

Abstract
Each year, millions of people worldwide contract tuberculosis (TB), the deadliest infection. The spread of infections with drug-resistant strains of Mycobacterium tuberculosis (Mtb) that are refractory to treatment poses a major global challenge. A major cause of resistance to antitubercular drugs of last resort, aminoglycosides, is overexpression of the Eis (enhanced intracellular survival) enzyme of Mtb, which inactivates aminoglycosides by acetylating them. We showed previously that this inactivation of aminoglycosides could be overcome by our recently reported Eis inhibitors that are currently in development as potential aminoglycoside adjunctive therapeutics against drug-resistant TB. To interrogate the robustness of the Eis inhibitors, we investigated the enzymatic activity of Eis and its inhibition by Eis inhibitors from three different structural families for nine single-residue mutants of Eis, including those found in the clinic. Three engineered mutations of the substrate binding site, D26A, W36A, and F84A, abolished inhibitor binding while compromising Eis enzymatic activity 2- to 3-fold. All other Eis mutants, including clinically observed ones, were potently inhibited by at least one inhibitor. This study helps position us one step ahead of Mtb resistance to Eis inhibitors as they are being developed for TB therapy.
Keywords: acetylation, enzyme inhibitor, kanamycin, mutations, mycobacteria
Graphical Abstract
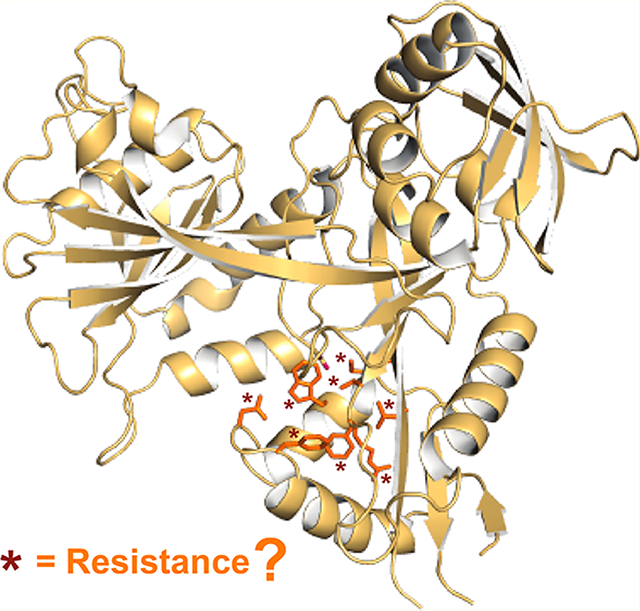
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), has been gaining resistance to antitubercular drugs due to the use of the same first-line antibiotics against this widespread disease over the course of 60 years.1 Some infections have become impossible to treat in all but the most sophisticated clinical settings.2,3 Currently, Mtb infections are classified into three clinical categories: drug-sensitive (responding to conventional first-line therapy), multidrug-resistant (MDR; defined as resistant to at least two first-line drugs, isoniazid and rifampin), and extensively drug-resistant (XDR; MDR-TB with additional resistance to a fluoroquinolone and one of the injectable drugs: kanamycin (KAN), capreomycin, or amikacin (AMK)).4 Pre-XDR-TB is defined as MDR-TB with additional resistance to either a fluoroquinolone or an injectable agent but not both.5 With increasing resistance of Mtb to current treatments, nearly 1.5 million deaths, and 9 million people falling ill with TB every year, new strategies are needed to combat this deadly global threat.6
Drug resistance in Mtb evolves and propagates exclusively through chromosomal mutations; no resistance vectors have been found.7,8 Specifically, resistance in Mtb emerges through single-nucleotide polymorphisms.9 Mtb undergoes a random mutation every 5000 replicated base pairs in vitro10 resulting in about 800 mutations for each replication of the Mtb genome. Therefore, as many as 800 genes and regulatory DNA elements can be affected each time Mtb undergoes cell division, excluding the mutations that inactivate the genes that are essential for bacterial survival. A common and well-characterized mechanism of resistance to a last-resort antitubercular drug, KAN, is its acetylation by the Eis (enhanced intracellular survival) protein upon its upregulation.11 In approximately one-third of the clinical isolates of KAN-resistant Mtb, Eis upregulation occurs due to one of several single mutations in the eis promoter.11,12 In another ~10%, Eis is overexpressed due to mutations in the 5′ untranslated region (5′ UTR) of the transcription factor whiB7.13 Eis is very versatile in its capacity to acetylate all clinically used aminoglycosides;14 therefore, the prospects of developing a novel aminoglycoside that would not be susceptible to inactivation by Eis are poor. Instead, we hypothesized that an Eis inhibitor, when used as a drug together with KAN, would have the potential to inactivate Eis and protect this antibiotic from inactivation, thus preventing or overcoming KAN resistance of TB. We discovered and synthesized several families of compounds with diverse structures that potently inhibited the Eis enzymatic activity15–20 and restored sensitivity to KAN of Mtb K204.16,17 Mtb K204 is a variant of a commonly used Mtb strain, H37Rv, which harbors an eis promoter mutation (C-14T) responsible for Eis upregulation and consequent KAN resistance of this strain and clinically encountered strains containing this or other mutations in the eis promoter.12 Our previous structural studies demonstrate that these Eis inhibitors function by noncovalent binding in the aminoglycoside binding pocket of Eis competitively with the aminoglycoside.15–17
