

Abstract
Granulomas are pathologically defined as focal aggregations of activated macrophages, Langerhans cells, and lymphocytes. Granulomas form in the lungs when the immune system barricades the substances it perceives as foreign but is unable to remove. Granulomas manifest with numerous imaging appearances in thoracic radiology, and their presence is a nonspecific finding. Granulomatous lung diseases comprise multiple entities with variable clinical manifestations and outcomes. Their imaging findings are rarely specific and can mimic malignancies, often triggering an extensive diagnostic workup. Radiologists must be familiar with the clinical manifestations and imaging findings of these entities to generate appropriate differential diagnoses. This review describes the imaging manifestations of various noninfectious, necrotizing, and nonnecrotizing granulomatous diseases that primarily affect the thorax.
©RSNA, 2020
SA-CME LEARNING OBJECTIVES
After completing this journal-based SA-CME activity, participants will be able to:
■ Recognize the imaging manifestations of thoracic noninfectious granulomatous processes.
■ Describe common clinical scenarios associated with thoracic noninfectious granulomatous processes.
■ Discuss the imaging and clinical approach to diagnosing thoracic noninfectious granulomatous processes.
Introduction
A granuloma is a collection of inflammatory cells, activated macrophages, Langerhans cells, and lymphocytes. A granuloma forms when the immune system cannot eliminate a foreign antigen and instead attempts to “wall it off.” Granulomatous lung diseases include a spectrum of infectious and noninfectious entities that manifest a variety of lung findings at imaging (1). The imaging appearance of some granulomatous diseases may mimic malignancy, leading to confirmatory biopsy (1). The radiologist has a role in identifying granulomatous disease processes that can be confidently diagnosed on the basis of imaging and the clinical context, which obviates the need for biopsy. The approach to the patient with a potential granulomatous disease must rely on correlating clinical, imaging, laboratory, and pathologic findings in a multidisciplinary fashion. The purposes of this article are to review the various noninfectious granulomatous entities and to show their pertinent imaging features so that the radiologist might consider this group of conditions in the differential diagnosis for appropriate cases. An algorithmic imaging approach that provides a framework for thinking about these conditions is presented (Fig 1).
Figure 1.

Flowchart categorization of various granulomatous diseases by imaging manifestations. BG = bronchocentric granulomatosis, CVID = common variable immunodeficiency, EGPA = eosinophilic granulomatosis with polyangiitis, GL-ILD = granulomatous and lymphocytic interstitial lung disease, GPA = granulomatosis with polyangiitis, IBD = inflammatory bowel disease, LG = lymphomatoid granulomatosis, pLCH = pulmonary Langerhans cell histiocytosis, RV = right ventricle.
Pathologic Mechanism of Granuloma Formation
The formation of a granuloma and the imaging appearance of its composition vary on the basis of its anatomic location and the triggering antigen, which is often unknown. The pathologic process also differs among diseases, and hence the explanation for granuloma formation for each of the diseases is beyond the scope of this article. The following narrative provides an example of granuloma formation in sarcoidosis (Fig 2). Guardian monocytes differentiate into specialized immune cells known as antigen-presenting cells, namely macrophages and dendritic cells, when they encounter an offending antigen (2). These antigens also trigger mast cells to release tissue necrosis factor (TNF)-α. TNF–α activates circulating neutrophils, which then activate the monocytes, creating a feedback loop (3). Tissue macrophages attempt to engulf the offending antigen and also release TNF-α, which initiates upregulation of activation and adhesion molecules. Activated macrophages produce chemokine ligands that help recruit other monocytes, regulatory T cells, and B cells. Natural killer cells help to further activate histiocytes and dendritic cells. These interactions result in a cascade of events that lead to recruitment of T cells to the site of inflammation. These T cells help macrophages in creating the granuloma (4,5). Over the course of days, a mature granuloma forms if the antigen persists (6).
Figure 2.

Diagram shows the simplified version of sarcoid granuloma formation in steps. Antigens trigger the mast cell to release TNF (step 1), which then activates the neutrophils in the bloodstream (step 2). These activated neutrophils activate the monocytes (step 3). In a separate pathway, antigens activate natural killer (NK) cells (step 4), and these antigens are also taken up by the tissue macrophages (step 5), which would release TNF (step 6). Activated macrophages release multiple ligands after NK cells stimulate interferon gamma (IFN-γ) (step 7); these multiple chemokine ligands (eg, monocyte chemoattractant protein 1) further attract multiple immune cells (Th1/17 cells, monocytes, regulatory T cells, and B cells) in the bloodstream (step 8). In addition, IFN-γ produced by local NK and gamma delta (γδ) T cells further activates resident tissue histiocytes and dendritic cells (step 9). Activated antigen-loaded dendritic cells then migrate to peripheral lymph nodes via the lymphatic channels (step 10), and under the influence of IL-1 T cells, mature into Th1 cells. Activated Th1 then produces IL-2 that expands the T cell population (step 11) and joins other immune cells in the bloodstream (step 12). Then, these Th1 cells and other immune cells go to sites of inflammation and help in formation of a granuloma by maturation of macrophages (step 13). Over the course of days, the mature granuloma is formed, with other cells if the antigen persists. The inflammatory deposits in the lymph nodes (step 11) and the formation of the granuloma itself (step 13) are what translate into the lymphadenopathy, and pulmonary nodules demonstrate, in this example of granulomatous disease, sarcoidosis.
Approach to Noninfectious Granulomatous Conditions of the Thorax
Although the imaging features of noninfectious granulomatous thoracic conditions are rarely specific, radiologists should consider them when conventional treatment of presumed infection is not effective or when biopsy results indicate a granulomatous process. A checklist for granulomatous disease should include consolidation, nodules, cavitation, airway involvement, vessel wall involvement, pleural effusion, and lymphadenopathy. At imaging, these processes can be categorized by their predominant feature and distribution including (a) macronodules, masses, or consolidation in a peribronchovascular distribution, (b) masses in a random distribution, (c) micronodules in a perilymphatic distribution, and (d) micronodules in a centrilobular distribution (Fig 1).
Macronodules, Masses, or Consolidation in a Peribronchovascular Distribution
Eosinophilic Granulomatosis with Polyangiitis
Eosinophilic granulomatosis with polyangiitis (EGPA, formerly known as Churg-Strauss syndrome) is vasculitis that demonstrates necrotizing granulomas at histopathologic evaluation. It is the least common antineutrophil cytoplasmic antibody–associated small-vessel vasculitis and has a prevalence rate of 17.8 cases per 1 000 000 inhabitants, as described in a Japanese survey (7). The mean age of onset is 38 years, with nearly all patients showing asthma and eosinophilia (8). Diagnosis is based on criteria set by the American College of Rheumatology in 1990 and is made when a positive biopsy for vasculitis is associated with at least four of the following: (a) asthma, (b) peripheral eosinophilia, (c) neuropathy (mono or poly), (d) nonfixed pulmonary infiltrates, (e) sinus abnormalities, and (f) extravascular eosinophils seen in biopsy specimen. These criteria yield sensitivity of 85% and specificity of 99.7% (9).
Chest radiograph abnormalities are nonspecific in EGPA. The most common radiographic abnormality is bilateral, nonsegmental, multifocal, and typically peripheral consolidation (10). Pleural effusion is seen in up to 50% of cases and may be secondary to eosinophilic cardiomyopathy or eosinophilic pleuritis (8). CT is better than other modalities for characterization of the parenchymal findings of EGPA. In one retrospective study (10), CT showed pulmonary parenchymal abnormalities with a bronchiolocentric pattern in approximately 76% of patients and with an airspace pattern in approximately 40% of patients. Findings in this study included small nodules (63%), ground-glass opacities (53%), bronchial wall thickening (53%), bronchial dilatation (53%), consolidation (42%), interlobular septal thickening (42%), and mosaic attenuation (47%).
Parenchymal opacities show a mixture of eosinophilic pneumonia, necrotizing granulomas, and granulomatous vasculitis at histologic evaluation. Small nodules correspond to eosinophilic bronchiolitis and peribronchiolar vasculitis, while bronchial wall thickening is usually from airway wall eosinophilic and lymphocytic infiltration (11). Mediastinal lymphadenopathy is uncommon and manifests in less than 25% of patients (9). Because of the eosinophilia, patients are more prone to thromboembolic phenomena, especially pulmonary embolism (12). Cavitary lesions are uncommon in EGPA, as opposed to in granulomatosis with polyangiitis (GPA), in which they are common (13). The imaging findings in EGPA are presented in Figures 3 and 4.
Figure 3a.

Manifestations of EGPA on axial CT images in four patients include discrete peripheral subpleural consolidation (a), micronodules (b), ground-glass opacity (c), and interlobular septal line thickening intermixed with ground-glass opacity and bronchiectasis (d).
Figure 4a.

EGPA in a 40-year-old woman. (a) Axial CT image of the chest shows peripheral consolidations in the right lower lobe (arrows), which were the patient’s pulmonary manifestation of EGPA.(b) Maxillofacial CT image shows pansinusitis that includes the left sphenoid sinus (orange arrow) and bilateral ethmoid sinuses (yellow arrows). (c) Axial CT image at 4-year follow-up shows consolidation with cavitation (arrow). Given the new development of cavitation, short-term-interval follow-up or biopsy was recommended, because EGPA consolidations rarely have cavitations. The patient was lost to follow-up and later presented with pelvic pain. CT showed diffuse lytic osseous lesions including a pathologic fracture of the left iliac bone, which was secondary to metastatic non–small cell lung cancer at biopsy.
Figure 3b.

Manifestations of EGPA on axial CT images in four patients include discrete peripheral subpleural consolidation (a), micronodules (b), ground-glass opacity (c), and interlobular septal line thickening intermixed with ground-glass opacity and bronchiectasis (d).
Figure 3c.

Manifestations of EGPA on axial CT images in four patients include discrete peripheral subpleural consolidation (a), micronodules (b), ground-glass opacity (c), and interlobular septal line thickening intermixed with ground-glass opacity and bronchiectasis (d).
Figure 3d.
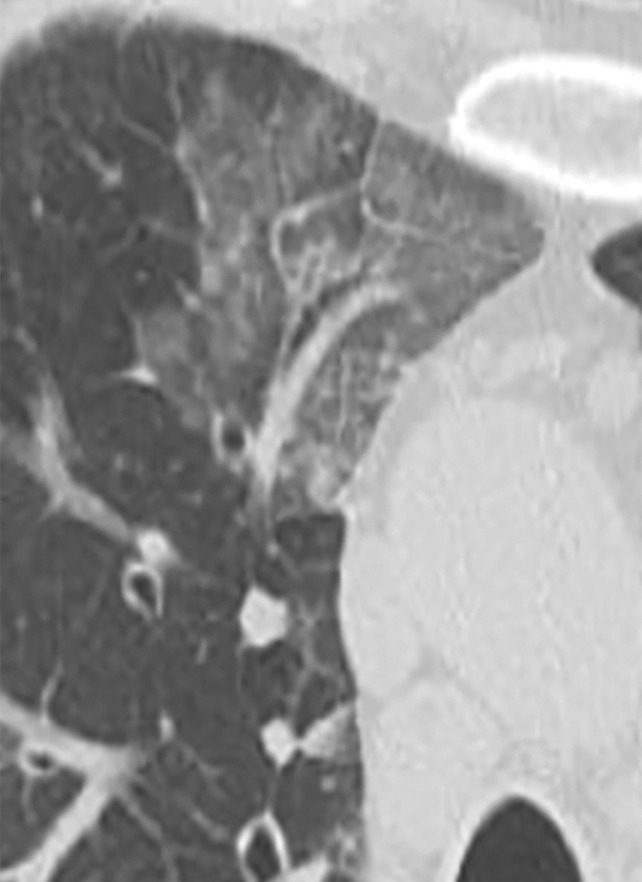
Manifestations of EGPA on axial CT images in four patients include discrete peripheral subpleural consolidation (a), micronodules (b), ground-glass opacity (c), and interlobular septal line thickening intermixed with ground-glass opacity and bronchiectasis (d).
Figure 4b.

