

Abstract
Objective:
The purpose of this paper is to review the typical cognitive and motor impairments seen in fragile X-associated tremor/ataxia syndrome (FXTAS), essential tremor (ET), Parkinson disease (PD), spinocerebellar ataxias (SCAs), multiple system atrophy (MSA), and progressive supranuclear palsy (PSP) in order to enhance diagnosis of FXTAS patients.
Method:
We compared the cognitive and motor phenotypes of FXTAS with each of these other movement disorders. Relevant neuropathological and neuroimaging findings are also reviewed. Finally, we describe the differences in age of onset, disease severity, progression rates and average lifespan in FXTAS compared to ET, PD, SCAs, MSA and PSP. We conclude with a flow chart algorithm to guide the clinician in the differential diagnosis of FXTAS.
Results:
By comparing the cognitive and motor phenotypes of FXTAS with the phenotypes of ET, PD, SCAs, MSA, and PSP we have clarified potential symptom overlap while elucidating factors that make these disorders unique from one another. In summary, the clinician should consider a FXTAS diagnosis and testing for the Fragile X mental retardation 1 (FMR1) gene premutation if a patient over the age of 50 (1) presents with cerebellar ataxia and/or intention tremor with mild parkinsonism, (2) has the middle cerebellar peduncle (MCP) sign, global cerebellar and cerebral atrophy, and/or subcortical white matter lesions on MRI, or (3) has a family history of fragile X related disorders, intellectual disability, autism, premature ovarian failure and has neurological signs consistent with FXTAS. Peripheral neuropathy, executive function deficits, anxiety, or depression are supportive of the diagnosis.
Conclusions:
Distinct profiles in the cognitive and motor domains between these movement disorders may guide practitioners in the differential diagnosis process and ultimately lead to better medical management of FXTAS patients.
Keywords: FXTAS, essential tremor, Parkinson disease, spinocerebellar ataxia, multiple system atrophy, progressive supranuclear palsy
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late onset neurodegenerative disorder that occurs in some individuals with a “premutation (PM) size” 55–200 CGG repeat expansion in the fragile X mental retardation 1 (FMR1) gene. Although core motor features include tremor and cerebellar ataxia, there is high phenotypic variability with some carriers demonstrating parkinsonism, peripheral neuropathy, executive function deficits, dementia, and neuropsychiatric problems (E. Berry-Kravis et al., 2007; J. Grigsby et al., 2014; R. J. Hagerman et al., 2001; M. A. Leehey, 2009; A. L. Seritan et al., 2008). Because FXTAS was first reported in the literature relatively recently (R. J. Hagerman et al., 2001) and has high phenotypic variability and overlap of symptoms with other more well-known movement disorders, it is frequently initially diagnosed as other diseases at the time of onset (D. A. Hall et al., 2005). This is especially true when patients are seen by a primary care physician or general neurologist, or at a non-fragile X-associated clinic where FXTAS may not be readily recognized. Thus, we aim to compare and contrast the cognitive and motor phenotype in FXTAS to the movement disorders which FXTAS patients are most frequently misdiagnosed with. These include essential tremor (ET), Parkinson disease (PD), spinocerebellar ataxias (SCAs), multiple system atrophy (MSA), and progressive supranuclear palsy (PSP). This information will guide practitioners in the differential diagnostic process and may provide some insight into potential common pathophysiological mechanisms for these movement disorders.
This review is divided into three major sections:
The cognitive phenotypes in these disorders with emphasis on the known similarities or differences with FXTAS. This section was further subdivided into A. Executive function, B. Global Cognition, Memory, Mild Cognitive Impairment (MCI) and Dementia, C. Other Cognitive Functions: Language and Visuospatial Processing, D. Neuropsychiatric Disturbances including depression, anxiety, emotions, hallucinations, and psychosis. Other genetic factors, neuroimaging, and neuropathological findings are included when relevant. The comparisons and known prevalence rates of each of these cognitive and neuropsychiatric disorders in FXTAS and each of these other movement disorders are presented in Table 1.
The motor phenotypes in these disorders with emphasis on the known similarities or differences with FXTAS. This section reviews the motor phenotype of FXTAS as it compares to the phenotypes seen in ET, PD, SCAs, MSA, and PSP. For each movement disorder, motor profiles are presented as a summary of the respective tremor, cerebellar ataxia, parkinsonism, and eye movement abnormality symptoms as they compare to those seen in FXTAS. Case reports are described in which patients given an initial diagnosis were later identified as being FMR1 PM carriers, a subset of which were given a new diagnosis of FXTAS. The epidemiological data is presented through a review of studies screening for the FMR1 premutation in these other disorders. Results of imaging studies have been reviewed when relevant. The known prevalence rates of motor symptoms and signs in each movement disorder are presented in Table 2.
A brief comparison of the age of onset, disease severity and progression, and average lifespan in these disorders which is summarized in Table 3.
Table 1.
Prevalence of Cognitive and Neuropsychiatric Findings in fragile X-associated tremor/ataxia syndrome (FXTAS), Essential Tremor (ET), Parkinson Disease (PD), Spinocerebellar Ataxias (SCAs), Multiple System Atrophy (MSA), and Progressive Supranuclear Palsy (PSP)
| Disease | Executive Function | Global Cognition, Memory, MCI | Dementia | Language Deficits | Visuospatial processing deficits | Depression | Anxiety | Hallucinations/psychosis |
|---|---|---|---|---|---|---|---|---|
| FXTAS | Prominent (+) (Brega, 2008; Grigsby, 2007; Yang, 2013, 2014), severity increases with advancing age (Bacalman, 2006; Bourgeois, 2006, 2007; Seritan, 2008) | MCI can be present (+), relative sparing of memory encoding and recognition | 37–42% men (Seritan, 2008, 2013), unknown in women | Mild dysnomia (+) (Grigsby, 2006, 2008) | Present (+) (Grigsby, 2007, 2008) | 43.5% (Bourgeois, 2011) | 52% (Bourgeois, 2011) | Very rare (Seritan, 2013) |
| ET | Present but mild (+) (Benito-Leon, 2006; Lombardi, 2001; Sinoff, 2014; Troster, 2002) | Up to 69% in middle aged patients (mean age 56 years) (Sinoff, 2014) | Up to 33% (Sinoff, 2014) 70% higher than controls in late onset ET (> 65 years) (Romero, Benito-Leon, 2012) | Mild dysnomia (+) (Lombardi, 2001) | Present (+) (Sahin, 2006; Troster, 2002 | 18% (Sinoff, 2014) | 25% (Sinoff, 2014) | _ |
| PD | Present (+) (prevalence is included in the MCI category) | Up to 42.5% (Yarnall, 2014) | 46–82% (Williams-Gray, 2013; Hely, 2008) | 50% in those with MCI (Pfeiffer, 2014) | 46% in those with MCI (Pfeiffer, 2014); also present (+) in those with GBA or E326K polymorphism (Mata, 2014) | 37–70% (Aarsland, 1999, 2009; Goldman, 2014) | 20–49% (Gallagher, 2011) | 30–50 % hallucinations (Zhu, 2013) 27–40% psychosis (70% in those living >20 years post diagnosis) (Levin, 2015) |
| SCAs | Present (+) in SCA1, 2, 3, 8, 14, 19 (Burk, 2001, 2003; Hernandez-Castillo, 2015; Braga-Neto, 2012; Radvany, 1993; Zawacki, 2002; Lilja, 2005; Torrens, 2008; Klebe, 2005; Schelhaas, 2003) | Rare in SCA3 (Braga-Neto, Pedroso, 2012; Kawai, 2004; Maruff, 1996; Radvany,1993; Zawacki, 2002), Present (+) in DRPLA (late onset form) (Naito, 1982; Tsuji, 2012; Vale, 2010), SCA1 (Donato,2012), SCA2 (Durr, 1995), SCA17 (Koutsis, 2014; Toyoshima, 1993; Zuhlke, 2007) | Rare in SCA3 Braga-Neto, 2012; Kawai, 2004; Maruff, 1996; Radvany,1993; Zawacki, 2002), Present (+) in DRPLA (late onset form) (Naito, 1982; Tsuji, 2012; Vale, 2010), SCA1 (Donato, 2012), SCA2 (Durr, 1995), SCA17 (Koutsis, 2014; Toyoshima, 1993; Zuhlke, 2007) | Present(+) in SCA 6 (van Gaalen, 2014) | Present (+) in SCA1, 2, 3 (Braga-Neto, 2012; Braga-Neto, Pedroso, 2012; Fancellu, 2013; Feng, 2014; Kawai, 2004; Orsi, 2011) | 17–26% (Lo, 2016; Schmitz-Hubsch, 2011) | Variably present (+) in SCA 6 (Suenaga, 2008) and 8 (Torrens, 2008) | Rare |
| MSA | Up to 54% (Auzou, 2015; Siri, 2013) | Present (+) (Balas, 2010; Brown, 2010; Burk, 2006; Hong, 2011) Kim, 2013, 2015; Lyoo, 2008; Siri, 2013) | Up to 30% (Brown, 2010; Kitayama, 2009) | Present (+) in demented patients (Kao, 2009; Lyoo, 2008) | Present (+) in demented patients (Brown, 2010; Kim, 2013) | 30–85% (Benrud-Larson, 2005; Schrag, 2006, 2010; Siri, 2013) | 37% (Schrag, 2010) and appears more prevalent in MSA-C (Balas, 2010) | Rare (Williams, 2008) |
| PSP | Up to 70–90% (Brown, 2010; Gerstenecker, 2013; Kaat, 2011) | Present (+) (Respondek, 2015) | 58% (Pillon, 1991) | (−) (Magherini, 2005) | Present (+) (Borroni, 2008; Esmonde, 1996; Ghosh, 2013) | 50% higher than controls (Bloise, 2014; Esmonde, 1996; Gerstenecker, 2013) | 18% (Litvan, 1996) | 5–11% (Gerstenecker, 2013) |
Key:MCI, mild cognitive impairment; GBA, glucocerebrosidase gene mutation; E326K, polymorphism in the GBA gene; DRPLA, dentatorubral-pallidoluysian atrophy; (−) not seen or reported in the literature; (+) present but unknown prevalence; “Up to” refers to lifetime prevalence rates
Table 2.
Prevalence of Motor Phenotypes in fragile X-associated tremor/ataxia syndrome (FXTAS), Essential Tremor (ET), Parkinson Disease (PD), Spinocerebellar Ataxias (SCAs), Multiple System Atrophy (MSA), and Progressive Supranuclear Palsy (PSP)
| Movement Disorder | Tremor | Cerebellar Ataxia | Parkinsonism | Eye Movement Abnormalities |
|---|---|---|---|---|
| FXTAS | 77% (men, mean age 65 ± 7) (Juncos, 2011) + Kinetic > rest tremor (Berry-Kravis, 2007) Hands >> head (Apartis 2012; Leehey, 2003; Peters, 2006) |
41–66% (men, mean age 65 ± 7; men and women, 66.6 ± 8.2) (Juncos, 2011; Niu, 2014) | 29–32% (men and women, mean age 66.6 ± 8.2; men, 65 ± 7) (Niu, 2014; Juncos, 2011) | May be present (+) |
| ET | Kinetic: 25–98% (mean ages 62.5 ± 14.6; 66.90 ± 12.35) (Deuschl, 2000; Ghika, 2015) Intention: 33–89% (mean ages 62.5 ± 14.6; 66.90 ± 12.35) (Deuschl, 2000; Ghika, 2015) Postural: 80% (mean age 66.90 ± 12.35) (Ghika, 2015) Rest: 1.9–46.4% (may emerge with advanced disease) (Louis, 2015) Hands >> head > voice (Jankovic, 2002; Deuschl, 2009) |
50% - Mild (mean age 69) (Singer, 1994) | 64% Typical (median age 65) (Jankovic, 1995) | 35–41% (mean ages 54.2 ± 4.1; 27.6 ± 17.3 years) (Schwartz, 1999; Helmchen, 2003) |
| PD | ++ Rest 38.9% TD (58.4 ± 10.9) (Wu, 2015) | − | ++ | Present (+) (Antoniades, 2015; Crawford, 1989; Shibasaki, 1979) (Age range 49–69) |
| SCAs | Postural and Intention present (+) in SCA 2, 12, 15, 20, 27 (Perlman, 2011; Storey, 2014) | ++ but may or may not be seen in SCA 17 and DRPLA (Matilla-Dueñas, 2012; Perlman, 2011; Storey, 2014) | Present (+) in SCA 2, 3, 17, 21 (Park, 2015; Perlman, 2011; Storey, 2014) | Present (+) in SCA 1, 2, 3, 5, 6, 7, 8, 14, 28, 37 (Storey, 2014) |
| MSA | MSA-P: Rest- Up to 60% Postural- Up to 60% Intention- Up to 37% (Low, 2015) MSA-C: Rest- Up to 22% Postural- Up to 45% Intention- Up to 94% (Low, 2015) Overall: Up to 80% (Kaindlstorfer, 2013) |
MSA-P- Up to 40% MSA-C- Up to 100% (Low, 2015) |
MSA-P: Up to 98% MSA-C: Up to 73% (Low, 2015) |
Present (+) (MSA-C mean age 63.2 ± 7.1) (MSA-P mean age 60.7 ± 7.6) (Terao, 2016) |
| PSP | Up to 42% (Fujioka, 2016) | Rare (Kanazawa, 2009; Iwasaki, 2013; Koga, 2016; Kanazawa, 2013) | Up to 19% typical Parkinsonism Up to 40% atypical Parkinsonism (Respondek, 2015) |
++ (Liscic, 2013; Williams, 2005) |
Key:TD, tremor-dominant; DRPLA, dentatorubral-pallidoluysian atrophy; MSA-P, multiple system atrophy parkinsonism subtype; MSA-C, multiple system atrophy cerebellar subtype, (−) not seen or reported in the literature, (+) present but unknown prevalence, (++) required for diagnosis, “Up to” refers to lifetime prevalence rates
Table 3.
Summary of Age of onset, disease severity and progression, and average lifespan in fragile X-associated tremor/ataxia syndrome (FXTAS), Essential Tremor (ET), Parkinson Disease (PD), Spinocerebellar Ataxias (SCAs), Multiple System Atrophy (MSA), and Progressive Supranuclear Palsy (PSP)
| Disease | Age of onset (AOO) | Disease Severity | Disease Progression | Average Survival Time |
|---|---|---|---|---|
| FXTAS | > 55 years (men) unknown in women | variable | slow | 21 years from age of onset* |
| ET | variable (55.8 ± 15.1 years) hereditary and sporadic ET: < 65 years senile ET: > 65 years | relatively mild | very slow | normal |
| PD | 58.4 ± 11.0 years | variable | moderate | 15 years from age of onset* |
| SCAs | variable SCA1–3: 30–40 years SCA6: 52 ± 12 years | variable | variable | variable; earlier death with earlier age of onset SCA 6: normal |
| MSA | 56 ± years | severe | rapid | 9–10 years from age of onset* |
| PSP | 60–70 years | severe | very rapid | 6–8 years from age of onset* |
Key:
estimated prevalence rate
Finally, we summarize the data with a flow chart suggesting the appropriate differential diagnosis of FXTAS (Figure 1).
Figure 1. Differential diagnosis of FXTAS.
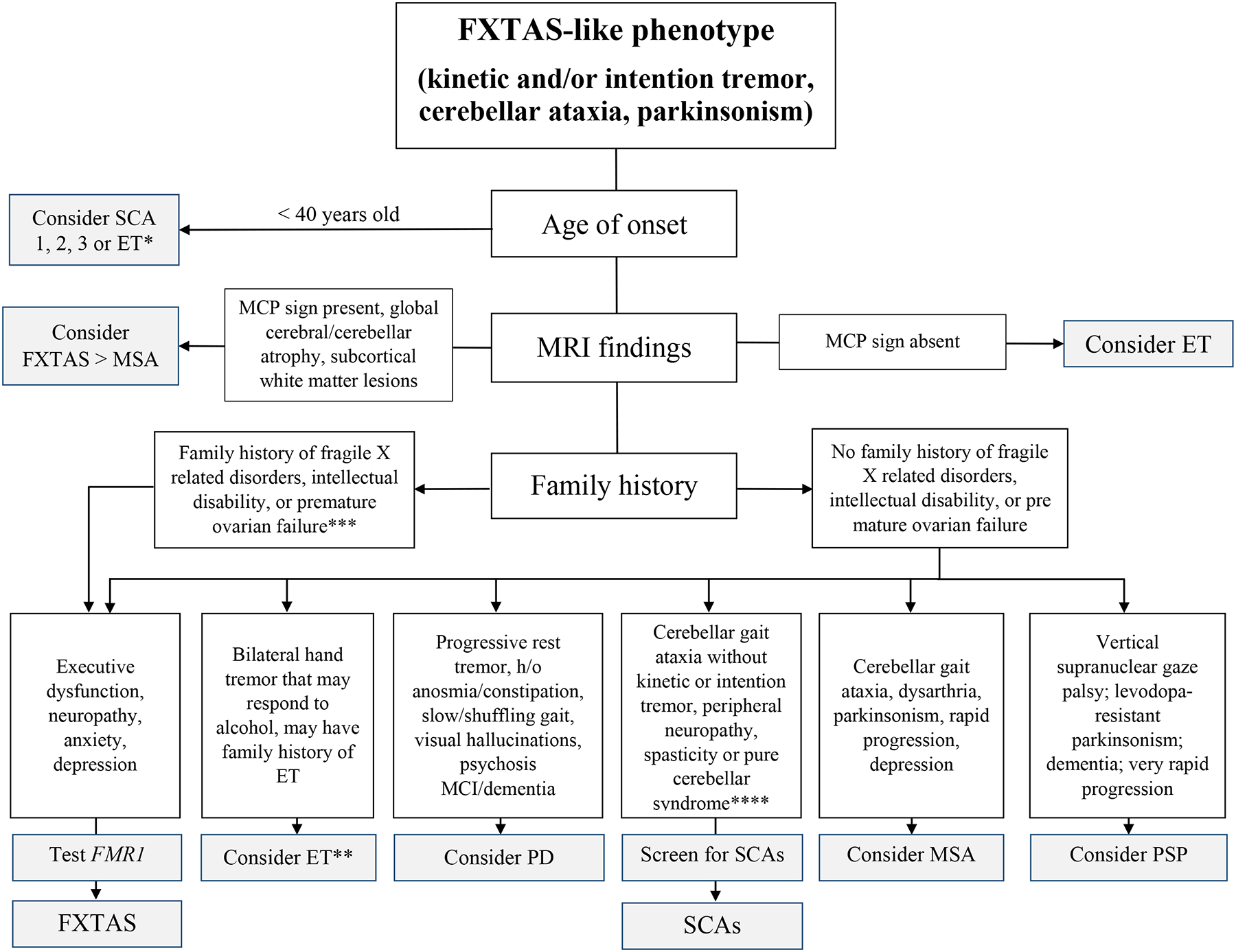
Key: FXTAS, fragile X associated tremor/ataxia syndrome; ET, essential tremor; PD, Parkinson’s syndrome; SCA, spinocerebellar ataxia; MSA, multiple system atrophy; PSP, progressive supranuclear palsy.
* This refers to the early-onset subtype of ET (Deuschl, 2009).
** This refers to the late-onset subtype of ET, which is more likely to be confused with FXTAS (Deuschl, 2009).
*** Also inquire regarding a FH of autism, developmental delay, or learning disabilities.
**** Cerebellar eye signs include nystagmus, saccadic pursuits, and slowed saccades. If the patient has ataxia and prominent kinetic tremor, consider SCA12.
Methods:
To identify relevant publications for this literature review, we performed a PubMed search using a combination of the following key search terms:
FXTAS, FMR1 premutation carriers, screening studies, essential tremor, Parkinson disease, spinocerebellar ataxias, multiple system atrophy, progressive supranuclear palsy, executive function, working memory, verbal fluency, dementia, cognitive impairment, visuospatial, language, neuropsychiatric symptoms, apathy, anxiety, depression, hallucinations, psychosis, age of onset, disease progression, longevity, life expectancy.
I. Cognitive Phenotypes
We begin by reviewing the cognitive features of FXTAS. We then review these same features in ET, PD, SCA and MSA, and PSP, concluding each section with a summary comparison between each of these other movement disorders and FXTAS. Table 1 contains a summary of these comparisons. Other genetic factors, neuroimaging, and neuropathological findings are included when relevant.
The term “cerebellar cognitive affective syndrome” was coined to describe the cognitive impairment and psychiatric symptoms in patients with various types of cerebellar disease (Bodranghien et al., 2015; Schmahmann & Sherman, 1998). This syndrome is characterized by deficits in executive function, working and verbal memory, visuospatial processing, and language, as well as emotional problems. It is postulated that this constellation of cognitive and affective signs are due to disruption in cerebro-cerebellar circuits (Bernard et al., 2012; Bernard et al., 2013; Ramnani, 2012), as well as subcortical structures that connect with the prefrontal cortex, including the basal ganglia and limbic structures (Heyder, Suchan, & Daum, 2004; Middleton & Strick, 2000b). Evidence supports the idea that the basal ganglia and cerebellum form reciprocally connected loops with the prefrontal cortex mediating cognitive functions (Middleton & Strick, 2000a). This anatomical and functional circuitry suggests that pathology in one region may cause dysfunction in the other.
Executive function comprises several cognitive capabilities including disinhibition, working memory, attentional regulation, and verbal fluency (Gilbert & Burgess, 2008). One aspect of executive function is the ability to self-regulate one’s behavior by initiating purposeful behaviors while inhibiting socially inappropriate or irrelevant behaviors (Beer, John, Scabini, & Knight, 2006; Jurado & Rosselli, 2007). Working memory is the capacity to temporarily store information in the brain and then manipulate that information to accomplish complex cognitive tasks (Baddeley, 2010). Attention is the ability to prioritize salient information while other extraneous information is competing for cognitive resources (Alvarez & Emory, 2006; Fan et al., 2009; Lavie, Hirst, de Fockert, & Viding, 2004). Verbal fluency is a higher ordered cognitive function that requires information retrieval from memory (Alvarez & Emory, 2006; Lavie et al., 2004). This retrieval requires executive functions including selective attention and self-monitoring, response generation and inhibition, and mental set shifting. The brain regions thought to mediate executive functions include the fronto- parietal attentional network (Fox et al., 2005; Markett et al., 2014) and fronto-cerebellar pathways (Bernard et al., 2012; Krienen & Buckner, 2009). More specific networks also exist which are devoted to working memory and verbal fluency and are modality specific. These include the temporal, parietal and occipital lobes for sensorimotor processing and subsequent multimodal areas in the frontal and parietal lobes which maintain and manipulate task specific information (Rama, 2008; Zimmer, 2008).
Mild cognitive impairment (MCI) is a diagnostic category for the “symptomatic, pre-dementia phase” of individuals with a trajectory of cognitive decline (Albert et al., 2011). Individuals with MCI, in contrast to those with dementia, have preserved activities of daily living and no significant deficits in social or occupational function (Petersen, 2011). Amnestic versus non-amnestic sub-categories of MCI have also been defined based on the number and types of cognitive domains affected and whether memory impairments exist (Petersen, 2011). MCI is typically diagnosed by cut-off scores on the Montreal cognitive assessment (MoCA) or the Mini-mental state examination (MMSE), although the MoCA is thought to have greater sensitivity for distinguishing MCI (Trzepacz et al., 2015).
Fragile X-Associated Tremor / Ataxia Syndrome (FXTAS)
Four recent reviews have extensively detailed the literature regarding the cognitive and neuropsychiatric phenotypes observed in PM carrier men and women, with and without FXTAS (Besterman et al., 2014; Birch, Cornish, Hocking, & Trollor, 2014; J. Grigsby et al., 2014; Wheeler et al., 2014). This paper provides a more general overview in order to compare these phenotypes with those seen in ET, PD, SCAs, MSA, and PSP. In addition, literature on PM carriers with FXTAS will be emphasized in order to allow for direct comparisons with other diagnosed movement disorders and not PM carriers without FXTAS because it is not yet known whether cognitive deficits in this population represent early prodromal signs of FXTAS or a neurodevelopmental effect of the PM. It is important to emphasize that detailed cognitive studies in FXTAS may be more sparse than these other movement disorders, especially PD, PSP and MSA because FXTAS was first discovered in 2001.
A. Executive Function
Many recent studies have documented and further refined the evidence for specific cognitive deficits in PM carriers with FXTAS. Men with FXTAS have been more extensively studied than women. The primary cognitive phenotype appears to be one of deficits in executive function (Brega et al., 2008; J. Grigsby et al., 2007; Yang et al., 2013; Yang et al., 2014) that may then progress to widespread cognitive deficits including dementia in advanced disease stages (Bacalman et al., 2006; Bourgeois et al., 2006; Bourgeois et al., 2007; A. L. Seritan et al., 2008). Working memory dysfunction (Cornish et al., 2009; J. Grigsby et al., 2007) and deficits in attentional control, response inhibition and self-regulation (Cornish et al., 2008; J. Grigsby et al., 2007), and verbal fluency (J. Grigsby et al., 2007) have all been reported in men with FXTAS. Reduced information processing speed has also been reported in men with FXTAS (J. Grigsby et al., 2007; A. Seritan, Cogswell, & Grigsby, 2013). Recent reports indicate that women with FXTAS also have deficits in executive function including areas of response inhibition and performance monitoring (Yang et al., 2013; Yang et al., 2014). Some women with FXTAS also have abnormal semantic processing and verbal learning skills (Yang et al., 2014).
Interestingly, men with FXTAS have reduced inferior frontal cortical activity while performing a working memory task (Hashimoto, Backer, Tassone, Hagerman, & Rivera, 2011). This cortical area appears to be important in encoding and memory formation (Blumenfeld & Ranganath, 2007). Indeed, neuroimaging studies have correlated dysfunctional frontal and cerebellar networks with alterations in cognitive function in PM carriers with FXTAS. Significant grey matter loss in the left inferior frontal cortex and anterior cingulate cortex in men with FXTAS has been associated with poor working memory performance (Hashimoto, Javan, Tassone, Hagerman, & Rivera, 2011). The anterior cingulate cortex appears to be important for executive function and working memory tasks that require attention, self-monitoring of performance, and cognitive effort (Paus, 2001). In addition, abnormal fronto-parietal attentional network dynamics appear to underlie some of the executive function deficits seen in FXTAS (Yang et al., 2013). Interestingly, frontal assessment battery scores were correlated with hyperintensities in the splenium of the corpus callosum in men and women with FXTAS (Apartis, Blancher, & Meissner, 2012). Significant reductions in connectivity of the superior and middle cerebellar peduncles (MCP), representing the outflow and inflow connections between the cerebellum and cerebral cortex, respectively, has been found in men with FXTAS (Hashimoto, Srivastava, Tassone, Hagerman, & Rivera, 2011). A recent neuroimaging study found that reduced functional connectivity in the MCP and the genu of the corpus callosum were associated with lower executive function and information processing speed in PM carriers with and without FXTAS (Filley et al., 2015). Lower microstructural integrity in the splenium of the corpus callosum was also correlated with reduced information processing speed. Since the MCP contains fronto-pontocerebellar fibers, its reduced connectivity supports the suggestion that the dysexecutive syndrome in FXTAS is mediated, at least in part, by abnormal fronto-cerebellar connections (J. Grigsby et al., 2014).