Because aminoglycoside-modifying enzymes (AMEs) have been known to be responsible for most of the clinically relevant aminoglycoside resistance in different bacterial pathogens, inhibitors of these enzymes have been previously sought, and this pursuit is ongoing.21 The ATP analogue 5′-[p-(fluorosulfonyl)benzoyl]adenosine22 and the kinase inhibitor wortmannin were shown to inhibit aminoglycoside kinases.23 Another example of an AME inhibitor is streptidine, which competes with the aminoglycoside streptomycin for binding aminoglycoside nucleotidyltransferase ANT(6), overcoming ANT(6)-mediated resistance to streptomycin of Escherichia coli overexpressing ANT(6).24 A modern chromatography-coupled mass spectrometry method was recently developed for discovery of AMEs.25
A major mode of resistance to an antibiotic is a mutation in its target. Such mutations significantly weaken the binding of this antibiotic to its target while not substantially affecting a function of this target. This mode of resistance, through Eis mutations, may occur in response to treatment with Eis inhibitors (in combination with KAN). Therefore, we set out to investigate whether our Eis inhibitors maintain their potency in the face of Eis mutagenesis.
RESULTS AND DISCUSSION
Crystal Structures of Eis−Inhibitor Complexes
This study is focused on the effect of Eis mutations on the potency of three Eis inhibitors from different structural families (compounds 1, 2, and 3 in Figure 1). These molecules were shown to potently inhibit Eis in vitro and abolish KAN resistance in a relevant Mtb strain where Eis was upregulated as a result of a single clinically observed mutation in the eis promoter.12 Resistance mutations in enzymes lie predominantly in the inhibitor binding sites. In order to determine which residues interact with the three Eis inhibitors, we determined crystal structures of Eis in complexes with inhibitors 1, 2, and 3. In all three cases, the inhibitors were well-defined by a strong difference electron density (Figure 2). All the inhibitors bound at a site partially overlapping with the aminoglycoside binding pocket, as defined by our previously reported structure of the Eis–tobramycin complex (Figure 2A),26 consistent with the competitive mode of inhibition. Our previously published crystal structures of Eis in complex with inhibitors from these and other families also show an inhibitor–aminoglycoside binding site overlap.15–17 Furthermore, kinetic data demonstrated that inhibitor 3 is, indeed, a competitive inhibitor.7
Figure 1.

Chemical structures of tobramycin (TOB) and the Eis inhibitors used in this study. Inhibitors 1 (SGT449), 2 (SGT335), and 3 (SGT416) in this study were numbered 46,17 2h*,16 and 37b,15 respectively, in our previous publications.
Figure 2.

Crystal structures of Eis monomer bound to TOB and the inhibitors. (A) The zoom-in view of the substrate binding site in the Eis–TOB complex (PDB ID: 4JD6).26 The Eis residues mutated in this study are shown as sticks in this panel. To simplify the view, the CoA was omitted in this figure. (B) The Eis–inhibitor 1 complex. A sulfate ion (the sulfur is shown in yellow and the oxygens in red) is also present. (C) The Eis–inhibitor 2 complex. (D) Eis–inhibitor 3 complex. Polder inhibitor omit maps27 contoured at 5σ are shown as a raspberry colored mesh in panels B, C, and D. The Eis residues interacting with TOB and the inhibitors are shown in orange. Oxygen, nitrogen, fluorine, and sulfur atoms are colored red, blue (light blue in inhibitors and Eis residues, dark blue in TOB), cyan, and pale yellow, respectively. Carbon atoms are colored light blue in TOB and dark blue, green, and purple in inhibitors 1, 2, and 3, respectively. The Eis C-terminus is labeled as C-ter.
Inhibitor 1 is bound to Eis making several electrostatic, π–π stacking, and hydrophobic interactions (Figure 2B). Specifically, the naphthalene moiety is stacked between Trp36 and Phe84. The naphthalene makes nonpolar contacts with Ala33, the aliphatic portion of Arg37, Val40, Leu63, and Met65. van der Waals interactions are observed between the Oγ of Ser83 and the naphthalene moiety. Hydrophobic contacts are observed between the N-methyl group of the sulfonamide moiety and Trp36. A sulfonamide oxygen of the inhibitor is at a hydrogen bonding distance from the main chain nitrogen of Ile28 (the O–N distance is 3.1 Å). The quinoxalinedione moiety of inhibitor 1 stacks in a parallel fashion against the phenyl ring of Phe24, while an NH group of the quinoxalinedione ring forms a hydrogen bond with the carboxyl group of Asp26 (the N–O distance is 2.7 Å).