EGPA in a 40-year-old woman. (a) Axial CT image of the chest shows peripheral consolidations in the right lower lobe (arrows), which were the patient’s pulmonary manifestation of EGPA.(b) Maxillofacial CT image shows pansinusitis that includes the left sphenoid sinus (orange arrow) and bilateral ethmoid sinuses (yellow arrows). (c) Axial CT image at 4-year follow-up shows consolidation with cavitation (arrow). Given the new development of cavitation, short-term-interval follow-up or biopsy was recommended, because EGPA consolidations rarely have cavitations. The patient was lost to follow-up and later presented with pelvic pain. CT showed diffuse lytic osseous lesions including a pathologic fracture of the left iliac bone, which was secondary to metastatic non–small cell lung cancer at biopsy.
Figure 4c.

EGPA in a 40-year-old woman. (a) Axial CT image of the chest shows peripheral consolidations in the right lower lobe (arrows), which were the patient’s pulmonary manifestation of EGPA.(b) Maxillofacial CT image shows pansinusitis that includes the left sphenoid sinus (orange arrow) and bilateral ethmoid sinuses (yellow arrows). (c) Axial CT image at 4-year follow-up shows consolidation with cavitation (arrow). Given the new development of cavitation, short-term-interval follow-up or biopsy was recommended, because EGPA consolidations rarely have cavitations. The patient was lost to follow-up and later presented with pelvic pain. CT showed diffuse lytic osseous lesions including a pathologic fracture of the left iliac bone, which was secondary to metastatic non–small cell lung cancer at biopsy.
Although it is not part of the diagnostic criteria, cardiac involvement is quite prevalent in EGPA, with a reported prevalence of 66% clinically and 92% at autopsy (14). Cardiac MRI findings of EGPA include reduced left ventricular ejection fraction, myocardial edema (shown as high T2 values), and late gadolinium subendocardial enhancement with or without a left ventricular apical thrombus. Unlike myocardial infarction, subendocardial late gadolinium enhancement in EGPA is not confined to one vascular territory (14).
Granulomatosis with Polyangiitis
Previously known as Wegener granulomatosis, granulomatosis with polyangiitis (GPA) is a necrotizing granulomatous vasculitis that typically manifests with a classic triad of upper airway symptoms (usually sinusitis), lower respiratory symptoms (such as hemoptysis), and glomerulonephritis (with hematuria) (8). Diagnosis of GPA is based on the American College of Rheumatology 1990 criteria and includes granulomatous vasculitis at biopsy, urinary sediment (the cellular prill that remains after urine centrifugation is cell- and crystal-free with a very low protein concentration; but in renal diseases such as GPA, it can contain red blood cells), abnormal chest radiograph, and oral or nasal inflammation. The presence of two or more of these four criteria yields sensitivity and specificity of 88.2% and 92%, respectively (15). However, if diagnosis does not fulfill the criteria of the American College of Rheumatology, then Watts and Robson (16) have proposed an alternative diagnostic scheme. Diagnosis of GPA can be made if (a) histologic results are compatible with the guidelines of the Chapel Hill Consensus Conference for GPA; (b) histologic results are compatible with microscopic polyangiitis, and the patient has surrogate markers of GPA; or (c) there are no histologic results, but there are positive surrogate markers and serologic results for antiprotease 3, myeloperoxidase, and antineutrophil cytoplasmic antibodies (ANCA). As with EGPA, CT is the mainstay in evaluating patients with GPA who have suspected thoracic involvement (17).
The incidence of thoracic involvement in GPA is up to 75% (17). Nodules or masses with or without central cavitation are the most frequent finding in GPA (Fig 5). These nodules tend to be bronchiolocentric or subpleural and are typically larger than those seen in EGPA. Ground-glass opacity and parenchymal consolidations, which occasionally cavitate, can also occur. Ground-glass opacity usually represents diffuse alveolar hemorrhage secondary to necrotizing capillaritis. Up to 10% of patients have centrilobular tree-in-bud nodules, which may be secondary to bronchiolar wall involvement or retained blood products in the distal airways. Bronchial wall thickening involving segmental and subsegmental bronchi is seen in 70% of patients with GPA, while more centrally located airway involvement is far less common (8). Most lung and airway abnormalities improve after treatment (8). Although the vasculitic component of GPA predominantly affects small vessels, large-vessel involvement is also possible, typically in conjunction with pulmonary and small-vessel findings. Pleural effusions can occur in GPA, but mediastinal lymphadenopathy is uncommon.
Figure 5a.

GPA in a 27-year-old woman. (a) Axial chest CT image shows a cavitary pulmonary nodule (arrow) in the lingula. (b) Axial maxillofacial CT image shows diffuse sinus thickening involving the bilateral maxillary sinuses (arrows) with septal perforation (★).
Figure 5b.

GPA in a 27-year-old woman. (a) Axial chest CT image shows a cavitary pulmonary nodule (arrow) in the lingula. (b) Axial maxillofacial CT image shows diffuse sinus thickening involving the bilateral maxillary sinuses (arrows) with septal perforation (★).
Necrobiotic Nodules
Necrobiotic lung nodules are sterile cavitary nodules that are commonly associated with rheumatoid arthritis, and rarely, with inflammatory bowel disease. There may be a single nodule or multiple nodules, and they may regress without treatment. Necrobiotic nodules are more common in male patients (18). The histologic appearance of a necrobiotic nodule is that of a granuloma, given the central area of necrosis surrounded by palisading macrophages and lymphocytes. The mechanism of formation is complex, but molecular profiling results have suggested that a destructive inflammatory process in the nodule is driven by lymphocytes, and this confirms it to be a granuloma. Further evidence comes from the fact that patients treated with methotrexate were found to have increased formation of rheumatoid lung nodules, because methotrexate activates adenosine A1 receptors, leading to enhanced cellular fusion and formation of multinucleated giant-cell granulomas (19,20).
Necrobiotic nodules predominately arise in subpleural locations and often cavitate (Fig 6). Typically, they are asymptomatic but can occasionally rupture, resulting in infection, pleural effusion, or bronchopleural fistulas (21). Rheumatoid nodules typically regress with treatment of rheumatoid arthritis, but methotrexate use can cause paradoxical enlargement (22). A rare association is Caplan syndrome, in which multiple necrobiotic nodules develop in patients with coal worker pneumoconiosis. Care must be taken to avoid confusing an atypical infection for a necrobiotic nodule, with the former being much more common in patients with rheumatoid arthritis, especially in the era of immunosuppressive therapy. As a general rule, tree-in-bud opacities or large cavitating masses should raise the concern for an atypical infection.
Figure 6a.

Necrobiotic nodules in a 55-year-old woman with rheumatoid arthritis and a history of smoking. (a, b) Coronal CT images show multiple cavitary nodules (red arrows) that ruptured into the right pleural space (yellow arrows in b) and then into the skin, forming a pleurocutaneous fistula (blue arrow in a). (c) Axial maximum intensity projection reconstruction CT image shows one of the peripheral cavitary necrobiotic nodules (green arrow).
Figure 6b.

Necrobiotic nodules in a 55-year-old woman with rheumatoid arthritis and a history of smoking. (a, b) Coronal CT images show multiple cavitary nodules (red arrows) that ruptured into the right pleural space (yellow arrows in b) and then into the skin, forming a pleurocutaneous fistula (blue arrow in a). (c) Axial maximum intensity projection reconstruction CT image shows one of the peripheral cavitary necrobiotic nodules (green arrow).
Figure 6c.
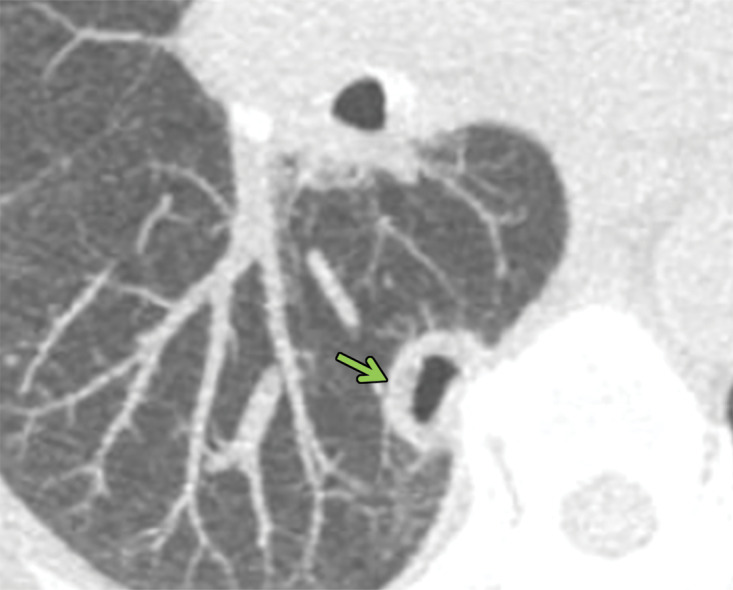
Necrobiotic nodules in a 55-year-old woman with rheumatoid arthritis and a history of smoking. (a, b) Coronal CT images show multiple cavitary nodules (red arrows) that ruptured into the right pleural space (yellow arrows in b) and then into the skin, forming a pleurocutaneous fistula (blue arrow in a). (c) Axial maximum intensity projection reconstruction CT image shows one of the peripheral cavitary necrobiotic nodules (green arrow).
Angiocentric Lymphoma or Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis, originally described by Liebow and colleagues in 1969 and 1972, is a rare form of non-Hodgkin lymphoma with an overlap of pulmonary lymphoma and GPA that is associated with Epstein-Barr virus (23,24). It primarily affects the lungs and consists of a polymorphous and atypical lymphoreticular infiltrate involving vessel walls with variable necrosis (23,24). The mortality rates of lymphomatoid granulomatosis vary between 53% and 63.5% of patients (23).
Imaging features of lymphomatoid granulomatosis overlap substantially with those of GPA (24,25). As with GPA, peribronchovascular nodules are the most common pattern at CT. These nodules may or may not cavitate and are usually hypermetabolic at fluorine 18 (18F) fluorodeoxyglucose PET (Fig 7) (23). Mediastinal lymphadenopathy and thin-walled cysts have also been described in some patients. In comparison to other lymphomas, lymphomatoid granulomatosis is considered extranodal, and lymphadenopathy is rare (26). Occasionally, lymphomatoid granulomatosis may involve the pulmonary artery wall and manifest as wall thickening or an intraluminal filling defect in the pulmonary artery. As with GPA, vascular findings are almost always seen in combination with parenchymal involvement.
Figure 7a.

Lymphomatoid granulomatosis in a 54-year-old man who presented with shortness of breath and skin nodules. (a) Frontal chest radiograph shows multiple pulmonary nodules of varying sizes (yellow boxes). (b, c) Axial CT image (b) and coronal maximum intensity projection CT image (c) show these nodules (yellow boxes in b and c) as solid and distributed throughout the lung. (d) Captured rotating maximum intensity projection 18F fluorodeoxyglucose PET/CT image shows diffuse hypermetabolic activity of these pulmonary nodules. Multiple hypermetabolic cutaneous nodules are also seen (purple boxes). Skin biopsy results confirmed the diagnosis of lymphomatoid granulomatosis.
Figure 7b.

