

Abstract
Classical homocystinuria is a recessive inborn error of metabolism caused by mutations in the cystathionine beta-synthase (CBS) gene. The highest incidence of CBS deficiency in the world is found in the country of Qatar due to the combination of high rates of consanguinity and the presence of a founder mutation, c.1006C>T (p.R336C). This mutation does not respond to pyridoxine and is considered severe. Here we describe the creation of a mouse that is null for the mouse Cbs gene and expresses human p.R336C CBS from a zinc-inducible transgene (Tg-R336C Cbs−/−). Zinc treated Tg-R336C Cbs−/− mice have extreme elevation in both serum tHcy and liver tHcy compared to control transgenic mice. Both the steady-state protein levels and CBS enzyme activity levels in liver lysates from Tg-R336C Cbs−/− mice are significantly reduced compared to that found in Tg-hCBS Cbs−/− mice expressing wild-type human CBS. Treatment of Tg-R336C Cbs−/− mice with the proteasome inhibitor bortezomib results in stabilization of liver CBS protein and an increase in activity to levels found in corresponding Tg-hCBS Cbs−/− wild type mice. Surprisingly, serum tHcy did not fully correct even though liver enzyme activity was as high as control animals. This discrepancy is explained by in vitro enzymatic studies of mouse liver extracts showing that p.R336C causes reduced binding affinity for the substrate serine by almost seven-fold and significantly increased dependence on pyridoxal phosphate in the reaction buffer. These studies demonstrate that the p.R336C alteration effects both protein stability and substrate/cofactor binding.
Keywords: inborn error, homocysteine, methionine, metabolism, mouse model, missense mutation
Introduction
Cystathionine β-synthase (CBS) deficiency (MIM# 613381) is a recessive genetic disorder that is the cause of classical homocystinuria, the most common inborn error of sulfur amino acid metabolism (Mudd 2011). CBS catalyzes the condensation of homocysteine with serine to form cystathionine and uses pyridoxal phosphate (PLP) as a key prosthetic group. It is most abundant in liver and kidney. The vast majority of CBS mutant alleles are predicted to encode for missense alterations in the CBS protein (Kraus et al 1999). CBS deficient patients are characterized by extreme elevations in plasma total homocysteine (tHcy), and patients suffer a variety of pathologies including thrombosis, osteoporosis, mental retardation, and dislocated lenses. The main treatment strategy is to reduce patient tHcy by a combination of B-vitamins, betaine supplementation and dietary methionine restriction.
The overall frequency of CBS deficiency in Western populations is about 0.5–1 per 100000, but this can vary widely between countries due to the presence of specific founder mutations (Moorthie et al 2014). By far the highest frequency of CBS deficiency is found in the country of Qatar, with an estimated incidence of 1:1800 (Zschocke et al 2009; Gan-Schreier et al 2010). This high prevalence is primarily attributed to a single founder mutation, p.R336C (c.1006C>T) (GenBank reference sequence NM_000071.2), and is further enhanced by a high rate of consanguineous marriages. Homozygous individuals for p.R336C have a severe vitamin B6 (pyridoxine) non-responsive phenotype and tend to have significant clinical complications (El-Said et al 2006; Zschocke et al 2009). Examination of p.R336C on the human CBS crystal structure indicates that this alteration is located in an alpha helix and that it forms hydrogen bonds with p.D388 and p.L386 (Supplemental Figure 1). Human p.R336C expressed in yeast appears stable and functionally inactive, while expression in HEK293T and HEPG2 cells suggest that the mutation causes a reduction in protein stability (Ismail et al 2019).
In previous work, the Kruger lab has created a number of genetically engineered humanized mouse models of CBS deficiency (Wang et al 2004; Wang et al 2005; Gupta et al 2008; Gupta et al 2017). These models utilize a transgene that expresses a cDNA encoding a specific human CBS allele under control of the zinc-inducible metallothionine promoter combined with homozygous deletion of the endogenous mouse Cbs gene. Interestingly, several mutant CBS proteins can behave quite differently in mice than in cell culture or microbiological expression systems (Kim et al 1997; Gupta et al 2008; Gupta et al 2017). In some cases, the behavior of mutant proteins in mice more closely resembles the behavior observed in CBS deficient patients compared to behavior in other expression systems (Gupta et al 2008; Gupta et al 2017). The goal of the experiments described here is to create and characterize a mouse model of p.R336C CBS in order to determine how this mutation affects CBS function in vivo.
Methods
DNA and constructs
All nucleotide numbering is based on the GenBank RefSeq NM_000071.1. Site-directed mutagenesis with the Quick Change XL site-directed mutagenesis kit from Agilent was used to introduce a c.1006 C>T (p.R336C) change into pLW2 (Wang et al 2004), which contains a hemagglutinin epitope tagged version of the human CBS cDNA. The entire open reading frame of the resulting clone, pLW2:R336C, was sequenced to verify no additional changes occurred due to the mutagenesis process. Plasmid pLW2:R336C was subsequently digested with MfeI and cloned into the EcoR1 site of MT-LCR expression vector 2999 (Palmiter et al 1993). The resulting plasmid was designated pLW3:R336C.
Mouse generation
Tg-R336C transgenic mice were created identically to Tg-hCBS mice as previously described (Wang et al 2004). Approximately 70 injected C57BL6/C3H F2 embryos were transferred to pseudopregnant mice, which resulted in the birth of 14 pups, of which only one positive animal (a male) was obtained. This DNA positive male was then backcrossed to a Cbs−/+ female (C57BL/6J background, (Watanabe et al 1995)) and germline transmission was confirmed. To create Tg-R336C Cbs−/− mice, Tg-R336C Cbs+/− siblings were mated with each other in cages with water containing 25 mM ZnSO4, to induce transgene expression. Although Tg-R336C Cbs−/− animals were born in approximately Mendelian ratios, the vast majority of them were dead at the time of weaning. Therefore, Tg-R336C Cbs+/− mice were backcrossed with C3H/HeJ mice for three generations before again intercrossing.
Genotyping of offspring was generally done between 10 and 14 days of age as previously described (Wang et al 2004). Animals were fed standard rodent chow (Teklad 2018SX). For weight studies, sex and age matched siblings between 66 and 160 days of age were used.
