

Abstract
Oxygen sensing is central to metazoan biology and has implications for human disease. Mammalian cells express multiple oxygen-dependent enzymes called 2-oxoglutarate (OG)-dependent dioxygenases (2-OGDDs), but they vary in their oxygen affinities and hence their ability to sense oxygen. The 2-OGDD histone demethylases control histone methylation. Hypoxia increases histone methylation, but whether this reflects direct effects on histone demethylases, or indirect effects caused by the hypoxic-induction of the HIF (Hypoxia-inducible Factor) transcription factor or the 2-OG antagonist 2-hydroxyglutarate (2-HG), is unclear. Here we report that hypoxia promotes histone methylation in a HIF- and 2-HG-independent manner. We found that the H3K27 histone demethylase KDM6A/UTX, but not its paralog KDM6B, is oxygen-sensitive. KDM6A loss, like hypoxia, prevented H3K27 demethylation and blocked cellular differentiation. Restoring H3K27 methylation homeostasis in hypoxic cells reversed these effects. Thus oxygen directly affects chromatin regulators to control cell fate.
Oxygen’s appearance in earth’s atmosphere was a watershed that created the evolutionary selection pressure for a conserved pathway used by metazoans to sense and respond to changes in ambient oxygen that converges on the heterodimeric HIF (Hypoxia-Inducible Factor) transcription factor. Under normoxic conditions, the HIFα subunit is prolyl hydroxylated by the EglN (Egg Laying-Defective Nine) isoenzymes of the 2-oxoglutarate (2-OG)-dependent dioxygenase family. Hydroxylated HIFα is earmarked for degradation by the von Hippel-Lindau (VHL) ubiquitin ligase complex. Hypoxia inactivates the EglNs and thereby stabilizes HIFα, which then associates with HIF1β [also called ARNT (Aryl Hydrocarbon Nuclear Translocator)] and transcriptionally activates genes that promote adaptation to inadequate oxygen (1).
The 2-OG-dependent dioxygenase family also includes the collagen prolyl hydroxylases, the JmjC (Jumonji C) domain histone Lysine Demethylases (KDMs), the Ten Eleven Translocation (TET) DNA hydroxylases, and ~50 other enzymes that are relatively understudied (2). In contrast to the high oxygen affinity (low KM) collagen prolyl hydroxylases, the EglNs exhibit low oxygen affinities (high KM) (1), which enables them to sense physiological changes in oxygen.
Several previous studies have suggested that oxygen regulates histone methylation. Certain KDMs display low oxygen affinities in vitro (3). Moreover, many KDMs are transcriptionally activated by hypoxia and HIF (4), perhaps to compensate for a decrease in their enzymatic specific activity. Finally, hypoxia can induce histone hypermethylation (5). However, in these previous studies, it was unclear whether histone hypermethylation reflected a direct effect of hypoxia on KDMs or was confounded by indirect consequences of hypoxia (and HIF). For example, in some cells hypoxia increases the L-enantiomer of 2-hydroxyglutarate (L-2HG), which is an endogenous inhibitor of 2-OG-dependent dioxygenases (6–8). Moreover, HIF can potentially affect chromatin in many ways, such as by altering KDM protein levels (vide supra), by inducing chromatin-modifying enzymes other than KDMs [e.g. TETs (9) and DNA methyltransferases (DNMTs) (10)], or by upregulating transcription factors that enforce an epithelial-mesenchymal transition (EMT) and accompanying epigenetic reprogramming (11).
To rigorously address whether hypoxia has a direct or indirect effect on histone methylation, we lentivirally transduced an Arnt-defective (HIF-inactive) mouse hepatoma cell line (mHepa-1 c4) to express either Green Fluorescent Protein (GFP), wild-type ARNT (WT), or a functionally inactive ARNT mutant (Δ414) that is missing 414 base pairs from its N-terminus, thereby eliminating its basic-Helix-Loop-Helix (bHLH) domain and ability to heterodimerize (Fig. 1A and fig. S1, A to C). mHepa-1 c4 did not tolerate prolonged growth in 1% oxygen, which is the oxygen concentration typically used to model hypoxia ex vivo. We therefore used more modest levels of hypoxia (2-5%) to study these cells. As expected, canonical HIF-target genes (e.g. Egln3 and Ndrg1) were transcriptionally induced by 5% oxygen in the cells expressing wild-type ARNT, but not in the cells expressing ARNT (Δ 414) or GFP (fig. S1D).
Fig. 1. Hypoxia Causes HIF-Independent Histone Hypermethylation.
