

Abstract
Background
Benign childhood epilepsy with centrotemporal spikes (BECTS) or benign rolandic epilepsy is the most common epileptic syndrome in school‐age children. Genetics is an important factor in BECTS pathogenesis, and <10 genes were associated with BECTS. This study aimed to identify novel genetic causes of BECTS.
Methods
We conducted whole‐exome sequencing on a patient with BECTS and validated the findings by Sanger sequencing in a pedigree with three patients.
Results
CHRNA4 c.1007G>A was identified in three patients with BECTS in a pedigree. Carbamazepine, which should be carefully used in BECTS, was observed to be effective in the treatment of our atypical BECTS proband based on the molecular diagnosis of CHRNA4.
Conclusion
This is the first study on CHRNA4 variant in BECTS, which widened the genetic spectrum of BECTS and contributed to precise medicine in BECTS.
Keywords: centrotemporal, CHRNA4, epilepsy, pedigree, rolandic
Our research is the first report of CHRNA4 variant in BECTS. Carbamazepine which should be carefully used in BECTS was observed to be effective in the treatment of the disease based on molecular diagnosis.

Abbreviations
- ACMG
American College of Medical Genetics
- BECTS
childhood with centrotemporal spikes
- BRE
benign rolandic epilepsy
- CADD
Combined Annotation Dependent Depletion
- CBZ
carbamazepine
- CSWS
continuous spike and waves during slow‐wave sleep
- EEG
electroencephalography
- LKS
Landau‐Kleffner syndrome
- MRI
magnetic resonance imaging
- PolyPhen‐2
Polymorphism Phenotyping v2
- SIFT
Sorting Intolerant From Tolerant
- SNVs
single‐nucleotide variants
- WES
whole‐exome sequencing
1. INTRODUCTION
Benign childhood epilepsy with centrotemporal spikes (BECTS) or benign rolandic epilepsy is the most common epileptic syndrome in school‐age children, with a prevalence of 15%–25% (Tovia et al., 2011). The seizures in typical BECTS are hemifacial motor seizures that occur during the sleeping period, accompanied by possible somatosensory symptoms in the face, tongue, lips, etc (Loiseau & Duche, 1992). The hallmark of BECTS is its manifestations in electroencephalography (EEG) with spikes in biphasic or triphasic appearance and high amplitude. These typical spikes were located in the centrotemporal areas (rolandic areas) at T3–C5 or T4–C6 in EEG (Kramer, 2008). BECTS is a benign seizure with spontaneous remission of seizures in adolescence (Gkampeta & Pavlou, 2012). Based on the abovementioned background, BECTS is a benign form of epilepsy characterized by age‐ and localization‐related seizures (Proposal for revised classification of epilepsies & epileptic syndromes. Commission on Classification & Terminology of the International League Against Epilepsy, 1989).
Genetics is an important factor in BECTS pathogenesis because a majority of patients with BECTS have a positive family history (up to 59%; Xiong & Zhou, 2017). The genetically determined features of BECTS have made genetic research particularly important in the disease. To date, <10 genes were associated with BECTS (GRIN2A, ELP4, BDNF, KCNQ2/3, DEPDC5, RBFOX1/3, and GABRG2; Dryzalowski, Jozwiak, Franckiewicz, & Strzelecka, 2018). Except for GRIN2A and ELP4, most genes were initially identified in other neurological diseases and then explored in BECTS.
In our study, we identified a novel CHRNA4 (OMIM 118504) variant in a pedigree with BECTS. CHRNA4 encodes neuronal nicotinic acetylcholine receptor α4 subunits and has been previously reported to be related to autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE; Hwang, Makita, Kurahashi, Cho, & Hirose, 2011; Wang, Liu, Wang, & Wu, 2014). Although carbamazepine (CBZ) is considered to be only “sometimes appropriate” in BECTS (Wheless, Clarke, Arzimanoglou, & Carpenter, 2007), we observed effective treatment of CBZ in our atypical BECTS proband based on the molecular diagnosis of CHRNA4. Our study widened the genetic spectrum of BECTS and contributed to the precise treatment of BECTS.