Liver pathology
Haematoxylin and eosin (H and E) staining was performed on livers fixed in 10% neutral buffered formalin as previously described (Wang et al 2005). Quantitation of mitotic figures was determined by examining liver sections from four Tg-R336C Cbs−/− mice and four Tg-R336C Cbs+/− under 200x magnification. Ten non-overlapping fields from each liver was examnined by a trained pathologist (Kathy Qi).
Metabolite measurements
Liver metabolite measurements and serum tCys (total cysteine) were performed using LC-MS/MS in the Blom lab as previously described (Esse et al 2014). Serum amino acids including tHcy and methionine were measured in the Kruger lab using a Biochrom 30 amino acid analyzer as previously described (Wang et al 2004).
Western blotting, CBS enzyme activity, and qRT-PCR
Liver homogenates were prepared as previously described (Shan et al 2001; Wang et al 2005). Immunoblot analysis was performed as described (Gupta et al 2008). CBS activity was analyzed in the presence and absence of AdoMet (250 μM) as previously described (Wang et al 2004). One unit of activity is defined as nmoles of cystathionine formed per milligram of protein per hour. In kinetic studies, a single substrate was titrated in, while all other components were held constant as previously described (Chen et al 2006). Kd values were determined by non-linear curve fitting using GraphPad Prism 6.0 software. It should be noted that addition of high concentrations of bortezomib (up to 52 μM) to in vitro enzyme activity reactions had no effect on activity.
For qRT-PCR, RNA was extracted from livers using TRIzol (Life Technologies) followed by cleanup using Qiagen RNeasy Mini kit. Gene expression analysis was done using Taq Man probes for human CBS (Hs00163925_m1, Applied Biosystems) and mouse β-actin (Mm01205647_g1). Quantification of signal was achieved using an Applied Biosystems 7900 HT detection system. Each sample was assayed in duplicate for both probes, and averages were used for statistical analysis. Relative signal strength was calculated using the ΔΔCt method (Livak and Schmittgen 2001).
Bortezomib studies
Tg-R336C Cbs−/− mice were induced by zinc water (25 mM ZnSO4) for 11 days, and bortezomib was given at a dose of 0.49 mg/kg/day using an Alzet microosmotic pump implanted subcutaneously as previously described (Gupta et al 2017). Serum was collected by retroorbital bleed after zinc pretreatment, but before pump implantation. Mice were sacrificed after 48 hours and tissues and serum were collected for further analysis. For pyridoxine and serine supplementation studies, water was supplemented with 0.4 g/l pyridoxine and 220 g/l L-serine for six days prior to pump implantation.
Statistics
All values cited in text and figures are arithmetic mean ± standard error. T-tests are all two-sided with a p<0.05 considered significant. One-way ANOVA (analysis of variance) followed by a post-hoc Tukey’s test was used in experiments that involved multiple comparisons. Statistics were all performed using GraphPad Prism 6.0 software.
Results
Generation of Tg-R366C Cbs−/− mice
We created a zinc-inducible regulated transgene for p.R336C using the same methodology as our lab has done for other CBS mutants (Wang et al 2004) (Supplemental Figure 2a). A single founder was obtained which was then backcrossed to a Cbs+/− heterozygote (C57BL6 background) to confirm Mendelian transmission and transgene expression in response to zinc (Supplemental Figure 2b). Resulting Tg-R336C Cbs+/− mice were then intercrossed to generate Tg-R336C Cbs−/− mice. Out of 72 pups generated, 8 Tg-R336C Cbs−/− mice were detected by genotyping at 10–14 days of age, but only 1 survived until the time of weaning. This finding indicated that the Tg-R336C transgene cannot rescue the high degree of neonatal lethality associated with homozygosity for Cbs− in C57BL/6J mice (Watanabe et al 1995). The one surviving mice was analyzed at four months of age. The mouse was smaller than its littermates and showed the characteristic facial alopecia observed in CBS deficient mice (Robert et al 2004). Total serum homocysteine was >400 μM and this did not decrease upon zinc treatment.
To circumvent the neonatal lethality, we decided to backcross the mice three times onto the onto the C3H background, as Cbs−/− mice survive the neonatal period at a significantly higher percentage in this background (Akahoshi et al 2008; Gupta et al 2017). After backcrossing, we found that 73% (54/74) Tg-R336C Cbs−/− mice survived to at least 5 weeks of age.
Physical and biochemical characterization of Tg-R336C Cbs−/− mice
Surviving Tg-R366C Cbs−/− mice on the C3H background were significantly smaller than sibling age-matched Tg-R336C Cbs+/− mice (Figure 1a). Unlike Cbs−/− mice in the C57BL6 background (Robert et al 2004; Gupta et al 2009), Tg-R366C Cbs−/− had only very mild facial hair loss (Figure 1b). However, we did observe that their overall coat color was significantly lighter than their Cbs+/+ and Cbs+/− siblings. Examination of H and E sections of Tg-R366C Cbs−/− livers showed moderate hepatocyte hypertrophy (Figure 2b). In addition, we observed that Tg-R366C Cbs−/− livers had a significant number of mitotic figures present (48 in 40 non-overlapping fields), while they were almost entirely absent in Tg-R336C Cbs+/− mice (2 in 40 non-overlapping fields, P<10−6). In No differences were observed in the kidneys.
Fig 1. Phenotypes of Tg-R336C Cbs−/− mice.
