

Abstract
Introduction:
Pulmonary arterial hypertension (PAH) is one of the leading causes of mortality in systemic sclerosis (SSc). We explored the impact of the updated haemodynamic definition of pulmonary hypertension (PH), as proposed by the 6th World Symposium on Pulmonary Hypertension.
Methods:
In this single-centre retrospective analysis, patients with SSc who had right heart catheterisation (RHC) were included. We compared the prior PH definition to the updated PH definition. The prior definition classified PH as mean pulmonary arterial pressure (mPAP) ⩾25 mmHg and further divided into pre-capillary PH (PAH and PH due to lung disease and/or hypoxia), post-capillary PH, and combined pre- and post-capillary PH groups. For the updated definition, PH was classified as mPAP >20 mmHg and further divided into the different groups. We validated our findings in the DETECT cohort.
Results:
Between 2005 and March 2019, 268 RHCs were performed in this single-centre cohort. Using the prior definition, 137 (51%) were diagnosed with PH, with 89 classified as pre-capillary PH (56 with PAH and 33 with PH due to lung disease and/or hypoxia), 29 as post-capillary PH, and 19 as combined pre- and post-capillary PH. When the updated definition was applied to the cohort, seven out of 131 (5%) with no PH were reclassified to pre-capillary PH (PAH (n=1), PH due to lung disease (n=3) and post-capillary PH (n=3)). In those with mPAP 21–24 mmHg, with no left heart or significant lung disease, one out of 28 (4%) in our cohort and four out of 36 (11%) in the DETECT cohort were reclassified as PAH
Conclusion:
The updated PH definition does not appear to have a significant impact on the diagnosis of PH in two different screening cohorts.
Introduction
Systemic sclerosis-related pulmonary arterial hypertension (SSc-PAH) is the one of the leading causes of mortality [1, 2] and accounts for up to 26% of SSc-related deaths [3]. Recent data from clinical trials and observational registries suggest better outcomes, including survival, are associated with uniform screening and early, aggressive combination therapies [4–6]. Previous World Symposia on Pulmonary Hypertension (WSPH) defined pulmonary hypertension (PH) as mean pulmonary arterial pressure (mPAP) ⩾25 mmHg and PAH is characterised haemodynamically by the presence of pre-capillary PH, including end-expiratory pulmonary arterial wedge pressure (PAWP) ⩽15 mmHg and pulmonary vascular resistance (PVR) >3 Wood Units (WU) [7–9].
Kovacs et al. [10] published a systematic review where they analysed available data obtained by right heart catheterisation (RHC) studies in healthy individuals and revealed that the mean±sd mPAP is 14.0±3.3 mmHg; 2 sd supports that mPAP >20 mmHg is above the upper limit of normal. In addition, data from various scleroderma cohorts suggest that patients with borderline elevations of mPAP (defined as mPAP 21–24 mmHg) are an intermediate step between normal PAP (mPAP ⩽20 mmHg) and PH (mPAP ⩾25 mmHg), associated with decreased exercise capacity and greater risk of developing resting PH [11–15]. Based on this and other data, the 2018 6th WSPH Task Force proposed an updated haemodynamic definition of PAH as mPAP >20 mmHg, PAWP ⩽15 mmHg and PVR ⩾3 WU (table 1) [16, 17]. The 6th WSPH Task Force recommended to include PVR ⩾3 WU for classification of pre-capillary PH to differentiate the elevation of mPAP due to other causes (driven by the contribution of cardiac output and/or PAWP).
TABLE 1.
Haemodynamic definitions of pulmonary hypertension (PH)
| Prior definition | New definition | |
|---|---|---|
| Group 1 (PAH) | mPAP ⩾25 mmHg and PVR >3 WU | mPAP >20 mmHg and PVR ⩾3 WU |
| PAWP ⩽15 mmHg | PAWP ⩽15 mmHg | |
| No/mild lung disease or FVC ⩾70% | No/mild lung disease or FVC ⩾70% | |
| Group 2 (post-capillary)# | mPAP ⩾25 mmHg | mPAP >20 mmHg |
| PAWP >15 mmHg | PAWP >15 mmHg | |
| PVR <3 WU | PVR <3 WU | |
| Group 3 (lung disease and/or hypoxia) | mPAP ⩾25 mmHg and PVR >3 WU | mPAP >20 mmHg and PVR ⩾3 WU |
| PAWP ⩽15 mmHg | PAWP ⩽15 mmHg | |
| Moderate/severe lung disease¶ or FVC <70% | Moderate/severe lung disease¶ or FVC <70% | |
| Group 4 (combined pre- and post-capillary) | mPAP ⩾25 mmHg | mPAP >20 mmHg |
| PAWP >15 mmHg | PAWP >15 mmHg | |
| PVR >3 WU | PVR ⩾3 WU |
PAH: pulmonary arterial hypertension; mPAP: mean pulmonary arterial pressure; PVR: pulmonary vascular resistance; WU: Wood Units; PAWP: pulmonary arterial wedge pressure; ILD: interstitial lung disease; FVC: forced vital capacity.
