

Abstract
Background
The urea cycle plays a key role in preventing the accumulation of toxic nitrogenous waste products, including two essential enzymes: ornithine transcarbamylase (OTC) and argininosuccinate lyase (ASL). Ornithine transcarbamylase deficiency (OTCD) results from mutations in the OTC. Meanwhile, argininosuccinate lyase deficiency (ASLD) is caused by mutations in the ASL.
Methods
Blood tandem mass spectrometric analysis and urea organic acidemia screening were performed on five Chinese cases, including three OTCD and two ASLD patients. Next‐generation sequencing was then used to make a definite diagnosis, and the related variants were validated by Sanger sequencing.
Results
The five patients exhibited severe clinical symptoms, with abnormal biochemical analysis and amino acids profile. Genetic analysis revealed two variants [c.77G>A (p.Arg26Gln); c.116G>T (p.Gly39Val)] in the OTC, as well as two variants [c.1311T>G (p.Tyr437*); c.961T>A (p.Tyr321Asn)] in the ASL. Conservation analysis showed that the amino acids of the two novel mutations were highly conserved in different species and were predicted to be possibly damaging with several in silico prediction programs. 3D‐modeling analysis indicated that the two novel missense variants might result in modest distortions of the OTC and ASL protein structures, respectively.
Conclusions
Two novel variants expand the mutational spectrums of the OTC and ASL. All the results may contribute to a better understanding of the clinical course and genetic characteristics of patients with urea cycle disorders.
Keywords: argininosuccinate lyase deficiency, next‐generation sequencing, ornithine transcarbamylase deficiency, urea cycle disorder
Firstly, we have demonstrated five Chinese urea cycle disorder cases in detail, including three ornithine transcarbamylase deficiency patients and two argininosuccinate lyase deficiency patients. Secondly, two novel missense variants including c.116G>T in OTC and c.961T>A in ASL are found and discussed fully.

1. INTRODUCTION
Urea cycle disorders (UCDs) is an inherited metabolic error in the nitrogen waste‐disposal system transforming ammonia to urea (Helman, Pacheco‐Colón, & Gropman, 2014). The entire urea cycle comprises six enzymes and two amino acid transporters. The six enzymes are N‐acetylglutamate synthase (NAGS, EC 2.3.1.1), carbamoyl‐phosphate synthase I (CPS1, EC 6.3.4.16), ornithine transcarbamylase (OTC, EC 2.1.3.3), argininosuccinate synthase (ASS, EC 6.3.4.5), argininosuccinate lyase (ASL, EC 4.3.2.1), and arginase (ARG, EC 3.5.3.1) (Nagata, Matsuda, & Oyanagi, 1991; Nakamura, Kido, Mitsubuchi, & Endo, 2014; Nassogne, Héron, Touati, Rabier, & Saudubray, 2005). The two amino acid transporters are ornithine transporter (ORNT1; ornithine/citrulline carrier) and citrin (aspartate/glutamate carrier) (Matsumoto et al., 2019). Ornithine transcarbamylase is a mitochondrial urea cycle enzyme that catalyzes the reaction between carbamyl phosphate and ornithine to form citrulline and phosphate, which is essential for the conversion of neurotoxic ammonia into nontoxic urea in mammals and other ureotelic animals (Chongsrisawat et al., 2018). Ornithine transcarbamylase deficiency (OTCD, MIM #311250) is the most common inherited defect of urea genesis because the OTC is located on X chromosome (Caldovic, Abdikarim, Narain, Tuchman, & Morizono, 2015; Lindgren, De Martinville, Horwich, Rosenberg, & Francke, 1984). The OTC is located on the short arm of the X chromosome within band Xp21.1 and encodes a 354‐residue polypeptide, including a leader sequence of 32 amino acids and a mature peptide of 322 amino acids, and the posttranscriptional modification takes place upon entry into mitochondria (Maestri, Lord, Glynn, Bale, & Brusilow, 1998). Caldovic et al. (2015) updated the disease‐causing mutations of the OTC to a total of 417, including 29 first‐reported mutations. In March 2018, 507 OTC mutations were reported in the Human Gene Mutation Database (HGMD) [http://www.hgmd.org/]. Most OTC mutations consist of single‐base substitutions (approximately 84%), while smaller proportions consist of small deletions or insertions (12%) and larger deletions (4%) (Horwich, Kalousek, Fenton, Pollock, & Rosenberg, 1986). As reported in the literature, the clinical symptoms of OTCD include recurrent vomiting, coma, cerebral edema, neurobehavioral changes, or even death (Choi et al., 2015; Storkanova et al., 2013). Since the onset of OTCD symptoms is extremely variable, it is essential to accumulate information on genetic analysis to determine genotype/phenotype correlation for an accurate diagnosis.
Argininosuccinate lyase, as the fourth step of UCD, catalyzes the hydrolytic cleavage of argininosuccinate into fumarate and arginine, which is essential for ammonia detoxification (Baruteau et al., 2017; Hu et al., 2015; Wen et al., 2016). Biochemically, ASL deficiency (ASLD, MIM #207900), the second most common UCD, frequently presents with the accumulation of argininosuccinate and citrulline, and the depletion of arginine in body fluids (Yankol, Mecit, Kanmaz, Acarli, & Kalayoglu, 2017). Argininosuccinate lyase deficiency follows an autosomal‐recessive trait and is caused by a defect of an enzyme ASL. The enzyme is expressed in many organs but the highest concentrations are in periportal hepatocytes, which are the only cells expressing the full urea cycle (O'Brien & Barr, 1981; Yu, Thompson, Yip, Howell, & Davidson, 2001). Clinically, ASLD patients are variable, from severe neonatal forms displaying serious brain damage and death to milder late‐onset forms displaying mental retardation, vomiting, failure to thrive, and behavioral disorders (Kim et al., 2018; Rostami, Haberle, Setoudeh, Zschocke, & Sayarifard, 2017). As reported in the literature, ASLD is the second most common UCD caused by pathogenic variants in the ASL located on chromosome 7 within band q11.2, which encodes a 464‐amino acid protein. Balmer et al. (2014) updated the number of mutations of the ASL to 134 in 2014. According to the HGMD, 154 mutations in the ASL have been reported up to March of 2018 (http://www.hgmd.org/). The clinical phenotype is highly heterogeneous and molecular genetic testing is useful for the diagnosis of ASLD.