2. MATERIALS AND METHODS
2.1. Ethical compliance
All participants provided informed consent, and the study was approved by the ethics committee of the maternal and child health hospital of Hunan province (2020–S003).
2.2. Family members
The family members of the BECTS pedigree were recruited from the maternal and child health hospitals of Hunan province. Leukocyte DNA was extracted from peripheral blood using the phenol chloroform method.
Information on clinical presentations was obtained by an experienced neurologist. EEG (asleep and awake) and magnetic resonance imaging (MRI) were performed in each patient. BECTS was diagnosed according to the International League Against Epilepsy diagnostic criteria for BECTS based on clinical and EEG features (Proposal for revised classification of epilepsies & epileptic syndromes. Commission on Classification & Terminology of the International League Against Epilepsy, 1989).
2.3. Whole‐exome sequencing and bioinformatic analysis
Whole‐exome sequencing (WES) was conducted on DNA from BECTS proband using a HiSeq 2500 system (Illumina) with a mean depth of 100×.
Initially, according to the protocols of (Ulintz, Wu, & Gates, 2019) WES data are processed preliminarily (data alignment and filtering; Ulintz et al., 2019). The ANNOVAR software was used to annotate the data (Wang, Li, & Hakonarson, 2010). A public database (ESP6500, 1000 Genomes Project, ExAC, gnomAD) was used to filter variants with frequencies >0.001. The pathogenicity of single‐nucleotide variants (SNVs) was predicted using five software programs (Polymorphism Phenotyping v2 [PolyPhen‐2] [http://genetics.bwh.harvard.edu/pph2/], Sorting Intolerant From Tolerant [https://sift.bii.a‐star.edu.sg/], Combined Annotation Dependent Depletion [CADD] [https://cadd.gs.washington.edu/], MutationTaster [http://www.mutationtaster.org/], and FATHMM [http://fathmm.biocompute.org]). The international guidelines of the American College of Medical Genetics (ACMG) were used to explain the pathogenicity of the identified variant (Richards et al., 2015). The variant location in the functional domain of CHRNA4 protein was predicted using the SMART database (http://smart.embl‐heidelberg.de/). The clinical features related to previously reported CHRNA4 variants were found by searching the OMIM database (https://www.omim.org/) and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
Sanger sequencing was performed on the DNA of the proband's parents, brother, and sister to validate the variant found in WES.
3. RESULTS
3.1. Genetic analyses
The WES results revealed a nonsynonymous heterozygous variant CHRNA4c.1007G>A (NM_000744.6) in the proband. Sanger sequencing validated the variant in the proband and his younger brother and mother (Figure 1a). The variant cosegregated with BECTS in the pedigree, as shown in Figure 1b. The variant was predicted to be disease‐causing by MutationTaster and damaging by FATHMM and CADD, but tolerable by SIFT and benign by PolyPhen‐2. The allele frequency was 0.0001 in the gnomAD database and 0 in the ExAC and 1000 Genomes Project databases in the East Asian population. The variant was classified as likely pathogenic according to the ACMG/AMP 2015 guideline.
FIGURE 1.
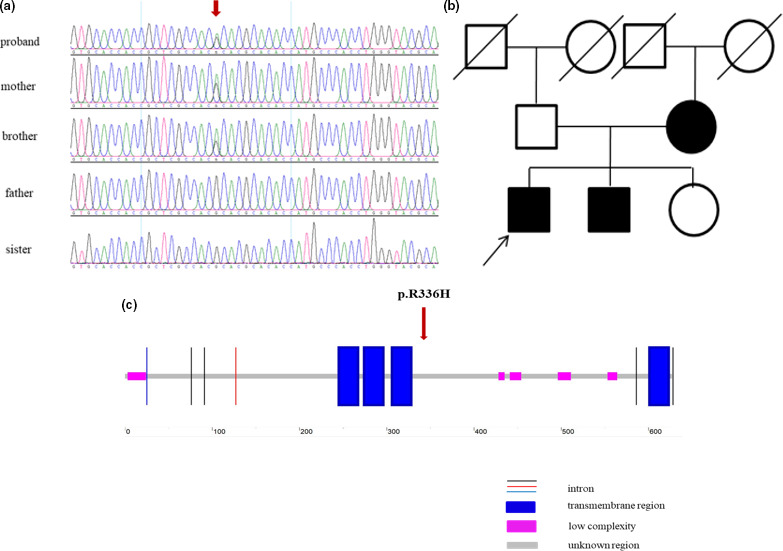
(a) Sanger sequencing analysis of the proband and his sibling and parents. Variant CHRNA4 c.1007G>A was detected in the proband and his younger brother and mother. (b) Pedigree chart of the family. (c) Schematic structure of CHRNA4 protein by searching the SMART database (http://smart.embl‐heidelberg.de/). The location of variant CHRNA4 c.1007G>A is indicated by a red arrow
When searching the SMART database (http://smart.embl‐heidelberg.de/) for the variant c.1007G>A (p.R336H), the variant was predicted to be located in an unknown region of the CHRNA4 protein (Figure 1c).
