

Abstract
Rationale:
Pathogenic variations in the lamin gene (LMNA) cause familial dilated cardiomyopathy (DCM). LMNA insufficiency caused by LMNA pathogenic variants is believed to be the basic mechanism underpinning LMNA-related DCM.
Objective:
To assess whether silencing of cardiac Lmna causes DCM and investigate the role of Yin Yang 1 (Yy1) in suppressing Lmna DCM.
Methods and Results:
We developed a Lmna DCM mouse model induced by cardiac specific Lmna shRNA. Silencing of cardiac Lmna induced DCM with associated cardiac fibrosis and inflammation. We demonstrated that upregulation of Yy1 suppressed Lmna DCM and cardiac fibrosis by inducing Bmp7 expression and preventing upregulation of Ctgf. Knockdown of upregulated Bmp7 attenuated the suppressive effect of Yy1 on DCM and cardiac fibrosis. However, upregulation of Bmp7 alone was not sufficient to suppress DCM and cardiac fibrosis. Importantly, upregulation of Bmp7 together with Ctgf silencing significantly suppressed DCM and cardiac fibrosis. Mechanistically, upregulation of Yy1 regulated Bmp7 and Ctgf reporter activities and modulated Bmp7 and Ctgf gene expression in cardiomyocytes. Downregulation of Ctgf inhibited TGFβ/Smad signaling in DCM hearts. Regulation of both Bmp7 and Ctgf further suppressed TGFβ/Smad signaling. In addition, co-modulation of Bmp7 and Ctgf reduced CD3+ T cell numbers in DCM hearts.
Conclusions:
Our findings demonstrate that upregulation of Yy1 or co-modulation of Bmp7 and Ctgf offer novel therapeutic strategies for the treatment of DCM caused by LMNA insufficiency.
Subject Terms: Animal Models of Human Disease, Basic Science Research, Fibrosis, Gene Therapy, Inflammation
Keywords: DCM, Cardiac fibrosis, Yy1, Bmp7, Ctgf, cardiomyopathy, gene therapy, transcription factors, fibrosis
Graphical Abstract
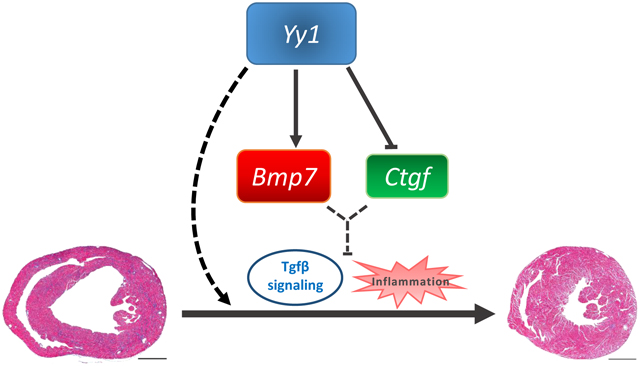
Introduction
Dilated cardiomyopathy (DCM) is one of the most common causes of heart failure, an increasingly pandemic condition characterized by impaired cardiac performance and high morbidity and mortality.1 DCM is defined by the presence of left ventricular (LV) enlargement and contractile dysfunction together with accumulation of interstitial fibrosis. DCM patients are also at high risk of ventricular arrhythmias and sudden death. As heart failure progresses, treatment options including evidence-based polypharmacy and cardiac resynchronisation therapy may become ineffective, leaving heart transplant as a final resort, but available to very few. Five year mortality after initial diagnosis of heart failure remains approximately 50%. Genetic variations in more than 50 genes have been implicated as causative in DCM2, 3. Among them, pathogenic variations in the lamin gene (LMNA), encoding A and C- type nuclear lamins (Lamin A/C), are the second most common cause of familial DCM and account for 5–8 % of all cases of autosomal dominant DCM4. LMNA insufficiency caused by pathogenic variants including missense and nonsense mutations is believed to be the basic mechanism underlying lamin-related DCM. Lmna knockout mice (Lmna −/−), generated previously, exhibit systemic defects including short lifespan, growth retardation, cardiac defects, muscular dystrophy, neuropathy and lipodystrophy5–7. Lmna −/− knockout or mutations result in aberrant signaling including autophagy/mTOR, MAPK, WNT/β-catenin and NF-κB signaling pathways8–12.Modulating activities of p38α, WNT/β-catenin and autophagy/mTOR partially suppresses DCM and/or cardiac fibrosis in Lmna animal models, indicating that regulation of aberrant signaling pathways could be beneficial to individuals with LMNA related DCM8, 11, 13.
LMNA is universally expressed in most cell types including cardiomyocytes (CMs) and noncardiomyocytes (non-CMs). It is not known whether silencing of cardiac LMNA causes DCM. To circumvent systemic non-cardiac defects or toxicity due to gene targeting, Cre expression or tamoxifen administration, we took advantage of the adeno-associated virus (AAV) system to modulate Lmna gene expression in vivo14–16. Cardiac specific shRNA based on a miRNA backbone and cardiac promoter can be used to reduce gene expression specifically in CMs. Recently, Lmna related DCM has been linked to the deregulation of cardiac cell cycle17, 18. Additionally, the transcription factor Yy1 is known to be associated with cell cycle progression19. It is not known whether upregulation of Yy1 could suppress DCM and cardiac fibrosis related to Lmna insufficiency. Moreover, it is unknown by what mechanisms and signaling pathways Yy1 regulates Lmna DCM and cardiac fibrosis. Here, we address these major gaps and identify therapeutic candidates that could be translated into treatment for DCM and cardiac fibrosis.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Animal protocols.
All mice were maintained and studied using protocols approved by the Institutional Animal Care and Use Committee (IACUC) of National University of Singapore. Animal work was undertaken in compliance with Singapore National Advisory Committee for Laboratory Animal Research guidelines. Relevant national and institutional guidelines and regulations were consulted before commencement of any animal work. All studies were conducted in male C57BL/6JINV (Jax) mice. Power calculations were performed to estimate sample sizes (error alpha 0.05, power 0.8). For virus injection, 50μl viruses were injected into the thoracic cavity of 1.5 week old pups via insulin syringe, avoiding the heart and lungs. We selected pups with body weight between 4.5 and 5 g. Overweight or underweight pups were excluded before animal grouping and virus administration. Pups were randomly assigned for injection. For heart harvesting, each mouse was anesthetized with 5% isoflurane and the heart was exposed by opening of the chest. 15% KCl was then injected into the inferior vena cava to achieve asystole at diastole, and the heart was rapidly isolated and flushed with D-PBS injected through the LV to wash out blood while clamping the aorta with Reynolds forceps. Half of the apex was isolated and immersed in RNALater (Qiagen, 76104) at room temperature for RNA extraction, while the other half was snap frozen in liquid nitrogen for protein extraction. The remaining section of the heart was fixed in 4% paraformaldehyde for 24 hours and subsequently embedded in paraffin.
Echocardiogram (Echo) and surface electrocardiogram (ECG).
Mouse cardiac dimensions and function were measured by echocardiography three and/or four weeks after virus transduction (VisualSonics, Vevo 2100, 40 Mhz-550S probe). Investigators were blinded to animal group identities during echo procedure and analysis. All mice were shaved to expose the chest area one day before measurement. During echo, 1.5% isoflurane mixed with oxygen were applied to each mouse, and cine of 300 frames of both B mode and M mode (left parasternal long and short axes) were recorded when heart rate was around 450–500 bpm. Measurements were processed by Vevo®LAB (VisualSonics Inc.). LV tracings were averaged from at least 3 consecutive heart beats of M-mode. LVDD (LV diastolic dimensions), LVWT (LV posterior wall thickness), EF (ejection fraction) and FS (fractional shortening) were obtained from short axis images.
Cell culture and transfection.
HEK293T (ATCC CRL-1573™) cells were cultured at 37 °C with 5% CO2, maintained in DMEM (Hyclone) supplemented with 10 % FBS (Hyclone), 1 mM sodium pyruvate, and 10 μg/ml gentamicin (Invitrogen 15750060). Transfection of shRNA constructs and other plasmids was performed using PEI (Polysciences. Inc, 24765–2) or Lipofectamine 3000 (Invitrogen L3000015) according to manufacturer’s instructions.
Recombinant adeno-associated virus 9 (rAAV9) production, purification and titration.
In brief, rAAVs were produced in HEK293T cells by transient triple plasmid transfection, including rAAV viral vector with gene to be delivered, helper plasmids pAdΔF6 and plasmid pAAV2/9 (Penn Vector Core). AAV constructs were propagated using Stbl3 competent cells (Thermo Fisher Scientific, C7373–03). Three days after transfection, viruses were collected from cell pellets and were purified by Optiprep density gradient medium (Sigma, D-1556). After concentration using centrifugal filters (Milipore, UFC910096), viruses were aliquoted and stored at −80°C. For virus titration, forward primer: gataaaagcagtctgggctttcaca and reverse primer: gagcccatataagcccaagctattg were designed to target the rAAV genome-containing particle, cTnT promoter region, and to determine the physical titers by qPCR.