PH due to left heart disease
high-resolution computed tomography showing >20% total lung involvement due to ILD or if the total lung involvement due to ILD was 10-20% but the patient had concomitant moderate-to-severe emphysema.
We analysed retrospective data in scleroderma spectrum disorders from a PAH screening database of the University of Michigan (Ann Arbor, MI, USA) cohort to assess the impact of the updated haemodynamic definition of PH, including reclassification of patients with no PH to PH, and validated our data in the DETECT cohort [1]. Our objectives were to investigate the impact of the updated clinical PH classification in scleroderma spectrum disorders and the impact of including PVR in the updated definition of PH.
Patients and methods
Patients were included in this retrospective analysis of a prospective cohort (referred to as the University of Michigan cohort from hereon) if they had scleroderma spectrum disorders (SSc and overlap syndrome with scleroderma spectrum) [18], were evaluated at the University of Michigan scleroderma and PH clinics, and had RHC at the University of Michigan. This population represents an ongoing cohort to validate the DETECT algorithm [11] and other screening algorithms in scleroderma spectrum disorders, including transthoracic ECG, pulmonary function tests and N-terminal pro-brain natriuretic peptide [18]; details have been published recently [19]. Diagnosis of SSc was confirmed by a rheumatologist with expertise in scleroderma. Chart review was performed to extract age, race, sex, subtype of SSc, disease duration (defined from initial non-Raynaud’s phenomenon sign or symptom), scleroderma-specific autoantibodies and pulmonary function test results. High-resolution computer tomography (HRCT) scans were reviewed by two thoracic radiologists who assessed the degree of total lung involvement in increments of 10% to up to 30% or >30% lung involvement and if there was concomitant emphysema. If emphysema was present, it was classified as mild, moderate or severe. RHCs had been performed by a cardiologist due to concern for PH based on a positive screening test [18] or clinical signs/symptoms of PH. The thermodilution method was used to calculate the cardiac output and PVR [7, 20].
We compared the prior PH definition to the updated PH definition. The prior definition classified PH as mPAP ⩾25 mmHg and further divided into Group 1 (PAH), Group 2 (post-capillary PH), Group 3 (PH due to lung disease and/or hypoxia: HRCT showing >20% total lung involvement due to interstitial lung disease (ILD) or if the total lung involvement due to ILD was 10–20% but the patient had concomitant moderate-to-severe emphysema; if HRCT is not available, then forced vital capacity (FVC) <70% predicted within a median of 2 months of the RHC) and Group 4 (combined pre- and post-capillary PH) (table 1) [21]. For the updated classification, we used the published definitions where the mPAP was changed from ⩾25 to >20 mmHg and PVR was changed from >3 to ⩾3 WU. The patients were then further classified into the four aforementioned subsets [17]. We validated our results in the DETECT cohort [1, 11]. Briefly, the DETECT study was a multicentre study that systematically evaluated 466 SSc patients at increased risk for development of SSc-PAH. DETECT was the first SSc-PAH detection study to evaluate all subjects using RHC. Patients (n=244) were included in the current analysis if they had: 1) PAWP ⩽15 mmHg by RHC, 2) no significant ILD (defined as FVC <60% predicted or FVC 60–70% predicted with moderate-to-severe ILD on HRCT), 3) no systemic hypertension (stage I hypertension defined as systolic blood pressure ⩾140 mmHg or diastolic blood pressure ⩾90 mmHg) and 4) no left atrial enlargement.
Descriptive statistics for baseline demographics were determined based on PH groups. For continuous variables that followed a normal distribution, means and standard deviations were compared across groups using the t-test. For continuous variables that did not follow a normal distribution, medians and ranges were compared using the Wilcoxon rank sum test. For categorical variables, counts and proportions were calculated and compared across groups using Chi-squared tests or Fisher’s exact test. A significance level of p<0.05 was used for all statistical tests. Missing data, if any, was not imputed. Analyses were conducted in SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Between 2005 and March 2019, 268 RHCs were performed at the University of Michigan in patients who were at risk for PH based on PAH screening algorithms and guidelines, and are included in this retrospective analysis (figure 1a and b). Of the 268 patients, 11 patients diagnosed with overlap syndrome also met the criteria for SSc according to the 2013 SSc classification criteria [22].
FIGURE 1.