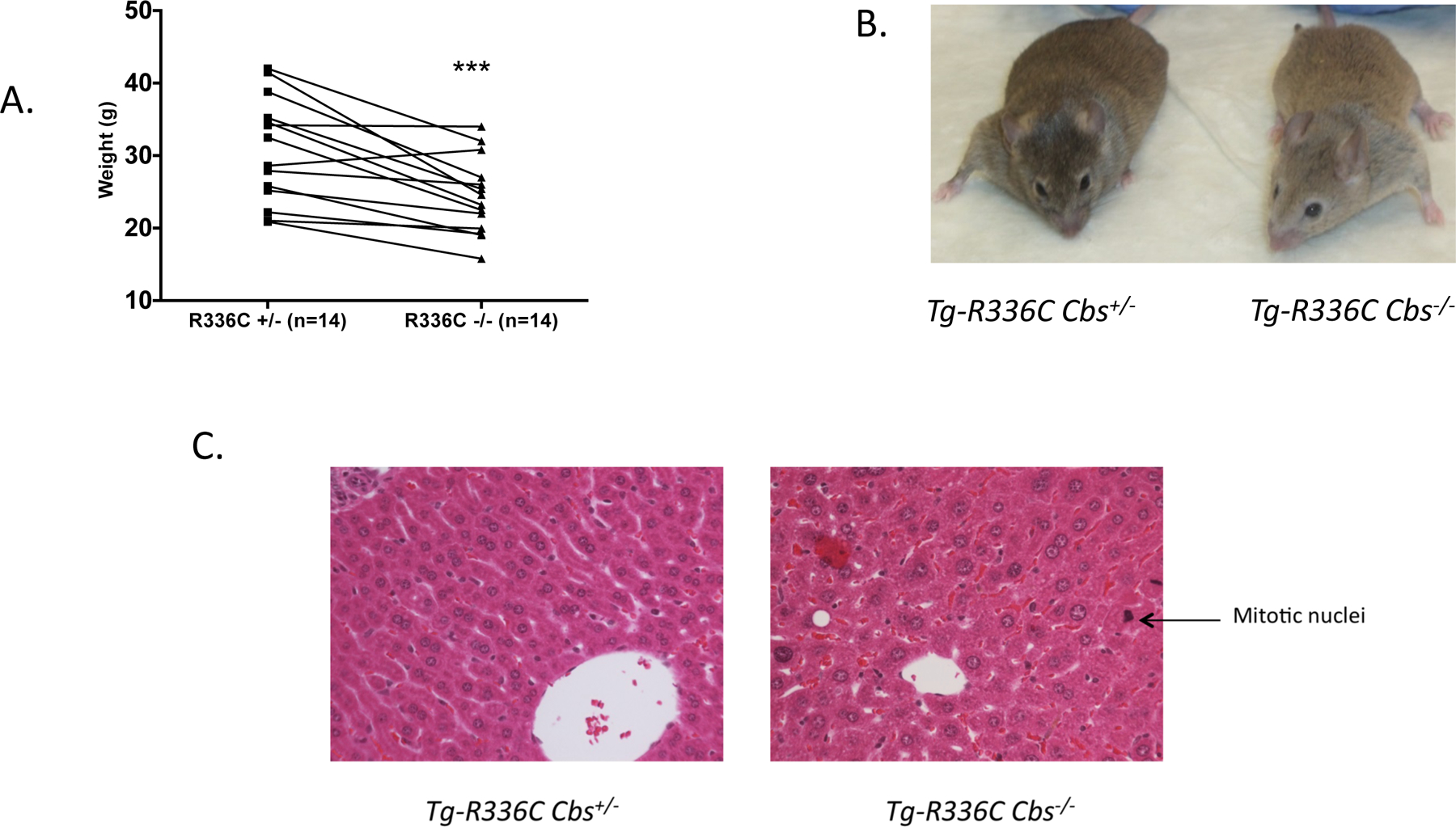
(A), Weight of age and sex matched Tg-R336C Cbs−/− mice in comparison to sibling control Tg-R366C Cbs+/−. Mice were aged between 2.2–5.3 months. *** is P<0.005 (paired t-test). (B), Photograph of a 4-month old male sibling pair of indicated genotypes. (C), Representative H & E stained liver sections of 4 month old sibling pair of the indicated genotypes at 40x magnification. Arrow indicates example of mitotic nuclei observed at a higher frequency in Tg-R366C Cbs−/− mice. Also note larger size of hepatocyte nuclei throughout section.
Fig 2. Homocysteine and methionine in Tg-R336C Cbs−/− mice.
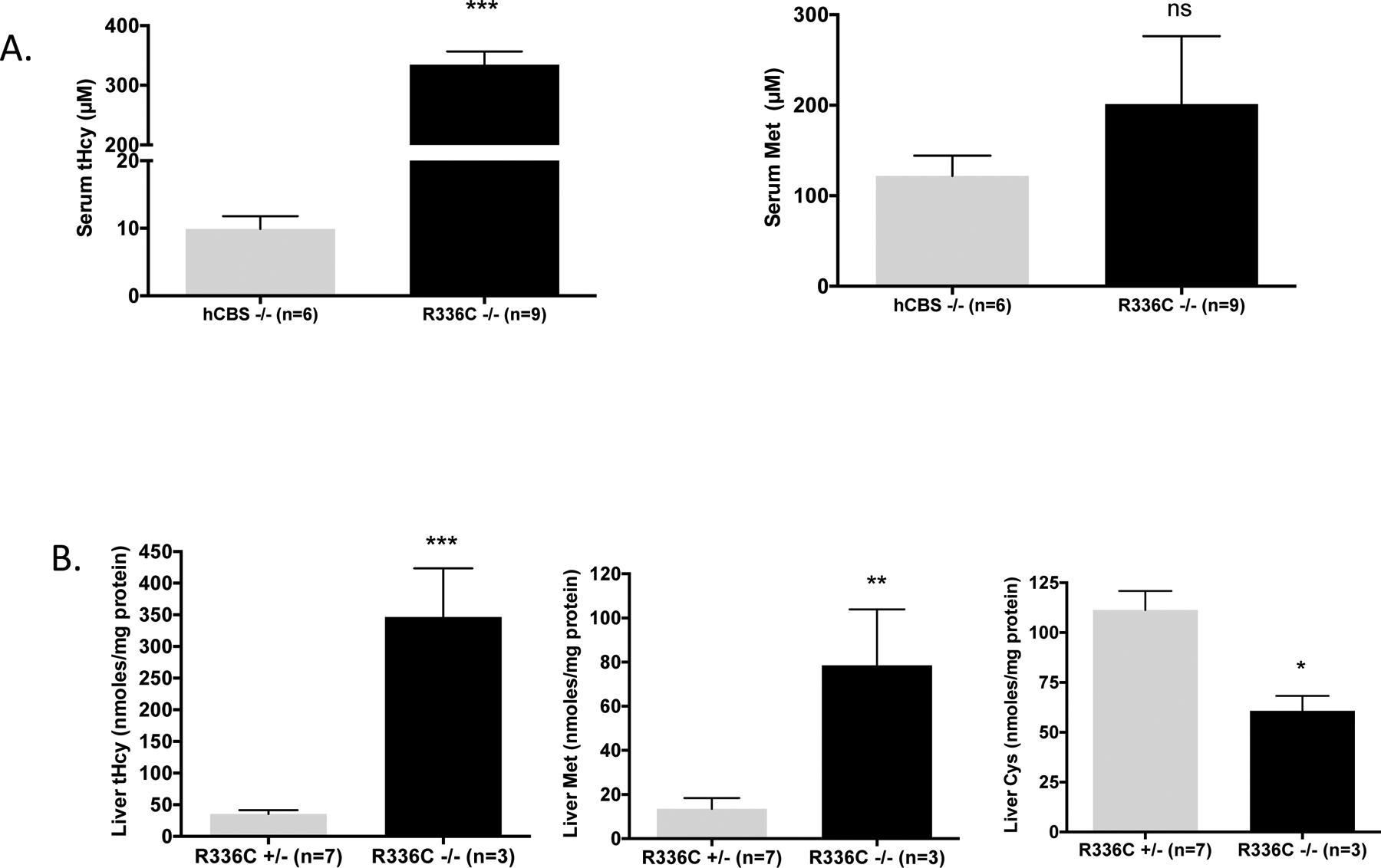
(A), Serum total homocysteine (tHcy) and methionine in μM in zinc-induced Tg-hCBS Cbs−/− and Tg-R336C Cbs−/− mice. Error bar shows standard error (SE). *** indicates P<0.001. (B), tHcy, methionine, and total cysteine (tCys) in liver lysates from zinc-induced Tg-R336C Cbs−/− and Tg-R336C Cbs+/− mice. *** indicates P<0.001, **P<0.01, *P<0.05.