Lymphomatoid granulomatosis in a 54-year-old man who presented with shortness of breath and skin nodules. (a) Frontal chest radiograph shows multiple pulmonary nodules of varying sizes (yellow boxes). (b, c) Axial CT image (b) and coronal maximum intensity projection CT image (c) show these nodules (yellow boxes in b and c) as solid and distributed throughout the lung. (d) Captured rotating maximum intensity projection 18F fluorodeoxyglucose PET/CT image shows diffuse hypermetabolic activity of these pulmonary nodules. Multiple hypermetabolic cutaneous nodules are also seen (purple boxes). Skin biopsy results confirmed the diagnosis of lymphomatoid granulomatosis.
Figure 7c.

Lymphomatoid granulomatosis in a 54-year-old man who presented with shortness of breath and skin nodules. (a) Frontal chest radiograph shows multiple pulmonary nodules of varying sizes (yellow boxes). (b, c) Axial CT image (b) and coronal maximum intensity projection CT image (c) show these nodules (yellow boxes in b and c) as solid and distributed throughout the lung. (d) Captured rotating maximum intensity projection 18F fluorodeoxyglucose PET/CT image shows diffuse hypermetabolic activity of these pulmonary nodules. Multiple hypermetabolic cutaneous nodules are also seen (purple boxes). Skin biopsy results confirmed the diagnosis of lymphomatoid granulomatosis.
Figure 7d.
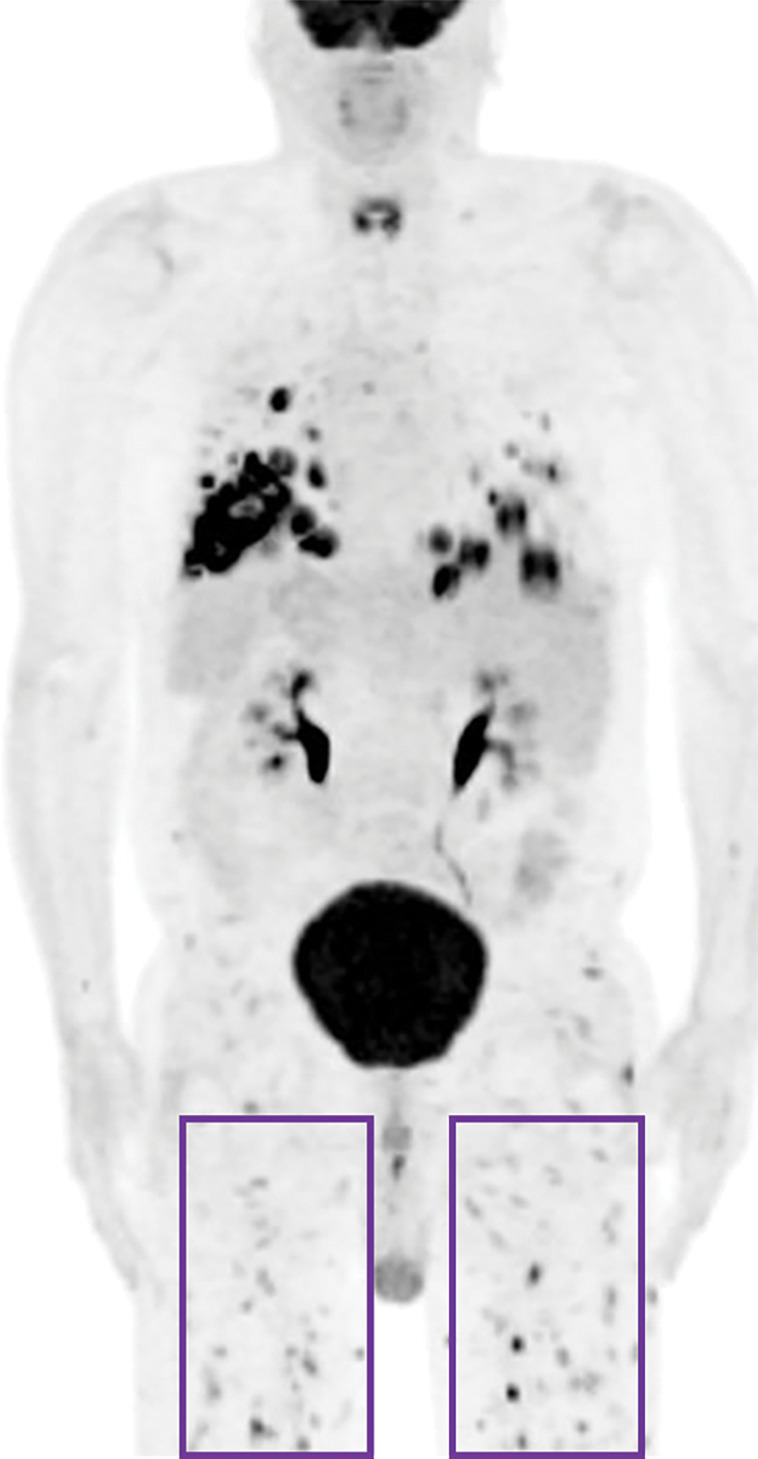
Lymphomatoid granulomatosis in a 54-year-old man who presented with shortness of breath and skin nodules. (a) Frontal chest radiograph shows multiple pulmonary nodules of varying sizes (yellow boxes). (b, c) Axial CT image (b) and coronal maximum intensity projection CT image (c) show these nodules (yellow boxes in b and c) as solid and distributed throughout the lung. (d) Captured rotating maximum intensity projection 18F fluorodeoxyglucose PET/CT image shows diffuse hypermetabolic activity of these pulmonary nodules. Multiple hypermetabolic cutaneous nodules are also seen (purple boxes). Skin biopsy results confirmed the diagnosis of lymphomatoid granulomatosis.
Necrotizing Sarcoid Granulomatosis
As described by Quaden et al (27), Liebow et al made reference to a sarcoid-like granulomatosis with vasculitis and necrosis and coined the term necrotizing sarcoid granulomatosis when describing the entities related to GPA. Many authors believe that this unusual diagnosis is a variant of sarcoidosis; however, its classification remains in doubt given overlapping features with hypersensitivity pneumonitis, EGPA, and GPA (28). In our experience, this is a diagnosis of exclusion that can only be made after multidisciplinary discussion. Extrapulmonary manifestations are common, and lack of upper airway and renal involvement can help differentiate necrotizing sarcoid from GPA. Antineutrophil cytoplasmic antibody levels tend to be normal, allowing differentiation from vasculitis (27).
CT studies show the typical sarcoid-like mediastinal and hilar lymphadenopathy. As with sarcoidosis, necrotizing sarcoid granulomatosis tends to manifest with nodules or masslike areas of consolidation (28,29) (Fig 8). Cavitation can sometimes help in differentiating necrotizing sarcoid granulomatosis from sarcoidosis (30). Pleural effusions may also be seen in some patients (28,29).
Figure 8.

Necrotizing sarcoidosis in a 56-year-old woman. Axial chest CT image shows multiple masslike solid pulmonary nodules with surrounding ground-glass halos (arrows). Percutaneous biopsy of one of the pulmonary nodules showed necrotizing sarcoidosis. These large nodules may cavitate, although cavitation is not present in this case.
Bronchocentric Granulomatosis
Bronchocentric granulomatosis is a rare condition in which airway granulomas form in response to a variety of insults. More than 50% of cases are associated with asthma and allergic bronchopulmonary aspergillosis. These granulomas have been postulated to be histopathologic manifestations of fungal hypersensitivity (31–33). Bronchocentric granulomatosis is characterized by peribronchiolar or peribronchial necrotizing granulomas that do not invade the pulmonary arteries. This is in contrast to GPA, necrotizing sarcoidosis, and lymphomatoid granulomatosis, all of which have an angiocentric component (32). The imaging manifestations of bronchocentric granulomatosis are nonspecific and include nodular or masslike lesions and pneumonic consolidation (33). The findings are usually unilateral, with preferential involvement of the upper lobes. In cases associated with allergic bronchopulmonary aspergillosis, mucoid impaction and atelectasis or consolidation can be seen (33). Diagnosis is almost never based on imaging alone and requires biopsy (Fig 9).
Figure 9a.

Bronchocentric granulomatosis in three patients. (a) Axial CT image in a 35-year-old man shows a spiculated nodule (arrow) in the left lower lobe. (b) Axial CT image in a 44-year-old woman shows peribronchovascular consolidation (white box) with tiny satellite nodules (arrow). (c) Axial CT image in a 51-year-old woman shows right lower lobe consolidation (dashed box) with peripheral bronchiectasis (purple arrow) and cystic bronchiectasis (blue arrow) with mucus plugging in the left lower lobe.
Figure 9b.
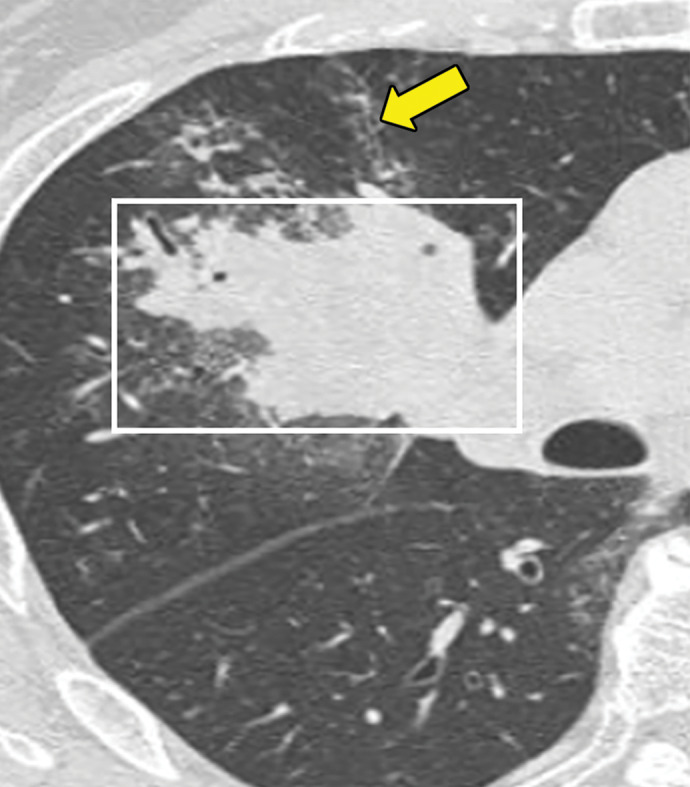
Bronchocentric granulomatosis in three patients. (a) Axial CT image in a 35-year-old man shows a spiculated nodule (arrow) in the left lower lobe. (b) Axial CT image in a 44-year-old woman shows peribronchovascular consolidation (white box) with tiny satellite nodules (arrow). (c) Axial CT image in a 51-year-old woman shows right lower lobe consolidation (dashed box) with peripheral bronchiectasis (purple arrow) and cystic bronchiectasis (blue arrow) with mucus plugging in the left lower lobe.
Figure 9c.

Bronchocentric granulomatosis in three patients. (a) Axial CT image in a 35-year-old man shows a spiculated nodule (arrow) in the left lower lobe. (b) Axial CT image in a 44-year-old woman shows peribronchovascular consolidation (white box) with tiny satellite nodules (arrow). (c) Axial CT image in a 51-year-old woman shows right lower lobe consolidation (dashed box) with peripheral bronchiectasis (purple arrow) and cystic bronchiectasis (blue arrow) with mucus plugging in the left lower lobe.
Macronodules, Masses, or Consolidation in Random Distribution: Hyalinizing Granuloma
Hyalinizing granuloma is an idiopathic condition that was originally described in 1977. It tends to manifest with pulmonary nodules and masses that show concentric hyaline lamellae, usually with perivascular collections of plasma cells and lymphocytes, at histologic evaluation. These masses and nodules are often multiple and bilateral (34). Several mechanisms have been proposed, including atypical immune response to mycobacterial or fungal antigens (akin to fibrosing mediastinitis) or burned-out myofibroblastic tumors. It is commonly associated with fibrosing mediastinitis and retroperitoneal fibrosis, with an underlying autoimmune mechanism (35). Patients are often asymptomatic and can present with vague chest-related symptoms such as shortness of breath, cough, fatigue, low-grade fever, pleuritic chest pain, and in rare cases, hemoptysis (36).
On radiographs, bilateral large nodules or masses are found. Pleural effusions and lymphadenopathy are rare. On CT images, these nodules or masses tend to be randomly distributed and often contain calcium (36,37) (Fig 10). The differential diagnosis for these nodules includes evaluation for amyloid deposition, hamartomas, and metastases.
Figure 10.