The executive dysfunction phenotype in FXTAS appears to be similar to that observed in many movement disorders, especially those that overlap with FXTAS phenomenology, including ET, PD, the SCAs, MSA, and PSP. These overlapping, non-motor symptoms make the differential diagnosis difficult, but information obtained from their severity and prevalence might help distinguish patients for placement into the appropriate diagnostic classification.
B. Global Cognition, Memory, MCI, and Dementia
Verbal and non-verbal learning and memory deficits including delayed recall (Moore et al., 2004; Yang et al., 2014) and reduced processing speed (J. Grigsby et al., 2007) have been reported in men with FXTAS. Older men with FXTAS have a higher incidence of dementia that may coincide with motor symptom onset or even precede it (Bacalman et al., 2006; Bourgeois et al., 2007; A. L. Seritan et al., 2008; Sevin et al., 2009). The reported frequency of dementia in older men (> 55 years) with FXTAS may be as high as 50% (A. Seritan et al., 2013; A. L. Seritan et al., 2008). The dementia observed in men with FXTAS is suggested to be similar to “white matter dementia” (J. Grigsby et al., 2014), given the similarities in cognitive dysfunction between patients with FXTAS and those with subcortical white matter lesions (Schmahmann, Smith, Eichler, & Filley, 2008). Others, however, have described the dementia in FXTAS to be one of mixed cortical and subcortical dementia (Besterman et al., 2014; A. Seritan et al., 2013; A. L. Seritan et al., 2008), which may be consistent with the global cerebellar and cortical gray matter loss (including the frontal cortex) (Brunberg et al., 2002; R. J. Hagerman et al., 2001; Jacquemont et al., 2003) and high densities of intranuclear inclusions in the hippocampus and cerebral and cerebellar cortex of post mortem FXTAS patients (Greco et al., 2002; Greco et al., 2006; Greco et al., 2008; Tassone et al., 2012), as well as subcortical white matter lesions (R. J. Hagerman et al., 2001; Jacquemont et al., 2003) seen in FXTAS. Additionally, the report of correlations between reduced IQ scores and reductions in cortical, cerebellar and hippocampal volume in men with FXTAS (Cohen et al., 2006) supports the role for these areas in the cognitive decline seen in the disease. The dementia in FXTAS patients may be different from that in Alzheimer disease (AD) because FXTAS patients have been shown to display less severe explicit memory and attentional deficits that those with MCI or early AD (A. L. Seritan et al., 2008; Yang et al., 2014). However, working memory and verbal fluency and language deficits may be similar in demented FXTAS patients and those with AD (A. L. Seritan et al., 2008).
Although the prevalence and severity of FXTAS symptoms in PM carrier women is significantly lower than that in men (Coffey et al., 2008; R. J. Hagerman et al., 2001; Jacquemont et al., 2003; M. A. Leehey, 2009), dementia has been reported in a small number of women with FXTAS (Al-Hinti, Nagan, & Harik, 2007; R. J. Hagerman et al., 2004; Karmon & Gadoth, 2008; L. Rodriguez-Revenga et al., 2010; A. L. Seritan et al., 2008; Tassone et al., 2012; Yang et al., 2014) and in a woman carrier who had parkinsonism, but no action tremor or cerebellar ataxia (Yachnis, Roth, & Heilman, 2010). In one specific case series, dementia was reported in four out of eight women with FXTAS suggesting the incidence might be higher than previously thought (Tassone et al., 2012). Post mortem neuropathological studies of these FXTAS women demonstrated increased intranuclear inclusions in the frontal and superior temporal gyrus and hippocampus, which was similar to the findings previously reported in men with FXTAS (Greco et al., 2002; Greco et al., 2006). However, three of the women with dementia also had cortical amyloid plaques and neurofibrillary tangles suggesting they may have had co-occurring AD, while the fourth had cortical Lewy bodies typically seen in Lewy body dementia. More studies examining the incidence and type of dementia in PM carrier women are needed. In addition, the examination and establishment of criteria for MCI versus dementia in FXTAS has not been rigorously examined like the PD criteria discussed below. Prospective studies regarding the age-dependent prevalence of MCI and dementia and rates of conversion in FXTAS are needed.
Intelligence quotient scores have been reported to be in the normal range in non-demented FXTAS patients (J. Grigsby et al., 2008; Loesch, Churchyard, & Brotchie, 2005) but global cognition is frequently impaired (J. Grigsby et al., 2007; J. Grigsby et al., 2008). Reduced cognitive event-related potentials (ERP) using verbal learning tasks as electrophysiological indices of verbal memory skills have been found in patients with mild FXTAS (Olichney et al., 2010). However, the exact pattern of ERP findings was found to be different from that in patients with MCI or early AD (Olichney et al., 2008; Olichney et al., 2013). Specifically, men and women with FXTAS showed relative sparing of memory encoding and recognition processes, while both groups had similar indices of poor semantic and word repetition priming (Yang et al., 2014). In general, verbal and implicit memory as determined by ERPs appears to be intact in women with FXTAS (Yang et al., 2014).
C. Other Cognitive Functions: Language and Visuospatial Processing
General expressive and receptive language appear to be intact in men with FXTAS (Brega et al., 2008; J. Grigsby et al., 2008), although mild dysnomia (J. Grigsby et al., 2006; J. Grigsby et al., 2008) has been reported. Visuospatial deficits have been reported in men with FXTAS (J. Grigsby, Brega, & Leehey, 2007; J. Grigsby et al., 2008). Specifically, men with FXTAS were found to perform significantly lower than controls on the Block Design subtest of the Wechsler Adult Intelligence Scale-Third Edition (WAIS-III).
D. Neuropsychiatric Disturbances
Studies suggest that PM carriers men and woman with FXTAS have high levels of depression, anxiety, and social phobia (Bourgeois et al., 2007; Bourgeois et al., 2011). Specifically, the lifetime prevalence rates of mood (65%) and anxiety (52%) disorders is significantly higher in FXTAS than in the general population (Bourgeois et al., 2011). The incidence of a major depressive disorder specifically has been reported to be 43.5%. It has been suggested that some of these affective symptoms in women may be impacted by the stress of raising a child with FXS (Hunter, Sherman, Grigsby, Kogan, & Cornish, 2012). The amygdala, hippocampus and medial prefrontal cortex all appear to play a role in learning and expressing fear and anxiety (Tovote, Fadok, & Luthi, 2015). One report found an association between reduced right hippocampal volume and anxiety in women with FXTAS and paranoid symptoms in men with FXTAS (P. E. Adams et al., 2010). Imaging studies have demonstrated significant grey matter loss in the amygdala and limbic cortical regions in FXTAS patients (Hashimoto, Javan et al., 2011) but other groups have not found reductions in amygdala volume in FXTAS (Selmeczy et al., 2011). Reduced left amygdala volume has been associated with depression and obsessive-compulsive behaviors in FXTAS patients (Hashimoto, Javan et al., 2011). Reduced connectivity in the fornix and stria terminalis previously reported in men with FXTAS (Hashimoto, Srivastava et al., 2011) could possibly be associated with the increased depression and anxiety observed in these patients. Similarly, the reduced amygdala volume reported in men with FXTAS (Hashimoto, Javan et al., 2011) and the reduced amygdala activation when viewing fearful faces seen in some PM carrier men (Hessl et al., 2007) may be linked to the lack of affect, apathy, (Bacalman et al., 2006), and/or social phobia (17.4% lifetime prevalence) (Bourgeois et al., 2011)shown by some PM carriers with FXTAS. However, frontal lobe deficits could also contribute to the apathy seen in FXTAS as this had been shown in patients with various types of frontal lobe degeneration (Barrash, Tranel, & Anderson, 2000; Chow, 2000; R. Levy & Dubois, 2006; R. Levy & Dubois, 2006; Niedermeyer, 1998). More research is needed on the prevalence of apathy in FXTAS because existing studies were done with very low subject numbers.
In general, psychotic symptoms are rare in FXTAS (A. Seritan et al., 2013). However, visual hallucinations have been reported in one woman PM carrier who had an atypical presentation with rapidly progressing dementia followed by hallucinations and paranoid delusions, loss of expressive language and then parkinsonian motor symptoms (Yachnis et al., 2010). Another case series of four PM carrier sisters reported hallucinations, delusions and psychosis in one sister with FXTAS (D. A. Hall et al., 2016).
Essential Tremor (ET)
ET is one of the most comment adult movement disorders and is characterized by symmetrical action tremor in the upper limbs and less commonly the head, face, jaw, voice, tongue, trunk, and lower limbs (Benito-Leon & Louis, 2011). Recently, ET has been categorized into subtypes based on age of onset: hereditary and sporadic ET, both with onset prior to age 65, and senile ET with onset over age 65 (G. Deuschl & Elble, 2009). ET patients that develop symptoms later in life are most likely to be confused with FXTAS patients who typically develop symptoms after the age of 50. Many investigators dispute the notion that ET is benign because accumulating evidence over the past decade suggests that it is a heterogeneous, progressive neurodegenerative disorder with cognitive and neuropsychological impairments including dementia, depression, and changes in personality (Benito-Leon & Louis, 2011; Jhunjhunwala & Pal, 2014). These cognitive profiles overlap significantly with FXTAS. While there are few reports of neuropathological changes in ET, those that exist report significant cerebellar atrophy, axonal swelling (also termed torpedo formation), loss of Purkinje cells, Bergmann gliosis, cortical gray matter atrophy, and ubiquinated, intranuclear inclusions in the cerebral cortex, hippocampus and Purkinje cells (E. D. Louis et al., 2007; E. D. Louis et al., 2012). Strikingly, all these neuropathological findings have been reported in FXTAS (Greco et al., 2002; Greco et al., 2006; Greco et al., 2008; Tassone et al., 2012).
A. Executive Function
ET may be associated with executive function deficits including attention, verbal memory and working memory, and verbal fluency impairments (Benito-Leon & Louis, 2006; Lombardi, Woolston, Roberts, & Gross, 2001; Sinoff & Badarny, 2014; Troster et al., 2002). These deficits were originally reported to be mild but new studies are emerging which demonstrate global cognitive function deficits that are associated with greater functional disability (E. D. Louis, Benito-Leon, Vega-Quiroga, Bermejo-Pareja, & Neurological Disorders in Central Spain (NEDICES) Study Group, 2010a). Moreover, the constellation of non-motor features of ET may occur in a prodromal phase (E. D. Louis, 2015), which is similarly seen in PM carriers. These executive function deficits are thought to be of the cortico-frontal type suggesting pathology in the cerebello-thalamic-frontal regions and interconnecting pathways (Walterfang & van de Warrenburg, 2014). This has been supported by: 1) diffusion tensor imaging (DTI) studies showing significant correlations between executive function test scores and integrity in the frontal white matter, cingulum, inferior superior longitudinal and uncinate fasciculi, anterior thalamic radiations, and posterior lobe of the cerebellum in ET patients (Bhalsing et al., 2015) and 2) grey matter loss in the cerebellum, medial frontal, anterior cingulate, and insular cortices which correlated significantly with neurocognitive and neuropsychological functioning (Bhalsing et al., 2014).
B. Global Cognition, Memory, MCI, and Dementia
Recently, the prevalence of MCI in middle aged ET patients (mean age 56 years) was shown to be as high as 69% with 8% converting to dementia within 2 years, while another 25% without MCI converted to dementia (Sinoff & Badarny, 2014). Previously the total incidence of both MCI and dementia in ET was reported to between 20–25% (Benito-Leon & Louis, 2011; Benito-Leon, Louis, Mitchell, & Bermejo-Pareja, 2011; Thawani, Schupf, & Louis, 2009). Moreover, a review of 18 ET studies demonstrated that those with elderly-onset ET (> 65 years) were 57% more likely to have MCI (Benito-Leon et al., 2011) and 70% more likely to have dementia than controls (Benito-Leon et al., 2011; Romero, Benito-Leon, & Bermejo-Pareja, 2012), suggesting an age-related neurodegenerative process. A prospective study of 135 non-demented patients with ET compared to over 2,000 controls showed a seven times faster rate of cognitive decline in ET patients over a three year period after adjusting for age and education (E. D. Louis, Benito-Leon, Vega-Quiroga, Bermejo-Pareja, & Neurological Disorders in Central Spain (NEDICES) Study Group, 2010b). Prospective studies regarding the age-dependent prevalence of MCI and dementia and rates of conversion in FXTAS are needed.
C. Other Cognitive Functions: Language and Visuospatial Processing
Reduced visuospatial functions (Sahin et al., 2006; Troster et al., 2002) and naming difficulties (Lombardi et al., 2001) have also been reported in ET patients. However, there are no reports demonstrating specific language deficits in ET.
D. Neuropsychiatric Disturbances
Depression and anxiety are now being recognized and studied in greater detail in ET (E. D. Louis, 2015). The prevalence of depression is ~18 % and that of anxiety is 25% (Sinoff & Badarny, 2014). A recent study suggested that higher rates of depression rather than the tremor severity in ET patients is associated with lower quality of life ratings (E. D. Louis, Huey, Gerbin, & Viner, 2012b). In fact, certain groups have suggested that the association of higher self-reported ratings of depression is associated with increased risk for ET, suggesting this may be a prodromal, non-motor feature of the disorder (E. D. Louis, Benito-Leon, Bermejo-Pareja, & Neurological Disorders in Central Spain (NEDICES) Study Group, 2007). Increased apathy is also seen in some ET patients, which appears to be independent of depressive symptoms and may occur in the prodromal phase (E. D. Louis, Huey, Gerbin, & Viner, 2012a). Increased apathy has also been reported in FXTAS (Bacalman et al., 2006). The presence of hallucinations and/or psychosis has not been reported in patients with ET.
E. Comparison Between FXTAS and ET
Studies suggest that executive function deficits may be milder and less prevalent in ET than FXTAS. However, the incidence of MCI is reportedly high in ET, but it is difficult to compare this data to FXTAS because to date MCI has not been specifically studied as a distinct entity in FXTAS. Studies of dementia indicate that its incidence may be lower in those with younger onset ET than in FXTAS but similar to those with late onset or senile ET (> 65 years), although more studies with aged matched populations are need to confirm this statement. Mild dysnomia occurs in both FXTAS and ET as does visuospatial processing difficulties. Rates of anxiety and depression appear to be much higher in FXTAS than in ET. Hallucinations are very rare in FXTAS and have not been reported in ET. Overall, the prevalence of executive function deficits, dementia, and anxiety and depression appear to be much higher in FXTAS than in typical, younger onset ET and examining these cognitive and psychiatric profiles may be helpful in distinguishing these two disorders.
Parkinson Disease (PD)
A. Executive Function
The cognitive phenotype in prodromal, early, and late PD is characterized by executive function deficits including those in the domains of attention and working memory (C. A. Antoniades, Demeyere, Kennard, Humphreys, & Hu, 2015; Goldman, Williams-Gray, Barker, Duda, & Galvin, 2014; Lanni et al., 2014; Ohta et al., 2014; Siepel et al., 2014; Weintraub et al., 2015). Numerous studies have also shown reduced verbal fluency in PD patients compared to healthy controls (Dadgar, Khatoonabadi, & Bakhtiyari, 2013; Pettit, McCarthy, Davenport, & Abrahams, 2013; Siepel et al., 2014; Weintraub et al., 2015), although not all groups have found this deficit in their cohorts (C. A. Antoniades et al., 2015; Lanni et al., 2014). Furthermore, it has been observed that the level and type of verbal fluency deficits are dependent on PD disease severity (Koerts et al., 2013). The APOE ε4 allele was also associated with impaired semantic verbal fluency in 645 PD patients without dementia (Mata et al., 2014). As noted above, all of these executive function impairments have been observed in PM carriers with and without FXTAS (Besterman et al., 2014; Birch et al., 2014; J. Grigsby et al., 2014; Wheeler et al., 2014), which could possibly represent prodromal markers. Significant associations between white matter integrity in the prefrontal cortex and executive functioning have been found in PD patients (Auning et al., 2014), similar to that described above in FXTAS. In PD, attentional deficits may result from low dopamine levels in the frontal cortex and abnormal dopaminergic frontal-striatal networks (Fallon, Hampshire, Williams-Gray, Barker, & Owen, 2013; Fallon, Williams-Gray, Barker, Owen, & Hampshire, 2013). It was previously thought that executive function deficits in PD were predictive for the development of dementia (Janvin, Aarsland, & Larsen, 2005; G. Levy et al., 2002). However, a recent ten year follow up study revealed that this was not the case and instead semantic memory and visuospatial processing deficits were predictive for later dementia (Williams-Gray et al., 2013).
B. Global Cognition, Memory, MCI, and Dementia
MCI, especially in the memory domain, may be as high as 42.5% in newly diagnosed PD patients (Yarnall et al., 2014), and higher rates of depression were also reported in this same group. Previous reports noted a lower prevalence of MCI (~ 19–36%) in early PD (Aarsland et al., 2010; I. Litvan et al., 2011), but this was before the adoption of a recently revised diagnostic criteria for MCI in PD (I. Litvan et al., 2012). Presently, the definition of MCI in PD includes executive function deficits as well as language, working memory, or visuospatial deficits. In FXTAS, this diagnostic distinction has not been established. Lower episodic memory, visuospatial function, semantic fluency and mental flexibility in PD patients with MCI is associated with greater conversion to dementia (Hobson & Meara, 2015). Dementia develops in up to 46–82% of PD patients that live 10 (Williams-Gray et al., 2013) to 20 years (Hely, Reid, Adena, Halliday, & Morris, 2008) after the initial diagnosis, and MCI is a predictor for its occurrence (Aarsland, Tandberg, Larsen, & Cummings, 1996; Aarsland et al., 2010). This is much higher than the prevalence of dementia in FXTAS, which ranges from 37–50% in men with FXTAS over the age of 55 (A. Seritan et al., 2013; A. L. Seritan et al., 2008). The dementia in PD may be of either the fronto-subcortical or cortical/hippocampal type (Janvin et al., 2006). It is presently thought that these cognitive deficits are due to Lewy body development, AD-like pathology, or dysfunction in non-dopaminergic mechanisms (Goldman et al., 2014) which has not been fully researched in the dementia seen in FXTAS.
C. Other Cognitive Functions: Language and Visuospatial Processing
Some studies have reported visuospatial impairments in PD (Tang et al., 2016; Williams-Gray et al., 2009; Williams-Gray et al., 2013), whereas others have not (Ohta et al., 2014). However, PD patients who carry the glucocerebrosidase (GBA) mutation or the E326K polymorphism within the GBA gene do show a significant reduction in visuospatial abilities (Mata et al., 2014). One study showed language/praxis deficits in 50% and visuospatial/constructional deficits in 46% of 26 PD-MCI patients compared to 54 PD patients without MCI (Pfeiffer, Løkkegaard, Zoetmulder, Friberg, & Werdelin, 2014). PD-MCI patients scored significantly lower than PD patients with normal cognition in domains of language, verbal fluency, and visuospatial function (Karrasch, Laatu, Martikainen, & Marttila, 2013), although another study found no significant differences in visuospatial abilities between these groups (Noh et al., 2014).
D. Neuropsychiatric Disturbances
Reports of the prevalence of depressive symptoms in PD patients range from 37–70% (Aarsland et al., 1999; Aarsland et al., 2009; Goldman et al., 2014) and can develop in the premotor stage (Aarsland, Pahlhagen, Ballard, Ehrt, & Svenningsson, 2011). Depression has been reported to be significantly higher (up to 58%) in those with dementia (Aarsland et al., 1996; Karantzoulis & Galvin, 2013; Klatka, Louis, & Schiffer, 1996). Apathy is also a common symptom (27–40%) as is anxiety (Aarsland et al., 2009; Aarsland, Marsh, & Schrag, 2009). The lifetime prevalence of anxiety in PD has been reported to be between 20 and 49% (Gallagher & Schrag, 2012). Anxiety may co-occur with depression in 40% of PD patients (Aarsland et al., 1999). Clinically significant neuropsychiatric symptoms were found to be associated with more severe parkinsonian symptoms (Aarsland et al., 2009) and impaired quality of life for patients and their family members (Aarsland & Kramberger, 2015).
Visual hallucinations are relatively common in PD patients (Aarsland et al., 1999; Aarsland, Larsen, Cummins, & Laake, 1999; Bertram & Williams, 2012) and were previously thought to be due to chronic dopaminergic therapy, but are now thought to be secondary to neuronal loss and Lewy body pathology in ventral and temporal regions of the brain (D. R. Williams & Lees, 2005). These hallucinations have recently been reported to occur in the premotor stage in 33% of PD patients (Pagonabarraga et al., 2015). They are present in approximately 30 to 42% of diagnosed patients, but prevalence rates have been shown to be as high as 50% in a 5 year longitudinal study (Zhu, van Hilten, Putter, & Marinus, 2013). Delusions have been reported in ~15 to 20% of PD patients and are even more common in demented PD patients, with prevalence rates up to 29% (Aarsland et al., 1999; Aarsland, Larsen, Cummins et al., 1999). Recently, the prevalence of psychosis in PD patients that live 20 or more years after initial diagnosis was reported to be as high as 70% (Levin, Hasan, & Hoglinger, 2015). Both visual hallucinations and delusional thoughts are significantly associated with age, stage and severity of the disease, and the presence and severity of cognitive dysfunction and depression (Aarsland, Larsen, Cummins et al., 1999).
E. Comparison Between FXTAS and PD
Cognitive deficits in the domains of executive function (which are included in the criteria for MCI in PD) are reportedly higher in PD than in FXTAS. Studies suggest that the incidence of dementia is also significantly higher in patients with PD than in men with FXTAS. In addition, the language deficits in PD patients with MCI have prevalence rates of up to 50%, while only mild dysnomia has been reported in FXTAS. There are conflicting reports of visuospatial processing deficits in PD patients except in those with MCI, these deficits do exist in FXTAS. Depression rates may be more prevalent in PD while anxiety is much higher in FXTAS than in PD. Hallucinations and psychosis are very rare in FXTAS but have very high prevalence rates in PD, especially in advanced disease stages. Thus, the presence or absence of language deficits and hallucinations and psychosis in patients presenting with a parkinsonian like movement disorder might help distinguish FXTAS from PD.
Spinocerebellar Ataxias (SCAs)
The spinocerebellar ataxias (SCAs) are a heterogeneous group of autosomal dominant genetic disorders characterized by progressive neurodegeneration of the cerebellum and its connections (Durr, 2010). Besides cerebellar ataxia and kinetic tremor, other neurological signs may include cognitive impairment, peripheral neuropathy, ophthalmoplegia, pyramidal, and extrapyramidal signs (Manto & Lorivel, 2011). Although over 40 SCAs have been identified, the most common SCAs are caused by CAG repeat expansions in a variety of genes. There are genetic similarities in many cases of SCAs where the underlying etiology is a polyglutamine trinucleotide repeat expansion and FXTAS. While expansions account for about 45% of SCA cases, up to 50% of SCAs are presently of unknown genetic etiology. Neurodegeneration in the SCAs typically includes the cerebellar cortex, Purkinje cells, dentate and inferior olivary nuclei, the pons, and their interconnections (C. Brenneis, Bösch, Schocke, Wenning, & Poewe, 2003; Estrada, Galarraga, Orozco, Nodarse, & Auburger, 1999; O’Hearn et al., 2015). Atrophy in several regions of the frontal lobe have also been reported (C. Brenneis et al., 2003; Estrada et al., 1999). Intra-nuclear inclusions containing polyglutamine are common in many of the degenerative brain regions involved in the SCAs (Legros & Manto, 1999), a finding that is similar to those found in portmortem brains of FXTAS patients (Greco et al., 2002; Greco et al., 2006; Greco et al., 2008; Tassone et al., 2012). In this review, we will limit our discussion to SCAs 1–3, 6, 8, 14, 17, 19, and dentatorubropallidoluysian atrophy (DRPLA) due to the presence of cerebellar ataxia and cognitive impairment documented in these diseases which overlap with the findings in FXTAS.
A. Executive Function
In SCA1, cognition is relatively spared early in the disease, but executive dysfunction and impaired verbal memory may develop in later stages (Burk et al., 2001; Bürk et al., 2003). SCA2 has a preclinical phase characterized by executive function deficits and imaging studies have demonstrated reduced functional connectivity between the cerebellum and frontal-parietal cortices which correlated with patient’s neuropsychological deficits (Hernandez-Castillo et al., 2015). As noted above, FXTAS may also have a similar stage of executive function deficits prior to the development of motor signs (Besterman et al., 2014; Birch et al., 2014; J. Grigsby et al., 2014; Wheeler et al., 2014). SCA3, also known as Machado-Joseph disease, has quite robust similarities in the cognitive and neuropsychiatric disturbances seen in FXTAS. These include executive dysfunction (Braga-Neto, Pedroso et al., 2012; Radvany, Camargo, Costa, Fonseca, & Nascimento, 1993; Zawacki, Grace, Friedman, & Sudarsky, 2002), abnormal visual attention and visual processing (Maruff et al., 1996), verbal fluency, and verbal and visual memory deficits (Braga-Neto, Pedroso et al., 2012; Braga-Neto, Pedroso, Barsottini, & Schmahmann, 2015; Y. Kawai et al., 2004). Like FXTAS patients, SCA3 patients may have global cortical atrophy in the frontal, temporal, parietal, occipital, and limbic lobes, but white matter atrophy is absent except in the cerebellum (D’Abreu et al., 2012). In SCA8 (Lilja, Hamalainen, Kaitaranta, & Rinne, 2005; Torrens et al., 2008), SCA14 (Klebe et al., 2005), and SCA19 (Schelhaas et al., 2003) there is relatively frequent loss of executive function which may be similar to the frontal-executive dysfunction seen in FXTAS.
Numerous studies have found that SCA patients, like some patients with FXTAS, have verbal fluency deficits as a form of impairment in executive function (Braga-Neto, Pedroso et al., 2012; Bürk et al., 2003; Fancellu et al., 2013; Feng et al., 2014; Y. Kawai et al., 2008a; Y. Kawai et al., 2004; Orsi et al., 2011; Rodríguez-Labrada et al., 2014; Suenaga et al., 2008; Zawacki et al., 2002). These include SCA1, SCA2, SCA3, and SCA6. However, two studies did not find differences in verbal fluency between SCA3 and SCA6 patients and healthy controls (Globas et al., 2003; Lopes et al., 2013).
B. Global Cognition, Memory, Mild Cognitive Impairment (MCI) and Dementia
In contrast to FXTAS, dementia is rarely seen in SCA3 (Braga-Neto, Pedroso et al., 2012; Y. Kawai et al., 2004; Maruff et al., 1996; Radvany et al., 1993; Zawacki et al., 2002), which could be partially explained by the lack of subcortical white matter loss despite widespread cortical atrophy seen in SCA3 patients (D’Abreu et al 2012). DRPLA is another spinocerebellar degenerative disease caused by a CAG trinucleotide expansion in the atrophin 1 gene (I. Kanazawa, 1998). Patients with the late onset (> 40 years of age) form of the disease tend to present with cerebellar ataxia, choreoathetosis, delusions, and dementia (Naito & Oyanagi, 1982; Tsuji, 2012; Vale et al., 2010). Mild to moderate dementia has also been reported in SCA1 (Donato, Mariotti, & Taroni, 2012), SCA17 (Koutsis et al., 2014; Toyoshima, Onodera, Yamada, Tsuji, & Takahashi, 1993; Zuhlke & Burk, 2007), and to a variable extent in SCA2 (Durr et al., 1995) and SCA12 patients (Dohlinger, Hauser, Borkert, Luft, & Schulz, 2008).