3.2. Clinical features of family members carrying the CHRNA4 variant
Proband: The proband is a 5‐year‐old boy. He was born uneventfully after the normal period of gestation (38 weeks). At the age of 3 years, the child had sudden seizures during the school nap. Hemiclonic seizures and hemifacial contraction accompanied with drooling were observed, and the seizures lasted for approximately 1–2 min with no loss of consciousness. No attention was paid until tonic‐clonic status epilepticus, starting with twitching of the left face, was observed once during wakefulness half a year ago. It lasted for approximately 3 min until spontaneous remission of the seizures. Absence seizures presenting with motor and speech arrest and loss of consciousness were also observed 5–10 times and lasted for a few seconds during the daytime. After hospitalization, EEG showed spike and slow waves in the right rolandic area during sleep, with a discharge index of 60% in non‐rapid eye movement sleep (electrical status epilepticus during slow‐wave sleep [ESES]; Figure 2a). No speech delay or intellectual disability was observed. Neurological examination and brain MRI findings were normal. The proband received antiepileptic treatment with sodium valproate dose of 20 mg kg−1 day−1. However, symptoms were not relieved.
FIGURE 2.
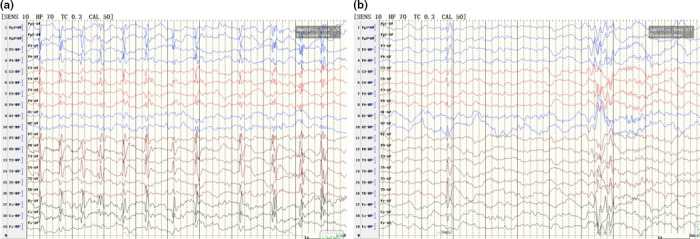
(a) Electroencephalography (EEG) in the proband showed spike and slow waves in the right rolandic area during sleep, with a discharge index of 60% in non‐rapid eye movement sleep (NREM) (electrical status epilepticus during slow‐wave sleep [ESES]). (b) EEG findings in the brother showed spike and slow waves in the right rolandic area during sleep
Brother: The younger brother is a 3‐year‐old boy. He was also born at full‐term, with no abnormalities during pregnancy. Half a year ago, the parents observed the left arm jerking and left mouth twitching accompanied by grunting shortly after falling asleep. They observed similar seizures approximately three times, which lasted for nearly 1 min. Interictal EEG showed spike and slow waves in the right rolandic area during sleep (Figure 2b). Neurological examination and brain MRI findings were normal.
Mother: The mother is a 30‐year‐old woman. She described that her mother noticed her arms and legs shaking during sleep at elementary school age. The seizures had spontaneously resolved during adolescence.
We searched CHRNA4‐related clinical features in the OMIM database and found that the gene was only reported to be associated with epilepsy, nocturnal frontal lobe, 1 (phenotype MIM number: 600513). Additionally, we searched for c.1007G>A in CHRNA4 in the ClinVar database and found that the specific variant was previously reported to be related to sporadic nocturnal frontal lobe epilepsy (Kurahashi & Hirose, 1993). CBZ was orally administered to the proband at 15 mg kg−1 day−1. After using the medication for 1 month, the frequency of seizures was reduced. EEG showed a reduction in the number of spikes and slow waves in the right rolandic area during sleep (Figure 3a). After 6 months, the seizures stopped, and EEG recordings showed no discharges during sleep (Figure 3b). CBZ was also administered to his brother, and the seizures were relieved.