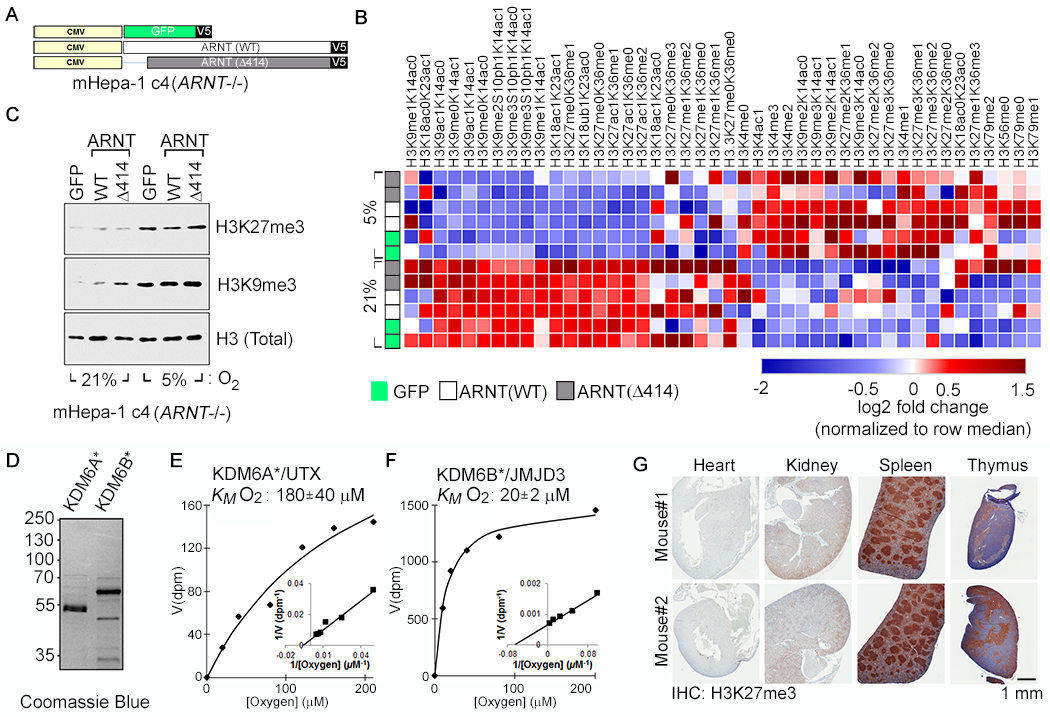
(A-C) Vector schematic (A), Histone modification profiling by mass spectrometry (B), and Histone immunoblot analysis (C) of Arnt-deficient mouse Hepatoma (mHepa-1 c4) cells that were lentivirally transduced to produce the indicated V5-tagged proteins and cultured at the indicated oxygen levels for 4 days. In (B), rows represent two biological replicates of the indicated samples and the color in each cell represents log2 fold change relative to all samples in the column, normalized for total histone using an internal control peptide (Histone H3: residues 41-49). (D-F) Coomassie blue staining (D) and biochemical analysis of baculovirally expressed and purified JumonjiC (JmjC) catalytic domains of KDM6A [KDM6A*] (E) and KDM6B [KDM6B*] (F). KM values are mean +/− SD (N=3). (G) Immunohistochemical analysis of the indicated tissues derived from representative male and female age-matched mice
We next used a multiplexed mass spectrometric assay (12) to quantify changes in histone methylation in response to hypoxia in the isogenic [GFP, ARNT(WT), ARNT(Δ 414)] mHepa-1 c4 cells. Unsupervised clustering of histone modification patterns revealed that the isogenic cell lines clustered primarily based on oxygen availability during growth and not HIF status (Fig. 1B). Consistent with prior reports (5), hypoxia promoted the dimethylation (me2) and trimethylation (me3) of H3K4 (histone 3, lysine 4), H3K9, and H3K27 (Fig. 1B). Hypermethylation of H3K9 and H3K27 was confirmed by immunoblot analysis (Fig. 1C). We also observed a concomitant decrease in hypomethylated H3K27 (me0/me1 states) and acetylated H3K27, in response to hypoxia (Fig. 1B), which is consistent with the knowledge that histone methylation and acetylation are reciprocally regulated. The H3K27 hypermethylation was not explained by increased expression of EZH2 (enhancer of zeste homolog 2), which controls bulk H3K27 methylation, or decreased expression of the primary H3K27 histone demethylases KDM6A and KDM6B (fig. S2).
Hypoxia also promoted histone hypermethylation in VHL−/− RCC4 human renal carcinoma cells (fig. S3). Thus hypoxia promotes histone hypermethylation both in cells that cannot mount a HIF response (mHepa-1 c4 cells) and in cells that constitutively overproduce HIF (RCC4 cells), arguing that hypoxia’s effects on histone methylation are not caused by changes in HIF activity.