In the present study, we report on five Chinese patients with UCDs, including three OTCD patients and two homozygous patients with ASLD. Their clinical manifestations and molecular genetic characteristics are also described in detail. Furthermore, two novel mutations of the OTC [c.116G>T (p.Gly39Val)] and the ASL [c.961T>A (p.Tyr321Asn)] are reported, which expand the mutational spectrum of the OTC and ASL.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This project was approved by the Ethics Committee of Quanzhou Women and Children's Hospital. The parents of these patients gave written informed consent for molecular study and publication.
2.2. Patients and laboratory tests
We retrospectively analyzed five patients (four males and one female) diagnosed with UCD between September 2013 and March 2018 at Quanzhou Women and Children's Hospital. Patients 1–3 were diagnosed with OTCD, and patients 4 and 5 were diagnosed with ASLD. Patient 2 is the elder sister of patent 3. She is a 17‐month‐old girl and has been hospitalized repeatedly due to liver problem. Amino acid levels on the dried blood spot were analyzed by liquid chromatography–tandem mass spectrometry (ACQUITY TQD; Waters, Milford, MA, USA). Urine samples were collected and prepared based on the method previously described by Fu, Iga, Kimura, and Yamaguchi (2000), and analyzed with gas chromatography–tandem mass spectrometry (7890B/5977A; Agilent Technologies, Santa Clara, CA, USA).
2.3. Next‐generation sequencing (NGS) and Sanger sequencing
Genomic DNA was extracted from dried blood spots of each patient. After library preparation, solution hybridization, and beads capture, the genomic DNA was sequenced for candidate inherited metabolic diseases‐related genes (NAGS [MIM 608300], OTC [MIM 300461], CPS1 [MIM 608307], ASS1 [MIM 603470], ASL [MIM 608310], ARG1 [MIM 608313], SLC25A13 [MIM 603859], ORNT1 [MIM 603861], OAT [MIM 613349]) with a high‐throughput sequencing instrument (Illumina Nextseq 500).
Sanger sequencing was used to validate the identified variants. The interest regions were amplified using custom oligonucleotide primers and standard PCR conditions. The primers were used as follows: OTC (c.116G>T) forward primer: 5′‐AAAGAATGCCTTATCAA‐3′, reverse primer: 5′‐TGTATGCCTGTATGCTC‐3′, product length 401 bp; ASL (c.961T>A) forward primer: 5′‐CCTCTGGGCTGATGGTGG‐3′, reverse primer: 5′‐GCAGCAGTTTCTGTCCTTTCC‐3′, product length 664 bp. The PCR cycle consisted of an initial denaturation step of 2 min at 95°C followed by 36 cycles of 30 s at 95°C, 1 min at 60°C, and 1 min at 72°C, and a final step at 72°C for 2 min. After purification of the PCR products, direct sequencing was performed using an ABI Prism 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
2.4. Bioinformatics analysis
We checked the identified variants in frequently used databases such as the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), dbSNP (https://www.ncbi.nlm.nih.gov/pro‐ jects/SNP/), ExAC consortium (http://exac.broadinstitue.org/), and the 1,000 Genome Project database (http://www.1000genomes.org/). The variants were further assessed for possible pathogenicity using several bioinformatic programs, including SIFT, PolyPhen‐2, PROVEAN, and MutationTaster. Multiple amino acid sequences of different species were extracted from the National Center for Biotechnology Information (NCBI) and aligned to evaluate the evolutionary conservation of the variants using DNAman software. To build three‐dimensional (3D) models of OTC and ASL, homology modeling was used utilizing Swiss Model Workspace, and PDB files were then submitted to Swiss‐PdbViewer 4.10 for the 3D‐structure.
3. RESULTS
3.1. Clinical characteristics and genotypes of three OTCD patients
Patient 1, a male infant, was hospitalized for severe jaundice, coma, and respiratory depression at 2 days of age. Biochemical analysis revealed hyperammonemia (400 μmol/L, reference: 10–47 μmol/L), hypoglycemia (1.6 mmol/L, reference: 3.8–6.1 mmol/L), and prolonged clotting time. Newborn screening testing revealed the amino acids levels: citrulline 2.66 μmol/L (reference: 5.5–38 μmol/L); methionine 106.63 μmol/L (reference: 7–45 μmol/L); tyrosine 464.05 μmol/L (reference: 33–300 μmol/L); alanine 2,558.06 μmol/L (reference: 110–644 μmol/L); and glycine 1,114.45 μmol/L (reference: 178–900 μmol/L). The urinary orotic acid level was 132.92 μmol/mmol creatinine (reference: 0.0–1.5 μmol/mmol creatinine). The NGS results revealed a single nucleotide change from G to A, c.77G>A in the OTC, and he was diagnosed as early‐onset OTCD. The nucleotide change resulted in a hemizygous missense mutation, p.Arg26Gln, which has been previously reported in the OTCD patients (Grompe, Caskey, & Fenwick, 1991).