Inhibitor 2 containing a pyrrolopyrazine core makes extensive hydrophobic interactions in the binding pocket (Figure 2C). The pyrrole portion interacts with Ile28 and, sterically, with Ser32, while the pyrazine portion contacts Phe27 and Ala33. The fluorophenyl moiety attached to the pyrazine ring is held sterically between Asp26 and Glu401. The cationic nitrogen of the pyrazine ring makes a cation−π system contact with Trp36 and, on the other face of the ring, its charge is neutralized by a bound sulfate ion from the crystallization solution. This fluorophenyl ring likely rotates, as the electron density for it is weaker than for the rest of the molecule. The acetophenone ring is sandwiched between Trp36 and Phe84, and its oxygen is at a distance consistent with a weak hydrogen bond (3.6 Å) from the hydroxyl group oxygen of Ser83. The fluoro-substituted acetophenone is snugly bound in a hydrophobic pocket formed by Trp13, the aliphatic stem of Arg37, Val40, Leu63, and Met65.
Inhibitor 3 is bound to Eis through extensive hydrophobic interactions (Figure 2D). Specifically, the large 1,2,4-triazino[5,6b]indole-3-thioether ring system is sandwiched between the side chains of Trp36 and Phe84. The ring system also interacts sterically with Ala33 and Phe27 and, through nonpolar contacts, with Leu63 and Met65. The fluorine substitution at the C8 position of the ring system projects into the hydrophobic pocket of the aliphatic portion of Trp13, Arg37, Val40, and Leu63. The N-methyl group shows steric interaction with the hydroxyl group of Ser83 and the C-terminal carboxyl group. Electrostatic interactions between the C-terminal carboxyl group and this nitrogen likely also contribute to the Eis–inhibitor binding. The flexible thioether containing substitution extends toward the opening of the binding pocket, appropriately positioning the diethyl-amino group for a salt bridge with Asp26 and a weak electrostatic interaction with Glu401. The thioether linker is sterically guided by the backbone of Phe27 and hydrophobic interactions with Ile28.
On the basis of these crystal structures, we chose to mutate seven residues (Asp26, Trp36, Arg37, Leu63, Met65, Ser83, and Phe84; Table S1) that were not directly involved in the acetyl transfer14 and were observed to interact either with the bound tobramycin26 (TOB; Figure 2A) or with the bound inhibitors (Figure 2B–D). The interactions between these residues and the inhibitors observed in the structures are summarized in Table 1. Residues Asp26, Trp36, Arg37, Leu63, Met65, Ser83, and Phe84 interact with all three inhibitors, whereas Phe24 interacts with inhibitor 1 and TOB, but not with inhibitors 2 or 3. We previously demonstrated that Phe24Ala abolished KAN acetylation activity of Eis.14 In the crystal structure of the Eis–TOB complex, the phenyl ring of Phe24 is stacked against one of the TOB sugar moieties (Figure 2A). Therefore, mutagenesis of this residue was not pursued in this study. Mutation in this residue would simply be disadvantageous to Mtb under KAN stress. On the other hand, residues Arg37, Leu63, and Met65 were observed to interact with the inhibitors, but not with TOB and, therefore, these residues were especially interesting to mutate.
Table 1.