Vector construction
shRNA candidates were designed to target 21 base-pair gene-specific regions (Invitrogen). The miR-155 backbone based shRNA cassette was inserted at the 3 prime end of the AAV-cTnT-EGFP vector. EGFP was replaced by Bmp7, Yy1 and Ccnd1 for gain of function studies. The sequences of shRNAs and cloning primers are as follows:
| Lmna shRNA-1 | agtctcgaatccgcattgaca |
| Lmna shRNA-2 | ggctaagaagcagcttcagga |
| LacZ shRNA | aaatcgctgatttgtgtagtc |
| Bmp7 shRNA | ctagtggaacatgacaaagaa |
| Ctgf shRNA | cctgtcaagtttgagctttct |
| Ccnd1 shRNA | cagatgtgaagttcatttcca |
| Bmp7-F | ccggctagcatgcacgtgcgctcgctgcgcg |
| Bmp7-R | ccgggtaccctagtggcagccacaggcccggac |
| Yy1-F | gggcaattgatggcctcgggcgacaccctctacat |
| Yy1-R | gggggtacctcactggttgtttttggctttagcg |
| Ccnd1-F | ccccaattgatggaacaccagctcctgtgctg |
| Ccnd1-R | cccggtacctcagatgtccacatctcgcacgtcg |
Promoter regions (~ 1 kb) of Bmp7 or Ctgf were amplified from C57BL/6JINV (Jax) mouse genomic DNA and cloned into pCAG-Cherry (Addgene plasmid # 73978) by replacement of the CAG promoter. Tgfb1, Bmp7 and Yy1 were amplified from mouse heart cDNA and cloned into pCAGIG (Addgene Plasmid #11159). Promoter cloning primers are as follows:
| Bmp7-F | atagctagcaaaaatccaagcctggacctcagta |
| Bmp7-R | tattctagagagtggatctggctgagtcttcttg |
| Ctgf-F | ataactagtaaaaatccaagcctggacctcagta |
| Ctgf-R | tatactagtgagtggatctggctgagtcttcttg |
RNAseq library preparation and next generation sequencing.
Total RNA from left ventricular tissue of male mice (n = 2 per group) was extracted to establish RNAseq libraries. RNA samples were pre-treated with Truseq Stranded Total RNA Library Prep kit (Illumina, RS-122–2201) to remove abundant cytoplasmic rRNA. The remaining intact RNA was fragmented using a chemical mix, followed by first- and second-strand cDNA synthesis using random hexamer primers. End-repaired fragments were ligated with a unique Illumina adapter. All individually indexed samples were subsequently pooled together and multiplexed for sequencing. Libraries were sequenced using the Illumina Hiseq 2000 sequencing system and paired-end 101bp reads were generated for analysis. Potential genetic candidates (log2 fold change < −1 or > 1, FDR < 0.05 and log2 CPM ≥ 3.32) were identified from DCM group (Lmna shRNA) compared to control group (Ctrl shRNA). Differentially expressed genes were uploaded to Morpheus for Hierarchical clustering and color-coded heat-map. Canonical pathways were analyzed by Gene Set Enrichment Analysis (GSEA, Broad Institute).
Quantitative real-time PCR (qPCR).
Transcription levels were quantified by qPCR. Total RNA was extracted using Trizol. cDNA was synthesized using Maxima First Strand kit (ThermoFisher, K1641) and qPCR was carried out by KAPA SYBR Fast qPCR Master Mix kit (KAPA Biosystems, KR0389). We used ΔCT to quantify relative expression levels and data were normalized to Ctcf expression. All qPCR primers are listed as follows:
| Primer | sequence | Primer | sequence |
|---|---|---|---|
| EGFP forward | cgaaggctacgtccaggagc | Tgfb1 forward | gaaggacctgggttggaagtggatc |
| EGFP reverse | cgatgttgtggcggatcttg | Tgfb1 reverse | tgtgttggttgtagagggcaaggac |
| Nppa forward | tttcaagaacctgctagaccacctg | Tgfb2 forward | attgctgccttcgccctctttacat |
| Nppa reverse | gcttttcaagagggcagatctatcg | Tgfb2 reverse | aggctgaggactttggtgtgttgag |
| Myh7 forward | agcattctcctgctgtttcctt | Tgfb3 forward | ccagatacttcgaccggatgagcac |
| Myh7 reverse | tgagccttggattctcaaacg | Tgfb3 reverse | tctccattgggctgaaaggtgtgac |
| Ctcf forward | atgtcacaccttacctttgcctgaa | Col1a1 forward | gagcctgagtcagcagattgagaac |
| Ctcf reverse | ccttcctgctgttcttcctcaaaat | Col1a1 reverse | cctgtctccatgttgcagtagacct |
| Bmp7 forward | agaatcgctccaagacgccaaagaa | Col1a2 forward | acccttctcactcctgaaggctcta |
| Bmp7 reverse | ctctccctcacagtagtaggcagca | Col1a2 reverse | tatgagttcttcgctggggtgttta |
| Ctgf forward | acacctaaaatcgccaagcctgtca | Postn forward | gccttctcttgatcgtcttctaggc |
| Ctgf reverse | aatggcaggcacaggtcttgatgaa | Postn reverse | cttgaggagtacgacgcagaagaag |
| Ccnd1 forward | atgagaacaagcagaccatccgcaa | Yy1 forward | cggggaataagaagtgggagcagaa |
| Ccnd1 reverse | cggtagcaggagaggaagttgttgg | Yy1 reverse | caggagggagtttcttgcctgtcat |
Histological and immunostaining analysis.
Heart samples were fixed in 4% paraformaldehyde for 24 hours, then embedded in paraffin and sectioned at 4 μm intervals. Paraffin samples were further treated with xylene (to remove paraffin) and re-hydrated. Hematoxylin and eosin (HE) was applied to observe myocyte architecture and Masson trichrome (MT) to identify cardiac fibrosis. Quantification of fibrosis was calculated as the blue-stained areas relative to total ventricular area, using ImageJ (NIH). For antigen retrieval of αSMA and CD3, samples were boiled in citrate buffer (pH 6.0) after the rehydration step. The following primary antibodies were used: PCM1 (SIGMA, HPA023374), Lamin A/C (Santa Cruz, sc-376248AF594), αSMA (Abcam, ab32575), CD3 (Agilent, A0452), Arginase-1(Cell Signaling Technology. Inc, 93668), Iba-1 (Wako Laboratory chemicals, 019–19741) and cTnI (Abcam, ab8295). DAPI was used for nuclear staining (ThermoFisher, D1306). Species-specific secondary antibody only controls were used to check for signal specificity. Total positive signals from three completed cross sections were counted for each heart sample by an investigator who was blinded to group identities, and data were normalized to total nucleus number or total ventricular area.
Western blots.
Frozen heart tissues were lysed in cold RIPA buffer with protease inhibitor (Sigma, 4693116001) and were homogenized with prechilled TissueLyser (Qiagen, 25/s, 2mins, 3 cycles) and metal beads. 20 μg of each sample was separated by SDS-page gel (10%) and transferred onto nitrocellulose membranes (0.2 μm, Biorad, 162–0112) for blotting. The blots were probed with a primary antibody followed by a secondary antibody conjugated to horseradish peroxidase. The following primary antibodies were used: phospho-Smad2 (Ser465/Ser467) (Cell Signaling Technology, 18338), Ctgf (Santa Cruz Biotechnology, Inc, sc-365970), Bmp7 (Proteintech Group, Inc 12221–1-AP), Ccnd1 (Abcam, ab16663) and Lamin B1 (Abcam, ab16048). The secondary antibody used was Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP (Thermofisher, A16035). Protein levels on the blots were detected using the enhanced chemiluminescence system (GE Healthcare, RPN2106) according to the manufacturer’s instructions. Protein band intensity was quantified using Image J (NIH, 1.52e) and protein levels were normalized to Lamin B1.
Statistical analyses.
Statistical analysis was performed by Prism 7.04 (GraphPad Software, La Jolla, California). Sample distribution was tested for normality using a Shapiro-Wilk normality test. For data that followed a normal Gaussian distribution, P value between two groups was obtained by two-tailed, unpaired T-test with Welch correction, and one-way ANOVA with Tukey’s multiple comparisons test was performed for comparison of multiple groups. For data that depart from normality, differences between groups were analyzed by Mann-Whitney test. Quantitative data are shown as mean ± SD.
Results
Cardiac Lmna shRNA induces dilated cardiomyopathy.