Biochemically, Tg-R366C Cbs−/− mice on zinc water (transgene expressed), had extreme elevation in serum tHcy compared to control Tg-hCBS Cbs−/− on zinc (334 μM vs. 9.9 μM, P<0.0001) (Figure 2a). Serum methionine levels were elevated compared to controls, but this was not statistically significant due to wide variation (201 μM vs. 122 μM, P=0.42). In liver lysates, Tg-R366C Cbs−/− mice had significant elevations in tHcy, methionine, and a significant reduction liver tCys (Figure 2b) compared to sibling Tg-R366C Cbs+/− control mice.
p.R336C CBS has reduced stability which is reversed by proteasome inhibition
We examined the steady state levels of p.R336C in the livers of induced Tg-R366C Cbs−/− mice using both denaturing and native gel electrophoresis followed by immunoblot analysis. In both assays the overall CBS protein level was significantly reduced compared to the amount of CBS observed in extracts from control Tg-hCBS Cbs−/− mice (Wang et al 2004) expressing wild-type human CBS (Figure 3a). This difference was also reflected in the lysates’ CBS activity. Under standard reaction conditions, we observed that Tg-hCBS Cbs−/− extracts had a mean activity of 993 units vs. 134 units (P<0.001) for the Tg-R366C Cbs−/− extracts (Figure 3b). It should be noted that the difference in CBS protein levels is not due to differences in the levels of CBS mRNA in the two lines (Figure 3c).
Fig 3. Analysis of p.R336C CBS protein and mRNA.
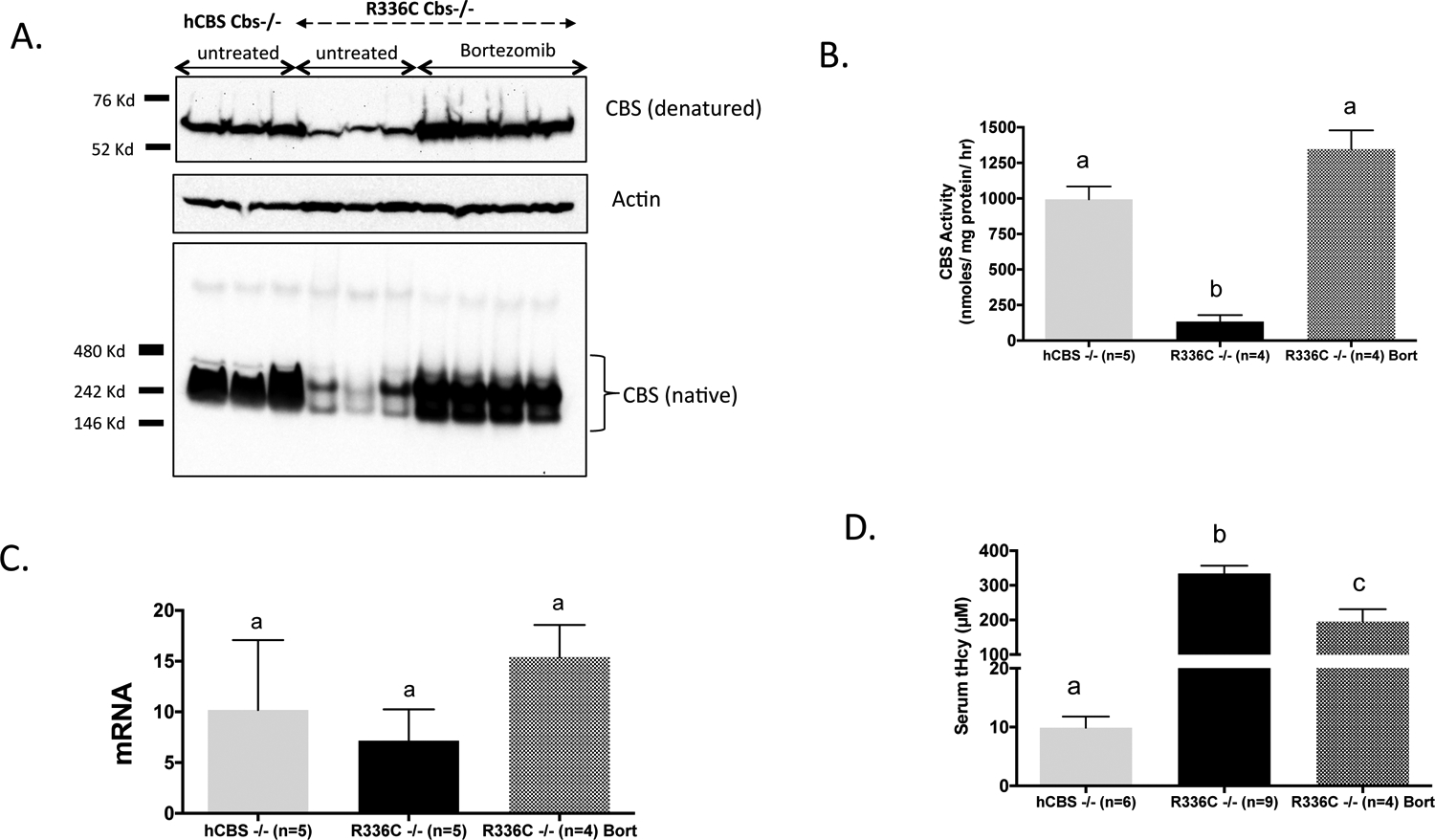
(A), Western blot of CBS expression in liver lysates of zinc-induced Tg-hCBS Cbs−/−, Tg-R336C Cbs−/−, and bortezomib treated Tg-R336C Cbs−/− mice run on denaturing (top) and native (bottom) gels. Actin was used as a loading control. Positions of molecular weight makers for each gel type is also shown. (B), CBS activity present in dialyzed crude lysates from liver of indicated genotype and treatment. Different letters at top indicate P<0.001 as judged by ANOVA followed by Tukey’s multiple comparison test. (C), Relative liver human CBS mRNA levels as determined by quantitative real time PCR using mouse actin expression as a normalizing control. (D), Serum tHcy homocysteine in indicated mice.