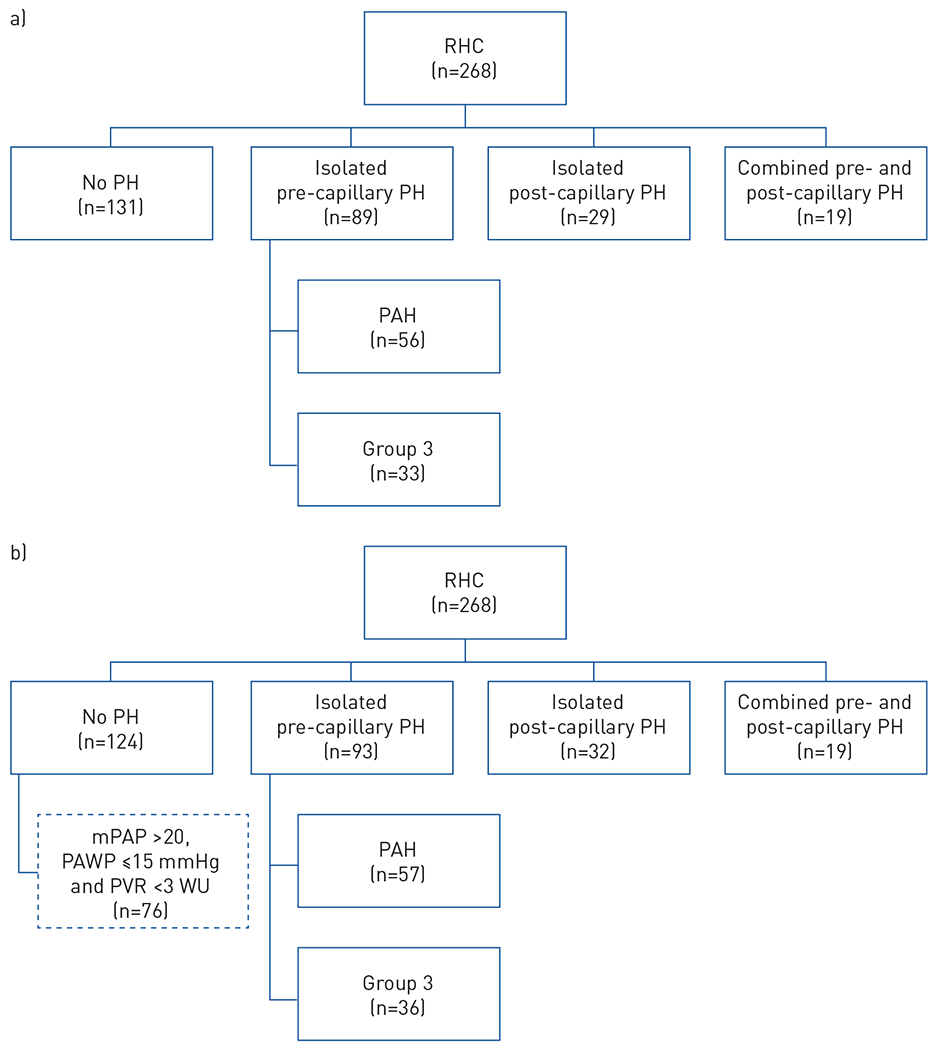
Classification according to a) the prior and b) the new haemodynamic definition of pulmonary hypertension (PH) in the University of Michigan cohort. RHC: right heart catheterisation; PAH: pulmonary arterial hypertension; mPAP: mean pulmonary arterial pressure; PAWP: pulmonary arterial wedge pressure; PVR: pulmonary vascular resistance; WU: Wood Units.
The mean±sd age of the University of Michigan cohort was 60.6±11.7 years, 85% were female, disease duration was 9.8±9.1 years, 35% had diffuse cutaneous SSc and 57% had limited cutaneous SSc. The mean±sd mPAP on RHC for the overall cohort was 30.6±11.9 mmHg, PAWP was 12.6±4.7 mmHg and PVR was 3.9±3.7 WU. In patients with PH based on the updated definition (n=144), the mean±sd age was 61.5±11.3 years, 85% were female, disease duration was 9.4±9.5 years and 53% had limited cutaneous SSc (table 2). The mean±sd mPAP on RHC was 37.9±11.2 mmHg, PAWP was 13.9±5.4 mmHg and PVR was 5.6±4.3 WU.
TABLE 2.