We next explored whether hypoxia’s effects on histone methylation in mHepa-1 c4 cells were caused by metabolic changes that can inhibit KDM activity, such as increased L-2HG or decreased 2-OG (6–8, 13). In the isogenic mHepa-1 c4 cells, 5% oxygen did not significantly induce either total 2-HG (fig. S4A), or enantiomer-specific 2-HG (fig. S4, B and C), and actually increased 2-OG levels (fig. S4D). 2-HG was modestly induced in parental mHepa-1 c4 cells by more profound levels of hypoxia (0.5-2% oxygen) (fig. S4, C and E), albeit as D-2HG rather than L-2HG (fig. S4C). The significance of the latter finding is unclear. Even under these more extreme conditions, the 2-HG levels achieved were ~100-fold below both the intracellular levels in mutant IDH1 (Isocitrate Dehydrogenase 1) cells (fig. S4F), wherein 2-HG serves as an oncometabolite (14), and the intracellular levels required to promote histone methylation in mHepa-1 c4 cells treated with cell-permeable versions of D-2HG or L-2HG (fig. S4, G and H). The latter observation is consistent with the biochemical D-2HG and L-2HG IC50 values for the KDMs (15).
Hypoxia can incite reactive oxygen species (ROS), which can inhibit 2-OG-dependent dioxygenases (16). Treating mHepa-1 c4 cells with the ROS-inducer tert-Butyl Hydroperoxide (tBHP) showed that intracellular ROS levels ~10-fold higher than those observed after exposure to 2% oxygen were required to induce histone methylation (fig. S5). These findings suggested that the HIF-independent effects of hypoxia on KDM activity were not caused by increased L-2HG, decreased 2-OG, or increased ROS, but instead were caused by a direct effect of hypoxia on the enzymatic activities of specific KDMs.
In support of this idea, we found that recombinant KDM4B, KDM5A, and KDM6A [also called Ubiquitously-Transcribed TPR Protein on the X chromosome (UTX)] have relatively low oxygen affinities that are comparable to the EglN family, whereas recombinant KDM4A, KDM5B, KDM5C, KDM5D, and KDM6B have high oxygen affinities (fig. S6 to S8). Finally, like full-length KDM6A, the isolated KDM6A catalytic domain also had low oxygen affinities compared to their KDM6B counterparts (Fig. 1, D to F, and fig. S8).
We focused our attention on the KDM6 H3K27 demethylases because hypoxia and histone H3K27 methylation have been independently linked to the control of cellular differentiation (17–19), because this histone mark can be manipulated with drugs, and because KDM6A has the lowest oxygen affinity of the KDMs tested to date. We first confirmed that hypoxia induced H3K27 methylation in additional human cell lines including 293T embryonic kidney cells, MCF7 breast cells, and SK-N-BE(2) neuroblastoma cells (fig. S9). Moreover, histological analysis showed elevated H3K27 methylation in mouse tissues that are known to be hypoxic, such as the kidney (20), splenic germinal centers (21), and thymus (22), but not in well-oxygenated tissues such as the heart (Fig. 1G). Similarly, H3K27 methylation is increased in hypoxic regions of mouse tumors (23–25) (fig. S10). Finally, Gene-Set Enrichment Analysis (26) of ~2000 human tumors that were previously annotated as “Normoxic” or “Hypoxic” based on their HIF signature (27) (Table S1), revealed that “Hypoxic” tumors had transcriptional signatures indicative of H3K27 hypermethylation (fig. S11 and Tables, S2 and S3).
To examine the effect of hypoxia on cell differentiation in vitro, we studied C2C12 murine myoblasts. C2C12 differentiate into Myosin Heavy Chain (MyHC)-positive multinucleated myotubes when shifted from serum-rich growth media (GM) to nutrient-poor differentiation media (DM). Hypoxia blocks C2C12 differentiation (fig. S12, A to D) (28, 29). Hypoxic C2C12 cells grown in DM entered a quiescence-like state, but more readily proliferated when returned to GM under normoxic conditions compared to their normoxic counterparts (fig. S12, E to G). Similarly, hypoxia blocked the myogenic differentiation of mouse embryo fibroblasts (MEFs) that conditionally express MyoD (fig. S13).
Eliminating ARNT in C2C12 cells using CRISPR/Cas9 blocked (rather than accentuating) their ability to differentiate under normoxic conditions (fig. S14, A to C) and did not rescue their ability to differentiate under hypoxia (fig. S14D). Moreover, expression of stabilized versions of HIF1α or HIF2α did not block normoxic C2C12 differentiation (fig. S14, E and F). Thus, the differentiation block exhibited by hypoxia in C2C12 cells is not due to HIF activation.
Total 2-HG was not induced, and L-2HG was induced only about 2-fold (fig. S15, A to C), in C2C12 cells by 2% oxygen. C2C12 cells that were pharmacologically or genetically manipulated to have intracellular L-2HG levels 3-5-fold higher than observed under hypoxia still differentiated in DM (fig. S15, D to G). Therefore, hypoxia’s effects on C2C12 differentiation were not caused by 2-HG.