Patient 2, a girl, started to present with hyperammonemia and liver dysfunction at 7 months of age. She was diagnosed as late‐onset female OTCD and started on arginine, sodium benzoate, and a low‐protein diet to reduce the level of serum ammonia, along with reduced glutathione, potassium magnesium aspartate, and compound glycyrrhizin tablets for liver protection. During the next 10 months, she had multiple hospital admissions for recurrent hyperammonemia and deteriorating liver function. At 17 months of age, biochemical analysis revealed hyperammonemia (>280 μmol/L, reference: 10–47 μmol/L), increased alanine aminotransferase (226 U/L, reference: 0–40 U/L), and elevated aspartate aminotransferase (214 U/L, reference: 0–40 U/L). The amino acid analysis revealed that citrulline was in the normal range (19.46 μmol/L, reference: 5.5–38 μmol/L). However, the urinary orotic acid level was significantly increased (35.54 μmol/mmol creatinine, reference: 0.0–1.5 μmol/mmol creatinine). A cranial magnetic resonance imaging scan revealed abnormal signals in the bilateral frontal lobe, which suggested the probability of brain injury, and the diffusion of cerebral edema in the bilateral telencephalon was also observed. She deteriorated rapidly afterward and died at the age of two‐and‐a‐half years. The results of the genetic analysis revealed that she carried a novel heterozygote c.116G>T mutation in exon 2 of the OTC (Figure 1b), which had not been previously reported in the literature or registered in the HGMD, NCBI, dbSNP, or 1,000 Genomes databases. The nucleotide change resulted in a missense mutation (p.Gly39Val), which was predicted to be possibly damaging with several in silico prediction programs (Table 1). The Gly residue at position 39 of OTC was assessed to be highly conserved among different species with DNAman software (Figure 1c). Additionally, 3D‐modeling analysis showed that the mutation c.116G>T (p.Gly39Val) resulted in a replacement of Gly with Val having a much larger side chain, and then induced an extension of amino‐acid side chain, which might trigger modest structural distortion of the OTC protein (Figure 2). Taken together, the c.116G>T (p.Gly39Val) variant was considered deleterious and likely to be pathogenic.
Figure 1.
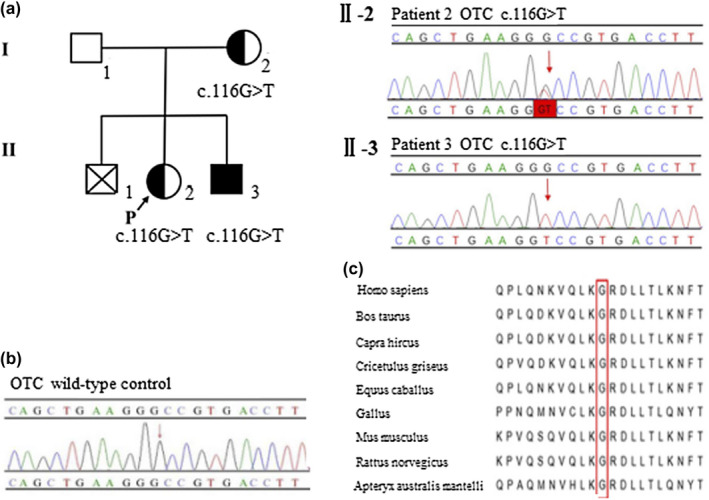
(a) Pedigree of patients 2 and 3. The arrow denotes proband with the OTCD family. Patient 2 (Ⅱ‐2) is heterozygous for c.116G>T (p.Gly39Val) and patient 3 (Ⅱ‐3) carries the hemizygous mutation c.116G>T (p.Gly39Val). (b) Validation of the novel OTC mutation by Sanger sequencing. Ⅱ‐2. Patient 2 has a heterozygous mutation c.116G>T (p.Gly39Val) in exon 2; Ⅱ‐3. Patient 3 is hemizygous for c.116G>T (the variant is indicated by a red arrow). Their mother has a heterozygous mutation c.116G>T, which was validated in another hospital. (c) Multiple sequence alignment using Clustal X. The glycine residue at position 39 (highlighted by a red box) is highly conserved among different species
Table 1.
Analysis and in silico prediction of the two novel OTC and ASL gene variants of UCD patients
| Patient | Gene | Location | Nucleotide change | Protein change | Parental origin | SIFT | PolyPhen‐2 | Mutation taster | PROVEAN | HGMD | ClinVar | Freq in 1,000 Genome | Freq in ExAC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2,3 | OTC | Exon 2 | c.116G>T | p.Gly39Val | Maternal | 0.01 | 1 | 1 | −5.7 | ND | ND | ND | ND |
| 5 | ASL | Exon 13 | c.961T > A | p.Tyr321Asn | Maternal | 0 | 0.933 | 1 | −8.43 | ND | ND | ND | ND |
The reference sequence used in this study was based on the NCBI37/hg19 assembly of the human genome. NM_000531.5 and NM_000048.3 were employed as reference sequence for OTC and ASL, respectively. ND, no data.
Figure 2.
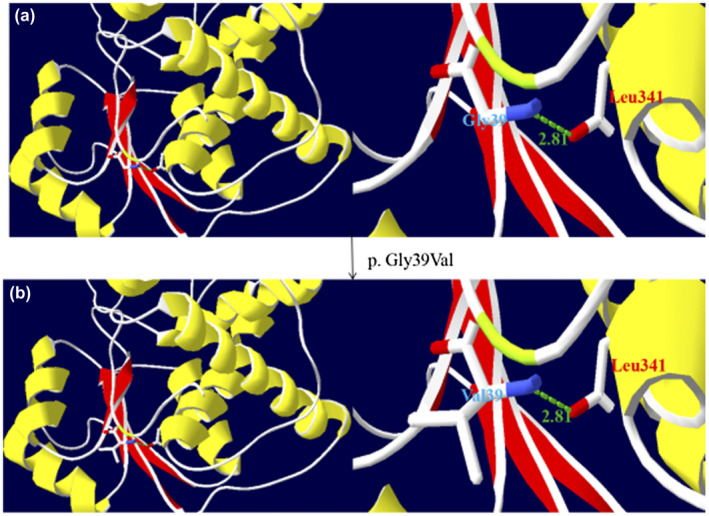
Three‐dimensional modeling structure analysis of wild‐type and mutant products of OTC. Green dashed lines represent hydrogen bonds and the green number shows the hydrogen bonds distances. (a) A segment of the OTC structure showing Gly39 hydrogen bonding with Leu341. (b) A segment of the OTC structure showing Val39 having much larger side chain and equally hydrogen bonding with Leu341. The larger side chain may induce an extension of the amino‐acid side chain, which might distort the structure of OTC protein. (The color in this figure is that selected by the Secondary Structure Succession of Swiss‐PdbViewer 4.10.)