Pulmonary hyalinizing granulomas in a 56-year-old man. Axial CT image shows multiple large well-circumscribed nodules and masses of different sizes with coarse irregular calcifications.
Micronodules in a Perilymphatic Pattern: Sarcoidosis
Sarcoidosis is a well-known multisystem disease that primarily affects the thorax. Although the etiology remains unknown, noncaseating granulomas are classically seen at histopathologic evaluation. With an incidence of 1–40 cases per 100 000 people per year, sarcoidosis has a significant predilection for black people and for women of all races (38). Sarcoidosis is a cell-mediated immune response to unidentified antigens in genetically susceptible patients. A recent meta-analysis (39) that included 58 studies involving more than 6000 patients with sarcoidosis concluded that Propionibacteria and mycobacteria may be associated with the development of sarcoidosis. The multistep process of sarcoid granuloma formation is depicted in Figure 2. Clinically, fatigue is the most common concern, but patients with thoracic sarcoidosis may present with additional nonspecific symptoms, including cough, dyspnea, and chest pain in 9%–19% of cases (40).
Chest radiographs are abnormal in more than 90% of patients with thoracic sarcoidosis (40). Mediastinal lymphadenopathy is commonly seen in the right paratracheal and aortopulmonary window regions. Symmetric hilar lymphadenopathy may also be seen (41) (Fig 11). Unlike metastasis or lymphoma, sarcoidosis tends to lack mass effect on the adjacent vasculature (41). While chest radiography frequently shows the lymphadenopathy, it is less sensitive in characterizing the pulmonary parenchymal abnormalities. Sarcoidosis can manifest with variable pulmonary parenchymal findings at CT, including perilymphatic micronodules, random micronodules, or larger nodules or masses with surrounding satellite nodules (“galaxy” sign). Peribronchovascular, often central, consolidation may also be seen. In more advanced cases, fibrosis may be present and tends to be more central in an axial distribution and more predominant in the upper lobe, allowing differentiation from usual interstitial pneumonia. Air trapping may also be seen with sarcoidosis and should not be confused with hypersensitivity pneumonitis (41).
Figure 11a.

Sarcoidosis in two patients. (a, b) Axial CT image (lung window) in a 49-year-old woman (a) shows many bilateral perilymphatic pulmonary nodules (red arrows), although no mediastinal lymphadenopathy was noted. (b) Coronal 18F fluorodeoxyglucose PET CT image in the same patient shows numerous hypermetabolic mediastinal and hilar lymph nodes (green arrows) that are in keeping with sarcoidosis. (c) Axial CT image (lung window) in a 51-year-old man shows multiple upper lobe–predominant central perilymphatic nodules (red boxes) and a few peripheral subpleural nodules (arrows).
Figure 11b.

Sarcoidosis in two patients. (a, b) Axial CT image (lung window) in a 49-year-old woman (a) shows many bilateral perilymphatic pulmonary nodules (red arrows), although no mediastinal lymphadenopathy was noted. (b) Coronal 18F fluorodeoxyglucose PET CT image in the same patient shows numerous hypermetabolic mediastinal and hilar lymph nodes (green arrows) that are in keeping with sarcoidosis. (c) Axial CT image (lung window) in a 51-year-old man shows multiple upper lobe–predominant central perilymphatic nodules (red boxes) and a few peripheral subpleural nodules (arrows).
Figure 11c.

Sarcoidosis in two patients. (a, b) Axial CT image (lung window) in a 49-year-old woman (a) shows many bilateral perilymphatic pulmonary nodules (red arrows), although no mediastinal lymphadenopathy was noted. (b) Coronal 18F fluorodeoxyglucose PET CT image in the same patient shows numerous hypermetabolic mediastinal and hilar lymph nodes (green arrows) that are in keeping with sarcoidosis. (c) Axial CT image (lung window) in a 51-year-old man shows multiple upper lobe–predominant central perilymphatic nodules (red boxes) and a few peripheral subpleural nodules (arrows).
Micronodules in a Centrilobular Pattern
Talcosis
Talc (magnesium silicate) is commonly used in industrial manufacturing and is also added as a filler in some oral medications. Talc deposition in lung tissue can result from either inhalational exposure to talc (ie, pneumoconiosis) or after intravenous administration. Intravenous administration is used most commonly in an illicit setting in which users may crush or melt talc-containing oral medications, dissolve them in water, and inject the solution intravenously. This results in innumerable tiny talc particles lodging in the pulmonary capillary bed in the secondary pulmonary nodule, from where they can migrate into the pulmonary interstitium and form talc granulomas (42). This anatomic deposition manifests with the typical centrilobular pattern. These patients experience progressive dyspnea and pulmonary function decline. Pulmonary hypertension can also develop from talc particles obstructing the pulmonary capillary bed.
Chest radiography is insensitive but may show a diffuse micronodular pattern. In talcosis due to injected material, CT typically shows innumerable submillimeter micronodules in a centrilobular pattern. Occasionally, the nodules may show high attenuation (42,43) (Fig 12). Conglomerate masses are uncommon and typically related to inhalation of talc particles. In cases of inhalational exposure, the involved areas are more likely to contain areas of high attenuation resembling calcium (43,44).
Figure 12a.

Talcosis in two patients. (a) Axial CT image in a 27-year-old man with inhalational talcosis shows many diffuse centrilobular ground-glass nodules with nodular opacity and internal high attenuation. (b) Axial CT image in a 31-year-old man with intravenous talcosis shows innumerable tree-in-bud nodules with an enlarged main pulmonary artery from resultant pulmonary hypertension. This tree-in-bud distribution is in part due to intravenous particles obstructing terminal pulmonary arteries.
Figure 12b.

Talcosis in two patients. (a) Axial CT image in a 27-year-old man with inhalational talcosis shows many diffuse centrilobular ground-glass nodules with nodular opacity and internal high attenuation. (b) Axial CT image in a 31-year-old man with intravenous talcosis shows innumerable tree-in-bud nodules with an enlarged main pulmonary artery from resultant pulmonary hypertension. This tree-in-bud distribution is in part due to intravenous particles obstructing terminal pulmonary arteries.
Pulmonary Langerhans Cell Histiocytosis
Pulmonary Langerhans cell histiocytosis (PLCH) (which should be distinguished from systemic Langerhans cell histiocytosis [LCH]) usually occurs in young adult smokers. PLCH does not have a predilection for individuals of either sex but primarily affects white patients. Patients with PLCH usually present with dyspnea or a nonproductive cough. In the cystic stage of this disease, patients may present with pneumothorax (45). Histologically, PLCH begins with the accumulation of Langerhans cells in the bronchial epithelium and the formation of granulomas. As these granulomas evolve, the fibrosis resulting from the process has been postulated to cause traction on the central bronchioles (46). Studies (47,48) suggest that PLCH may be a myeloid neoplasm with inflammatory properties, as cells express surface CD1a proteins and up to 50% of the cells harbor activating BRAF and MAPK mutations (47). PLCH is associated with acute myeloid leukemia (48).
Chest radiography shows bilateral and mostly symmetric upper lobe–predominant disease with sparing of the costophrenic angles. Findings may include small nodules and/or findings of the cystic component of the disease, which often appear on chest radiographs as multiple lines where the cyst walls are tangential to the imaging projection. Coexistent emphysema may also be seen (45,46). High-resolution CT is superior to conventional radiography in delineating the findings of PLCH, which include nodules that may or not be cavitary and mid- to upper-lung–predominant thin-walled cysts that often coalesce and form unusual shapes. Distribution is the key in differentiating PLCH from other cystic lung diseases, because PLCH preferentially spares the subpleural region of the costophrenic recess, the anterior right middle lobe, and the lingula (47,48) (Fig 13).
Figure 13a.

PLCH in a 30-year-old male smoker. Axial CT images show bizarrely shaped cysts (yellow box in a) predominantly in the upper lobe and multiple centrilobular nodules (red box in a). The lung bases are relatively spared (b).
Figure 13b.

PLCH in a 30-year-old male smoker. Axial CT images show bizarrely shaped cysts (yellow box in a) predominantly in the upper lobe and multiple centrilobular nodules (red box in a). The lung bases are relatively spared (b).
Granulomatous and Lymphocytic Interstitial Lung Disease
Granulomatous and lymphocytic interstitial lung disease (GL-ILD) is a relatively recently described granulomatous lung disease that is almost always associated with common variable immunodeficiency. The British Lung Foundation and the United Kingdom Primary Immunodeficiency Network consensus statement defined GL-ILD as “a distinct clinico-radio-pathological ILD occurring in patients with common variable immunodeficiency disorders, associated with a lymphocytic infiltrate and/or granuloma in the lung, and in whom other conditions have been considered and where possible excluded” (49). Bates et al (50) described GL-ILD in patients with common variable immunodeficiency who had overlap between lymphoproliferative and granulomatous histologic results of lung biopsies. The pathogenesis remains unknown. In one cohort, the overall prevalence of interstitial lung disease among patients with combined variable immunodeficiency was 26%, with more than two-thirds of these patients showing GL-ILD in surgical lung biopsies. This study showed the presence of dyspnea, splenomegaly, and deficiencies in circulating CD3+ and CD8+ cells. In a large European registry, lymphoma and GL-ILD were the most common predictors of mortality among patients with common variable immunodeficiency (51).
The most common imaging finding of GL-ILD is diffuse lower lobe–predominant micronodules (52). These are often only seen with CT. Most cases also demonstrate mid- to lower-lung–predominant interlobular septal line thickening. Other imaging manifestations include mild bronchiectasis, multifocal pulmonary consolidation, and lymphadenopathy (Fig 14). Splenomegaly is almost always seen in GL-ILD.
Figure 14a.