Interactions of Inhibitors with Eis Residues of Interest
| Eis residue/ligand | TOB | inhibitor 1 | inhibitor 2 | inhibitor 3 |
|---|---|---|---|---|
| F24 | hydrophobic | π–π | ||
| D26 | salt bridge | H-bond | steric | salt bridge |
| W36 | H-bond/steric | π–π stacking | π–π stacking | π–π stacking |
| R37 | hydrophobic | hydrophobic | hydrophobic | |
| L63 | hydrophobic | hydrophobic | hydrophobic | |
| M65 | hydrophobic | hydrophobic | hydrophobic | |
| S83 | H-bond | steric | steric | steric |
| F84 | hydrophobic | π–π stacking | π–π stacking | π–π stacking |
To determine the effects of mutating the other seven residues on aminoglycoside acetylation and inhibitor binding, we individually mutated them to alanine. Furthermore, a search among all published genomic sequences of Mtb strains yielded mutations in some of these residues in clinical isolates, specifically Trp36Arg (in Mtb BTB10–295),28 as well as Arg37Gly (in Mtb XTB13–167) and Ser83Gly (in Mtb XTB13100);29 therefore, we also included these Eis mutants in this study. A role of these or any other mutations in the coding region of eis in resistance to KAN or other anti-TB drugs is unknown. These previous studies showed that Mtb BTB10–295 and XTB13–100 were pre-XDR strains and Mtb XTB13–167 was an XDR strain, and all three strains were KAN-resistant. Strain BTB10–295 harbored mutation A1400G in the 16S rRNA (rrs) that was observed to be strongly associated with a high level of KAN and AMK resistance in Mtb,28,30 likely due to disruption of aminoglycoside binding to the ribosome. Therefore, inhibition of Eis in this context would not abolish KAN resistance. Nevertheless, these results raise the possibility of the presence of Trp36Arg mutation in the absence of ribosome mutations in other strains. Strain XTB13–167 did not contain any rrs, eis promoter, or whiB7 5′ UTR mutations; therefore, upregulation of Eis as a cause of KAN resistance could not be ruled out. Strain XTB13–100 contained a C-12T mutation in the eis promoter, which was shown to increase expression of Eis to the level that may cause clinically relevant resistance to KAN.12
It should be noted that we were not attempting to evaluate the propensity of Mtb for mutagenesis of the eis gene nor predict or model the most likely mutations that the Eis inhibitors may select for upon their clinical use. Instead, this study was aimed to assess how Eis acetylation activity responded to mutagenesis and, importantly, if so, whether our inhibitors were robust and versatile in their capacity to inhibit various mutants. For example, if a mutation were found to abolish inhibition by one of the inhibitors, could another inhibitor inactivate the mutant enzyme?
Effect of Mutation on Steady-State Kinetics of Acetylation of Kanamycin by Eis
We first examined the effect of Eis mutations on the steady-state kinetics of acetylation of the clinically relevant aminoglycoside, kanamycin (KAN) (Table 2 and Figure S1). Three mutations, Asp26Ala, Arg37Ala, and clinically identified Arg37Gly, modestly reduced the binding affinity of Eis to KAN. Eis_D26A displayed the largest, 2.3-fold, increase in the Km value of KAN from that for the wild-type enzyme Km (wt) = 551 ± 134 μM to Km (D26A) = 1280 ± 360 μM, while the kcat values were more similar to each other, 14.3 ± 1.9 min−1 (wt) and 14.1 ± 2.5 min−1 (D26A). Asp26 forms a salt bridge with one of the amino groups on the deoxystreptamine ring of the bound TOB in the crystal structure (Figure 1A). This effect of the mutation underscores an important contribution to the binding affinity of salt bridges between acidic residues of Eis and the amino groups of aminoglycosides. Two of the mutants, Trp36Arg and Met65Ala, displayed modestly increased affinity of Eis to KAN, with Km = 352 ± 77 μM and Km = 247 ± 19 μM, respectively. The 1.6-fold increase of the binding affinity to KAN for the clinically identified Trp36Arg was, however, offset by a significant (3.3-fold) decrease in the maximum turnover rate for this mutant, kcat = 4.4 ± 0.4 min−1, the lowest rate of all tested mutants. These observations indicate that the increased binding affinity of the Trp36Arg mutant is accompanied by mispositioning of the KAN amino groups that are acetylated or, less likely, by a reduction of the product release rate or by both. In contrast, the maximum turnover rate of the Met65Ala mutant was 2-fold higher, resulting in a 4.6-fold increase in the catalytic efficiency from that of the wild-type enzyme. It is possible that, even though Met65 does not directly interact with the bound TOB and, by inference, with bound KAN, the smaller Ala residue at this position allows for more adaptability of the Eis active site to KAN binding and acetylation. The only other mutation that statistically significantly increased (2-fold) the catalytic efficiency of the enzyme was the clinically identified Ser83Gly, solely by an increase in kcat. As with the Met65Ala mutant, more binding site space and backbone flexibility must result in somewhat improved positioning of the KAN molecule in the mutant enzyme active site for acetylation. A loss of a potential hydrogen bond with the hydroxyl of the Ser and KAN can likely be compensated by a hydrogen bond with a water molecule that can fill the void formed by the removal of the side chain in the mutant. Mutants Leu63Ala and a clinically observed Arg37Gly displayed comparable steady-state kinetics to those of the wild-type enzyme. These observations were consistent with the lack of direct interactions of these residues with the bound TOB and, likely, with KAN. The kinetic parameters for the other mutants were generally unaffected or modestly (less than 2-fold) negatively affected.
Table 2.