The reduction in steady state p.R336C CBS protein relative to WT hCBS, despite having similar levels of mRNA, suggested that p.R336C may have increased propensity for protein misfolding, which often results in degradation of the protein by the proteasome (Balch et al 2008). Previous studies from our lab have shown that treatment of mice with proteasome inhibitors like bortezomib can functionally rescue at least three different patient-derived mutant proteins (Singh et al 2010; Gupta et al 2013; Gupta et al 2017). Therefore, we examined the effect of short-term bortezomib treatment on the stability and activity of p.R366C CBS. Treatment of Tg-R336C Cbs−/− mice on zinc water with bortezomib resulted in a dramatic increase in liver CBS protein and activity (Figure 3a and 3b). Mean liver CBS activity in treated animals was 1346 units compared to 134 units in untreated mice (P<0.001). In fact, the level of CBS enzyme activity detected in treated animals was not statistically different compared to Tg-hCBS Cbs−/− mice (994 vs. 1346 units). Surprisingly, serum tHcy in Tg-R336C Cbs−/− animals was only moderately reduced by bortezomib treatment (196 versus 334 μM, P<0.01) and was much higher than that observed in control Tg-hCBS Cbs−/− mice (9.9 μM) (Figure 3d).
To explore this further, we plotted serum tHcy vs CBS enzyme activity in 140 mice expressing various human CBS alleles (both untreated and proteasome treated) that our lab had studied over the years (Wang et al 2004; Chen et al 2006; Gupta et al 2008; Singh et al 2010; Gupta et al 2013; Gupta et al 2017) (Supplemental Figure 3). The plot shows a negative correlation between tHcy and in vitro liver CBS activity and indicates that the samples from the Tg-R336C Cbs−/− mice (especially the bortezomib treated samples) tend to have relatively high serum tHcy relative compared to their liver CBS activity. These data suggest that other factors are may be impacting catalytic efficiency of R336C CBS enzyme in vivo.
p.R336C has altered affinity for pyridoxine, AdoMet, and serine
One possible explanation for the reduced catalytic efficiency of bortezomib-treated p.R336C protein may be related to its enzymatic properties such as affinity for Hcy, serine, PLP, or response to the allosteric activator AdoMet. We tested each of these parameters using liver lysates from zinc-induced Tg-hCBS Cbs−/−, Tg-R336C Cbs−/−, and Tg-R336C Cbs−/− mice treated with bortezomib. Extracts expressing p.R336C from untreated or bortezomib treated mice had a 5-fold increase in Km values for serine compared to extracts from mice expressing WT hCBS (Table 1; Supplemental Figure 4). The Km for Hcy decreased 31–60% in p.R336C samples, although the difference in Km was only significant for the bortezomib-treated sample. The Kact (concentration of AdoMet needed for 50% maximal stimulation) revealed that the p.R336C extracts had higher affinity for AdoMet compared to the hCBS sample, i.e. they achieved maximal activation at a lower concentration of AdoMet. With regards to PLP affinities, we found p.R336C expressing extracts required exogenous PLP in the reaction buffer for maximal enzyme activity, while WT hCBS extracts were just as active in the absence of PLP (Supplemental Figure 5). These results indicate that p.R336C has significantly reduced PLP affinity compared to wild type CBS protein.
Table 1.
Binding affinities for CBS alleles.
| CBS allele | Km-ser (CI) | Km-Hcy (CI) | Kact-adoMet (CI) |
|---|---|---|---|
| Wt hCBS | 0.9 mM (0.6–1.2) | 3.5 mM (2.7–4.2) | 33 μM (21–46) |
| p.R336C | 5.1 mM (3.6–6.7) | 1.4 mM (0.9–1.9) | 15 μM (10–19) |
| p.R336C+bortez | 4.5 mM (2.9–6.0) | 2.4 mM (1.0–3.8) | 13 μM (7.7–18) |
CI=95% confidence interval
Given these findings, we wondered if a combination of pyridoxine, serine, and bortezomib might be more effective than bortezomib alone in lowering tHcy in Tg-R336C Cbs−/− mice. To test this idea, Tg-R366C Cbs−/− mice were given zinc water containing 0.4 g/L pyridoxine and 220g/L serine, which would result in a 15-fold and 29-fold increase in each substance over the basal diet. After one week on this diet, mice were then implanted with osmotic pumps delivering bortezomib. Despite the supplementation with pyridoxine and serine, we failed to observe any difference in either serum tHcy, liver CBS enzyme activity levels, or levels of liver CBS protein in the supplemented animals (Supplemental Figure 6).