C. Other Cognitive Functions: Language and Visuospatial Processing
Language impairments, especially in writing and comprehension, have been reported in patients with SCA6(van Gaalen et al., 2014). However, language impairments were not observed in a study of SCA3 patients (Zawacki et al., 2002). Visuospatial deficits have been reported in some of the SCAs including SCA1, SCA2 and SCA3 (Braga-Neto et al., 2012; Braga-Neto, Pedroso et al., 2012; Fancellu et al., 2013; Feng et al., 2014; Y. Kawai et al., 2004; Orsi et al., 2011). However, other studies found no differences in visuospatial processing between SCA1, SCA2, SCA3, and SCA6 patients and controls (Bürk et al., 2003; Garrard, Martin, Giunti, & Cipolotti, 2008; Globas et al., 2003; Y. Kawai et al., 2008a; Lopes et al., 2013).
D. Neuropsychiatric Disturbances
Depression and anxiety are common psychiatric features in the SCAs (Braga-Neto, Pedroso et al., 2012; Braga-Neto, Pedroso et al., 2012; Cecchin et al., 2007; Klinke et al., 2010; Lopes et al., 2013; McMurtray, Clark, Flood, Perlman, & Mendez, 2006; O’Hearn et al., 2015; Pedroso et al., 2013; Saute et al., 2010; Schmitz-Hubsch et al., 2008; Schmitz-Hubsch et al., 2011; Silva, Marques, Lourenço, Hallak, & Osório, 2015), with an overall depression prevalence rate of 17–26% (Lo et al., 2016; Schmitz-Hubsch et al., 2011). However, some studies have not observed increases in depression or anxiety in SCA2 and SCA3 patients (Feng et al., 2014; Roeske et al., 2013), and one study reported increased anxiety but not depression in SCA6 (Suenaga et al., 2008). Increased apathy has also been reported by the caregivers of SCA3 patients (Zawacki et al., 2002). Suicidal ideation appears to be more frequently observed in SCA3 patients (65% prevalence) than in the normal population (Lo et al., 2016). Mood disturbances, including depression, anxiety, irritability and changes in personality are also common in SCA8(Torrens et al., 2008).
E. Comparison Between FXTAS and SCAs
Executive function deficits appear to be relatively common in many of the SCAs but prevalence rates have not been detailed in the literature which is also the case in FXTAS. Thus, it is difficult at present to compare these cognitive deficits between SCAs and FXTAS. MCI and dementia are rare in SCA 3 but have been reported in late onset DRPLA, SCA1, 2 and 17. Visuospatial processing deficits exist in SCAs 1 to 3, similar to that seen in FXTAS. Depression has been reported in ~ 20% of patients with SCA but this is much lower than that in FXTAS. Prevalence rates of anxiety in a few of the SCAs have been reported and appear to be of much lower magnitude than in FXTAS. Like FXTAS, hallucinations and psychosis are rare in the majority of the SCAs. These findings suggest that low rates of anxiety and depression in many of the SCAs may be helpful in distinguishing patients with these disorders from FXTAS.
Multiple System Atrophy (MSA)
Multiple system atrophy (MSA) is an idiopathic, adult onset, progressive synucleinopathy characterized by parkinsonism, cerebellar ataxia, autonomic failure, and corticospinal signs with neurodegeneration in striatonigral, olivopontocerebellar and autonomic brain regions (Gilman et al., 2008; Konagaya, Sakai, Matsuoka, Konagaya, & Hashizume, 1999; Konagaya, Konagaya, Sakai, Matsuoka, & Hashizume, 2002). Two subtypes of MSA exist: a rigid parkinsonism (MSA-P) type and a cerebellar type (MSA-C) characterized by progressive cerebellar ataxia (Stankovic et al., 2014). Cognitive impairment in the form of executive function deficits is common in both types of MSA (Stankovic et al., 2014). The cognitive deficits in MSA appear to overlap significantly with both FXTAS and parkinsonian disorders. Additionally, these deficits may precede the motor impairments in MSA (Kitayama, Wada-Isoe, Irizawa, & Nakashima, 2009), a finding that may also exist in FXTAS (Besterman et al., 2014; Birch et al., 2014; J. Grigsby et al., 2014; Wheeler et al., 2014). Imaging and neuropathological findings suggest that cognitive impairments in MSA originate from loss of striatal connections to the frontal cortex, with additional contributions from cortical and cerebellar and subcortical white matter degeneration (Stankovic et al., 2014).
A. Executive Function
Executive function deficits occur in up to 54% of MSA patients (Auzou et al., 2015; Siri et al., 2013) and therefore may be as prevalent as in FXTAS. This will require further study as the prevalence rates have not yet been reported in FXTAS, although the literature suggests it is a prominent finding and is a minor diagnostic criterion. The specific deficits are similar in both disorders and include problems with working memory and attention (Balas, Balash, Giladi, & Gurevich, 2010; J. S. Kim et al., 2015), problem solving, response inhibition (Dujardin, Defebvre, Krystkowiak, Degreef, & Destee, 2003; Kao et al., 2009), and verbal fluency (Balas et al., 2010; Burk, Daum, & Rub, 2006; Hong, Song, Lee, Sohn, & Lee, 2011; J. S. Kim et al., 2015; Walterfang & van de Warrenburg, 2014). These impairments have been attributed to degeneration in the frontal and temporal cortex, cerebellum, striatum, and thalamic structures and their interconnections (J. S. Kim et al., 2015). Patients with MSA show significant cortical thinning in the fronto-temporo-parietal regions with greater atrophy in frontal areas (Konagaya et al., 2002) and atrophy of the thalamus and cerebellum (M. J. Lee et al., 2015). The severity of atrophic changes in the bilateral striatum, thalamus, cerebellum, left pericalcarine gyrus, and the neocortex in general were significantly correlated with attentional, executive, and visuospatial dysfunctions in MSA patients (J. S. Kim et al., 2015; M. J. Lee et al., 2015). There is some evidence that basal ganglia atrophy is one of the earliest sign in MSA, which then drives widespread cortical atrophy (C. Brenneis et al., 2007). However, other groups have shown that hypometabolism begins in the cerebellum and frontal cortex and then progresses to the caudate nucleus and other cortical areas in those with a mixed type of MSA (Lyoo et al., 2008).
B. Global Cognition, Memory, MCI, and Dementia
MSA patients frequently have impairments in encoding, verbal learning, long term memory, immediate and delayed recall, and recognition (Balas et al., 2010; Brown et al., 2010; Burk et al., 2006; Hong et al., 2011; H. J. Kim et al., 2013; J. S. Kim et al., 2015; Lyoo et al., 2008; Siri et al., 2013). The degree of motor impairment in MSA predicts the severity of cognitive deficits (Brown et al., 2010; Kawamura et al., 2010). Dementia is not included in the primary diagnostic criteria for MSA (Gilman et al., 2008), and severe dementia is presently an exclusion criteria, but its prevalence in the disorder is reported to be as high as 30% (Brown et al., 2010; Kitayama et al., 2009). In contrast, memory and executive function deficits are considered to be clinical criteria for establishing a FXTAS diagnosis in PM carriers (Jacquemont et al., 2003). There also have been reports of patients presenting with dementia who later met criteria for MSA (Jang, Lee, Jang, Kim, & Chung, 2012). Some have suggested that if MSA were not more rapidly progressive than PD, the rates of dementia might be the same in both disorders (80%) (Stankovic et al., 2014). Neuroimaging findings in MSA patients with dementia include reduced cortical thickness in the precuneus/cuneus, uncus, and posterior cingulate cortices compared to those without dementia and controls (J. S. Kim et al., 2013). Cerebellar and prefrontal, limbic and temporal lobe degeneration and atrophy have also been reported in demented MSA patients (J. S. Kim et al., 2013). A longitudinal neuroimaging study in MSA patients that develop dementia reported progressive frontal and temporal lobe degeneration (C. Brenneis et al., 2007). Moreover, neuropathological findings include significant neuronal loss, glial cytoplasmic inclusions and astroglios in the frontal and temporal lobes in MSA patients who were demented at death (Konagaya et al., 1999).
C. Other Cognitive Functions: Language and Visuospatial Processing
Visuospatial impairments (Hong et al., 2011; Y. Kawai et al., 2008b) similar to those seen in FXTAS (J. Grigsby et al., 2008; Yang et al., 2014) have been reported in MCA patients, but others have only found these deficits in demented MSA patients (Brown et al., 2010; H. J. Kim et al., 2013). General expressive and receptive language functions, like that in men and women with FXTAS (Brega et al., 2008; J. Grigsby et al., 2008), appear to be intact in non-demented MSA patients (Kao et al., 2009; Lyoo et al., 2008).
D. Neuropsychiatric Disturbances
Moderate to severe depression has been reported in at least 30% of MSA patients with total rates of depression as high as 85% (Benrud-Larson, Sandroni, Schrag, & Low, 2005; Schrag et al., 2006; Schrag et al., 2010; Siri et al., 2013). Depression severity has been associated with significant reductions in dorsolateral prefrontal cortex glucose metabolism in MSA patients (Herting et al., 2007). Anxiety is also reported to affect 37% of MSA patients (Schrag et al., 2010), and appears more prevalent in MSA-C (Balas et al., 2010). Hallucinations and psychosis are rare in MSA (D. R. Williams, Warren, & Lees, 2008), unlike the much higher frequencies (30–70 %; see above) reported in PD.
E. Comparison Between FXTAS and MSA
The executive function deficits appear to be similar in MSA and FXTAS. However, MSA patients have more significant impairments in memory and recognition than FXTAS patients. Rates of dementia are slightly higher in men with FXTAS than in MSA, but this may be due to a longer lifespan in FXTAS. Depression and anxiety are very common in both FXTAS and MSA while hallucinations and psychosis are rare in both disorders. Due to the high overlap in cognitive profiles for these two disorders, further clinical information is needed to distinguish one from the other.
Progressive Supranuclear Palsy (PSP)
PSP is a sporadic, rapidly progressive neurodegenerative disorder characterized by supranuclear vertical gaze palsy, parkinsonism with symmetrical rigidity and bradykinesia, postural instability, frontal and subcortical dementia, and pseudobulbar palsy (Colosimo, Bak, Bologna, & Berardelli, 2014). PSP is now known to be a tauopathy which causes numerous neurofibrillary tangles to develop in the brainstem and basal ganglia (Josephs, 2015). PSP, like FXTAS, has high phenotypic variability which also results in its frequent misdiagnosis (Respondek et al., 2014; Respondek & Hoglinger, 2015).
A. Executive Function
PSP patients are known to have early and severe deficits in executive functions especially planning, problem solving, abstract reasoning and concept formation (Magherini & Litvan, 2005). Deficits in attention (Bak, Crawford, Hearn, Mathuranath, & Hodges, 2005; Esmonde, Giles, Gibson, & Hodges, 1996; Ghosh, Carpenter, & Rowe, 2013; Grafman, Litvan, Gomez, & Chase, 1990; Kaat, Chiu, Boon, & van Swieten, 2011; Millar, Griffiths, Zermansky, & Burn, 2006), disinhibition(Gerstenecker, Duff, Mast, Litvan, & ENGENE-PSP Study Group, 2013), verbal fluency (Bak et al., 2005; Cotelli et al., 2006; Daniele et al., 2013; Esmonde, Giles, Xuereb, & Hodges, 1996), and working memory (Maher, Smith, & Lees, 1985; Pillon, Dubois, & Agid, 1991) have all been reported in patients with PSP. Severe impairments on tests of verbal fluency, particularly letter fluency, have been reported in patients with PSP (Esmonde, Giles, Gibson et al., 1996). In fact, verbal fluency deficits are included in the supportive criterion for diagnosing PSP (I. Litvan, Agid, & Calne, 1996).
Prevalence rates for executive function deficits in PSP patients are as high as 70 to 90% (Brown et al., 2010; Gerstenecker et al., 2013; Kaat et al., 2011). Therefore, these appear more significant and frequent than in patients with FXTAS or PD (Pillon et al., 1995). Moreover, patients with PSP may be more severely impaired than patients with PD with dementia, dementia with Lewy bodies, and AD on the Dementia Rating Scale Initiation/Perseveration subscale (Rosser & Hodges, 1994), which is a commonly used screening instrument for executive dysfunction (Aarsland et al., 2003). A two year longitudinal study found greater decline in executive functioning and higher rates of conversion to dementia in PSP compared to PD patients (Soliveri et al., 2000).
B. Global Cognition, Memory, MCI, and Dementia
Non-demented PSP patients appear to have normal short term memory and long term recognition (I. Litvan, Grafman, Gomez, & Chase, 1989; van der Hurk & Hodges, 1995), but they can have other memory impairments including those in the domain of verbal learning (I. Litvan et al., 1989), and mild to moderate deficits in processing stored information necessary for recall (Magherini & Litvan, 2005). Patients with PSP eventually develop severe slowness in information processing speed for global cognitive functions (Respondek & Hoglinger, 2015). Impaired learning and memory deficits in demented PSP patients has been postulated to result from degeneration in striato-frontal areas, similar to that seen in PD and Huntington disease (Pillon et al., 1994; Pillon et al., 1995). The general, progressive cognitive decline observed in PSP is thought to be greater than that observed in PD and MSA patients with or without dementia (Magherini & Litvan, 2005; Monza et al., 1998). PSP typically includes frontal and subcortical dementia (Respondek & Hoglinger, 2015), and one report found that approximately 58% of PSP patients develop dementia (Pillon et al., 1991). However, demented PSP patients have been reported to have higher memory subscores than demented PD patients (Aarsland et al., 2003). While the cognition in FXTAS patients has not been compared to that in PSP, it is likely that PSP cognitive deficits may be greater than those seen in FXTAS given the research presented above.
C. Other Cognitive Functions: Language and Visuospatial Processing
PSP patients generally have intact language functions (Magherini & Litvan, 2005), although cases of aphasia, speech apraxia, reduced spontaneous speech initiation, and echolalia have been reported (Esmonde, Giles, Xuereb et al., 1996). There have been several cases reported in the literature of progressive non-fluent aphasia and speech apraxia as a presenting feature of PSP, with later development of the cognitive and motor phenotypic features (Boeve et al., 2003; Esmonde, Giles, Gibson et al., 1996; Mochizuki et al., 2003; Spaccavento, Del Prete, Craca, & Loverre, 2014).
PSP patients frequently have visuospatial deficits (Borroni et al., 2008; Esmonde, Giles, Gibson et al., 1996; Ghosh et al., 2013). In association with these deficits are difficulties in orienting visual attention in the vertical visual field (Rafal, Posner, Friedman, Inhoff, & Bernstein, 1988), which may not be surprising given the presence of vertical gaze palsy seen in 75 to 90% of PSP patients (Esmonde, Giles, Gibson et al., 1996; I. Litvan et al., 1996; Vidailhet et al., 1994).
D. Neuropsychiatric Disturbances
Apathy is a distinguishing behavioral abnormality in patients with PSP, with the first report describing its occurrence in 90% of patients and this was not related to cognitive impairment or disease duration (I. Litvan, Mega, Cummings, & Fairbanks, 1996). In fact, high apathy scores with relatively low anxiety scores (18%) have been found to be helpful in the diagnosis of PSP (I. Litvan, Mega et al., 1996). Depression has been reported be over 50% higher in PSP patients than healthy controls (Bloise et al., 2014; Esmonde, Giles, Gibson et al., 1996; Gerstenecker et al., 2013), and may even precede the motor impairments (W. H. Kim et al., 2009; Quante, Jakob, & Wolf, 2008). Hallucinations, psychosis, and delusions were thought to be rare in PSP (H. F. Chiu, 1995). However, a recent multi-site study of 154 PSP patients reported the incidence of these behavioral abnormalities to range from 5 to 11% of patients. This contrasts to the high prevalence of these disturbances in PD (Aarsland, Litvan, & Larsen, 2001; Magherini & Litvan, 2005).
E. Comparison Between FXTAS and PSP
Executive function deficits and cognitive decline are more severe and prevalent in PSP than FXTAS. The rates of dementia are also higher in PSP. Depression rates appear to be similar in both disorders but apathy is much higher and anxiety is lower in PSP than FXTAS. Hallucinations and psychosis are extremely rare in FXTAS but are now known to be more prevalent in PSP than previously thought. However, there is enough cognitive phenotype overlap between FXTAS and PSP to warrant further clinical examination before making a diagnosis.
II. Motor Phenotypes
We begin this section by reviewing the motor features of FXTAS. We then review these same features in ET, PD, SCA and MSA, and PSP, concluding each section with a summary comparison between each of these other movement disorders and FXTAS. Table 2 contains a summary of these comparisons. Case reports, epidemiological data, and results of imaging studies are included in this review.
Fragile X-Associated Tremor / Ataxia Syndrome (FXTAS)
A. Tremor
Studies have observed tremor in approximately 77% of men with FXTAS (Juncos et al., 2011). The tremor is typically a bilateral postural or kinetic tremor, and although rest tremor may be seen in some patients it is often accompanied by intention tremor (E. Berry-Kravis, Abrams, & Coffey, 2007). Pure resting tremor is rare in FXTAS (Apartis et al., 2012; Baba & Uitti, 2005). The tremor is typically present in the upper extremities, although head tremor has been seen in some cases (Apartis et al., 2012; M. Leehey et al., 2003; Peters et al., 2006); voice tremor has not been reported in FXTAS. Furthermore, FXTAS tremor may or may not respond to alcohol (Gorman, Fairgrieve, Birchall, & Chinnery, 2008; M. Leehey et al., 2003; Peters et al., 2006). Three distinct tremor patterns were identified in a study of 17 FXTAS patients using tremor recordings from a Neuropack device (Apartis et al., 2012). These consisted of an action tremor resembling the tremor of ET in 35% of the patients, a cerebellar intention tremor and postural tremor in 29%, and a unilateral upper limb rest tremor in 12%. The CATSYS system has also been used to quantitatively characterize the tremor in FXTAS (Aguilar, Sigford, & Soontarapornchai, 2008; Juncos et al., 2011; Narcisa, Aguilar, & Nguyen, 2011). A study of 16 men with FXTAS showed increased intention tremor in both hands (Aguilar et al., 2008). Postural hand tremor was not detected in this group. Another study of 23 FXTAS women did find significantly increased postural hand tremor but not intention or writing tremor compared to controls (Narcisa et al., 2011).
B. Cerebellar Ataxia
Cerebellar gait ataxia and progressive loss of motor coordination is one of the primary features of FXTAS (E. Berry-Kravis et al., 2007; Matilla-Dueñas, 2012), seen in 41–66% of patients (Juncos et al., 2011; Niu et al., 2014). Recently our group quantitatively characterized the balance and gait deficits in PM carriers with FXTAS and found abnormalities similar to previous studies in various types of cerebellar disorders (O’Keefe et al., 2015; O’Keefe, Robertson-Dick, Hall, & Berry-Kravis, 2015). Multiple studies have demonstrated significant cerebellar ataxia in men with FXTAS as measured by the International Cooperative Ataxia Rating Scale ICARS (Jacquemont et al., 2003; Loesch et al., 2005; Trouillas, Takayanagi, & Hallet, 1997).
C. Parkinsonism
One common feature of FXTAS is a form of parkinsonism (E. Berry-Kravis et al., 2007) seen in 29–32% of patients (Juncos et al., 2011; Niu et al., 2014), which mimics, and is often indistinguishable from idiopathic PD (D. Hall, Howard, & Hagerman, 2009). However, the parkinsonism in FXTAS may be milder than that of PD with a lower degree of bradykinesia than in typical parkinsonism (E. Berry-Kravis, Lewin, & Wuu, 2003). Likewise, the prevalence of a parkinsonian gait pattern is low in FXTAS (Cilia, Kraff, & Canesi, 2009; D. Hall et al., 2009; Yachnis et al., 2010).
A screening study in 56 FXTAS patients revealed PD was the most frequent initial diagnosis (D. Hall, Berry-Kravis, & Jacquemont, 2005) and it has been suggested that there may be a PD phenotype of the disease present in a subset of patients (D. Hall, Tassone, Klepitskaya, & Leehey, 2012). Furthermore, the parkinsonism in PM carriers may show a good response to levodopa, making it difficult to recognize as atypical (Cilia et al., 2009; D. Hall et al., 2009; Hedrich, Pramstaller, & Stübke, 2005). Evidence also suggests that persons with lower size FMR1 premutation expansions and gray zone alleles (45–54 CGG repeats) may display a parkinsonian phenotype (D. Hall, Berry-Kravis, & Zhang, 2011; D. Hall & O’keefe, 2012; Trost et al., 2014).
D. Eye Movement Abnormalities
Specific oculomotor deficits have been understudied in FXTAS. However, in one case study, abnormalities including dysmetric saccades, saccadic pursuits, transient endgaze nystagmus, vertical optokinetic nystagmus, slowed vertical saccades, square wave jerks, and impaired vertical gaze were observed in FXTAS patients (Fraint, Vittal, & Szewka, 2014). However, these patients were described due to their interesting eye movement abnormalities and do not represent the typical FXTAS patient. While abnormal saccades and nystagmus are common in cerebellar disorders (Cogan, Chu, & Reingold, 1982), vertical gaze deficits suggest that a PSP-like phenotype may be present in some individuals with FXTAS. The prevalence of nystagmus and PSP-like abnormalities in the FXTAS population as a whole is unknown and is likely to be low.
Essential Tremor (ET):
A. Tremor
The tremor of ET is typically characterized as postural and kinetic tremor (80 and 25–98% prevalence, respectively) (G. Deuschl, Wenzelburger, Loffler, Raethjen, & Stolze, 2000; Ghika, Kyrozis, Potagas, & Louis, 2015) of the upper extremities that is predominantly bilateral (J. Jankovic, 2002; Zappia, Albanese, & Bruno, 2003). Another core criteria for diagnosing ET is isolated head tremor without dystonia (G. Deuschl, Bain, & Brin, 1998; J. Jankovic, 2002). Approximately 33–89% of ET patients also display an intention tremor suggestive of cerebellar dysfunction (G. Deuschl et al., 2000; Ghika et al., 2015), and the kinetic tremor in ET has been shown to be more severe than the postural tremor using clinical rating scales (E. D. Louis, 2013). Rest tremor in the arms has also been reported in ET, with highly variable prevalence rates ranging from 1–46% (E. Louis, Hernandez, & Michalec, 2015). The tremor in ET is seen less frequently in the head (34–53%), voice (20%), tongue (20%), face/jaw (7–18%), lower extremities (10%), and trunk (5%) (Benito-Leon & Louis, 2006; G. Deuschl & Elble, 2009; Elble, 2000; Whaley, Putzke, Baba, Wszolek, & Uitti, 2007). ET tremor improves with alcohol in 46–96% of cases (Ghika et al., 2015; Hopfner et al., 2015).
B. Cerebellar Ataxia
Studies have found mild gait deficits in ET patients, including abnormalities in tandem gait in approximately half of patients (Hoskovcová et al., 2013; Hubble, Busenbark, Pahwa, Lyons, & Koller, 1997; M. Kronenbuerger et al., 2009; Singer, Sanchez-Ramos, & Weiner, 1994; Stolze, Petersen, Raethjen, Wenzelburger, & Deuschl, 2001). Other studies have noted mild postural instability in ET (Bove, Marinelli, Avanzino, Marchese, & Abbruzzese, 2006; Hoskovcová et al., 2013). However, regular bipedal gait in ET patients appears to be normal (Stolze et al., 2001).
C. Parkinsonism
There appears to be a possible association between ET and PD, such that individuals with a family history of ET are more likely to develop PD and vice versa (J. Jankovic, Beach, Schwartz, & Contant, 1995; Lang, Kierans, & Blair, 1987). Given the overlap in symptoms and increased prevalence of parkinsonism features in ET, there may be a subset of ET patients who have preclinical PD (Schwartz, Badarny, Gofman, & Hocherman, 1999). One study found that 64% of ET patients displayed a typical parkinsonian phenotype (J. Jankovic et al., 1995), with others specifically observing akinesia or bradykinesia similar to that in PD (Jiménez-Jiménez et al., 2010; Montgomery, Baker, Lyons, & Koller, 2000). There also is some evidence of reduced nigrostriatal function in ET patients (M. S. Lee et al., 1999). However, it is unclear whether the parkinsonism seen in ET is attributable to PD or constitutes some other form of parkinsonism (J. Jankovic, 2002).
D. Eye Movement Abnormalities
Oculomotor abnormalities have been reported in several studies of ET. One study found impairments in smooth pursuit initiation and suppressed vestibulo-ocular reflexes in 41% of ET patients (Helmchen et al., 2003). These findings were significantly greater in patients who had intention tremor versus postural tremor, indicating possible cerebellar dysfunction as the cause of the abnormal eye movements. Other studies reported visuomotor tracking deficits (Schwartz et al., 1999), abnormal eye-head coordination (Trillenberg et al., 2006), and absent or delayed eye blink reflexes (M. Kronenbuerger, Gerwig, Brol, Block, & Timmann, 2007) in ET patients.
E. Case Reports
There are numerous instances of patients who received an initial diagnosis of ET that were later discovered to be PM carriers, some of whom met clinical diagnostic criteria for FXTAS (Jacquemont et al., 2003). Frequently these individuals remained misdiagnosed for years until symptoms progressed, prompting the consideration of an alternative diagnosis. Seven of these cases have been described in the literature, illustrating the commonalities between FXTAS and ET (Gorman et al., 2008; Ishii, Hosaka, Adachi, Nanba, & Tamaoka, 2010; M. Leehey et al., 2003; Peters et al., 2006; Seixas, Vale, & Jorge, 2011). All were men over 50 years of age, six of whom presented with postural tremor and eight with kinetic tremor. The response to alcohol was varied, with definite improvement in only two cases. Cerebellar gait ataxia was seen in three cases with an additional three demonstrating impaired tandem gait. Five cases showed hyperintensities in the MCP, which has never been described in ET. Four patients had global cortical atrophy and five had cerebellar atrophy, which has been reported in ET. Four patients had a family history of ET, but all seven had a fragile X PM carrier or individual with fragile X syndrome (FXS), a neurodevelopmental disorder caused by the full mutation (>200 CGG repeats), in their families.
F. Epidemiologic Data
Despite multiple case reports of patients with FXTAS presenting with tremor mimicking that of ET, screening of ET populations has surprisingly yielded virtually no cases of the FMR1 premutation (Arocena, Louis, & Tassone, 2004; Clark, Ye, & Liu, 2015; Deng, Le, & Jankovic, 2004; Tan, Zhao, & Puong, 2004). However, the exclusion criteria for these studies might have eliminated some individuals with FXTAS, as they typically exclude subjects with parkinsonism and ataxia. Thus, there may be underestimation of the FMR1 premutation in the ET population.