Kinetic Values of Wild-Type Eis (Eis_wt) and Its Mutants
| enzyme | Km (μM) | kcat (min−1) | kcat/Km (min−1 μM−1) |
|---|---|---|---|
| Eis_wta | 551 ± 134 | 14.3 ± 1.9 | 0.026 ± 0.007 |
| Eis_D26A | 1280 ± 360 | 14.1 ± 2.5 | 0.011 ± 0.004 |
| Eis_W36A | 556 ± 148 | 8.3 ± 1.0 | 0.015 ± 0.004 |
| Eis_W36R | 352 ± 77 | 4.4 ± 0.4 | 0.013 ± 0.003 |
| Eis_R37Aa,c | 971 ± 166 | 9.5 ± 0.3 | 0.010 ± 0.002 |
| Eis_R37Gb,c | 696 ± 44 | 12.9 ± 0.1 | 0.019 ± 0.001 |
| Eis_L63Ab,c | 484 ± 93 | 19.4 ± 1.5 | 0.040 ± 0.008 |
| Eis_M65Ac | 247 ± 19 | 29.7 ± 0.9 | 0.122 ± 0.015 |
| Eis_S83Gb | 432 ± 48 | 21.9 ± 0.8 | 0.051 ± 0.006 |
| Eis_F84A | 569 ± 94 | 8.5 ± 0.7 | 0.015 ± 0.003 |
Effect of Mutants on Inhibitor Potency and Binding
Having examined the effects of the mutations on the activity of Eis, we next tested their effect on the inhibition of this enzyme by inhibitors 1, 2, and 3 (Table 3 and Figure S2). Because the effects of the mutations on Km values were relatively small, IC50 measurements accurately reflect the trend of binding affinities of the inhibitors. Mutations Asp26Ala, Trp36Ala, and Phe84Ala all rendered the enzyme insensitive to all three inhibitors up to 200 μM. Mutations Trp36Ala and Phe84Ala significantly compromise major hydrophobic interfaces between Eis and the aromatic moieties of the inhibitor molecules (Figure 1B–D). In a relative sense, the deleterious effect of these mutations on the catalytic efficiency of the enzyme is much smaller (~2-fold), consistent with a likely less significant relative contribution of the interfaces between these residues and the larger and less hydrophobic KAN. Similarly, the relative contribution of the salt bridge between Asp26 and an amino group of the aminoglycoside substrate to Eis–aminoglycoside binding is much smaller than the contribution of similar Asp26–inhibitor interactions, where a more fixed position of the inhibitor may preclude replacing these interactions by another acidic residue of Eis. Nevertheless, these mutations are predicted to impose a fitness penalty, as the ~2-fold lower catalytic efficiency of Eis_D26A, Eis_W36A, and Eis_F84A as compared to the wild-type enzyme is predicted to result in a 2-fold reduction of the MIC of KAN, on the basis of the relationship between the Eis expression levels in different mutant strains and KAN MIC values.12 It should also be noted that, while an Asp can be mutated to an Ala by a single nucleotide substitution, a Trp and a Phe cannot. Both a Trp and a Phe can nevertheless be mutated to a Ser by a single nucleotide substitution. We were pleased to observe that, even though a clinically found Trp36Arg mutation rendered Eis insensitive to compounds 2 and 3, compound 1 displayed a 2-fold improvement in the IC50 value from that observed with the wild-type Eis. It appears that the cationic guanidinyl group of the Arg residue at this position can maintain a strong cation–aromatic ring interaction only with the heteroatom-free naphthalene substituent of compound 1, but not with a somewhat more conformationally flexible parafluorophenyl group of compound 2 or the nitrogen-rich triazinoindole ring of compound 3. Eis_R37A, in accord with a relatively minor hydrophobic interaction between the aliphatic stem of Arg37 and the inhibitors observed, displayed modestly increased IC50 values (1.5- to 4.4-fold) for all compounds, thereby not significantly perturbing the high inhibitor potency. A more severely structurally perturbing clinically found mutation, Arg37Gly, while insignificantly altering KAN acetylation (in agreement with the lack of interactions with TOB; Figure 2A, Table 1), had a more disruptive effect on inhibitor binding and inhibition. While compound 1 no longer inhibited the enzyme and compound 3 displayed an 18.5-fold increase in IC50, the IC50 value of compound 2 increased by only 5-fold. Even for this disruptive mutant, compounds 2 and 3 retained considerable potency, with an IC50 of ~1 μM. Arg37 is one of the most distant residues from the catalytic site among the residues that interact with the inhibitors; its mutation to a Gly likely has a conformational effect on the backbone and may destabilize several highly optimized Eis–inhibitor interactions. For the remaining clinically relevant Ser83Gly mutation, the structural variability of the inhibitor scaffolds was highly advantageous: while compound 2 did not inhibit Eis_S83G and compound 1 exhibited a 10-fold increase in IC50 value, the IC50 value for compound 3 was unaffected. We propose that the triple ring system unique to inhibitor 3 is held largely by extensive stacking interactions, whereas the steric contribution of the N-methyl-Ser83 interaction is relatively minor compared to the relative contribution of this residue to the stability of the complexes of Eis with inhibitors 1 and 2.
Table 3.
