

Abstract
Objectives:
Alterations of the gut microbiota have been implicated in many forms of arthritis, but an examination of cartilage microbial patterns have not been performed. The objective of this study was to characterize the microbial DNA profile of articular cartilage and determine changes associated with osteoarthritis (OA).
Methods:
16s rRNA gene deep sequencing was performed on eroded and intact cartilage samples from knee and hip OA patients and cadaveric controls. Microbial DNA diversity was assessed, groups compared, and metagenomic profiles reconstructed. Confirmation was performed in an independent cohort by clade-specific qPCR. Human results were compared to cartilage from OA-susceptible C57BL6 and OA-resistant MRL/MpJ mice. Germ-free C57BL6 mouse cartilage was analyzed as a methodological control.
Results:
Alpha diversity was reduced in human OA vs. control (p<0.0001), and in hip samples vs. knees (p<0.0001). Numerous clades were different in human OA vs. controls, similar findings were noted in murine B6 vs. MRL comparisons. Hip samples were microbiologically distinct from knee samples. OA microbial DNA demonstrated increased Gram-negative constituents (p=0.02). Functional analysis demonstrated increases in lipopolysaccharide production (p=9.9E-3), phosphatidylinositol signaling (p=4.2E-4), and nitrogen metabolism (p=8E-3) and decreases in sphingolipid metabolism (p=7.7E-4) associated with OA.
Conclusions:
Our study reveals a microbial DNA signature in human and mouse cartilage. We find alterations in this signature, including increases in Gram-negative constituents, during the development and progression of human OA. Furthermore, we identified strain-specific signatures within mouse cartilage that mirror human patterns. The establishment and potential pathogenic role of these DNA signatures deserve further study.
Introduction:
Osteoarthritis (OA) is a chronic musculoskeletal disease characterized by progressive loss of function of joints leading to pain, mobility loss, significant morbidity, and early mortality. It is the leading cause of chronic disability in the US, affecting roughly half of adults over 65 years of age(1). Despite its impact, there are no disease-modifying drug therapies available, in no small part due to our incomplete understanding of OA pathogenesis. OA development involves the interplay of at least three major components: genetic predisposition, aging, and environmental factors. Genetic risk, particularly for knee OA, is relatively small, estimated in twin studies to be less than 50%(2). Thus, there has recently been significant interest in identifying non-genetic risk factors contributing to OA pathogenesis. Among these, innate immune activation, macrophage inflammatory responses, toll-like receptor (TLR) activation, and complement activation(3–6) have been recently described.
One potential driver of chronic innate immune activation in OA is the microbiome. The gut microbiome in both humans and mice changes with aging and obesity, the two strongest non-genetic risk factors for OA(7,8). In mice, induction of obesity by feeding a high fat diet leads to a proinflammatory shift in gut microbial contents and accelerates OA development following destabilization of the medial meniscus (DMM) surgery. Intriguingly, supplementation of the mouse diet with the fiber oligofructose can reverse obesity-related gut microbial changes and protect mice from OA development(9). Furthermore, compared to mice housed conventionally, germ-free mice have reduced OA pathology following DMM surgery(10).
Microbial products are strongly immunogenic, and several have been previously linked with OA development. The innate immune receptors TLR2 and TLR4 are upregulated in OA cartilage(11,12) and are stimulated by lipopolysaccharide (LPS), a constituent of the Gram-negative bacterial cell wall. Serum LPS levels have been associated with osteophyte development in human OA, and synovial fluid LPS levels are associated with osteophytes, radiographic joint space narrowing, and pain / functional severity scores(13). Bacterial DNA is a potent stimulator of innate immunity (14,15), which has mainly been studied in the context of the gastrointestinal(16) and respiratory systems(17), although recent reports have suggested a role of both circulating and synovial fluid bacterial DNA in rheumatoid arthritis patients(18,19). The first examination of bacterial DNA using next-generation sequencing in synovial fluid and synovial biopsies from knees of OA and RA patients was published in 2018, and described a number of bacterial DNA clades which were characteristic of both disease states(20). However, there have to date been no evaluations of a cartilage microbial DNA signature in either humans or mice. In this study, we hypothesized that shifts in microbial diversity and/or composition would be associated with the development and progression of OA.
Materials and Methods:
Ethics Statement:
The institutional review boards of all involved institutions approved this study.
Human cartilage samples:
Eroded and intact sections of hip and knee cartilage were obtained from patients undergoing arthroplasty for end-stage primary OA at the Oklahoma Joint Reconstruction Institute. Specimens were placed into sterile containers in the operating room and transferred to the laboratory. Cadaveric control cartilage was obtained from the National Disease Research Interchange (NDRI) from patients without a history of OA (histologically confirmed, see Table 1), rheumatoid arthritis, systemic lupus erythematosus, or other autoimmune diseases. NDRI personnel obtained sterile knee and hip articular cartilage within 24 hours of donor death, flash froze in liquid nitrogen and shipped on dry ice to our laboratory. Our final discovery cohort analysis included 34 eroded hip specimens, 33 intact hip specimens (one specimen removed due to failed amplification), 21 eroded knee specimens, 21 intact knee specimens, 10 control hip specimens and 10 control knee specimens.
Table 1:
Patient sample demographic characteristics. Presented in mean ± SEM.
| Age | %Female | BMI | Eroded cartilage OARSI score | Intact cartilage OARSI score | |
|---|---|---|---|---|---|
| 16s Deep-sequencing cohort | |||||
| Hip | |||||
| OA (n=34) | 65 ± 2 | 58% | 29 ± 1* | 13 ± 1 | 3.3 ± 0.6 |
| Control (n=10) | 66 ± 3 | 40% | 27 ± 2 | n/a | 1.5 ± 0.5 |
| Knee | |||||
| OA (n=21) | 59 ± 2* | 57% | 34 ± 1* | 15 ± 1* | 2.9 ± 0.6 |
| Control (n=10) | 68 ± 4* | 40% | 30 ± 3 | n/a | 0.0 ± 0.0 |
| qPCR confirmation cohort | |||||
| Hip | |||||
| OA (n=5) | 67 ± 5 | 60% | 25 ± 1 | 9 ± 1 | 1.0 ± 0.3 |
| Control (n=5) | 54 ± 4 | 20% | 33±3 | n/a | 0.0 ± 0.0 |
| Knee | |||||
| OA (n=5) | 60 ± 4 | 20% | 35 ± 1 | 10 ± 2* | 2.0 ± 1.7 |
| Control (n=5) | 59 ± 4 | 20% | 32 ± 1 | n/a | 0.0 ± 0.0 |
Statistically significant differences:
Knee OA vs. knee control age: p=0.03
Hip OA vs. knee OA BMI: p=0.01
Knee 16s cohort eroded vs. knee qPCR confirmation cohort eroded OARSI score: p=0.05
Mouse cartilage samples:
Adult male (11 week-old) C57BL/6 (n=8) and adult male MRL/MpJ (11 week-old) (n=8) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed at the Oklahoma Medical Research Foundation (OMRF). Husbandry adhered to the NIH Guide for the Care and Use of Laboratory Animals. At 12 weeks of age, mice were sacrificed and mouse knee joints dissected in a biosafety cabinet using sterilized instruments following skin and synovial capsule decontamination with chlorhexidine. Articular cartilage was removed from the tibia and femur and flash frozen in liquid nitrogen. Germ-free adult male (12 week-old) C57BL/6 mice (n=7) were obtained from the Gnotobiotic Mouse Core Facility at OMRF and dissected similarly.