G. Comparison Between FXTAS and ET
As with the cognitive phenotype, ET patients that develop motor symptoms later in life are most likely to be confused with FXTAS patients due to similar age of symptom onset. The tremor in both FXTAS and ET is typically characterized by postural and kinetic tremor of the upper extremities, although ET tremor can also be seen in the head, voice, tongue, jaw/face, lower extremities, and trunk (Benito-Leon & Louis, 2006; G. Deuschl & Elble, 2009; Elble, 2000; Whaley et al., 2007). Furthermore, the tremor is predominantly bilateral in ET whereas FXTAS tremor may be asymmetrical (Apartis et al., 2012). Rest tremor may be present in both FXTAS and ET but is rarely seen in isolation in either disorder. FXTAS tremor may or may not respond to alcohol (Gorman et al., 2008; M. Leehey et al., 2003; Peters et al., 2006), while it appears to respond more frequently in ET (Ghika et al., 2015; Hopfner et al., 2015). Both disorders may have cerebellar gait ataxia and/or postural instability, although this is usually significantly milder in ET. Parkinsonism may be present in both disorders, but prevalence rates for this are lower in FXTAS than ET. Oculomotor deficits are very common in ET but have not been specifically studied in much detail in FXTAS. Both ET and FXTAS patients may have a positive family history for ET. However, there is typically additional family history of fragile X related disorders and/or intellectual disability in FXTAS.
Parkinson Disease (PD):
A. Tremor
The classical tremor in PD is a rest tremor that is typically asymmetrical at disease onset (Pagano, Ferrara, Brooks, & Pavese, 2016). However, the percentage of patients with symmetrical tremor rises as age of onset increases. Roughly half of PD patients have a tremor that is predominantly localized to a specific body part (Pagano et al., 2016). Studies have shown that approximately 38% of PD patients have a tremor dominant form of the disease (Wu et al., 2015).
B. Cerebellar Ataxia
Typical PD patients do not show signs of cerebellar gait ataxia, but rather a slow, shuffling gait pattern, with freezing of gait. Approximately 51% of PD patients present with a postural instability gait disorder form of the disease (Wu et al., 2015). The gait deficits in PD involve trouble with gait initiation rather than the wide-based gait pattern seen in ataxic patients. Furthermore, PD patients are likely to show reduced gait variability (Fernandez-Lago et al., 2015) as opposed to the increased variability usually seen in FXTAS and other types of ataxia (O’Keefe, Robertson-Dick et al., 2015).
C. Parkinsonism
The characteristic parkinsonism in PD includes rest tremor, bradykinesia, hypokinesia, rigidity and postural instability. Bradykinesia and hypokinesia are slowness of movement and a lack of movement, respectively, caused by dysfunction of dopaminergic neurons in the basal ganglia of the brain. These result in lack of facial expressions, known as “masked facies,” absent reciprocal arm swing during walking, difficulty with movement initiation, and a slow shuffling gait. The rigidity can be either lead-pipe rigidity, with increased muscle tone throughout range of motion caused by co-contraction of muscles on both sides of joint, or cogwheel rigidity, with rigidity interrupted by series of relaxations creating a movement like cogs on a wheel. Postural instability results from deficits in postural reflexes and results in gait abnormalities and balance problems.
D. Eye Movement Abnormalities
PD patients have deficits in saccadic movements (C. Antoniades & Kennard, 2015). Delayed reaction time, reduced maximum saccadic velocity, and hypometria of saccadic eye movements have all been reported in PD patients (Crawford, Henderson, & Kennard, 1989; Shibasaki, Tsuji, & Kuroiwa, 1979). Smooth pursuits may be interrupted by additional, short saccades (Shibasaki et al., 1979).
E. Case Reports
There are multiple case reports of patients who were given an initial diagnosis of PD and were later found to be PM carriers. Eleven of these have been detailed in the literature (Cilia et al., 2009; D. Hall et al., 2009; Healy et al., 2009; Hedrich et al., 2005; Pablo-Fernandez, Doherty, & Holton, 2015; Yachnis et al., 2010). Rest tremor was present in ten of these patients and the majority displayed both rigidity and bradykinesia. Six patients demonstrated typical PD like gait while cerebellar gait ataxia was only reported in two of the cases. Unified Parkinson Disease Rating Scale (UPDRS) (Fahn, Elton, & UPDRS Program Members., 1987) scores and Hoehn & Yahr (H & Y) (Hoehn & Yahr, 1967) stages reported in two studies were in the mild severity range of parkinsonian symptoms. Seven of the cases showed a good response to levodopa, whereas three failed to respond. Two cases had a family history of PD, and five had the fragile X premutation or full mutation in their families.
Several neuroimaging studies have been performed in order to better understand the nigrostrial function of FXTAS patients with parkinsonism. Single Photon Emission Computer Tomography (SPECT) imaging, a technique that allows visual examination of the integrity of the brain’s dopaminergic pathways, found moderate presynaptic dopaminergic nigrostriatal terminal loss and bilateral, asymmetric reduced tracer uptake and transport in four of the previously described cases (Cilia et al., 2009; Healy et al., 2009; Pablo-Fernandez et al., 2015). Likewise, a study of five men with FXTAS and parkinsonism showed midbrain and striatal neurodegeneration in all five patients and asymmetrical decreased striatal dopamine uptake in three patients (Scaglione, Ginestroni, & Vella, 2008). However, studies using [123I]FP-CIT (DaTSCAN, a radiopharmaceutical that binds to striatal presynaptic dopamine transporters) found preserved presynaptic nigrostriatal function in FXTAS patients (Ceravolo, Antonini, & Volterrani, 2005). Thus, the role for abnormal nigrostrial function in the parkinsonian phenotype of FXTAS requires further investigation.
F. Epidemiologic Data
Similar to ET, screening studies of populations with parkinsonism and/or PD have provided little to no evidence of FMR1 PM carriers. A few studies have found a significant increase in gray zone alleles in patients with PD and/or parkinsonism (Annesi et al., 2004; D. Hall et al., 2011; Loesch, Tassone, & Lo, 2013), suggesting that these alleles may play a role in the development of parkinsonism/PD. However, this finding has not been consistent in the literature (Costa, Gao, & Carrillo, 2011). While studies thus far suggest that there is not an increase in FMR1 premutation frequency in PD/parkinsonism, many PM carriers would be eliminated from participation in many of these studies due to recruitment methods and inclusion criteria. For example, PD DNA repositories frequently exclude patients with cerebellar signs based on the UK PD Brain Bank Criteria, which would screen out PM carriers with cerebellar gait ataxia (D. Hall et al., 2011).
G. Comparison Between FXTAS and PD
The parkinsonism of FXTAS is typically milder than that seen in PD, and may be accompanied by other symptoms, such as peripheral neuropathy, which may distinguish it from PD (E. Berry-Kravis et al., 2007; Klein, Schneider, & Lang, 2009). A rest tremor similar to that in PD may be seen in FXTAS but is usually accompanied by intention and/or postural tremor. Most FXTAS patients display an ataxic gait pattern, while patients with PD typically have a slowed, shuffling gait. The percentage of FXTAS patients that do show a parkinsonian gait pattern is low.
Spinocerebellar Ataxias (SCAs) and Multiple System Atrophy (MSA):
A. Tremor
Postural and action tremor in various body parts may be seen in many of the SCAs (Perlman, 2011; E. Storey, 2014), and up to 67% of MSA patients experience tremor (G. K. Wenning, Ben Shlomo, Magalhaes, Daniel, & Quinn, 1994; Yabe et al., 2006). The tremor in MSA is heterogeneous with approximately half showing a postural tremor, one third showing a rest tremor, and others showing a cerebellar intention tremor (Kaindlstorfer, Granata, & Wenning, 2013). The prevalence of rest and postural tremors within the MSA subtypes have been reported to be 38 and 60% in the multiple system atrophy parkinsonism subtype (MSA-P) and 22 and 45% in the multiple system atrophy cerebellar subtype (MSA-C), respectively (Low et al., 2015).
B. Cerebellar Ataxia
Cerebellar gait ataxia and loss of coordination due to cerebellar degeneration is a characteristic feature of all the SCAs (Matilla-Dueñas, 2012; Perlman, 2011; E. Storey, 2014). Likewise, MSA is characterized by a mid to late-onset cerebellar ataxia (Kamm, Healy, & Quinn, 2005). It is one of the core phenotypic features in both MSA-P and MSA-C with frequencies of 40% and 100%, respectively (Gilman et al., 2008; Low et al., 2015).
C. Parkinsonism
Parkinsonism has been reported in multiple SCAs, including SCA 2, 3, 6, 8, 17, and 21 (Park, Kim, & Jeon, 2015; Perlman, 2011; E. Storey, 2014), and both levodopa-responsive and atypical parkinsonism have been reported (Park et al., 2015). Likewise, one of the core diagnostic motor features in MSA is a rapidly progressive parkinsonism that is more symmetrical and less responsive to levodopa than in PD (Gilman et al., 2008; Levin, Kurz, Arzberger, Giese, & Hoglinger, 2016; G. K. Wenning & Stefanova, 2009). Postural instability develops earlier and progresses more rapidly than in PD (Gilman et al., 2008). The prevalence rates of parkinsonism in the MSA subtypes are reported to be 98% in MSA-P and 73% in MSA-C (Low et al., 2015).
D. Eye Movement Abnormalities
Oculomotor abnormalities have been observed in several of the SCAs including SCA 1, 2, 3, 5, 6, 7, 8, 14, 28, 37 (E. Storey, 2014). Nystagmus is a common feature in many of the SCAs (Perlman, 2011), and supranuclear ophthalmaplegy is seen in some of the autosomal dominant SCAs (Harding, 1993; Schöls, Bauer, Schmidt, Schulte, & Riess, 2004). For example, patients with SCA3 may experience slowed saccades, restriction of upward gaze, and disconjugate eye movements (Matilla-Dueñas, Corral-Juan, Volpini, & Sanchez, 2012). In MSA, oculomotor dysfunction is frequent and can include dysmetric saccades (Terao et al., 2016), square wave jerks (70%), mild vertical supranuclear gaze palsy (26%), positioning downbeat nystagmus (40%), saccadic hypometria (73%), impaired smooth pursuit (93%), reduced VOR suppression (66%), and gaze-evoked nystagmus (40%) (Anderson, 2008). The prevalence of nystagmus in the MSA subtypes is reported to be 17% in MSA-P and 41% in MSA-C, with an overall prevalence of 23% (Low et al., 2015).
E. Case Reports
Screening studies for cerebellar ataxia in patients with suspected SCA or MSA identified 32 PM carriers (Biancalana, Toft, & Le Ber, 2005; Brussino, Gellera, & Saluto, 2005; Cellini, Forleo, & Ginestroni, 2006; Faruq, Srivastava, & Suroliya, 2014; Kamm et al., 2005; L. Rodriguez-Revenga, Gómez-Anson, & Muñoz, 2007; Seixas, Maurer, & Lin, 2005; Seixas et al., 2011; Van Esch, Dom, & Bex, 2005; Wardle, Majounie, & Muzaimi, 2009; Zühlke et al., 2004). The cases included seven PM carrier women and 25 PM carrier men with symptoms beginning between the ages of 40 and 72. Cerebellar ataxia was documented in 29 of the cases and gait instability in 13. Intention, rest, postural and/or other kinetic tremors were present in the majority of cases. Oculomotor dysfunction was also present in nine cases. Autonomic dysfunction was seen in 17 patients and peripheral neuropathy in ten. Cerebral and/or cerebellar atrophy was found on MRI in nearly all patients, with white matter lesions in nine patients and the MCP sign in 15. Family history was not available for the majority of cases; however, two patients had relatives who were PM carriers and two had relatives with FXS or a learning disability.
F. Epidemiological Data
Despite numerous case reports of PM carriers identified through cerebellar ataxia screenings, the overall data from screening studies is mixed as to whether there is a higher incidence of the FMR1 PM among cerebellar ataxia patient populations. Of the studies in populations that had already tested negative for standard SCA genetic panels, low frequencies (0.06 to 2.2%) of the PM were detected (S. A. Adams, Steenblock, Thibodeau, & Lindor, 2008; Brussino et al., 2005; Milunsky & Maher, 2004; Rajkiewicz, Sułek-Piatkowska, & Krysa, 2008; L. Rodriguez-Revenga et al., 2007; Seixas et al., 2005; Zühlke et al., 2004). However, two other SCA screenings found slightly higher PM frequencies of 4.1 and 5.1%, respectively (Macpherson, Waghorn, & Hammans, 2003; Van Esch et al., 2005), and another screening found an even higher frequency of 9% PM carriers who had a SCA12-like phenotype (Faruq et al., 2014). Of the studies in populations not yet genetically tested for the SCAs, three found no PM carriers (Kerber, Jen, & Perlman, 2005; Kraft, Furtado, & Ranawaya, 2005; Tan et al., 2004), and frequencies of the premutation were consistently low in two other cerebellar ataxia screenings, at 1.1 and 2.1%, respectively (Cellini et al., 2006; Wardle et al., 2009).
Screening studies in MSA patients have found FMR1 gray zone alleles ranging from 41–53 CGG repeats in 4.6 to 7% of patients (Biancalana et al., 2005; Garland, Vnencak-Jones, & Biaggioni, 2004), suggesting that these alleles have low prevalence rates in populations with the MSA phenotype (Garland et al., 2004). Likewise, two additional screening studies of MSA patients identified few to no PM carriers (Kamm et al., 2005; Zhang, Gu, & Wang, 2013), suggesting that the FMR1 premutation is not common in MSA populations.
G. Comparison Between FXTAS and SCA and MSA
The cerebellar ataxia seen in FXTAS is similar to that seen in SCA, making accurate diagnosis of patients who display this characteristic challenging. Both FXTAS and SCA are characterized by a progressive lack of motor coordination, including an ataxic gait pattern and difficulty with tandem walking. SCA patients may also exhibit additional clinical findings that are common in FXTAS (P. J. Hagerman & Hagerman, 2015; Tassone & Hagerman, 2012), such as autonomic dysfunction (Yeh, Lu, & Chou, 2005), peripheral neuropathy (Rosenberg, 1992), and/or parkinsonism (Matilla-Dueñas, 2012). There is also significant clinical and radiological overlap between men with FXTAS and MSA-C patients (Jacquemont et al., 2003). Both disorders are characterized by a mid to late-onset cerebellar ataxia, and cases have been reported of MSA-C patients with MCP hyperintensities, cerebellar and cerebral atrophy, and white matter lesions on MRI (E. Storey & Billimoria, 2005), which are characteristic in FXTAS.
One distinguishing motor feature that may help to differentiate these patients is the moderate to severe kinetic tremor characteristic of FXTAS but rare in the SCAs and MSA. One exception is SCA12, which is the only SCA for which kinetic tremor is the presenting sign and most frequent symptom (Pulst, 2003). Given that the tremor in SCA12 may resemble the tremor of FXTAS, there is the potential for FXTAS tremor to be mistaken as SCA12 (Faruq et al., 2014). However, SCA12 has a very low prevalence rate, except in Northern India where it causes 7% of the dominant ataxias (E. Storey & Billimoria, 2005). Another distinguishing factor is the autosomal dominant inheritance pattern for most SCAs, which would be lacking in FXTAS families.
Progressive Supranuclear Palsy (PSP):
A. Tremor
Up to 42% of PSP patients present with some form of tremor, including postural/action, rest, intention, or a combination of tremor types (Fujioka et al., 2016). PSP-parkinsonism (PSP-P), is characterized by asymmetric onset of rest tremor that is moderately responsive to levodopa (Liscic, Srulijes, Gröger, Maetzler, & Berg, 2013; D. R. Williams et al., 2005).
B. Cerebellar Ataxia
Postural instability with falls is the most common initial symptom in classical PSP (I. Litvan et al., 1996; D. R. Williams et al., 2005), and there are a few case reports of PSP patients having cerebellar ataxia (Iwasaki et al., 2013; M. Kanazawa et al., 2009; Koga et al., 2016). A recent study identified a rare subtype of PSP with predominant cerebellar ataxia as the initial and primary symptom of the disease (M. Kanazawa et al., 2013). However, the overall prevalence of cerebellar ataxia appears to be extremely low in PSP.
C. Parkinsonism
Atypical parkinsonian features occur in approximately 40% of PSP cases, including levodopa-resistant symmetric akinesia and axial rigidity (Levin et al., 2016; Respondek & Hoglinger, 2015; STEELE, RICHARDSON, & OLSZEWSKI, 1964). However, PSP-P occurring in approximately 20% of cases presents as an asymmetric levodopa-responsive parkinsonism which is difficult to distinguish from idiopathic PD early in the disease (Levin et al., 2016; D. R. Williams & Lees, 2010; D. R. Williams et al., 2005).
D. Eye Movement Abnormalities
Vertical supranuclear gaze palsy is the main characteristic feature of PSP (Liscic et al., 2013; D. R. Williams et al., 2005), and is typically preceded by a slowing of vertical saccades (I. Litvan et al., 1996). Diplopia, photophobia, and eyelid apraxia may also manifest early in the disease course (D. R. Williams et al., 2005). Patients with PSP also display deficits in their saccades, optokinetic reflexes, and smooth pursuit (Amtage et al., 2014).
E. Case Reports
Four patients from a larger case report of 19 FXTAS patients were described who had presented with motor symptoms, including oculomotor abnormalities, resembling those seen in PSP (Fraint et al., 2014). Two demonstrated absent vertical optokinetic nystagmus, one of which had additional decreased lateral optokinetic nystagmus, and another showed decreased vertical optokinetic nystagmus. Four of the patients had slowed saccades and one also had dysmetria of saccades. One patient also demonstrated square wave jerks. These oculomotor findings along with other overlapping motor symptoms characteristic of PSP including postural instability and parkinsonism, (I. Litvan et al., 1996) suggest that there may a PSP-like variation of FXTAS (Fraint et al., 2014). However, as these abnormal eye movements are not typical in FXTAS, it is possible that these patients had dual pathology which will need to be elucidated in future studies.
F. Comparison Between FXTAS and PSP
Although both FXTAS and PSP patients may present with similar tremor types and/or a parkinsonism which is similar to that seen in idiopathic PD, the cerebellar ataxia signs that are characteristic of FXTAS is rare in PSP. Eye movement abnormalities have not been well studied in FXTAS but appear to be much less common than in PSP, which has a classic vertical supranuclear gaze palsy. However, there may be a PSP-like phenotype in a small subset of patients with FXTAS.
III. Age of onset, disease severity and progression, and average lifespan in FXTAS compared to ET, PD, SCAs, MSA, and PSP
All of these movement disorders are chronic and progressive, with some symptoms that may be managed through pharmacological, surgical, and rehabilitative treatments but there are currently no curative treatments for any of these neurodegenerative disorders.
Disease onset in FXTAS typically begins after 55 years of age (R. J. Hagerman et al., 2001). Functional disability is largely correlated with gait and balance abnormalities (M. A. Leehey, 2009; O’Keefe et al., 2015; O’Keefe, Robertson-Dick et al., 2015). Women tend to experience a much milder disease phenotype than men (Coffey et al., 2008; R. J. Hagerman et al., 2001; Jacquemont et al., 2003; M. A. Leehey, 2009) due to the presence of the normal FMR1 allele on the second X chromosome, as well as possible skewed x-inactivation (E. Berry-Kravis, Potanos, Weinberg, Zhou, & Goetz, 2005; D. A. Hall et al., 2016; M. Leehey et al., 2008; O’Keefe et al., 2015; L. Rodriguez-Revenga et al., 2010). Mean survival time is quite variable in FXTAS, but time from motor symptom onset and death has been reported to be 21 years (M. A. Leehey et al., 2007).
ET is one of the most common movement disorders, with a prevalence rate of approximately 5% in the general population which increases to over 20% in the elderly (Sinoff & Badarny, 2014). The average onset of ET in one study of 52 ET patients was 55.8 ± 15.1 years of age (Sinoff & Badarny, 2014). Furthermore, as noted previously, ET has been categorized into hereditary and sporadic ET, both with onset prior to age 65, and senile ET with onset over age 65 (G. Deuschl & Elble, 2009). Tremor severity increases over time, which may lead to significant disability including physical, psychological, and social impairment (Auff, Doppelbauer, & Fertl, 1991). Life expectancy appears to be normal in ET, but this finding is not universally accepted and requires further study(G. Deuschl & Elble, 2009; E. D. Louis, Benito-Leon, Ottman, Bermejo-Pareja, & Neurological Disorders in Central Spain (NEDICES) Study Group, 2007; Romero et al., 2012).
Age is the greatest risk factor for the development of idiopathic PD, with an average age of onset reported to be 58.4 ± 11.0 years (Sidransky et al., 2009; Wang et al., 2014), which is similar to that in FXTAS. However, there recently has been an increase in diagnosis of PD in individuals younger than 50 years of age (Gopalakrishna & Alexander, 2015). Non-motor symptoms such as constipation, rapid eye movement (REM) sleep behavior disorder, anosmia, and cognitive and behavioral problems usually present first with motor symptoms developing over time (Goldman et al., 2014; Goldman, Aggarwal, & Schroeder, 2015). Symptoms eventually progress to interfere with activities of daily living. The mean survival time following motor onset in PD is approximately 15 years (Forsaa et al., 2010).
Symptom onset in the SCAs, including the most common SCAs (1, 2, 3, and 6) is quite variable and can range from 15 to over 70 years of age (Schols et al., 1998). SCA1, SCA2, and SCA3 usually present between 30 and 40 years of age (Jacobi et al., 2015; Schols et al., 1998), whereas SCA6 was reported to have an average age of onset of 52 ± 12 years in a German cohort of 21 patients (Schols et al., 1998). Disease progression has been shown to be greatest in SCA1 followed by SCA 3 and SCA2, with SCA 6 showing the slowest progression (Jacobi et al., 2015). Faster progression is associated with younger age of onset and longer repeat expansions in SCA1, SCA2 and SCA6. In SCA1, patients are usually wheelchair-bound within 15–20 years of disease onset. Disease severity is highly variable in SCA2 and SCA3, with patients showing a wide spectrum of disability. SCA6 typically has the least disease severity allowing for a normal lifespan in most cases (Paulson, 2009).
MSA has a mean onset of approximately 56 years of age and is characterized by severe progression of disability and poor quality of life (Schrag et al., 2006). Mean survival time from symptom onset is 9–10 years (Schrag, Wenning, Quinn, & Ben-Shlomo, 2008), and nocturnal sudden death is a primary cause of death in MSA patients (Shimohata et al., 2008).
The average age of PSP onset is between 60 and 70 years of age, with motor symptoms appearing first followed by cognitive and behavioral and systemic problems (Arena et al., 2015). The mean survival time in patients with PSP is 6 to 8 years (W. Z. Chiu et al., 2010; Respondek & Hoglinger, 2015).
In summary, MSA and PSP have the most severe and rapid disease progression of these movement disorders. Although PD appears to be more severe than FXTAS based on studies of disease progression and survival time in PD, no prospective studies on the progression and survival time of FXTAS have been performed. However, the disability in advanced FXTAS is likely to be as severe as in advanced PD. The SCAs generally have earlier onset but have variable disease progression. ET is the least severe of all these disorders and may not have a reduced lifespan. This data is summarized in Table 3.
A flow chart to assist in the differential diagnosis of FXTAS is provided in Figure 1.
Conclusion
The purpose of this paper was to review FXTAS in the context of the movement disorders for which it is most frequently confused in order to better understand the similarities and differences among these disorders and identify potential markers which may be helpful for clinicians when diagnosing patients in the clinic. Increasing the accuracy of FXTAS diagnosis will allow for earlier and more targeted treatments and lead to better outcomes for these individuals.
Failure to correctly identify carriers of the FMR1 premutation and provide the necessary genetic counseling may have serious detrimental consequences for future generations of the patient’s family. In addition to FXTAS, women PM carriers are at risk for developing fragile X-associated Primary Ovarian Insufficiency, which may result in premature ovarian failure leading to early menopause and infertility, making early reproductive consultation essential for family planning (Sherman et al., 2014). Furthermore, PM carrier women are at risk for having children with FXS a neurodevelopmental disorder caused by the full mutation (>200 CGG repeats). Therefore, identifying PM carrier men and women as early as possible can yield life-changing benefits for all members of the family.
By comparing the cognitive and motor phenotypes of FXTAS with each of these other movement disorders we hope to have clarified potential symptom overlap while elucidating factors that make these disorders unique from one another. Although family history may provide key clues for diagnosing FXTAS, alone it is not sufficient as many PM carriers do not have, or are not aware of, family members with known fragile X mutations and/or associated disorders. In order to reduce misdiagnoses, it is essential that clinicians learn to recognize the subtle but distinguishing cognitive and motor features of FXTAS.
In summary, the clinician should consider a FXTAS diagnosis and testing for the Fragile X mental retardation 1 (FMR1) gene premutation if a patient over the age of 50: (1) presents with cerebellar ataxia and/or intention tremor with mild parkinsonism, (2) has the middle cerebellar peduncle (MCP) sign, global cerebellar and cerebral atrophy, and/or subcortical white matter lesions on MRI, or (3) has a family history of fragile X related disorders, intellectual disability, autism, premature ovarian failure and has neurological signs consistent with FXTAS. Peripheral neuropathy, executive function deficits, anxiety, or depression are supportive of the diagnosis.
Acknowledgements
This work was supported in part by awards from the Rush Translational Science Consortium (JO), NFXF Research Fellowship award (ER), and NINDS R01 NS082416 (DAH).