IC50 Values for the Inhibitors of Interest Measured for Eis_wt and Its Mutants
| enzyme | inhibitor | IC50 (μM) | Ki (μM) |
|---|---|---|---|
| Eis_wt | 1 | 0.088 ± 0.012 | 0.048 ± 0.003 |
| 2 | 0.197 ± 0.025 | 0.095 ± 0.005 | |
| 3 | 0.093 ± 0.02 | 0.096 ± 0.010 | |
| Eis_D26A | 1 | >200 | –a |
| 2 | >200 | – | |
| 3 | >200 | – | |
| Eis_W36A | 1 | >200 | – |
| 2 | >200 | – | |
| 3 | >200 | – | |
| Eis_W36R | 1 | 0.046 ± 0.016 | 0.037 ± 0.004 |
| 2 | >200 | – | |
| 3 | >200 | – | |
| Eis_R37A | 1 | 0.388 ± 0.071 | 0.175 ± 0.037 |
| 2 | 0.301 ± 0.047 | 0.197 ± 0.017 | |
| 3 | 0.252 ± 0.024 | 0.185 ± 0.015 | |
| Eis_R37G | 1 | >200 | – |
| 2 | 0.975 ± 0.080 | 0.351 ± 0.047 | |
| 3 | 1.72 ± 0.21 | 0.349 ± 0.039 | |
| Eis_L63A | 1 | >200 | – |
| 2 | 0.227 ± 0.018 | 0.147 ± 0.014 | |
| 3 | 0.148 ± 0.016 | 0.063 ± 0.006 | |
| Eis_M65A | 1 | 0.643 ± 0.099 | 0.122 ± 0.015 |
| 2 | >200 | – | |
| 3 | >200 | – | |
| Eis_S83G | 1 | 0.880 ± 0.080 | 1.03 ± 0.09 |
| 2 | >200 | – | |
| 3 | 0.118 ± 0.017 | 0.060 ± 0.006 | |
| Eis_F84A | 1 | >200 | – |
| 2 | >200 | – | |
| 3 | >200 | – |
Ki measurements were not carried out for IC50 > 200 μM due to very weak inhibitor binding.
Mutations Leu63Ala and Met65Ala also had varied effects on inhibition by the three compounds, underscoring the high value of the differences in inhibitor structures and the resulting differences in their interactions with Eis. Even though these residues appear to have minor individual contributions to nonpolar surface contacts with the inhibitors, together with the neighboring hydrophobic residues, they form extensive hydrophobic interfaces. The relative extent of contacts of these surfaces with nonpolar moieties of the inhibitors differs among the three inhibitors due to their differences in chemical structure. Eis_L63A was not inhibited by compound 1, whereas the potency of compounds 2 and 3 against this mutant was not significantly affected. It appears the presence of the fluorophenyl in inhibitors 2 and 3, which lies in the vicinity of Leu63 and Met65, contributes significantly to the stability of Eis complexes with these inhibitors. Analogously, Eis_M65A was not inhibited by compounds 2 and 3, and it showed a 3.5-fold higher IC50 value for compound 1.
Our previous steady-state kinetic analysis of inhibition kinetics, in agreement with the structural observations, showed that inhibitor 3 was competitive with KAN15 and that an analogue of inhibitor 2 displayed a mixed mode of inhibition with a strong competitive contribution.20 Here, we have performed similar kinetic studies for inhibitors 1, 2, and 3 both with the wild-type Eis and its mutants (Figure S1, Table S2). The data indicated competitive inhibition kinetics in all cases, as visualized by the Lineweaver–Burk plots (Figure S3). For all combinations of enzyme and inhibitor, where inhibition was observed, the Ki value was determined by nonlinear regression analysis (Table 3). As expected on the basis of a relative insensitivity of Km values to mutagenesis, the IC50 values described above faithfully represented the trend in enzyme binding affinities of these inhibitors, with most Ki values being at most 2-fold smaller than IC50 (Km for most mutants was equal or larger than the concentration of KAN in the IC50 measurements).
CONCLUDING REMARKS
In this study, we have presented mutations of seven residues of Eis that are in the aminoglycoside/inhibitor binding site. Mutation of six of these residues had significant effects on the inhibition activity of at least one of the families of inhibitors tested. Three of the mutations, Asp26Ala, Trp36Ala, and Phe84Ala, even though not likely to happen naturally, were shown to render all three inhibitors inactive while only modestly perturbing the KAN acetylating activity of Eis. Nevertheless, this knowledge alerts us to a potential disruptive effect of mutagenesis of these residues. With this knowledge in hand, we can take steps to design inhibitors with functionalities that do not depend on interactions with these residues for binding Eis or that accommodate alterations to these residues. We were delighted to learn that, for the other six mutations, including all three clinically identified Eis mutations, one or more of the inhibitors maintained their potency. This reassures us in the prospects of developing Eis inhibitors that could become a staple of MDR- and XDR-TB treatments as a cocktail with KAN. Identifying possible routes of resistance for these agents remains a key step to increasing the lifetime and usability of these molecules once they enter the clinic. This study illustrates how bringing together genomics with basic bench science can guide us in the challenging pursuit of future medicine.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by a National Institutes of Health (NIH) grant AI090048 (to S.G.-T.) and startup funds from the College of Pharmacy at the University of Kentucky (to S.G.-T. and O.V.T.). We thank the staff of sector 22 (SER-CAT) of the Advanced Photon Source at the Argonne National Laboratories for their assistance with the remote X-ray diffraction data collection. The beamline use was supported, in part, by the Center for Structural Biology at the University of Kentucky.