Sample processing:
Full-thickness cartilage sections were dissected from the femoral head (human hip samples) or tibial plateau (human and mouse knee samples). A representative section was saved in 4% paraformaldehyde for histopathological scoring. Approximately 200mg of cartilage tissue was cryogenically ground using a grinder mill (Spex CertiPrep, Middlesex, UK), murine samples were cryogenically ground using a Precellys Cryolys (Bertin, Bretonneux, France). DNA was isolated using a DNeasy kit (Qiagen). Plasticware and reagents were decontaminated by a 30-minute UV exposure as previously described(21,22). PCR master mixes were decontaminated with double-stranded DNAse treatment (PCR decontamination kit, Arcticzymes, Tromsø, Norway). Sterile water was processed using the same (murine) procedure as a negative control.
Histology:
Following fixation in 4% paraformaldehyde, samples were embedded in paraffin, placed on slides and stained for hematoxylin and eosin and Safranin-O staining. OA cases (eroded and intact specimens) and disease-free controls were scored using the Osteoarthritis Research Society International (OARSI) scale(23) to confirm gross categorization (Table 1).
16S ribosomal RNA (rRNA) gene sequencing:
Microbial profiles were determined by sequencing a ~460bp region including the V3 and V4 variable region of bacterial 16s rRNA genes. The gene fragment was amplified from approximately 30ng of DNA in each sample (primers in Supplementary Table 1) using a high-fidelity polymerase (NEB Q5, New England Biolabs)(24) and confirmed by 1% agarose gel electrophoresis. Illumina Nextera XT indices were attached (Illumina), pooled in equimolar amounts, and sequenced on an Illumina miSeq sequencer using a 250bp paired-end sequencing protocol by the Clinical Genomics Center at OMRF.
16S rRNA OTU classification:
Quality filtering, operational taxonomic unit (OTU) classification and microbial diversity analysis was performed using the Quantitative Insights into Microbial Ecology (QIIME) software package, version 1.9.1(25). Sequences were assigned to OTUs using the UCLUST algorithm(26) using a 97% pairwise identity threshold and taxonomy assigned using the GreenGenes 13_8 database(27).
Diversity analyses:
Alpha diversity was characterized using the observed OTUs method following rarefaction to the lowest number of OTUs present per group. Beta diversity was evaluated on a variance-adjusted, weighted unifrac model. Principal component analysis was performed and an Adonis (permuted analysis of variance, a multi-factor PERMANOVA) test with 999 permutations was used to calculate statistical significance of group differences(28,29).
Group analyses:
Group analyses were performed using the linear discriminant analysis effect size (LEfSe) pipeline(30). LEfSe performs a non-parametric Kruskal-Wallis sum-rank test(31) to detect features with significant differential abundance between groups, p-values≤0.01 were considered significant. Next, it uses a linear discriminant analysis (LDA)(32) to estimate the effect size of each differentially abundant feature. An LDA threshold of ≥2 were considered significant(33). QIIME was used to calculate group Benjamini-Hochberg FDR-corrected q-values; q≤0.01 was chosen as the ‘FDR-corrected’ significance threshold. For Gram status comparisons, differences were evaluated by Student t-tests, p≤0.05 was considered statistically significant.
Human confirmation cohort and clade-specific qPCR:
An independent confirmation cohort including 10 eroded hip, 10 intact hip, 10 eroded knee, 10 intact knee, 5 cadaveric control hip, and 5 cadaveric control knee specimens was obtained as above. Clade-specific quantitative PCR (qPCR) analysis was performed to calculate the relative presence of Alphaproteobacteria(34,35), Firmicutes(36), Betaproteobacteria(36), Bacteroidetes(36), Pseudomonas(37), and Burkholderiales(38) in each sample compared to a universal bacterial primer set (Supplementary Table 1), using the Luna universal probe qPCR kit (New England Biolabs) on a RotorGeneQ (Qiagen) instrument. PCR cycling conditions were: 95°C for 10 mins, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Relative clade composition was calculated using the delta-delta CT method(39). Group differences were calculated with a Student t-test, p≤0.05 was considered statistically significant.
Prediction of metagenome content and imputed bacterial functional classification
The Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) software package(40) was used to impute bacterial metagenomes from our 16S deep sequencing microbial DNA data, and functional annotation applied using the Kyoto Encyclopedia of Gene and Genomes (KEGG) catalog(41). Statistical analysis was performed using the Statistical Analysis of Metagenomic Profiles (STAMP) package(42). Statistical significance and effect sizes among three groups (human OA-eroded, OA-intact, and control) were calculated using ANOVA. Statistical significance was defined as p≤0.05 and Benjamini-Hochberg FDR corrected q≤0.1.
Results:
Human OA cartilage demonstrates reduced constituent microbial DNA diversity compared to control specimens.
We performed 16s deep sequencing analysis of human cartilage microbial DNA in 34 eroded hip specimens, 33 intact hip specimens (one specimen removed due to failed amplification), 21 eroded knee specimens, 21 intact knee specimens, 10 control hip specimens and 10 control knee specimens. There were no statistically significant differences in the number of raw OTU counts from each of the groups (eroded: 21000±4000 OTUs, mean±SEM vs. intact specimens: 11000±2000 counts vs. control: 10000±2000 counts, control vs. eroded p=0.24, control vs. intact p=0.59, eroded vs. intact p=0.06). Comparing hips to knees, there was no difference in raw samples from OA specimens (knee: 16000±3000 vs. hip: 15000±3000, p=0.8), although there was a reduction in raw counts from hip control samples compared to knee controls (knee: 17000±4000 vs. hip: 3300±600, p=0.002).
Differences in bacterial alpha diversity were seen among groups (Figure 1A) following rarefaction, where disease-free controls were more diverse than both OA-eroded and OA-intact sections (control 124±6 vs. OA-eroded 99±4 mean±SEM, p=0.0005; control vs. OA-intact 97±3, p<0.0001). No differences in alpha diversity were seen in eroded vs. intact cartilage (p=0.57). Hip samples demonstrated lower alpha diversity than knees, both in OA (hip: 89±3 vs. knee: 113±4 mean±SEM, p<0.0001) and control sections (106±6 vs. 151±6, p<0.0001) (Figure 1C). Differences were also seen in beta diversity when comparing eroded to control tissue (p=0.001) and intact to control tissue (p=0.001), but not when comparing eroded to intact tissue (p=0.47) (Figure 1B). Knee and hip samples demonstrated differences in beta diversity when comparing knee OA vs. hip OA (p=0.001) and knee control vs. hip control (p=0.002) (Figure 1D).