References
- Aarsland D, Bronnick K, Alves G, Tysnes OB, Pedersen KF, Ehrt U, & Larsen JP (2009). The spectrum of neuropsychiatric symptoms in patients with early untreated parkinson’s disease. Journal of Neurology, Neurosurgery, and Psychiatry, 80(8), 928–930. doi: 10.1136/jnnp.2008.166959 [doi] [DOI] [PubMed] [Google Scholar]
- Aarsland D, Bronnick K, Williams-Gray C, Weintraub D, Marder K, Kulisevsky J, … Emre M (2010). Mild cognitive impairment in parkinson disease: A multicenter pooled analysis. Neurology, 75(12), 1062–1069. doi: 10.1212/WNL.0b013e3181f39d0e [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsland D, & Kramberger MG (2015). Neuropsychiatric symptoms in parkinson’s disease. Journal of Parkinson’s Disease, 5(3), 659–667. doi: 10.3233/JPD-150604 [doi] [DOI] [PubMed] [Google Scholar]
- Aarsland D, Larsen JP, Cummins JL, & Laake K (1999). Prevalence and clinical correlates of psychotic symptoms in parkinson disease: A community-based study. Archives of Neurology, 56(5), 595–601. [DOI] [PubMed] [Google Scholar]
- Aarsland D, Larsen JP, Lim NG, Janvin C, Karlsen K, Tandberg E, & Cummings JL (1999). Range of neuropsychiatric disturbances in patients with parkinson’s disease. Journal of Neurology, Neurosurgery, and Psychiatry, 67(4), 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsland D, Litvan I, & Larsen JP (2001). Neuropsychiatric symptoms of patients with progressive supranuclear palsy and parkinson’s disease. The Journal of Neuropsychiatry and Clinical Neurosciences, 13(1), 42–49. [DOI] [PubMed] [Google Scholar]
- Aarsland D, Litvan I, Salmon D, Galasko D, Wentzel-Larsen T, & Larsen JP (2003). Performance on the dementia rating scale in parkinson’s disease with dementia and dementia with lewy bodies: Comparison with progressive supranuclear palsy and alzheimer’s disease. Journal of Neurology, Neurosurgery, and Psychiatry, 74(9), 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsland D, Marsh L, & Schrag A (2009). Neuropsychiatric symptoms in parkinson’s disease. Movement Disorders : Official Journal of the Movement Disorder Society, 24(15), 2175–2186. doi: 10.1002/mds.22589 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsland D, Pahlhagen S, Ballard CG, Ehrt U, & Svenningsson P (2011). Depression in parkinson disease--epidemiology, mechanisms and management. Nature Reviews.Neurology, 8(1), 35–47. doi: 10.1038/nrneurol.2011.189 [doi] [DOI] [PubMed] [Google Scholar]
- Aarsland D, Tandberg E, Larsen JP, & Cummings JL (1996). Frequency of dementia in parkinson disease. Archives of Neurology, 53(6), 538–542. [DOI] [PubMed] [Google Scholar]
- Adams PE, Adams JS, Nguyen DV, Hessl D, Brunberg JA, Tassone F, … Hagerman RJ (2010). Psychological symptoms correlate with reduced hippocampal volume in fragile X premutation carriers. American Journal of Medical Genetics.Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics, 153B(3), 775–785. doi: 10.1002/ajmg.b.31046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams SA, Steenblock KJ, Thibodeau SN, & Lindor NM (2008). Premutations in the FMR1 gene are uncommon in men undergoing genetic testing for spinocerebellar ataxia. Journal of Neurogenetics, 22(1), 77–92. doi: 10.1080/01677060701686242 [DOI] [PubMed] [Google Scholar]
- Aguilar D, Sigford K, & Soontarapornchai K (2008). A quantitative assessment of tremor and ataxia in FMR1 premutation carriers using CATSYS. Am J Med Genet A, 146A, 629–635. [DOI] [PubMed] [Google Scholar]
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, … Phelps CH (2011). The diagnosis of mild cognitive impairment due to alzheimer’s disease: Recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for alzheimer’s disease. Alzheimer’s & Dementia : The Journal of the Alzheimer’s Association, 7(3), 270–279. doi: 10.1016/j.jalz.2011.03.008 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hinti JT, Nagan N, & Harik SI (2007). Fragile X premutation in a woman with cognitive impairment, tremor, and history of premature ovarian failure. Alzheimer Disease and Associated Disorders, 21(3), 262–264. doi: 10.1097/WAD.0b013e31811ec130 [DOI] [PubMed] [Google Scholar]
- Alvarez JA, & Emory E (2006). Executive function and the frontal lobes: A meta-analytic review. Neuropsychology Review, 16(1), 17–42. doi: 10.1007/s11065-006-9002-x [doi] [DOI] [PubMed] [Google Scholar]
- Amtage F, Maurer C, Hellwig S, Tüscher O, Kreft A, Weiller C, … Meyer PT (2014). Functional correlates of vertical gaze palsy and other ocular motor deficits in PSP: An FDG-PET study. Parkinsonism & Related Disorders, 20(8), 898–906. [DOI] [PubMed] [Google Scholar]
- Annesi G, Nicoletti G, Tarantino P, Cutuli N, Annesi F, Marco EV, … Quattrone A (2004). FRAXE intermediate alleles are associated with parkinson’s disease. Neuroscience Letters, 368(1), 21–24. doi: 10.1016/j.neulet.2004.06.049 [doi] [DOI] [PubMed] [Google Scholar]
- Antoniades C, & Kennard C (2015). Ocular motor abnormalities in neurodegenerative disorders. Eye, 29(2), 200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniades CA, Demeyere N, Kennard C, Humphreys GW, & Hu MT (2015). Antisaccades and executive dysfunction in early drug-naive parkinson’s disease: The discovery study. Movement Disorders : Official Journal of the Movement Disorder Society, 30(6), 843–847. doi: 10.1002/mds.26134 [doi] [DOI] [PubMed] [Google Scholar]
- Apartis E, Blancher A, & Meissner W (2012). FXTAS: New insights and the need for revised diagnostic criteria. Neurology, 79(18), 1898–1907. [DOI] [PubMed] [Google Scholar]
- Arena JE, Weigand SD, Whitwell JL, Hassan A, Eggers SD, Hoglinger GU, … Josephs KA (2015). Progressive supranuclear palsy: Progression and survival. Journal of Neurology, doi: 10.1007/s00415-015-7990-2 [doi] [DOI] [PubMed] [Google Scholar]
- Arocena D, Louis E, & Tassone F (2004). Screen for expanded FMR1 alleles in patients with essential tremor. Mov Disord, 19(8), 930–933. [DOI] [PubMed] [Google Scholar]
- Auff E, Doppelbauer A, & Fertl E (1991). Essential tremor: Functional disability vs. subjective impairment. J Neural Transm Suppl, 33, 105–110. [DOI] [PubMed] [Google Scholar]
- Auning E, Kjaervik VK, Selnes P, Aarsland D, Haram A, Bjornerud A, … Fladby T (2014). White matter integrity and cognition in parkinson’s disease: A cross-sectional study. BMJ Open, 4(1), e003976-2013-003976. doi: 10.1136/bmjopen-2013-003976 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auzou N, Dujardin K, Biundo R, Foubert-Samier A, Barth C, Duval F, … Meissner WG (2015). Diagnosing dementia in multiple system atrophy by applying movement disorder society diagnostic criteria for parkinson’s disease dementia. Parkinsonism & Related Disorders, 21(10), 1273–1277. doi: 10.1016/j.parkreldis.2015.08.013 [doi] [DOI] [PubMed] [Google Scholar]
- Baba Y, & Uitti R (2005). Fragile X-associated tremor/ataxia syndrome and movement disorders Curr Opin Neurol, 393–398. [DOI] [PubMed] [Google Scholar]
- Bacalman S, Farzin F, Bourgeois JA, Cogswell J, Goodlin-Jones BL, Gane LW, … Hagerman RJ (2006). Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: Newly described fronto-subcortical dementia. The Journal of Clinical Psychiatry, 67(1), 87–94. [DOI] [PubMed] [Google Scholar]
- Baddeley A (2010). Working memory. Current Biology : CB, 20(4), R136–40. doi: 10.1016/j.cub.2009.12.014 [doi] [DOI] [PubMed] [Google Scholar]
- Bak TH, Crawford LM, Hearn VC, Mathuranath PS, & Hodges JR (2005). Subcortical dementia revisited: Similarities and differences in cognitive function between progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and multiple system atrophy (MSA). Neurocase, 11(4), 268–273. doi:W202735U669J62H2 [pii] [DOI] [PubMed] [Google Scholar]
- Balas M, Balash Y, Giladi N, & Gurevich T (2010). Cognition in multiple system atrophy: Neuropsychological profile and interaction with mood. Journal of Neural Transmission (Vienna, Austria : 1996), 117(3), 369–375. doi: 10.1007/s00702-009-0365-z [doi] [DOI] [PubMed] [Google Scholar]
- Barrash J, Tranel D, & Anderson SW (2000). Acquired personality disturbances associated with bilateral damage to the ventromedial prefrontal region. Developmental Neuropsychology, 18(3), 355–381. doi: 10.1207/S1532694205Barrash [doi] [DOI] [PubMed] [Google Scholar]
- Beer JS, John OP, Scabini D, & Knight RT (2006). Orbitofrontal cortex and social behavior: Integrating self-monitoring and emotion-cognition interactions. Journal of Cognitive Neuroscience, 18(6), 871–879. doi: 10.1162/jocn.2006.18.6.871 [doi] [DOI] [PubMed] [Google Scholar]
- Benito-Leon J, & Louis ED (2006). Essential tremor: Emerging views of a common disorder. Nature Clinical Practice.Neurology, 2(12), 666–78; quiz 2p following 691. doi:ncpneuro0347 [pii] [DOI] [PubMed] [Google Scholar]
- Benito-Leon J, & Louis ED (2011). Update on essential tremor. Minerva Medica, 102(6), 417–440. doi:R10113266 [pii] [PubMed] [Google Scholar]
- Benito-Leon J, Louis ED, Mitchell AJ, & Bermejo-Pareja F (2011). Elderly-onset essential tremor and mild cognitive impairment: A population-based study (NEDICES). Journal of Alzheimer’s Disease : JAD, 23(4), 727–735. doi: 10.3233/JAD-2011-101572 [doi] [DOI] [PubMed] [Google Scholar]
- Benrud-Larson LM, Sandroni P, Schrag A, & Low PA (2005). Depressive symptoms and life satisfaction in patients with multiple system atrophy. Movement Disorders : Official Journal of the Movement Disorder Society, 20(8), 951–957. doi: 10.1002/mds.20450 [doi] [DOI] [PubMed] [Google Scholar]
- Bernard JA, Peltier SJ, Wiggins JL, Jaeggi SM, Buschkuehl M, Fling BW, … Seidler RD (2013). Disrupted cortico-cerebellar connectivity in older adults. Neuroimage, 83, 103–119. doi: 10.1016/j.neuroimage.2013.06.042 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JA, Seidler RD, Hassevoort KM, Benson BL, Welsh RC, Wiggins JL, … Peltier SJ (2012). Resting state cortico-cerebellar functional connectivity networks: A comparison of anatomical and self-organizing map approaches. Frontiers in Neuroanatomy, 6, 31. doi: 10.3389/fnana.2012.00031 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Abrams L, & Coffey S (2007). Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Mov Disord, 22(14), 2018–2030. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Lewin F, & Wuu J (2003). Tremor and ataxia in fragile X premutation carriers: Blinded videotape study. Ann Neurol, 53(5), 616–623. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, … Leehey MA (2007). Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Movement Disorders : Official Journal of the Movement Disorder Society, 22(14), 2018–30, quiz 2140. doi: 10.1002/mds.21493 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Potanos K, Weinberg D, Zhou L, & Goetz CG (2005). Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Annals of Neurology, 57(1), 144–147. doi: 10.1002/ana.20360 [DOI] [PubMed] [Google Scholar]
- Bertram K, & Williams DR (2012). Visual hallucinations in the differential diagnosis of parkinsonism. Journal of Neurology, Neurosurgery, and Psychiatry, 83(4), 448–452. doi: 10.1136/jnnp-2011-300980 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besterman AD, Wilke SA, Mulligan TE, Allison SC, Hagerman R, Seritan AL, & Bourgeois JA (2014). Towards an understanding of neuropsychiatric manifestations in fragile X premutation carriers. Future Neurology, 9(2), 227–239. doi: 10.2217/fnl.14.11 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalsing KS, Kumar KJ, Saini J, Yadav R, Gupta AK, & Pal PK (2015). White matter correlates of cognitive impairment in essential tremor. AJNR.American Journal of Neuroradiology, 36(3), 448–453. doi: 10.3174/ajnr.A4138 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalsing KS, Upadhyay N, Kumar KJ, Saini J, Yadav R, Gupta AK, & Pal PK (2014). Association between cortical volume loss and cognitive impairments in essential tremor. European Journal of Neurology : The Official Journal of the European Federation of Neurological Societies, 21(6), 874–883. doi: 10.1111/ene.12399 [doi] [DOI] [PubMed] [Google Scholar]
- Biancalana V, Toft M, & Le Ber I (2005). FMR1 premutations associated with fragile X-associated tremor/ataxia syndrome in multiple system atrophy. Arch Neurol, 62(6), 962–966. [DOI] [PubMed] [Google Scholar]
- Birch RC, Cornish KM, Hocking DR, & Trollor JN (2014). Understanding the neuropsychiatric phenotype of fragile X-associated tremor ataxia syndrome: A systematic review. Neuropsychology Review, 24(4), 491–513. doi: 10.1007/s11065-014-9262-9 [doi] [DOI] [PubMed] [Google Scholar]
- Bloise MC, Berardelli I, Roselli V, Pasquini M, Stirpe P, Colosimo C, … Fabbrini G (2014). Psychiatric disturbances in patients with progressive supranuclear palsy: A case-control study. Parkinsonism & Related Disorders, 20(9), 965–968. doi: 10.1016/j.parkreldis.2014.05.015 [doi] [DOI] [PubMed] [Google Scholar]
- Blumenfeld RS, & Ranganath C (2007). Prefrontal cortex and long-term memory encoding: An integrative review of findings from neuropsychology and neuroimaging. The Neuroscientist : A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 13(3), 280–291. doi:13/3/280 [pii] [DOI] [PubMed] [Google Scholar]
- Bodranghien F, Bastian A, Casali C, Hallett M, Louis ED, Manto M, … van Dun K (2015). Consensus paper: Revisiting the symptoms and signs of cerebellar syndrome. Cerebellum (London, England), doi: 10.1007/s12311-015-0687-3 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve B, Dickson D, Duffy J, Bartleson J, Trenerry M, & Petersen R (2003). Progressive nonfluent aphasia and subsequent aphasic dementia associated with atypical progressive supranuclear palsy pathology. European Neurology, 49(2), 72–78. doi:68502 [doi] [DOI] [PubMed] [Google Scholar]
- Borroni B, Turla M, Bertasi V, Agosti C, Gilberti N, & Padovani A (2008). Cognitive and behavioral assessment in the early stages of neurodegenerative extrapyramidal syndromes. Archives of Gerontology and Geriatrics, 47(1), 53–61. doi:S0167–4943(07)00144–6 [pii] [DOI] [PubMed] [Google Scholar]
- Bourgeois JA, Cogswell JB, Hessl D, Zhang L, Ono MY, Tassone F, … Hagerman RJ (2007). Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. General Hospital Psychiatry, 29(4), 349–356. doi: 10.1016/j.genhosppsych.2007.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois JA, Farzin F, Brunberg JA, Tassone F, Hagerman P, Zhang L, … Hagerman R (2006). Dementia with mood symptoms in a fragile X premutation carrier with the fragile X-associated tremor/ataxia syndrome: Clinical intervention with donepezil and venlafaxine. The Journal of Neuropsychiatry and Clinical Neurosciences, 18(2), 171–177. doi: 10.1176/appi.neuropsych.18.2.171 [DOI] [PubMed] [Google Scholar]
- Bourgeois JA, Seritan AL, Casillas EM, Hessl D, Schneider A, Yang Y, … Hagerman RJ (2011). Lifetime prevalence of mood and anxiety disorders in fragile x premutation carriers. The Journal of Clinical Psychiatry, 72(2), 175–82. doi: 10.4088/JCP.09m05407blu [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove M, Marinelli L, Avanzino L, Marchese R, & Abbruzzese G (2006). Posturographic analysis of balance control in patients with essential tremor. Movement Disorders, 21(2), 192–198. [DOI] [PubMed] [Google Scholar]
- Braga-Neto P, Felicio AC, Hoexter MQ, Pedroso JL, Dutra LA, Alessi H, … Barsottini OG (2012). Cognitive and olfactory deficits in machado-joseph disease: A dopamine transporter study. Parkinsonism & Related Disorders, 18(7), 854–858. doi: 10.1016/j.parkreldis.2012.04.015 [doi] [DOI] [PubMed] [Google Scholar]
- Braga-Neto P, Pedroso JL, Alessi H, Dutra LA, Felicio AC, Minett T, … Barsottini OG (2012). Cerebellar cognitive affective syndrome in machado joseph disease: Core clinical features. Cerebellum (London, England), 11(2), 549–556. doi: 10.1007/s12311-011-0318-6 [doi] [DOI] [PubMed] [Google Scholar]
- Braga-Neto P, Pedroso JL, Barsottini OG, & Schmahmann JD (2015). Cognition in SCA21 reflects developmental and adult onset cerebellar cognitive affective syndrome. Brain : A Journal of Neurology, 138(Pt 7), e364. doi: 10.1093/brain/awu382 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brega AG, Goodrich G, Bennett RE, Hessl D, Engle K, Leehey MA, … Grigsby J (2008). The primary cognitive deficit among males with fragile X-associated tremor/ataxia syndrome (FXTAS) is a dysexecutive syndrome. Journal of Clinical and Experimental Neuropsychology, 30(8), 853–869. doi: 10.1080/13803390701819044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis C, Bösch S, Schocke M, Wenning G, & Poewe W (2003). Atrophy pattern in SCA2 determined by voxel-based morphometry. Neuroreport, 14(14), 1799–1802. [DOI] [PubMed] [Google Scholar]
- Brenneis C, Egger K, Scherfler C, Seppi K, Schocke M, Poewe W, & Wenning GK (2007). Progression of brain atrophy in multiple system atrophy. A longitudinal VBM study. Journal of Neurology, 254(2), 191–196. doi: 10.1007/s00415-006-0325-6 [doi] [DOI] [PubMed] [Google Scholar]
- Brown RG, Lacomblez L, Landwehrmeyer BG, Bak T, Uttner I, Dubois B, … NNIPPS Study Group. (2010). Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain : A Journal of Neurology, 133(Pt 8), 2382–2393. doi: 10.1093/brain/awq158 [doi] [DOI] [PubMed] [Google Scholar]
- Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, … Hagerman PJ (2002). Fragile X premutation carriers: Characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR.American Journal of Neuroradiology, 23(10), 1757–1766. [PMC free article] [PubMed] [Google Scholar]
- Brussino A, Gellera C, & Saluto A (2005). FMR1 gene premutation is a frequent genetic cause of late-onset sporadic cerebellar ataxia. Neurology, 64(1), 145–147. [DOI] [PubMed] [Google Scholar]
- Bürk K, Globas C, Bösch S, Klockgether T, Zühlke C, Daum I, & Dichgans J (2003). Cognitive deficits in spinocerebellar ataxia type 1, 2, and 3. J Neurol, 250(2), 207–211. [DOI] [PubMed] [Google Scholar]
- Burk K, Bosch S, Globas C, Zuhlke C, Daum I, Klockgether T, & Dichgans J (2001). Executive dysfunction in spinocerebellar ataxia type 1. European Neurology, 46(1), 43–48. doi:50755 [pii] [DOI] [PubMed] [Google Scholar]
- Burk K, Daum I, & Rub U (2006). Cognitive function in multiple system atrophy of the cerebellar type. Movement Disorders : Official Journal of the Movement Disorder Society, 21(6), 772–776. doi: 10.1002/mds.20802 [doi] [DOI] [PubMed] [Google Scholar]
- Cecchin CR, Pires AP, Rieder CR, Monte TL, Silveira I, Carvalho T, … Jardim LB (2007). Depressive symptoms in machado-joseph disease (SCA3) patients and their relatives. Community Genetics, 10(1), 19–26. doi:000096276 [pii] [DOI] [PubMed] [Google Scholar]
- Cellini E, Forleo P, & Ginestroni A (2006). Fragile X premutation with atypical symptoms at onset. Arch Neurol, 63(8), 1135–1138. [DOI] [PubMed] [Google Scholar]
- Ceravolo R, Antonini A, & Volterrani D (2005). Dopamine transporter imaging study in parkinsonism occurring in fragile x premutation carriers. Neurology. 2005 Dec 27;65(12):1971–3., 65(12), 1971–1973. [DOI] [PubMed] [Google Scholar]
- Chiu HF (1995). Psychiatric aspects of progressive supranuclear palsy. General Hospital Psychiatry, 17(2), 135–143. doi:016383439400103K [pii] [DOI] [PubMed] [Google Scholar]
- Chiu WZ, Kaat LD, Seelaar H, Rosso SM, Boon AJ, Kamphorst W, & van Swieten JC (2010). Survival in progressive supranuclear palsy and frontotemporal dementia. Journal of Neurology, Neurosurgery, and Psychiatry, 81(4), 441–445. doi: 10.1136/jnnp.2009.195719 [doi] [DOI] [PubMed] [Google Scholar]
- Chow TW (2000). Personality in frontal lobe disorders. Current Psychiatry Reports, 2(5), 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilia R, Kraff J, & Canesi M (2009). Screening for the presence of FMR1 premutation alleles in women with parkinsonism. Arch Neurol, 66(2), 244–249. [DOI] [PubMed] [Google Scholar]
- Clark L, Ye X, & Liu X (2015). Genetic analysis of FMR1 repeat expansion in essential tremor. Neurosci Lett, 593, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, … Hagerman RJ (2008). Expanded clinical phenotype of women with the FMR1 premutation. American Journal of Medical Genetics.Part A, 146A(8), 1009–1016. doi: 10.1002/ajmg.a.32060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogan DG, Chu FC, & Reingold DB (1982). Ocular signs of cerebellar disease. Archives of Ophthalmology (Chicago, Ill.: 1960), 100(5), 755–760. [DOI] [PubMed] [Google Scholar]
- Cohen S, Masyn K, Adams J, Hessl D, Rivera S, Tassone F, … Hagerman R (2006). Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology, 67(8), 1426–1431. doi: 10.1212/01.wnl.0000239837.57475.3a [DOI] [PubMed] [Google Scholar]
- Colosimo C, Bak TH, Bologna M, & Berardelli A (2014). Fifty years of progressive supranuclear palsy. Journal of Neurology, Neurosurgery, and Psychiatry, 85(8), 938–944. doi: 10.1136/jnnp-2013-305740 [doi] [DOI] [PubMed] [Google Scholar]
- Cornish KM, Kogan CS, Li L, Turk J, Jacquemont S, & Hagerman RJ (2009). Lifespan changes in working memory in fragile X premutation males. Brain and Cognition, 69(3), 551–558. doi: 10.1016/j.bandc.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish KM, Li L, Kogan CS, Jacquemont S, Turk J, Dalton A, … Hagerman PJ (2008). Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex; a Journal Devoted to the Study of the Nervous System and Behavior, 44(6), 628–636. doi: 10.1016/j.cortex.2006.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Gao L, & Carrillo F (2011). Intermediate alleles at the FRAXA and FRAXE loci in parkinson’s disease. Parkinsonism Relat Disord, 17(4), 281–284. [DOI] [PubMed] [Google Scholar]
- Cotelli M, Borroni B, Manenti R, Alberici A, Calabria M, Agosti C, … Cappa SF (2006). Action and object naming in frontotemporal dementia, progressive supranuclear palsy, and corticobasal degeneration. Neuropsychology, 20(5), 558–565. doi:2006-10978-006 [pii] [DOI] [PubMed] [Google Scholar]
- Crawford TJ, Henderson L, & Kennard C (1989). Abnormalities of nonvisually-guided eye movements in parkinson’s disease. Brain : A Journal of Neurology, 112 (Pt 6)(Pt 6), 1573–1586. [DOI] [PubMed] [Google Scholar]
- D’Abreu A, Franca MC Jr, Yasuda CL, Campos BA, Lopes-Cendes I, & Cendes F (2012). Neocortical atrophy in machado-joseph disease: A longitudinal neuroimaging study. Journal of Neuroimaging : Official Journal of the American Society of Neuroimaging, 22(3), 285–291. doi: 10.1111/j.1552-6569.2011.00614.x [doi] [DOI] [PubMed] [Google Scholar]
- Dadgar H, Khatoonabadi A, & Bakhtiyari J (2013). Verbal fluency performance in patients with non-demented parkinson’s disease. Iran J Psychiatry, 8(1), 55–58. [PMC free article] [PubMed] [Google Scholar]
- Daniele A, Barbier A, Di Giuda D, Vita MG, Piccininni C, Spinelli P, … Gainotti G (2013). Selective impairment of action-verb naming and comprehension in progressive supranuclear palsy. Cortex; a Journal Devoted to the Study of the Nervous System and Behavior, 49(4), 948–960. doi: 10.1016/j.cortex.2012.03.024 [doi] [DOI] [PubMed] [Google Scholar]
- Deng H, Le W, & Jankovic J (2004). Premutation alleles associated with parkinson disease and essential tremor. Jama, 292(14), 1685–1686. [DOI] [PubMed] [Google Scholar]
- Deuschl G, Bain P, & Brin M (1998). Consensus statement of the movement disorder society on tremor. Movement Disorders, 13(S3), 2–23. [DOI] [PubMed] [Google Scholar]
- Deuschl G, & Elble R (2009). Essential tremor--neurodegenerative or nondegenerative disease towards a working definition of ET. Movement Disorders : Official Journal of the Movement Disorder Society, 24(14), 2033–2041. doi: 10.1002/mds.22755 [doi] [DOI] [PubMed] [Google Scholar]
- Deuschl G, Wenzelburger R, Loffler K, Raethjen J, & Stolze H (2000). Essential tremor and cerebellar dysfunction clinical and kinematic analysis of intention tremor. Brain : A Journal of Neurology, 123 (Pt 8)(Pt 8), 1568–1580. [DOI] [PubMed] [Google Scholar]
- Dohlinger S, Hauser TK, Borkert J, Luft AR, & Schulz JB (2008). Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum (London, England), 7(2), 204–214. doi: 10.1007/s12311-008-0025-0 [doi] [DOI] [PubMed] [Google Scholar]
- Donato SD, Mariotti C, & Taroni F (2012). Spinocerebellar ataxia type 1. Handbook of Clinical Neurology, 103, 399–421. doi: 10.1016/B978-0-444-51892-7.00025-5 [doi] [DOI] [PubMed] [Google Scholar]
- Dujardin K, Defebvre L, Krystkowiak P, Degreef JF, & Destee A (2003). Executive function differences in multiple system atrophy and parkinson’s disease. Parkinsonism & Related Disorders, 9(4), 205–211. doi:S1353802002000500 [pii] [DOI] [PubMed] [Google Scholar]
- Durr A (2010). Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. The Lancet.Neurology, 9(9), 885–894. doi: 10.1016/S1474-4422(10)70183-6 [doi] [DOI] [PubMed] [Google Scholar]
- Durr A, Brice A, Lepage-Lezin A, Cancel G, Smadja D, Vernant JC, & Agid Y (1995). Autosomal dominant cerebellar ataxia type I linked to chromosome 12q (SCA2: Spinocerebellar ataxia type 2). Clinical Neuroscience (New York, N.Y.), 3(1), 12–16. [PubMed] [Google Scholar]
- Elble RJ (2000). Essential tremor frequency decreases with time. Neurology, 55(10), 1547–1551. [DOI] [PubMed] [Google Scholar]
- Esmonde T, Giles E, Gibson M, & Hodges JR (1996). Neuropsychological performance, disease severity, and depression in progressive supranuclear palsy. Journal of Neurology, 243(9), 638–643. [DOI] [PubMed] [Google Scholar]