Similar to hypoxia, treatment of C2C12 cells with the KDM6 family inhibitor GSK-J4, promoted H3K27 hypermethylation and blocked myogenic differentiation (fig. S16). Similar results were obtained with MEFs expressing MyoD (fig. S13). In contrast, KDM-C70, a KDM5 family inhibitor, did not block C2C12 differentiation (fig. S17).
The differential oxygen affinities of KDM6A and KDM6B suggested that the ability of hypoxia to promote H3K27 methylation and block differentiation is caused specifically by a loss of KDM6A activity. Indeed, downregulating KDM6A, but not KDM6B, with different shRNAs, phenocopied the effects of hypoxia on differentiation (Fig. 2, A and B, and fig. S18, A to D) (30). Moreover, eliminating KDM6A in C2C12 cells with CRISPR/Cas9 blocked their ability to differentiate unless they were rescued with an sgRNA-resistant KDM6A cDNA (Fig. 2, C to E). In contrast, eliminating KDM6B had no effect (fig. S18, E and F) and eliminating KDM5A promoted differentiation (fig. S19). Notably, bulk H3K27me3 was oxygen-insensitive in the Kdm6a-deficient C2C12 cells, consistent with KDM6A being the primary oxygen sensor amongst the KDM6 paralogs (Fig. 2F).
Fig. 2. Regulation of Myogenic Differentiation by the KDM6A H3K27 Demethylase.
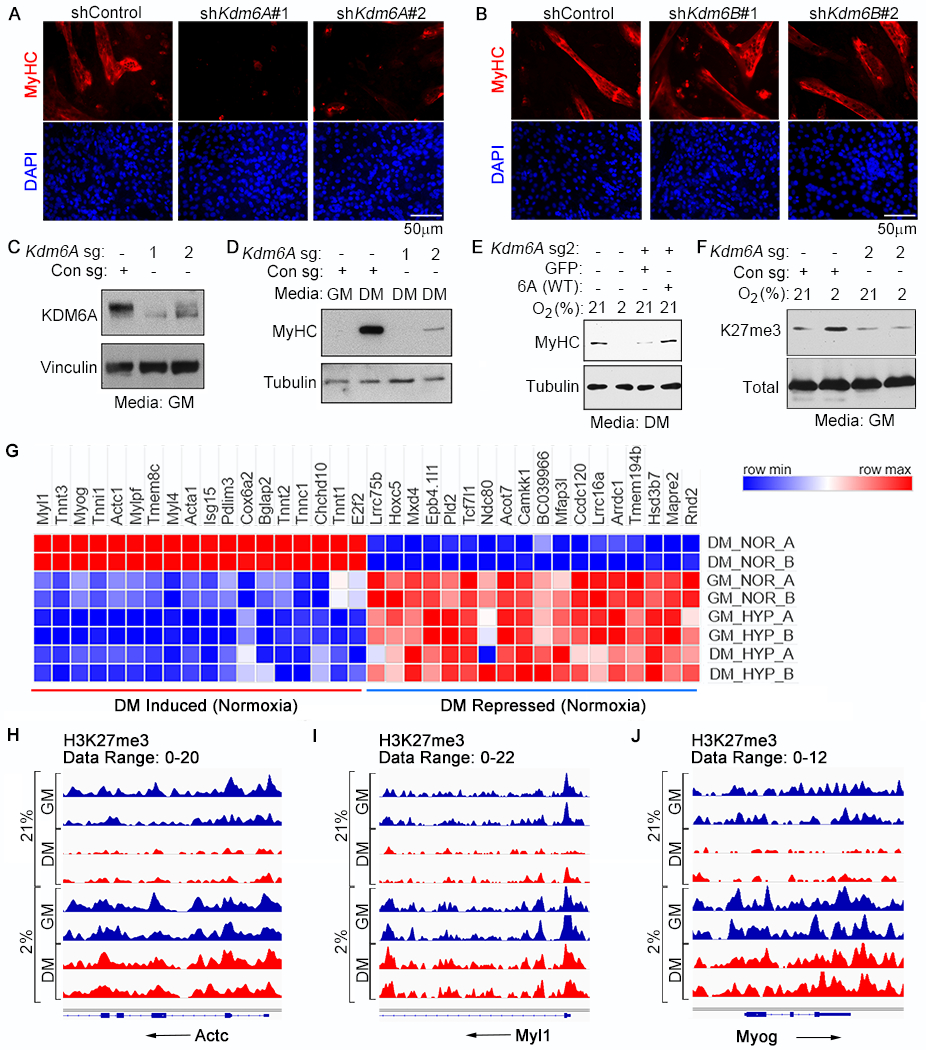
(A-B) Immunofluorescence analysis of C2C12 cells that were lentivirally transduced to express the shRNAs targeting Kdm6A (A) or Kdm6B (B) and then cultured in differentiation media for 4 days. (C-E) Immunoblot analysis of C2C12 cells lentivirally transduced to express the indicated sgRNAs and cultured for 4 days either in growth media (GM) or differentiation media (DM), as indicated. (E) Immunoblot analysis of C2C12 cells expressing, where indicated, Kdm6a sg2 [described in (C) and (D)] that were lentivirally transduced to produce either GFP (control) or wild-type human KDM6A [6A(WT)] and then cultured in DM at the indicated oxygen concentrations for 4 days. The mouse Kdm6A sg2 target sequence is not conserved in human KDM6A. (F) Immunoblot analysis of histones from C2C12 cells expressing the indicated sgRNA that were cultured at the indicated oxygen concentrations for 3 days. (G-J) Heatmap representing mRNA levels determined by RNA-Seq (G) and H3K27me3 levels determined by ChIP-Seq analysis at the Actc1 (H), Myl1 (I), and the Myog (J) loci from two biological replicates (A and B) of C2C12 cells cultured in the indicated media for 4 days at the indicated oxygen concentration.