Patient 3, the younger brother of patient 2, was brought to our hospital for jaundice, hypotonia, and poor responses at the age of 1 day. The serum ammonia was 160 μmol/L (reference: 10–47 μmol/L), which then increased to >280 μmol/L after 2 days. Prolonged clotting time was also observed. Tandem mass spectrometry revealed low citrulline (2.55 μmol/L, reference: 5.5–38 μmol/L) and several acylcarnitine increases. His parents refused treatment and a urinary organic acids profile was not detected. Genetic analysis revealed a hemizygous mutation c.116G>T (Figure 1b). He (Ⅱ‐3) was diagnosed as early‐onset OTCD. Sanger sequencing confirmed that the hemizygous mutation was the same with his elder sister (patient 2, Ⅱ‐2). They had a male sibling (Ⅱ‐1, deceased), also suspected to be, but not proven, OTCD. His mother (Ⅰ‐2) was confirmed to carry the heterozygous mutation c.116G>T (p.Gly39Val) in another hospital, and she had a normal phenotype until now. The novel mutation was inherited from his mother. The pedigree was shown in Figure 1a.
3.2. Clinical characteristics and genotypes of two patients with ASLD
Patient 4, a premature male infant, was hospitalized for poor response, convulsions, and hypotonia at 7 days of age. Laboratory tests revealed hyperammonemia (>280 μmol/L, reference: 10–47 μmol/L), high alpha‐fetoprotein (AFP, 40,589.44 ng/ml, reference: 0–7 ng/ml), greatly increased total bilirubin (TBIL, 218.6 μmol/L, reference: 5.1–19.0 μmol/L), elevated direct bilirubin (DBIL, 106.6 μmol/L, reference: 0–6.8 μmol/L), and high total bile acid (206.4 μmol/L, reference: 0–10 μmol/L). Metabolic acidosis and prolonged clotting time were also observed. Amino acid analysis revealed increased citrulline (266.87 μmol/L, reference: 5.5–38 μmol/L), methionine (66.34 μmol/L, reference: 7–45 μmol/L), and alanine (656.08 μmol/L, reference: 110–644 μmol/L). Urinary organic acid analysis revealed that orotic acid was 13.67 μmol/mmol creatinine (reference: 0.0–1.5 μmol/mmol creatinine), and uracil was 33.6 μmol/mmol creatinine (reference: 0.0–7.0 μmol/mmol creatinine). Orotic acid is a feature that may be observed in patients with ASLD. The impaired recycling of ornithine causes an accumulation of carbamyl phosphate, leading to the overproduction of orotic acid and pyrimidine in the absence of a source of ornithine (Erez, Nagamani, & Lee, 2011; Parsons, Scott, Pinto, Carter, & Snyder, 1987). Argininosuccinate in urine and plasma was not measured for the lack of equipment in our hospital. Genetic analysis revealed that he had a homozygous nonsense mutation c.1311T>G in exon 17 of the ASL. He was diagnosed with ASLD. The nucleotide change resulted in a termination codon at position 437 (p.Tyr437*), which has been reported in ASLD patients (Lin, Yu, Li, Zheng, & Fu, 2017; Wen et al., 2016).
Patient 5, a male infant, was born to consanguineous parents (maternal cousins) as the first child. He was hospitalized for convulsions and poor feeding at 1 day of age. Laboratory tests indicated hyperammonemia (>280 μmol/L, reference: 10–47 μmol/L), high TBIL (170.7 μmol/L, reference: 5.1–19.0 μmol/L), and slightly increased DBIL (13.0 μmol/L, reference: 0.0–6.8 μmol/L). Tandem mass spectrometry revealed increased citrulline (172.18 μmol/L, reference: 5.5–38 μmol/L). Argininosuccinate in urine and plasma was not measured. Genetic analysis revealed that he had a homozygous novel mutation c.961T>A (p.Tyr321Asn) located in exon 13 of the ASL (Figure 3b), which has not been reported previously in the literature or registered in HGMD, NCBI dbSNP, or the 1,000 Genomes databases. The pedigree is shown in Figure 3a. The Tyr residue at position 321 of ASL was also assessed to be highly conserved (Figure 3c). For mutation p.Tyr321Asn, the disappearance of the bulky and rigid phenyl side chain should trigger some mild steric structural distortion of the ASL protein (Figure 4). Furthermore, the mutation p.Tyr321Asn led to some distant interactions of the amino acid with an adjacent domain and made new hydrogen bonds with the side chain of Asn114, backbone of Ala203, and side chain of Thr233, which might be expected to further cause distortion of the ASL architecture (Figure 4). The variant was predicted to be possibly damaging by in silico prediction programs (Table 1). Overall, the missense variant was likely deleterious. He was diagnosed as ASLD and treated with low protein diet and arginine hydrochloride. Serum ammonia was decreased to 43 μmol/L (reference: 10–47 μmol/L) after 4 days. However, he had multiple hospital admissions for limb weakness, liver damage, vomit, and convulsions during the next 2 years.
Figure 3.