FIGURE 3.
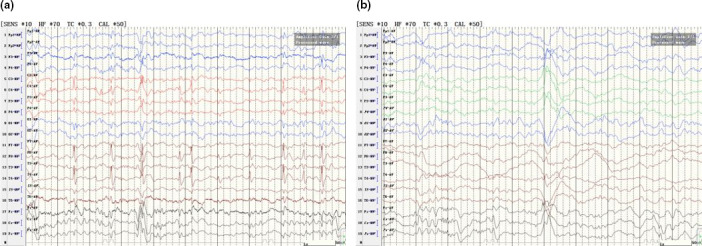
(a) Electroencephalography (EEG) findings in the proband who was treated with carbamazepine (CBZ) for 1 month showed a reduction in the number of spikes and slow waves in the right rolandic area during sleep. (b) EEG findings in the proband who was treated with CBZ for 6 months showed no discharges during sleep
4. DISCUSSION
Benign childhood epilepsy with centrotemporal spikes is the most common focal benign epilepsy in childhood, and genetic factors contribute to nearly 60% of BECTS (Xiong & Zhou, 2017). To date, <10 genes were identified to be associated with BECTS (Dryzalowski et al., 2018). We initially identified a likely pathogenic CHRNA4 variant in a Chinese pedigree with BECTS. Moreover, we observed effective treatment of CBZ in our BECTS proband and his brother based on the molecular diagnosis of CHRNA4, although CBZ is often considered to be used cautiously in BECTS. Our research widened the genetic spectrum of BECTS and contributed to understanding the molecular mechanisms of the disease.
The neuronal nicotinic acetylcholine receptor CHRNA4 protein consists of four transmembrane helical regions (M1‐M4), five low complexity regions, and some unknown regions (http://smart.embl‐heidelberg.de/). Functional researchers have found that the M2 transmembrane region is located in the central pore of the receptor and determines the ion selectivity of the receptor (Unwin, 1995). The typical clinical feature of CHRNA4 variant carriers is nocturnal frontal lobe epilepsy, and the reported variants in CHRNA4 in nocturnal frontal lobe epilepsy are located in or close to the M2 region of the receptor (Hwang et al., 2011). The variants in the M2 region have a gain‐of‐function effect, which could increase the receptor sensitivity to acetylcholine (Bertrand et al., 2002; Hoda et al., 2008). Our study found a new clinical syndrome—BECTS—in CHRNA4 carriers. The variant in CHRNA4 is located in an unknown region of CHRNA4 protein as predicted by the SMART database (Figure 1c). Although the mechanisms underlying CHRNA4 variant‐related BECTS are unknown, we speculated that different structural domain dysfunctions caused by different variants are the cause of various clinical manifestations.
It has been reported that the antiepileptic drug CBZ causes seizures with EEG deteriorations in BECTS. Using the drug could induce partial epilepsy to generalized seizures in BECTS (Nanba & Maegaki, 1999), precipitate seizures atypically (frequency increase, atypical absences, EEG discharges diffusion, etc.; Corda, Gelisse, Genton, Dravet, & Baldy‐Moulinier, 2001), and even lead to the development of epilepsy with a deteriorating EEG (continuous spike and wave during slow‐wave sleep) in patients with benign BECTS (Perucca, Gram, Avanzini, & Dulac, 1998). Moreover, drug‐induced seizure exacerbation is more likely to occur in patients with BECTS with atypical EEG manifestations other than typical EEG features in the rolandic region (Guerrini, Belmonte, & Genton, 1998). It has been reported that seizures and ESES in patients with Landau‐Kleffner syndrome are aggravated by CBZ treatment (Perucca et al., 1998). Therefore, European experts hold the opinion that CBZ is only “sometimes appropriate” for BECTS treatment, and some critics consider other medications for BECTS (Wheless et al., 2007).