Baseline characteristics of the University of Michigan cohort
| Total | No PH | PH | p-value | |
|---|---|---|---|---|
| Subjects | 268 | 124 | 144 | |
| Age years | 60.6±11.7 | 59.6±12.1 | 61.5±11.3 | 0.323 |
| Female | 228 (85.07) | 106 (85.48) | 122 (84.72) | 0.862 |
| Race | ||||
| Caucasian | 212 (79.10) | 98 (79.03) | 114 (79.17) | 0.112 |
| African-American | 38 (14.18) | 14 (11.29) | 24 (16.67) | |
| Other | 18 (6.72) | 12 (9.68) | 6 (4.17) | |
| Type of SSc | ||||
| Limited cutaneous SSc | 154 (57.46) | 77 (62.10) | 77 (53.47) | 0.174 |
| Diffuse cutaneous SSc | 94 (35.07) | 42 (33.87) | 52 (36.1 1) | |
| Sine scleroderma | 9 (3.36) | 3 (2.42) | 6 (4.17) | |
| MCTD | 11 (4.10) | 2 (1.61) | 9 (6.25) | |
| Disease duration# years | 9.8±9.1 | 10.3±8.8 | 9.4±9.5 | 0.152 |
| Autoantibodies | ||||
| Antinuclear antibody (n=236) | 213 (90.25) | 99 (89.19) | 114 (91.20) | 0.603 |
| Anticentromere (n=181) | 44 (24.31) | 18 (21.43) | 26 (26.80) | 0.401 |
| Anti-RNA polymerase 3 (n=84) | 17 (20.24) | 8 (19.51) | 9 (20.93) | 0.872 |
| Anti-Scl-70 (n=225) | 32 (14.22) | 21 (20.79) | 11 (8.87) | 0.011 |
| Anti-U1 ribonucleoprotein (n=218) | 32 (14.68) | 12 (11.65) | 20 (17.39) | 0.232 |
| HRCT showing ILD (n=226) | 164 (72.57) | 80 (77.67) | 84 (68.29) | 0.116 |
| Pulmonary function tests | ||||
| FVC % pred | 76.4±20.3 | 80.2±18.7 | 73.1±21.0 | 0.004 |
| Dlco % pred (n=253) | 50.0±18.5 | 57.1±17.2 | 43.8±17.4 | <0.0001 |
| Right heart catheterisation | ||||
| mPAP mmHg | 30.6±11.9 | 22.0±5.0 | 37.9±11.2 | <0.0001 |
| PAWP mmHg | 12.6±4.7 | 11.1±3.0 | 13.9±5.4 | <0.0001 |
| TPG mmHg | 18.0±11.5 | 10.9±4.0 | 24.0±12.3 | <0.0001 |
| Cardiac output L·min−1 | 5.5±1.6 | 5.9±1.5 | 5.0±1.5 | <0.0001 |
| PVR WU | 3.9±3.7 | 1.9±0.6 | 5.6±4.3 | <0.0001 |
Data are presented as n, mean±sd or n (%), unless otherwise stated. SSc: systemic sclerosis; MCTD: mixed connective tissue disease; HRCT: high-resolution computed tomography; ILD: interstitial lung disease; FVC: forced vital capacity; Dlco: diffusing capacity of the lung for carbon monoxide; mPAP: mean pulmonary arterial pressure; PAWP: pulmonary arterial wedge pressure; TPG: transpulmonary gradient; PVR: pulmonary vascular resistance; WU: Wood Units.
disease duration calculated from date of first non-Raynaud’s symptom to date of RHC.
Impact of updated classification
Based on the haemodynamics data, 131 patients within the University of Michigan cohort did not have PH based on the prior PH definition (figure 1a). In the updated definition, seven patients were reclassified from no PH to pre-capillary PH (PAH (n=1), Group 3 (n=3) or post-capillary PH (n=3)) (figure 1b and table 3). The one patient who was reclassified as having PAH according to the updated definition had stable disease with no signs/symptoms of progression of PAH (7 years after the RHC) (table 3). Also, for those subjects who were reclassified as Group 2 or 3 according to the new definition, one patient each with Group 2 PH and Group 3 PH died, primarily due to severe malabsorption due to gastrointestinal dysmotility.
TABLE 3.