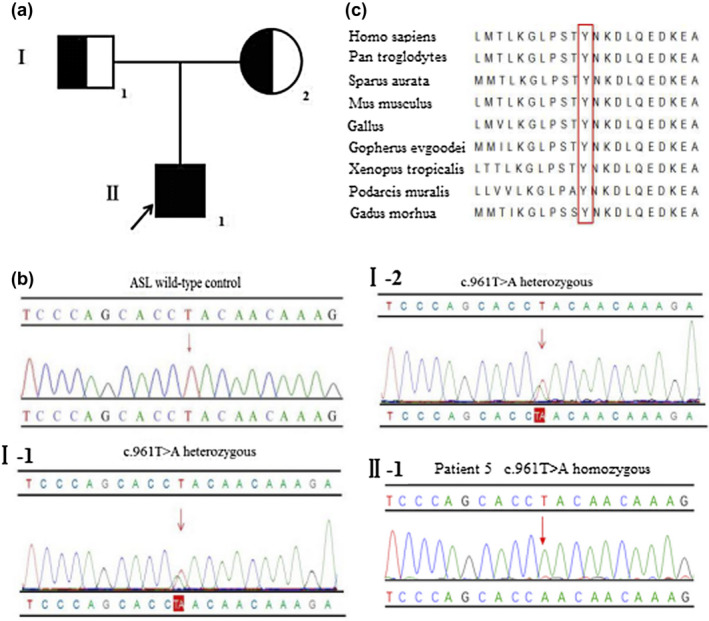
(a) Pedigree of patient 5. The arrow denotes the proband. (b) Sanger sequencing analysis of the ASL, respectively, identified the mutation c.961T>A (p.Tyr321Asn) in exon 13 (Ⅰ‐1) heterozygous in his father, (Ⅰ‐2) heterozygous in his mother, and (Ⅱ‐1) homozygous in patient 5 (the variant is indicated by a red arrow). (c) Multiple sequence alignment using Clustal X. The tyrosine residue at position 321 (highlighted by a red box) is highly conserved among different species
Figure 4.
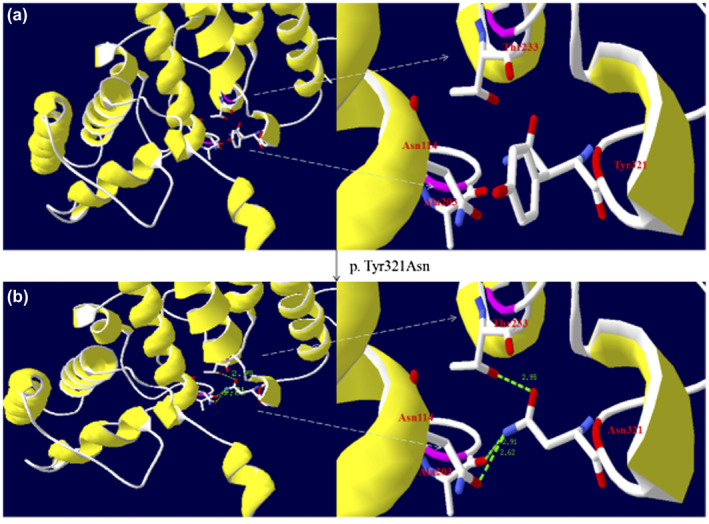
Three‐dimensional modeling structure analysis of wild‐type and mutant products of ASL. Green dashed lines represent hydrogen bonds and the green number shows the hydrogen bonds distances. (a) A segment of the ASL structure showing Tyr321 has a side chain of benzene and it has no hydrogen bonds with the adjacent domain. (b) A segment of the ASL structure showing that Asn321 has a side chain without benzene and that its backbone makes new hydrogen bonds with the side chain of Asn114, the backbone of Ala203, and the side chain of Thr233. The disappearance of the bulky and rigid benzene side chain and new hydrogen bonds may induce a distortion of the ASL protein structure. (The color in this figure is that selected by the Secondary Structure Succession of Swiss‐PdbViewer 4.10.)
Clinical features, phenotypes, and mutations of the five UCD patients are, respectively, summarized in Tables 2 and 3. The mutations in all patients were validated by direct DNA sequencing. The Gly residue at position 39 of OTC and the Tyr residue at position 321 of ASL were both assessed to be highly conserved among different species with DNAman software. Additionally, the two novel variants were predicted to be possibly damaging in several in silico prediction programs.
Table 2.
Clinical features identified in the five Chinese UCD patients
| No. | Age at onset | Type | Sex | Clinical presentation | Clinical outcome | Family history of similar disease | CIT (reference: 5.5–38 μmol/L) | AMON level (reference: 10–47 μmol/L) |
|---|---|---|---|---|---|---|---|---|
| 1 | 2 days | N | M | Severe jaundice, coma, respiratory depression | Death | No | 2.66 | 400 |
| 2 | 7 months | L | F | Hyperammonemia, liver dysfunction | Brain injury, cerebral edema | Yes, younger brother (patient 3) | ND | >280 |
| 3 | 1 day | N | M | Jaundice, hypotonia, poor responses | Death | Yes, elder sister (patient 2) | 2.55 | >280 |
| 4 | 7 days | N | M | Poor response, convulsions, hypotonia | Death | No | 266.87 | >280 |
| 5 | 1 day | N | M | Convulsions, poor feeding | Limb weakness, liver damage, vomit, convulsion | No | 172.18 | >280 |
Abbreviations: AMON, ammonia; CIT, citrulline; F, female; L, late‐onset phenotype; M, male; N, neonatal‐onset phenotype; ND, not detected.
Table 3.
Details of OTC and ASL gene mutations in the five UCD patients
| Patient no. | Gene | Reference sequence | Exon position | Nucleotide change | Amino acid change | Mutation type | Type of change | Novelty | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | OTC | NM_000531.5 | 1 | c.77G>A | p.Arg26Gln | Missense | Hemizygous | Reported | [1] |
| 2 | OTC | NM_000531.5 | 2 | c.116G>T | p.Gly39Val | Missense | Heterozygous | Novel | This study |
| 3 | OTC | NM_000531.5 | 2 | c.116G>T | p.Gly39Val | Missense | Hemizygous | Novel | This study |
| 4 | ASL | NM_000048.3 | 17 | c.1311T>G | p.Tyr437* | Stopgain | Homozygous | Reported | [24] |
| 5 | ASL | NM_000048.3 | 13 | c.961T>A | p.Tyr321Asn | Missense | Homozygous | Novel | This study |
The reference sequence used in this study was based on the NCBI37/hg19 assembly of the human genome.