We developed a Lmna DCM mouse model induced by either of two independent Lmna shRNAs delivered specifically to cardiomyocytes by AAV as previously described (Figure 1a, Table 1)14. Each Lmna shRNA caused impaired cardiac contraction, enlarged left ventricle (LV) heart chamber size and reduced LV wall thickness, coupled with interstitial fibrosis. Immunostaining indicated that most CM nuclei marked by PCM-1 in Lmna shRNA hearts had reduced Lamin A/C signal (Figure 1b). Lamin A/C was significantly reduced by ~ 75% in CMs derived from Lmna shRNA hearts (Figure 1c). To examine pathways regulated by Lmna shRNA, we profiled control and Lmna shRNA hearts by RNAseq (Online Figure Ia, b). Hierarchical clustering uncovered genes significantly dysregulated in the Lmna shRNA group compared to the control shRNA group. We analyzed this gene list by Gene Set Enrichment Analysis (GSEA, Broad Institute). Top significantly enriched upregulated gene sets identified “Matrisome”, “Core Matrisome” and “ECM Glycoproteins” that were common in two independent Lmna shRNA groups by canonical pathway analysis. Downregulated gene sets were enriched in “TCA cycle and respiratory electron transport”, “respiratory electron transport ATP synthesis” and “Oxidative Phosphorylation”. Furthermore, common dysregulated gene sets identified hallmark signature associated with fibrosis and inflammation (Online Figure Ic). The results of pathway analysis were concordant with Lmna DCM phenotypes including impaired cardiac performance with fibrosis/extracellular matrix production (ECM). Consistent with cardiac fibrosis and activation of matrisome/ECM pathways, Lmna DCM hearts showed a significant upregulation of phosphorylated Smad2 (p-Smad2) and the myofibroblast marker αSMA (Figure 1d, e). Additionally, Lmna DCM was also associated with cardiac inflammation indicated by upregulation of macrophage marker Iba-1 and T cell marker CD3 (Figure 1f, g). The two independent Lmna shRNAs both induced similar DCM, cardiac fibrosis and cardiac inflammation, indicating the phenotype is not due to a non-specific effect of shRNA. We selected Lmna DCM induced by Lmna shRNA-1 for subsequent assessment. Taken together, these results suggest Lmna DCM activates TGFβ signaling and induces cardiac fibrosis coupled with inflammation.
Figure 1. Cardiac specific Lmna shRNA induces DCM in mice.
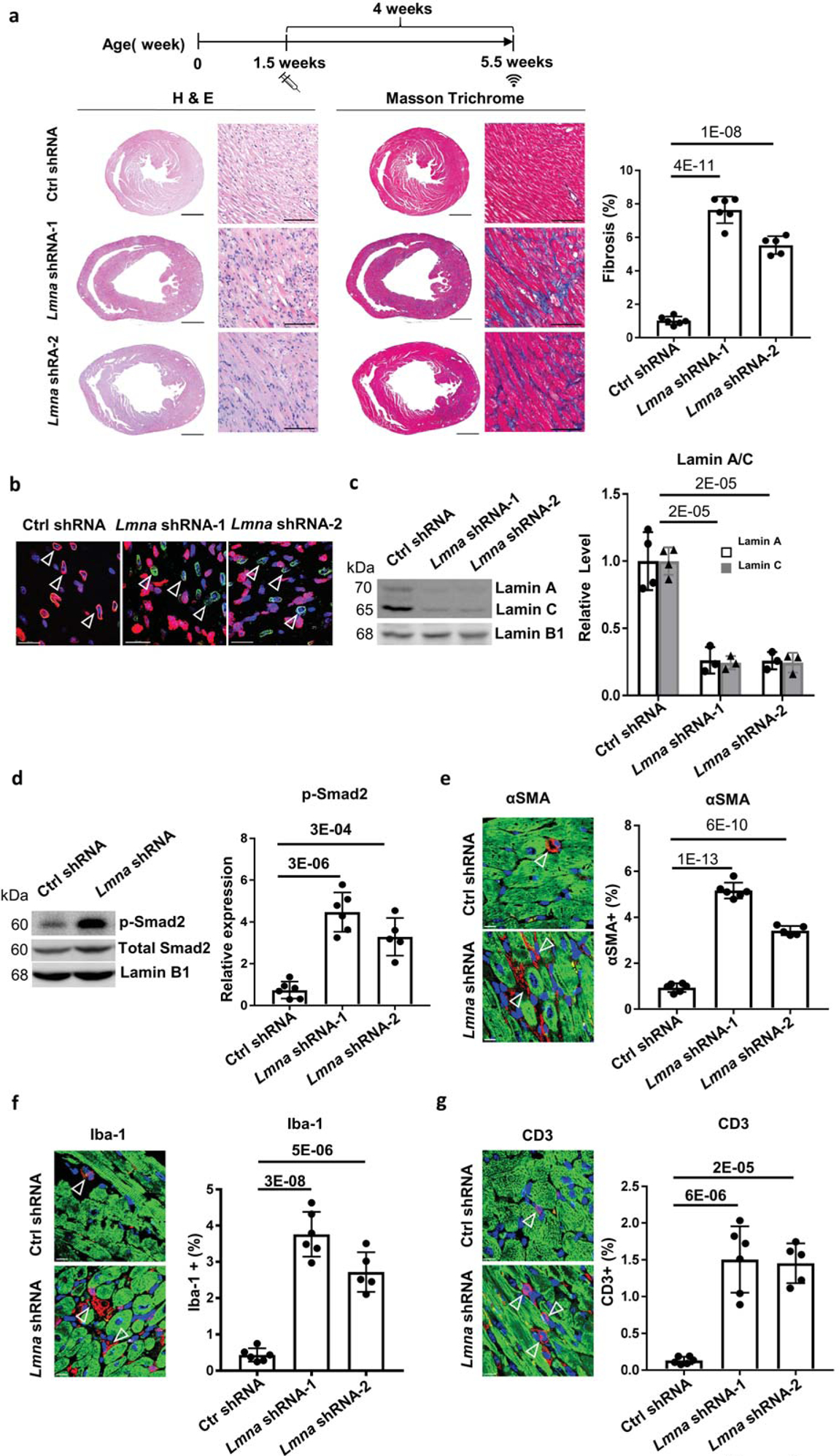
(a) Experimental timeline showing timepoints of virus injection and echocardiogram. Cardiac performance was assessed by echocardiogram at 5.5 week old. H&E and Masson Trichrome (MT) staining was performed on paraffin heart sections taken 4 weeks after control shRNA and Lmna shRNA transduction. Quantification of myocardial fibrosis of MT sections is shown, virus dose, 2.0E+13 vg/kg, n ≥ 5. Scale bars: 1000 μm for complete heart images; 100 μm for enlarged images. (b) Paraffin heart section immunostained for Lamin A/C (red), PCM1 (green) and DAPI (blue) in mice transduced with control shRNA and Lmna shRNA, scale bar = 100 μm. (c, d) Western blot and quantitative analysis of Lamin A/C protein levels in isolated cardiomyocytes (c) and phospho-Smad2 protein levels in mouse heart tissues (d), n ≥ 3. Data were normalized to Lamin B1. (e-g) Paraffin heart sections (left) and quantifications (right) of (e) αSMA (red), (f) Iba-1 (red) and (g) CD3 (red), cTnI (green) and DAPI (blue) positive cells in mice transduced with control shRNA (top) and Lmna shRNA (bottom), n ≥ 5, scale bar = 50 μm.
Table 1. Effect of Lmna shRNA on cardiac function in mice.
Effect of Lmna shRNA-1 and Lmna shRNA-2 versus Ctrl shRNA on mice at a dose of 2.0E+13 vg/kg assessed at 5.5 weeks. P values represent comparisons to mice transduced with control shRNA at respective age. LVDD, left ventricular diastolic dimension; LVWT, LV wall thickness; EF, ejection fraction; FS, fractional shortening.
| shRNA | Age (weeks) | n | LVDD (mm) | P | LVWT (mm) | P | EF (%) | P | FS (%) | P |
|---|---|---|---|---|---|---|---|---|---|---|
| Ctrl shRNA | 5.5 | 6 | 3.81 ± 0.10 | 0.68 ± 0.10 | 57.80 ± 4.81 | 29.97 ± 3.20 | ||||
| Lmna shRNA-1 | 6 | 4.25 ± 0.11 | 2E-06 | 0.45 ± 0.08 | 2E-05 | 12.13 ± 3.16 | 4E-11 | 5.33 ± 1.79 | 8E-11 | |
| Lmna shRNA-2 | 5 | 4.30 ± 0.32 | 5E-04 | 0.43 ± 0.12 | 4E-03 | 11.66 ± 10.66 | 1E-05 | 5.42 ± 5.01 | 1E-05 |
Yy1 suppresses Lmna DCM and cardiac fibrosis
To assess whether Yy1 could suppress Lmna DCM, we treated Lmna DCM mice with either Yy1 or EGFP delivered by AAV at 1.5 weeks old. Four weeks later, ejection fraction (EF) was assessed by echocardiography. EF of Lmna DCM mice treated with Yy1 was significantly improved compared to those treated with EGFP, suggesting that Yy1 suppresses DCM induced by Lmna insufficiency (Table 2). Importantly, cardiac fibrosis was significantly reduced by Yy1 in Lmna DCM hearts (Figure 2a). Fibrosis markers Col1a1 and Col1a2 expression were significantly suppressed by Yy1 (Figure 2b). Consistent with this data, αSMA-positive myofibroblast numbers were significantly reduced in Lmna DCM hearts treated with Yy1 (Figure 2c).