Individual data on seven patients reclassified from nopulmonary hypertension (PH) to PH based on the updated definition proposed by the 6th World Symposium on Pulmonary Hypertension
| Patient | Age years | Updated classification | Sex | Antibody | Type of SSc | Disease duration years# | FVC % pred | Dlco % pred | ILD and severity | mPAP mmHg | PAWP mmHg | Cardiac output L·min−1 | PVR WU | Management | Current status |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 68 | PAH | Female | Nucleolar pattern on ANA | Limited | 16 | 105 | 56 | Emphysema, no ILD | 22 | 8 | 4.1 | 3.41 | Sildenafil 20 mg 3 times a day | Alive |
| 2 | 59 | Group 2 PH | Female | Anticentromere+ | Limited | 18 | 95 | 80 | HRCT not performed | 21 | 16 | 5.7 | 0.87 | Diuretics | Alive |
| 3 | 51 | Group 2 PH | Male | Nucleolar pattern on ANA | Limited | 1 | 97 | 85 | No ILD on HRCT | 21 | 16 | 5.6 | 0.89 | Diuretics | Alive |
| 4 | 61 | Group 2 PH | Female | Anti-Scl-70+ | Diffuse | 7 | 73 | 33 | NSIP pattern, <20% ILD | 23 | 17 | 3.95 | 1.52 | NA | Died due to recurrent aspiration pneumonia and GI dysmotility |
| 5 | 70 | Group 3 PH | Male | Negative scleroderma antibody | Diffuse | 5 | 63 | 33 | CPFE, NSIP pattern, >30% ILD, mild emphysema | 22 | 10 | 3.55 | 3.38 | NA | Died due to failure to thrive (severe pseudo-obstruction) |
| 6 | 81 | Group 3 PH | Female | Anticentromere+ | Sine | 1 | 108 | 52 | CPFE, UIP pattern, 20–30% ILD | 23 | 6 | 4.37 | 3.89 | No PAH- or scleroderma-specific therapy | Alive |
| 7 | 61 | Group 3 PH | Female | Anti-Scl-70+ | Diffuse | 2 | 43 | 19 | CPFE, UIP pattern, >30% ILD, severe emphysema | 23 | 6 | 3.43 | 4.96 | Mycophenolate mofetil | Lost to follow-up in 2012 |
SSc: systemic sclerosis; FVC: forced vital capacity; Dlco: diffusing capacity of the lung for carbon monoxide; ILD: interstitial lung disease; mPAP: mean pulmonary arterial pressure; PAWP: pulmonary arterial wedge pressure; PVR: pulmonary vascular resistance; WU: Wood Units; PAH: pulmonary arterial hypertension; ANA: antinuclear antibody; HRCT: high-resolution computed tomography; NSIP: nonspecific interstitial pneumonia; NA: not available; GI: gastrointestinal; CPFE: combined pulmonary fibrosis and emphysema; UIP: usual interstitial pneumonia.
from onset of symptoms to RHC.
Of the 124 patients not diagnosed with PH according to the new haemodynamic definition, 76 had mPAP >20 mmHg, PAWP ⩽15 mmHg and PVR <3 WU (figure 1b). Of these, 45 had mPAP 21–24 mmHg, PAWP <15 mmHg and PVR <3 WU. 19 patients out of the 45 had lung disease; seven with PVR <2 WU and 11 with PVR ⩾2–<3 WU.
Impact of addition of PVR in the updated definition
Previous publications in SSc have defined pre-capillary PH as mPAP ⩾25 mmHg, PAWP ⩽15 mmHg and have not uniformly included PVR as part of the definition [11–15]. We explored the impact of excluding PVR on the pre-capillary PH in the University of Michigan cohort. With the updated classification, there were 169 patients who had mPAP >20 and PAWP ⩽15 mmHg. Of these patients, 87 had no/minimal lung disease (defined as <20% total lung involvement due to lung disease) (figure 2a). In the updated classification, there were 47 patients who had mPAP 21–24 mmHg and PAWP ⩽15 mmHg. Of these patients, 28 had no/minimal lung disease (figure 2a) and only one patient (3%) had PVR ⩾3 WU.
FIGURE 2.