4. DISCUSSION
Pediatric rare diseases are often rapid deterioration with high mortality, which can be improved by early diagnosis and treatment. Ornithine transcarbamylase deficiency is a rare inborn error caused by mutation in the OTC. In this report, two missense mutations were identified in three OTCD patients. Patients 1 and 3 were, respectively, hemizygous missense mutation of c.77G>A and c.116G>T in the OTC, and both presented severe early‐onset forms. The late‐onset patient 2, the elder sister of patient 3, carried a heterozygote mutation c.116G>T. The mutation c.77G>A was first reported in 1989 in a 5‐year‐old Hispanic boy who was mildly mentally retarded. He presented at 2 weeks of age with hyperammonemic coma (Grompe, Muzny, & Caskey, 1989). The single nucleotide variant c.77G>A resulted in an Arg26Gln change and then decreased OTCase mRNA through disruption of the leader sequence, which was required for directing mitochondrial localization of the OTC precursor. In China, c.77G>A has also been reported in an OTCD boy exhibiting poor suck at 11 days, who ultimately died at 13 days (Shao et al., 2017). In our study, patient 1 presented with hyperammonemic coma and respiratory depression at 2 days of age. Thus, we speculated that the c.77G>A missense mutation was associated with the early‐onset type of OTCD. Furthermore, the mutation c.116G>T (p.Gly39Val) was first reported in our study. c.116G>A (p.Gly39Asp) has been reported to affect an evolutionarily conserved amino acid, and the possibility of polymorphism has been excluded (Shchelochkov et al., 2009). Another mutation, c.115G>T (p.Gly39Cys), having been detected in either neonatal or late‐onset OTCD patients, was predicted to cause a potential turn in a poorly conserved domain of protein (Calvas et al., 1998). Therefore, the glycine was predicted to be located in a conserved position of the OTC polypeptide, which means the variant at this site might be deleterious. Several in silico prediction programs predicted the variant to be possibly damaging. 3D‐modeling analysis demonstrated that the mutation c.116G>T (p.Gly39Val) induced an extension of the amino‐acid side chain and might distort the structure of the OTC protein. Overall, the missense variant was likely deleterious based on the above results of various function‐prediction algorithms.
As reported in the literature, early‐onset patients of OTCD usually show no residual OTC enzyme activity and null alleles (McCullough et al., 2000), while female late‐onset patients are unfavorably lyonized (Grompe et al., 1991). For the reason, the early‐onset patients 1 and 3 exhibited acute phenotype and ultimately died. However, the late‐onset patient 2 had less serious clinical symptoms, probably because of skewed lyonization. As reported in the literature, about 20% of female carriers of OTC mutations present symptomatic OTCD (Caldovic et al., 2015; Maestri et al., 1998), and, unfortunately, patient 2 is one of these 20%. However, her mother had been normal in phenotype until now, which may be explained by random X‐chromosome inactivation. When the inactivation is skewed to express more of the mutated allele, an X‐linked recessive disorder may affect female carriers (Yorifuji et al., 1998), and the skewed X‐inactivation possibly led to the different phenotypes of patient 2 and her mother.
In this study, patients 4 and 5 both had unique homozygous mutations in the ASL, including the nonsense mutation c.1311T>G (p.Tyr437*) in patient 4 and the missense mutation c. 961T>A (p.Tyr321Asn) in patient 5. The c.1311T>G mutation was first reported in an ASLD patient in 2013 (Maestri et al., 1998), and was first identified in a Chinese patient in 2017 (Lin et al., 2017). A conservation analysis indicated that the amino acid was highly conserved in different species, suggesting that the variant might be deleterious. Furthermore, this nonsense variant resulted in premature termination of the protein, decreased by 28 amino acids, which might affect the stability of the protein and lead to ASLD (Lin et al., 2017). The missense mutation c.961T>A (p.Tyr321Asn) was first determined in this study. A conservation analysis in different species showed that this amino acid was highly conserved among different species. Several in silico prediction programs predicted the variant to be possibly damaging. 3D‐modeling analysis revealed that the mutation c.961T>A (p.Tyr321Asn) changed the amino acid side chain and that the introduction of hydrogen bonds might further distort the structure of ASL protein. Taken together, the missense mutant is likely deleterious based on the above results.
Herein, three OTCD patients and two ASLD patients all presented with hyperammonemia, and the ultimate death of patients 1, 3, and 4 emphasizes the significance of early diagnosis and monitoring in the early stages of life. Children with UCD are usually only recognized after symptoms of hyperammonemia, such as lethargy and coma, which means a significant delay in diagnosis. Furthermore, some patients never experience symptomatic hyperammonemias but nevertheless develop severe psychomotor retardation. A delay in diagnosis results in either death or neurodisability (Nassogne et al., 2005; Picca et al., 2001). Therefore, an accurate genetic diagnosis is essential not only for the appropriate management of the patients, but also for genetic counseling in the context of subsequent pregnancies and carrier testing.
5. CONCLUSIONS
We have characterized the clinical presentations, biochemical manifestations, and mutation analysis of five Chinese UCD patients, including three OTCD patients and two patients with ASLD. Two novel mutations (c.116G>T [p.Gly39Val]; c.961T>A [p.Tyr321Asn]) have been identified by NGS and then validated by Sanger sequencing, which enrich the mutational spectrums of the OTC and ASL. Conservation analysis showed that these amino acids are highly conserved across a broad range of species, and 3D‐modeling analysis showed that the two novel missense variants may, respectively, distort the OTC and ASL protein structures. More information about the biological relevance of the mutations will hopefully be available from the genotype database. In particular, knowledge of genotypes associated with clinical and biochemical phenotypes would be beneficial for patients and their families.
CONSENT FOR PUBLICATION
We confirm that the five patients’ parents have given their written consent for publication of their medical data.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
ZZ, YL, and WL cared for patients and collected the clinical data. LZ, MJ, and WW performed the mutation analysis. ZZ and WW participated in study design, data analysis, and the first draft of the manuscript. Finally, QF guided the whole research process. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank the patients' families for their cooperation.