Discussion
In this paper, we have analyzed the p.R336C mutation in the mouse model of CBS deficiency. This mutation is interesting because it is the predominant allele found in CBS deficient patients from Qatar, which has the highest rate of CBS deficiency in the world (Gan-Schreier et al 2010). In humans, this allele is associated with a relatively severe disease that is non-responsive to pyridoxine treatment. Here, we created a transgenic mouse that expresses human p.R336C hCBS under control of the zinc inducible MT-I promoter and then crossed in the Cbs− allele to generate mice that only express p.R336C CBS. The characterization of these mice revealed several important observations. First, p.R336C CBS cannot rescue the neonatal lethal phenotype associated with Cbs−/− in a C57BL6/J background, but on a C3H background Tg-R336C Cbs−/− mice survive into adulthood and have severely elevated serum tHcy levels. Second, the p.R336C mutation results in lower steady state levels of CBS protein due to increased proteolysis by the proteasome. Third, independent of the stability effect, the p.R336C mutation lowers the binding affinity of the enzyme to its substrate, serine and its co-factor PLP. Lastly, treatment with the proteasome inhibitor bortezomib can partially decrease, but could not fully normalize the hyperhomocysteinemia in Tg-R336C Cbs−/− mice.
The p.R336C mutation is the fifth different CBS allele that our lab has examined for neonatal rescue in the C57BL6 strain background. WT, p.I278T, and P.S466L CBS rescue lethality, while p.R266K and p.R336C do not (Wang et al 2004; Wang et al 2005; Gupta et al 2008; Gupta et al 2017). This difference in rescue does not seem to correlate with levels of residual enzyme activity. Liver extracts from zinc induced Tg-R336C Cbs−/− mice actually have significantly more residual enzyme activity that those of Tg-I278T Cbs−/− or Tg-S466L Cbs−/− mice, even though the latter two alleles entirely rescue the neonatal lethality. Therefore, it seems likely that the neonatal lethality may be related to some non-enzymatic function of the mutant CBS proteins that is differentially affected by each mutation. However, it is important to note that this neonatal lethality is far less penetrant on the C3H background, so it is possible to get viable adult Tg-R336C Cbs−/− mice (Akahoshi et al 2008) (Gupta et al 2017). Adult Tg-R336C Cbs−/− mice have extremely elevated tHcy (>300 μM) and are significantly smaller than their Cbs+/− and Cbs+/+ siblings. These elevated tHcy levels are similar to the tHcy levels observed in C57BL6 models of Cbs deficiency (Kruger 2017). Other noticeable differences include a slightly lighter coat color, mild facial alopecia, hypertrophy of liver cells, and a noticeable increase in cells undergoing mitosis. These phenotypes have all been observed in other mouse models of Cbs deficiency and probably reflect the toxic effects of highly elevated serum tHcy.
Adult Tg-R336C Cbs−/− mice on zinc have low steady state levels of liver CBS protein and activity levels compared to Tg-hCBS Cbs−/− mice. This difference reflects decreased protein stability as treatment with the proteasome inhibitor bortezomib increases both p.R336C CBS protein and activity to wild-type levels. Based on our previously published work, the simplest explanation is that the p.R336C alteration affects the ability of CBS to fold-properly and that the misfolded protein is ubiquitinated and sent to the proteasome for degradation. This behavior is not unique and similar behavior has been observed for mice expressing three other patient derived missense CBS mutations, specifically p.I278T, p.R266K, and p.S466L (Gupta et al 2008; Gupta et al 2013; Gupta et al 2017). However, an unexpected finding in this study was that even though bortezomib treatment fully corrected in vitro liver CBS activity, it failed to fully correct mouse serum tHcy in vivo. This discrepancy led us to examine if p.R336C might be affecting kinetic parameters of the CBS. We found that even in bortezomib stabilized samples, the binding affinity for serine, homocysteine, PLP, and AdoMet were all significantly altered. The Km for serine was increased approximately five-fold, while the Km for homocysteine was slightly decreased.
A striking observation was that mouse liver expressed p.R336C CBS did not exhibit enzyme activity unless exogenous PLP added to the reaction mixture. This finding was unexpected because p.R336C patients do not respond to pyridoxine. However, it has proven extremely difficult to model pyridoxine response in any model of CBS deficiency. For example, mice expressing two known pyridoxine-responsive patient derived CBS alleles (p.I278T and p.R266K) fail to show any increased sensitivity to PLP in vitro or in vivo (Chen et al 2006; Gupta et al 2017). Lysates derived from human cell lines derived from pyridoxine responsive patients also fail to show any consistent increase in CBS activity in reponse to pyridoxine in the reaction mixture (Fowler et al 1978; Kluijtmans et al 1999). One can speculate that the inability of invesigators to successfully mimic pyridoxine response in mice or other systems may have to do with either a unique folding environment in human liver or that pharmacologic rescue of PLP in humans may be working via some indirect mechanism.
How does changing p.R336 to p.C336 cause these dramatic effects? Examination of the CBS crystal structure (Supplemental Fig 1) shows that the R336 forms hydrogen bonds that would help orient and stabilize the region of CBS between L386 and D388 relative to the alpha helix that contains R366. A substitution to cysteine would disrupt these bonds and allow this region to no longer be anchored in this position. While the p.L386 to p.D388 region is not adjacent to the active-site and the catalytic PLP, there are some key PLP stabilizing residues located just downstream (p.Y381, p.D376, p.P375). One could imagine that breaking hydrogen bonds in this region could affect the positioning of these key residues.
The results described here are somewhat, but not entirely consistent with the cellular expression studies of p.R336C in HEK and HepG2 cells (Ismail et al 2019). Like the current studies, this group also found that p.R336C alteration caused reduced steady state levels of CBS protein. However, unlike the mouse liver, there was no residual CBS activity detected in cell lysates. Also, although Ismail et al. observed that treatment of cells with the chemical chaperone betaine appeared to increase p.R336C protein concentration, this stabilized protein was inactive. This is in contrast to the results reported here, where we observed a strong increase in enzyme activity when mice were treated with proteasome inhibitor. It is unknown if treatment of the HEK or HepG2 cells treated with proteasome inhibitor might mimic our findings in mice.
Supplementary Material
Supplemental Figure 1. Location of R336 on CBS structure. R336 is shown in orange with dotted lines showing potential hydrogen bonds to D388 and L386 (green). Yellow space fill shows catalytic PLP and red space fill shows heme. Blue residues (Y380, D376, P375) show potential link on how loss of R336 hydrogen bonds may affect active site. Model was made using Pymol and CBS structure 4PCU (Ereno-Orbea et al 2014).