- Esmonde T, Giles E, Xuereb J, & Hodges J (1996). Progressive supranuclear palsy presenting with dynamic aphasia. Journal of Neurology, Neurosurgery, and Psychiatry, 60(4), 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada R, Galarraga J, Orozco G, Nodarse A, & Auburger G (1999). Spinocerebellar ataxia 2 (SCA2): Morphometric analyses in 11 autopsies. Acta Neuropathol, 97(3), 306–310. [DOI] [PubMed] [Google Scholar]
- Fahn S, Elton R, & UPDRS Program Members. (1987). Unified parkinson’s disease rating scale In Fahn S, Marsden C, Goldstein M & Calne D (Eds.), Recent developments in parkinson’s disease, vol. 2 (2nd ed., pp. 153–163- 293–304). Florham Park, NJ: Macmillan Healthcare Information. [Google Scholar]
- Fallon SJ, Hampshire A, Williams-Gray CH, Barker RA, & Owen AM (2013). Putative cortical dopamine levels affect cortical recruitment during planning. Neuropsychologia, 51(11), 2194–2201. doi: 10.1016/j.neuropsychologia.2013.07.016 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon SJ, Williams-Gray CH, Barker RA, Owen AM, & Hampshire A (2013). Prefrontal dopamine levels determine the balance between cognitive stability and flexibility. Cerebral Cortex (New York, N.Y.: 1991), 23(2), 361–369. doi: 10.1093/cercor/bhs025 [doi] [DOI] [PubMed] [Google Scholar]
- Fan J, Gu X, Guise KG, Liu X, Fossella J, Wang H, & Posner MI (2009). Testing the behavioral interaction and integration of attentional networks. Brain and Cognition, 70(2), 209–220. doi: 10.1016/j.bandc.2009.02.002 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancellu R, Paridi D, Tomasello C, Panzeri M, Castaldo A, Genitrini S, … Girotti F (2013). Longitudinal study of cognitive and psychiatric functions in spinocerebellar ataxia types 1 and 2. J Neurol, 260(12), 3134–3143. [DOI] [PubMed] [Google Scholar]
- Faruq M, Srivastava A, & Suroliya V (2014). Identification of FXTAS presenting with SCA 12 like phenotype in india. Parkinsonism Relat Disord, 20(10), 1089–1093. [DOI] [PubMed] [Google Scholar]
- Feng L, Chen D, Hou L, Huang L, Lu S, Liang X, & Li X (2014). Cognitive impairment in native chinese with spinocerebellar ataxia type 3. Eur Neurol, 71(5–6), 262–270. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lago H, Bello O, Lopez-Alonso V, Sanchez JA, Morenilla L, & Fernandez-del-Olmo MA (2015). Gait pattern and cognitive performance during treadmill walking in parkinson disease. American Journal of Physical Medicine & Rehabilitation / Association of Academic Physiatrists, 94(11), 931–940. doi: 10.1097/PHM.0000000000000392 [doi] [DOI] [PubMed] [Google Scholar]
- Filley CM, Brown MS, Onderko K, Ray M, Bennett RE, Berry-Kravis E, & Grigsby J (2015). White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology, 84(21), 2146–2152. doi: 10.1212/WNL.0000000000001612 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsaa EB, Larsen JP, Wentzel-Larsen T, Goetz CG, Stebbins GT, Aarsland D, & Alves G (2010). A 12-year population-based study of psychosis in parkinson disease. Archives of Neurology, 67(8), 996–1001. doi: 10.1001/archneurol.2010.166 [doi] [DOI] [PubMed] [Google Scholar]
- Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, & Raichle ME (2005). The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proceedings of the National Academy of Sciences of the United States of America, 102(27), 9673–9678. doi:0504136102 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraint A, Vittal P, & Szewka A (2014). New observations in the fragile X-associated tremor/ataxia syndrome (FXTAS) phenotype. Front Genet, 5, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka S, Algom AA, Murray ME, Sanchez-Contreras MY, Tacik P, Tsuboi Y, … Ross OA (2016). Tremor in progressive supranuclear palsy. Parkinsonism & Related Disorders, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher DA, & Schrag A (2012). Psychosis, apathy, depression and anxiety in parkinson’s disease. Neurobiology of Disease, 46(3), 581–589. doi: 10.1016/j.nbd.2011.12.041 [doi] [DOI] [PubMed] [Google Scholar]
- Garland E, Vnencak-Jones C, & Biaggioni I (2004). Fragile X gene premutation in multiple system atrophy. J Neurol Sci, 227(1), 115–118. [DOI] [PubMed] [Google Scholar]
- Garrard P, Martin N, Giunti P, & Cipolotti L (2008). Cognitive and social cognitive functioning in spinocerebellar ataxia : A preliminary characterization. J Neurol, 255(3), 398–405. [DOI] [PubMed] [Google Scholar]
- Gerstenecker A, Duff K, Mast B, Litvan I, & ENGENE-PSP Study Group. (2013). Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Research, 210(3), 1205–1210. doi: 10.1016/j.psychres.2013.08.045 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstenecker A, Mast B, Duff K, Ferman TJ, Litvan I, & ENGENE-PSP Study Group. (2013). Executive dysfunction is the primary cognitive impairment in progressive supranuclear palsy. Archives of Clinical Neuropsychology : The Official Journal of the National Academy of Neuropsychologists, 28(2), 104–113. doi: 10.1093/arclin/acs098 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghika A, Kyrozis A, Potagas C, & Louis ED (2015). Motor and non-motor features: Differences between patients with isolated essential tremor and patients with both essential tremor and Parkinson’s Disease. Tremor and Other Hyperkinetic Movements, 5, 335. doi: 10.7916/D83777WK [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh BC, Carpenter RH, & Rowe JB (2013). A longitudinal study of motor, oculomotor and cognitive function in progressive supranuclear palsy. PloS One, 8(9), e74486. doi: 10.1371/journal.pone.0074486 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SJ, & Burgess PW (2008). Executive function. Current Biology : CB, 18(3), R110–4. doi: 10.1016/j.cub.2007.12.014 [doi] [DOI] [PubMed] [Google Scholar]
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, … Vidailhet M (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71(9), 670–676. doi: 10.1212/01.wnl.0000324625.00404.15 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globas C, Bösch S, Zühlke C, Daum I, Dichgans J, & Bürk K (2003). The cerebellum and cognition. intellectual function in spinocerebellar ataxia type 6 (SCA6). J Neurol, 250(12), 1482–1487. [DOI] [PubMed] [Google Scholar]
- Goldman JG, Aggarwal NT, & Schroeder CD (2015). Mild cognitive impairment: An update in parkinson’s disease and lessons learned from alzheimer’s disease. Neurodegenerative Disease Management, doi: 10.2217/nmt.15.34 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JG, Williams-Gray C, Barker RA, Duda JE, & Galvin JE (2014). The spectrum of cognitive impairment in lewy body diseases. Movement Disorders : Official Journal of the Movement Disorder Society, 29(5), 608–621. doi: 10.1002/mds.25866 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishna A, & Alexander SA (2015). Understanding parkinson disease: A complex and multifaceted illness. The Journal of Neuroscience Nursing : Journal of the American Association of Neuroscience Nurses, 47(6), 320–326. doi: 10.1097/JNN.0000000000000162 [doi] [DOI] [PubMed] [Google Scholar]
- Gorman G, Fairgrieve S, Birchall D, & Chinnery P (2008). Fragile X premutation presenting as essential tremor. J Neurol Neurosurg Psychiatry, 79(10), 1195–1196. [DOI] [PubMed] [Google Scholar]
- Grafman J, Litvan I, Gomez C, & Chase TN (1990). Frontal lobe function in progressive supranuclear palsy. Archives of Neurology, 47(5), 553–558. [DOI] [PubMed] [Google Scholar]
- Greco CM, Berman RF, Martin RM, Tassone F, Schwartz PH, Chang A, … Hagerman PJ (2006). Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain : A Journal of Neurology, 129(Pt 1), 243–255. doi: 10.1093/brain/awh683 [DOI] [PubMed] [Google Scholar]
- Greco CM, Hagerman RJ, Tassone F, Chudley AE, Del Bigio MR, Jacquemont S, … Hagerman PJ (2002). Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain : A Journal of Neurology, 125(Pt 8), 1760–1771. [DOI] [PubMed] [Google Scholar]
- Greco CM, Tassone F, Garcia-Arocena D, Tartaglia N, Coffey SM, Vartanian TK, … Hagerman RJ (2008). Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Archives of Neurology, 65(8), 1114–1116. doi: 10.1001/archneur.65.8.1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigsby J, Brega A, & Leehey M (2007). Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Mov Disord, 22(5), 645–650. [DOI] [PubMed] [Google Scholar]
- Grigsby J, Brega AG, Engle K, Leehey MA, Hagerman RJ, Tassone F, … Reynolds A (2008). Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology, 22(1), 48–60. doi: 10.1037/0894-4105.22.1.48 [DOI] [PubMed] [Google Scholar]
- Grigsby J, Brega AG, Jacquemont S, Loesch DZ, Leehey MA, Goodrich GK, … Hagerman PJ (2006). Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS). Journal of the Neurological Sciences, 248(1–2), 227–233. doi: 10.1016/j.jns.2006.05.016 [DOI] [PubMed] [Google Scholar]
- Grigsby J, Brega AG, Leehey MA, Goodrich GK, Jacquemont S, Loesch DZ, … Hagerman RJ (2007). Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Movement Disorders : Official Journal of the Movement Disorder Society, 22(5), 645–650. doi: 10.1002/mds.21359 [DOI] [PubMed] [Google Scholar]
- Grigsby J, Cornish K, Hocking D, Kraan C, Olichney JM, Rivera SM, … Yang JC (2014). The cognitive neuropsychological phenotype of carriers of the FMR1 premutation. Journal of Neurodevelopmental Disorders, 6(1), 28-1955-6-28. Epub 2014 Jul 30. doi: 10.1186/1866-1955-6-28 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ, & Hagerman RJ (2015). Fragile X-associated tremor/ataxia syndrome. Annals of the New York Academy of Sciences, 1338(1), 58–70. doi: 10.1111/nyas.12693 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, … Hagerman PJ (2004). Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. American Journal of Human Genetics, 74(5), 1051–1056. doi: 10.1086/420700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, … Hagerman PJ (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology, 57(1), 127–130. [DOI] [PubMed] [Google Scholar]
- Hall D, Tassone F, Klepitskaya O, & Leehey M (2012). Fragile X-associated tremor ataxia syndrome in FMR1 gray zone allele carriers. Mov Disord, 27(2), 296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Robertson-Dick E, O’Keefe JA, Hadd A, Zhou L, & Berry-Kravis E (2016). X-inactivation in the clinical phenotype of fragile X premutation carrier sisters Neurol Genet, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D, Berry-Kravis E, & Jacquemont S (2005). Initial diagnoses given to persons with the fragile X associated tremor/ataxia syndrome (FXTAS). Neurology, 65(2), 299–301. [DOI] [PubMed] [Google Scholar]
- Hall D, Berry-Kravis E, & Zhang W (2011). FMR1 gray-zone alleles: Association with parkinson’s disease in women?. Mov Disord, 26(10), 1900–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D, Howard K, & Hagerman R (2009). Parkinsonism in FMR1 premutation carriers may be indistinguishable from parkinson disease. Parkinsonism Relat Disord, 15(2), 156–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D, & O’keefe J (2012). Fragile x-associated tremor ataxia syndrome: The expanding clinical picture, pathophysiology, epidemiology, and update on treatment. Tremor Other Hyperkinet Mov (N Y), 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Berry-Kravis E, Jacquemont S, Rice CD, Cogswell J, Zhang L, … Leehey MA (2005). Initial diagnoses given to persons with the fragile X associated tremor/ataxia syndrome (FXTAS). Neurology, 65(2), 299–301. doi: 10.1212/01.wnl.0000168900.86323.9c [DOI] [PubMed] [Google Scholar]
- Harding AE (1993). Clinical features and classification of inherited ataxias. Advances in Neurology, 61, 1–14. [PubMed] [Google Scholar]
- Hashimoto R, Backer KC, Tassone F, Hagerman RJ, & Rivera SM (2011). An fMRI study of the prefrontal activity during the performance of a working memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Journal of Psychiatric Research, 45(1), 36–43. doi: 10.1016/j.jpsychires.2010.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Javan AK, Tassone F, Hagerman RJ, & Rivera SM (2011). A voxel-based morphometry study of grey matter loss in fragile X-associated tremor/ataxia syndrome. Brain : A Journal of Neurology, 134(Pt 3), 863–878. doi: 10.1093/brain/awq368; 10.1093/brain/awq368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Srivastava S, Tassone F, Hagerman RJ, & Rivera SM (2011). Diffusion tensor imaging in male premutation carriers of the fragile X mental retardation gene. Movement Disorders : Official Journal of the Movement Disorder Society, 26(7), 1329–1336. doi: 10.1002/mds.23646; 10.1002/mds.23646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Bressman S, Dickson J, Silveira-Moriyama L, Schneider SA, Sullivan SS, … Bomanji J (2009). Evidence for pre and postsynaptic nigrostriatal dysfunction in the fragile X tremor–Ataxia syndrome. Movement Disorders, 24(8), 1245–1247. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Pramstaller P, & Stübke K (2005). Premutations in the FMR1 gene as a modifying factor in parkin-associated parkinson’s disease?. Mov Disord, 20(8), 1060–1062. [DOI] [PubMed] [Google Scholar]
- Helmchen C, Hagenow A, Miesner J, Sprenger A, Rambold H, Wenzelburger R, … Deuschl G (2003). Eye movement abnormalities in essential tremor may indicate cerebellar dysfunction. Brain : A Journal of Neurology, 126(Pt 6), 1319–1332. [DOI] [PubMed] [Google Scholar]
- Hely MA, Reid WG, Adena MA, Halliday GM, & Morris JG (2008). The sydney multicenter study of parkinson’s disease: The inevitability of dementia at 20 years. Movement Disorders : Official Journal of the Movement Disorder Society, 23(6), 837–844. doi: 10.1002/mds.21956 [doi] [DOI] [PubMed] [Google Scholar]
- Hernandez-Castillo CR, Galvez V, Mercadillo RE, Diaz R, Yescas P, Martinez L, … Fernandez-Ruiz J (2015). Functional connectivity changes related to cognitive and motor performance in spinocerebellar ataxia type 2. Movement Disorders : Official Journal of the Movement Disorder Society, 30(10), 1391–1399. doi: 10.1002/mds.26320 [doi] [DOI] [PubMed] [Google Scholar]
- Herting B, Beuthien-Baumann B, Pottrich K, Donix M, Triemer A, Lampe JB, … Holthoff VA (2007). Prefrontal cortex dysfunction and depression in atypical parkinsonian syndromes. Movement Disorders : Official Journal of the Movement Disorder Society, 22(4), 490–497. doi: 10.1002/mds.21237 [doi] [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera S, Koldewyn K, Cordeiro L, Adams J, Tassone F, … Hagerman RJ (2007). Amygdala dysfunction in men with the fragile X premutation. Brain : A Journal of Neurology, 130(Pt 2), 404–416. doi: 10.1093/brain/awl338 [DOI] [PubMed] [Google Scholar]
- Heyder K, Suchan B, & Daum I (2004). Cortico-subcortical contributions to executive control. Acta Psychologica, 115(2–3), 271–289. doi: 10.1016/j.actpsy.2003.12.010 [doi] [DOI] [PubMed] [Google Scholar]
- Hobson P, & Meara J (2015). Mild cognitive impairment in parkinson’s disease and its progression onto dementia: A 16-year outcome evaluation of the denbighshire cohort. International Journal of Geriatric Psychiatry, 30(10), 1048–1055. doi: 10.1002/gps.4261 [doi] [DOI] [PubMed] [Google Scholar]
- Hoehn M, & Yahr M (1967). Parkinsonism: Onset, progression and mortality. Neurology, 17, 427–442. [DOI] [PubMed] [Google Scholar]
- Hong HJ, Song SK, Lee PH, Sohn YH, & Lee JE (2011). Cognitive impairments in multiple system atrophy of the cerebellar type. Journal of Movement Disorders, 4(1), 41–45. doi: 10.14802/jmd.11007 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner F, Erhart T, Knudsen K, Lorenz D, Schneider SA, Zeuner KE, … Kuhlenbaumer G (2015). Testing for alcohol sensitivity of tremor amplitude in a large cohort with essential tremor. Parkinsonism & Related Disorders, 21(8), 848–851. doi: 10.1016/j.parkreldis.2015.05.005 [doi] [DOI] [PubMed] [Google Scholar]
- Hoskovcová M, Ulmanová O, Šprdlík O, Sieger T, Nováková J, Jech R, & Růžička E (2013). Disorders of balance and gait in essential tremor are associated with midline tremor and age. The Cerebellum, 12(1), 27–34. [DOI] [PubMed] [Google Scholar]
- Hubble J, Busenbark K, Pahwa R, Lyons K, & Koller W (1997). Clinical expression of essential tremor: Effects of gender and age. Movement Disorders, 12(6), 969–972. [DOI] [PubMed] [Google Scholar]
- Hunter JE, Sherman S, Grigsby J, Kogan C, & Cornish K (2012). Capturing the fragile X premutation phenotypes: A collaborative effort across multiple cohorts. Neuropsychology, 26(2), 156–164. doi: 10.1037/a0026799; 10.1037/a0026799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K, Hosaka A, Adachi K, Nanba E, & Tamaoka A (2010). A japanese case of fragile-X-associated tremor/ataxia syndrome (FXTAS). Internal Medicine (Tokyo, Japan), 49(12), 1205–1208. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Mori K, Ito M, Tatsumi S, Mimuro M, & Yoshida M (2013). An autopsied case of progressive supranuclear palsy presenting with cerebellar ataxia and severe cerebellar involvement. Neuropathology, 33(5), 561–567. [DOI] [PubMed] [Google Scholar]
- Jacobi H, du Montcel ST, Bauer P, Giunti P, Cook A, Labrum R, … Klockgether T (2015). Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: A longitudinal cohort study. The Lancet.Neurology, 14(11), 1101–1108. doi: 10.1016/S1474-4422(15)00202-1 [doi] [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman R, Leehey M, Grigsby J, Zhang L, & Brunberg J (2003). Fragile X premutation tremor/ataxia syndrome: Molecular, clinical, and neuroimaging correlates. Am J Hum Genet, 72(869–878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang EH, Lee JK, Jang HJ, Kim MJ, & Chung SJ (2012). A case of multiple system atrophy-cerebellar type preceded by dementia. Journal of Movement Disorders, 5(2), 48–52. doi: 10.14802/jmd.12011 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J (2002). Essential tremor: A heterogenous disorder. Mov Disord, 17(4), 638–644. [DOI] [PubMed] [Google Scholar]
- Jankovic J, Beach J, Schwartz K, & Contant C (1995). Tremor and longevity in relatives of patients with parkinson’s disease, essential tremor, and control subjects. Neurology, 45(4), 645–648. [DOI] [PubMed] [Google Scholar]
- Janvin CC, Aarsland D, & Larsen JP (2005). Cognitive predictors of dementia in parkinson’s disease: A community-based, 4-year longitudinal study. Journal of Geriatric Psychiatry and Neurology, 18(3), 149–154. doi:18/3/149 [pii] [DOI] [PubMed] [Google Scholar]
- Janvin CC, Larsen JP, Salmon DP, Galasko D, Hugdahl K, & Aarsland D (2006). Cognitive profiles of individual patients with parkinson’s disease and dementia: Comparison with dementia with lewy bodies and alzheimer’s disease. Movement Disorders : Official Journal of the Movement Disorder Society, 21(3), 337–342. doi: 10.1002/mds.20726 [doi] [DOI] [PubMed] [Google Scholar]
- Jhunjhunwala K, & Pal PK (2014). The non-motor features of essential tremor: A primary disease feature or just a secondary phenomenon? Tremor and Other Hyperkinetic Movements (New York, N.Y.), 4, 255. doi: 10.7916/D8D798MZ [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Jiménez F, Rubio L, Alonso-Navarro H, Calleja M, Pilo-de-la-Fuente B, Plaza-Nieto J, … Agúndez J (2010). Impairment of rapid repetitive finger movements and visual reaction time in patients with essential tremor. European Journal of Neurology, 17(1), 152–159. [DOI] [PubMed] [Google Scholar]
- Josephs KA (2015). Key emerging issues in progressive supranuclear palsy and corticobasal degeneration. Journal of Neurology, 262(3), 783–788. doi: 10.1007/s00415-015-7682-y [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncos J, Lazarus J, Graves-Allen E, Shubeck L, Rusin M, Novak G, … Sherman S (2011). New clinical findings in the fragile X-associated tremor ataxia syndrome (FXTAS) Neurogenetics, 12(2), 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado MB, & Rosselli M (2007). The elusive nature of executive functions: A review of our current understanding. Neuropsychology Review, 17(3), 213–233. doi: 10.1007/s11065-007-9040-z [doi] [DOI] [PubMed] [Google Scholar]
- Kaat DL, Chiu WZ, Boon AJ, & van Swieten JC (2011). Recent advances in progressive supranuclear palsy: A review. Current Alzheimer Research, 8(3), 295–302. doi:BSP/CAR /0120 [pii] [DOI] [PubMed] [Google Scholar]
- Kaindlstorfer C, Granata R, & Wenning GK (2013). Tremor in multiple system atrophy - a review. Tremor and Other Hyperkinetic Movements (New York, N.Y.), 3, tre-03-165-4252-1. eCollection 2013. doi:tre-03-165-4252-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamm C, Healy D, & Quinn N (2005). The fragile X tremor ataxia syndrome in the differential diagnosis of multiple system atrophy: Data from the EMSA study group. Brain, 128(Pt 8), 1855–1860. [DOI] [PubMed] [Google Scholar]
- Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T, … Nishizawa M (2009). Cerebellar involvement in progressive supranuclear palsy: A clinicopathological study. Movement Disorders, 24(9), 1312–1318. [DOI] [PubMed] [Google Scholar]
- Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, & Shimohata T (2013). Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism & Related Disorders, 19(12), 1149–1151. [DOI] [PubMed] [Google Scholar]
- Kanazawa I (1998). Dentatorubral-pallidoluysian atrophy or naito-oyanagi disease. Neurogenetics, 2(1), 1–17. doi:9800056 [pii] [DOI] [PubMed] [Google Scholar]
- Kao AW, Racine CA, Quitania LC, Kramer JH, Christine CW, & Miller BL (2009). Cognitive and neuropsychiatric profile of the synucleinopathies: Parkinson disease, dementia with lewy bodies, and multiple system atrophy. Alzheimer Disease and Associated Disorders, 23(4), 365–370. doi: 10.1097/WAD.0b013e3181b5065d [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzoulis S, & Galvin JE (2013). Update on dementia with lewy bodies. Current Translational Geriatrics and Experimental Gerontology Reports, 2(3), 196–204. doi: 10.1007/s13670-013-0053-6 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmon Y, & Gadoth N (2008). Fragile X associated tremor/ataxia syndrome (FXTAS) with dementia in a female harbouring FMR1 premutation. Journal of Neurology, Neurosurgery, and Psychiatry, 79(6), 738–739. doi: 10.1136/jnnp.2007.139642 [DOI] [PubMed] [Google Scholar]
- Karrasch M, Laatu S, Martikainen K, & Marttila R (2013). CERAD test performance and cognitive impairment in parkinson’s disease. Acta Neurol Scand, 128(6), 409–413. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Suenaga M, Watanabe H, Ito M, Kato K, Kato T, … Sobue G (2008a). Prefrontal hypoperfusion and cognitive dysfunction correlates in spinocerebellar ataxia type 6. J Neurol Sci, 15(271(1–2)), 68–74. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Suenaga M, Takeda A, Ito M, Watanabe H, Tanaka F, … Sobue G (2008b). Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology, 70(16 Pt 2), 1390–1396. doi: 10.1212/01.wnl.0000310413.04462.6a [doi] [DOI] [PubMed] [Google Scholar]
- Kawai Y, Takeda A, Abe Y, Washimi Y, Tanaka F, & Sobue G (2004). Cognitive impairments in machado-joseph disease. Archives of Neurology, 61(11), 1757–1760. doi:61/11/1757 [pii] [DOI] [PubMed] [Google Scholar]
- Kawamura K, Shimohata T, Nakayama H, Tomita M, Ozawa T, & Nishizawa M (2010). Factors influencing the cognitive function in patients with multiple system atrophy. Movement Disorders : Official Journal of the Movement Disorder Society, 25(16), 2891–2892. doi: 10.1002/mds.23260 [doi] [DOI] [PubMed] [Google Scholar]
- Kerber K, Jen J, & Perlman S (2005). Late-onset pure cerebellar ataxia: Differentiating those with and without identifiable mutations. J Neurol Sci, 238(1–2), 41–45. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jeon BS, Kim YE, Kim JY, Kim YK, Sohn CH, … Lee JY (2013). Clinical and imaging characteristics of dementia in multiple system atrophy. Parkinsonism & Related Disorders, 19(6), 617–621. doi: 10.1016/j.parkreldis.2013.02.012 [doi] [DOI] [PubMed] [Google Scholar]
- Kim JS, Yang JJ, Lee DK, Lee JM, Youn J, & Cho JW (2015). Cognitive impairment and its structural correlates in the parkinsonian subtype of multiple system atrophy. Neuro-Degenerative Diseases, 15(5), 294–300. doi: 10.1159/000430953 [doi] [DOI] [PubMed] [Google Scholar]
- Kim JS, Youn J, Yang JJ, Lee DK, Lee JM, Kim ST, … Cho JW (2013). Topographic distribution of cortical thinning in subtypes of multiple system atrophy. Parkinsonism & Related Disorders, 19(11), 970–974. doi: 10.1016/j.parkreldis.2013.06.012 [doi] [DOI] [PubMed] [Google Scholar]
- Kim WH, Lee YS, Jung SH, Choi HJ, Lee MJ, Kang MH, … Bae JN (2009). Major depressive disorder preceding the onset of progressive supranuclear palsy. Psychiatry Investigation, 6(2), 112–114. doi: 10.4306/pi.2009.6.2.112 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama M, Wada-Isoe K, Irizawa Y, & Nakashima K (2009). Assessment of dementia in patients with multiple system atrophy. European Journal of Neurology, 16(5), 589–594. doi: 10.1111/j.1468-1331.2009.02544.x [doi] [DOI] [PubMed] [Google Scholar]
- Klatka LA, Louis ED, & Schiffer RB (1996). Psychiatric features in diffuse lewy body disease: A clinicopathologic study using alzheimer’s disease and parkinson’s disease comparison groups. Neurology, 47(5), 1148–1152. [DOI] [PubMed] [Google Scholar]
- Klebe S, Durr A, Rentschler A, Hahn-Barma V, Abele M, Bouslam N, … Stevanin G (2005). New mutations in protein kinase cgamma associated with spinocerebellar ataxia type 14. Annals of Neurology, 58(5), 720–729. doi: 10.1002/ana.20628 [doi] [DOI] [PubMed] [Google Scholar]
- Klein C, Schneider S, & Lang A (2009). Hereditary parkinsonism: Parkinson disease look-alikes--an algorithm for clinicians to “PARK” genes and beyond. Mov Disord, 24(14), 2042–2058. [DOI] [PubMed] [Google Scholar]