Zheng Z, Lin Y, Lin W, et al. Clinical and genetic analysis of five Chinese patients with urea cycle disorders. Mol Genet Genomic Med. 2020;8:e1301 10.1002/mgg3.1301
Funding information
This study was supported by the Quanzhou Science and Technology Plan Project in China (no. 2019N049S).
Contributor Information
Wenjun Wang, Email: wangwenjun@biosan.cn.
Qingliu Fu, Email: zhyetyy@163.com.
DATA AVAILABILITY STATEMENT
The data generated or analyzed during this study are included in this article. The data are available from the corresponding author on reasonable request.
REFERENCES
- Balmer, C. , Pandey, A. V. , Rüfenacht, V. , Nuoffer, J. M. , Fang, P. , Wong, L. J. , & Häberle, J. (2014). Mutations and polymorphisms in the human Argininosuccinate Lyase (ASL) gene. Human Mutation, 35, 27–35. 10.1002/humu.22469 [DOI] [PubMed] [Google Scholar]
- Baruteau, J. , Jameson, E. , Morris, A. A. , Chakrapani, A. , Santra, S. , Vijay, S. , … Davison, J. E. (2017). Expanding the phenotype in argininosuccinic aciduria: Need for new therapies. Journal of Inherited Metabolic Disease, 40, 357–368. 10.1007/s10545-017-0022-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldovic, L. , Abdikarim, I. , Narain, S. , Tuchman, M. , & Morizono, H. (2015). Genotype‐phenotype correlations in ornithine transcarbamylase deficiency: A mutation update. Journal of Genetics and Genomics, 42, 181–194. 10.1016/j.jgg.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvas, P. , Segues, B. , Rozet, J. M. , Rabier, D. , Bonnefond, J. P. , & Munnich, A. (1998). Novel intragenic deletions and point mutations of the ommithine transcarbamylase gene in congenital hyperammcsnermia. Human Mutation, S1, S81–S84. 10.1002/humu.1380110128 [DOI] [PubMed] [Google Scholar]
- Choi, J.‐H. , Lee, B. H. , Kim, J. H. , Kim, G.‐H. , Kim, Y.‐M. , Cho, J. , … Yoo, H.‐W. (2015). Clinical outcomes and the mutation spectrum of the OTC gene in patients with ornithine transcarbamylase deficiency. Journal of Human Genetics, 60, 501–507. 10.1038/jhg.2015.54 [DOI] [PubMed] [Google Scholar]
- Chongsrisawat, V. , Damrongphol, P. , Ittiwut, C. , Ittiwut, R. , Suphapeetiporn, K. , & Shotelersuk, V. (2018). The phenotypic and mutational spectrum of Thai female patients with ornithine transcarbamylase deficiency. Gene, 679, 377–381. 10.1016/j.gene.2018.09.026 [DOI] [PubMed] [Google Scholar]
- Erez, A. , Nagamani, S. C. , & Lee, B. (2011). Argininosuccinate lyase deficiency‐argininosuccinic aciduria and beyond. American Journal of Medical Genetics Part C Seminars in Medical Genetics, 157, 45–53. 10.1002/ajmg.c.30289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, X. W. , Iga, M. , Kimura, M. , & Yamaguchi, S. (2000). Simplified screening for organic acidemia using GC/MS and dried urine filter paper: A study on neonatal mass screening. Early Human Development, 58, 41–55. 10.1016/S0378-3782(00)00053-0 [DOI] [PubMed] [Google Scholar]
- Grompe, M. , Caskey, C. T. , & Fenwick, R. G. (1991). Improved molecular diagnostics for ornithine transcarbamylase deficiency. The American Journal of Human Genetics, 48, 212–222. 10.1016/0378-1119(91)90191-D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grompe, M. , Muzny, D. M. , & Caskey, C. T. (1989). Scanning detection of mutations in human ornithine transcarbamoylase by chemical mismatch cleavage. Proceedings of the National Academy of Sciences of the United States of America, 86, 5888–5892. 10.2307/34226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman, G. , Pacheco‐Colón, I. , & Gropman, A. L. (2014). The urea cycle disorders. Seminars in Neurology, 34, 341–349. 10.1055/s-0034-1386771 [DOI] [PubMed] [Google Scholar]
- Horwich, A. L. , Kalousek, F. , Fenton, W. A. , Pollock, R. A. , & Rosenberg, L. E. (1986). Targeting of pre‐ornithine transcarbamylase to mitochondria: Definition of critical regions and residues in the leader peptide. Cell, 44, 451–459. 10.1016/0092-8674(86)90466-6 [DOI] [PubMed] [Google Scholar]
- Hu, L. Y. , Pandey, A. V. , Balmer, C. , Eggimann, S. , Rüfenacht, V. , Nuoffer, J. M. , & Häberle, J. (2015). Unstable argininosuccinate lyase in variant forms of the urea cycle disorder argininosuccinic aciduria. Journal of Inherited Metabolic Disease, 38, 815–827. 10.1007/s10545-014-9807-3 [DOI] [PubMed] [Google Scholar]
- Kim, D. , Ko, J. M. , Kim, Y. M. , Seo, G. H. , Kim, G. H. , Lee, B. H. , & Yoo, H. W. (2018). Low prevalence of argininosuccinate lyase deficiency among inherited urea cycle disorders in Korea. Journal of Human Genetics, 63, 911–917. 10.1038/s10038-018-0467-2 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Yu, K. , Li, L. , Zheng, Z. , & Fu, Q. (2017). Mutational analysis of ASS1, ASL and SLC25A13 genes in six Chinese patients with citrullinemia. Chinese Journal of Medical Genetics, 34, 676–679. 10.3760/cma.j.issn.1003-9406.2017.05.012 [DOI] [PubMed] [Google Scholar]