Previous work showed that KDM6A is directly recruited to myogenic targets during differentiation (30). We therefore investigated whether differentiation programs driven by KDM6A activity involve transcriptional changes that depend on H3K27me3 elimination. Comparison of transcriptional signatures of normoxic C2C12 cells grown in GM versus DM revealed profound differences in transcriptional output, particularly of muscle-specific target genes (Fig. 2G, fig. S20, and Table S5). Hypoxia (and the consequent differentiation block), however, blunted the transcriptional differences between these two conditions (fig. S20A), which was associated with a failure of these cells to induce muscle-specific markers in DM (Fig. 2G and S20B). H3K27me3 status typically represses transcription. The inability of C2C12 cells grown in DM to activate late myogenic genes (e.g. Actc, Myl1, and Myog) under hypoxia correlated with a failure to erase H3K27me3 at those loci (Fig 2, H to J and fig. S21), presumably due to inactivation of KDM6A. This was specific because H3K4me3 decreased at late myogenic genes under hypoxia (fig. S21).
Loss of cellular differentiation is a hallmark of cancer and KDM6A is a human tumor suppressor gene that is inactivated in a variety of cancers, including leukemia, kidney cancer, and bladder cancer (31). Remarkably, Gene Set Enrichment Analysis showed that a myogenic differentiation gene set, which presumably also contains genes linked to differentiation in other contexts, is more highly expressed in KDM6A wild-type bladder cancers compared to KDM6A mutant tumors (fig. S22 and Table S6).
These data suggest that KDM6A inactivation by hypoxia promotes the persistence of H3K27me3 and prevents the transcriptional reprogramming required for differentiation. If true, the effects of hypoxia on differentiation should be redressed by inhibiting H3K27 methyltransferase activity (Fig. 3A). Like mHepa-1 c4 cells, hypoxia did not alter the protein levels of the EZH H3K27 methyltransferases in C2C12 cells (fig. S23A). Inhibiting the H3K27 methyltransferase EZH2 with CRISPR/Cas9 (Fig. 3B and fig. S23B) or with the drug GSK126 (fig. S23, C and D) reduced H3K27me3 levels and partially rescued the ability of C2C12 cells to differentiate under hypoxic conditions. By contrast, the G9a/GLP methyltransferase inhibitor UNC638 was ineffective (fig. S23, C and D). Finally, GSK126 rescued the hypoxia-induced differentiation block in human primary myoblasts and in MEFs conditionally expressing MyoD (fig. S23, E and F).
Fig. 3. Restoring the Balance of H3K27 Methyltransferase Activity to H3K27 Demethylase Activity Rescues Myogenic Differentiation Under Hypoxic Conditions.