Table 2. Effect of Yy1 on Lmna DCM in mice.
Effect of Yy1 or EGFP at a dose of 0.5E+13 vg/kg on Lmna DCM mice assessed at 5.5 weeks. P values represent comparisons to Lmna DCM mice treated with EGFP. LVDD, left ventricular diastolic dimension; LVWT, LV wall thickness; EF, ejection fraction; FS, fractional shortening.
| Lmna DCM Treatment | Age (weeks) | n | LVDD (mm) | P | LVWT (mm) | P | EF (%) | P | FS (%) | P |
|---|---|---|---|---|---|---|---|---|---|---|
| EGFP | 5.5 | 5 | 4.23 ± 0.10 | 0.40 ± 0.07 | 14.98 ± 4.39 | 6.64 ± 2.03 | ||||
| Yy1 | 6 | 3.97 ± 0.18 | 0.02 | 0.51 ± 0.06 | 0.03 | 27.40 ± 2.55 | 0.0002 | 12.54 ± 1.27 | 0.0002 |
Figure 2. Yy1 suppresses Lmna DCM and cardiac fibrosis.
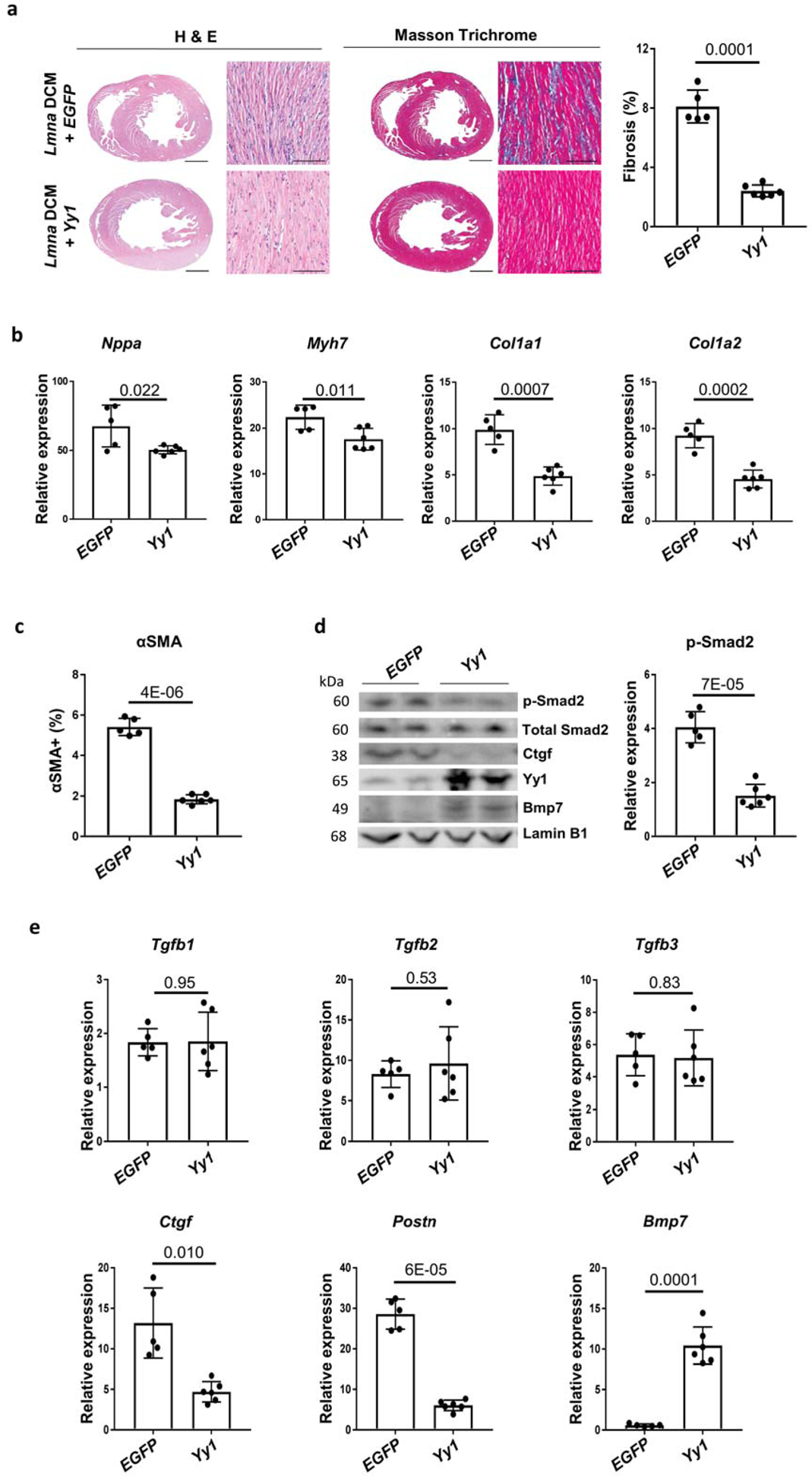
(a) H&E and MT staining of paraffin heart sections of Lmna DCM mice treated with EGFP or Yy1. Quantification of myocardial fibrosis of MT sections is shown, n ≥ 5. Scale bars: 1000 μm for complete heart images; 100 μm for enlarged images. (b) Quantitative real-time PCR analyses of Nppa, Myh7, Col1a1 and Col1a2 expressions in EGFP and Yy1 treated groups. Mouse hearts were harvested 4 weeks after transduction, n ≥ 5. (c) Quantification of αSMA positive cells in paraffin heart sections from Lmna DCM mice treated with EGFP and Yy1, n ≥ 5. (d) Western blot and quantitative analysis of phospho-Smad2 (p-Smad2) protein levels in mouse heart tissue of Lmna DCM mice treated with EGFP and Yy1, n ≥ 5. Data were normalized to Lamin B1. (e) Quantitative real-time PCR analyses of Tgfb1, Tgfb2, Tgfb3, Ctgf, Postn and Bmp7 in Lmna DCM mice treated with EGFP and Yy1, n ≥ 5.
To assess whether Yy1 suppresses Lmna DCM through regulating the cardiac cell cycle, we constructed an AAV vector co-expressing the Yy1 gene and Ccnd1 shRNA (designated as Yy1-Ccnd1 shRNA). Ccnd1 is a cell cycle related gene regulated by Yy120. Although upregulation of Ccnd1 by Yy1 was abolished in the group treated with Yy1-Ccnd1 shRNA, rescue of cardiac performance and fibrosis was not compromised by the Ccnd1 shRNA (Online Figure IIa, b, Online Table I). Col1a1 and Col1a2 expression remained significantly suppressed by Yy1-Ccnd1 shRNA (Online Figure IIc). Next we examined whether upregulation of Ccnd1 could suppress Lmna DCM. EF of Lmna DCM mice treated with Ccnd1 was not improved compared to those treated with EGFP (Online Table II). Cardiac fibrosis was not affected by upregulation of Ccnd1 in Lmna DCM hearts (Online Figure IId, e, f). Consistent with this, Col1a1 and Col1a2 expression were not suppressed upon upregulation of Ccnd1 (Online Figure IIg).
We therefore hypothesized that other downstream targets of Yy1, especially secreted factors, might regulate Lmna DCM and cardiac fibrosis. TGFβ/Smad signaling is believed to play a key role in cardiac performance and fibrosis21. p-Smad2 was elevated in Lmna DCM hearts, and this was significantly reduced ~ 65% by Yy1, indicating that TGF-β/Smad signaling is suppressed by Yy1 (Figure 2d). Although the expression of secreted TGFβ cytokines (Tgfb1/2/3) was not significantly affected by Yy1, Ctgf (connective tissue growth factor) and Postn (periostin) were significantly suppressed by Yy1 (Figure 2e). Ctgf and periostin are secreted ECM proteins, which are known to be responsive to TGFβ signaling22, 23. A member of the TGF-β superfamily, Bmp7 is dysregulated in the mouse transverse aortic constriction (TAC) model and clinical aortic stenosis24, 25. Importantly, Bmp7 was significantly upregulated by ~ 10 fold in Lmna DCM hearts treated with Yy1.