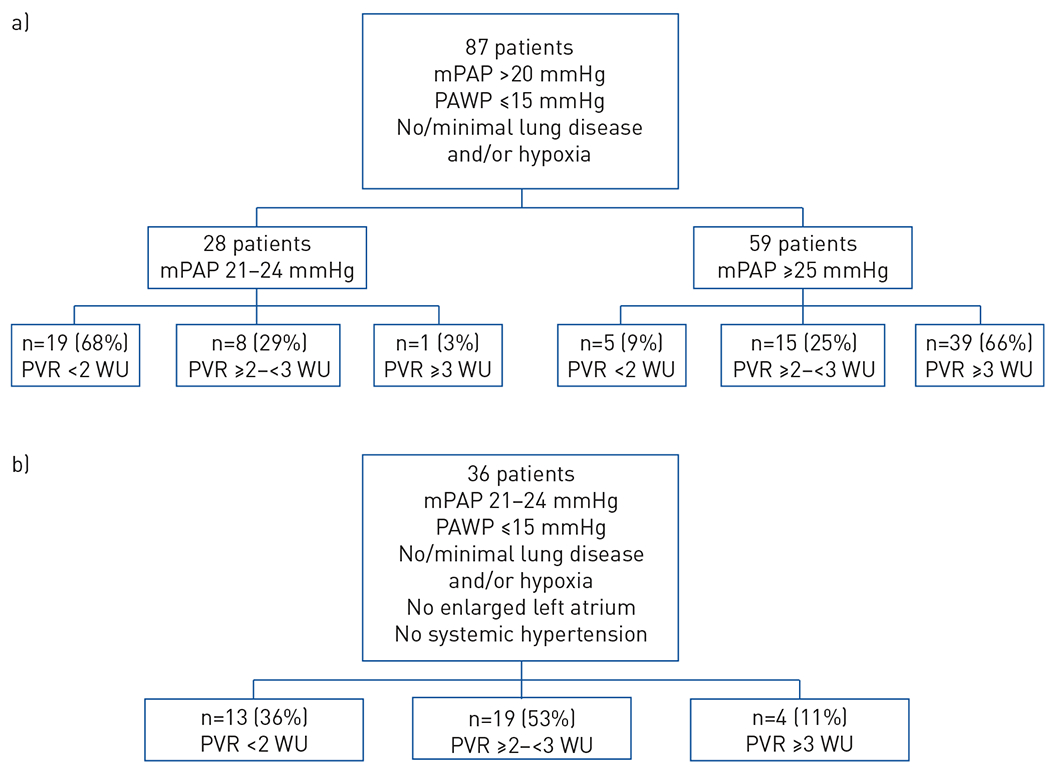
Distribution of borderline mean pulmonary arterial pressure [mPAP] in a) the University of Michigan cohort (mPAP >20 mmHg) and b] the DETECT study cohort (mPAP 21-24 mmHg), both stratified by pulmonary vascular resistance (PVR).
Validation in the DETECT cohort
We had previously shown that 36 out of 244 (14.75%) patients in the DETECT cohort had mPAP 21–24 mmHg (patients with PAWP ⩾15 mmHg, significant ILD, enlarged left atrium and systemic hypertension were excluded [11]). Based on the new classification, four out of 36 (11%) of the patients met the new PAH criteria. Of the remaining 32 patients, 19 (53%) had PVR ⩾2–<3 WU and 13 (36%) had PVR <2 WU (figure 2b).
Discussion
The updated haemodynamic definition of PH was proposed by the 6th WSPH based on growing evidence in the literature, especially in high-risk groups such as SSc [11–15]. Our data suggest that the updated definition did not have a significant impact on reclassification, with only seven patients (5%) being classified as PH in the University of Michigan cohort. Of these patients, four belong to pre-capillary PH group, with one classified as Group 1 PH and three as Group 3 PH. In those with mPAP 21–24 mmHg, no left heat disease or clinically meaningful lung disease, one out of 28 (4%) in the University of Michigan cohort and four out of 36 (11%) in the DETECT cohort were reclassified as PAH.
Previous data from different scleroderma cohorts suggest that patients with SSc and borderline mPAP (mPAP 21–24 mmHg) have a decreased exercise capacity and an increased risk of developing resting PH. Using the DETECT cohort, Visovatti et al. [11] showed that borderline mPAP is an intermediate stage, and may be a continuum between normal mPAP and PAH. Of 244 patients, 36 (15%) had borderline mPAP. Univariable logistic regression showed the mean tricuspid regurgitation velocity in patients with borderline PAP (mean 2.7 m·s−1) to be intermediate between normal mPAP (mean 2.3 m·s−1) and PAH (mean 3.0 m·s−1). When comparing borderline PAP with PAH, the statistically significant differences included less likelihood to be in World Health Organization functional class III/IV, lower percentage with telangiectasia, lower FVC % pred/Dlco % pred ratio, lower percentage with anticentromere antibody and lower right atrial pressure (all p<0.05). Coghlan et al. [14] published follow-up on cohorts from two centres in Europe using the DETECT inclusion criteria and showed that a greater proportion of patients converted to PH at a median follow-up of 3 years in the borderline mPAP group (33.3%) compared with 22% in the normal mPAP group. There was no difference in survival between the two groups. Valerio et al. [15] reviewed data at a large scleroderma centre in the UK and showed a hazard ratio of 3.7 for the diagnosis of PH on subsequent RHC in the group with borderline mPAP compared with the group with normal mPAP (mPAP ⩽20 mmHg) (p<0.001). Within the borderline mPAP group, 18.5% developed PAH within 3 years and 27.1% developed PAH within 5 years. There was no difference in survival in those with normal mPAP versus borderline mPAP. Bae et al. [13] reviewed the PHAROS registry and, after excluding patients with significant iLD, compared SSc patients with normal mPAP and borderline mPAP, showing the latter group to have significantly higher right ventricular systolic pressures on echocardiography, higher PVR and a higher transpulmonary gradient. Follow-up data involving 24 patients who underwent repeat RHC, based on signs and symptoms, at mean follow-up of 13.7 months found that 32% of patients with normal mPAP and 55% of patients with borderline mPAP developed resting PH. Finally, Kovacs et al. [12] showed that patients with SSc who have borderline mPAP had lower exercise capacity, as measured by the 6-min walk test and peak oxygen uptake on cardiopulmonary exercise. All of these studies highlight the importance of borderline mPAP in the SSc population.