Figure 1:
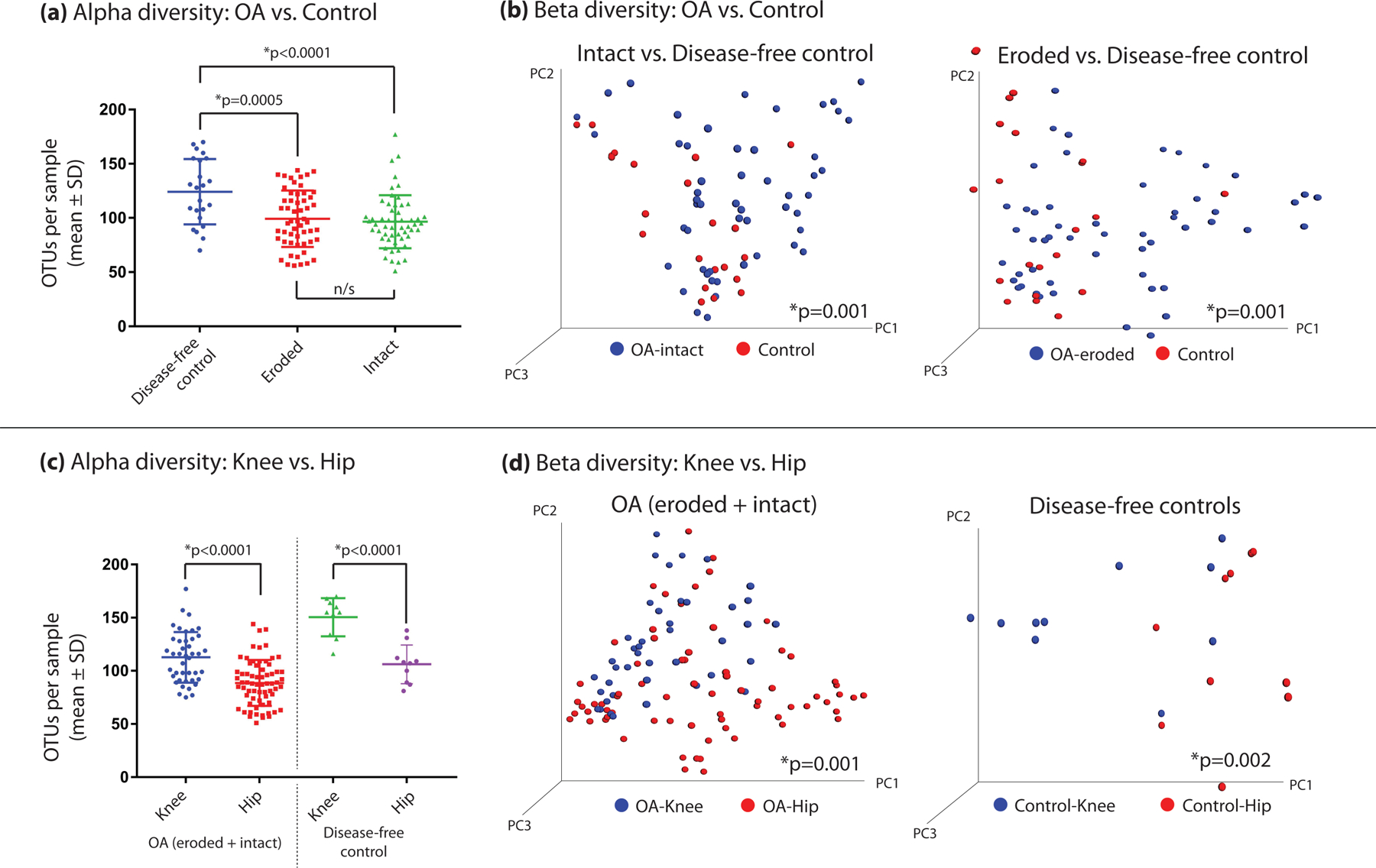
(a,b) Differences in both alpha and beta diversity among human cartilage microbial DNA clades comparing OA tissues to controls and (c,d) comparing knee tissues to hip tissues. Alpha diversity was compared using the observed OTUs method, whereas beta diversity is represented by a 3-dimensional principal component analysis, with statistical significance calculated using the Adonis method.
Human OA and control cartilage have distinct microbial DNA signatures.
We next examined clade differences among all OA samples (intact+eroded, from both knees and hips) and all control samples (knee and hip). LEfSe demonstrated 63 clades which were significantly different among the two groups (Figure 2A,B, Supplementary Table 2). Among these, 35 passed FDR correction with q≤0.01. Among OA specimens, members of the phylum Proteobacteria were enriched (LDA effect size 3.9, p=0.02, q=0.06); specifically, class Betaproteobacteria (LDA ES 3.9, p=7E-6, q=0.06). Enriched among control samples were members of the phylum Actinobacteria (LDA ES 3.6, p=0.004, q=0.01) class Clostridia (LDA ES 3.5, p=0.03, q=0.06). When OA specimens were subdivided into OA-eroded vs. OA-intact and compared to controls, statistical power was reduced but a clear pattern emerged, with control specimens being characterized by members of the phylum Bacteroidetes and the class Alphaproteobacteria, and both eroded and intact specimens by members of the class Betaproteobacteria. LDA analysis demonstrated 42 clades as significantly different (Figure 2B, Supplementary Table 3).
Figure 2:

(a) Statistically significant differences in microbial DNA clades among human OA and control specimens, (b) Cladogram comparing OA-eroded vs. OA-intact vs. control specimens. (c) Clade comparison among hip OA and control specimens. (d) Clade comparison among knee OA and control specimens. Bars represent linear discriminant analysis (LDA) effect size scores. Asterisk indicates clade passed FDR correction. Cladogram of differences among eroded, intact, and control cartilage (b).
We then considered knee and hip samples separately. In hip samples, 3 clades were significantly different when comparing eroded to intact cartilage (Figure 2C, Supplementary Table 4): order Vibrionales (LDA ES 2.1, p=0.05) in hip-intact cartilage and family Pseudomonadaceae (LDA ES 2.1, p=0.02) and genus Pseudomonas (LDA ES 2.04, p=0.03) in hip-eroded cartilage. Comparing hip-OA specimens to control hips, 16 clades were significantly different, with 7 meeting our FDR threshold (Figure 2C, Supplementary Table 5). These included class Alphaproteobacteria (LDA ES 2.4, p=0.03, q=0.05) and genus Mycobacterium (LDA ES 2.6, p=0.002, q=0.05), increased in controls, and class Betaproteobacteria (LDA ES 2.9, p=0.007, q=0.05) increased in OA. In knee samples, no statistically significant differences were seen between eroded and intact cartilage. Comparing knee OA to knee controls, 41 clades were different (Figure 2D, Supplementary Table 6). These included class Clostridia (LDA ES 3.1, p=2E-4) and phylum Bacteroidetes (LDA ES 2.3, p=0.003), both increased in control cartilage, and class Betaproteobacteria (LDA ES 3, p=2E-4) and order Burkholderiales (LDA ES 3, p=6E-4), both increased in OA cartilage.