GL-ILD in a 50-year-old woman with common variable immunodeficiency. (a) Axial CT image at initial presentation shows diffuse mosaic attenuation. (b) Axial CT image at follow-up a few months later shows improving mosaic attenuation with new micronodules (yellow oval). Follow-up CT after treatment with systemic steroids (not shown) showed improvement of both the diffuse mosaic attenuation and micronodules.
Figure 14b.
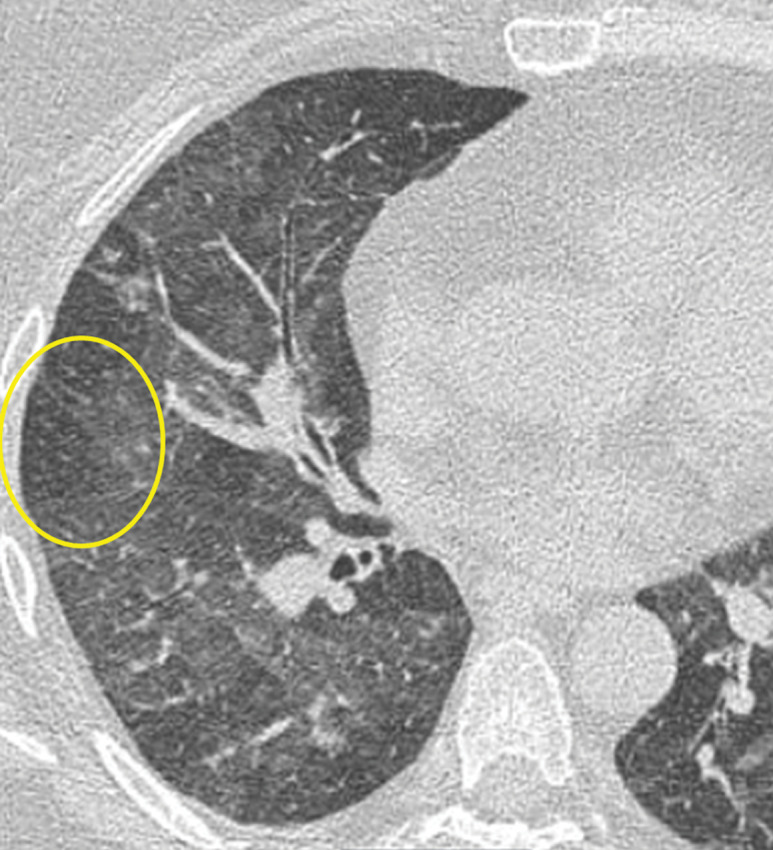
GL-ILD in a 50-year-old woman with common variable immunodeficiency. (a) Axial CT image at initial presentation shows diffuse mosaic attenuation. (b) Axial CT image at follow-up a few months later shows improving mosaic attenuation with new micronodules (yellow oval). Follow-up CT after treatment with systemic steroids (not shown) showed improvement of both the diffuse mosaic attenuation and micronodules.
Aspiration
Aspiration, the spillage of ingested solid or liquid material into the airways and lungs, is frequently encountered at CT. Isolated occurrences of aspiration are common and frequently without any adverse clinical outcomes. Aspiration is more frequent and consequential in patients with neurologic and swallowing disorders, oropharyngeal cancers, those under sedation, and those who have experienced trauma (53). The particulate matter in aspiration functions as a foreign antigen to trigger granuloma formation and is helpful in the definitive diagnosis of aspiration at histopathologic evaluation (54). Exogenous lipoid pneumonia was the most common pattern in patients with chronic aspiration at histopathologic analysis (55).
At CT, aspiration can have a variable appearance based on duration, the material that was aspirated, the amount of material aspirated, and whether the aspiration has caused an underlying pneumonia, which may or may not be present (56). Simple aspiration is typically associated with centrilobular, often tree-in-bud, nodules due to impaction of material in the distal airways. More central airways may show evidence of focal plugging or partial occlusion with mucus, fluid, or debris. The surrounding parenchyma may also demonstrate findings of inflammation, which may manifest as surrounding ground glass. When aspiration is occlusive, atelectasis, evidenced by enhancing parenchymal opacification with volume loss, may be present. In patients with associated aspiration pneumonia, nonenhancing airspace consolidation can be seen. Findings may also be present in the airways themselves, with bronchial wall thickening and bronchiectasis often seen, particularly in patients with chronic or repeated bouts of aspiration. Finally, the location of suggestive findings is helpful in patients with suspected aspiration (56,57). Aspiration is commonly seen in dependent areas of the lung. Thus, the inferior and posterior regions of the lungs are more likely to be involved. Because of the more vertical anatomy of the right main stem bronchus and bronchus intermedius, aspiration also often favors the right side (57). In the majority of the cases of chronic occult aspiration, imaging demonstrates the predominance of bronchial wall thickening, centrilobular nodules, and lower lung–predominant tree-in-bud opacities. The other pattern can be chronic unclassifiable interstitial pneumonia, in which imaging may show ground-glass opacities, reticulations, traction bronchiectasis, and honeycombing in nonclassical distribution (55) (Fig 15).
Figure 15.
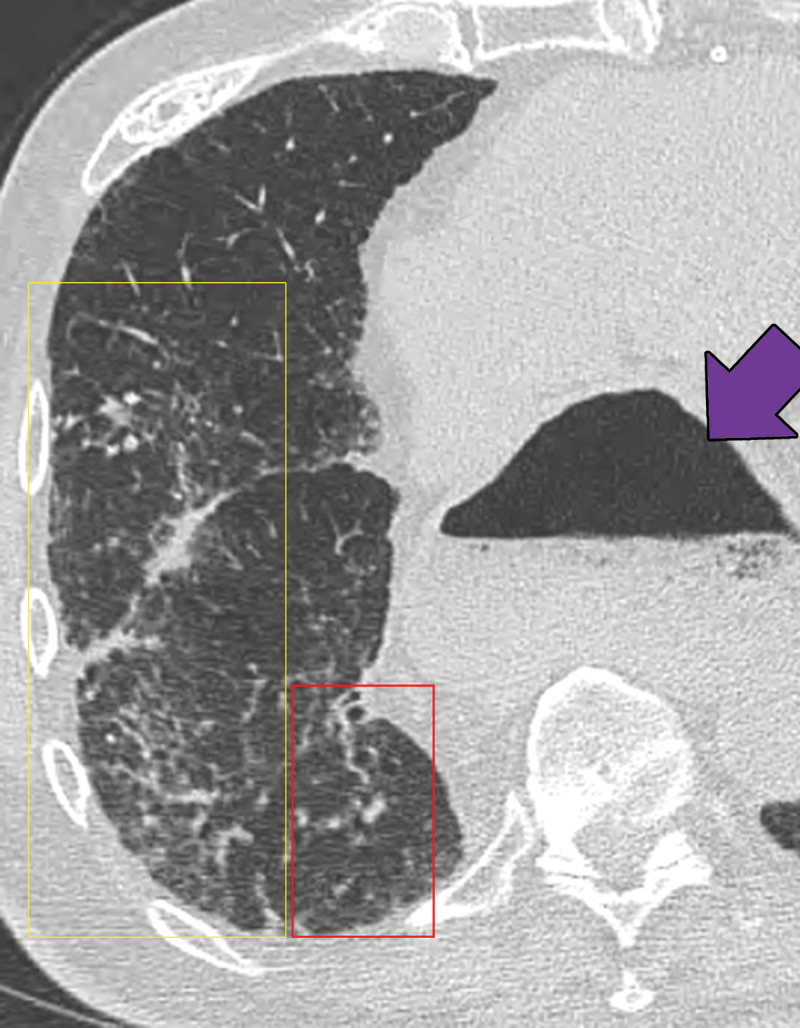
Sequelae of chronic aspiration in a 75-year-old man who underwent esophagectomy and gastric pull-through 9 years previously for treatment of esophageal cancer. Axial CT image shows lower lung–predominant ground-glass opacity and reticulation in the right middle and lower lobes (yellow box) and mild bronchiectasis (red box). A dilated gastric conduit is also present (arrow). These findings were stable from a prior examination and were most consistent with sequelae of chronic aspiration.
Other Nonnecrotizing Granulomatous Diseases
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis, formerly referred to as extrinsic allergic alveolitis, is an inflammatory response of the lungs to an inhaled antigen or chemical that is characterized in part by noncaseating granulomas at pathologic examination. More common subtypes of hypersensitivity pneumonitis include hot tub lung, bird fancier’s lung, and farmer’s lung. Diagnosis can be challenging, because the offending antigen is found in less than 50% of cases. Acute, subacute, and chronic subtypes of hypersensitivity pneumonitis have been described. The definition varies according to different authors (57–59), and the clinical onset and radiologic features also vary. The lack of a clinical consensus for separating these forms of hypersensitivity pneumonitis has resulted in some authors suggesting the use of alternative terms such as active versus fibrosing hypersensitivity pneumonitis on the basis of imaging findings.
The CT imaging manifestations of hypersensitivity pneumonitis vary from ground-glass opacities to frank fibrosis. In patients in whom hypersensitivity pneumonitis has not progressed to fibrosis, the predominant features include centrilobular ground-glass nodules and/or lobular air trapping; the latter is best seen with expiratory examination. The combination of ground-glass opacities and areas of air trapping interspersed with normal lung tissue at CT has been described as the “head cheese” sign (Fig 16). When hypersensitivity pneumonitis progresses to fibrosis, its imaging pattern can be extremely variable, mimicking general features of both usual interstitial pneumonia and nonspecific interstitial pneumonia. Fibrotic changes in unusual, particularly upper lobe, distributions should raise suspicion for chronic hypersensitivity pneumonitis. Air trapping seen in three or more lobes is also suggestive of hypersensitivity pneumonitis (57).
Figure 16a.
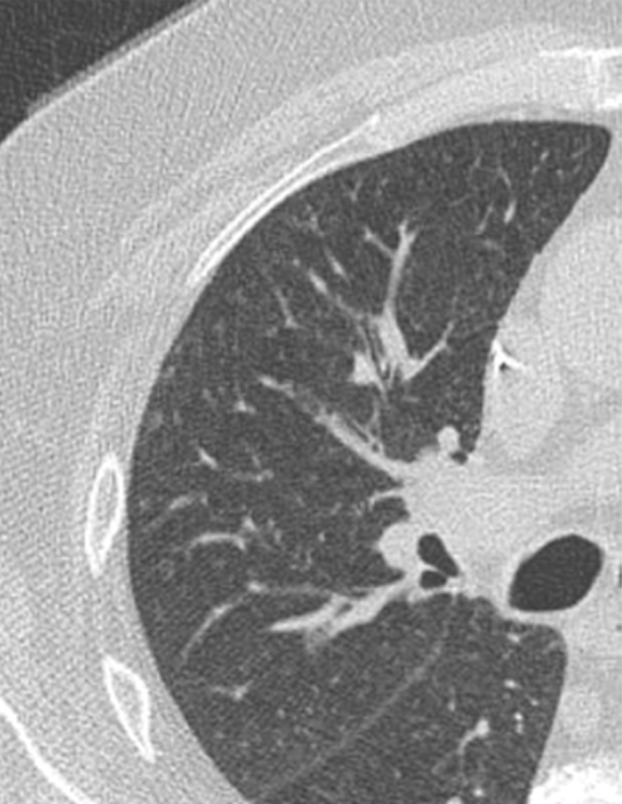
Variable CT imaging appearance of hypersensitivity pneumonitis in three patients. (a) Axial CT image in a 35-year-old man shows multiple centrilobular nodules. (b) Axial CT image in a 56-year-old woman shows mixed ground-glass opacity and mosaic attenuation interspersed with normal lung parenchyma (head cheese sign). (c, d) Axial CT images in a 46-year-old woman with a long history of using a hot tub shows areas of ground-glass attenuation predominantly in the left upper lobe (white oval in c). Evidence of air trapping appears on the expiratory image (d).
Figure 16b.

Variable CT imaging appearance of hypersensitivity pneumonitis in three patients. (a) Axial CT image in a 35-year-old man shows multiple centrilobular nodules. (b) Axial CT image in a 56-year-old woman shows mixed ground-glass opacity and mosaic attenuation interspersed with normal lung parenchyma (head cheese sign). (c, d) Axial CT images in a 46-year-old woman with a long history of using a hot tub shows areas of ground-glass attenuation predominantly in the left upper lobe (white oval in c). Evidence of air trapping appears on the expiratory image (d).
Figure 16c.

Variable CT imaging appearance of hypersensitivity pneumonitis in three patients. (a) Axial CT image in a 35-year-old man shows multiple centrilobular nodules. (b) Axial CT image in a 56-year-old woman shows mixed ground-glass opacity and mosaic attenuation interspersed with normal lung parenchyma (head cheese sign). (c, d) Axial CT images in a 46-year-old woman with a long history of using a hot tub shows areas of ground-glass attenuation predominantly in the left upper lobe (white oval in c). Evidence of air trapping appears on the expiratory image (d).
Figure 16d.