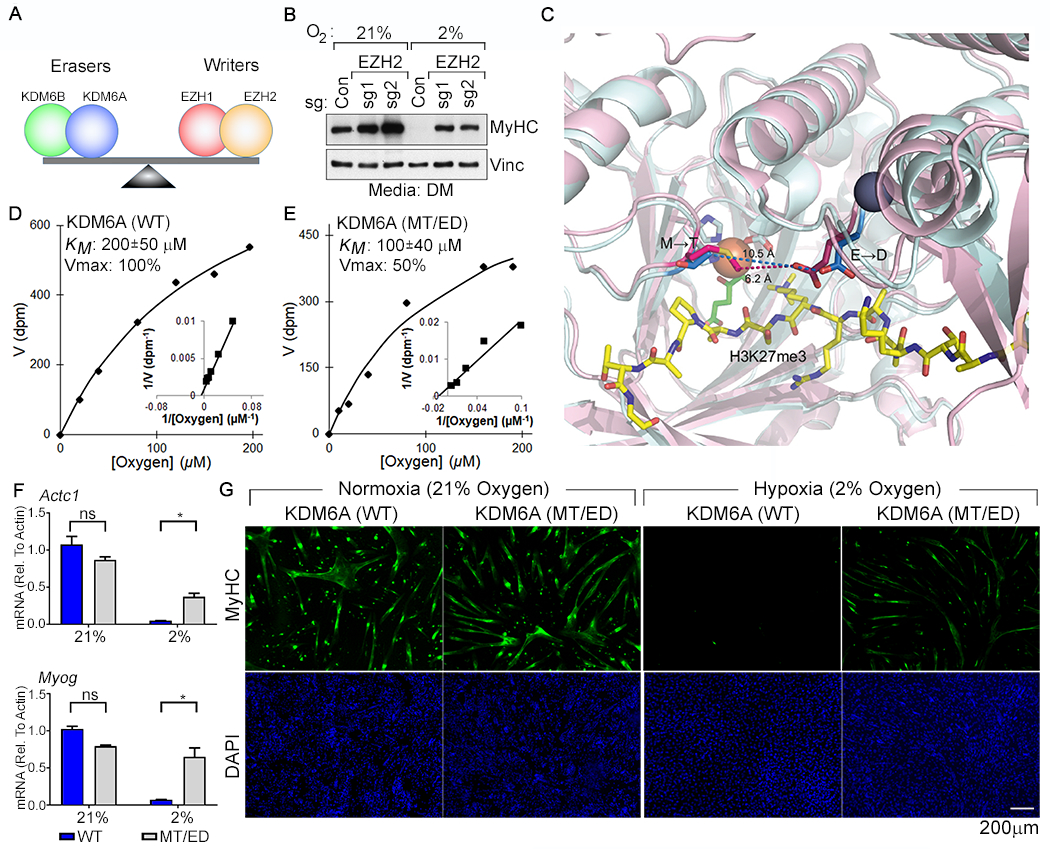
(A) Model for control of H3K27 methylation by the indicated opposing demethylases (“erasers”) and methyltransferases (“writers”). (B) Immunoblot analysis of C2C12 cells lentivirally transduced to express the indicated sgRNAs and cultured under the indicated conditions. (C) Structural models of the KDM6A (pink) and KDM6B (cyan) catalytic pockets. The non-conserved M1190 (KDM6A) → T1434 (KDM6B) and the E1335 (KDM6A) → D1579 (KDM6B) are highlighted. Peptidic H3K27me3 substrate (yellow), Fe+2 (orange), 2-oxoglutarate (green), and Zn+2 (grey) are shown. (D-E) Michaelis-Menten plots (inset Lineweaver-Burk plot) and measured oxygen KM and Vmax values (mean +/− SD, N=3) of recombinant KDM6A wild-type and the MT/ED mutant. (F-G) Real-Time qPCR analysis (mean +/− SD, N =3) of the indicated genes (F) and immunofluorescence analysis (G) of C2C12 cells transduced to produce wild-type human KDM6A or the KDM6A MT/ED variant and then cultured in DM at the indicated oxygen concentrations for 4 days.
Hypoxia can also, in a HIF-independent manner, alter the differentiation of human mammary epithelial (HMLE) cells (25), causing an EMT (fig. S24, A and B) and upregulation of the cancer stem-like marker CD44 (fig. S24C). Similar to our findings with C2C12 cells, these hypoxia-associated changes were phenocopied by pharmacologic (GSK-J4) (fig. S24, A to C) or genetic (CRISPR/Cas9) disruption of KDM6A (fig. S24, D to F) and rescued by the EZH inhibitor GSK-126 (fig. S24, A to C).
Finally, we tried to directly increase KDM6A’s oxygen affinity. We reasoned that certain non-conserved residues within the catalytic domains of KDM6A and KDM6B caused their vastly different oxygen affinities. We overlaid published models of the catalytic JmjC domains of KDM6A with KDM6B (32, 33) and noted two non-conserved residues, M1190 (KDM6A) → T1434 (KDM6B) and E1335 (KDM6A) → D1579 (KDM6B), lining the 2-OG- and Fe+2 binding-pocket (Fig. 3C). As predicted, a KDM6A variant that harbored these two “KDM6B-like” changes [MT/ED: M1190 → T and E1335→ D], displayed a 2-fold increased affinity for oxygen in vitro, albeit at the cost of a decreased Vmax (Fig. 3, D and E). Wild-type KDM6A and the MT/ED variant were comparably insensitive to L-2HG and to ROS (fig. S25, A to D), which was induced less than 2-fold by 2% oxygen in C2C12 cells (fig. S25E). Both wild-type KDM6A and the MT/ED rescued the ability of KDM6A-deficient C2C12 cells to differentiate under normoxia (fig. S26A). The double mutant, however, was superior to wild-type KDM6A at rescuing differentiation under hypoxic conditions in both parental and KDM6A-deficient C2C12 cells, presumably due to its enhanced oxygen affinity (Fig. 3, F and G, and fig. S26, B to D).