- Klinke I, Minnerop M, Schmitz-Hübsch T, Hendriks M, Klockgether T, Wüllner U, & Helmstaedter C (2010). Neuropsychological features of patients with spinocerebellar ataxia (SCA) types 1, 2, 3, and 6. Cerebellum, 9(3), 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerts J, Meijer H, Colman K, Tucha L, Lange K, & Tucha O (2013). What is measured with verbal fluency tests in parkinson’s disease patients at different stages of the disease?. J Neural Transm (Vienna), 120(3), 403–411. [DOI] [PubMed] [Google Scholar]
- Koga S, Josephs KA, Ogaki K, Labbé C, Uitti RJ, Graff-Radford N, … Rademakers R (2016). Cerebellar ataxia in progressive supranuclear palsy: An autopsy study of PSP-C. Movement Disorders, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konagaya M, Konagaya Y, Sakai M, Matsuoka Y, & Hashizume Y (2002). Progressive cerebral atrophy in multiple system atrophy. Journal of the Neurological Sciences, 195(2), 123–127. doi:S0022510X0100692X [pii] [DOI] [PubMed] [Google Scholar]
- Konagaya M, Sakai M, Matsuoka Y, Konagaya Y, & Hashizume Y (1999). Multiple system atrophy with remarkable frontal lobe atrophy. Acta Neuropathologica, 97(4), 423–428. [DOI] [PubMed] [Google Scholar]
- Koutsis G, Panas M, Paraskevas GP, Bougea AM, Kladi A, Karadima G, & Kapaki E (2014). From mild ataxia to huntington disease phenocopy: The multiple faces of spinocerebellar ataxia 17. Case Reports in Neurological Medicine, 2014, 643289. doi: 10.1155/2014/643289 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft S, Furtado S, & Ranawaya R (2005). Adult onset spinocerebellar ataxia in a canadian movement disorders clinic. Can J Neurol Sci, 32(4), 450–458. [DOI] [PubMed] [Google Scholar]
- Krienen FM, & Buckner RL (2009). Segregated fronto-cerebellar circuits revealed by intrinsic functional connectivity. Cerebral Cortex (New York, N.Y.: 1991), 19(10), 2485–2497. doi: 10.1093/cercor/bhp135 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenbuerger M, Konczak J, Ziegler W, Buderath P, Frank B, Coenen VA, … Timmann D (2009). Balance and motor speech impairment in essential tremor. The Cerebellum, 8(3), 389–398. [DOI] [PubMed] [Google Scholar]
- Kronenbuerger M, Gerwig M, Brol B, Block F, & Timmann D (2007). Eyeblink conditioning is impaired in subjects with essential tremor. Brain : A Journal of Neurology, 130(Pt 6), 1538–1551. doi:awm081 [pii] [DOI] [PubMed] [Google Scholar]
- Lang AE, Kierans C, & Blair RD (1987). Family history of tremor in parkinson’s disease compared with those of controls and patients with idiopathic dystonia. Advances in Neurology, 45, 313–316. [PubMed] [Google Scholar]
- Lanni K, Ross J, Higginson C, Dressler E, Sigvardt K, Zhang L, … Disbrow E (2014). Perceived and performance-based executive dysfunction in parkinson’s disease. J Clin Exp Neuropsychol, 36(4), 342–355. [DOI] [PubMed] [Google Scholar]
- Lavie N, Hirst A, de Fockert JW, & Viding E (2004). Load theory of selective attention and cognitive control. Journal of Experimental Psychology.General, 133(3), 339–354. doi: 10.1037/0096-3445.133.3.339 [doi] [DOI] [PubMed] [Google Scholar]
- Lee MJ, Shin JH, Seoung JK, Lee JH, Yoon U, Oh JH, … Kim EJ (2015). Cognitive impairments associated with morphological changes in cortical and subcortical structures in multiple system atrophy of the cerebellar type. European Journal of Neurology : The Official Journal of the European Federation of Neurological Societies, doi: 10.1111/ene.12796 [doi] [DOI] [PubMed] [Google Scholar]
- Lee MS, Kim YD, Im JH, Kim HJ, Rinne JO, & Bhatia KP (1999). 123I-IPT brain SPECT study in essential tremor and parkinson’s disease. Neurology, 52(7), 1422–1426. [DOI] [PubMed] [Google Scholar]
- Leehey M, Berry-Kravis E, Goetz C, Zhang L, Hall D, Li L, … Hagerman P (2008). FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology, 70(16 Pt 2), 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leehey M, Munhoz R, Lang A, Brunberg J, Grigsby J, Greco C, … Hagerman R (2003). The fragile X premutation presenting as essential tremor. Arch Neurol, 60(1), 117–121. [DOI] [PubMed] [Google Scholar]
- Leehey MA (2009). Fragile X-associated tremor/ataxia syndrome: Clinical phenotype, diagnosis, and treatment. Journal of Investigative Medicine : The Official Publication of the American Federation for Clinical Research, 57(8), 830–836. doi: 10.231/JIM.0b013e3181af59c4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leehey MA, Berry-Kravis E, Min SJ, Hall DA, Rice CD, Zhang L, … Hagerman PJ (2007). Progression of tremor and ataxia in male carriers of the FMR1 premutation. Movement Disorders : Official Journal of the Movement Disorder Society, 22(2), 203–206. doi: 10.1002/mds.21252 [DOI] [PubMed] [Google Scholar]
- Legros B, & Manto MU (1999). Autosomal dominant spinocerebellar ataxia. [Les ataxies spino-cerebelleuses (SCA) autosomiques dominantes] Revue Medicale De Bruxelles, 20(6), 495–503. [PubMed] [Google Scholar]
- Levin J, Hasan A, & Hoglinger GU (2015). Psychosis in parkinson’s disease: Identification, prevention and treatment. Journal of Neural Transmission (Vienna, Austria : 1996), doi: 10.1007/s00702-015-1400-x [doi] [DOI] [PubMed] [Google Scholar]
- Levin J, Kurz A, Arzberger T, Giese A, & Hoglinger GU (2016). The differential diagnosis and treatment of atypical parkinsonism. Deutsches Arzteblatt International, 113(5), 61–69. doi: 10.3238/arztebl.2016.0061 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy G, Jacobs DM, Tang MX, Cote LJ, Louis ED, Alfaro B, … Marder K (2002). Memory and executive function impairment predict dementia in parkinson’s disease. Movement Disorders : Official Journal of the Movement Disorder Society, 17(6), 1221–1226. doi: 10.1002/mds.10280 [doi] [DOI] [PubMed] [Google Scholar]
- Levy R, & Dubois B (2006). Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cerebral Cortex (New York, N.Y.: 1991), 16(7), 916–928. doi:bhj043 [pii] [DOI] [PubMed] [Google Scholar]
- Lilja A, Hamalainen P, Kaitaranta E, & Rinne R (2005). Cognitive impairment in spinocerebellar ataxia type 8. Journal of the Neurological Sciences, 237(1–2), 31–38. doi:S0022–510X(05)00191–7 [pii] [DOI] [PubMed] [Google Scholar]
- Liscic R, Srulijes K, Gröger A, Maetzler W, & Berg D (2013). Differentiation of progressive supranuclear palsy: Clinical, imaging and laboratory tools. Acta Neurologica Scandinavica, 127(5), 362–370. [DOI] [PubMed] [Google Scholar]
- Litvan I, Agid Y, & Calne D (1996). Clinical research criteria for the diagnosis of progressive supranuclear palsy (steele-richardson-olszewski syndrome): Report of the NINDS-SPSP international workshop. Neurology, 47(1), 1–9. [DOI] [PubMed] [Google Scholar]
- Litvan I, Aarsland D, Adler CH, Goldman JG, Kulisevsky J, Mollenhauer B, … Weintraub D (2011). MDS task force on mild cognitive impairment in parkinson’s disease: Critical review of PD-MCI. Movement Disorders : Official Journal of the Movement Disorder Society, 26(10), 1814–1824. doi: 10.1002/mds.23823 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Goldman JG, Troster AI, Schmand BA, Weintraub D, Petersen RC, … Emre M (2012). Diagnostic criteria for mild cognitive impairment in parkinson’s disease: Movement disorder society task force guidelines. Movement Disorders : Official Journal of the Movement Disorder Society, 27(3), 349–356. doi: 10.1002/mds.24893 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Grafman J, Gomez C, & Chase TN (1989). Memory impairment in patients with progressive supranuclear palsy. Archives of Neurology, 46(7), 765–767. [DOI] [PubMed] [Google Scholar]
- Litvan I, Mangone CA, McKee A, Verny M, Parsa A, Jellinger K, … Pearce RK (1996). Natural history of progressive supranuclear palsy (steele-richardson-olszewski syndrome) and clinical predictors of survival: A clinicopathological study. Journal of Neurology, Neurosurgery, and Psychiatry, 60(6), 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Mega MS, Cummings JL, & Fairbanks L (1996). Neuropsychiatric aspects of progressive supranuclear palsy. Neurology, 47(5), 1184–1189. [DOI] [PubMed] [Google Scholar]
- Lo RY, Figueroa KP, Pulst SM, Perlman S, Wilmot G, Gomez C, … Kuo SH (2016). Depression and clinical progression in spinocerebellar ataxias. Parkinsonism & Related Disorders, 22, 87–92. doi: 10.1016/j.parkreldis.2015.11.021 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch D, Churchyard A, & Brotchie P (2005). Evidence for, and a spectrum of, neurological involvement in carriers of the fragile X pre-mutation: FXTAS and beyond. Clin Genet, 67(5), 412–417. [DOI] [PubMed] [Google Scholar]
- Loesch D, Tassone F, & Lo J (2013). New evidence for, and challenges in, linking small CGG repeat expansion FMR1 alleles with parkinson’s disease. Clin Genet, 84(4), 382–385. [DOI] [PubMed] [Google Scholar]
- Lombardi WJ, Woolston DJ, Roberts JW, & Gross RE (2001). Cognitive deficits in patients with essential tremor. Neurology, 57(5), 785–790. [DOI] [PubMed] [Google Scholar]
- Lopes T, D’Abreu A, França MJ, Yasuda C, Betting L, Samara A, … Cendes F (2013). Widespread neuronal damage and cognitive dysfunction in spinocerebellar ataxia type 3. J Neurol, 260(9), 2370–2379. [DOI] [PubMed] [Google Scholar]
- Louis E, Hernandez N, & Michalec M (2015). Prevalence and correlates of rest tremor in essential tremor: Cross-sectional survey of 831 patients across four distinct cohorts. European Journal of Neurology, 22(6), 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED (2013). The primary type of tremor in essential tremor is kinetic rather than postural: Cross-sectional observation of tremor phenomenology in 369 cases. European Journal of Neurology, 20(4), 725–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED (2015). Non-motor symptoms in essential tremor: A review of the current data and state of the field. Parkinsonism & Related Disorders, doi:S1353-8020(15)00373-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED, Benito-Leon J, Bermejo-Pareja F, & Neurological Disorders in Central Spain (NEDICES) Study Group. (2007). Self-reported depression and anti-depressant medication use in essential tremor: Cross-sectional and prospective analyses in a population-based study. European Journal of Neurology : The Official Journal of the European Federation of Neurological Societies, 14(10), 1138–1146. doi:ENE1923 [pii] [DOI] [PubMed] [Google Scholar]
- Louis ED, Benito-Leon J, Ottman R, Bermejo-Pareja F, & Neurological Disorders in Central Spain (NEDICES) Study Group. (2007). A population-based study of mortality in essential tremor. Neurology, 69(21), 1982–1989. doi:69/21/1982 [pii] [DOI] [PubMed] [Google Scholar]
- Louis ED, Benito-Leon J, Vega-Quiroga S, Bermejo-Pareja F, & Neurological Disorders in Central Spain (NEDICES) Study Group. (2010a). Cognitive and motor functional activity in non-demented community-dwelling essential tremor cases. Journal of Neurology, Neurosurgery, and Psychiatry, 81(9), 997–1001. doi: 10.1136/jnnp.2009.202838 [doi] [DOI] [PubMed] [Google Scholar]
- Louis ED, Benito-Leon J, Vega-Quiroga S, Bermejo-Pareja F, & Neurological Disorders in Central Spain (NEDICES) Study Group. (2010b). Faster rate of cognitive decline in essential tremor cases than controls: A prospective study. European Journal of Neurology : The Official Journal of the European Federation of Neurological Societies, 17(10), 1291–1297. doi: 10.1111/j.1468-1331.2010.03122.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED, Faust PL, Vonsattel JP, Honig LS, Rajput A, Robinson CA, … Hernandez N (2007). Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain : A Journal of Neurology, 130(Pt 12), 3297–3307. doi:awm266 [pii] [DOI] [PubMed] [Google Scholar]
- Louis ED, Huey ED, Gerbin M, & Viner AS (2012a). Apathy in essential tremor, dystonia, and parkinson’s disease: A comparison with normal controls. Movement Disorders : Official Journal of the Movement Disorder Society, 27(3), 432–434. doi: 10.1002/mds.24049 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED, Huey ED, Gerbin M, & Viner AS (2012b). Depressive traits in essential tremor: Impact on disability, quality of life, and medication adherence. European Journal of Neurology : The Official Journal of the European Federation of Neurological Societies, 19(10), 1349–1354. doi: 10.1111/j.1468-1331.2012.03774.x [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis ED, Mazzoni P, Ma KJ, Moskowitz CB, Lawton A, Garber A, & Vonsattel JP (2012). Essential tremor with ubiquitinated intranuclear inclusions and cerebellar degeneration. Clinical Neuropathology, 31(3), 119–126. doi:9607 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, … Wooten F (2015). Natural history of multiple system atrophy in the USA: A prospective cohort study. The Lancet Neurology, 14(7), 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyoo CH, Jeong Y, Ryu YH, Lee SY, Song TJ, Lee JH, … Lee MS (2008). Effects of disease duration on the clinical features and brain glucose metabolism in patients with mixed type multiple system atrophy. Brain : A Journal of Neurology, 131(Pt 2), 438–446. doi: 10.1093/brain/awm328 [doi] [DOI] [PubMed] [Google Scholar]
- Macpherson J, Waghorn A, & Hammans S (2003). Observation of an excess of fragile-X premutations in a population of males referred with spinocerebellar ataxia. Hum Genet, 112(5–6), 619–20. [DOI] [PubMed] [Google Scholar]
- Magherini A, & Litvan I (2005). Cognitive and behavioral aspects of PSP since steele, richardson and olszewski’s description of PSP 40 years ago and albert’s delineation of the subcortical dementia 30 years ago. Neurocase, 11(4), 250–262. doi:K66V403265787766 [pii] [DOI] [PubMed] [Google Scholar]
- Maher ER, Smith EM, & Lees AJ (1985). Cognitive deficits in the steele-richardson-olszewski syndrome (progressive supranuclear palsy). Journal of Neurology, Neurosurgery, and Psychiatry, 48(12), 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto M, & Lorivel T (2011). Cognitive repercussions of hereditary cerebellar disorders. Cortex; a Journal Devoted to the Study of the Nervous System and Behavior, 47(1), 81–100. doi: 10.1016/j.cortex.2009.04.012 [doi] [DOI] [PubMed] [Google Scholar]
- Markett S, Reuter M, Montag C, Voigt G, Lachmann B, Rudorf S, … Weber B (2014). Assessing the function of the fronto-parietal attention network: Insights from resting-state fMRI and the attentional network test. Human Brain Mapping, 35(4), 1700–1709. doi: 10.1002/hbm.22285 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruff P, Tyler P, Burt T, Currie B, Burns C, & Currie J (1996). Cognitive deficits in machado-joseph disease. Annals of Neurology, 40(3), 421–427. doi: 10.1002/ana.410400311 [doi] [DOI] [PubMed] [Google Scholar]
- Mata I, Leverenz J, Weintraub D, Trojanowski J, Hurtig H, Van Deerlin V, … Zabetian C (2014). APOE, MAPT, and SNCA genes and cognitive performance in parkinson disease. JAMA Neurol, 71(11), 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Dueñas A (2012). Machado-joseph disease and other rare spinocerebellar ataxias. Adv Exp Med Biol, 724, 172–188. [DOI] [PubMed] [Google Scholar]
- Matilla-Dueñas A, Corral-Juan M, Volpini V, & Sanchez I (2012). The spinocerebellar ataxias: Clinical aspects and molecular genetics Neurodegenerative diseases (pp. 351–374) Springer. [DOI] [PubMed] [Google Scholar]
- McMurtray A, Clark D, Flood M, Perlman S, & Mendez M (2006). Depressive and memory symptoms as presenting features of spinocerebellar ataxia. J Neuropsychiatry Clin Neurosci, 18(3), 420–422. [DOI] [PubMed] [Google Scholar]
- Middleton FA, & Strick PL (2000a). Basal ganglia and cerebellar loops: Motor and cognitive circuits. Brain Research.Brain Research Reviews, 31(2–3), 236–250. doi:S0165017399000405 [pii] [DOI] [PubMed] [Google Scholar]
- Middleton FA, & Strick PL (2000b). Basal ganglia output and cognition: Evidence from anatomical, behavioral, and clinical studies. Brain and Cognition, 42(2), 183–200. doi: 10.1006/brcg.1999.1099 [doi] [DOI] [PubMed] [Google Scholar]
- Millar D, Griffiths P, Zermansky AJ, & Burn DJ (2006). Characterizing behavioral and cognitive dysexecutive changes in progressive supranuclear palsy. Movement Disorders : Official Journal of the Movement Disorder Society, 21(2), 199–207. doi: 10.1002/mds.20707 [doi] [DOI] [PubMed] [Google Scholar]
- Milunsky J, & Maher T (2004). Fragile X carrier screening and spinocerebellar ataxia in older males. Am J Med Genet A, 125A(3), 320. [DOI] [PubMed] [Google Scholar]
- Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, & Shoji S (2003). Progressive supranuclear palsy presenting with primary progressive aphasia--clinicopathological report of an autopsy case. Acta Neuropathologica, 105(6), 610–614. doi: 10.1007/s00401-003-0682-5 [doi] [DOI] [PubMed] [Google Scholar]
- Montgomery EB, Baker KB, Lyons K, & Koller WC (2000). Motor initiation and execution in essential tremor and parkinson’s disease. Movement Disorders, 15(3), 511–515. [DOI] [PubMed] [Google Scholar]
- Monza D, Soliveri P, Radice D, Fetoni V, Testa D, Caffarra P, … Girotti F (1998). Cognitive dysfunction and impaired organization of complex motility in degenerative parkinsonian syndromes. Arch Neurol, 55(3), 372–378. [DOI] [PubMed] [Google Scholar]
- Moore CJ, Daly EM, Schmitz N, Tassone F, Tysoe C, Hagerman RJ, … Murphy DG (2004). A neuropsychological investigation of male premutation carriers of fragile X syndrome. Neuropsychologia, 42(14), 1934–1947. doi: 10.1016/j.neuropsychologia.2004.05.002 [doi] [DOI] [PubMed] [Google Scholar]
- Naito H, & Oyanagi S (1982). Familial myoclonus epilepsy and choreoathetosis: Hereditary dentatorubral-pallidoluysian atrophy. Neurology, 32(8), 798–807. [DOI] [PubMed] [Google Scholar]
- Narcisa V, Aguilar D, & Nguyen D (2011). A quantitative assessment of tremor and ataxia in female FMR1 premutation carriers using CATSYS. Curr Gerontol Geriatr Res, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermeyer E (1998). Frontal lobe functions and dysfunctions. Clinical EEG (Electroencephalography), 29(2), 79–90. [DOI] [PubMed] [Google Scholar]
- Niu Y, Yang J, Hall DA, Leehey MA, Tassone F, Olichney JM, … Zhang L (2014). Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism & Related Disorders, 20(4), 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh S, Han Y, Mun C, Chung E, Kim E, Ji K, … Kim S (2014). Analysis among cognitive profiles and gray matter volume in newly diagnosed parkinson’s disease with mild cognitive impairment. J Neurol Sci, 347(1–2), 210–213. [DOI] [PubMed] [Google Scholar]
- O’Hearn EE, Hwang HS, Holmes SE, Rudnicki DD, Chung DW, Seixas AI, … Margolis RL (2015). Neuropathology and cellular pathogenesis of spinocerebellar ataxia type 12. Movement Disorders : Official Journal of the Movement Disorder Society, doi: 10.1002/mds.26348 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K, Takahashi K, Gotoh J, Yamaguchi K, Seki M, Nihei Y, … Keio Parkinson’s Disease Database. (2014). Screening for impaired cognitive domains in a large parkinson’s disease population and its application to the diagnostic procedure for parkinson’s disease dementia. Dementia and Geriatric Cognitive Disorders Extra, 4(2), 147–159. doi: 10.1159/000362124 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe JA, Robertson-Dick E, Dunn EJ, Li Y, Deng Y, Fiutko AN, … Hall DA (2015). Characterization and early detection of balance deficits in fragile X premutation carriers with and without fragile X-associated Tremor/Ataxia syndrome (FXTAS). Cerebellum (London, England), doi: 10.1007/s12311-015-0659-7 [doi] [DOI] [PubMed] [Google Scholar]
- O’Keefe JA, Robertson-Dick EE, Hall DA, & Berry-Kravis E (2015). Gait and functional mobility deficits in fragile X-associated Tremor/Ataxia syndrome. Cerebellum (London, England), doi: 10.1007/s12311-015-0714-4 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Chan S, Wong LM, Schneider A, Seritan A, Niese A, … Hagerman R (2010). Abnormal N400 word repetition effects in fragile X-associated tremor/ataxia syndrome. Brain : A Journal of Neurology, 133(Pt 5), 1438–1450. doi: 10.1093/brain/awq077 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Pak J, Salmon DP, Yang JC, Gahagan T, Nowacki R, … Iragui-Madoz VJ (2013). Abnormal P600 word repetition effect in elderly persons with preclinical alzheimer’s disease. Cognitive Neuroscience, 4(3–4), 143–151. doi: 10.1080/17588928.2013.838945 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichney JM, Taylor JR, Gatherwright J, Salmon DP, Bressler AJ, Kutas M, & Iragui-Madoz VJ (2008). Patients with MCI and N400 or P600 abnormalities are at very high risk for conversion to dementia. Neurology, 70(19 Pt 2), 1763–1770. doi:01.wnl.0000281689.28759.ab [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi L, D’Agata F, Caroppo P, Franco A, Caglio M, Avidano F, … Mortara P (2011). Neuropsychological picture of 33 spinocerebellar ataxia cases. J Clin Exp Neuropsychol, 33(3), 315–325. [DOI] [PubMed] [Google Scholar]
- Pablo-Fernandez E, Doherty K, & Holton J (2015). Concomitant fragile X-associated tremor ataxia syndrome and parkinson’s disease: A clinicopathological report of two cases. J Neurol Neurosurg Psychiatry, 86(8), 934–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano G, Ferrara N, Brooks DJ, & Pavese N (2016). Age at onset and parkinson disease phenotype. Neurology, doi: 10.1212/WNL.0000000000002461 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagonabarraga J, Martinez-Horta S, Fernandez de Bobadilla R, Perez J, Ribosa-Nogue R, Marin J, … Kulisevsky J (2015). Minor hallucinations occur in drug-naive parkinson’s disease patients, even from the premotor phase. Movement Disorders : Official Journal of the Movement Disorder Society, doi: 10.1002/mds.26432 [doi] [DOI] [PubMed] [Google Scholar]
- Park H, Kim H, & Jeon BS (2015). Parkinsonism in spinocerebellar ataxia. BioMed Research International, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL (2009). The spinocerebellar ataxias. Journal of Neuro-Ophthalmology : The Official Journal of the North American Neuro-Ophthalmology Society, 29(3), 227–237. doi: 10.1097/WNO0b013e3181b416de [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus T (2001). Primate anterior cingulate cortex: Where motor control, drive and cognition interface. Nature Reviews.Neuroscience, 2(6), 417–424. doi: 10.1038/35077500 [doi] [DOI] [PubMed] [Google Scholar]
- Pedroso JL, Franca MC Jr, Braga-Neto P, D’Abreu A, Saraiva-Pereira ML, Saute JA, … Barsottini OG (2013). Nonmotor and extracerebellar features in machado-joseph disease: A review. Movement Disorders : Official Journal of the Movement Disorder Society, 28(9), 1200–1208. doi: 10.1002/mds.25513 [doi] [DOI] [PubMed] [Google Scholar]
- Perlman SL (2011). Spinocerebellar degenerations. Handbook of Clinical Neurology, 100, 113–140. doi: 10.1016/B978-0-444-52014-2.00006-9 [doi] [DOI] [PubMed] [Google Scholar]
- Peters N, Kamm C, Asmus F, Holinski-Feder E, Kraft E, Dichgans M, … Bötzel K (2006). Intrafamilial variability in fragile x-associated tremor/ataxia syndrome. Mov Disord, 21(1), 98–102. [DOI] [PubMed] [Google Scholar]
- Petersen RC (2011). Clinical practice. mild cognitive impairment. The New England Journal of Medicine, 364(23), 2227–2234. doi: 10.1056/NEJMcp0910237 [doi] [DOI] [PubMed] [Google Scholar]
- Pettit L, McCarthy M, Davenport R, & Abrahams S (2013). Heterogeneity of letter fluency impairment and executive dysfunction in parkinson’s disease. J Int Neuropsychol Soc, 19(9), 986–994. [DOI] [PubMed] [Google Scholar]
- Pfeiffer H, Løkkegaard A, Zoetmulder M, Friberg L, & Werdelin L (2014). Cognitive impairment in early-stage non-demented parkinson’s disease patients. Acta Neurol Scand, 129(5), 307–318. [DOI] [PubMed] [Google Scholar]
- Pillon B, Deweer B, Michon A, Malapani C, Agid Y, & Dubois B (1994). Are explicit memory disorders of progressive supranuclear palsy related to damage to striatofrontal circuits? comparison with alzheimer’s, parkinson’s, and huntington’s diseases. Neurology, 44(7), 1264–1270. [DOI] [PubMed] [Google Scholar]
- Pillon B, Dubois B, & Agid Y (1991). Severity and specificity of cognitive impairment in alzheimer’s, huntington’s, and parkinson’s diseases and progressive supranuclear palsy. Annals of the New York Academy of Sciences, 640, 224–227. [DOI] [PubMed] [Google Scholar]
- Pillon B, Gouider-Khouja N, Deweer B, Vidailhet M, Malapani C, Dubois B, & Agid Y (1995). Neuropsychological pattern of striatonigral degeneration: Comparison with parkinson’s disease and progressive supranuclear palsy. Journal of Neurology, Neurosurgery, and Psychiatry, 58(2), 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst S (2003). In Pulst S (Ed.), Genetics of movement disorders (1st ed.). London, UK: Academic Press. [Google Scholar]
- Quante A, Jakob F, & Wolf J (2008). Depression preceding the onset of progressive supranuclear paralysis: A case report. The Journal of Neuropsychiatry and Clinical Neurosciences, 20(2), 247–248. doi: 10.1176/appi.neuropsych.20.2.247-a [doi] [DOI] [PubMed] [Google Scholar]
- Radvany J, Camargo CH, Costa ZM, Fonseca NC, & Nascimento ED (1993). Machado-joseph disease of azorean ancestry in brazil: The catarina kindred. neurological, neuroimaging, psychiatric and neuropsychological findings in the largest known family, the “catarina” kindred. Arquivos De Neuro-Psiquiatria, 51(1), 21–30. [DOI] [PubMed] [Google Scholar]