Variable CT imaging appearance of hypersensitivity pneumonitis in three patients. (a) Axial CT image in a 35-year-old man shows multiple centrilobular nodules. (b) Axial CT image in a 56-year-old woman shows mixed ground-glass opacity and mosaic attenuation interspersed with normal lung parenchyma (head cheese sign). (c, d) Axial CT images in a 46-year-old woman with a long history of using a hot tub shows areas of ground-glass attenuation predominantly in the left upper lobe (white oval in c). Evidence of air trapping appears on the expiratory image (d).
Progressive Massive Fibrosis
Progressive massive fibrosis can be an end-stage manifestation of silicosis or coal worker’s pneumoconiosis. In fact, the presence of progressive massive fibrosis defines complicated silicosis. Progressive massive fibrosis results from the confluence of nodules leading to large conglomerate masses, typically in an upper lobe distribution. Histologically, these conglomerate masses are composed of multiple foci of central hyalinized collagen surrounded by a rim of macrophages, which are suggestive of granulomatous inflammation (60). These masses eventually become fibrotic and retract the pleura, leading to parenchymal scarring and bullous disease. Macrophages in granulomas mediate fibrosis by releasing cytokines such as fibronectin and alveolar macrophage–derived growth factors (60).
At imaging, progressive massive fibrosis manifests with bilateral upper lobe opacities with irregular margins. The masses of progressive massive fibrosis measure greater than 1 cm (60–63). The lateral margins of these masses often parallel the chest wall. CT better shows these conglomerate masses or masslike consolidations, usually with adjacent perilymphatic micronodules, and mediastinal lymph nodes, all of which can develop coarse internal calcifications (Fig 17). Occasionally, the areas of progressive massive fibrosis can undergo necrosis and cavitation (61); in this situation, evaluation for a superimposed tuberculosis infection, which can complicate up to 25% of silicosis cases, should be performed (63).
Figure 17a.

Progressive massive fibrosis in a 61-year-old man. Frontal chest radiograph (a) shows large bilateral upper lobe–predominant masses with irregular margins (arrows) and upper lobe volume loss, indicated by a juxtaphrenic peak (arrowhead). (b) Axial CT image shows bilateral large calcified conglomerate masses with adjacent fibrosis and calcified mediastinal lymph nodes.
Figure 17b.

Progressive massive fibrosis in a 61-year-old man. Frontal chest radiograph (a) shows large bilateral upper lobe–predominant masses with irregular margins (arrows) and upper lobe volume loss, indicated by a juxtaphrenic peak (arrowhead). (b) Axial CT image shows bilateral large calcified conglomerate masses with adjacent fibrosis and calcified mediastinal lymph nodes.
Xanthogranulomatous Diseases of the Chest
Xanthogranulomatous diseases are rare aggressive inflammatory conditions that can be caused by inflammatory, infectious, histiocytic, or inherited lysosomal disorders. The presence of lipid-laden macrophages and histiocytes that accompany these granulomas makes them histopathologically related to other granulomatous disorders (64). Xanthogranulomatous cholecystitis and xanthogranulomatous pyelonephritis are well-recognized conditions in the abdomen, but isolated xanthogranulomatous pulmonary disorders are rare. Pulmonary involvement is usually secondary to systemic disorders such as Rosai-Dorfman disease and Erdheim-Chester disease.
Erdheim-Chester Disease
Erdheim-Chester disease is a rare multisystem non-Langerhans cell histiocytic disorder primarily affecting middle-aged to older men (65). Histologic evaluation in Erdheim-Chester disease shows fibrosis with foamy histiocytic infiltration. These histiocytic cells stain positive for CD68 and are negative for S-100, CD1a, and Birebeck granules, which differentiates them from Langerhans type histiocytosis (66). The most common clinical symptom at presentation is bone pain, with imaging showing multifocal metaphyseal-diaphyseal medullary osteosclerotic lesions of the long bones, with sparing of the epiphysis (67,68). Extraskeletal disease develops in about 50% of patients and may involve the retroperitoneum, kidneys, lungs, central nervous system, orbits, skin, heart, breasts, sinonasal mucosa, and skeletal muscle. Thoracic involvement is seen in only 20%–30% of cases (67–69). The foamy histiocytes can involve the lung, pleura, pericardium, myocardium, and periaortic soft tissues. Pulmonary involvement most commonly manifests as a cough with progressive dyspnea. Neurologic and cardiac involvement portends a worse prognosis (67–69).
In the lungs, Erdheim-Chester disease usually manifests with smooth septal line thickening, fissural thickening, centrilobular nodules, or ground-glass opacities (67). Periaortic soft-tissue thickening that may extend around the atria and epicardial fat may also be present. These findings, particularly when seen with the osseous manifestations, should trigger concern for Erdheim-Chester disease (Fig 18).
Figure 18a.

Erdheim-Chester disease in a 55-year-old man. (a) Axial CT image shows interlobular and intralobular smooth septal line thickening (arrows) with bilateral pleural effusions (★). (b) Sagittal reconstruction CT image (soft-tissue window) shows a rind of soft tissue encasing the heart (purple arrow) and aorta (yellow arrow). (c) Axial CT image (bone window) also shows sclerotic lesions, which are hallmark findings of this disease (blue arrows).
Figure 18b.
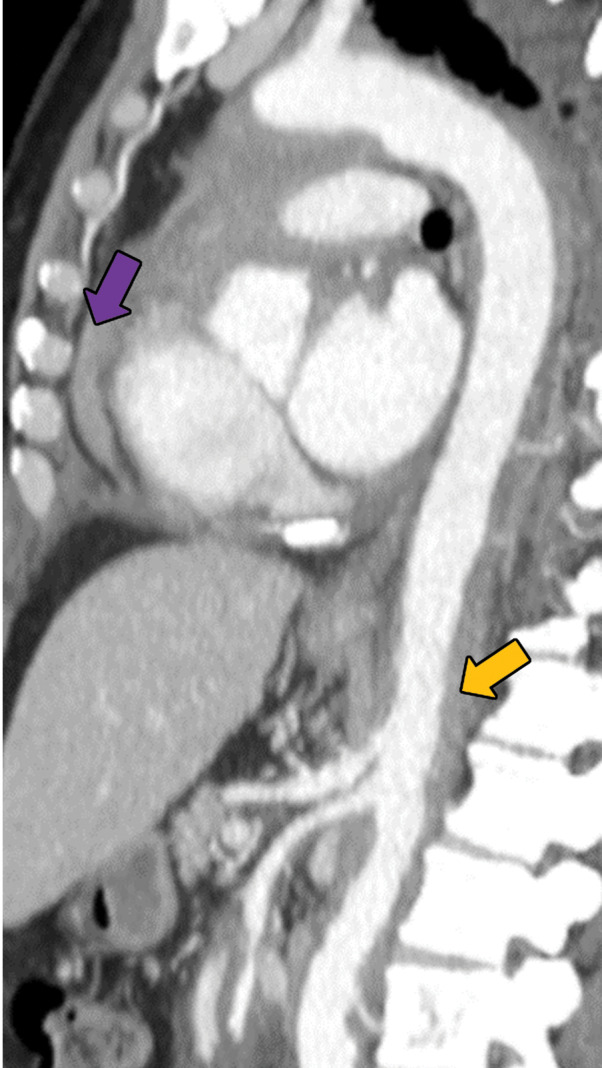
Erdheim-Chester disease in a 55-year-old man. (a) Axial CT image shows interlobular and intralobular smooth septal line thickening (arrows) with bilateral pleural effusions (★). (b) Sagittal reconstruction CT image (soft-tissue window) shows a rind of soft tissue encasing the heart (purple arrow) and aorta (yellow arrow). (c) Axial CT image (bone window) also shows sclerotic lesions, which are hallmark findings of this disease (blue arrows).
Figure 18c.