Bmp7 serves as a key downstream target of Yy1 in suppressing Lmna DCM and cardiac fibrosis
To assess whether Bmp7 is an important downstream target of Yy1, we generated an AAV vector co-expressing the Yy1 gene and Bmp7 shRNA (designated as Yy1-Bmp7 shRNA). We treated Lmna DCM mice with AAV expressing Yy1-Bmp7 shRNA, Yy1-Ctrl shRNA or EGFP-Ctrl shRNA. Cardiac performance and fibrosis was assessed by echocardiography and histology (Table 3, Figure 3a). Importantly, rescue of cardiac performance and fibrosis was compromised by Bmp7 reduction, suggesting that Bmp7 serves as a key downstream target of Yy1 in suppressing Lmna DCM and fibrosis. Yy1-Bmp7 shRNA significantly abolished upregulation of Bmp7 and attenuated the downregulation of fibrosis markers Col1a1 and Col1a2 compared to Yy1-Ctrl shRNA (Figure 3b).
Table 3. Effect of Yy1-Bmp7 shRNA on Lmna DCM in mice.
Effect of Yy1-Bmp7 shRNA, Yy1-Ctrl shRNA or EGFP-Ctrl shRNA at a dose of 0.5E+13 vg/kg on Lmna DCM assessed at 5.5 weeks. P values represent comparisons to Lmna DCM mice treated with EGFP-Ctrl shRNA. LVDD, left ventricular diastolic dimension; LVWT, LV wall thickness; EF, ejection fraction; FS, fractional shortening.
| Lmna DCM Treatment | Age (weeks) | n | LVDD (mm) | P | LVWT (mm) | P | EF (%) | P | FS (%) | P |
|---|---|---|---|---|---|---|---|---|---|---|
| EGFP-Ctrl shRNA | 5.5 | 7 | 4.34 ± 0.17 | 0.41 ± 0.05 | 13.55 ±3.53 | 5.99 ± 1.63 | ||||
| Yy1-Ctrl shRNA | 8 | 4.09 ± 0.13 | 0.005 | 0.48 ± 0.06 | 0.03 | 28.98 ± 3.66 | 6E-07 | 13.38 ± 1.89 | 1E-06 | |
| Yy1-Bmp7 shRNA | 8 | 4.58 ± 0.17 | 0.02 | 0.51 ± 0.07 | 0.01 | 14.07 ±2.78 | 0.76 | 6.25 ± 1.29 | 0.74 |
Figure 3. Co-regulation of Bmp7 and Ctgf suppresses Lmna DCM and cardiac fibrosis.
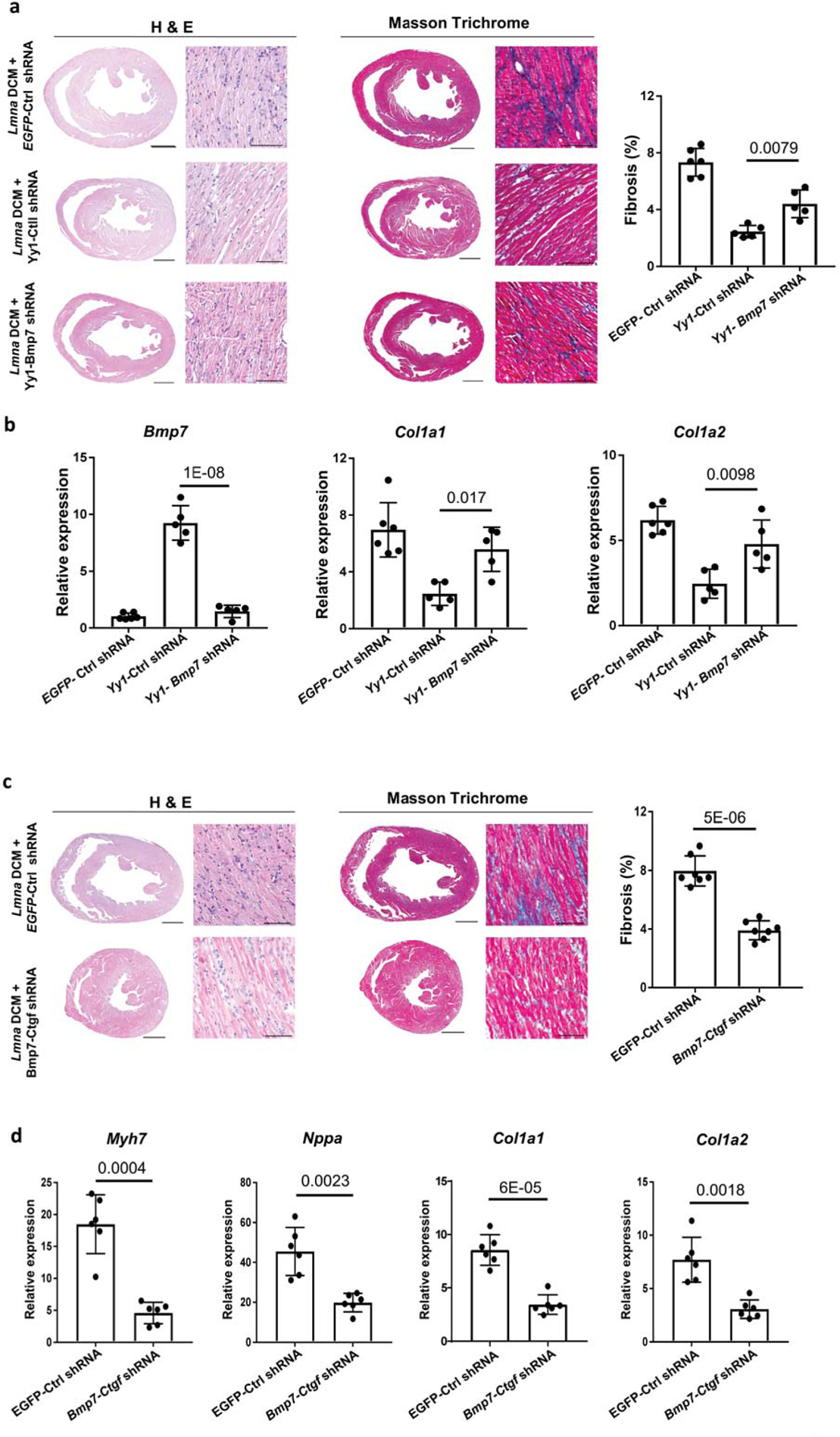
(a and c) H&E (left panel) and MT (middle panel) staining of paraffin heart sections of Lmna DCM mice 4 weeks after (a) EGFP-Ctrl shRNA, Yy1-Ctrl shRNA or Yy1-Bmp7 shRNA transduction and (c) EGFP-Ctrl shRNA or Bmp7-Ctgf shRNA transduction. Quantification of myocardial fibrosis (right panel) from MT-stained sections, n ≥ 6. Scale bars: 1000 μm for complete heart images; 100 μm for enlarged images. (b) Quantitative real-time PCR analyses of Bmp7, Col1a1 and Col1a2 expression in Lmna DCM groups treated with EGFP-Ctrl shRNA, Yy1-Ctrl shRNA or Yy1-Bmp7 shRNA, n ≥ 5. (d) Quantitative real-time PCR analyses of Myh7, Nppa, Col1a1 and Col1a2 expressions in Lmna DCM groups treated with EGFP-Ctrl shRNA or Bmp7-Ctgf shRNA groups, n ≥ 6.
Co-modulation of Bmp7 and Ctgf suppresses Lmna DCM and cardiac fibrosis.
