1845–1850. doi: 10.1001/jama.1994.03520230055038. [DOI] [PubMed] [Google Scholar]
- 49.Zenger F, Russmann S, Junker E, Wüthrich C, Bui MH, Lauterburg BH. Decreased glutathione in patients with anorexia nervosa. Risk factor for toxic liver injury? Eur J Clin Nutr. 2004;58:238–243. doi: 10.1038/sj.ejcn.1601772. [DOI] [PubMed] [Google Scholar]
- 50.Michna E, Duh MS, Korves C, Dahl JL. Removal of opioid/acetaminophen combination prescription pain medications: assessing the evidence for hepatotoxicity and consequences of removal of these medications. Pain Med. 2010;11:369–378. doi: 10.1111/j.1526-4637.2010.00811.x. [DOI] [PubMed] [Google Scholar]
- 51.Rumore MM, Blaiklock RG. Influence of age-dependent pharmacokinetics and metabolism on acetaminophen hepatotoxicity. J Pharm Sci. 1992;81:203–207. doi: 10.1002/jps.2600810302. [DOI] [PubMed] [Google Scholar]
- 52.Wilkes JM, Clark LE, Herrera JL. Acetaminophen overdose in pregnancy. South Med J. 2005;98:1118–1122. doi: 10.1097/01.smj.0000184792.15407.51. [DOI] [PubMed] [Google Scholar]
- 53.Lucena MI, Andrade RJ, Kaplowitz N, García-Cortes M, Fernández MC, Romero-Gomez M, Bruguera M, Hallal H, Robles-Diaz M, Rodriguez-González JF, Navarro JM, Salmeron J, Martinez-Odriozola P, Pérez-Alvarez R, Borraz Y, Hidalgo R Spanish Group for the Study of Drug-Induced Liver Disease. Phenotypic characterization of idiosyncratic drug-induced liver injury: the influence of age and sex. Hepatology. 2009;49:2001–2009. doi: 10.1002/hep.22895. [DOI] [PubMed] [Google Scholar]
- 54.Rochon J, Protiva P, Seeff LB, Fontana RJ, Liangpunsakul S, Watkins PB, Davern T, McHutchison JG Drug-Induced Liver Injury Network (DILIN) Reliability of the Roussel Uclaf Causality Assessment Method for assessing causality in drug-induced liver injury. Hepatology. 2008;48:1175–1183. doi: 10.1002/hep.22442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takikawa H, Takamori Y, Kumagi T, Onji M, Watanabe M, Shibuya A, Hisamochi A, Kumashiro R, Ito T, Mitsumoto Y, Nakamura A, Sakaguchi T. Assessment of 287 Japanese cases of drug induced liver injury by the diagnostic scale of the International Consensus Meeting. Hepatol Res. 2003;27:192–195. doi: 10.1016/s1386-6346(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 56.Temple R. Hy's law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15:241–243. doi: 10.1002/pds.1211. [DOI] [PubMed] [Google Scholar]
- 57.Jones AL. Mechanism of action and value of N-acetylcysteine in the treatment of early and late acetaminophen poisoning: a critical review. J Toxicol Clin Toxicol. 1998;36:277–285. doi: 10.3109/15563659809028022. [DOI] [PubMed] [Google Scholar]
- 58.Kerr F, Dawson A, Whyte IM, Buckley N, Murray L, Graudins A, Chan B, Trudinger B. The Australasian Clinical Toxicology Investigators Collaboration randomized trial of different loading infusion rates of N-acetylcysteine. Ann Emerg Med. 2005;45:402–408. doi: 10.1016/j.annemergmed.2004.08.040. [DOI] [PubMed] [Google Scholar]
- 59.Fontana RJ. Acute liver failure including acetaminophen overdose. Med Clin North Am. 2008;92:761–794, viii. doi: 10.1016/j.mcna.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rumack BH. Acetaminophen hepatotoxicity: the first 35 years. J Toxicol Clin Toxicol. 2002;40:3–20. doi: 10.1081/clt-120002882. [DOI] [PubMed] [Google Scholar]
- 61.Food and Drug Administration. Acetadote (acetylcysteine) injection package insert. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/021539s004lbl.pdf. [Google Scholar]