Human knee and hip samples have distinct microbial DNA signatures, which are shared among both OA and disease-free tissues.
We next examined differences in cartilage microbiome composition based on joint location. First, we compared knee-OA to hip-OA specimens, where LEfSe identified 36 differential clades, 8 of which met our FDR threshold (Figure 3A, 3B, Supplementary Table 7). Hip-OA samples were characterized by members of the Proteobacteria family, including family Rhodocyclaceae (LDA ES 2.1, p=0.0002, q=0.08), whereas knee samples included members of the phylum Actinobacteria, including family Micrococcaceae (LDA ES 2.4, p=0.009, q=0.06), and multiple members of phyla Firmicutes, including genus Exiguobacterium (LDA ES 2.5, p=3E-11, q=8E-5).
Figure 3:
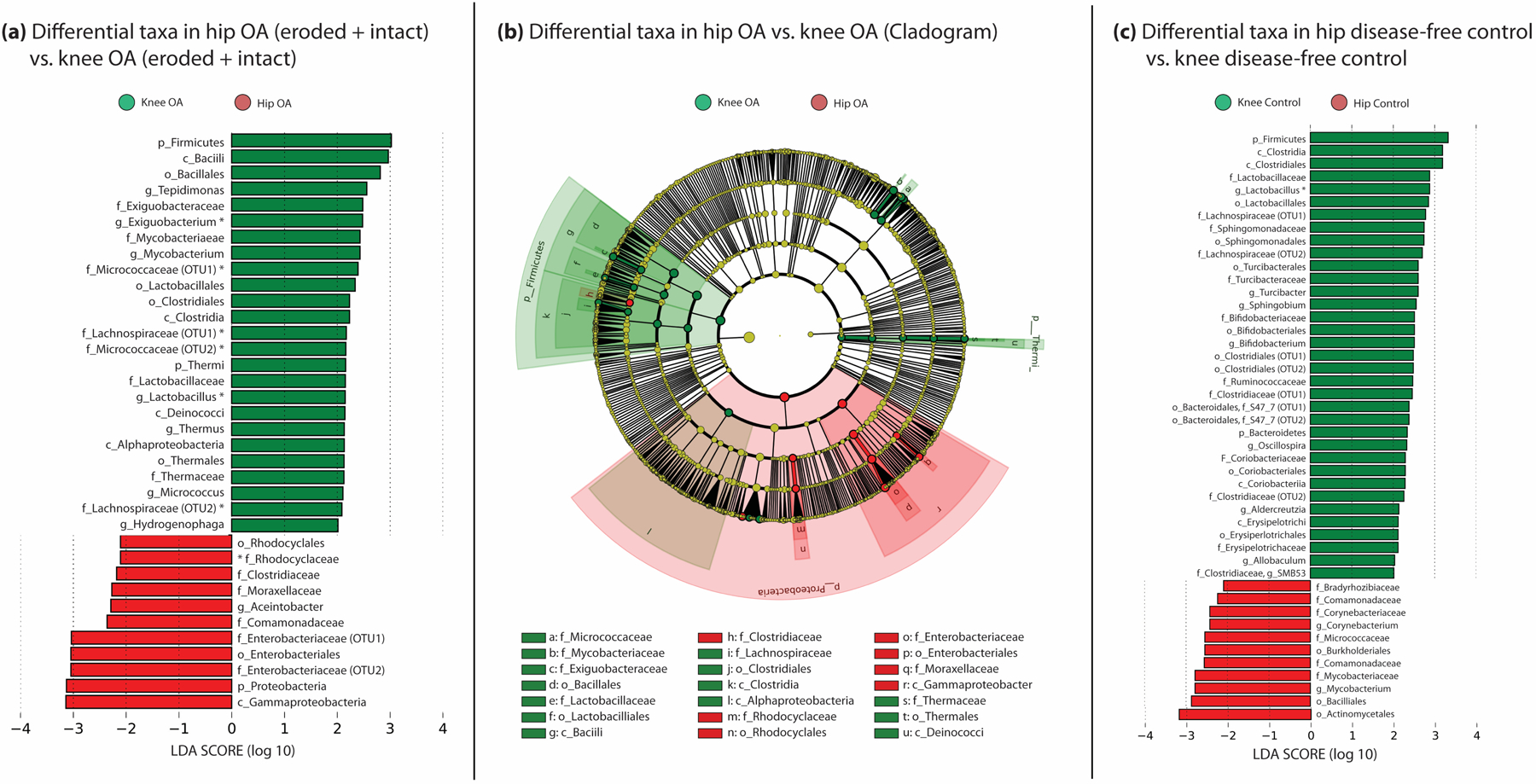
(a) Statistically significant differences in human microbial DNA clades comparing knee and hip specimens among OA specimens. (b) Cladogram comparing hip OA to knee OA clades. (c) Statistically significant differences in clades between hip and knee control specimens. Bars represent LDA effect size scores. Asterisk indicates clade passed FDR correction.
Comparing disease-free control sections, we identified 46 differences, including many of the same clades found in the above OA analysis; one (genus Lactobacillus) met FDR criteria (Figure 3C, Supplementary Table 8). Hips were characterized by members of the phylum Actinobacteria, including order Actinomycetales (LDA ES 3.2, p=0.03), and class Betaproteobacteria, including order Burkholderiales (p=0.02) and family Comamonadaceae (p=0.01). Knees were characterized by phylum Firmicutes, including genus Lactobacillus (LDA ES 2.9, p=7E05, q=0.07), class Clostridia (LDA ES 3.2, p=0.0004) and order Clostridiales (LDA ES 3.2, p=0.0004).
Clade-specific qPCR confirmed 16S sequencing findings in an independent cohort.
We next confirmed our findings in a separate cohort of OA and disease-free control cartilage using clade-specific qPCR (Figure 4). All confirmation results were similar to our deep-sequencing analysis. Specifically, we confirmed both class Alphaproteobacteria (intact vs. control p=0.007, eroded vs. control p=0.006) and phylum Bacteroidetes (intact vs. control p=0.02, eroded vs. control p=0.002). Comparing hip-OA to hip-control, we confirmed class Alphaproteobacteria (p=0.0002) and class Betaproteobacteria (p=0.02). Comparing hip-eroded to hip-intact, we confirmed genus Pseudomonas (p=0.04). Comparing knee-OA to knee-control, we confirmed class Betaproteobacteria (p=0.03) and order Burkholderiales (p=0.05). Finally, comparing hip-OA to knee-OA, we confirmed phylum Firmicutes (p=0.03).