Bmp7 antagonizes fibrogenesis in renal and pulmonary disease26, 27. To assess whether upregulation of Bmp7 is sufficient to suppress Lmna DCM and cardiac fibrosis, we treated Lmna DCM mice with AAV expressing Bmp7 (Online Table III, Online Figure IIIa). Unexpectedly, we did not observe a rescue effect of Bmp7 on cardiac performance and fibrosis in Lmna DCM mice, suggesting upregulation of Bmp7 alone is not sufficient to suppress Lmna DCM and cardiac fibrosis (Online Figure IIIb). We deduced that additional factors are required to work together with Bmp7. Ctgf and Postn, the downstream responsive genes of TGFβ, were not affected by upregulation of Bmp7, suggesting that upregulation of Bmp7 does not regulates TGFβ signaling in Lmna DCM (Online Figure IIIc). Ctgf is highly expressed in CMs and upregulated in response to cardiac injury upon TGF-β stimulation28, 29. To assess whether Ctgf is involved in suppressing Lmna DCM, we reduced Ctgf expression in Lmna DCM mice by Ctgf shRNA. Similar to upregulation of Bmp7, downregulation of Ctgf did not significantly restore cardiac performance, or reduce fibrosis and related markers in Lmna DCM mice (Online Table IV, Online Figure IIIc, IIId). To assess whether modulating both Bmp7 and Ctgf could work cooperatively to suppress Lmna DCM and cardiac fibrosis, we generated an AAV vector expressing both the Bmp7 gene and Ctgf shRNA (designated as Bmp7-Ctgf shRNA). Lmna DCM mice were treated with either Bmp7-Ctgf shRNA or EGFP-control shRNA. Importantly, Bmp7-Ctgf shRNA significantly restored cardiac performance in Lmna DCM mice compared to those treated with EGFP-control shRNA (Table 4). Cardiac fibrosis was significantly reduced in Lmna DCM mice treated with Bmp7-Ctgf shRNA (Figure 3c). Furthermore, markers of heart failure and cardiac fibrosis including Myh7, Nppa, Col1a1 and Col1a2 were also significantly suppressed by Bmp7-Ctgf shRNA (Figure 3d). We therefore hypothesize that Yy1 suppresses Lmna DCM through upregulation of a member of the TGF-β superfamily (Bmp7) and downregulation of a downstream responsive gene of TGFβ signaling pathway (Ctgf).
Table 4. Effect of Bmp7-Ctgf shRNA on Lmna DCM in mice.
Effect of Bmp7-Ctgf shRNA at a dose of 2.0E+13 vg/kg on Lmna DCM mice assessed at 5.5 weeks. values represent comparisons to Lmna DCM mice treated with EGFP-Ctrl shRNA. LVDD, left ventricular diastolic dimension; LVWT, LV wall thickness; EF, ejection fraction; FS, fractional shortening.
| Lmna DCM Treatment | Age (weeks) | n | LVDD (mm) | P | LVWT (mm) | P | EF (%) | P | FS (%) | P | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EGFP-Ctrl shRNA | 5.5 | 7 | 4.25 ± 0.15 | 0.44 ±0.09 | 14.31 ± 4.43 | 6.34 ± 2.04 | |||||
| Bmp7-Ctgf shRNA | 7 | 3.72 ± 0.06 | 2E-06 | 0.61 ±0.07 | 0.003 | 46.05 ± 4.89 | 2E-08 | 22.43 ± 2.62 | 2E-08 |
Upregulation of Yy1 induces Bmp7 and suppresses Ctgf in cardiomyocytes.
Bmp7 was induced, while Ctgf was downregulated by upregulation of Yy1 in Lmna DCM mice. To assess whether Yy1 regulates Bmp7 and Ctgf promoter activity, we cloned Bmp7 and Ctgf reporters harboring the promoter regions of Bmp7 or Ctgf before a Cherry reporter gene (Online Figure IVa). When co-transfected with Bmp7-Cherry reporter in 293T cells, Yy1 significantly enhanced the activity of Bmp7-Cherry reporter, suggesting Yy1 can directly regulate Bmp7 promoter activity (Online Figure IVb). Yy1 did not modulate the activity of Ctgf-Cherry reporter, indicating Yy1 does not regulate basal promoter activity of Ctgf (Online Figure IVc). To assess whether Yy1 prevents upregulation of Ctgf promoter activity, we used Tgfb1 to activate the Ctgf-Cherry reporter. Importantly, the enhanced activity of Ctgf reporter following Tgfb1 treatment was abolished by Yy1, suggesting Yy1 can also regulate the promoter activity of Ctgf (Online Figure IVd). To examine whether Yy1 regulates Bmp7 and Ctgf expression in CMs, we isolated CMs from control, Lmna DCM or Lmna DCM mice treated with Yy1 as described (Online Figure IVe)30. Bmp7 expression was reduced in CMs isolated from Lmna DCM mice. Upregulation of Yy1 significantly induced Bmp7 expression. Conversely, Ctgf was significantly induced in CMs isolated from Lmna DCM mice, and this upregulation was abolished by Yy1 (Online Figure IVf).
Bmp7 and Ctgf modulate TGFβ signaling and cardiac inflammation.
To dissect the mechanisms of Bmp7 and Ctgf in suppressing DCM, we compared TGFβ signaling and cardiac inflammation in Lmna DCM hearts treated with Bmp7, Ctgf shRNA or Bmp7-Ctgf shRNA. Upregulation of p-Smad2 in Lmna DCM hearts was not significantly affected by Bmp7, suggesting that upregulation of Bmp7 does not suppress TGFβ signaling (Figure 4a). p-Smad2 elevation was however significantly reduced ~ 30% by Ctgf shRNA (Figure 4b). Similar to the suppressive effect of Yy1, Bmp7-Ctgf shRNA significantly decreased p-Smad2 further by ~ 60%, suggesting that profound inhibition of TGFβ/Smad signaling is responsible for suppressing Lmna DCM and cardiac fibrosis (Figure 4c). Immunostaining of macrophage marker Iba-1 showed infiltration of macrophages in Lmna DCM hearts (Figure 1f, 4d). The increased Iba-1 signal in Lmna DCM hearts was not significantly affected by Bmp7 or Ctgf shRNA, suggesting that total macrophage numbers are not affected by Bmp7 or Ctgf. On a simplified basis, macrophages may be polarized into two broad types, M1 (classically activated macrophages) and M2 (alternatively activated macrophages)31. The expression of M2 macrophages markers Arg-1 and Chil3 were induced more in Lmna DCM hearts treated with Bmp7 compared to Ctgf shRNA (Figure 4e). However, the number of Arg-1 positive cells was not significantly increased in Lmna DCM hearts treated with Bmp7 compared to EGFP, suggesting Bmp7 has a limited role in regulating cardiac macrophage polarization (Figure 4f). Interestingly, CD3+ T cell numbers in Lmna DCM hearts were reduced by Bmp7-Ctgf shRNA, but not Bmp7 or Ctgf shRNA individually (Figure 4g). Consistent with previous results, modulation of Bmp7 or Ctgf alone is not sufficient to suppress DCM, cardiac fibrosis and inflammation. Taken together, these results suggest that modulation of both Bmp7 and Ctgf suppresses DCM and cardiac fibrosis by inhibiting the TGFβ/Smad signaling pathway and regulating cardiac inflammation.
Figure 4. Bmp7 and Ctgf regulate cardiac inflammation and TGFβ signaling.
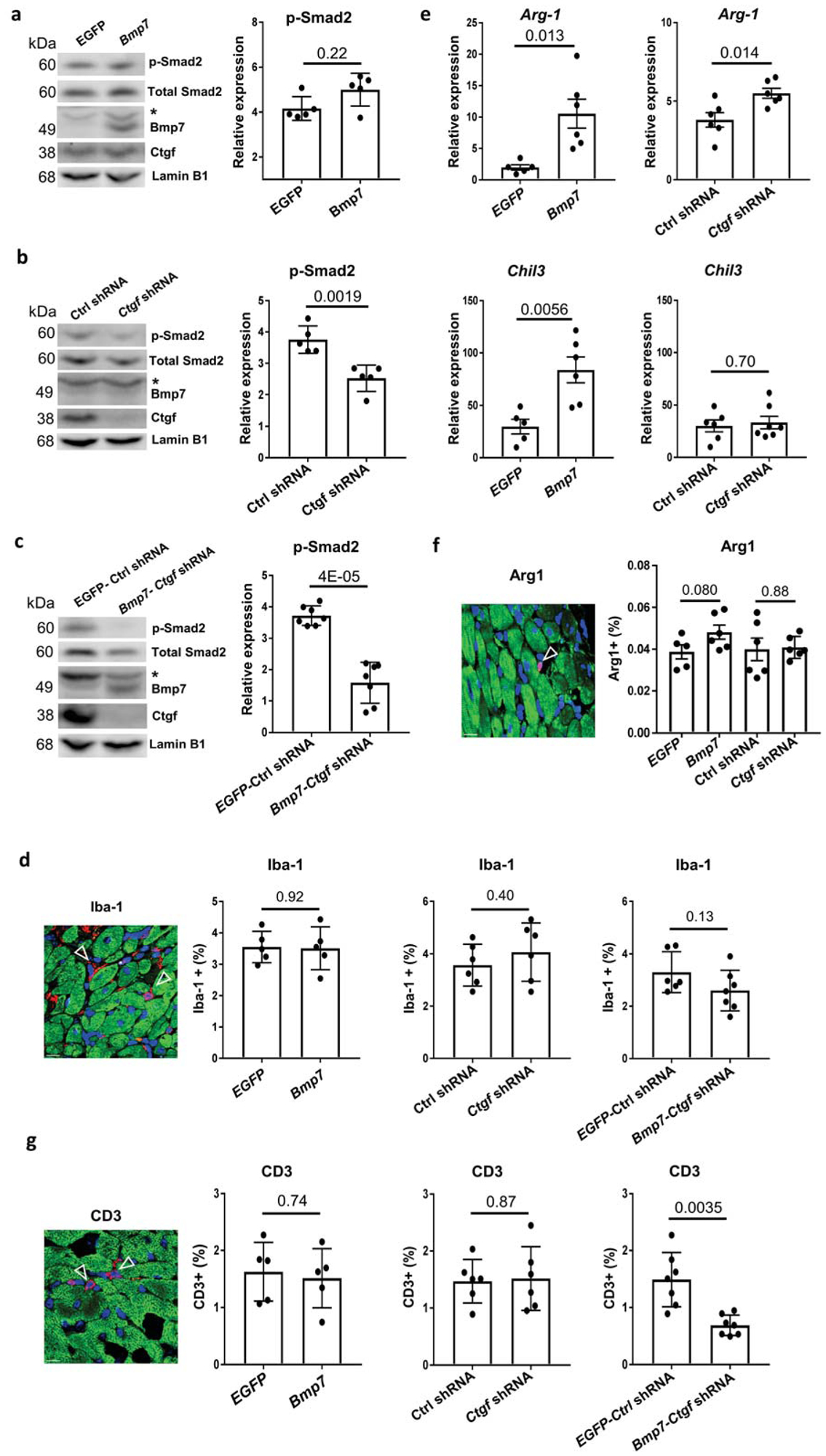
(a-c) Western blot (left panel) and quantitative analysis (right panel) of phospho-Smad2 (p-Smad2) protein levels in mouse heart tissue of Lmna DCM groups treated with (a) EGFP or Bmp7; (b) Ctrl shRNA or Ctgf shRNA or (c) EGFP Ctrl shRNA or Bmp7-Ctgf shRNA, n ≥ 5. Data were normalized to Lamin B1.* indicates the non-specific band. (d, f and g) Paraffin heart section staining (left panel) and quantifications (right pane) of (d) Iba-1 (red), (f) Arginase 1 (red) and (g) CD3 (red), cTnI (green) and DAPI (blue) positive cells in Lmna DCM groups treated with EGFP, Bmp7, Ctrl shRNA, Ctgf shRNA, EGFP-Ctrl shRNA or Bmp7-Ctgf shRNA, scale bar = 50 μm, n ≥5. (e) Quantitative real-time PCR analyses of Arg-1 and Chl3 expression in Lmna DCM groups treated with EGFP, Bmp7, Ctrl shRNA, Ctgf shRNA, n ≥ 5.