- Rafal RD, Posner MI, Friedman JH, Inhoff AW, & Bernstein E (1988). Orienting of visual attention in progressive supranuclear palsy. Brain : A Journal of Neurology, 111 (Pt 2)(Pt 2), 267–280. [DOI] [PubMed] [Google Scholar]
- Rajkiewicz M, Sułek-Piatkowska A, & Krysa W (2008). Screening for premutation in the FMR1 gene in male patients suspected of spinocerebellar ataxia. Neurol Neurochir Pol, 42(6), 497–504. [PubMed] [Google Scholar]
- Rama P (2008). Domain-dependent activation during spatial and nonspatial auditory working memory. Cognitive Processing, 9(1), 29–34. doi: 10.1007/s10339-007-0182-y [doi] [DOI] [PubMed] [Google Scholar]
- Ramnani N (2012). Frontal lobe and posterior parietal contributions to the cortico-cerebellar system. Cerebellum (London, England), 11(2), 366–383. doi: 10.1007/s12311-011-0272-3 [doi] [DOI] [PubMed] [Google Scholar]
- Respondek G, & Hoglinger GU (2015). The phenotypic spectrum of progressive supranuclear palsy. Parkinsonism & Related Disorders, doi:S1353-8020(15)00432-0 [pii] [DOI] [PubMed] [Google Scholar]
- Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, … Movement Disorder Society-endorsed PSP Study Group. (2014). The phenotypic spectrum of progressive supranuclear palsy: A retrospective multicenter study of 100 definite cases. Movement Disorders : Official Journal of the Movement Disorder Society, 29(14), 1758–1766. doi: 10.1002/mds.26054 [doi] [DOI] [PubMed] [Google Scholar]
- Rodríguez-Labrada R, Velázquez-Pérez L, Aguilera-Rodríguez R, Seifried-Oberschmidt C, Peña-Acosta A, Canales-Ochoa N, … Laffita Mesa J (2014). Executive deficit in spinocerebellar ataxia type 2 is related to expanded CAG repeats: Evidence from antisaccadic eye movements. Brain Cogn, 91, 28–34. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Gómez-Anson B, & Muñoz E (2007). FXTAS in spanish patients with ataxia: Support for female FMR1 premutation screening. Mol Neurobiol, 35(3), 324–328. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Pagonabarraga J, Gomez-Anson B, Lopez-Mourelo O, Madrigal I, Xuncla M, … Mila M (2010). Motor and mental dysfunction in mother-daughter transmitted FXTAS. Neurology, 75(15), 1370–1376. doi: 10.1212/WNL.0b013e3181f73660 [DOI] [PubMed] [Google Scholar]
- Roeske S, Filla I, Heim S, Amunts K, Helmstaedter C, Wüllner U, … Minnerop M (2013). Progressive cognitive dysfunction in spinocerebellar ataxia type 3. Mov Disord, 28(10), 1435–1438. [DOI] [PubMed] [Google Scholar]
- Romero JP, Benito-Leon J, & Bermejo-Pareja F (2012). The NEDICES study: Recent advances in the understanding of the epidemiology of essential tremor. Tremor and Other Hyperkinetic Movements (New York, N.Y.), 2, tre-02-70-346-2. Epub 2012 Jun 15. doi:tre-02-70-346-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg R (1992). Machado-joseph disease: An autosomal dominant motor system degeneration. Mov Disord, 7(3), 193–203. [DOI] [PubMed] [Google Scholar]
- Rosser AE, & Hodges JR (1994). The dementia rating scale in alzheimer’s disease, huntington’s disease and progressive supranuclear palsy. Journal of Neurology, 241(9), 531–536. [DOI] [PubMed] [Google Scholar]
- Sahin HA, Terzi M, Ucak S, Yapici O, Basoglu T, & Onar M (2006). Frontal functions in young patients with essential tremor: A case comparison study. The Journal of Neuropsychiatry and Clinical Neurosciences, 18(1), 64–72. doi:18/1/64 [pii] [DOI] [PubMed] [Google Scholar]
- Saute JA, da Silva AC, Donis KC, Vedolin L, Saraiva-Pereira ML, & Jardim LB (2010). Depressive mood is associated with ataxic and non-ataxic neurological dysfunction in SCA3 patients. Cerebellum (London, England), 9(4), 603–5; author reply 606–7. doi: 10.1007/s12311-010-0205-6 [doi] [DOI] [PubMed] [Google Scholar]
- Scaglione C, Ginestroni A, & Vella A (2008). MRI and SPECT of midbrain and striatal degeneration in fragile X-associated tremor/ataxia syndrome J Neurol, 255(1), 144–146. [DOI] [PubMed] [Google Scholar]
- Schelhaas HJ, van de Warrenburg BP, Hageman G, Ippel EE, van Hout M, & Kremer B (2003). Cognitive impairment in SCA-19. Acta Neurologica Belgica, 103(4), 199–205. [PubMed] [Google Scholar]
- Schmahmann JD, & Sherman JC (1998). The cerebellar cognitive affective syndrome. Brain : A Journal of Neurology, 121 (Pt 4)(Pt 4), 561–579. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Smith EE, Eichler FS, & Filley CM (2008). Cerebral white matter: Neuroanatomy, clinical neurology, and neurobehavioral correlates. Annals of the New York Academy of Sciences, 1142, 266–309. doi: 10.1196/annals.1444.017 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Hubsch T, Coudert M, Bauer P, Giunti P, Globas C, Baliko L, … Klockgether T (2008). Spinocerebellar ataxia types 1, 2, 3, and 6: Disease severity and nonataxia symptoms. Neurology, 71(13), 982–989. doi: 10.1212/01.wnl.0000325057.33666.72 [doi] [DOI] [PubMed] [Google Scholar]
- Schmitz-Hubsch T, Coudert M, Tezenas du Montcel S, Giunti P, Labrum R, Durr A, … Klockgether T (2011). Depression comorbidity in spinocerebellar ataxia. Movement Disorders : Official Journal of the Movement Disorder Society, 26(5), 870–876. doi: 10.1002/mds.23698 [doi] [DOI] [PubMed] [Google Scholar]
- Schöls L, Bauer P, Schmidt T, Schulte T, & Riess O (2004). Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. The Lancet Neurology, 3(5), 291–304. [DOI] [PubMed] [Google Scholar]
- Schols L, Kruger R, Amoiridis G, Przuntek H, Epplen JT, & Riess O (1998). Spinocerebellar ataxia type 6: Genotype and phenotype in german kindreds. Journal of Neurology, Neurosurgery, and Psychiatry, 64(1), 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag A, Geser F, Stampfer-Kountchev M, Seppi K, Sawires M, Kollensperger M, … European MSA-Study Group. (2006). Health-related quality of life in multiple system atrophy. Movement Disorders : Official Journal of the Movement Disorder Society, 21(6), 809–815. doi: 10.1002/mds.20808 [doi] [DOI] [PubMed] [Google Scholar]
- Schrag A, Sheikh S, Quinn NP, Lees AJ, Selai C, Mathias C, … Jahanshahi M (2010). A comparison of depression, anxiety, and health status in patients with progressive supranuclear palsy and multiple system atrophy. Movement Disorders : Official Journal of the Movement Disorder Society, 25(8), 1077–1081. doi: 10.1002/mds.22794 [doi] [DOI] [PubMed] [Google Scholar]
- Schrag A, Wenning GK, Quinn N, & Ben-Shlomo Y (2008). Survival in multiple system atrophy. Movement Disorders : Official Journal of the Movement Disorder Society, 23(2), 294–296. doi: 10.1002/mds.21839 [doi] [DOI] [PubMed] [Google Scholar]
- Schwartz M, Badarny S, Gofman S, & Hocherman S (1999). Visuomotor performance in patients with essential tremor. Movement Disorders, 14(6), 988–993. [DOI] [PubMed] [Google Scholar]
- Seixas A, Maurer M, & Lin M (2005). FXTAS, SCA10, and SCA17 in american patients with movement disorders. Am J Med Genet A, 136(1), 87–89. [DOI] [PubMed] [Google Scholar]
- Seixas A, Vale J, & Jorge P (2011). FXTAS is rare among portuguese patients with movement disorders: FMR1 premutations may be associated with a wider spectrum of phenotypes. Behav Brain Funct, 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmeczy D, Koldewyn K, Wang JM, Lee A, Harvey D, Hessl DR, … Rivera SM (2011). Investigation of amygdala volume in men with the fragile X premutation. Brain Imaging and Behavior, 5(4), 285–294. doi: 10.1007/s11682-011-9132-5; 10.1007/s11682-011-9132-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seritan A, Cogswell J, & Grigsby J (2013). Cognitive dysfunction in premutation carriers. Current Psychiatry Reviews, 9(1), 78–84. doi: 10.2174/157340013805289635 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seritan AL, Nguyen DV, Farias ST, Hinton L, Grigsby J, Bourgeois JA, & Hagerman RJ (2008). Dementia in fragile X-associated tremor/ataxia syndrome (FXTAS): Comparison with alzheimer’s disease. American Journal of Medical Genetics.Part B, Neuropsychiatric Genetics : The Official Publication of the International Society of Psychiatric Genetics, 147B(7), 1138–1144. doi: 10.1002/ajmg.b.30732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevin M, Kutalik Z, Bergman S, Vercelletto M, Renou P, Lamy E, … Jacquemont S (2009). Penetrance of marked cognitive impairment in older male carriers of the FMR1 gene premutation. Journal of Medical Genetics, 46(12), 818–824. doi: 10.1136/jmg.2008.065953 [DOI] [PubMed] [Google Scholar]
- Sherman SL, Curnow EC, Easley CA, Jin P, Hukema RK, Tejada MI, … Usdin K (2014). Use of model systems to understand the etiology of fragile X-associated primary ovarian insufficiency (FXPOI). Journal of Neurodevelopmental Disorders, 6(1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki H, Tsuji S, & Kuroiwa Y (1979). Oculomotor abnormalities in parkinson’s disease. Archives of Neurology, 36(6), 360–364. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Ozawa T, Nakayama H, Tomita M, Shinoda H, & Nishizawa M (2008). Frequency of nocturnal sudden death in patients with multiple system atrophy. Journal of Neurology, 255(10), 1483–1485. doi: 10.1007/s00415-008-0941-4 [doi] [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, … Ziegler SG (2009). Multicenter analysis of glucocerebrosidase mutations in parkinson’s disease. The New England Journal of Medicine, 361(17), 1651–1661. doi: 10.1056/NEJMoa0901281 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel FJ, Bronnick KS, Booij J, Ravina BM, Lebedev AV, Pereira JB, … Aarsland D (2014). Cognitive executive impairment and dopaminergic deficits in de novo parkinson’s disease. Movement Disorders : Official Journal of the Movement Disorder Society, 29(14), 1802–1808. doi: 10.1002/mds.26051 [doi] [DOI] [PubMed] [Google Scholar]
- Silva U, Marques W, Lourenço C, Hallak J, & Osório F (2015). Psychiatric disorders, spinocerebellar ataxia type 3 and CAG expansion. J Neurol, 262(7), 1777–1779. [DOI] [PubMed] [Google Scholar]
- Singer C, Sanchez-Ramos J, & Weiner WJ (1994). Gait abnormality in essential tremor. Movement Disorders, 9(2), 193–196. [DOI] [PubMed] [Google Scholar]
- Sinoff G, & Badarny S (2014). Mild cognitive impairment, dementia, and affective disorders in essential tremor: A prospective study. Tremor and Other Hyperkinetic Movements (New York, N.Y.), 4, 227. doi: 10.7916/D85B00KN [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siri C, Duerr S, Canesi M, Delazer M, Esselink R, Bloem BR, … Antonini A (2013). A cross-sectional multicenter study of cognitive and behavioural features in multiple system atrophy patients of the parkinsonian and cerebellar type. Journal of Neural Transmission (Vienna, Austria : 1996), 120(4), 613–618. doi: 10.1007/s00702-013-0997-x [doi] [DOI] [PubMed] [Google Scholar]
- Soliveri P, Monza D, Paridi D, Carella F, Genitrini S, Testa D, & Girotti F (2000). Neuropsychological follow up in patients with parkinson’s disease, striatonigral degeneration-type multisystem atrophy, and progressive supranuclear palsy. Journal of Neurology, Neurosurgery, and Psychiatry, 69(3), 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaccavento S, Del Prete M, Craca A, & Loverre A (2014). A case of atypical progressive supranuclear palsy. Clinical Interventions in Aging, 9, 31–39. doi: 10.2147/CIA.S51640 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic I, Krismer F, Jesic A, Antonini A, Benke T, Brown RG, … Movement Disorders Society MSA (MODIMSA) Study Group. (2014). Cognitive impairment in multiple system atrophy: A position statement by the neuropsychology task force of the MDS multiple system atrophy (MODIMSA) study group. Movement Disorders : Official Journal of the Movement Disorder Society, 29(7), 857–867. doi: 10.1002/mds.25880 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JC, Richardson JC, & Olszewski J (1964). Progressive supranuclear palsy: A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Archives of Neurology, 10(4), 333–359. [DOI] [PubMed] [Google Scholar]
- Stolze H, Petersen G, Raethjen J, Wenzelburger R, & Deuschl G (2001). The gait disorder of advanced essential tremor. Brain : A Journal of Neurology, 124(Pt 11), 2278–2286. [DOI] [PubMed] [Google Scholar]
- Storey E (2014). Genetic cerebellar ataxias. Semin Neurol, 34(3), 280–292. [DOI] [PubMed] [Google Scholar]
- Storey E, & Billimoria P (2005). Increased T 2 signal in the middle cerebellar peduncles on MRI is not specific for fragile X premutation syndrome. Journal of Clinical Neuroscience, 12(1), 42–43. [DOI] [PubMed] [Google Scholar]
- Suenaga M, Kawai Y, Watanabe H, Atsuta N, Ito M, Tanaka F, … Sobue G (2008). Cognitive impairment in spinocerebellar ataxia type 6. J Neurol Neurosurg Psychiatry, 79(5), 496–499. [DOI] [PubMed] [Google Scholar]
- Tan E, Zhao Y, & Puong K (2004). Fragile X premutation alleles in SCA, ET, and parkinsonism in an asian cohort. Neurology, 63(2), 362–363. [DOI] [PubMed] [Google Scholar]
- Tang H, Huang J, Nie K, Gan R, Wang L, Zhao J, … Wang L (2016). Cognitive profile of parkinson’s disease patients: A comparative study between early-onset and late-onset parkinson’s disease. The International Journal of Neuroscience, 126(3), 227–234. doi: 10.3109/00207454.2015.1010646 [doi] [DOI] [PubMed] [Google Scholar]
- Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, … Hagerman RJ (2012). Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes, Brain, and Behavior, 11(5), 577–585. doi: 10.1111/j.1601-183X.2012.00779.x; 10.1111/j.1601–183X.2012.00779.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, & Hagerman R (2012). The fragile X-associated tremor ataxia syndrome. Results and Problems in Cell Differentiation, 54, 337–357. doi: 10.1007/978-3-642-21649-7_18; 10.1007/978-3-642-21649-7_18 [DOI] [PubMed] [Google Scholar]
- Terao Y, Fukuda H, Tokushige S, Inomata-Terada S, Yugeta A, Hamada M, … Ugawa Y (2016). Is multiple system atrophy with cerebellar ataxia (MSA-C) like spinocerebellar ataxia and multiple system atrophy with parkinsonism (MSA-P) like Parkinson’s disease?–A saccade study on pathophysiology. Clinical Neurophysiology, 127(2), 1491–1502. [DOI] [PubMed] [Google Scholar]
- Thawani SP, Schupf N, & Louis ED (2009). Essential tremor is associated with dementia: Prospective population-based study in new york. Neurology, 73(8), 621–625. doi: 10.1212/WNL.0b013e3181b389f1 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens L, Burns E, Stone J, Graham C, Wright H, Summers D, … Zeman A (2008). Spinocerebellar ataxia type 8 in scotland: Frequency, neurological, neuropsychological and neuropsychiatric findings. Acta Neurologica Scandinavica, 117(1), 41–48. doi:ANE904 [pii] [DOI] [PubMed] [Google Scholar]
- Tovote P, Fadok JP, & Luthi A (2015). Neuronal circuits for fear and anxiety. Nature Reviews.Neuroscience, 16(6), 317–331. doi: 10.1038/nrn3945 [doi] [DOI] [PubMed] [Google Scholar]
- Toyoshima Y, Onodera O, Yamada M, Tsuji S, & Takahashi H (1993). Spinocerebellar ataxia type 17 In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, … Stephens K (Eds.), Genereviews(r) (). Seattle (WA): University of Washington, Seattle; doi:NBK1438 [bookaccession] [Google Scholar]
- Trillenberg P, Führer J, Sprenger A, Hagenow A, Kömpf D, Wenzelburger R, … Helmchen C (2006). Eye–hand coordination in essential tremor. Movement Disorders, 21(3), 373–379. [DOI] [PubMed] [Google Scholar]
- Trost N, Cook M, Hammersley E, Bui M, Brotchie P, Burgess T, … Loesch D (2014). White matter changes in patients with parkinson’s disease carrying small CGG expansion FMR1 alleles: A pilot study. Neurodegener Dis, 14(2), 67–76. [DOI] [PubMed] [Google Scholar]
- Troster AI, Woods SP, Fields JA, Lyons KE, Pahwa R, Higginson CI, & Koller WC (2002). Neuropsychological deficits in essential tremor: An expression of cerebello-thalamo-cortical pathophysiology? European Journal of Neurology, 9(2), 143–151. [DOI] [PubMed] [Google Scholar]
- Trouillas P, Takayanagi T, & Hallet M (1997). International cooperative ataxia rating scale for pharmacological assessment of the cerebellar syndrome. J Neurol Sci, 45, 205–211. [DOI] [PubMed] [Google Scholar]
- Trzepacz PT, Hochstetler H, Wang S, Walker B, Saykin AJ, & Alzheimer’s Disease Neuroimaging Initiative. (2015). Relationship between the montreal cognitive assessment and mini-mental state examination for assessment of mild cognitive impairment in older adults. BMC Geriatrics, 15, 107-015-0103-3. doi: 10.1186/s12877-015-0103-3 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji S (2012). Dentatorubral-pallidoluysian atrophy. Handbook of Clinical Neurology, 103, 587–594. doi: 10.1016/B978-0-444-51892-7.00041-3 [doi] [DOI] [PubMed] [Google Scholar]
- Vale J, Bugalho P, Silveira I, Sequeiros J, Guimaraes J, & Coutinho P (2010). Autosomal dominant cerebellar ataxia: Frequency analysis and clinical characterization of 45 families from portugal. European Journal of Neurology, 17(1), 124–128. doi: 10.1111/j.1468-1331.2009.02757.x [doi] [DOI] [PubMed] [Google Scholar]
- van der Hurk PR, & Hodges JR (1995). Episodic and semantic memory in alzheimer’s disease and progressive supranuclear palsy: A comparative study. Journal of Clinical and Experimental Neuropsychology, 17(3), 459–471. doi: 10.1080/01688639508405137 [doi] [DOI] [PubMed] [Google Scholar]
- Van Esch H, Dom R, & Bex D (2005). Screening for FMR-1 premutations in 122 older flemish males presenting with ataxia. Eur J Hum Genet, 13(1), 121–123. [DOI] [PubMed] [Google Scholar]
- van Gaalen J, de Swart B, Oostveen J, Knuijt S, van de Warrenburg B, & Kremer B (2014). Language impairment in cerebellar ataxia. Mov Disord, 29(10), 1307–1312. [DOI] [PubMed] [Google Scholar]
- Vidailhet M, Rivaud S, Gouider-Khouja N, Pillon B, Bonnet AM, Gaymard B, … Pierrot-Deseilligny C (1994). Eye movements in parkinsonian syndromes. Annals of Neurology, 35(4), 420–426. doi: 10.1002/ana.410350408 [doi] [DOI] [PubMed] [Google Scholar]
- Walterfang M, & van de Warrenburg BP (2014). Cognitive impairment in “other” movement disorders: Hidden defects and valuable clues. Movement Disorders : Official Journal of the Movement Disorder Society, 29(5), 694–703. doi: 10.1002/mds.25849 [doi] [DOI] [PubMed] [Google Scholar]
- Wang C, Cai Y, Gu Z, Ma J, Zheng Z, Tang BS, … Chinese Parkinson Study Group. (2014). Clinical profiles of parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in chinese individuals. Neurobiology of Aging, 35(3), 725.e1–725.e6. doi: 10.1016/j.neurobiolaging.2013.08.012 [doi] [DOI] [PubMed] [Google Scholar]
- Wardle M, Majounie E, & Muzaimi M (2009). The genetic aetiology of late-onset chronic progressive cerebellar ataxia. A population-based study J Neurol, 256(3), 343–348. [DOI] [PubMed] [Google Scholar]
- Weintraub D, Simuni T, Caspell-Garcia C, Coffey C, Lasch S, Siderowf A, … Parkinson’s Progression Markers Initiative. (2015). Cognitive performance and neuropsychiatric symptoms in early, untreated parkinson’s disease. Movement Disorders : Official Journal of the Movement Disorder Society, 30(7), 919–927. doi: 10.1002/mds.26170 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenning GK, & Stefanova N (2009). Recent developments in multiple system atrophy. Journal of Neurology, 256(11), 1791–1808. [DOI] [PubMed] [Google Scholar]
- Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, & Quinn NP (1994). Clinical features and natural history of multiple system atrophy. an analysis of 100 cases. Brain : A Journal of Neurology, 117 (Pt 4)(Pt 4), 835–845. [DOI] [PubMed] [Google Scholar]
- Whaley N, Putzke JD, Baba Y, Wszolek ZK, & Uitti RJ (2007). Essential tremor: Phenotypic expression in a clinical cohort. Parkinsonism & Related Disorders, 13(6), 333–339. [DOI] [PubMed] [Google Scholar]
- Wheeler AC, Bailey DB Jr, Berry-Kravis E, Greenberg J, Losh M, Mailick M, … Hagerman R (2014). Associated features in females with an FMR1 premutation. Journal of Neurodevelopmental Disorders, 6(1), 30-1955-6-30. Epub 2014 Jul 30. doi: 10.1186/1866-1955-6-30 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, & Lees AJ (2010). What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Movement Disorders, 25(3), 357–362. [DOI] [PubMed] [Google Scholar]
- Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, … Lees AJ (2005). Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain : A Journal of Neurology, 128(Pt 6), 1247–1258. doi:awh488 [pii] [DOI] [PubMed] [Google Scholar]
- Williams DR, & Lees AJ (2005). Visual hallucinations in the diagnosis of idiopathic parkinson’s disease: A retrospective autopsy study. The Lancet.Neurology, 4(10), 605–610. doi:S1474-4422(05)70146-0 [pii] [DOI] [PubMed] [Google Scholar]
- Williams DR, Warren JD, & Lees AJ (2008). Using the presence of visual hallucinations to differentiate parkinson’s disease from atypical parkinsonism. Journal of Neurology, Neurosurgery, and Psychiatry, 79(6), 652–655. doi:jnnp.2007.124677 [pii] [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, … Barker RA (2009). The distinct cognitive syndromes of parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain : A Journal of Neurology, 132(Pt 11), 2958–2969. doi: 10.1093/brain/awp245 [doi] [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Mason SL, Evans JR, Foltynie T, Brayne C, Robbins TW, & Barker RA (2013). The CamPaIGN study of parkinson’s disease: 10-year outlook in an incident population-based cohort. Journal of Neurology, Neurosurgery, and Psychiatry, 84(11), 1258–1264. doi: 10.1136/jnnp-2013-305277 [doi] [DOI] [PubMed] [Google Scholar]
- Wu Y, Guo X, Wei Q, Ou R, Song W, Cao B, … Shang H (2015). Non-motor symptoms and quality of life in tremor dominant vs postural instability gait disorder parkinson′ s disease patients. Acta Neurologica Scandinavica, [DOI] [PubMed] [Google Scholar]
- Yabe I, Soma H, Takei A, Fujiki N, Yanagihara T, & Sasaki H (2006). MSA-C is the predominant clinical phenotype of MSA in japan: Analysis of 142 patients with probable MSA. Journal of the Neurological Sciences, 249(2), 115–121. [DOI] [PubMed] [Google Scholar]
- Yachnis AT, Roth HL, & Heilman KM (2010). Fragile X dementia parkinsonism syndrome (FXDPS). Cognitive and Behavioral Neurology : Official Journal of the Society for Behavioral and Cognitive Neurology, 23(1), 39–43. doi: 10.1097/WNN.0b013e3181b6e1b9 [DOI] [PubMed] [Google Scholar]
- Yang JC, Chan SH, Khan S, Schneider A, Nanakul R, Teichholtz S, … Olichney JM (2013). Neural substrates of executive dysfunction in fragile X-associated tremor/ataxia syndrome (FXTAS): A brain potential study. Cerebral Cortex (New York, N.Y.: 1991), 23(11), 2657–2666. doi: 10.1093/cercor/bhs251 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Chi L, Teichholtz S, Schneider A, Nanakul R, Nowacki R, … Olichney JM (2014). ERP abnormalities elicited by word repetition in fragile X-associated tremor/ataxia syndrome (FXTAS) and amnestic MCI. Neuropsychologia, 63, 34–42. doi: 10.1016/j.neuropsychologia.2014.08.001 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Simon C, Niu YQ, Bogost M, Schneider A, Tassone F, … Olichney JM (2013). Phenotypes of hypofrontality in older female fragile x premutation carriers. Annals of Neurology, doi: 10.1002/ana.23933; 10.1002/ana.23933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Simon C, Schneider A, Seritan AL, Hamilton L, Hagerman PJ, … Olichney JM (2014). Abnormal semantic processing in females with fragile X-associated tremor/ataxia syndrome. Genes, Brain, and Behavior, 13(2), 152–162. doi: 10.1111/gbb.12114 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnall AJ, Breen DP, Duncan GW, Khoo TK, Coleman SY, Firbank MJ, … ICICLE-PD Study Group. (2014). Characterizing mild cognitive impairment in incident parkinson disease: The ICICLE-PD study. Neurology, 82(4), 308–316. doi: 10.1212/WNL.0000000000000066 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh T, Lu C, & Chou Y (2005). Autonomic dyfunction in machado-joseph disease. Arch Neurol, 62(4), 630–636. [DOI] [PubMed] [Google Scholar]
- Zappia M, Albanese A, & Bruno E (2003). Treatment of essential tremor: A systematic review of evidence and recommendations from the italian movement disorders association. J Neurol, 260(3), 714–740. [DOI] [PubMed] [Google Scholar]
- Zawacki T, Grace J, Friedman J, & Sudarsky L (2002). Executive and emotional dysfunction in machado-joseph disease. Mov Disord, 17(5), 1004–1010. [DOI] [PubMed] [Google Scholar]
- Zhang L, Gu W, & Wang G (2013). Analysis of fragile X mental retardation 1 gene premutation in multiple system atrophy patients. Zhonghua Yi Xue Za Zhi, 93(47), 3744–3747. [PubMed] [Google Scholar]
- Zhu K, van Hilten JJ, Putter H, & Marinus J (2013). Risk factors for hallucinations in parkinson’s disease: Results from a large prospective cohort study. Movement Disorders : Official Journal of the Movement Disorder Society, 28(6), 755–762. doi: 10.1002/mds.25389 [doi] [DOI] [PubMed] [Google Scholar]
- Zimmer HD (2008). Visual and spatial working memory: From boxes to networks. Neuroscience and Biobehavioral Reviews, 32(8), 1373–1395. doi: 10.1016/j.neubiorev.2008.05.016 [doi] [DOI] [PubMed] [Google Scholar]
- Zühlke C, Budnik A, Gehlken U, Dalski A, Purmann S, Naumann M, … Schwinger E (2004). FMR1 premutation as a rare cause of late onset ataxia--evidence for FXTAS in female carriers. J Neurol, 251(11), 1418–1419. [DOI] [PubMed] [Google Scholar]
- Zuhlke C, & Burk K (2007). Spinocerebellar ataxia type 17 is caused by mutations in the TATA-box binding protein. Cerebellum (London, England), 6(4), 300–307. doi: 10.1080/14734220601136177 [doi] [DOI] [PubMed] [Google Scholar]