Erdheim-Chester disease in a 55-year-old man. (a) Axial CT image shows interlobular and intralobular smooth septal line thickening (arrows) with bilateral pleural effusions (★). (b) Sagittal reconstruction CT image (soft-tissue window) shows a rind of soft tissue encasing the heart (purple arrow) and aorta (yellow arrow). (c) Axial CT image (bone window) also shows sclerotic lesions, which are hallmark findings of this disease (blue arrows).
Extranodal Rosai-Dorfman Disease
Rosai-Dorfman disease, also known as sinus histiocytosis with massive lymphadenopathy, is a rare xanthogranulomatous disorder associated with cutaneous and lymph node involvement (70). The most common intrathoracic manifestation of Rosai-Dorfman disease is mediastinal lymphadenopathy, while the most common extrathoracic manifestation is cervical lymphadenopathy. In one cohort, patients who presented with pulmonary manifestations (44%) had shortness of breath (70). In one registry of 423 patients, lung involvement, which has a worse prognosis, was seen in less than 3% of patients (71).
Thoracic Rosai-Dorfman disease most often involves the trachea, bronchi, and associated lymph nodes. CT findings may include solitary or multiple polypoid masses in the airways (70). Imaging may show additional mediastinal lymphadenopathy or pleural effusions (72). When the lung parenchyma is involved, a nonspecific interstitial pneumonia pattern is most common. Diffuse interstitial or airspace involvement occurs less frequently. When there is extensive parenchymal involvement, the pleura may also be involved (71,72). Rosai-Dorfman disease can, in rare cases, manifest with cystic lung disease (73). These imaging findings are presented in Figure 19.
Figure 19a.
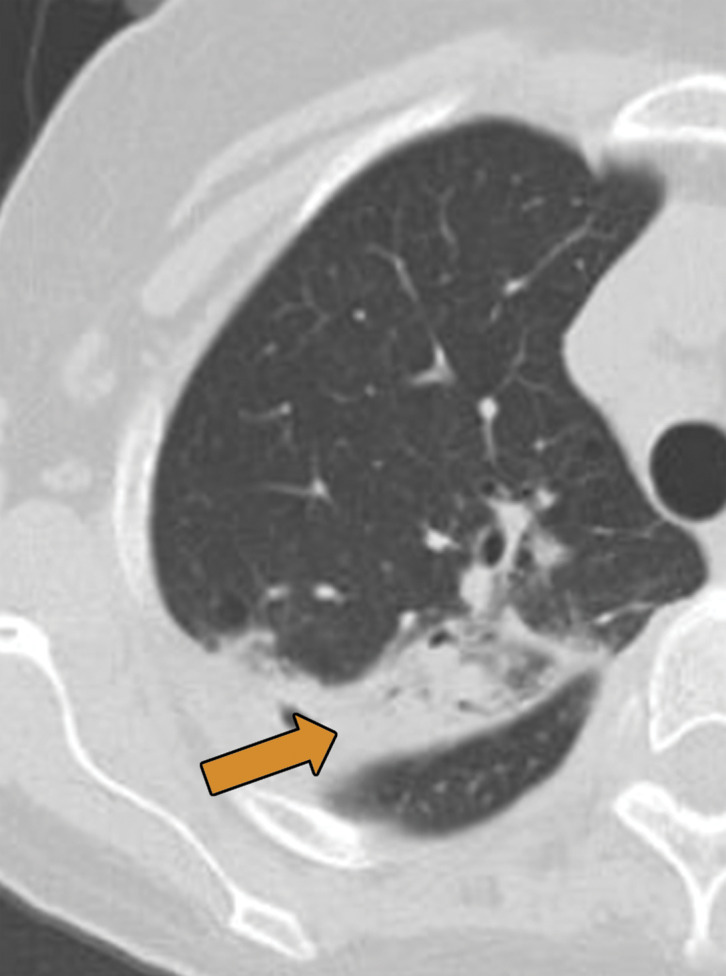
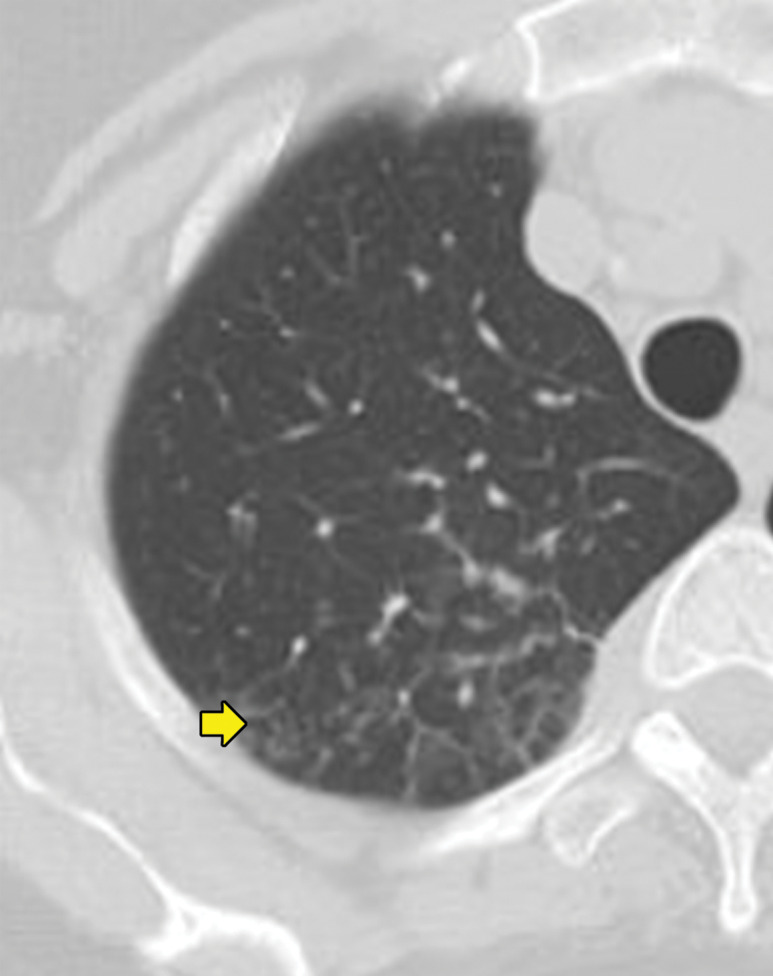
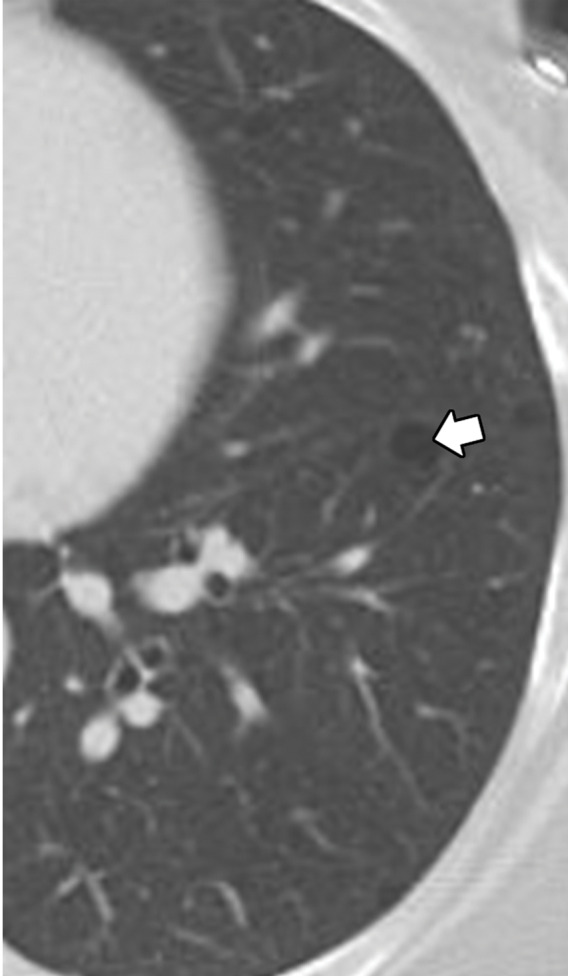
Biopsy-proven extranodal Rosai-Dorfman disease in a 65-year-old woman. (a–c) Axial CT images (lung window) show a patchy consolidation in the right upper lobe (arrow in a) with septal line thickening (arrow in b). Also note the tiny thin-walled cysts in the left lung (arrow in c). (d) Axial CT image (mediastinal windows) shows the axillary and mediastinal adenopathy (green arrows) and a soft-tissue chest wall mass (pink arrow). The most common finding in Rosai-Dorfman disease is lymphadenopathy. Most of these findings resolved with treatment (not shown).
Figure 19b.
Biopsy-proven extranodal Rosai-Dorfman disease in a 65-year-old woman. (a–c) Axial CT images (lung window) show a patchy consolidation in the right upper lobe (arrow in a) with septal line thickening (arrow in b). Also note the tiny thin-walled cysts in the left lung (arrow in c). (d) Axial CT image (mediastinal windows) shows the axillary and mediastinal adenopathy (green arrows) and a soft-tissue chest wall mass (pink arrow). The most common finding in Rosai-Dorfman disease is lymphadenopathy. Most of these findings resolved with treatment (not shown).
Figure 19c.
Biopsy-proven extranodal Rosai-Dorfman disease in a 65-year-old woman. (a–c) Axial CT images (lung window) show a patchy consolidation in the right upper lobe (arrow in a) with septal line thickening (arrow in b). Also note the tiny thin-walled cysts in the left lung (arrow in c). (d) Axial CT image (mediastinal windows) shows the axillary and mediastinal adenopathy (green arrows) and a soft-tissue chest wall mass (pink arrow). The most common finding in Rosai-Dorfman disease is lymphadenopathy. Most of these findings resolved with treatment (not shown).
Figure 19d.