Discussion
Pathogenic variants in LMNA are common causes of familial DCM with penetrance exceeding 90%32, 33. LMNA insufficiency due to pathogenic missense or nonsense mutations is suggested as a causal mechanism. Lmna knockout mice (−/−) exhibited systemic defects in addition to DCM6. Lmna +/−heterozygote mice developed late onset of DCM, suggesting a dose dependent effect in Lmna related DCM34. In addition, DCM mice induced by mutated Lmna (nPLAO/nPLAO) was less severe compared to Lmna (nPLAO/-) DCM mice, suggesting that certain LMNA related DCM is caused by insufficient LMNA, rather than toxic mutant LMNA35. LMNA is expressed in most cell types including CMs and non-CMs in heart tissues. It was previously unclear whether CM specific silencing of LMNA is sufficient to induce DCM. Here, without multiple crossing of compound genetic modified mice or overexpression of Cre or tamoxifen administration, we generated a new DCM mouse model by silencing Lmna specifically in CMs. This Lmna model resulted in a typical DCM phenotype including enlarged LV, reduced LV wall thickness and markedly impaired systolic function. Our Lmna DCM model showed a significant increase of cardiac interstitial fibrosis accompanied by cardiac inflammation. By reducing Lmna expression specifically in CMs, we provide a new Lmna DCM model complementary to genetic models currently available to study Lmna related diseases.
We uncovered that upregulation of Yy1 suppressed Lmna DCM and cardiac fibrosis by upregulating Bmp7 and downregulating Ctgf gene expression. Bmp7 was one important downstream target of Yy1 in suppressing DCM and cardiac fibrosis in the Lmna DCM model. Bmp7 is believed to suppress fibrosis by activating Smad1/5/8 and counteracting the TGFβ/Smad2/3 signaling pathway36, 37. TGFβ signaling is known to suppress cardiac Bmp7 expression24. In addition, upregulation of Bmp7 inhibits endothelial mesenchymal transition (EndMT) and cardiac fibrosis by opposing TGF-β signaling38. However, upregulation of Bmp7 in our model was not sufficient to suppress Lmna DCM/cardiac fibrosis and upregulation of TGFβ/Smad signaling. Instead, TGFβ/Smad signaling was dramatically suppressed by co-modulation of Bmp7 and Ctgf. Lmna DCM hearts showed infiltration of inflammatory cells including macrophages and T cells. Upregulation of Bmp7 alone did not affect macrophage infiltration, macrophage polarization or T cell infiltration in Lmna DCM hearts. Consequently, Bmp7 did not suppress DCM and cardiac fibrosis. Recently, blocking of activated T cells was shown to prevent progressive left ventricular dilatation in myocardial infarction models39, 40. Therefore, the observed reduction of T cell infiltration following co-modulation of Bmp7 and Ctgf may also play a role in attenuating Lmna DCM.
Ctgf is a known downstream gene of the TGFβ signaling pathway and is highly induced in various cardiovascular diseases28. Ctgf is believed to be a pro-fibrotic factor in many organs including the heart29. In our model, silencing of Ctgf reduced p-Smad2, indicating a positive feed backup loop between Ctgf and the TGFβ/Smad signaling pathway. Again, silencing of Ctgf alone did not significantly suppress Lmna DCM and cardiac fibrosis or inflammation. Thus, modulating individual downstream targets of the Yy1 gene may be insufficient to recapitulate its suppressive role in Lmna DCM. We reveal that Yy1 indeed played a yin yang role in transcriptional regulation of Bmp7 and Ctgf, reflecting its capacity as either an activator or repressor in gene regulation41. Importantly, we reveal that Bmp7 and Ctgf control multiple key modalities of Lmna DCM pathology, suggesting that combination treatment may be beneficial to individuals with Lmna DCM and possibly other cardiomyopathies.
AAV is a relatively safe, and increasingly used delivery tool for gene therapy42. Bmp7 delivered by AAV could overcome issues with the relatively short half-life of Bmp7 by intravenous or intraperitoneal administration43. Silencing of Ctgf by shRNA also represents an alternative to anti-CTGF antibodies which have been shown to improve outcomes in animal models of fibrotic disease44. Indeed, an anti-CTGF antibody is under evaluation in a clinical trial for treatment of idiopathic pulmonary fibrosis (IPF)45. It will be of great interest to know whether anti-CTGF works together with Bmp7 to suppress Lmna DCM and cardiac fibrosis. Taken together, our findings provide substantial supporting data for translational research to treat LMNA related DCM.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
LMNA mutations causes many defects including DCM.
Lmna knockout mice exhibit systemic defects in addition to DCM
What New Information Does This Article Contribute?
Cardiac Lmna insufficiency causes DCM coupled with cardiac fibrosis.
Upregulation of Yy1 suppresses Lmna DCM and cardiac fibrosis independent of cardiac cell cycle regulation.
Yy1 regulates its downstream targets Bmp7 and Ctgf.
Bmp7 serves as an important downstream gene of Yy1 in suppression of Lmna DCM and cardiac fibrosis.
Simultaneous upregulation of Bmp7 and suppression of Ctgf suppresses Lmna DCM and cardiac fibrosis.
LMNA mutants causes many defects including dilated cardiomyopathy (DCM). There is a lack of information on whether cardiac specific modulation of LMNA induces DCM. Herein, we reveal that reduction of cardiac Lmna causes DCM coupled with cardiac fibrosis in mice. Moreover, we demonstrate that upregulation of a transcription factor Yin Yang 1 (Yy1) suppresses Lmna DCM and cardiac fibrosis through regulation of its downstream genes, Bmp7 and Ctgf. Interestingly, modulation of Bmp7 or Ctgf individually was not sufficient to suppress DCM and cardiac fibrosis. Importantly, upregulation of Bmp7 together with Ctgf silencing significantly suppressed DCM and cardiac fibrosis through inhibition of TGFβ/Smad signaling. Our findings uncover a novel role of Yy1 and its downstream genes, and provide a solid foundation for the development of Yy1, Bmp7 or Ctgf as therapeutic targets for DCM and cardiac fibrosis.
Acknowledgments
We thank Ms. Zenia Tiang (Genome Institute of Singapore, Singapore) for RNAseq technical assistance. We thank Dr. Kenji Onoue (Nara Medical University, Japan) and Dr. Mark Richards (NUHS, Singapore) for discussion and comments on the manuscript.