Figure 4:
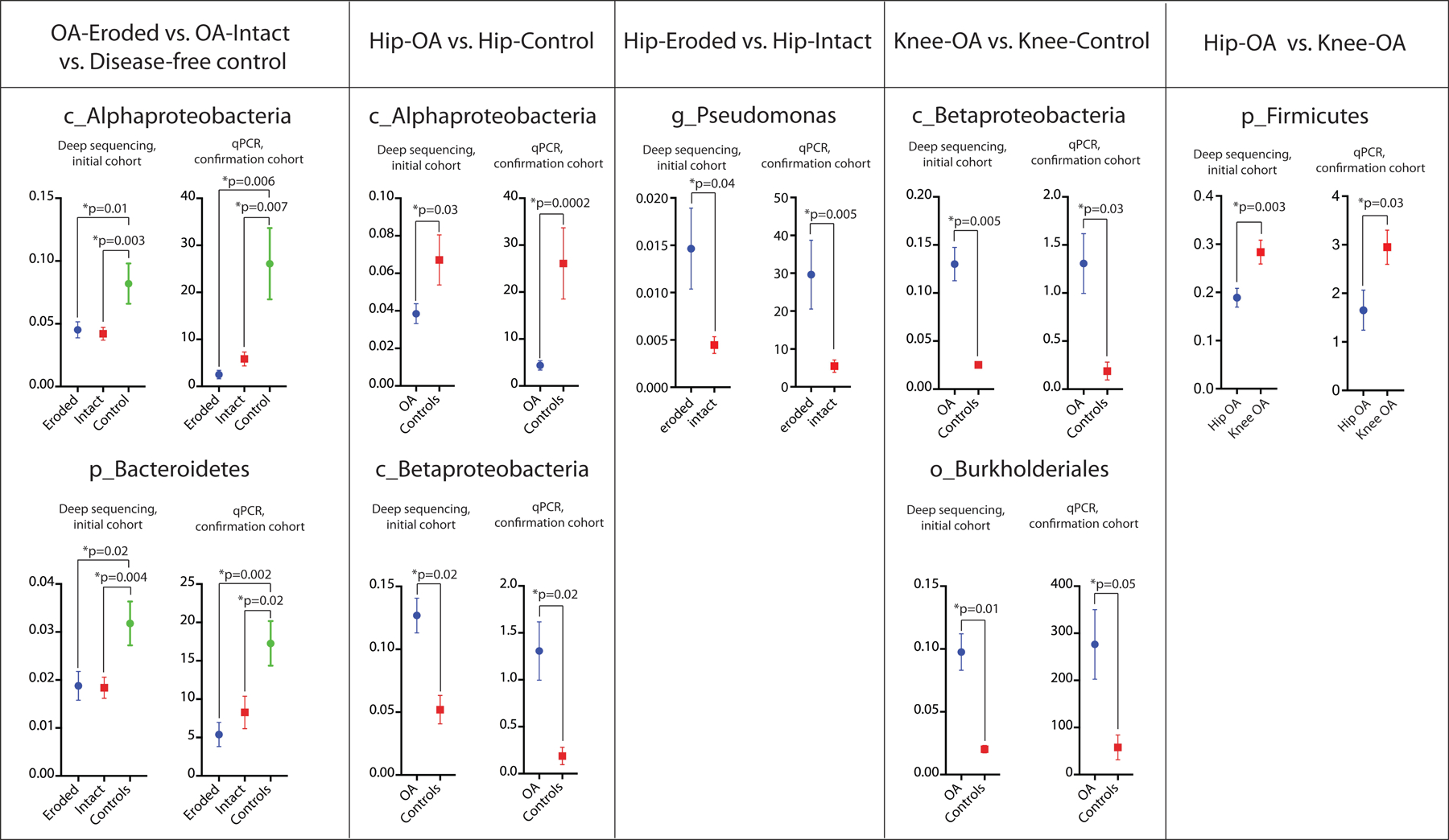
Clade-specific quantitative PCR confirmation in a separate confirmation cohort of differential clades among OA, control, knee, and hip samples. Initial deep sequencing cohort data, y-axis represents the fraction of total group clades, y-axis of confirmation cohort represents arbitrary units vs. universal bacterial primer set (delta-delta CT method).
Human OA cartilage demonstrates a shift towards Gram-negative constituents.
Next, we identified substantial increases in the proportion of constituent microbial DNA from Gram-negative organisms in OA patients compared to disease-free controls (37%±2% vs. 27%±2% mean±SEM, p=0.02). These differences persisted when eroded and intact sections were considered separately (OA-intact: 37%±3% vs. control: 27%±2%, p=0.03; OA-eroded 38%±3% vs. control 27%±2%, p=0.02), whereas OA-eroded and OA-intact specimens were not significantly different (p=0.84), Supplementary Figure 1.
Human cartilage imputed metagenomes suggest alterations in several canonical bacterial pathways.
Given the significant changes in cartilage microbial DNA patterns we found in human cartilage samples, we then sought to determine alterations in imputed metagenome function using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) package(40). Using PICRUSt, we found alterations in 37 Kyoto Encyclopedia of Gene and Genomes (KEGG) pathways (Figure 5), including reductions in phenylpropanoid biosynthesis (p=9.9E-5, q=0.03) and sphingolipid metabolism (p=7.7E-4, q=0.05) in OA samples, increases in phosphatidylinositol signaling in OA (p=4.2E-4, q=0.05), increases in nitrogen metabolism in OA (p=8.0E-3, q=0.08), and increases in lipopolysaccharide biosynthesis in OA (p=9.9E-3, q=0.1), among others (Supplementary Table 9).
Figure 5:

Differences in functional classification of bacterial species in human OA-eroded, OA-intact, and disease-free control cartilage identified through predictive metagenomic analysis using PICRUSt. Bars represent mean±S.D., stars represent mean. P-values were calculated using ANOVA, q-values are Benjamini-Hochberg FDR corrected
Mouse knee cartilage sample microbial DNA signatures vary by strain and demonstrate changes similar to human knee cartilage samples. Germ-free mouse knee samples lack a cartilage microbial DNA signature.
We next evaluated mouse knee cartilage samples from adult male OA-susceptible C57BL/6J (B6, n=8) and OA-protected MRL/MpJ mice (MRL, n=8). OTU raw counts were lower than in human samples, likely owing to the smaller tissue mass in mice (OTU counts 4214±913, mean±SEM). There were no differences in alpha diversity comparing B6 to MRL mouse samples (MRL 100±27 vs. B6 90±27, p=0.47). Beta diversity between the groups was not quite statistically significantly different (Adonis p=0.056).
LEfSe identified 32 clade group differences comparing MRL to B6 mice (Supplementary Figure 2, Supplementary Table 10). None met FDR criteria, likely due to small group sizes. Increased in MRL cartilage were members of the family Lactobacillaceae (LDA ES 3.4, p=0.003), family Turicibacteraceae (LDA ES 2.98,p=0.01), genus Actinomyces (LDA ES 2.86, p=0.01), various members of class Verrucomicrobiae (LDA ES 3.27, p=0.03). Characteristic of B6 cartilage were nucleic acid signatures from family Comamonadaceae from within family Burkholderiales (LDA ES 3.6, p=0.009), class Betaproteobacteria (LDA ES 3.7, p=0.01), and family Tissierellaceae from order Clostridiales (LDA ES 2.9, p=0.01).