Review of the above published data suggests that the definition of PAH was based on mPAP and PAWP without inclusion of a PVR cut-off. When applied in the University of Michigan cohort, 28 patients had mPAP ⩾21–24 mmHg, PAWP ⩽15 mmHg and no significant lung disease. Addition of PVR did not have a large effect, with only one patient (3%) being reclassified as PAH, and 11% in the DETECT cohort (four out of 36) met the new definition. Indeed, the addition of PVR is important as PH in SSc is often multifactorial and pulmonary artery vasculopathy, ILD, left heart disease or a combination of these can contributed to PH [23, 24]. In addition, combined pulmonary fibrosis/emphysema and pulmonary veno-occlusive disease also play a role in the differential diagnosis of these complex patients [24]. In the University of Michigan cohort, out of the seven patients who were reclassified from no PH to PH, three had combined pulmonary fibrosis/emphysema.
One of the hypotheses of the 6th WSPH Task Force was that a lower mPAP threshold will capture patients with early and milder pulmonary vascular disease in the hope of initiating earlier treatment, especially in patients who are at risk of progressive pulmonary vascular disease. Our data suggest that a large proportion of the University of Michigan and DETECT cohorts had milder haemodynamic parameters (mPAP 21–24 mmHg and PVR <3 WU) at the time of RHC. The proposal for PVR ⩾3 WU was consensus based during the 6th WSPH meeting and we believe that it may be too conservative. A systematic review by Kovacs et al. [10] supports this assertion: they showed that the mean±sd resting PVR in healthy subjects is 0.86±0.35 and 1.1±0.19 WU in those aged 24–50 and 51–69 years, respectively. Lowering the PVR to ⩾2 WU, which is >1 sd for healthy adults (based on Kovacs et al. [10]), we would have captured eight out of 28 (29%) additional patients in the University of Michigan cohort and 23 additional patients (64%) in the DETECT cohort. It is currently unknown if mPAP >20 mmHg and PVR ⩾2 WU represents a phenotype with risk of progressive pulmonary vascular disease or reflects an incidental haemodynamic finding where these patients would have done well without developing progressive PH but were diagnosed due to a uniform screening algorithm, especially due to the high prevalence of pulmonary vascular disease in scleroderma autopsy studies [25, 26]. Long-term follow-up is necessary to answer this important question.
Our study has many strengths. First, our patients in the University of Michigan cohort had a thorough evaluation and prospective data collection in a well-characterised cohort of patients with scleroderma spectrum and we validated our data in another international screening cohort (DETECT). Second, all RHCs were performed at the University of Michigan by an experienced cardiology team. Third, in the University of Michigan cohort, we had the HRCT scans reviewed and scored by thoracic radiologists and classified PAH versus Group 3 based on these findings. Finally, all patients underwent standardised screening for PH, including DETECT and other algorithms proposed after 2012 [18].
Although this study has many strengths, it is not without limitations. Both the University of Michigan and DETECT cohorts are screening cohorts, and the data may not be generalisable if this is not instituted uniformly in other cohorts. In addition, the University of Michigan cohort is a retrospective analysis of a prospective cohort and is subject to entry selection. Because the University of Michigan cohort is a detection cohort, RHC was not performed in a systematic manner, except after a positive screening test or due to signs or symptoms attributable to pulmonary vascular disease. However, the analysis of the DETECT cohort showed similar findings and provides confidence in our analysis.
Conclusions
In conclusion, the updated haemodynamic definition of PH does not appear to have a significant impact on the diagnosis of PAH in two screening cohorts of scleroderma spectrum disorders. Further analyses are needed to see the impact of the updated definition on long-term outcomes, including survival.
Acknowledgments
Conflict of interest: S. Jaafar has nothing to disclose. S. Visovatti has nothing to disclose. A. Young has nothing to disclose. S. Huang has nothing to disclose. P. Cronin has nothing to disclose. D. Vummidi has nothing to disclose. V. McLaughlin reports grants and personal fees from Actelion, Acceleron, Arena and Bayer, personal fees from Caremark and United Therapeutics, grants from Gilead and Sonovie. D. Khanna reports personal fees for consultancy from Actelion, AstraZeneca, Chemomab, Corbus, CSL Behring, Cytori, Horizon and UCB Pharma, grants and personal fees for consultancy from Bayer, BMS, Boehringer Ingelheim, EMD Serono, Genetech/Roche, GSK, Pfizer and Sanofi-Aventis/Genzy, employment fees from Civi Biopharma, an ownership interest in Eicos, grants from NIH NIAID and NIH NIAMS, during the conduct of the study; personal fees from AstraZeneca, outside the submitted work.