In our study, we considered a novel CHRNA4 variant in BECTS for molecular diagnosis. CHRNA4 encodes neuronal nicotinic acetylcholine receptor α4 subunits and is the first gene identified in ADNFLE (Steinlein et al., 1995). CBZ blocks heteromeric nicotinic acetylcholine receptors (nAChRs) and is particularly effective in the treatment of ADNFLE (Becchetti, Aracri, Meneghini, Brusco, & Amadeo, 2015). nAChR antagonists have been proven to be effective in CHRNA4‐related epileptic encephalopathy by molecular and cellular studies (Becchetti et al., 2015; Ghasemi & Hadipour‐Niktarash, 2015). After treating our proband and his brother with CHRNA4‐targeted drug CBZ, we observed remission in seizures. Our study emphasized the importance of therapeutic options for BECTS based on molecular diagnosis and contributed to precise medicine in epilepsies.
5. CONCLUSION
This is the first study on CHRNA4 variant in BECTS, which widened the genetic spectrum of BECTS and contributed to precise medicine in BECTS.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any conflict of interest.
AUTHOR CONTRIBUTIONS
NX provided the clinical data from patients and family members. XM did the genetic test and analyzed the data. YLC and QYL revised the manuscript. LS wrote the manuscript. ZYS provided case review. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant number 81801136), by the Natural Science Foundation of Hunan, China (grant number 2017JJ3142) and by the research project of Chenzhou first people's Hospital (N2019‐020). Thanks to the support of Berry Genomics for the sequencing analysis. We thank the patients and their family members for their cooperation.
Neng X, Xiao M, Yuanlu C, Qinyan L, Li S, Zhanyi S. Novel variant in CHRNA4 with benign childhood epilepsy with centrotemporal spikes and contribution to precise medicine. Mol Genet Genomic Med. 2020;8:e1264 10.1002/mgg3.1264
Li Shu and Zhanyi Song co‐corresponding authors
DATA AVAILABILITY STATEMENT
The datasets supporting the conclusions of this article are included within the article and its additional files.
REFERENCES
- Becchetti, A. , Aracri, P. , Meneghini, S. , Brusco, S. , & Amadeo, A. (2015). The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Frontiers in Physiology, 6, 22 10.3389/fphys.2015.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, D. , Picard, F. , Le Hellard, S. , Weiland, S. , Favre, I. , Phillips, H. , … Mulley, J. (2002). How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia, 43(Suppl 5), 112–122. 10.1046/j.1528-1157.43.s.5.16.x [DOI] [PubMed] [Google Scholar]
- Corda, D. , Gelisse, P. , Genton, P. , Dravet, C. , & Baldy‐Moulinier, M. (2001). Incidence of drug‐induced aggravation in benign epilepsy with centrotemporal spikes. Epilepsia, 42(6), 754–759. 10.1046/j.1528-1157.2001.30000.x [DOI] [PubMed] [Google Scholar]
- Dryzalowski, P. , Jozwiak, S. , Franckiewicz, M. , & Strzelecka, J. (2018). Benign epilepsy with centrotemporal spikes–Current concepts of diagnosis and treatment. Neurologia i Neurochirurgia Polska, 52(6), 677–689. 10.1016/j.pjnns.2018.08.010 [DOI] [PubMed] [Google Scholar]
- Ghasemi, M. , & Hadipour‐Niktarash, A. (2015). Pathologic role of neuronal nicotinic acetylcholine receptors in epileptic disorders: Implication for pharmacological interventions. Reviews in the Neurosciences, 26(2), 199–223. 10.1515/revneuro-2014-0044 [DOI] [PubMed] [Google Scholar]
- Gkampeta, A. , & Pavlou, E. (2012). Emerging genetic influences in benign epilepsy with centro‐temporal spikes‐BECTS. Epilepsy Research, 101(3), 197–201. 10.1016/j.eplepsyres.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Guerrini, R. , Belmonte, A. , & Genton, P. (1998). Antiepileptic drug‐induced worsening of seizures in children. Epilepsia, 39(Suppl 3), S2–10. 10.1111/j.1528-1157.1998.tb05118.x [DOI] [PubMed] [Google Scholar]