Independent lines of research have shown that oxygen and H3K27 methylation each regulate embryological development, cellular differentiation, stemness, and malignant transformation (17–19). We propose that these observations are linked. Specifically, we argue that oxygen has both direct and indirect effects on chromatin and that the former involves enzymes such as KDM6A, which couple changes in oxygen availability to changes in H3K27 methylation and the transcriptional control of cell fate.
The observed H3K4 hypermethylation and H3K9 hypermethylation in hypoxic Arnt-deficient mHepa-1 c4 cells, together with our biochemical studies, suggests that at least one H3K4 and one H3K9 histone demethylase also act as oxygen sensors. For example, our biochemical data, together with the data in the accompanying manuscript (34), argue that KDM5A plays such a role. Profound hypoxia can also inhibit other 2-OG-dependent enzymes, including TETs, leading to DNA hypermethylation (27).
Mammalian embryological development occurs in a hypoxic environment and mammalian stem cells are maintained in hypoxic niches. It is well established that hypoxia can affect stemness and cellular differentiation by activating HIF and HIF-target genes such as Oct4 (19). Such effects are not mutually exclusive with direct effects of oxygen on histone methylation and might serve to reinforce one another. Hypoxia promotes stemness in both metazoans and plants, but the HIF pathway is only present in the former (35). It is possible that oxygen sensing by histone demethylases evolutionarily preceded the emergence of oxygen-sensitive transcription factors.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Zhao, D. Lambrechts, and P. Carmeliet for sharing their TCGA tumor annotations. We thank R. Looper for synthesizing and sharing the esterified 2-HG. M. K. Koski and R. Wierenga for help with structural modeling, and T. Aatsinki and E. Lehtimaki for technical assistance. We thank the Broad GDAC group for help with the GDAC-GSEA source code. We thank M. Oser for sharing mouse KDM5A sgRNAs and V. Koduri for assistance in acquiring microscopic images.
Funding: WGK was supported by grants from the NIH (R01CA068490, P50CA101942, and R35CA210068). AAC was supported by grants from the ‘Friends of Dana-Farber’ and the NIH (Cancer Biology Training grant: T32CA009361 and the DF/HCC Kidney SPORE CEP and DRP award: P50CA101942). PK was supported by Academy of Finland Grants 266719 and 308009, the S. Juselius Foundation, the Jane and Aatos Erkko Foundation, and the Finnish Cancer Organizations. TL was supported by the Finnish Medical Foundation and, with PK, the Emil Aaltonen Foundation. S.K.M. is supported by an American Cancer Society postdoctoral fellowship (PF-14-144-01-TBE) and by a Career Enhancement Project award from the Dana-Farber/Harvard Cancer Center Brain SPORE. WGK is a HHMI Investigator.
Competing Interests: W.G.K. has financial interests related to Lilly Pharmaceuticals (Board of Directors), Agio Pharmaceuticals [Scientific Advisory Board (SAB)], Cedilla Therapeutics (Founder), Fibrogen (SAB), Nextech Invest (SAB), Peloton Therapeutics (SAB), Tango Therapeutics (Founder), and Tracon Pharmaceuticals (SAB). W.G.K. is a coinventor on a patent entitled “Pharmaceuticals and Methods for Treating Hypoxia and Screening Methods Therefor” that has been licensed to Fibrogen. S. S. has a consulting or advisory role in AstraZeneca/MedImmune, Merck, AACR, and NCI; patents, royalties, other intellectual property from Biogenex Laboratories; and research funding from AstraZeneca, Exelixis, Bristol-Myers Squibb. R.B receives research funding from Novartis.
Footnotes
List of Supplementary Materials:
Data and materials availability: All data are available in the manuscript or the supplementary material. RNA-Seq and ChIP-Seq data have been uploaded to the GEO (GSE114086).