Support statement: Dinesh Khanna is supported by NIH/NIAMS K24 AR063120. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Coghlan JG, Denton CP, Grunig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73: 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khanna D, McLaughlin V. Screening and early detection of pulmonary arterial hypertension in connective tissue diseases. It is time to institute it! Am J Respir Crit Care Med 2015; 192: 1032–1033. [DOI] [PubMed] [Google Scholar]
- 3.Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69: 1809–1815. [DOI] [PubMed] [Google Scholar]
- 4.Kolstad KD, Li S, Steen V, et al. Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). Chest 2018; 154: 862–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young A, Nagaraja V, Basilious M, et al. Update of screening and diagnostic modalities for connective tissue disease-associated pulmonary arterial hypertension. Semin Arthritis Rheum 2019; 48: 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan J, Lei L, Zhao C. Comparison between the efficacy of combination therapy and monotherapy in connective tissue disease associated pulmonary arterial hypertension: a systematic review and meta-analysis. Clin Exp Rheumatol 2018; 36: 1095–1102. [PubMed] [Google Scholar]
- 7.Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J 2019; 53: 1801904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 12 Suppl. S, 40S–47S. [DOI] [PubMed] [Google Scholar]
- 9.Badesch DB, Champion HC, Sanchez MA, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009; 54: 1 Suppl., S55–S66. [DOI] [PubMed] [Google Scholar]
- 10.Kovacs G, Berghold A, Scheidl S, et al. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009; 34: 888–894. [DOI] [PubMed] [Google Scholar]
- 11.Visovatti SH, Distler O, Coghlan JG, et al. Borderline pulmonary arterial pressure in systemic sclerosis patients: a post-hoc analysis of the DETECT study. Arthritis Res Ther 2014; 16: 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kovacs G, Maier R, Aberer E, et al. Borderline pulmonary arterial pressure is associated with decreased exercise capacity in scleroderma. Am J Respir Crit Care Med 2009; 180: 881–886. [DOI] [PubMed] [Google Scholar]
- 13.Bae S, Saggar R, Bolster MB, et al. Baseline characteristics and follow-up in patients with normal haemodynamics versus borderline mean pulmonary arterial pressure in systemic sclerosis: results from the PHAROS registry. Ann Rheum Dis 2012; 71: 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coghlan JG, Wolf M, Distler O, et al. Incidence of pulmonary hypertension and determining factors in patients with systemic sclerosis. Eur Respir J 2018; 51: 1701197. [DOI] [PubMed] [Google Scholar]
- 15.Valerio CJ, Schreiber BE, Handler CE, et al. Borderline mean pulmonary artery pressure in patients with systemic sclerosis: transpulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum 2013; 65: 1074–1084. [DOI] [PubMed] [Google Scholar]
- 16.Galiè N, McLaughlin VV, Rubin LJ, et al. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J 2019; 53: 1802148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khanna D, Gladue H, Channick R, et al. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65: 3194–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young A, Vummidi D, Visovatti S, et al. Prevalence, treatment and outcomes of coexistent pulmonary hypertension and interstitial lung disease in systemic sclerosis. Arthritis Rheum 2019; 71: 1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoeper MM, Maier R, Tongers J, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am J Respir Crit Care Med 1999; 160: 535–541. [DOI] [PubMed] [Google Scholar]
- 21.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015; 46: 903–975. [DOI] [PubMed] [Google Scholar]
- 22.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Ann Rheum Dis 2013; 72: 1747–1755. [DOI] [PubMed] [Google Scholar]
- 23.Gargani L, Voilliot D, D’Alto M, et al. Pulmonary circulation on the crossroads between the left and right heart in systemic sclerosis: a clinical challenge for cardiologists and rheumatologists. Heart Fail Clin 2018; 14: 271–281 [DOI] [PubMed] [Google Scholar]
- 24.Launay D, Sobanski V, Hachulla E, et al. Pulmonary hypertension in systemic sclerosis: different phenotypes. Eur Respir Rev 2017; 26: 170056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Angelo WA, Fries JF, Masi AT, et al. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46: 428–440. [DOI] [PubMed] [Google Scholar]
- 26.al-Sabbagh MR, Steen VD, Zee BC, et al. Pulmonary arterial histology and morphometry in systemic sclerosis: a case-control autopsy study. J Rheumatol 1989; 16: 1038–1042. [PubMed] [Google Scholar]