- Hoda, J. C. , Gu, W. , Friedli, M. , Phillips, H. A. , Bertrand, S. , Antonarakis, S. E. , … Bertrand, D. (2008). Human nocturnal frontal lobe epilepsy: Pharmocogenomic profiles of pathogenic nicotinic acetylcholine receptor beta‐subunit mutations outside the ion channel pore. Molecular Pharmacology, 74(2), 379–391. [DOI] [PubMed] [Google Scholar]
- Hwang, S. K. , Makita, Y. , Kurahashi, H. , Cho, Y. W. , & Hirose, S. (2011). Autosomal dominant nocturnal frontal lobe epilepsy: A genotypic comparative study of Japanese and Korean families carrying the CHRNA4 Ser284Leu mutation. Journal of Human Genetics, 56(8), 609–612. 10.1038/jhg.2011.69 [DOI] [PubMed] [Google Scholar]
- Kramer, U. (2008). Atypical presentations of benign childhood epilepsy with centrotemporal spikes: A review. Journal of Child Neurology, 23(7), 785–790. 10.1177/0883073808316363 [DOI] [PubMed] [Google Scholar]
- Kurahashi, H. , & Hirose, S. (1993). Autosomal Dominant Nocturnal Frontal Lobe Epilepsy Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., & Amemiya A. (Eds.), GeneReviews(R). Seattle, WA: University of Washington, Seattle. [PubMed] [Google Scholar]
- Loiseau, P. , & Duche, B. (1992). Benign rolandic epilepsy. Advances in Neurology, 57, 411–417. [PubMed] [Google Scholar]
- Nanba, Y. , & Maegaki, Y. (1999). Epileptic negative myoclonus induced by carbamazepine in a child with BECTS. Benign childhood epilepsy with centrotemporal spikes. Pediatric Neurology, 21(3), 664–667. 10.1016/S0887-8994(99)00054-5 [DOI] [PubMed] [Google Scholar]
- Perucca, E. , Gram, L. , Avanzini, G. , & Dulac, O. (1998). Antiepileptic drugs as a cause of worsening seizures. Epilepsia, 39(1), 5–17. 10.1111/j.1528-1157.1998.tb01268.x [DOI] [PubMed] [Google Scholar]
- Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia, 30(4), 389–399. 10.1111/j.1528-1157.1989.tb05316.x [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein, O. K. , Mulley, J. C. , Propping, P. , Wallace, R. H. , Phillips, H. A. , Sutherland, G. R. , … Berkovic, S. F. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics, 11(2), 201–203. [DOI] [PubMed] [Google Scholar]
- Tovia, E. , Goldberg‐Stern, H. , Ben Zeev, B. , Heyman, E. , Watemberg, N. , Fattal‐Valevski, A. , & Kramer, U. (2011). The prevalence of atypical presentations and comorbidities of benign childhood epilepsy with centrotemporal spikes. Epilepsia, 52(8), 1483–1488. 10.1111/j.1528-1167.2011.03136.x [DOI] [PubMed] [Google Scholar]
- Ulintz, P. J. , Wu, W. , & Gates, C. M. (2019). Bioinformatics Analysis of Whole Exome Sequencing Data. Methods in Molecular Biology, 1881, 277–318. [DOI] [PubMed] [Google Scholar]
- Unwin, N. (1995). Acetylcholine receptor channel imaged in the open state. Nature, 373(6509), 37–43. 10.1038/373037a0 [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. Y. , Liu, X. Z. , Wang, J. , & Wu, L. W. (2014). A novel mutation of the nicotinic acetylcholine receptor gene CHRNA4 in a Chinese patient with non‐familial nocturnal frontal lobe epilepsy. Epilepsy Research, 108(10), 1927–1931. 10.1016/j.eplepsyres.2014.08.024 [DOI] [PubMed] [Google Scholar]
- Wheless, J. W. , Clarke, D. F. , Arzimanoglou, A. , & Carpenter, D. (2007). Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disorders, 9(4), 353–412. [DOI] [PubMed] [Google Scholar]
- Xiong, W. , & Zhou, D. (2017). Progress in unraveling the genetic etiology of rolandic epilepsy. Seizure, 47, 99–104. 10.1016/j.seizure.2017.02.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.