Eleven of the clades which differentiated human knee control from knee OA cartilage also differentiated OA-protected MRL cartilage from OA-susceptible B6. These included members of order Lactobacillales, family Lachnospiraceae, genus Lactobacillus, order Turicibacterales, class Bacteroidia, and class Betaproteobacteria, among others. In every case, OTUs which overlapped shared a similar pattern: clades increased in human control cartilage were also increased in OA-protected MRL mice, whereas clades increased in human OA were also increased in OA-susceptible B6 cartilage. Also mirroring our human data, we found an enrichment in Gram-negative organism nucleic acid in B6 compared to MRL (B6: 50%±0.06% vs. MRL: 35%±0.03%, p=0.028, Figure 5).
Finally, as methodological controls, we examined cartilage samples sourced from germ-free C57BL/6 mice. We failed to identify a significant constituent microbial DNA signature in these mice; 16s PCR amplification bands were not seen on agarose gel electrophoresis and deep sequencing demonstrated very low amplification (GF OTU raw counts 196±44, mean±SEM, n=7). Indeed, germ-free samples were indistinguishable from water controls (water OTU raw counts 270±94, mean±SEM, n=3, comparison p=0.44).
Discussion:
In this study, we provide the first evidence of microbial nucleic acid signatures in human and mouse cartilage tissue. We identified shifts in this signature including reduced diversity following rarefaction and an increase in Gram negative constituents in OA compared to controls. We identified a number of clades which were characteristic of human OA-eroded, OA-intact, and control cartilage. Furthermore, we demonstrated that knee and hip cartilage are distinct, both with and without OA. We identified cartilage microbial DNA signatures in mouse knee tissues, which varied depending on mouse strain; OA-protected MRL mice had bacterial DNA signatures similar to disease-free control human tissue, whereas OA-susceptible B6 mice had signatures similar to human OA tissue. We did not find a significant cartilage microbial DNA signature in germ-free mice.
Our data add to the growing body of literature which have uncovered constituent microbial DNA signatures in a variety of human tissues previously thought sterile, including the central nervous system(43). The reduction in microbial alpha diversity seen in OA mirrors findings in other rheumatic diseases, where reduced diversity has been associated with reactive arthritis(44), ankylosing spondylitis(45), and rheumatoid arthritis(46). Our findings are in line with previous reports that members of the phylum Proteobacteria, specifically Betaproteobacteria, are markers of patients with obesity in humans and diet-induced obesity in mice(47), both of which are strongly associated with OA.
Few studies have examined gut microbiome perturbations in OA. Among these, a recent paper by Boer et al investigated the gut microbiome composition of 867 adults within the Dutch Rotterdam (RSIII) and LifeLines-DEEP cohorts (48). They identified, using 16S rRNA deep sequencing, four bacterial clades associated with knee pain as measured by WOMAC score. These included class Bacilli, order Lactobacillales, family Streptococcaceae, and genus Streptococcus. We found Lactobacillales to be associated with control human and MRL cartilage, although the families driving this association were not Streptococcaceae, but Aerococcaceae and Carnobacteriaceae. Interestingly though, we did find that members of Lactobacillales were strongly associated with knee samples when compared to hips. In 2018, Schott demonstrated that prebiotic supplementation (oligofructose) protected against both cartilage degeneration and synovial hyperplasia in disruption of the medial meniscus (DMM)-induced OA in mice fed a high-fat diet(9). Supplementation was associated with a shift in the gut microbiome towards an increase in Actinobacteria, several clades of which we identified as associated with disease-free control cartilage (i.e. Acidimicrobia, Friedmanniela, Coriobacteriia, Rubrobacteria, Figure 2, Supplementary Table 2). Indeed, the most-altered clade in oligofructose-supplemented mice was genus Bifidobacterium, which we found strongly associated with human control cartilage (FDR q-value 0.003). In 2018, Zhao and colleagues published the first deep sequencing-based evaluation of synovial fluid and synovial tissue from knees of human OA and RA patients(20). Many of the bacterial DNA clades they found characteristic of knee OA synovial tissue we found associated with control cartilage, including family Lanchospiraceae, order Coriobacteriales, and family Clostridiaceae, although this discordance is likely related to study design. Considering knees and hips together, we found that Proteobacteria, specifically Betaproteobacteria, were associated with OA. Increases in Proteobacteria has been linked in mice fed a high fat diet(49).
In our study, knee samples were characterized by nucleic acid sequences from multiple members of the phylum Firmicutes, one of the major constituents of the normal human gut microbiome (50). Among OA samples, geographic differences were similar to what we encountered in controls, with knee microbial DNA again being characterized by Firmicutes, and hips by Proteobacteria, although why these joints seem to have conserved geographic speciation is not clear. Increases in the Firmicutes:Bacteroidetes ratio in the gut have been linked to obesity (51). Among our samples, knee patients had a higher BMI than hip patients (29±1 vs. 34±1, p=0.01), but we did not find a statistically significant difference in the Firmicutes:Bacteroidetes DNA ratio in knees vs. hips (p=0.22). Knee OA samples were persistently characterized by Firmicutes, whereas hip OA samples were enriched in Proteobacteria, specifically, Beta- and Gammaproteobacteria. It should be noted that eight of the ten control knee/hip specimens were matched; that is, knee and hip cartilage specimens were from the same individual. This strengthens the argument that knee and hip microbial signature differences do indeed exist, and were not simply due to variation between individuals.
Intriguingly, we found significant overlap when comparing mouse and human cartilage microbial DNA patterns. For example, family Comamonadaceae and the class Betaproteobacteria were found more frequently in OA-susceptible B6 mice and both were among the most highly significant clades increased in human knee OA. Conversely, genus Lactobacillus, order Burkholderiales, family Turcibateraceae, order Erysipelotrichales, and order Bacteroidales sequences were increased in OA-protected MRL mice and were increased in disease-free human cartilage. In total, 11 clade overlaps were found between murine and human knee cartilage microbial DNA samples, representing one third of the total number of statistically significant clades in mice and including each of the top six most statistically significant clades in murine cartilage. In every case, clades characteristic of human control cartilage were increased in MRL samples, whereas clades characteristic of human OA cartilage were increased in B6 samples. Although speculative, this raises the intriguing possibility that OA-associated cartilage microbial DNA patterns may precede OA development.
Recent studies have highlighted the role of chronic, low-level inflammation in OA, including innate immune activation(5), macrophage-predominant inflammatory responses(50), toll-like receptor (TLR) activation(5), and complement activation(6). TLR2 and TLR4, upregulated in OA cartilage(51), are stimulated by lipopolysaccharide (LPS), a constituent of the Gram-negative bacterial cell wall. Serum LPS levels have been associated with osteophyte severity in human OA, and synovial fluid LPS has been associated with osteophyte severity, joint space narrowing, and total pain / functional severity scores(13). The source of LPS in OA patients is thought to be increased permeability from the gut, a feature of the diet-induced-obesity OA model(52); indeed, LPS elevation in obese patients has been suggested as a potential contributor to OA development(53). In agreement with these studies, we show herein that both human OA cartilage and OA-susceptible mouse cartilage contain an increased fraction of Gram-negative constituent bacterial DNA compared to human controls and OA-protected mice, respectively.