Biopsy-proven extranodal Rosai-Dorfman disease in a 65-year-old woman. (a–c) Axial CT images (lung window) show a patchy consolidation in the right upper lobe (arrow in a) with septal line thickening (arrow in b). Also note the tiny thin-walled cysts in the left lung (arrow in c). (d) Axial CT image (mediastinal windows) shows the axillary and mediastinal adenopathy (green arrows) and a soft-tissue chest wall mass (pink arrow). The most common finding in Rosai-Dorfman disease is lymphadenopathy. Most of these findings resolved with treatment (not shown).
Conclusion
Noninfectious thoracic granulomatous diseases are varied conditions that show granulomas at imaging but have a wide variety of clinical manifestations that can mimic other conditions. An organized approach to these conditions can be helpful in including or excluding them from the imaging differential diagnosis. Granulomatous diseases pose a unique challenge, given their myriad clinical manifestations, but narrowing the differential diagnosis on the basis of common imaging findings can help clinicians and pathologists come to a more conclusive diagnosis.
D.H.B. supported by the National Institutes of Health TOP-TIER grant T32-EB021955.
Recipient of a Certificate of Merit award for an education exhibit at the 2018 RSNA Annual Meeting.
For this journal-based SA-CME activity, the authors, editor, and reviewers have disclosed no relevant relationships.
Abbreviations:
- EGPA
- eosinophilic granulomatosis with polyangiitis
- GL-ILD
- granulomatous and lymphocytic interstitial lung disease
- GPA
- granulomatosis with polyangitis
- PLCH
- pulmonary Langerhans cell histiocytosis
- TNF
- tissue necrosis factor
References
- 1.Ohshimo S, Guzman J, Costabel U, Bonella F. Differential diagnosis of granulomatous lung disease: clues and pitfalls: Number 4 in the series “Pathology for the clinician”edited by Peter Dorfmüller and Alberto Cavazza. Eur Respir Rev 2017;26(145):170012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rivera A, Siracusa MC, Yap GS, Gause WC. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol 2016;17(4):356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017;9(6):7204–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guirado E, Schlesinger LS, Kaplan G. Macrophages in tuberculosis: friend or foe. Semin Immunopathol 2013;35(5):563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekkens MJ, Shedlock DJ, Jung E, et al. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun 2007;75(5): 2291–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarraf P, Kay J, Friday RP, Reginato AM. Wegener’s granulomatosis: is biologic therapy useful? Curr Rheumatol Rep 2006;8(4):303–311. [DOI] [PubMed] [Google Scholar]
- 7.Sada KE, Amano K, Uehara R, et al. A nationwide survey on the epidemiology and clinical features of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) in Japan. Mod Rheumatol 2014;24(4):640–644. [DOI] [PubMed] [Google Scholar]
- 8.Chung MP, Yi CA, Lee HY, Han J, Lee KS. Imaging of pulmonary vasculitis. Radiology 2010;255(2):322–341. [DOI] [PubMed] [Google Scholar]
- 9.Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33(8):1094–1100. [DOI] [PubMed] [Google Scholar]
- 10.Price M, Gilman MD, Carter BW, Sabloff BS, Truong MT, Wu CC. Imaging of Eosinophilic Lung Diseases. Radiol Clin North Am 2016;54(6):1151–1164. [DOI] [PubMed] [Google Scholar]
- 11.Kim YK, Lee KS, Chung MP, et al. Pulmonary involvement in Churg-Strauss syndrome: an analysis of CT, clinical, and pathologic findings. Eur Radiol 2007;17(12):3157–3165. [DOI] [PubMed] [Google Scholar]
- 12.Groh M, Pagnoux C, Baldini C, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 2015;26(7):545–553. [DOI] [PubMed] [Google Scholar]
- 13.Jeong YJ, Kim KI, Seo IJ, et al. Eosinophilic lung diseases: a clinical, radiologic, and pathologic overview. RadioGraphics 2007;27(3):617–637; discussion 637–639. [DOI] [PubMed] [Google Scholar]
- 14.d’Ersu E, Ribi C, Monney P, et al. Churg-Strauss syndrome with cardiac involvement: case illustration and contribution of CMR in the diagnosis and clinical follow-up. Int J Cardiol 2018;258:321–324. [DOI] [PubMed] [Google Scholar]
- 15.Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum 1990;33(8):1101–1107. [DOI] [PubMed] [Google Scholar]
- 16.Watts RA, Robson J. Introduction, epidemiology and classification of vasculitis. Best Pract Res Clin Rheumatol 2018;32(1):3–20. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Li C, Li J. Thoracic manifestation of Wegener’s granulomatosis: Computed tomography findings and analysis of misdiagnosis. Exp Ther Med 2018;16(1):413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warwick G, Leecy T, Silverstone E, Rainer S, Feller R, Yates DH. Pulmonary necrobiotic nodules: a rare extraintestinal manifestation of Crohn’s disease. Eur Respir Rev 2009;18(111):47–50. [DOI] [PubMed] [Google Scholar]
- 19.Merrill JT, Shen C, Schreibman D, et al. Adenosine A1 receptor promotion of multinucleated giant cell formation by human monocytes: a mechanism for methotrexate-induced nodulosis in rheumatoid arthritis. Arthritis Rheum 1997;40(7):1308–1315. [DOI] [PubMed] [Google Scholar]
- 20.Athanasou NA, Quinn J, Woods CG, Mcgee JO. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis 1988;47(5):398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis-associated lung disease. Eur Respir Rev 2015;24(135):1–16.25726549 [Google Scholar]
- 22.Combe B, Didry C, Gutierrez M, Anaya JM, Sany J. Accelerated nodulosis and systemic manifestations during methotrexate therapy for rheumatoid arthritis. Eur J Med 1993;2(3):153–156. [PubMed] [Google Scholar]
- 23.Lee JS, Tuder R, Lynch DA. Lymphomatoid granulomatosis: radiologic features and pathologic correlations. AJR Am J Roentgenol 2000;175(5):1335–1339. [DOI] [PubMed] [Google Scholar]
- 24.Wechsler RJ, Steiner RM, Israel HL, Patchefsky AS. Chest radiograph in lymphomatoid granulomatosis: comparison with Wegener granulomatosis. AJR Am J Roentgenol 1984;142(1):79–83. [DOI] [PubMed] [Google Scholar]
- 25.Hicken P, Dobie JC, Frew E. The radiology of lymphomatoid granulomatosis in the lung. Clin Radiol 1979;30(6):661–664. [DOI] [PubMed] [Google Scholar]
- 26.Song JY, Pittaluga S, Dunleavy K, et al. Lymphomatoid granulomatosis: a single institute experience—pathologic findings and clinical correlations. Am J Surg Pathol 2015;39(2):141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quaden C, Tillie-Leblond I, Delobbe A, et al. Necrotising sarcoid granulomatosis: clinical, functional, endoscopical and radiographical evaluations. Eur Respir J 2005;26(5):778–785. [DOI] [PubMed] [Google Scholar]
- 28.Lazzarini LCO, de Fatima do Amparo Teixeira M, Souza Rodrigues R, Marcos Nunes Valiante P. Necrotizing sarcoid granulomatosis in a family of patients with sarcoidosis reinforces the association between both entities. Respiration 2008;76(3):356–360. [DOI] [PubMed] [Google Scholar]
- 29.Chittock DR, Joseph MG, Paterson NA, McFadden RG. Necrotizing sarcoid granulomatosis with pleural involvement. Clinical and radiographic features. Chest 1994;106(3):672–676. [DOI] [PubMed] [Google Scholar]
- 30.Karpathiou G, Batistatou A, Boglou P, Stefanou D, Froudarakis ME. Necrotizing sarcoid granulomatosis: A distinctive form of pulmonary granulomatous disease. Clin Respir J 2018;12(4):1313–1319. [DOI] [PubMed] [Google Scholar]
- 31.Clee MD, Lamb D, Clark RA. Bronchocentric granulomatosis: a review and thoughts on pathogenesis. Br J Dis Chest 1983;77(3):227–234. [PubMed] [Google Scholar]
- 32.Katzenstein AL, Liebow AA, Friedman PJ. Bronchocentric granulomatosis, mucoid impaction, and hypersensitivity reactions to fungi. Am Rev Respir Dis 1975;111(4): 497–537. [DOI] [PubMed] [Google Scholar]
- 33.Ward S, Heyneman LE, Flint JD, Leung AN, Kazerooni EA, Müller NL. Bronchocentric granulomatosis: computed tomographic findings in five patients. Clin Radiol 2000;55(4):296–300. [DOI] [PubMed] [Google Scholar]
- 34.Engleman P, Liebow AA, Gmelich J, Friedman PJ. Pulmonary hyalinizing granuloma. Am Rev Respir Dis 1977;115(6):997–1008. [DOI] [PubMed] [Google Scholar]
- 35.Schlosnagle DC, Check IJ, Sewell CW, Plummer A, York RM, Hunter RL. Immunologic abnormalities in two patients with pulmonary hyalinizing granuloma. Am J Clin Pathol 1982;78(2):231–235. [DOI] [PubMed] [Google Scholar]
- 36.Colen RR, Nagle JA, Wittram C. Radiologic-pathologic conference of the Massachusetts General Hospital. Pulmonary hyalinizing granuloma. AJR Am J Roentgenol 2007;188(1):W15–W16. [DOI] [PubMed] [Google Scholar]
- 37.Ren Y, Raitz EN, Lee KR, Pingleton SK, Tawfik O. Pulmonary small lymphocytic lymphoma (mucosa-associated lymphoid tissue type) associated with pulmonary hyalinizing granuloma. Chest 2001;120(3):1027–1030. [DOI] [PubMed] [Google Scholar]
- 38.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014;383(9923): 1155–1167. [DOI] [PubMed] [Google Scholar]
- 39.Esteves T, Aparicio G, Garcia-Patos V. Is there any association between Sarcoidosis and infectious agents?: a systematic review and meta-analysis. BMC Pulm Med 2016;16(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999;160(2):736–755. [DOI] [PubMed] [Google Scholar]
- 41.Ganeshan D, Menias CO, Lubner MG, Pickhardt PJ, Sandrasegaran K, Bhalla S. Sarcoidosis from Head to Toe: What the Radiologist Needs to Know. RadioGraphics 2018;38(4):1180–1200. [DOI] [PubMed] [Google Scholar]
- 42.Ward S, Heyneman LE, Reittner P, Kazerooni EA, Godwin JD, Müller NL. Talcosis associated with IV abuse of oral medications: CT findings. AJR Am J Roentgenol 2000;174(3):789–793. [DOI] [PubMed] [Google Scholar]
- 43.Akira M, Kozuka T, Yamamoto S, Sakatani M, Morinaga K. Inhalational talc pneumoconiosis: radiographic and CT findings in 14 patients. AJR Am J Roentgenol 2007;188(2):326–333. [DOI] [PubMed] [Google Scholar]
- 44.Marchiori E, Souza Júnior AS, Müller NL. Inhalational pulmonary talcosis: high-resolution CT findings in 3 patients. J Thorac Imaging 2004;19(1):41–44. [DOI] [PubMed] [Google Scholar]
- 45.Abbott GF, Rosado-de-Christenson ML, Franks TJ, Frazier AA, Galvin JR. From the archives of the AFIP: pulmonary Langerhans cell histiocytosis. RadioGraphics 2004;24(3):821–841. [DOI] [PubMed] [Google Scholar]
- 46.Brauner MW, Grenier P, Tijani K, Battesti JP, Valeyre D. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology 1997;204(2):497–502. [DOI] [PubMed] [Google Scholar]
- 47.Vassallo R, Harari S, Tazi A. Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax 2017;72(10):937–945. [DOI] [PubMed] [Google Scholar]
- 48.Blakley MP, Dutcher JP, Wiernik PH. Pulmonary Langerhans cell histiocytosis, acute myeloid leukemia, and myelofibrosis in a large family and review of the literature. Leuk Res 2018;67:39–44. [DOI] [PubMed] [Google Scholar]
- 49.Hurst JR, Verma N, Lowe D, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement on the Definition, Diagnosis, and Management of Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract 2017;5(4):938–945. [DOI] [PubMed] [Google Scholar]
- 50.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004;114(2):415–421. [DOI] [PubMed] [Google Scholar]
- 51.Odnoletkova I, Kindle G, Quinti I, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis 2018;13(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torigian DA, LaRosa DF, Levinson AI, Litzky LA, Miller WT, Jr. Granulomatous-lymphocytic interstitial lung disease associated with common variable immunodeficiency: CT findings. J Thorac Imaging 2008;23(3):162–169. [DOI] [PubMed] [Google Scholar]
- 53.Marom EM, McAdams HP, Erasmus JJ, Goodman PC. The many faces of pulmonary aspiration. AJR Am J Roentgenol 1999;172(1):121–128. [DOI] [PubMed] [Google Scholar]
- 54.Mukhopadhyay S, Gal AA. Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med 2010;134(5):667–690. [DOI] [PubMed] [Google Scholar]
- 55.Cardasis JJ, MacMahon H, Husain AN. The spectrum of lung disease due to chronic occult aspiration. Ann Am Thorac Soc 2014;11(6):865–873. [DOI] [PubMed] [Google Scholar]
- 56.Franquet T, Giménez A, Rosón N, Torrubia S, Sabaté JM, Pérez C. Aspiration diseases: findings, pitfalls, and differential diagnosis. RadioGraphics 2000;20(3):673–685. [DOI] [PubMed] [Google Scholar]
- 57.Komiya K, Ishii H, Umeki K, et al. Computed tomography findings of aspiration pneumonia in 53 patients. Geriatr Gerontol Int 2013;13(3):580–585. [DOI] [PubMed] [Google Scholar]
- 58.Hirschmann JV, Pipavath SNJ, Godwin JD. Hypersensitivity pneumonitis: a historical, clinical, and radiologic review. RadioGraphics 2009;29(7):1921–1938. [DOI] [PubMed] [Google Scholar]
- 59.Miller R, Allen TC, Barrios RJ, et al. Hypersensitivity Pneumonitis A Perspective From Members of the Pulmonary Pathology Society. Arch Pathol Lab Med 2018;142(1):120–126. [DOI] [PubMed] [Google Scholar]
- 60.Chong S, Lee KS, Chung MJ, Han J, Kwon OJ, Kim TS. Pneumoconiosis: comparison of imaging and pathologic findings. RadioGraphics 2006;26(1):59–77. [DOI] [PubMed] [Google Scholar]
- 61.Rosen Y. Pathology of sarcoidosis. Semin Respir Crit Care Med 2007;28(1):36–52. [DOI] [PubMed] [Google Scholar]
- 62.Remy-Jardin M, Degreef JM, Beuscart R, Voisin C, Remy J. Coal worker’s pneumoconiosis: CT assessment in exposed workers and correlation with radiographic findings. Radiology 1990;177(2):363–371. [DOI] [PubMed] [Google Scholar]
- 63.Cowie RL. The epidemiology of tuberculosis in gold miners with silicosis. Am J Respir Crit Care Med 1994;150(5 Pt 1):1460–1462. [DOI] [PubMed] [Google Scholar]
- 64.Bourm KS, Menias CO, Ali K, Alhalabi K, Elsayes KM. Spectrum of Xanthogranulomatous Processes in the Abdomen and Pelvis: A Pictorial Review of Infectious, Inflammatory, and Proliferative Responses. AJR Am J Roentgenol 2017;208(3):475–484. [DOI] [PubMed] [Google Scholar]
- 65.Benoist N, Mikail N, Deschamps L, et al. Erdheim-Chester disease as assessed by modern multimodality imaging. Int J Cardiol 2016;207:235–237. [DOI] [PubMed] [Google Scholar]
- 66.Ahuja J, Kanne JP, Meyer CA, et al. Histiocytic disorders of the chest: imaging findings. RadioGraphics 2015;35(2):357–370. [DOI] [PubMed] [Google Scholar]
- 67.Zaveri J, La Q, Yarmish G, Neuman J. More than just Langerhans cell histiocytosis: a radiologic review of histiocytic disorders. RadioGraphics 2014;34(7):2008–2024. [DOI] [PubMed] [Google Scholar]
- 68.Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al. Erdheim-Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996;75(3):157–169. [DOI] [PubMed] [Google Scholar]
- 69.Shamburek RD, Brewer HB, Jr, Gochuico BR. Erdheim-Chester disease: a rare multisystem histiocytic disorder associated with interstitial lung disease. Am J Med Sci 2001;321(1):66–75. [DOI] [PubMed] [Google Scholar]
- 70.Cartin-Ceba R, Golbin JM, Yi ES, Prakash UBS, Vassallo R. Intrathoracic manifestations of Rosai-Dorfman disease. Respir Med 2010;104(9):1344–1349. [DOI] [PubMed] [Google Scholar]
- 71.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7(1):19–73. [PubMed] [Google Scholar]
- 72.Ohori NP, Yu J, Landreneau RJ, Thaete FL, Kane K. Rosai-Dorfman disease of the pleura: a rare extranodal presentation. Hum Pathol 2003;34(11):1210–1211. [DOI] [PubMed] [Google Scholar]
- 73.Goupil de Bouillé J, de Muret A, Diot E, et al. Pulmonary manifestations revealing Rosai-Dorfman disease. Sarcoidosis Vasc Diffuse Lung Dis 2015;32(3):275–277. [PubMed] [Google Scholar]