- 62.Bateman DN, Carroll R, Pettie J, Yamamoto T, Elamin ME, Peart L, Dow M, Coyle J, Cranfield KR, Hook C, Sandilands EA, Veiraiah A, Webb D, Gray A, Dargan PI, Wood DM, Thomas SH, Dear JW, Eddleston M. Effect of the UK's revised paracetamol poisoning management guidelines on admissions, adverse reactions and costs of treatment. Br J Clin Pharmacol. 2014;78:610–618. doi: 10.1111/bcp.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bond GR, Caravati EM, Dart RC, Heard KJ, Hoffman RS, Rumack BH, Snodgrass WR. Guidelines for the Management of Acetaminophen Overdose. Available from: https://www.tylenolprofessional.com/sites/tylenol_hcp_us/files/acetaminphen_overdose_treatment_info.pdf. [Google Scholar]
- 64.Rumack BH, Peterson RC, Koch GG, Amara IA. Acetaminophen overdose. 662 cases with evaluation of oral acetylcysteine treatment. Arch Intern Med. 1981;141:380–385. doi: 10.1001/archinte.141.3.380. [DOI] [PubMed] [Google Scholar]
- 65.Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985) N Engl J Med. 1988;319:1557–1562. doi: 10.1056/NEJM198812153192401. [DOI] [PubMed] [Google Scholar]
- 66.Duffull SB, Isbister GK. Predicting the requirement for N-acetylcysteine in paracetamol poisoning from reported dose. Clin Toxicol (Phila) 2013;51:772–776. doi: 10.3109/15563650.2013.830733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maher SZ, Schreibman IR. The Clinical Spectrum and Manifestations of Acute Liver Failure. Clin Liver Dis. 2018;22:361–374. doi: 10.1016/j.cld.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 68.Heard KJ. Acetylcysteine for acetaminophen poisoning. N Engl J Med. 2008;359:285–292. doi: 10.1056/NEJMct0708278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harrison PM, Keays R, Bray GP, Alexander GJ, Williams R. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335:1572–1573. doi: 10.1016/0140-6736(90)91388-q. [DOI] [PubMed] [Google Scholar]
- 70.Prescott LF, Illingworth RN, Critchley JA, Stewart MJ, Adam RD, Proudfoot AT. Intravenous N-acetylcystine: the treatment of choice for paracetamol poisoning. Br Med J. 1979;2:1097–1100. doi: 10.1136/bmj.2.6198.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGovern AJ, Vitkovitsky IV, Jones DL, Mullins ME. Can AST/ALT ratio indicate recovery after acute paracetamol poisoning? Clin Toxicol (Phila) 2015;53:164–167. doi: 10.3109/15563650.2015.1006399. [DOI] [PubMed] [Google Scholar]
- 72.Spiller HA, Winter ML, Klein-Schwartz W, Bangh SA. Efficacy of activated charcoal administered more than four hours after acetaminophen overdose. J Emerg Med. 2006;30:1–5. doi: 10.1016/j.jemermed.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 73.Spiller HA, Krenzelok EP, Grande GA, Safir EF, Diamond JJ. A prospective evaluation of the effect of activated charcoal before oral N-acetylcysteine in acetaminophen overdose. Ann Emerg Med. 1994;23:519–523. doi: 10.1016/s0196-0644(94)70071-0. [DOI] [PubMed] [Google Scholar]
- 74.Vulcano LA, Confalonieri O, Franci R, Tapia MO, Soraci AL. Efficacy of free glutathione and niosomal glutathione in the treatment of acetaminophen-induced hepatotoxicity in cats. Open Vet J. 2013;3:56–63. [PMC free article] [PubMed] [Google Scholar]
- 75.Khayyat A, Tobwala S, Hart M, Ercal N. N-acetylcysteine amide, a promising antidote for acetaminophen toxicity. Toxicol Lett. 2016;241:133–142. doi: 10.1016/j.toxlet.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 76.Shen F, Coulter CV, Isbister GK, Duffull SB. A dosing regimen for immediate N-acetylcysteine treatment for acute paracetamol overdose. Clin Toxicol (Phila) 2011;49:643–647. doi: 10.3109/15563650.2011.604034. [DOI] [PubMed] [Google Scholar]
- 77.Chan TY, Critchley JA. Adverse reactions to intravenous N-acetylcysteine in Chinese patients with paracetamol (acetaminophen) poisoning. Hum Exp Toxicol. 1994;13:542–544. doi: 10.1177/096032719401300806. [DOI] [PubMed] [Google Scholar]
- 78.Bailey B, McGuigan MA. Management of anaphylactoid reactions to intravenous N-acetylcysteine. Ann Emerg Med. 1998;31:710–715. doi: 10.1016/s0196-0644(98)70229-x. [DOI] [PubMed] [Google Scholar]