An interesting question is the timing and route of inoculation of the microbial DNA we describe, e.g., whether it is trafficked from the gastrointestinal tract via live bacterial organisms or carried into the articular space within immune cells. Another possibility is inoculation from subchondral bone via the osteochondral junction(54). We are also unable to comment on whether cartilage microbial DNA diversity decreases with age, as has been noted with the gut microbiome, although this may be an intriguing contributor to the increased OA risk seen with aging(55). Assuming that the cartilage bacterial DNA patterns we found in our study are indeed sourced from the systemic circulation, it is indeed a curious finding that these patterns vary within the same joint (i.e. eroded vs. intact areas). There are several potential explanations for this. Perhaps eroded regions of cartilage are more exposed to products of systemic circulation, either via inflammatory cells or blood contact directly, and therefore experience more rapid deposition and/or change of bacterial DNA compared to intact regions.
Our functional analysis, performed by reconstructing metagenomes using PICRUSt, identified several bacterial functional pathways predicted to be altered in association with OA. Several of these pathways are consistent with previous OA reports. For example, phenylpropanoids, reduced in OA-associated constituents, have reported anti-inflammatory effects(56). Phosphatidylinositol signaling is intimately linked with oxidative stress in OA(57). Bone mineral resorption, is a feature of late OA(58). There is a large volume of published data on the role of increased nitric oxide production and OA, which may be related to the increased nitrogen metabolism pathway seen in our functional analysis(59). Finally, we saw evidence for increased LPS production in our functional analysis, which is consistent with previous studies regarding LPS and OA, discussed in more detail above.
Our study does have several limitations. The first is the potential for contamination leading to false positive results. We undertook several steps to identify and prevent potential contamination as outlined previously, including processing samples in a sterile environment, running water controls, and decontaminating PCR reagents and plasticware. Critically, we did not find a microbial DNA signature when germ-free mouse cartilage was examined. Our analyses relied on detection and classification of microorganisms by nucleic acid analysis; therefore, we have not confirmed the presence of living bacteria within cartilage tissue. Given the inflammatory effects of bacterial DNA our findings nonetheless suggest a potential driver of local inflammation in OA. Our study may suffer from potential confounding, most likely in the form of comorbidities including obesity and aging (although neither of these risk factors was statistically significantly different among groups). We did perform separate analyses to determine if any cartilage bacterial DNA clades were strongly correlated with either obesity or aging independent of disease state but did not find any statistically significant associations. However, our study is likely underpowered to definitively state whether such correlations exist; future studies, particularly in mouse models, may be better equipped to answer these questions. Finally, we are unable to draw conclusions regarding the direct role of any specific bacterial DNA in the development or progression of OA, this sort of hypothesis-driven research should be a focus of future investigations, particularly in mouse models of OA.
In summary, herein we characterized a cartilage microbial DNA signature in human hip and knee cartilage and B6 and MRL mouse cartilage. Using a deep sequencing approach, we identified a variety of microbial clades that are associated with disease-free control, OA-intact, and OA-eroded tissue, and identified reductions in alpha diversity in OA tissue compared to disease-free controls. In addition, we identified several clades that were differentially present in knee and hip cartilage. Further, we identified a shift in the composition of OA samples towards Gram-negative constituents. Our human findings were reflected in a comparison of cartilage from OA-susceptible and OA-resistant mouse strains.
Confirmation of our findings will require further studies, and expansion to include examination of bacterial metabolites will allow better understanding of potential shifts in cartilage bacterial enzymes and their byproducts as a novel contributor to OA pathogenesis. An analysis of when and how the cartilage is inoculated with its microbiome should be undertaken in mouse models, as well as an evaluation of age-related shifts in the cartilage microbiome, which may offer a novel insight into age-related increases in OA susceptibility. Further studies examining the cartilage microbial DNA landscape in other, particularly inflammatory, arthropathies including rheumatoid arthritis are also in order. Finally, integrated longitudinal studies in animal models of OA, defining both cecal and cartilage microbiota may also enlighten the potential pathogenic role of certain bacterial species in the development of OA, and may provide an avenue for the development of novel therapeutics.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements and Support:
This work was supported by NIH grants K08AR070891 and P20GM125528. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding source had no involvement in the writing of this article.
We acknowledge the use of tissues procured by the National Disease Research Interchange (NDRI) with support from NIH grant U42OD011158.
This work was supported by a Physician Scientist Development Award (PDSA) from the Presbyterian Health Foundation, and an Oklahoma Center for the Advancement of Science and Technology (OCAST) Oklahoma Health Research Grant.
Footnotes
COI: The authors declare no conflicts of interest.
References:
- 1.Centers for Disease Control and Prevention (CDC). Prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation--United States, 2010–2012. MMWR Morb Mortal Wkly Rep 2013;62:869–873. [PMC free article] [PubMed] [Google Scholar]
- 2.Magnusson K, Scurrah K, Ystrom E, Ørstavik RE, Nilsen T, Steingrímsdóttir ÓA, et al. Genetic factors contribute more to hip than knee surgery due to osteoarthritis - a population-based twin registry study of joint arthroplasty. Osteoarthritis Cartilage 2017;25:878–884. [DOI] [PubMed] [Google Scholar]
- 3.Synovium Liu-Bryan R. and the innate inflammatory network in osteoarthritis progression. Curr Rheumatol Rep 2013;15:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraus VB, McDaniel G, Huebner JL, Stabler TV, Pieper CF, Shipes SW, et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage 2016. Available at: 10.1016/j.joca.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol 2008;20:565–572. [DOI] [PubMed] [Google Scholar]
- 6.Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, et al. Identification of a central role for complement in osteoarthritis. Nat Med 2011;17:1674–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Toole PW, Jeffery IB. Gut microbiota and aging. Science 2015;350:1214–1215. [DOI] [PubMed] [Google Scholar]
- 8.John GK, Mullin GE. The Gut Microbiome and Obesity. Curr Oncol Rep 2016;18:45. [DOI] [PubMed] [Google Scholar]
- 9.Schott EM, Farnsworth CW, Grier A, Lillis JA, Soniwala S, Dadourian GH, et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight 2018;3 Available at: 10.1172/jci.insight.95997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulici V, Kelley KL, Azcarate-Peril MA, Cleveland RJ, Sartor RB, Schwartz TA, et al. Osteoarthritis induced by destabilization of the medial meniscus is reduced in germ-free mice. Osteoarthritis Cartilage 2018;26:1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller RE, Miller RJ, Ishihara S, Belmadani A, Golub SB, Fosang AJ, et al. A 32-mer fragment of aggrecan promotes knee hyperalgesia in the DMM model of osteoarthritis through direct nociceptor activation via TLR2. Osteoarthritis Cartilage 2016;24:S452. [Google Scholar]