Supplemental Figure 2. Tg-R336C construct and expression. (A), Schematic showing Tg-R366C construct. Lines below show regions that were part of indicated subclones (see materials and methods). (B), Western blot showing expression of human and mouse CBS in offspring of Tg-R366C founder with Cbs+/− mouse. All mice were on zinc water to induce expression.
Supplemental Figure 3. Relationship between serum tHcy and liver CBS activity in various humanized transgenic Cbs−/− mice. Each dot represents a single Tg Cbs−/− mouse for which we have both tHcy and liver CBS enzyme activity. All mice were zinc induced. Red dots show Tg-R336C Cbs−/− mice treated with bortezomib. Green dots show Tg-R336C Cbs−/− in the absence of bortezomib. Black dots are derived from Tg-hCBS, Tg-I278T, Tg-R266K, and Tg-S466L Cbs−/− mice either untreated or treated with bortezomib that were previously described from our lab.
Supplemental Figure 4. Enzyme kinetics of CBS from dialyzed liver lysates. Dialyzed liver lysates from Tg-hCBS Cbs−/−, Tg-R336C Cbs−/−, and bortezomib-treated Tg-R366C Cbs−/− mice were used. (A), Enzyme activity vs. serine concentrations. Reactions were done in the presence of saturating concentration of DL-homocysteine (20 mM), 50 μM PLP, and 250 μM AdoMet. (B), Enzyme activity vs. DL-homocysteine concentrations. Reactions were done in the presence of saturating concentration of L-serine (10 mM), 50 μM PLP, and 250 μM AdoMet. (C), Enzyme activity vs. AdoMet concentration. Reactions were done in the presence of saturating concentration of L-serine and DL-homocysteine.
Supplemental Figure 5. Effect of oral pyridoxine (Pyr) and serine (Ser) supplementation on bortezomib rescue in Tg-R336C Cbs−/− mice. Tg-R336C Cbs−/− mice were given either 25 mM ZnSO4 water alone with an implanted osmotic pump containing saline, an osmotic pump containing bortezomib (0.49 mg/kg/day) in combination with ZnSO4 water, or a bortezomib osmotic pump with water containing ZnSO4, 0.4 g/L pyridoxine and 220 g/L L-serine. (A), Serum tHcy of indicated groups. (B), Liver CBS activity from indicated groups. (C), Denaturing gel immunoblot showing CBS protein from indicated groups. (D), Concentrations of selected serum amino acids in the presence or absence of pyridoxine and serine supplemented water.
Fig 6. Response of CBS activity to PLP in vitro. Dialyzed liver lysates from Tg-hCBS Cbs−/−, Tg-R336C Cbs−/− and Tg-R336C Cbs−/− mice treated with bortezomib were assayed for enzyme activity in the absence of PLP in the buffer (-PLP) or in the presences of 50 μM or 200 μM PLP. * indicates P<0.05, *** indicates P<0.001 compared to -PLP column.
Synopsis:
The common Qatari mutation p.R336C causes decreased protein stability and substrate binding in a humanized mouse model of CBS deficiency.
Acknowledgments
This work was funded in parts by grants from the National Institutes of Health (R01 DK101404; P30 CA006927) and NPRP grant # (NPR7-355-3-088) from the Qatar national research fund (QNRF, a member of Qatar foundation). We acknowledge the help of the Fox Chase Cancer Center Molecular Modeling facility for assistance in structural analysis and the Fox Chase Cancer Center Experimental Histopathology Core Facilty under the direction of Dr. Cathy Qi for mouse liver pathology studies.
Footnotes
Competing Interest Statement: Authors have no competing interests.
Ethics Approval: The study was approved by the Fox Chase Cancer Center IACUC (#99–26).
Patients Consent Statement: Not applicable.
Compliance with Ethical Standards: All animal studies were done in accordance with IACUC standards.
Informed consent: No patients were involved.
REFERENCES
- Akahoshi N, Kobayashi C, Ishizaki Y, et al. (2008) Genetic background conversion ameliorates semi-lethality and permits behavioral analyses in cystathionine beta-synthase-deficient mice, an animal model for hyperhomocysteinemia. Hum Mol Genet 17: 1994–2005. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319: 916–919. [DOI] [PubMed] [Google Scholar]
- Chen X, Wang L, Fazlieva R, Kruger WD (2006) Contrasting behaviors of mutant cystathionine beta-synthase enzymes associated with pyridoxine response. Hum Mutat 27: 474–482. [DOI] [PubMed] [Google Scholar]
- El-Said MF, Badii R, Bessisso MS, et al. (2006) A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum Mutat 27: 719. [DOI] [PubMed] [Google Scholar]
- Ereno-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martinez-Cruz LA (2014) Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by S-adenosylmethionine. Proc Natl Acad Sci U S A 111: E3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esse R, Imbard A, Florindo C, et al. (2014) Protein arginine hypomethylation in a mouse model of cystathionine beta-synthase deficiency. FASEB J 28: 2686–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler B, Kraus J, Packman S, Rosenberg LE (1978) Homocystinuria. Evidence for three distinct classes of cystathionine beta-synthase mutants in cultured fibroblasts. J Clin Invest 61: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan-Schreier H, Kebbewar M, Fang-Hoffmann J, et al. (2010) Newborn population screening for classic homocystinuria by determination of total homocysteine from Guthrie cards. J Pediatr 156: 427–432. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kühnisch J, Mustafa A, et al. (2009) Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J 23: 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Wang L, Anderl J, Slifker MJ, Kirk C, Kruger WD (2013) Correction of Cystathionine beta-Synthase Deficiency in Mice by Treatment with Proteasome Inhibitors. Hum Mutat 34: 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Wang L, Hua X, Krijt J, Kozich V, Kruger WD (2008) Cystathionine β-synthase p.S466L mutation causes hyperhomocysteinemia in mice. Hum Mutat 29: 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Wang L, Kruger WD (2017) The c.797 G>A (p.R266K) cystathionine beta-synthase mutation causes homocystinuria by affecting protein stability. Hum Mutat 38: 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail HM, Krishnamoorthy N, Al-Dewik N, et al. (2019) In silico and in vivo models for Qatari-specific classical homocystinuria as basis for development of novel therapies. Hum Mutat 40: 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CE, Gallagher PM, Guttormsen AB, et al. (1997) Functional modeling of vitamin responsiveness in yeast: a common pyridoxine-responsive cystathionine beta-synthase mutation in homocystinuria. Hum Mol Genet 6: 2213–2221. [DOI] [PubMed] [Google Scholar]