Sources Of Funding
This work was supported by grants from NMRC (J. J, R.F, NMRC/CIRG/1431/2015, NMRC/OFIRG/0056/2017 and J.W.W, NMRC/OFYIRG/0081/2018), MOE T1 (J.J, NUHS O-CRG 2016 and J.W.W NUHS O-CRG 2016 Oct-23) and Startup (NUHS J.J)
Nonstandard Abbreviations and Acronyms:
- αSMA
α-smooth muscle actin
- AAV
Adeno-associated virus
- Bmp7
Bone morphogenetic protein 7
- Ccnd1
Cyclin D1
- Ctgf
Connective tissue growth factor
- CM
Cardiomyocytes
- DCM
Dilated cardiomyopathy
- ECM
Extracellular matrix
- shRNA
Short hairpin RNA
- Lmna
Lamin A/C
- Myh7
Myosin heavy chain 7
- Nppa
Natriuretic peptide A
- PCM1
Pericentriolar material 1
- Postn
Periostin
- Tgfβ
Transforming growth factor beta
- Yy1
Yin Yang 1
Footnotes
Disclosures
None.
References
- 1.Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390:400–414 [DOI] [PubMed] [Google Scholar]
- 2.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547 [DOI] [PubMed] [Google Scholar]
- 3.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu JT, Muchir A, Nagy PL, Worman HJ. Lmna cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4:562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of a-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin a/c-deficient mice. J Clin Invest. 2004;113:357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubben N, Voncken JW, Konings G, van Weeghel M, van den Hoogenhof MM, Gijbels M, van Erk A, Schoonderwoerd K, van den Bosch B, Dahlmans V, Calis C, Houten SM, Misteli T, Pinto YM. Post-natal myogenic and adipogenic developmental: Defects and metabolic impairment upon loss of a-type lamins. Nucleus. 2011;2:195–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin a/c gene mutation. Sci Transl Med. 2012;4:144ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao CY, Anderson SS, Chicoine NH, Mayfield JR, Academia EC, Wilson JA, Pongkietisak C, Thompson MA, Lagmay EP, Miller DM, Hsu YM, McCormick MA, O’Leary MN, Kennedy BK. Rapamycin reverses metabolic deficits in lamin a/c-deficient mice. Cell Rep. 2016;17:2542–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ. Activation of mapk pathways links lmna mutations to cardiomyopathy in emery-dreifuss muscular dystrophy. J Clin Invest. 2007;117:1282–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Dour C, Macquart C, Sera F, Homma S, Bonne G, Morrow JP, Worman HJ, Muchir A. Decreased wnt/beta-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/c gene. Hum Mol Genet. 2017;26:333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, Lopez-Otin C. Nuclear lamina defects cause atm-dependent nf-kappab activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ. Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin a/c gene mutation. Hum Mol Genet. 2012;21:4325–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang J, Burgon PG, Wakimoto H, Onoue K, Gorham JM, O’Meara CC, Fomovsky G, McConnell BK, Lee RT, Seidman JG, Seidman CE. Cardiac myosin binding protein c regulates postnatal myocyte cytokinesis. Proc Natl Acad Sci U S A. 2015;112:9046–9051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bersell K, Choudhury S, Mollova M, Polizzotti BD, Ganapathy B, Walsh S, Wadugu B, Arab S, Kuhn B. Moderate and high amounts of tamoxifen in alphamhc-mercremer mice induce a DNA damage response, leading to heart failure and death. Dis Model Mech. 2013;6:1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, Gurha P, Marian AJ. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res. 2019;124:1198–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen SN, Lombardi R, Karmouch J, Tsai JY, Czernuszewicz G, Taylor MRG, Mestroni L, Coarfa C, Gurha P, Marian AJ. DNA damage response/tp53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with lmna (lamin a/c) mutations. Circ Res. 2019;124:856–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor yy1: Structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142 [DOI] [PubMed] [Google Scholar]
- 20.Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, Teti D, Bresciani F, Perillo B, Weisz A. Estrogens and progesterone promote persistent ccnd1 gene activation during g1 by inducing transcriptional derepression via c-jun/cfos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin d1 to its own gene promoter. Mol Cell Biol. 2004;24:7260–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (tgf)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. Ctgf expression is induced by tgf- beta in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819 [DOI] [PubMed] [Google Scholar]
- 23.Zhao S, Wu H, Xia W, Chen X, Zhu S, Zhang S, Shao Y, Ma W, Yang D, Zhang J. Periostin expression is upregulated and associated with myocardial fibrosis in human failing hearts. J Cardiol. 2014;63:373–378 [DOI] [PubMed] [Google Scholar]
- 24.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte tgf-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merino D, Villar AV, Garcia R, Tramullas M, Ruiz L, Ribas C, Cabezudo S, Nistal JF, Hurle MA. Bmp-7 attenuates left ventricular remodelling under pressure overload and facilitates reverse remodelling and functional recovery. Cardiovasc Res. 2016;110:331–345 [DOI] [PubMed] [Google Scholar]
- 26.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. Bmp-7 counteracts tgf-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968 [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Zhu Z, Wang Y, Gao A, Niu P, Tian L. Bone morphogenetic protein-7 inhibits silica-induced pulmonary fibrosis in rats. Toxicol Lett. 2013;220:103–108 [DOI] [PubMed] [Google Scholar]
- 28.Lipson KE, Wong C, Teng Y, Spong S. Ctgf is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5:S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorn LE, Petrosino JM, Wright P, Accornero F. Ctgf/ccn2 is an autocrine regulator of cardiac fibrosis. J Mol Cell Cardiol. 2018;121:205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ackers-Johnson M, Li PY, Holmes AP, O’Brien SM, Pavlovic D, Foo RS. A simplified, langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ Res. 2016;119:909–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized m2 mononuclear phagocytes. Trends Immunol. 2002;23:549–555 [DOI] [PubMed] [Google Scholar]
- 32.Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, Agozzino M, Campana C, Gavazzi A, Febo O, Marini M, Landolina M, Mortara A, Piccolo G, Vigano M, Tavazzi L, Arbustini E. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250–1260 [DOI] [PubMed] [Google Scholar]
- 33.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of lmna gene mutations: Do lamin a/c mutations portend a high risk of sudden death? J Mol Med (Berl). 2005;83:79–83 [DOI] [PubMed] [Google Scholar]
- 34.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, Sherwood M, Branco DM, Wakimoto H, Fishman GI, See V, Stewart CL, Conner DA, Berul CI, Seidman CE, Seidman JG. Lamin a/c haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008;44:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies BS, Barnes RH 2nd, Tu Y, Ren S, Andres DA, Spielmann HP, Lammerding J, Wang Y, Young SG, Fong LG. An accumulation of non-farnesylated prelamin a causes cardiomyopathy but not progeria. Hum Mol Genet. 2010;19:2682–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meng XM, Chung AC, Lan HY. Role of the tgf-beta/bmp-7/smad pathways in renal diseases. Clin Sci (Lond). 2013;124:243–254 [DOI] [PubMed] [Google Scholar]
- 37.Flevaris P, Khan SS, Eren M, Schuldt AJT, Shah SJ, Lee DC, Gupta S, Shapiro AD, Burridge PW, Ghosh AK, Vaughan DE. Plasminogen activator inhibitor type i controls cardiomyocyte transforming growth factor-beta and cardiac fibrosis. Circulation. 2017;136:664–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-tomesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961 [DOI] [PubMed] [Google Scholar]
- 39.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N. Cd4+ t cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124 [DOI] [PubMed] [Google Scholar]
- 40.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated t lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail. 2017;10:e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng Z, Cao P, Wan MM, Sui G. Yin yang 1: A multifaceted protein beyond a transcription factor. Transcription. 2010;1:81–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359. [DOI] [PubMed] [Google Scholar]
- 43.Vukicevic S, Basic V, Rogic D, Basic N, Shih MS, Shepard A, Jin D, Dattatreyamurty B, Jones W, Dorai H, Ryan S, Griffiths D, Maliakal J, Jelic M, Pastorcic M, Stavljenic A, Sampath TK. Osteogenic protein-1 (bone morphogenetic protein-7) reduces severity of injury after ischemic acute renal failure in rat. J Clin Invest. 1998;102:202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Usinger W, Nichols B, Gray J, Xu L, Seeley TW, Brenner M, Guo G, Zhang W, Oliver N, Lin A, Yeowell D. Cooperative interaction of ctgf and tgf-beta in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raghu G, Scholand MB, de Andrade J, Lancaster L, Mageto Y, Goldin J, Brown KK, Flaherty KR, Wencel M, Wanger J, Neff T, Valone F, Stauffer J, Porter S. Fg-3019 anti-connective tissue growth factor monoclonal antibody: Results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur Respir J. 2016;47:1481–1491 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.