- 12.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 2009;15:774–780. [DOI] [PubMed] [Google Scholar]
- 13.Huang Z, Stabler T, Pei F, Kraus V. Both systemic and local lipopolysaccharide (LPS) burden is associated with knee osteoarthritis (OA). Osteoarthritis Cartilage 2016;24:S329–S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itagaki K, Adibnia Y, Sun S, Zhao C, Sursal T, Chen Y, et al. Bacterial DNA Induces Pulmonary Damage Via TLR-9 Through Cross-talk With Neutrophils. Shock 2011;36:548–552. Available at: 10.1097/shk.0b013e3182369fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci U S A 2001;98:9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gómez-Moreno R, Robledo IE, Baerga-Ortiz A. Direct Detection and Quantification of Bacterial Genes Associated with Inflammation in DNA Isolated from Stool. Adv Microbiol 2014;4:1065–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz DA, Quinn TJ, Thorne PS, Sayeed S, Yi AK, Krieg AM. CpG motifs in bacterial DNA cause inflammation in the lower respiratory tract. J Clin Invest 1997;100:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Martinez RE, Abud-Mendoza C, Patiño-Marin N, Rizo-Rodríguez JC, Little JW, Loyola-Rodríguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol 2009;36:1004–1010. [DOI] [PubMed] [Google Scholar]
- 19.Pretorius E, Akeredolu O-O, Soma P, Kell DB. Major involvement of bacterial components in rheumatoid arthritis and its accompanying oxidative stress, systemic inflammation and hypercoagulability. Exp Biol Med 2017;242:355–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Chen B, Li S, Yang L, Zhu D, Wang Y, et al. Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients. Sci Rep 2018;8:14305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stinson LF, Keelan JA, Payne MS. Identification and removal of contaminating microbial DNA from PCR reagents: impact on low-biomass microbiome analyses. Lett Appl Microbiol 2019;68:2–8. [DOI] [PubMed] [Google Scholar]
- 22.Champlot S, Berthelot C, Pruvost M, Bennett EA, Grange T, Geigl E-M. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS One 2010;5 Available at: 10.1371/journal.pone.0013042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pritzker KPH, Gay S, Jimenez SA, Ostergaard K, Pelletier J-P, Revell PA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 2006;14:13–29. [DOI] [PubMed] [Google Scholar]
- 24.Anon. 16S Sample Preparation Guide. Available at: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf. [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26:2460–2461. [DOI] [PubMed] [Google Scholar]
- 27.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012;6:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang Q, Luan Y, Sun F. Variance adjusted weighted UniFrac: a powerful beta diversity measure for comparing communities based on phylogeny. BMC Bioinformatics 2011;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamady M, Knight R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res 2009;19:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruskal WH, Wallis WA. Use of Ranks in One-Criterion Variance Analysis. J Am Stat Assoc 1952;47:583–621. [Google Scholar]
- 32.Fisher RA. THE USE OF MULTIPLE MEASUREMENTS IN TAXONOMIC PROBLEMS. Ann Eugen 1936;7:179–188. [Google Scholar]
- 33.Battaglia T LEfSe · An Introduction to QIIME 1.9.1 Available at: https://twbattaglia.gitbooks.io/introduction-to-qiime/content/lefse.html. Accessed February 14, 2018. [Google Scholar]
- 34.Blackwood CB, Oaks A, Buyer JS. Phylum- and class-specific PCR primers for general microbial community analysis. Appl Environ Microbiol 2005;71:6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mühling M, Woolven-Allen J, Murrell JC, Joint I. Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J 2008;2:379–392. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y-W, Chen M-K, Yang B-Y, Huang X-J, Zhang X-R, He L-Q, et al. Use of 16S rRNA Gene-Targeted Group-Specific Primers for Real-Time PCR Analysis of Predominant Bacteria in Mouse Feces. Appl Environ Microbiol 2015;81:6749–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan IUH, Yadav JS. Real-time PCR assays for genus-specific detection and quantification of culturable and non-culturable mycobacteria and pseudomonads in metalworking fluids. Mol Cell Probes 2004;18:67–73. [DOI] [PubMed] [Google Scholar]
- 38.Miller TR, Franklin MP, Halden RU. Bacterial community analysis of shallow groundwater undergoing sequential anaerobic and aerobic chloroethene biotransformation. FEMS Microbiol Ecol 2007;60:299–311. [DOI] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 40.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 2014;30:3123–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emery DC, Shoemark DK, Batstone TE, Waterfall CM, Coghill JA, Cerajewska TL, et al. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer’s Post-Mortem Brain. Front Aging Neurosci 2017;9:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manasson J, Shen N, Garcia Ferrer HR, Ubeda C, Iraheta I, Heguy A, et al. Gut Microbiota Perturbations in Reactive Arthritis and Postinfectious Spondyloarthritis. Arthritis Rheumatol 2018;70:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Y-Y, Tan X, He Y-T, Zhou Y-Y, He X-H, Huang R-Y. Role of gut microbiome in ankylosing spondylitis: an analysis of studies in literature. Discov Med 2016;22:361–370. [PubMed] [Google Scholar]
- 46.Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Öckinger J, Kullberg S, et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 2016;4:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin N-R, Whon TW, Bae J-W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol 2015;33:496–503. [DOI] [PubMed] [Google Scholar]
- 48.Boer CG, Radjabzadeh D, Medina-Gomez C, Garmaeva S, Schiphof D, Arp P, et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat Commun 2019;10:4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen Y-Y, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009;137:1716–24.e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bondeson J, Blom AB, Wainwright S, Hughes C, Caterson B, van den Berg WB. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum 2010;62:647–657. [DOI] [PubMed] [Google Scholar]
- 51.Kim HA, Cho M-L, Choi HY, Yoon CS, Jhun JY, Oh HJ, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum 2006;54:2152–2163. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Tang H, Zhang C, Zhao Y, Derrien M, Rocher E, et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J 2015;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metcalfe D, Harte AL, Aletrari MO, Al Daghri NM, Al Disi D, Tripathi G, et al. Does endotoxaemia contribute to osteoarthritis in obese patients? Clin Sci 2012;123:627–634. [DOI] [PubMed] [Google Scholar]
- 54.Berthelot J-M, Sellam J, Maugars Y, Berenbaum F. Cartilage-gut-microbiome axis: a new paradigm for novel therapeutic opportunities in osteoarthritis. RMD Open 2019;5:e001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saraswati S, Sitaraman R. Aging and the human gut microbiota-from correlation to causality. Front Microbiol 2014;5:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korkina LG, Mikhal’Chik E, Suprun MV, Pastore S, Dal Toso R. Molecular mechanisms underlying wound healing and anti-inflammatory properties of naturally occurring biotechnologically produced phenylpropanoid glycosides. Cell Mol Biol 2007;53:84–91. [PubMed] [Google Scholar]
- 57.Yin W, Park J-I, Loeser RF. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem 2009;284:31972–31981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Findlay DM, Atkins GJ. Osteoblast-chondrocyte interactions in osteoarthritis. Curr Osteoporos Rep 2014;12:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abramson SB. Osteoarthritis and nitric oxide. Osteoarthritis Cartilage 2008;16 Suppl 2:S15–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.