- 79.Chiew AL, Gluud C, Brok J, Buckley NA. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst Rev. 2018;2:CD003328. doi: 10.1002/14651858.CD003328.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wittebole X, Hantson P. Use of the molecular adsorbent recirculating system (MARS™) for the management of acute poisoning with or without liver failure. Clin Toxicol (Phila) 2011;49:782–793. doi: 10.3109/15563650.2011.624102. [DOI] [PubMed] [Google Scholar]
- 81.Hopkins RE, Dobbin M, Pilgrim JL. Unintentional mortality associated with paracetamol and codeine preparations, with and without doxylamine, in Australia. Forensic Sci Int. 2018;282:122–126. doi: 10.1016/j.forsciint.2017.11.026. [DOI] [PubMed] [Google Scholar]
- 82.Germani G, Theocharidou E, Adam R, Karam V, Wendon J, O'Grady J, Burra P, Senzolo M, Mirza D, Castaing D, Klempnauer J, Pollard S, Paul A, Belghiti J, Tsochatzis E, Burroughs AK. Liver transplantation for acute liver failure in Europe: outcomes over 20 years from the ELTR database. J Hepatol. 2012;57:288–296. doi: 10.1016/j.jhep.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 83.Bernal W, Hyyrylainen A, Gera A, Audimoolam VK, McPhail MJ, Auzinger G, Rela M, Heaton N, O'Grady JG, Wendon J, Williams R. Lessons from look-back in acute liver failure? A single centre experience of 3300 patients. J Hepatol. 2013;59:74–80. doi: 10.1016/j.jhep.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 84.Simões C, Santos S, Vicente M, Sousa Cardoso F. Epidemiology of Acute Liver Failure from a Regional Liver Transplant Center in Portugal. GE Port J Gastroenterol. 2018;26:33–39. doi: 10.1159/000487312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hadem J, Tacke F, Bruns T, Langgartner J, Strnad P, Denk GU, Fikatas P, Manns MP, Hofmann WP, Gerken G, Grünhage F, Umgelter A, Trautwein C, Canbay A Acute Liver Failure Study Group Germany. Etiologies and outcomes of acute liver failure in Germany. Clin Gastroenterol Hepatol. 2012;10:664–9.e2. doi: 10.1016/j.cgh.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 86.Gulmez SE, Larrey D, Pageaux GP, Lignot S, Lassalle R, Jové J, Gatta A, McCormick PA, Metselaar HJ, Monteiro E, Thorburn D, Bernal W, Zouboulis-Vafiadis I, de Vries C, Perez-Gutthann S, Sturkenboom M, Bénichou J, Montastruc JL, Horsmans Y, Salvo F, Hamoud F, Micon S, Droz-Perroteau C, Blin P, Moore N. Transplantation for acute liver failure in patients exposed to NSAIDs or paracetamol (acetaminophen): the multinational case-population SALT study. Drug Saf. 2013;36:135–144. doi: 10.1007/s40264-012-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gulmez SE, Larrey D, Pageaux GP, Bernuau J, Bissoli F, Horsmans Y, Thorburn D, McCormick PA, Stricker B, Toussi M, Lignot-Maleyran S, Micon S, Hamoud F, Lassalle R, Jové J, Blin P, Moore N. Liver transplant associated with paracetamol overdose: results from the seven-country SALT study. Br J Clin Pharmacol. 2015;80:599–606. doi: 10.1111/bcp.12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gulmez SE, Unal US, Lassalle R, Chartier A, Grolleau A, Moore N. Risk of hospital admission for liver injury in users of NSAIDs and nonoverdose paracetamol: Preliminary results from the EPIHAM study. Pharmacoepidemiol Drug Saf. 2018;27:1174–1181. doi: 10.1002/pds.4640. [DOI] [PubMed] [Google Scholar]
- 89.Cooper SC, Aldridge RC, Shah T, Webb K, Nightingale P, Paris S, Gunson BK, Mutimer DJ, Neuberger JM. Outcomes of liver transplantation for paracetamol (acetaminophen)-induced hepatic failure. Liver Transpl. 2009;15:1351–1357. doi: 10.1002/lt.21799. [DOI] [PubMed] [Google Scholar]
- 90.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, Lee WM Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 91.Larson AM. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11:525–548, vi. doi: 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]