ABBREVIATIONS
- Eis
enhanced intracellular survival
- AMK
amikacin
- KAN
kanamycin
- MDR
multidrug-resistant
- Mtb
Mycobacterium tuberculosis
- TOB
tobramycin
- TB
tuberculosis
- XDR
extensively drug-resistant
Footnotes
The authors declare no competing financial interest.
Accession Codes
6P3T, 6P3U, and 6P3V.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.9b00228.
Experimental procedures; tables of primers used for cloning (Table S1), kinetic parameters for each triplicate experiment for Eis enzymes with varied concentrations of inhibitors (Table S2), and X-ray diffraction data collection and structure refinement statistics for Eis–inhibitor complexes (Table S3); Michaelis–Menten curves (Figure S1), IC50 curves (Figure S2), and representative Lineweaver−Burk plots (Figure S3) (PDF)
REFERENCES
- (1).Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, and Bayona J (2010) Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet 375 (9728), 1830–1843. [DOI] [PubMed] [Google Scholar]
- (2).Udwadia ZF, Amale RA, Ajbani KK, and Rodrigues C (2012) Totally drug-resistant tuberculosis in India. Clin. Infect. Dis 54 (4), 579–581. [DOI] [PubMed] [Google Scholar]
- (3).Caminero JA, Sotgiu G, Zumla A, and Migliori GB (2010) Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis 10 (9), 621–629. [DOI] [PubMed] [Google Scholar]
- (4).World Health Organization. (2018) Global tuberculosis report 2018, WHO, Geneva, Switzerland. [Google Scholar]
- (5).Banerjee R, Allen J, Westenhouse J, Oh P, Elms W, Desmond E, Nitta A, Royce S, and Flood J (2008) Extensively drug-resistant tuberculosis in California, 1993–2006. Clin. Infect. Dis 47 (4), 450–457. [DOI] [PubMed] [Google Scholar]
- (6).Green KD, and Garneau-Tsodikova S (2013) Resistance in tuberculosis: what do we know and where can we go? Front. Microbiol 4, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huitric E, Verhasselt P, Koul A, Andries K, Hoffner S, and Andersson DI (2010) Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother 54 (3), 1022–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gillespie SH (2002) Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob. Agents Chemother 46 (2), 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rosales-Klintz S, Jureen P, Zalutskayae A, Skrahina A, Xu B, Hu Y, Pineda-Garcia L, Merza MA, Muntean I, Bwanga F, Joloba M, and Hoffner SE (2012) Drug resistance-related mutations in multidrug-resistant Mycobacterium tuberculosis isolates from diverse geographical regions. Int. J. Mycobacteriol 1 (3), 124–130. [DOI] [PubMed] [Google Scholar]
- (10).McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, and Warner DF (2014) Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother 69 (2), 292–302. [DOI] [PubMed] [Google Scholar]
- (11).Campbell PJ, Morlock GP, Sikes RD, Dalton TL, Metchock B, Starks AM, Hooks DP, Cowan LS, Plikaytis BB, and Posey JE (2011) Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 55 (5), 2032–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zaunbrecher MA, Sikes RD Jr., Metchock B, Shinnick TM, and Posey JE (2009) Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 106 (47), 20004–20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Reeves AZ, Campbell PJ, Sultana R, Malik S, Murray M, Plikaytis BB, Shinnick TM, and Posey JE (2013) Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob. Agents Chemother. 57 (4), 1857–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen W, Biswas T, Porter VR, Tsodikov OV, and Garneau-Tsodikova S (2011) Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc. Natl. Acad. Sci. U. S. A 108 (24), 9804–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ngo HX, Green KD, Gajadeera CS, Willby MJ, Holbrook SYL, Hou C, Garzan A, Mayhoub AS, Posey JE, Tsodikov OV, and Garneau-Tsodikova S (2018) Potent 1,2,4-triazino[5,6 b]indole-3-thioether inhibitors of the kanamycin resistance enzyme Eis from Mycobacterium tuberculosis. ACS Infect. Dis 4 (6), 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Garzan A, Willby MJ, Ngo HX, Gajadeera CS, Green KD, Holbrook SY, Hou C, Posey JE, Tsodikov OV, and Garneau-Tsodikova S (2017) Combating enhanced intracellular survival Eis-mediated kanamycin resistance of Mycobacterium