- Kluijtmans LA, Boers GH, Kraus JP, et al. (1999) The molecular basis of cystathionine beta-synthase deficiency in Dutch patients with homocystinuria: effect of CBS genotype on biochemical and clinical phenotype and on response to treatment. Am J Hum Genet 65: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus JP, Janosik M, Kozich V, et al. (1999) Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat 13: 362–375. [DOI] [PubMed] [Google Scholar]
- Kruger WD (2017) Cystathionine beta-synthase deficiency: Of mice and men. Mol Genet Metab 121: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Moorthie S, Cameron L, Sagoo GS, Bonham JR, Burton H (2014) Systematic review and meta-analysis to estimate the birth prevalence of five inherited metabolic diseases. J Inherit Metab Dis 37: 889–898. [DOI] [PubMed] [Google Scholar]
- Mudd SH (2011) Hypermethioninemias of genetic and non-genetic origin: A review. American journal of medical genetics Part C, Seminars in medical genetics 157: 3–32. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Sandgren EP, Koeller DM, Brinster RL (1993) Distal regulatory elements from the mouse metallothionein locus stimulate gene expression in transgenic mice. Mol Cell Biol 13: 5266–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert K, Maurin N, Ledru A, Delabar J, Janel N (2004) Hyperkeratosis in cystathionine beta synthase-deficient mice: an animal model of hyperhomocysteinemia. The anatomical record Part A, Discoveries in molecular, cellular, and evolutionary biology 280: 1072–1076. [DOI] [PubMed] [Google Scholar]
- Shan X, Dunbrack RL Jr, Christopher SA Jr., Kruger WD (2001) Mutations in the regulatory domain of cystathionine beta-synthase can functionally suppress patient-derived mutations in cis. Hum Mol Genet 10: 635–643. [DOI] [PubMed] [Google Scholar]
- Singh LR, Gupta S, Honig NH, Kraus JP, Kruger WD (2010) Activation of mutant enzyme function in vivo by proteasome inhibitors and treatments that induce Hsp70. PLoS Genet 6: e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chen X, Tang B, Hua X, Klein-Szanto A, Kruger WD (2005) Expression of mutant human cystathionine beta-synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum Mol Genet 14: 2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jhee KH, Hua X, DiBello PM, Jacobsen DW, Kruger WD (2004) Modulation of cystathionine beta-synthase level regulates total serum homocysteine in mice. Circ Res 94: 1318–1324. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, et al. (1995) Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A 92: 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Kebbewar M, Gan-Schreier H, et al. (2009) Molecular neonatal screening for homocystinuria in the Qatari population. Hum Mutat 30: 1021–1022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Location of R336 on CBS structure. R336 is shown in orange with dotted lines showing potential hydrogen bonds to D388 and L386 (green). Yellow space fill shows catalytic PLP and red space fill shows heme. Blue residues (Y380, D376, P375) show potential link on how loss of R336 hydrogen bonds may affect active site. Model was made using Pymol and CBS structure 4PCU (Ereno-Orbea et al 2014).
Supplemental Figure 2. Tg-R336C construct and expression. (A), Schematic showing Tg-R366C construct. Lines below show regions that were part of indicated subclones (see materials and methods). (B), Western blot showing expression of human and mouse CBS in offspring of Tg-R366C founder with Cbs+/− mouse. All mice were on zinc water to induce expression.
Supplemental Figure 3. Relationship between serum tHcy and liver CBS activity in various humanized transgenic Cbs−/− mice. Each dot represents a single Tg Cbs−/− mouse for which we have both tHcy and liver CBS enzyme activity. All mice were zinc induced. Red dots show Tg-R336C Cbs−/− mice treated with bortezomib. Green dots show Tg-R336C Cbs−/− in the absence of bortezomib. Black dots are derived from Tg-hCBS, Tg-I278T, Tg-R266K, and Tg-S466L Cbs−/− mice either untreated or treated with bortezomib that were previously described from our lab.
Supplemental Figure 4. Enzyme kinetics of CBS from dialyzed liver lysates. Dialyzed liver lysates from Tg-hCBS Cbs−/−, Tg-R336C Cbs−/−, and bortezomib-treated Tg-R366C Cbs−/− mice were used. (A), Enzyme activity vs. serine concentrations. Reactions were done in the presence of saturating concentration of DL-homocysteine (20 mM), 50 μM PLP, and 250 μM AdoMet. (B), Enzyme activity vs. DL-homocysteine concentrations. Reactions were done in the presence of saturating concentration of L-serine (10 mM), 50 μM PLP, and 250 μM AdoMet. (C), Enzyme activity vs. AdoMet concentration. Reactions were done in the presence of saturating concentration of L-serine and DL-homocysteine.
Supplemental Figure 5. Effect of oral pyridoxine (Pyr) and serine (Ser) supplementation on bortezomib rescue in Tg-R336C Cbs−/− mice. Tg-R336C Cbs−/− mice were given either 25 mM ZnSO4 water alone with an implanted osmotic pump containing saline, an osmotic pump containing bortezomib (0.49 mg/kg/day) in combination with ZnSO4 water, or a bortezomib osmotic pump with water containing ZnSO4, 0.4 g/L pyridoxine and 220 g/L L-serine. (A), Serum tHcy of indicated groups. (B), Liver CBS activity from indicated groups. (C), Denaturing gel immunoblot showing CBS protein from indicated groups. (D), Concentrations of selected serum amino acids in the presence or absence of pyridoxine and serine supplemented water.
Fig 6. Response of CBS activity to PLP in vitro. Dialyzed liver lysates from Tg-hCBS Cbs−/−, Tg-R336C Cbs−/− and Tg-R336C Cbs−/− mice treated with bortezomib were assayed for enzyme activity in the absence of PLP in the buffer (-PLP) or in the presences of 50 μM or 200 μM PLP. * indicates P<0.05, *** indicates P<0.001 compared to -PLP column.