- Lindgren, V. , De Martinville, B. , Horwich, A. , Rosenberg, L. , & Francke, U. (1984). Human ornithine transcarbamylase locus mapped to band Xp21.1 near the Duchenne muscular dystrophy locus. Science, 226, 698–700. 10.1126/science.6494904 [DOI] [PubMed] [Google Scholar]
- Maestri, N. E. , Lord, C. , Glynn, M. , Bale, A. , & Brusilow, S. W. (1998). The phenotype of ostensibly healthy women who are carriers for ornithine transcarbamylase deficiency. Medicine (Baltimore), 77, 389–397. 10.1097/00005792-199811000-00005 [DOI] [PubMed] [Google Scholar]
- Matsumoto, S. , Häberle, J. , Kido, J. , Mitsubuchi, H. , Endo, F. , & Nakamura, K. (2019). Urea cycle disorders‐update. Journal of Human Genetics, 64, 833–847. 10.1038/s10038-019-0614-4 [DOI] [PubMed] [Google Scholar]
- McCullough, B. A. , Yudkoff, M. , Batshaw, M. L. , Wilson, J. M. , Raper, S. E. , & Tuchman, M. (2000). Genotype spectrum of ornithine transcarbamylase deficiency: Correlation with the clinical and biochemical phenotype. American Journal of Medical Genetics, 93, 313–319. [DOI] [PubMed] [Google Scholar]
- Nagata, N. , Matsuda, I. , & Oyanagi, K. (1991). Estimated frequency of urea cycle enzymopathies in Japan. American Journal of Medical Genetics, 39, 228–229. 10.1002/ajmg.1320390226 [DOI] [PubMed] [Google Scholar]
- Nakamura, K. , Kido, J. , Mitsubuchi, H. , & Endo, F. (2014). Diagnosis and treatment of urea cycle disorder of Japan. Pediatrics International, 56, 506–509. 10.1111/ped.12439 [DOI] [PubMed] [Google Scholar]
- Nassogne, M. C. , Héron, B. , Touati, G. , Rabier, D. , & Saudubray, J. M. (2005). Urea cycle defects: Management and outcome. Journal of Inherited Metabolic Disease, 28, 407–414. 10.1007/s10545-005-0303-7 [DOI] [PubMed] [Google Scholar]
- O'Brien, W. E. , & Barr, R. H. (1981). Argininosuccinate lyase: Purification and characterization from human liver. Biochemistry, 20, 2056–2060. 10.1021/bi00510a049 [DOI] [PubMed] [Google Scholar]
- Parsons, H. G. , Scott, R. B. , Pinto, A. , Carter, R. J. , & Snyder, F. F. (1987). Argininosuccinic aciduria: Long‐term treatment with arginine. Journal of Inherited Metabolic Disease, 10, 152–161. 10.1007/bf01800042 [DOI] [PubMed] [Google Scholar]
- Picca, S. , Dionisi‐Vici, C. , Abeni, D. , Pastore, A. , Rizzo, C. , Orzalesi, M. , … Bartuli, A. (2001). Extracorporeal dialysis in neonatal hyperammonemia: Modalities and prognostic indicators. Pediatric Nephrology, 16, 862–867. 10.1007/s004670100702 [DOI] [PubMed] [Google Scholar]
- Rostami, P. , Haberle, J. , Setoudeh, A. , Zschocke, J. , & Sayarifard, F. (2017). Two novel mutations in the argininosuccinate lyase gene in Iranian patients and literature review. Iranian Journal of Pediatrics, 27, e7666 10.5812/ijp.7666 [DOI] [Google Scholar]
- Shao, Y. , Jiang, M. , Lin, Y. , Mei, H. , Zhang, W. , Cai, Y. , … Liu, L. (2017). Clinical and mutation analysis of 24 Chinese patients with ornithine transcarbamylase deficiency. Clinical Genetics, 92, 318–322. 10.1111/cge.13004 [DOI] [PubMed] [Google Scholar]
- Shchelochkov, O. A. , Li, F.‐Y. , Geraghty, M. T. , Gallagher, R. C. , Van Hove, J. L. , Lichter‐Konecki, U. , … Wong, L.‐J. (2009). High‐frequency detection of deletions and variable rearrangements at the ornithine transcarbamylase (OTC) locus by oligonucleotide array CGH. Molecular Genetics & Metabolism, 96, 97–105. 10.1016/j.ymgme.2008.11.167 [DOI] [PubMed] [Google Scholar]
- Storkanova, G. , Vlaskova, H. , Chuzhanova, N. , Zeman, J. , Stranecky, V. , Majer, F. , … Dvorakova, L. (2013). Ornithine carbamoyltransferase deficiency: Molecular characterization of 29 families. Clinical Genetics, 84, 552–559. 10.1111/cge.12085 [DOI] [PubMed] [Google Scholar]
- Wen, W. , Yin, D. , Huang, F. , Guo, M. , Tian, T. , Zhu, H. , & Yang, Y. (2016). NGS in argininosuccinic aciduria detects a mutation (D145G) which drives alternative splicing of ASL: A case report study. BMC Medical Genetics, 17, 9 10.1186/s12881-016-0273-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankol, Y. , Mecit, N. , Kanmaz, T. , Acarli, K. , & Kalayoglu, M. (2017). Argininosuccinic Aciduria‐A rare indication for liver transplant: Report of two cases. Experimental & Clinical Transplantation, 15, 581–584. 10.6002/ect.2015.0078 [DOI] [PubMed] [Google Scholar]
- Yorifuji, T. , Muroi, J. , Uematsu, A. , Tanaka, K. , Kiwaki, K. , Endo, F. , … Furusho, K. (1998). X‐inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clinical Genetics, 54, 349–353. 10.1034/j.1399-0004.1998.5440415.x [DOI] [PubMed] [Google Scholar]
- Yu, B. , Thompson, G. D. , Yip, P. , Howell, P. L. , & Davidson, A. R. (2001). Mechanisms for intragenic complementation at the human argininosuccinate lyase locus. Biochemistry, 40, 15581–15590. 10.1021/bi011526e [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data generated or analyzed during this study are included in this article. The data are available from the corresponding author on reasonable request.