tuberculosis by novel pyrrolo[1,5-a]pyrazine-based Eis inhibitors. ACS Infect. Dis 3 (4), 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Garzan A, Willby MJ, Green KD, Gajadeera CS, Hou C, Tsodikov OV, Posey JE, and Garneau-Tsodikova S (2016) Sulfonamide-based inhibitors of aminoglycoside acetyltransferase Eis abolish resistance to kanamycin in Mycobacterium tuberculosis. J. Med. Chem 59 (23), 10619–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Garzan A, Willby MJ, Green KD, Tsodikov OV, Posey JE, and Garneau-Tsodikova S (2016) Discovery and optimization of two Eis inhibitor families as kanamycin adjuvants against drug-resistant M. tuberculosis. ACS Med. Chem. Lett 7 (12), 1219–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Willby MJ, Green KD, Gajadeera CS, Hou C, Tsodikov OV, Posey JE, and Garneau-Tsodikova S (2016) Potent inhibitors of acetyltransferase Eis overcome kanamycin resistance in Mycobacterium tuberculosis. ACS Chem. Biol 11 (6), 1639–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Green KD, Chen W, and Garneau-Tsodikova S (2012) Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. ChemMedChem 7 (1), 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zarate SG, De la Cruz Claure ML, Benito-Arenas R, Revuelta J, Santana AG, and Bastida A (2018) Overcoming aminoglycoside enzymatic resistance: Design of novel antibiotics and inhibitors. Molecules 23 (2), 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McKay GA, Robinson RA, Lane WS, and Wright GD (1994) Active-site labeling of an aminoglycoside antibiotic phosphotransferase (APH(3′)-IIIa). Biochemistry 33 (47), 14115–14120. [DOI] [PubMed] [Google Scholar]
- (23).Boehr DD, Lane WS, and Wright GD (2001) Active site labeling of the gentamicin resistance enzyme AAC(6′)-APH(2″) by the lipid kinase inhibitor wortmannin. Chem. Biol 8 (8), 791–800. [DOI] [PubMed] [Google Scholar]
- (24).Latorre M, Penalver P, Revuelta J, Asensio JL, GarciaJunceda E, and Bastida A (2007) Rescue of the streptomycin antibiotic activity by using streptidine as a ″decoy acceptor″ for the aminoglycoside-inactivating enzyme adenyl transferase. Chem. Commun No. 27, 2829–2831. [DOI] [PubMed] [Google Scholar]
- (25).Zhu L, Liu R, Liu T, Zou X, Xu Z, and Guan H (2019) A novel strategy to screen inhibitors of multiple aminoglycoside-modifying enzymes with ultra-high performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. J. Pharm. Biomed. Anal 164, 0520–527. [DOI] [PubMed] [Google Scholar]
- (26).Houghton JL, Biswas T, Chen W, Tsodikov OV, and Garneau-Tsodikova S (2013) Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis. ChemBioChem 14 (16), 2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, and Adams PD (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol 73 (2), 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Manson AL, Cohen KA, Abeel T, Desjardins CA, Armstrong DT, Barry CE 3rd, Brand J, Consortium TBGG, Chapman SB, Cho SN, Gabrielian A, Gomez J, Jodals AM, Joloba M, Jureen P, Lee JS, Malinga L, Maiga M, Nordenberg D, Noroc E, Romancenco E, Salazar A, Ssengooba W, Velayati AA, Winglee K, Zalutskaya A, Via LE, Cassell GH, Dorman SE, Ellner J, Farnia P, Galagan JE, Rosenthal A, Crudu V, Homorodean D, Hsueh PR, Narayanan S, Pym AS, Skrahina A, Swaminathan S, Van der Walt M, Alland D, Bishai WR, Cohen T, Hoffner S, Birren BW, and Earl AM (2017) Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat. Genet 49 (3), 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wollenberg KR, Desjardins CA, Zalutskaya A, Slodovnikova V, Oler AJ, Quinones M, Abeel T, Chapman SB, Tartakovsky M, Gabrielian A, Hoffner S, Skrahin A, Birren BW, Rosenthal A, Skrahina A, and Earl AM (2017) Wholegenome sequencing of Mycobacterium tuberculosis provides insight into the evolution and genetic composition of drug-resistant tuberculosis in Belarus. J. Clin. Microbiol 55 (2), 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Alangaden GJ, Kreiswirth BN, Aouad A, Khetarpal M, Igno FR, Moghazeh SL, Manavathu EK, and Lerner SA (1998) Mechanism of resistance to amikacin and kanamycin in Mycobacterium tuberculosis. Antimicrob. Agents Chemother 42 (5), 1295–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.