References and Notes
- 1.Kaelin WG Jr., The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nature reviews. Cancer 8, 865 (2008). [DOI] [PubMed] [Google Scholar]
- 2.McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ, Structural studies on human 2-oxoglutarate dependent oxygenases. Current opinion in structural biology 20, 659 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Hancock RL, Dunne K, Walport LJ, Flashman E, Kawamura A, Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 7, 791 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Melvin A, Rocha S, Chromatin as an oxygen sensor and active player in the hypoxia response. Cellular signalling 24, 35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shmakova A, Batie M, Druker J, Rocha S, Chromatin and oxygen sensing in the context of JmjC histone demethylases. The Biochemical journal 462, 385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Intlekofer AM et al. , Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism 22, 304 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oldham WM, Clish CB, Yang Y, Loscalzo J, Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism 22, 291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyrakis PA et al. , S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mariani CJ et al. , TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell reports 7, 1343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson CJ et al. , Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Human molecular genetics 23, 2176 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Higgins DF et al. , Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. The Journal of clinical investigation 117, 3810 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creech AL et al. , Building the Connectivity Map of epigenetics: chromatin profiling by quantitative targeted mass spectrometry. Methods 72, 57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB, Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losman JA, Kaelin WG Jr., What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes & development 27, 836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laukka T, Myllykoski M, Looper RE, Koivunen P, Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. Journal of molecular biology 430, 3081 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Kaelin WG Jr., ROS: really involved in oxygen sensing. Cell metabolism 1, 357 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Conway E, Healy E, Bracken AP, PRC2 mediated H3K27 methylations in cellular identity and cancer. Current opinion in cell biology 37, 42 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Manna S et al. , Histone H3 Lysine 27 demethylases Jmjd3 and Utx are required for T-cell differentiation. Nature communications 6, 8152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A, Oxygen in stem cell biology: a critical component of the stem cell niche. Cell stem cell 7, 150 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Safran M et al. , Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proceedings of the National Academy of Sciences of the United States of America 103, 105 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho SH et al. , Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature 537, 234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hale LP, Braun RD, Gwinn WM, Greer PK, Dewhirst MW, Hypoxia in the thymus: role of oxygen tension in thymocyte survival. American journal of physiology. Heart and circulatory physiology 282, H1467 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Adriaens ME et al. , Quantitative analysis of ChIP-seq data uncovers dynamic and sustained H3K4me3 and H3K27me3 modulation in cancer cells under hypoxia. Epigenetics & chromatin 9, 48 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prickaerts P et al. , Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenetics & chromatin 9, 46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Beucken T et al. , Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nature communications 5, 5203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thienpont B et al. , Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majmundar AJ et al. , O(2) regulates skeletal muscle progenitor differentiation through phosphatidylinositol 3-kinase/AKT signaling. Molecular and cellular biology 32, 36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yun Z, Lin Q, Giaccia AJ, Adaptive myogenesis under hypoxia. Molecular and cellular biology 25, 3040 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seenundun S et al. , UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. The EMBO journal 29, 1401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ler LD et al. , Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Science translational medicine 9, (2017). [DOI] [PubMed] [Google Scholar]
- 32.Kruidenier L et al. , A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488, 404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengoku T, Yokoyama S, Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes & development 25, 2266 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Batie M et al. , Hypoxia induces rapid changes to histone methylation reprogramming chromatin for the cellular response, Science 363, 1222 (2019) [DOI] [PubMed] [Google Scholar]
- 35.Considine MJ et al. , Learning To Breathe: Developmental Phase Transitions in Oxygen Status. Trends in plant science 22, 140 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Rohle D et al. , An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koivunen P et al. , Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakimoto H et al. , Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 2898 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varaljai R et al. , Increased mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB-deficient cells. Genes & development 29, 1817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Intlekofer AM et al. , L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nature chemical biology 13, 494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J, Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. The Journal of biological chemistry 278, 30772 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Kivirikko KI, Myllyla R, Posttranslational enzymes in the biosynthesis of collagen: intracellular enzymes. Methods in enzymology 82 Pt A, 245 (1982). [DOI] [PubMed] [Google Scholar]
- 43.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C et al. , Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cornwell M et al. , VIPER: Visualization Pipeline for RNA-seq, a Snakemake workflow for efficient and complete RNA-seq analysis. BMC bioinformatics 19, 135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin Q et al. , ChiLin: a comprehensive ChIP-seq and DNase-seq quality control and analysis pipeline. BMC bioinformatics 17, 404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson JT et al. , Integrative genomics viewer. Nature biotechnology 29, 24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kharchenko PV, Tolstorukov MY, Park PJ, Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature biotechnology 26, 1351 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casper J et al. , The UCSC Genome Browser database: 2018 update. Nucleic acids research 46, D762 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mi H et al. , PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic acids research 45, D183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.The Gene Ontology C, Expansion of the Gene Ontology knowledgebase and resources. Nucleic acids research 45, D331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng M, Bragelmann J, Kryukov I, Saraiva-Agostinho N, Perner S, FirebrowseR: an R client to the Broad Institute’s Firehose Pipeline. Database: the journal of biological databases and curation 2017, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Numayama-Tsuruta K, Kobayashi A, Sogawa K, Fujii-Kuriyama Y, A point mutation responsible for defective function of the aryl-hydrocarbon-receptor nuclear translocator in mutant Hepa-1c1c7 cells. European journal of biochemistry 246, 486 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Kim WY et al. , Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. The EMBO journal 25, 4650 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.